User login
Painful facial abscess
A 35-year-old woman presented to our clinic with a purple-red cyst on her right cheek that had been present for about 4 years but had worsened over the prior 2 weeks (FIGURE 1). She said she was experiencing excruciating pain and that the cyst had purulent drainage. She denied any history of diabetes, dental problems, recent trauma, or an inciting event.
On physical examination, there was no cervical lymphadenopathy, and her vital signs were normal. An incision and drainage procedure was performed. About 2 mL of purulent fluid was extracted and sent for aerobic and anaerobic cultures.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Cervicofacial actinomycosis
Direct Gram stain showed gram-positive cocci, so the patient was started on a 7-day course of cephalexin 500 mg tid. Five days later, the anaerobic culture grew Actinomyces neuii, revealing the diagnosis as cervicofacial actinomycosis; the patient stopped taking cephalexin. The patient was then switched to a 3-month course of amoxicillin 875 mg bid.
Actinomyces are natural inhabitants of the human oropharynx and gastrointestinal and genitourinary tracts.1-4 They are filamentous, gram-positive rods with characteristic sulfur granules (although these are not always present).1-4 It is believed that actinomycosis is endogenously acquired from deep tissue either through dental trauma, penetrating wounds, or compound fractures.2,4
The most common presentations of actinomycosis include cervicofacial (sometimes referred to as “lumpy jaw syndrome”), followed by abdominopelvic and thoracic/pulmonary, manifestations.2-4 Primary cutaneous actinomycosis is rare.5-9 Actinomycosis infection often manifests with indolent constitutional symptoms such as fatigue and anorexia.1 Most cases occur in men ages 20 to 60 years, although cases in women are increasingly being reported.2-4
Risk factors include poor dental hygiene or dental procedures, alcoholism, intrauterine device use, immunosuppression, appendicitis, and diverticulitis.2-4 The exact cause of this patient’s actinomycosis was unknown, as she did not have any known risk factors.
Furunculosis and sporotrichosis are part of the differential
Actinomycosis is often called a “great mimicker” due to its ability to masquerade as infection, malignancy, or fungus.1 The differential diagnosis for this patient’s presentation included bacterial soft-tissue infection (eg, furunculosis), infected epidermoid cyst, cutaneous tuberculosis, sporotrichosis, deep fungal infection, and nocardiosis.
Continue to: Furunculosis was initially suspected
Furunculosis was initially suspected, but the original wound culture demonstrated actinomycoses instead of traditional gram-positive bacteria.
A clinical diagnosis
The diagnosis of actinomycosis is usually made clinically, but definitive confirmation requires culture, which can be challenging with a slow-growing facultative or strict anaerobe that may take up to 14 days to appear.2-4 A Gram stain can aid in the diagnosis, but overall, there is a high false-negative rate in identifying actinomycosis.1,3,4
Treatment time can be lengthy, but prognosis is favorable
Unfortunately, there are no randomized controlled studies for treatment of actinomycosis. The majority of evidence for treatment comes from in vitro and clinical case studies.2-4,10 In general, prognosis of actinomycosis is favorable with low mortality, but chronic infection without complete resolution of symptoms can occur.1-4,7,8,10
First-line therapy for actinomycosis is a beta-lactam antibiotic, typically penicillin G or amoxicillin.2-4,10 High doses of prolonged intravenous (IV) and oral antibiotic therapy (2 to 12 months) based on location and complexity are standard.3,11 However, if there is minimal bone involvement and the patient shows rapid improvement, treatment could be shortened to a 4 to 6–week oral regimen.1,11 Surgical intervention can also shorten the required length of antibiotic duration.1,10
Cutaneous actinomycosis Tx. Amoxicillin/clavulanic acid has been shown to be an effective treatment for cutaneous actinomycosis, especially if polymicrobial infection is suspected.5,6 Individualized regimens for cutaneous actinomycosis—based on severity, location, and treatment response—are acceptable with close monitoring.1,2,11
Continue to: A lengthy recovery for our patient
A lengthy recovery for our patient
Seven weeks after the initial visit, the patient reported that she had taken only 20 days’ worth of the recommended 3-month course of amoxicillin. Fortunately, the lesion appeared to be healing well with no apparent fluid collection (FIGURE 2).

The patient was then prescribed, and completed, a 3-month course of amoxicillin/clavulanic acid
Nineteen months after initial treatment, the lesion reappeared as a painless cyst in a similar location (FIGURE 3). Plastic Surgery incised and drained the lesion and Infectious Diseases continued her on 3 months of amoxicillin/clavulanic acid 875 mg/125 mg bid, which she did complete.

Due to the continued presence of the lesion, a computed tomography scan of the face was ordered 2 years after the initial visit and demonstrated a superficial skin lesion with no mandibular involvement (FIGURE 4). She was then treated with 3 more months of amoxicillin/clavulanic acid 875 mg/125 mg bid, with the possibility of deep debridement if not improved. However, debridement was unnecessary as the cyst did not recur.

We believe that the course of this patient’s treatment was protracted because she never took oral antibiotics for more than 3 months at a time, and thus, her infection never completely resolved. In retrospect, we would have treated her more aggressively from the outset.
1. Najmi AH, Najmi IH, Tawhari MMH, et al. Cutaneous actinomycosis and long-term management through using oral and topical antibiotics: a case report. Clin Pract. 2018;8:1102. doi: 10.4081/ cp.2018.1102
2. Sharma S, Hashmi MF, Valentino ID. Actinomycosis. StatPearls Publishing; 2021.
3. Valour F, Sénécha A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-97. doi: 10.2147/IDR.S39601
4. Wong VK, Turmezei TD, Weston VC. Actinomycosis. BMJ. 2011;343:d6099. doi: 10.1136/bmj.d6099
5. Akhtar M, Zade MP, Shahane PL, et al. Scalp actinomycosis presenting as soft tissue tumour: a case report with literature review. Int J Surg Case Rep. 2015;16:99-101. doi: 10.1016/ j.ijscr.2015.09.030
6. Bose M, Ghosh R, Mukherjee K, et al. Primary cutaneous actinomycosis:a case report. J Clin Diagn Res. 2014;8:YD03-5. doi: 10.7860/JCDR/2014/8286.4591
7. Cataño JC, Gómez Villegas SI. Images in clinical medicine. Cutaneous actinomycosis. N Engl J Med. 2016;374:1773. doi: 10.1056/ NEJMicm1511213
8. Mehta V, Balachandran C. Primary cutaneous actinomycosis on the chest wall. Dermatol Online J. 2008;14:13.
9. Piggott SA, Khodaee M. A bump in the groin: cutaneous actinomycosis. J Family Community Med. 2017;24:203. doi: 10.4103/jfcm.JFCM_79_17
10. Bonifaz A, Tirado-Sánchez A, Calderón L, et al. Treatment of cutaneous actinomycosis with amoxicillin/clavulanic acid. J Dermatolog Treat. 2017;28:59-64. doi: 10.1080/09546634.2016.1178373
11. Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;;7:183-197. doi: 10.2147/IDR.S39601
A 35-year-old woman presented to our clinic with a purple-red cyst on her right cheek that had been present for about 4 years but had worsened over the prior 2 weeks (FIGURE 1). She said she was experiencing excruciating pain and that the cyst had purulent drainage. She denied any history of diabetes, dental problems, recent trauma, or an inciting event.
On physical examination, there was no cervical lymphadenopathy, and her vital signs were normal. An incision and drainage procedure was performed. About 2 mL of purulent fluid was extracted and sent for aerobic and anaerobic cultures.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Cervicofacial actinomycosis
Direct Gram stain showed gram-positive cocci, so the patient was started on a 7-day course of cephalexin 500 mg tid. Five days later, the anaerobic culture grew Actinomyces neuii, revealing the diagnosis as cervicofacial actinomycosis; the patient stopped taking cephalexin. The patient was then switched to a 3-month course of amoxicillin 875 mg bid.
Actinomyces are natural inhabitants of the human oropharynx and gastrointestinal and genitourinary tracts.1-4 They are filamentous, gram-positive rods with characteristic sulfur granules (although these are not always present).1-4 It is believed that actinomycosis is endogenously acquired from deep tissue either through dental trauma, penetrating wounds, or compound fractures.2,4
The most common presentations of actinomycosis include cervicofacial (sometimes referred to as “lumpy jaw syndrome”), followed by abdominopelvic and thoracic/pulmonary, manifestations.2-4 Primary cutaneous actinomycosis is rare.5-9 Actinomycosis infection often manifests with indolent constitutional symptoms such as fatigue and anorexia.1 Most cases occur in men ages 20 to 60 years, although cases in women are increasingly being reported.2-4
Risk factors include poor dental hygiene or dental procedures, alcoholism, intrauterine device use, immunosuppression, appendicitis, and diverticulitis.2-4 The exact cause of this patient’s actinomycosis was unknown, as she did not have any known risk factors.
Furunculosis and sporotrichosis are part of the differential
Actinomycosis is often called a “great mimicker” due to its ability to masquerade as infection, malignancy, or fungus.1 The differential diagnosis for this patient’s presentation included bacterial soft-tissue infection (eg, furunculosis), infected epidermoid cyst, cutaneous tuberculosis, sporotrichosis, deep fungal infection, and nocardiosis.
Continue to: Furunculosis was initially suspected
Furunculosis was initially suspected, but the original wound culture demonstrated actinomycoses instead of traditional gram-positive bacteria.
A clinical diagnosis
The diagnosis of actinomycosis is usually made clinically, but definitive confirmation requires culture, which can be challenging with a slow-growing facultative or strict anaerobe that may take up to 14 days to appear.2-4 A Gram stain can aid in the diagnosis, but overall, there is a high false-negative rate in identifying actinomycosis.1,3,4
Treatment time can be lengthy, but prognosis is favorable
Unfortunately, there are no randomized controlled studies for treatment of actinomycosis. The majority of evidence for treatment comes from in vitro and clinical case studies.2-4,10 In general, prognosis of actinomycosis is favorable with low mortality, but chronic infection without complete resolution of symptoms can occur.1-4,7,8,10
First-line therapy for actinomycosis is a beta-lactam antibiotic, typically penicillin G or amoxicillin.2-4,10 High doses of prolonged intravenous (IV) and oral antibiotic therapy (2 to 12 months) based on location and complexity are standard.3,11 However, if there is minimal bone involvement and the patient shows rapid improvement, treatment could be shortened to a 4 to 6–week oral regimen.1,11 Surgical intervention can also shorten the required length of antibiotic duration.1,10
Cutaneous actinomycosis Tx. Amoxicillin/clavulanic acid has been shown to be an effective treatment for cutaneous actinomycosis, especially if polymicrobial infection is suspected.5,6 Individualized regimens for cutaneous actinomycosis—based on severity, location, and treatment response—are acceptable with close monitoring.1,2,11
Continue to: A lengthy recovery for our patient
A lengthy recovery for our patient
Seven weeks after the initial visit, the patient reported that she had taken only 20 days’ worth of the recommended 3-month course of amoxicillin. Fortunately, the lesion appeared to be healing well with no apparent fluid collection (FIGURE 2).
The patient was then prescribed, and completed, a 3-month course of amoxicillin/clavulanic acid
Nineteen months after initial treatment, the lesion reappeared as a painless cyst in a similar location (FIGURE 3). Plastic Surgery incised and drained the lesion and Infectious Diseases continued her on 3 months of amoxicillin/clavulanic acid 875 mg/125 mg bid, which she did complete.
Due to the continued presence of the lesion, a computed tomography scan of the face was ordered 2 years after the initial visit and demonstrated a superficial skin lesion with no mandibular involvement (FIGURE 4). She was then treated with 3 more months of amoxicillin/clavulanic acid 875 mg/125 mg bid, with the possibility of deep debridement if not improved. However, debridement was unnecessary as the cyst did not recur.
We believe that the course of this patient’s treatment was protracted because she never took oral antibiotics for more than 3 months at a time, and thus, her infection never completely resolved. In retrospect, we would have treated her more aggressively from the outset.
A 35-year-old woman presented to our clinic with a purple-red cyst on her right cheek that had been present for about 4 years but had worsened over the prior 2 weeks (FIGURE 1). She said she was experiencing excruciating pain and that the cyst had purulent drainage. She denied any history of diabetes, dental problems, recent trauma, or an inciting event.
On physical examination, there was no cervical lymphadenopathy, and her vital signs were normal. An incision and drainage procedure was performed. About 2 mL of purulent fluid was extracted and sent for aerobic and anaerobic cultures.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Cervicofacial actinomycosis
Direct Gram stain showed gram-positive cocci, so the patient was started on a 7-day course of cephalexin 500 mg tid. Five days later, the anaerobic culture grew Actinomyces neuii, revealing the diagnosis as cervicofacial actinomycosis; the patient stopped taking cephalexin. The patient was then switched to a 3-month course of amoxicillin 875 mg bid.
Actinomyces are natural inhabitants of the human oropharynx and gastrointestinal and genitourinary tracts.1-4 They are filamentous, gram-positive rods with characteristic sulfur granules (although these are not always present).1-4 It is believed that actinomycosis is endogenously acquired from deep tissue either through dental trauma, penetrating wounds, or compound fractures.2,4
The most common presentations of actinomycosis include cervicofacial (sometimes referred to as “lumpy jaw syndrome”), followed by abdominopelvic and thoracic/pulmonary, manifestations.2-4 Primary cutaneous actinomycosis is rare.5-9 Actinomycosis infection often manifests with indolent constitutional symptoms such as fatigue and anorexia.1 Most cases occur in men ages 20 to 60 years, although cases in women are increasingly being reported.2-4
Risk factors include poor dental hygiene or dental procedures, alcoholism, intrauterine device use, immunosuppression, appendicitis, and diverticulitis.2-4 The exact cause of this patient’s actinomycosis was unknown, as she did not have any known risk factors.
Furunculosis and sporotrichosis are part of the differential
Actinomycosis is often called a “great mimicker” due to its ability to masquerade as infection, malignancy, or fungus.1 The differential diagnosis for this patient’s presentation included bacterial soft-tissue infection (eg, furunculosis), infected epidermoid cyst, cutaneous tuberculosis, sporotrichosis, deep fungal infection, and nocardiosis.
Continue to: Furunculosis was initially suspected
Furunculosis was initially suspected, but the original wound culture demonstrated actinomycoses instead of traditional gram-positive bacteria.
A clinical diagnosis
The diagnosis of actinomycosis is usually made clinically, but definitive confirmation requires culture, which can be challenging with a slow-growing facultative or strict anaerobe that may take up to 14 days to appear.2-4 A Gram stain can aid in the diagnosis, but overall, there is a high false-negative rate in identifying actinomycosis.1,3,4
Treatment time can be lengthy, but prognosis is favorable
Unfortunately, there are no randomized controlled studies for treatment of actinomycosis. The majority of evidence for treatment comes from in vitro and clinical case studies.2-4,10 In general, prognosis of actinomycosis is favorable with low mortality, but chronic infection without complete resolution of symptoms can occur.1-4,7,8,10
First-line therapy for actinomycosis is a beta-lactam antibiotic, typically penicillin G or amoxicillin.2-4,10 High doses of prolonged intravenous (IV) and oral antibiotic therapy (2 to 12 months) based on location and complexity are standard.3,11 However, if there is minimal bone involvement and the patient shows rapid improvement, treatment could be shortened to a 4 to 6–week oral regimen.1,11 Surgical intervention can also shorten the required length of antibiotic duration.1,10
Cutaneous actinomycosis Tx. Amoxicillin/clavulanic acid has been shown to be an effective treatment for cutaneous actinomycosis, especially if polymicrobial infection is suspected.5,6 Individualized regimens for cutaneous actinomycosis—based on severity, location, and treatment response—are acceptable with close monitoring.1,2,11
Continue to: A lengthy recovery for our patient
A lengthy recovery for our patient
Seven weeks after the initial visit, the patient reported that she had taken only 20 days’ worth of the recommended 3-month course of amoxicillin. Fortunately, the lesion appeared to be healing well with no apparent fluid collection (FIGURE 2).
The patient was then prescribed, and completed, a 3-month course of amoxicillin/clavulanic acid
Nineteen months after initial treatment, the lesion reappeared as a painless cyst in a similar location (FIGURE 3). Plastic Surgery incised and drained the lesion and Infectious Diseases continued her on 3 months of amoxicillin/clavulanic acid 875 mg/125 mg bid, which she did complete.
Due to the continued presence of the lesion, a computed tomography scan of the face was ordered 2 years after the initial visit and demonstrated a superficial skin lesion with no mandibular involvement (FIGURE 4). She was then treated with 3 more months of amoxicillin/clavulanic acid 875 mg/125 mg bid, with the possibility of deep debridement if not improved. However, debridement was unnecessary as the cyst did not recur.
We believe that the course of this patient’s treatment was protracted because she never took oral antibiotics for more than 3 months at a time, and thus, her infection never completely resolved. In retrospect, we would have treated her more aggressively from the outset.
1. Najmi AH, Najmi IH, Tawhari MMH, et al. Cutaneous actinomycosis and long-term management through using oral and topical antibiotics: a case report. Clin Pract. 2018;8:1102. doi: 10.4081/ cp.2018.1102
2. Sharma S, Hashmi MF, Valentino ID. Actinomycosis. StatPearls Publishing; 2021.
3. Valour F, Sénécha A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-97. doi: 10.2147/IDR.S39601
4. Wong VK, Turmezei TD, Weston VC. Actinomycosis. BMJ. 2011;343:d6099. doi: 10.1136/bmj.d6099
5. Akhtar M, Zade MP, Shahane PL, et al. Scalp actinomycosis presenting as soft tissue tumour: a case report with literature review. Int J Surg Case Rep. 2015;16:99-101. doi: 10.1016/ j.ijscr.2015.09.030
6. Bose M, Ghosh R, Mukherjee K, et al. Primary cutaneous actinomycosis:a case report. J Clin Diagn Res. 2014;8:YD03-5. doi: 10.7860/JCDR/2014/8286.4591
7. Cataño JC, Gómez Villegas SI. Images in clinical medicine. Cutaneous actinomycosis. N Engl J Med. 2016;374:1773. doi: 10.1056/ NEJMicm1511213
8. Mehta V, Balachandran C. Primary cutaneous actinomycosis on the chest wall. Dermatol Online J. 2008;14:13.
9. Piggott SA, Khodaee M. A bump in the groin: cutaneous actinomycosis. J Family Community Med. 2017;24:203. doi: 10.4103/jfcm.JFCM_79_17
10. Bonifaz A, Tirado-Sánchez A, Calderón L, et al. Treatment of cutaneous actinomycosis with amoxicillin/clavulanic acid. J Dermatolog Treat. 2017;28:59-64. doi: 10.1080/09546634.2016.1178373
11. Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;;7:183-197. doi: 10.2147/IDR.S39601
1. Najmi AH, Najmi IH, Tawhari MMH, et al. Cutaneous actinomycosis and long-term management through using oral and topical antibiotics: a case report. Clin Pract. 2018;8:1102. doi: 10.4081/ cp.2018.1102
2. Sharma S, Hashmi MF, Valentino ID. Actinomycosis. StatPearls Publishing; 2021.
3. Valour F, Sénécha A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-97. doi: 10.2147/IDR.S39601
4. Wong VK, Turmezei TD, Weston VC. Actinomycosis. BMJ. 2011;343:d6099. doi: 10.1136/bmj.d6099
5. Akhtar M, Zade MP, Shahane PL, et al. Scalp actinomycosis presenting as soft tissue tumour: a case report with literature review. Int J Surg Case Rep. 2015;16:99-101. doi: 10.1016/ j.ijscr.2015.09.030
6. Bose M, Ghosh R, Mukherjee K, et al. Primary cutaneous actinomycosis:a case report. J Clin Diagn Res. 2014;8:YD03-5. doi: 10.7860/JCDR/2014/8286.4591
7. Cataño JC, Gómez Villegas SI. Images in clinical medicine. Cutaneous actinomycosis. N Engl J Med. 2016;374:1773. doi: 10.1056/ NEJMicm1511213
8. Mehta V, Balachandran C. Primary cutaneous actinomycosis on the chest wall. Dermatol Online J. 2008;14:13.
9. Piggott SA, Khodaee M. A bump in the groin: cutaneous actinomycosis. J Family Community Med. 2017;24:203. doi: 10.4103/jfcm.JFCM_79_17
10. Bonifaz A, Tirado-Sánchez A, Calderón L, et al. Treatment of cutaneous actinomycosis with amoxicillin/clavulanic acid. J Dermatolog Treat. 2017;28:59-64. doi: 10.1080/09546634.2016.1178373
11. Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;;7:183-197. doi: 10.2147/IDR.S39601

Painful lumps in the axilla
A 30-year-old man presented to the clinic with a complaint of small painful lumps in his armpit. He stated that he initially experienced some itching and discomfort, but after a while he noticed some red, tender, swollen areas. He also mentioned an odorous yellow fluid that would sometimes drain from the lumps. Since first noticing them 2 years earlier, he reported that the nodules had disappeared and reappeared on their own several times.
On physical exam, several small red subcutaneous nodules were present in the axilla and tender to palpation (FIGURE 1A). The patient also had comedonal acne on his back (FIGURE 1B). The patient’s body mass index was 31, and he was a nonsmoker.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Hidradenitis suppurativa
The characteristic location and morphology of the lesions, along with the chronicity and odor, were critical in arriving at a diagnosis of hidradenitis suppurativa (HS).
HS is a chronic, inflammatory skin condition that normally manifests in areas of apocrine sweat glands, including the axilla, groin, and perianal, perineal, and inframammary locations.1 It begins when an abnormal hair follicle gets occluded and ruptures, spilling keratin and bacteria into the dermis. An inflammatory response can ensue with surrounding neutrophils and lymphocytes, which leads to abscess formation and destruction of the pilosebaceous unit. Sinus tracts form between the lesions, and a cycle of scarring, fistulas, and contractures can occur.
In this case, the comedones from acne conglobata on the patient’s back indicated a more global follicular occlusion disorder. The characteristic triad is hidradenitis suppurativa, acne conglobata, and dissecting cellulitis of the scalp—of which the patient had 2.
Other potential causes of the pathology include abnormal secretion of apocrine glands, abnormal antimicrobial peptides, deficient numbers of sebaceous glands, and abnormal invaginations of the epidermis.2 Increased levels of tumor necrosis factor alpha and other cytokines have been detected in HS lesions and are a potential target for therapy.
The prevalence of HS in the United States is approximately 0.1%.3 The condition typically begins between the ages of 18 and 39 years. The ratio of women to men affected by the condition is 3:1.2 There is no evident racial or ethnic predilection. There is an association with diabetes and Crohn disease.3 Obesity and smoking are risk factors.1
Continue to: The differential includes an array of common skin conditions
The differential includes an array of common skin conditions
The differential diagnosis in this case included carbuncles, cysts, acne, and abscesses.
A furuncle or carbuncle can result from an infection of hair follicles that can manifest as individual (furuncle) or clusters of (carbuncle) red, painful boils. They form on parts of the skin where hair grows, including the face, neck, armpits, shoulders, and buttocks. They respond well to treatment with antibiotics and incision and drainage. They can be recurrent but usually don’t cluster together in apocrine-rich areas, as seen with HS.
Epidermal inclusion cysts are keratin-filled inclusion cysts with epithelial-lined cyst walls. The cysts are subcutaneous and occasionally more superficial. They can occur almost anywhere but are most often found on the back, scalp, neck, face, and chest. They are usually solitary; however, when there are multiple cysts, they are not linked by sinus tracts as found in HS.
Inflammatory acne lesions tend to form on the face, neck, back, chest, and shoulders, while HS lesions appear most often in apocrine-rich intertriginous areas.
Skin abscesses are local deep infections of the skin caused by bacterial pathogens. The most common agent is Staphylococcus aureus (frequently methicillin resistant). Injection drug use and immunosuppression are risk factors. Although bacteria do not cause HS lesions, bacteria can exacerbate HS through colonization.
Continue to: No lab test needed to diagnosis hidradenitis suppurativa
No lab test needed to diagnose hidradenitis suppurativa
Diagnosis of HS is largely clinical and based on a patient’s history and physical exam findings.2 No specific laboratory test is needed.
Although the patient in this case did have comedonal acne on his back, the lesions that prompted his visit were in an apocrine-rich area, were recurrent, and broke open on their own to release foul-smelling contents—all typical characteristics of HS.
Treatment depends on the severity of the condition
There are 3 stages of HS: Hurley stage I involves abscess formation without tracts or scars. Hurley stage II involves recurrent abscesses with sinus tracts and scarring. Hurley stage III has diffuse involvement with multiple interconnected sinus tracts and abscesses across an entire area.2 Our patient fits into Hurley stage III.
Evidence-based treatment of mild disease (Hurley stage I) includes topical clindamycin 1% solution/gel bid or doxycycline 100 mg bid for widespread disease (Hurley stage II or resistant stage I).2 Chlorhexidine and benzoyl peroxide washes are also often recommended.3 If a patient does not respond to this treatment or the condition is moderate to severe, then clindamycin 300 po bid (with or without rifampin 600/d po) for 10 weeks should be considered.4,5 In a randomized placebo-controlled trial that compared the efficacy of oral clindamycin vs clindamycin plus rifampin in patients with HS, both therapeutic options were statistically equivalent.5 One small, randomized controlled study of patients with mild-to-moderate HS showed that tetracycline 500 mg bid for 3 months resulted in fewer abscesses and nodules but was not superior to topical clindamycin.3
If the patient doesn’t show improvement (Hurley stage III), then adalimumab is an option, as follows: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly, if needed.4 Adalimumab is currently the only FDA-approved treatment for HS. Infliximab by IV infusion can be effective in improving pain, disease severity, and quality of life in patients with moderate-to-severe HS.3 This patient was also a candidate for treatment with systemic retinoids (isotretinoin or acitretin), which could have helped both the HS and the acne conglobata.
Continue to: Intralesional steroid injectiosn with triamcinolone
Intralesional steroid injections with triamcinolone 10 mg/mL can reduce local pain and inflammation rapidly. Pain management is also critical, as HS is painful. First-line therapy includes nonsteroidal anti-inflammatory drugs, acetaminophen, atypical anticonvulsants, and serotonin and norepinephrine reuptake inhibitors.2 Opiate analgesics may be needed for breakthrough pain in patients with severe disease. Avoiding tight clothing, harsh products, and adhesive dressings, as well as using clear petroleum jelly, can prevent skin trauma and help with healing. Weight loss and smoking cessation are also associated with better outcomes.6,7
If medical management fails …
If there is no improvement with medical management, it may be time to consider local procedures such as unroofing/deroofing, punch debridement, skin-tissue-sparing excision with electrosurgical peeling, and laser excision. Incision and drainage may be necessary for acutely inflamed, painful abscesses but should not be routinely performed because lesions can recur.3
Referral to a plastic surgeon is necessary when patients are considering wide excisions of largely affected areas. Even when surgical excisions are performed, medical treatment is needed to prevent new lesions and recurrences.
Our patient was treated initially with oral doxycycline 100 mg bid and intralesional triamcinolone (10 mg/mL) in the most tender lesions. He was also provided with a prescription for ibuprofen 800 mg tid to be taken with meals. The family physician encouraged the patient to lose weight. The patient derived some benefit from treatment but continued to experience new painful lesions.
The physician prescribed oral clindamycin 300 mg bid at a follow-up visit to replace the oral doxycycline. When this failed, the patient was sent for labs to determine if he would be a candidate for adalimumab. When the screening labs were normal, a prescription for adalimumab was provided: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly.4
1. Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi: 10.2147/CCID.S111019
2. Ballard K, Shuman VL. Hidradenitis Suppurativa. StatPearls Publishing; 2021.
3. Wipperman J, Bragg DA, Litzner B. Hidradenitis suppurativa: rapid evidence review. Am Fam Physician. 2019;100:562-569.
4. Alikhan A, Lymch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561. doi:10.10126/j.jaad.2008.11.911
5. Caro RDC, Cannizzaro MV, Botti E, et al. Clindamycin versus clindamycin plus rifampicin in hidradenitis suppurativa treatment: clinical and ultrasound observations. J Am Acad Dermatol. 2019;80:1314-1321. doi: 10.1016/j.jaad.2018.11.035
6. Hendricks AJ, Hirt PA, Sekhon S, et al. Non-pharmacologic approaches for hidradenitis suppurativa - a systematic review. J Dermatolog Treat. 2021;32:11-18. doi: 10.1080/09546634.2019.1621981
7. Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi: 10.1016/j.jaad.2019.02.067
A 30-year-old man presented to the clinic with a complaint of small painful lumps in his armpit. He stated that he initially experienced some itching and discomfort, but after a while he noticed some red, tender, swollen areas. He also mentioned an odorous yellow fluid that would sometimes drain from the lumps. Since first noticing them 2 years earlier, he reported that the nodules had disappeared and reappeared on their own several times.
On physical exam, several small red subcutaneous nodules were present in the axilla and tender to palpation (FIGURE 1A). The patient also had comedonal acne on his back (FIGURE 1B). The patient’s body mass index was 31, and he was a nonsmoker.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Hidradenitis suppurativa
The characteristic location and morphology of the lesions, along with the chronicity and odor, were critical in arriving at a diagnosis of hidradenitis suppurativa (HS).
HS is a chronic, inflammatory skin condition that normally manifests in areas of apocrine sweat glands, including the axilla, groin, and perianal, perineal, and inframammary locations.1 It begins when an abnormal hair follicle gets occluded and ruptures, spilling keratin and bacteria into the dermis. An inflammatory response can ensue with surrounding neutrophils and lymphocytes, which leads to abscess formation and destruction of the pilosebaceous unit. Sinus tracts form between the lesions, and a cycle of scarring, fistulas, and contractures can occur.
In this case, the comedones from acne conglobata on the patient’s back indicated a more global follicular occlusion disorder. The characteristic triad is hidradenitis suppurativa, acne conglobata, and dissecting cellulitis of the scalp—of which the patient had 2.
Other potential causes of the pathology include abnormal secretion of apocrine glands, abnormal antimicrobial peptides, deficient numbers of sebaceous glands, and abnormal invaginations of the epidermis.2 Increased levels of tumor necrosis factor alpha and other cytokines have been detected in HS lesions and are a potential target for therapy.
The prevalence of HS in the United States is approximately 0.1%.3 The condition typically begins between the ages of 18 and 39 years. The ratio of women to men affected by the condition is 3:1.2 There is no evident racial or ethnic predilection. There is an association with diabetes and Crohn disease.3 Obesity and smoking are risk factors.1
Continue to: The differential includes an array of common skin conditions
The differential includes an array of common skin conditions
The differential diagnosis in this case included carbuncles, cysts, acne, and abscesses.
A furuncle or carbuncle can result from an infection of hair follicles that can manifest as individual (furuncle) or clusters of (carbuncle) red, painful boils. They form on parts of the skin where hair grows, including the face, neck, armpits, shoulders, and buttocks. They respond well to treatment with antibiotics and incision and drainage. They can be recurrent but usually don’t cluster together in apocrine-rich areas, as seen with HS.
Epidermal inclusion cysts are keratin-filled inclusion cysts with epithelial-lined cyst walls. The cysts are subcutaneous and occasionally more superficial. They can occur almost anywhere but are most often found on the back, scalp, neck, face, and chest. They are usually solitary; however, when there are multiple cysts, they are not linked by sinus tracts as found in HS.
Inflammatory acne lesions tend to form on the face, neck, back, chest, and shoulders, while HS lesions appear most often in apocrine-rich intertriginous areas.
Skin abscesses are local deep infections of the skin caused by bacterial pathogens. The most common agent is Staphylococcus aureus (frequently methicillin resistant). Injection drug use and immunosuppression are risk factors. Although bacteria do not cause HS lesions, bacteria can exacerbate HS through colonization.
Continue to: No lab test needed to diagnosis hidradenitis suppurativa
No lab test needed to diagnose hidradenitis suppurativa
Diagnosis of HS is largely clinical and based on a patient’s history and physical exam findings.2 No specific laboratory test is needed.
Although the patient in this case did have comedonal acne on his back, the lesions that prompted his visit were in an apocrine-rich area, were recurrent, and broke open on their own to release foul-smelling contents—all typical characteristics of HS.
Treatment depends on the severity of the condition
There are 3 stages of HS: Hurley stage I involves abscess formation without tracts or scars. Hurley stage II involves recurrent abscesses with sinus tracts and scarring. Hurley stage III has diffuse involvement with multiple interconnected sinus tracts and abscesses across an entire area.2 Our patient fits into Hurley stage III.
Evidence-based treatment of mild disease (Hurley stage I) includes topical clindamycin 1% solution/gel bid or doxycycline 100 mg bid for widespread disease (Hurley stage II or resistant stage I).2 Chlorhexidine and benzoyl peroxide washes are also often recommended.3 If a patient does not respond to this treatment or the condition is moderate to severe, then clindamycin 300 po bid (with or without rifampin 600/d po) for 10 weeks should be considered.4,5 In a randomized placebo-controlled trial that compared the efficacy of oral clindamycin vs clindamycin plus rifampin in patients with HS, both therapeutic options were statistically equivalent.5 One small, randomized controlled study of patients with mild-to-moderate HS showed that tetracycline 500 mg bid for 3 months resulted in fewer abscesses and nodules but was not superior to topical clindamycin.3
If the patient doesn’t show improvement (Hurley stage III), then adalimumab is an option, as follows: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly, if needed.4 Adalimumab is currently the only FDA-approved treatment for HS. Infliximab by IV infusion can be effective in improving pain, disease severity, and quality of life in patients with moderate-to-severe HS.3 This patient was also a candidate for treatment with systemic retinoids (isotretinoin or acitretin), which could have helped both the HS and the acne conglobata.
Continue to: Intralesional steroid injectiosn with triamcinolone
Intralesional steroid injections with triamcinolone 10 mg/mL can reduce local pain and inflammation rapidly. Pain management is also critical, as HS is painful. First-line therapy includes nonsteroidal anti-inflammatory drugs, acetaminophen, atypical anticonvulsants, and serotonin and norepinephrine reuptake inhibitors.2 Opiate analgesics may be needed for breakthrough pain in patients with severe disease. Avoiding tight clothing, harsh products, and adhesive dressings, as well as using clear petroleum jelly, can prevent skin trauma and help with healing. Weight loss and smoking cessation are also associated with better outcomes.6,7
If medical management fails …
If there is no improvement with medical management, it may be time to consider local procedures such as unroofing/deroofing, punch debridement, skin-tissue-sparing excision with electrosurgical peeling, and laser excision. Incision and drainage may be necessary for acutely inflamed, painful abscesses but should not be routinely performed because lesions can recur.3
Referral to a plastic surgeon is necessary when patients are considering wide excisions of largely affected areas. Even when surgical excisions are performed, medical treatment is needed to prevent new lesions and recurrences.
Our patient was treated initially with oral doxycycline 100 mg bid and intralesional triamcinolone (10 mg/mL) in the most tender lesions. He was also provided with a prescription for ibuprofen 800 mg tid to be taken with meals. The family physician encouraged the patient to lose weight. The patient derived some benefit from treatment but continued to experience new painful lesions.
The physician prescribed oral clindamycin 300 mg bid at a follow-up visit to replace the oral doxycycline. When this failed, the patient was sent for labs to determine if he would be a candidate for adalimumab. When the screening labs were normal, a prescription for adalimumab was provided: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly.4
A 30-year-old man presented to the clinic with a complaint of small painful lumps in his armpit. He stated that he initially experienced some itching and discomfort, but after a while he noticed some red, tender, swollen areas. He also mentioned an odorous yellow fluid that would sometimes drain from the lumps. Since first noticing them 2 years earlier, he reported that the nodules had disappeared and reappeared on their own several times.
On physical exam, several small red subcutaneous nodules were present in the axilla and tender to palpation (FIGURE 1A). The patient also had comedonal acne on his back (FIGURE 1B). The patient’s body mass index was 31, and he was a nonsmoker.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Hidradenitis suppurativa
The characteristic location and morphology of the lesions, along with the chronicity and odor, were critical in arriving at a diagnosis of hidradenitis suppurativa (HS).
HS is a chronic, inflammatory skin condition that normally manifests in areas of apocrine sweat glands, including the axilla, groin, and perianal, perineal, and inframammary locations.1 It begins when an abnormal hair follicle gets occluded and ruptures, spilling keratin and bacteria into the dermis. An inflammatory response can ensue with surrounding neutrophils and lymphocytes, which leads to abscess formation and destruction of the pilosebaceous unit. Sinus tracts form between the lesions, and a cycle of scarring, fistulas, and contractures can occur.
In this case, the comedones from acne conglobata on the patient’s back indicated a more global follicular occlusion disorder. The characteristic triad is hidradenitis suppurativa, acne conglobata, and dissecting cellulitis of the scalp—of which the patient had 2.
Other potential causes of the pathology include abnormal secretion of apocrine glands, abnormal antimicrobial peptides, deficient numbers of sebaceous glands, and abnormal invaginations of the epidermis.2 Increased levels of tumor necrosis factor alpha and other cytokines have been detected in HS lesions and are a potential target for therapy.
The prevalence of HS in the United States is approximately 0.1%.3 The condition typically begins between the ages of 18 and 39 years. The ratio of women to men affected by the condition is 3:1.2 There is no evident racial or ethnic predilection. There is an association with diabetes and Crohn disease.3 Obesity and smoking are risk factors.1
Continue to: The differential includes an array of common skin conditions
The differential includes an array of common skin conditions
The differential diagnosis in this case included carbuncles, cysts, acne, and abscesses.
A furuncle or carbuncle can result from an infection of hair follicles that can manifest as individual (furuncle) or clusters of (carbuncle) red, painful boils. They form on parts of the skin where hair grows, including the face, neck, armpits, shoulders, and buttocks. They respond well to treatment with antibiotics and incision and drainage. They can be recurrent but usually don’t cluster together in apocrine-rich areas, as seen with HS.
Epidermal inclusion cysts are keratin-filled inclusion cysts with epithelial-lined cyst walls. The cysts are subcutaneous and occasionally more superficial. They can occur almost anywhere but are most often found on the back, scalp, neck, face, and chest. They are usually solitary; however, when there are multiple cysts, they are not linked by sinus tracts as found in HS.
Inflammatory acne lesions tend to form on the face, neck, back, chest, and shoulders, while HS lesions appear most often in apocrine-rich intertriginous areas.
Skin abscesses are local deep infections of the skin caused by bacterial pathogens. The most common agent is Staphylococcus aureus (frequently methicillin resistant). Injection drug use and immunosuppression are risk factors. Although bacteria do not cause HS lesions, bacteria can exacerbate HS through colonization.
Continue to: No lab test needed to diagnosis hidradenitis suppurativa
No lab test needed to diagnose hidradenitis suppurativa
Diagnosis of HS is largely clinical and based on a patient’s history and physical exam findings.2 No specific laboratory test is needed.
Although the patient in this case did have comedonal acne on his back, the lesions that prompted his visit were in an apocrine-rich area, were recurrent, and broke open on their own to release foul-smelling contents—all typical characteristics of HS.
Treatment depends on the severity of the condition
There are 3 stages of HS: Hurley stage I involves abscess formation without tracts or scars. Hurley stage II involves recurrent abscesses with sinus tracts and scarring. Hurley stage III has diffuse involvement with multiple interconnected sinus tracts and abscesses across an entire area.2 Our patient fits into Hurley stage III.
Evidence-based treatment of mild disease (Hurley stage I) includes topical clindamycin 1% solution/gel bid or doxycycline 100 mg bid for widespread disease (Hurley stage II or resistant stage I).2 Chlorhexidine and benzoyl peroxide washes are also often recommended.3 If a patient does not respond to this treatment or the condition is moderate to severe, then clindamycin 300 po bid (with or without rifampin 600/d po) for 10 weeks should be considered.4,5 In a randomized placebo-controlled trial that compared the efficacy of oral clindamycin vs clindamycin plus rifampin in patients with HS, both therapeutic options were statistically equivalent.5 One small, randomized controlled study of patients with mild-to-moderate HS showed that tetracycline 500 mg bid for 3 months resulted in fewer abscesses and nodules but was not superior to topical clindamycin.3
If the patient doesn’t show improvement (Hurley stage III), then adalimumab is an option, as follows: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly, if needed.4 Adalimumab is currently the only FDA-approved treatment for HS. Infliximab by IV infusion can be effective in improving pain, disease severity, and quality of life in patients with moderate-to-severe HS.3 This patient was also a candidate for treatment with systemic retinoids (isotretinoin or acitretin), which could have helped both the HS and the acne conglobata.
Continue to: Intralesional steroid injectiosn with triamcinolone
Intralesional steroid injections with triamcinolone 10 mg/mL can reduce local pain and inflammation rapidly. Pain management is also critical, as HS is painful. First-line therapy includes nonsteroidal anti-inflammatory drugs, acetaminophen, atypical anticonvulsants, and serotonin and norepinephrine reuptake inhibitors.2 Opiate analgesics may be needed for breakthrough pain in patients with severe disease. Avoiding tight clothing, harsh products, and adhesive dressings, as well as using clear petroleum jelly, can prevent skin trauma and help with healing. Weight loss and smoking cessation are also associated with better outcomes.6,7
If medical management fails …
If there is no improvement with medical management, it may be time to consider local procedures such as unroofing/deroofing, punch debridement, skin-tissue-sparing excision with electrosurgical peeling, and laser excision. Incision and drainage may be necessary for acutely inflamed, painful abscesses but should not be routinely performed because lesions can recur.3
Referral to a plastic surgeon is necessary when patients are considering wide excisions of largely affected areas. Even when surgical excisions are performed, medical treatment is needed to prevent new lesions and recurrences.
Our patient was treated initially with oral doxycycline 100 mg bid and intralesional triamcinolone (10 mg/mL) in the most tender lesions. He was also provided with a prescription for ibuprofen 800 mg tid to be taken with meals. The family physician encouraged the patient to lose weight. The patient derived some benefit from treatment but continued to experience new painful lesions.
The physician prescribed oral clindamycin 300 mg bid at a follow-up visit to replace the oral doxycycline. When this failed, the patient was sent for labs to determine if he would be a candidate for adalimumab. When the screening labs were normal, a prescription for adalimumab was provided: 160 mg subcutaneously at Week 0, 80 mg at Week 2, and then 40 mg weekly.4
1. Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi: 10.2147/CCID.S111019
2. Ballard K, Shuman VL. Hidradenitis Suppurativa. StatPearls Publishing; 2021.
3. Wipperman J, Bragg DA, Litzner B. Hidradenitis suppurativa: rapid evidence review. Am Fam Physician. 2019;100:562-569.
4. Alikhan A, Lymch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561. doi:10.10126/j.jaad.2008.11.911
5. Caro RDC, Cannizzaro MV, Botti E, et al. Clindamycin versus clindamycin plus rifampicin in hidradenitis suppurativa treatment: clinical and ultrasound observations. J Am Acad Dermatol. 2019;80:1314-1321. doi: 10.1016/j.jaad.2018.11.035
6. Hendricks AJ, Hirt PA, Sekhon S, et al. Non-pharmacologic approaches for hidradenitis suppurativa - a systematic review. J Dermatolog Treat. 2021;32:11-18. doi: 10.1080/09546634.2019.1621981
7. Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi: 10.1016/j.jaad.2019.02.067
1. Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi: 10.2147/CCID.S111019
2. Ballard K, Shuman VL. Hidradenitis Suppurativa. StatPearls Publishing; 2021.
3. Wipperman J, Bragg DA, Litzner B. Hidradenitis suppurativa: rapid evidence review. Am Fam Physician. 2019;100:562-569.
4. Alikhan A, Lymch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561. doi:10.10126/j.jaad.2008.11.911
5. Caro RDC, Cannizzaro MV, Botti E, et al. Clindamycin versus clindamycin plus rifampicin in hidradenitis suppurativa treatment: clinical and ultrasound observations. J Am Acad Dermatol. 2019;80:1314-1321. doi: 10.1016/j.jaad.2018.11.035
6. Hendricks AJ, Hirt PA, Sekhon S, et al. Non-pharmacologic approaches for hidradenitis suppurativa - a systematic review. J Dermatolog Treat. 2021;32:11-18. doi: 10.1080/09546634.2019.1621981
7. Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi: 10.1016/j.jaad.2019.02.067

‘Multimorbidity’ more commonly seen in people with lupus
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
Facing Up to the Diagnosis
ANSWER
All of these items were rightly considered to be in the differential for this lesion, so the answer is choice “f.”
DISCUSSION
Individuals with type IV skin are less likely than those with types II and III to develop skin cancer. So, although cancer was definitely in the differential, the other items were considered just as, if not more, likely in this case.
The only way to sort through these diagnostic possibilities was to perform a biopsy. In this case, the entire lesion was removed by saucerization technique, under local anesthesia. The specimen provided would be adequate to detect any cancer, which a smaller specimen could easily miss.
The pathology results showed pigmented basal cell carcinoma. Given the patient’s extensive history of sun exposure, and the steady growth of the lesion, this was hardly a surprise. But prior to the biopsy, one could just as easily imagine the lesion to be, for example, a wart.
The take-home message is obvious: Nothing can take the place of biopsy in establishing a precise diagnosis. With that information in hand, the patient was referred for consultation with a Mohs surgeon. Surgical removal and closure would likely set him back several thousand dollars and leave a considerable scar.
ANSWER
All of these items were rightly considered to be in the differential for this lesion, so the answer is choice “f.”
DISCUSSION
Individuals with type IV skin are less likely than those with types II and III to develop skin cancer. So, although cancer was definitely in the differential, the other items were considered just as, if not more, likely in this case.
The only way to sort through these diagnostic possibilities was to perform a biopsy. In this case, the entire lesion was removed by saucerization technique, under local anesthesia. The specimen provided would be adequate to detect any cancer, which a smaller specimen could easily miss.
The pathology results showed pigmented basal cell carcinoma. Given the patient’s extensive history of sun exposure, and the steady growth of the lesion, this was hardly a surprise. But prior to the biopsy, one could just as easily imagine the lesion to be, for example, a wart.
The take-home message is obvious: Nothing can take the place of biopsy in establishing a precise diagnosis. With that information in hand, the patient was referred for consultation with a Mohs surgeon. Surgical removal and closure would likely set him back several thousand dollars and leave a considerable scar.
ANSWER
All of these items were rightly considered to be in the differential for this lesion, so the answer is choice “f.”
DISCUSSION
Individuals with type IV skin are less likely than those with types II and III to develop skin cancer. So, although cancer was definitely in the differential, the other items were considered just as, if not more, likely in this case.
The only way to sort through these diagnostic possibilities was to perform a biopsy. In this case, the entire lesion was removed by saucerization technique, under local anesthesia. The specimen provided would be adequate to detect any cancer, which a smaller specimen could easily miss.
The pathology results showed pigmented basal cell carcinoma. Given the patient’s extensive history of sun exposure, and the steady growth of the lesion, this was hardly a surprise. But prior to the biopsy, one could just as easily imagine the lesion to be, for example, a wart.
The take-home message is obvious: Nothing can take the place of biopsy in establishing a precise diagnosis. With that information in hand, the patient was referred for consultation with a Mohs surgeon. Surgical removal and closure would likely set him back several thousand dollars and leave a considerable scar.

“Like a berry,” was how a 38-year-old Hispanic man described the lesion that had been slowly growing on his face for 4 years. His family was alarmed by it, but he reasoned that since it didn’t hurt and the surrounding area wasn’t especially red, it likely wasn’t much of a problem.
The patient worked as roofer up to 7 days per week when the weather was good and had been doing so since he was old enough to work. He had no insurance and was not inclined to spend money on a health care visit. When his wife finally convinced him to go to the urgent care clinic, he paid $100 just to be told he needed to see a dermatologist. He was so disgusted he almost refused to wait the 6 weeks it took to get into the dermatology office.
The patient, who had type IV skin with little evidence of sun damage, had an obvious, large, coarsely mamillated nodule on the left upper nasolabial area. The lesion measured 1.8 cm and was reddish blue. It was moderately firm, but no increased warmth could be detected. No nodes could be felt in the area. His skin elsewhere was free of any notable changes.

Adalimumab biosimilar Cyltezo gets interchangeability designation
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
European agency supports marketing of abrocitinib for atopic dermatitis
The
The full indication is for the treatment of moderate to severe AD in adults who are candidates for systemic therapy, according to a summary of the opinion, made on Oct. 14. It will be available as 50-mg, 100-mg, and 200-mg tablets, will be marketed under the name Cibinqo, and “should be prescribed by physicians experienced in the treatment of atopic dermatitis,” the statement said.
“The benefits of Cibinqo are its ability to improve the skin condition as measured by improvements in the Investigator’s Global Assessment 0/1 and Eczema Area and Severity Index 75 response and to reduce itching in patients with atopic dermatitis,” according to the opinion. The most common side effects of abrocitinib are nausea, headache, acne, herpes simplex, increased blood creatine phosphokinase, vomiting, dizziness, and upper abdominal pain, the statement said, and infections are the most serious.
Abrocitinib was first approved for AD in the United Kingdom and in Japan in September, and is under review at the Food and Drug Administration for this indication. The first JAK inhibitor approved for AD in the United States is topical ruxolitinib (Opzelura), approved in September, for the short-term, noncontinuous chronic treatment of mild to moderate AD in nonimmunocompromised patients aged 12 years and older whose disease is not adequately controlled with topical prescription treatments, “or when those therapies are not advisable.”
The
The full indication is for the treatment of moderate to severe AD in adults who are candidates for systemic therapy, according to a summary of the opinion, made on Oct. 14. It will be available as 50-mg, 100-mg, and 200-mg tablets, will be marketed under the name Cibinqo, and “should be prescribed by physicians experienced in the treatment of atopic dermatitis,” the statement said.
“The benefits of Cibinqo are its ability to improve the skin condition as measured by improvements in the Investigator’s Global Assessment 0/1 and Eczema Area and Severity Index 75 response and to reduce itching in patients with atopic dermatitis,” according to the opinion. The most common side effects of abrocitinib are nausea, headache, acne, herpes simplex, increased blood creatine phosphokinase, vomiting, dizziness, and upper abdominal pain, the statement said, and infections are the most serious.
Abrocitinib was first approved for AD in the United Kingdom and in Japan in September, and is under review at the Food and Drug Administration for this indication. The first JAK inhibitor approved for AD in the United States is topical ruxolitinib (Opzelura), approved in September, for the short-term, noncontinuous chronic treatment of mild to moderate AD in nonimmunocompromised patients aged 12 years and older whose disease is not adequately controlled with topical prescription treatments, “or when those therapies are not advisable.”
The
The full indication is for the treatment of moderate to severe AD in adults who are candidates for systemic therapy, according to a summary of the opinion, made on Oct. 14. It will be available as 50-mg, 100-mg, and 200-mg tablets, will be marketed under the name Cibinqo, and “should be prescribed by physicians experienced in the treatment of atopic dermatitis,” the statement said.
“The benefits of Cibinqo are its ability to improve the skin condition as measured by improvements in the Investigator’s Global Assessment 0/1 and Eczema Area and Severity Index 75 response and to reduce itching in patients with atopic dermatitis,” according to the opinion. The most common side effects of abrocitinib are nausea, headache, acne, herpes simplex, increased blood creatine phosphokinase, vomiting, dizziness, and upper abdominal pain, the statement said, and infections are the most serious.
Abrocitinib was first approved for AD in the United Kingdom and in Japan in September, and is under review at the Food and Drug Administration for this indication. The first JAK inhibitor approved for AD in the United States is topical ruxolitinib (Opzelura), approved in September, for the short-term, noncontinuous chronic treatment of mild to moderate AD in nonimmunocompromised patients aged 12 years and older whose disease is not adequately controlled with topical prescription treatments, “or when those therapies are not advisable.”
Itchy vesicles
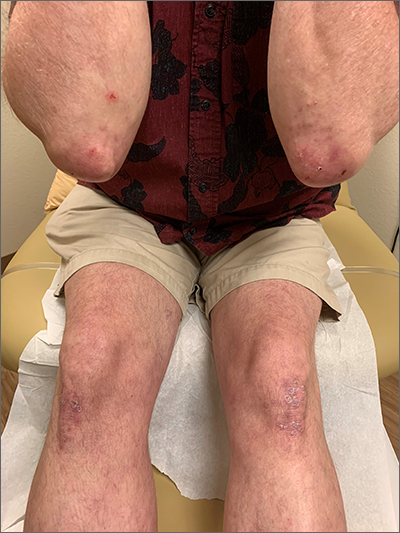
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The patient’s history of celiac disease and the presence of vesicular lesions affecting primarily extensor surfaces pointed to a diagnosis of dermatitis herpetiformis (DH).
DH is an autoimmune skin condition that is associated with gluten sensitivity. It is more frequent in individuals of northern European heritage.1
The lesions associated with DH are intensely pruritic grouped papules, vesicles, and tense blisters that appear more commonly on extensor areas of the lower limbs, elbows, buttocks, and sacral region. Involvement of the oral mucosa is rare and not all patients with DH experience the intestinal symptoms of gluten sensitivity.
Direct immunofluorescence of perilesional skin is the gold standard to confirm the diagnosis. Histology of the lesions is also performed, but findings may vary depending on the age of the lesion. Additionally, serology can help to confirm the diagnosis. Both tissue and epidermal transglutaminase antibodies are often present in the serum but may be negative if the patient is following a gluten-free diet. In this case, biopsy was not ordered because the patient already had a biopsy-confirmed diagnosis of celiac disease and a classic presentation of lesions.
Treatment of DH consists of a strict gluten-free diet and dapsone as first-line pharmacologic therapy. Typically, dapsone is started at doses of 25 to 50 mg daily and increased, as needed and tolerated, to 100 to 200 mg per day. Improvement is usually seen within 2 days of treatment initiation. Dapsone has multiple potential adverse effects; the most common is hemolysis. Since individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop severe hemolysis if treated with dapsone, it is necessary to screen for this condition prior to initiation of treatment. A complete blood count and liver and renal function testing are typically done before, and during, treatment with dapsone.1
This patient had normal levels of G6PD and his screening labs were also normal. He was started on dapsone orally 25 mg/d and follow-up was pending.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Marcella Colom, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
1. Mendes FBR, Hissa-Elian A, de Abreu MAMM, et al. Review: dermatitis herpetiformis. An Bras Dermatol. 2013;88:594-599. doi:10.1590/abd1806-4841.20131775
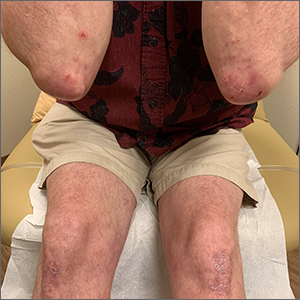
Fecal microbiota transplants may improve resistance to melanoma immunotherapy
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their

Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”

Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.

“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”

Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their
Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”
Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.
“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”
Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
In the fall of 2020, Hassane M. Zarour, MD, and colleagues began to pore over raw data from their
Preclinical mouse studies have demonstrated that the gut microbiota could influence the response of tumors to anti–PD-1 immunotherapy, but FMT had not been previously evaluated in human patients with malignant melanoma whose disease persisted or progressed after medical therapy. Only 30%-40% of melanoma patients respond to anti–PD-1 immunotherapy, so the researchers’ sense of anticipation was palpable. “It’s a high-risk, high-reward study, so you never know,” Dr. Zarour, a dermatologist and immunologist who is coleader of the melanoma program at the University of Pittsburgh Medical Center’s Hillman Cancer Center, said in an interview.
For the study, which was funded by the National Institutes of Health and published in Science, Dr. Zarour and a team of colleagues, including Diwakar Davar, MD, a medical oncologist/hematologist at UPMC and Giorgio Trinchieri, MD, head of the cancer immunology section at the National Cancer Institute, enrolled 16 patients with advanced melanoma whose disease had persisted or progressed with anti-PD-1 drugs; donors were 7 patients with advanced melanoma who had responded to pembrolizumab, 4 with a complete response and 3 with a partial response, with a median progression-free survival of 56 months.
After donors and patients underwent serial stool sampling and studies to stamp out the potential for transmitting infectious agents, the researchers administered the donor-derived FMT to patients via colonoscopy every 14 days for 3 weeks, followed by pembrolizumab. To their delight, 6 of the 15 evaluable recipients responded to treatment, with a reduction in tumor or long-term disease stabilization. Moreover, responders also showed increased abundance of taxa that were previously associated with response to immunotherapy, increased activation of CD8+ T cells, and decreased frequency of interleukin-8–expressing myeloid cells.
“This opens new doors for the future,” Dr. Zarour said. “It’s very encouraging, but I don’t want to overstate the data. It’s a small, nonrandomized trial, but one has to keep in mind that people were skeptical about this work; they didn’t think FMT would work. Now we see many people coming into the field to investigate the role of the microbiome as a therapeutic tool, which is great.”
Teri Greiling, MD, characterized the finding as a key development in understanding the microbiome’s potential to influence the course of melanoma and other diseases. “What’s emerging over the last decade of research is that our immune system has a close, back-and-forth relationship with our microbiota,” said Dr. Greiling, associate professor of dermatology at Oregon Health & Science University, Portland. “From day 1 of birth, we’re colonized by microbes that train our immune system how to function. In response, your immune system keeps those microbes in check and shapes which ones are allowed to colonize, and which ones are a target for attack. Thus, inflammatory responses are generated. Similarly, the goal of immunotherapy is to activate the immune system to fight cancer. This study shows that the immune system continues to need the colonizing microbes in our body to function optimally.”
Immunotherapy with checkpoint inhibitors was not an option for malignant melanoma patients until 2011, she noted, so the potential for FMT to further improve outcomes is welcome news for patients and their families. “We went from a less than 5% chance of survival with metastatic melanoma to now, with the right combination of checkpoint inhibitors, we’re up over 50%, which is amazing in a decade,” Dr. Greiling said. “Still, we’re losing half of our patients. If [FMT provides] a 30% improvement over that, that would be great, but it’s hard to extrapolate from such small numbers.”
Positive results in an Israeli study
Results from a similar, smaller phase 1 trial of 2 FMT donors and 10 recipients with metastatic melanoma who had progressed on anti-PD-1 therapy, from the Ella Lemelbaum Institute for Immuno-Oncology at Sheba Medical Center in Tel HaShomer, Israel, yielded similar results. The FMT protocol in this study included colonoscopy and oral stool capsules, followed by the reintroduction of anti–PD-1 therapy with nivolumab. The two FMT donors had previously been treated with anti–PD-1 monotherapy for metastatic melanoma and had achieved a clinical response for at least 1 year. Of the 10 FMT recipients, 1 had a complete response and 2 had a partial response.
“We expected changes in the immune system but did not expect that 3 out of the 10 patients in our study would be turned from nonresponders to responders,” the study’s lead author, Erez N. Baruch, MD, PhD, told this news organization. “Since this was a first-in-human study, we were aiming to assess safety and not clinical responses. [We found] that microbiota modulation can change the immune infiltration within melanoma tumors and by this affect response to immunotherapy.”
Dr. Baruch, an internal medicine resident in the physician-scientist track program at the University of Texas, Houston, said that the findings create a potential new therapeutic paradigm, or a new “playing ground” for drug development that can support existing immunotherapies. “It is important for dermatologists to understand that disruptions of the gut microbiota, mainly by antibiotics, may be harmful to melanoma patients,” he said. “Antibiotics in cancer patients should be used judiciously but of course should not be avoided when there’s an indication.”
As for next steps, Dr. Zarour and colleagues are recruiting more patients to boost their sample size and conducting sequential analysis of the microbiome of study participants “to better determine what the good and bad bugs are,” he said. “There are so many variables, including diet and geography. We need more data.” The hope is to develop a “microbiome signature” to identify patients likely to respond to FMT, and maybe one day, a probiotic capsule that patients take to optimize their response to immunotherapy.
“We don’t want to say that the microbiome is responsible for everything, but it’s responsible for some of the response and some of the resistance to treatment,” Dr. Zarour said. “So, we want to identify what candidate nonresponders are more likely to respond to FMT and be able to stick the right stool in the donor. This goes to better education of the microbiome signature. We are working hard on that.”
Dr. Baruch added that performing FMT for melanoma patients requires tight collaborations between oncologists, dermatologists, GI, and infectious disease experts. “These usually can be done in the setting of large cancer centers and will probably not be available in any hospital,” he said. “This is why understanding the mechanisms and developing an FMT-like drug is important. We are focusing on studying the mechanisms behind the clinical effect in order to develop a drug with an FMT-like effect without the safety and logistic issues related to FMTs.”
Tamia A. Harris-Tryon, MD, PhD, whose lab at the University of Texas Southwestern Medical Center at Dallas is studying how diet and the microbiota impact skin immunity, underscored the importance of evaluating the characteristics of the diet of patients as trials of FMT in melanoma patients carry on. “We know that the diet impacts the repertoire of microbes that colonize the gut,” said Dr. Harris-Tryon, assistant professor in the department of dermatology at the medical center. “The diet of the recipient likely has an impact” on the success of donor FMT.
She also noted that other skin conditions have been linked to a disrupted gut microbiome, such as psoriasis. “Given the safety of FMT in both of these studies, trials of FMT in psoriasis and other systemic skin conditions should be considered,” she said.
According to Dr. Zarour, mounting data from separate studies show that some gut microbiota play a role in adverse events experienced by melanoma patients on immunotherapy. “That is very important, especially with combination therapy,” he said. “There are also microbes involved in resistance to treatment, so the idea would be to identify these microbes.”
Studies raise more questions
In the opinion of Dr. Greiling, results from these two studies raise more questions than they answer. “The big question ... is why and how does FMT work, and how can we make the response better?” she said. “Is there one particular gene product from one microbe that is the key magic ingredient, and we can harness this as a drug? More likely it’s a complex interplay between multiple bacterial species needed to direct the immune response. Is there a group of microbes that is the same from person to person, or is it more complex?”
Then there are pending regulatory concerns. “We know that FMT works for [Clostridioides] difficile colitis but it’s not officially [Food and Drug Administration] approved,” Dr. Greiling said. “The FDA is really struggling with how to approve or regulate using bacteria as a drug. Where is that crossover? That inhibits things moving forward, for good reason. You want to balance safety with live microbes.”
The UPMC clinical trial was supported by Merck. Dr. Zarour disclosed that he is supported by grants from the National Cancer Institute and the James W. and Frances G. McGlothlin Chair in Melanoma Immunotherapy Research at UPMC. The Israeli study was funded by the Ella Lemelbaum Institute for Immuno-Oncology. Dr. Baruch was supported by the Allen Berg Fund for Excellence in Immuno-Oncology Research. Dr. Greiling and Dr. Harris-Tryon reported having no relevant financial disclosures.
FDA OKs iPLEDGE change for gender-neutral language
The Food and Drug Administration has approved a modification to the isotretinoin risk-mitigation program to make it more inclusive for transgender patients.
Beginning on Dec. 13, 2021, Previously, there were three risk categories: females of reproductive potential, females not of reproductive potential, and males.
In recent years, dermatologists and others have advocated for the change, hoping to make the process more inclusive and less intrusive for their transgender patients.
Isotretinoin (Accutane, Absorica, Amnesteem, Claravis, others) has a high risk of severe birth defects, and has been linked with other health issues, making it crucial for those with the ability to become pregnant to take contraceptive precautions while on the medication. Under the iPLEDGE program, physicians, patients, and pharmacies prescribing, using, or dispensing the drug must all be registered, with requirements that include the use of two forms of an effective contraceptive and regular pregnancy testing for patients who can become pregnant.
The FDA had given notification in June 2018 that the REMS modification and labeling change would be required, replacing the gender-specific language with gender-neutral language, according to an FDA spokesperson. The change was based on feedback that the gender-specific language can be a barrier to access for some patients. The FDA approved the modification on Oct. 8.
Expert reactions
“This is an exciting and welcome change from the FDA on iPLEDGE that many dermatologists, myself included, have advocated for quite a few years,” Howa Yeung, MD, MSc, assistant professor of dermatology at Emory University, Atlanta, said in an interview.
In a report on the dermatologic care for lesbian, gay, bisexual, and transgender persons published in the Journal of the American Academy of Dermatology, Dr. Yeung and his colleagues noted that more than 10 million lesbian, gay, bisexual and transgender people live in the United States and that improving their health is a public health priority.
“For cisgender patients, nothing has changed – patients will continue to receive appropriate educational material related to isotretinoin based on their pregnancy potential,” Dr. Yeung said. “For transgender and gender diverse patients, this is a huge step forward.”
Under the previous system, doctors were asked to register patients using gender binary categories, “which were confusing when they did not reflect reality” for these patients, Dr. Yeung said. The new system, Dr. Yeung added, “will make my job easier. I no longer have to struggle between respecting the patient’s gender identity and providing medically necessary care for patients with severe acne.”
“The new terminology is not just respectful, it also is simpler and makes more sense,” agreed Joshua D. Safer, MD, executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System and professor of medicine at the Icahn School of Medicine at Mount Sinai, New York. “As it stood, a transgender man with his uterus and ovaries in place might be missed in the pregnancy surveillance system because he could simply be labeled a man and not followed further. At the same time, both transgender women and cisgender women who were at no risk of pregnancy could be subject to more medical scrutiny that might have been consider intrusive.”
The change “validates the important point that pregnancy potential is not exclusively defined by sociocultural constructs of gender and allow dermatologists to focus purely on what matters when prescribing isotretinoin – whether an individual is able to become pregnant or not, regardless of their gender identity,” Klint Peebles, MD, a dermatologist at Kaiser Permanente in Washington, D.C., and suburban Maryland, who has also advocated for the change, said in an interview.
FDA elaborates
The modification includes important changes for doctors, pharmacists, and patients alike, according to the FDA.
Health care providers must assign and confirm their currently enrolled patient’s risk category when they first log in to the IPLEDGE REMS website on or after Dec. 13, the effective date. They should be sure any patient whose prescription RMA (iPLEDGE authorization) expires on Dec. 11-12 is told to obtain their prescription before midnight, Eastern time, Dec. 10.
Pharmacists will be affected, too, since the iPLEDGE REMS changed to a new platform vendor and the current “switch” pharmacy management system will be removed as a method to verify authorization to dispense isotretinoin. With these changes, as of Dec. 13, pharmacists can’t use the switch system to obtain a predispense authorization, or RMA (risk management authorization). They will need to obtain an RMA online by accessing the iPLEDGE REMS website or via telephone to the PLEDGE REMS center, 866-495-0654, before dispensing the prescription.
Patients, beginning Dec. 13, will have the option of presenting a unique QR code at the pharmacy on their smartphone rather than providing the iPLEDGE identification number. The code can be accessed by logging into their account on the iPLEDGE REMS website.
Patients with an isotretinoin prescription RMA that expires Dec. 11-12, must obtain the prescription before 11:59 p.m. Eastern time on Dec. 10. If the RMA expires before the prescription is picked up, the patient must begin the authorization process all over again.
Dr. Safer, Dr. Yeung, and Dr. Peebles have no relevant disclosures.
More information on the update and the isotretinoin REMS program is available on the FDA website.
The Food and Drug Administration has approved a modification to the isotretinoin risk-mitigation program to make it more inclusive for transgender patients.
Beginning on Dec. 13, 2021, Previously, there were three risk categories: females of reproductive potential, females not of reproductive potential, and males.
In recent years, dermatologists and others have advocated for the change, hoping to make the process more inclusive and less intrusive for their transgender patients.
Isotretinoin (Accutane, Absorica, Amnesteem, Claravis, others) has a high risk of severe birth defects, and has been linked with other health issues, making it crucial for those with the ability to become pregnant to take contraceptive precautions while on the medication. Under the iPLEDGE program, physicians, patients, and pharmacies prescribing, using, or dispensing the drug must all be registered, with requirements that include the use of two forms of an effective contraceptive and regular pregnancy testing for patients who can become pregnant.
The FDA had given notification in June 2018 that the REMS modification and labeling change would be required, replacing the gender-specific language with gender-neutral language, according to an FDA spokesperson. The change was based on feedback that the gender-specific language can be a barrier to access for some patients. The FDA approved the modification on Oct. 8.
Expert reactions
“This is an exciting and welcome change from the FDA on iPLEDGE that many dermatologists, myself included, have advocated for quite a few years,” Howa Yeung, MD, MSc, assistant professor of dermatology at Emory University, Atlanta, said in an interview.
In a report on the dermatologic care for lesbian, gay, bisexual, and transgender persons published in the Journal of the American Academy of Dermatology, Dr. Yeung and his colleagues noted that more than 10 million lesbian, gay, bisexual and transgender people live in the United States and that improving their health is a public health priority.
“For cisgender patients, nothing has changed – patients will continue to receive appropriate educational material related to isotretinoin based on their pregnancy potential,” Dr. Yeung said. “For transgender and gender diverse patients, this is a huge step forward.”
Under the previous system, doctors were asked to register patients using gender binary categories, “which were confusing when they did not reflect reality” for these patients, Dr. Yeung said. The new system, Dr. Yeung added, “will make my job easier. I no longer have to struggle between respecting the patient’s gender identity and providing medically necessary care for patients with severe acne.”
“The new terminology is not just respectful, it also is simpler and makes more sense,” agreed Joshua D. Safer, MD, executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System and professor of medicine at the Icahn School of Medicine at Mount Sinai, New York. “As it stood, a transgender man with his uterus and ovaries in place might be missed in the pregnancy surveillance system because he could simply be labeled a man and not followed further. At the same time, both transgender women and cisgender women who were at no risk of pregnancy could be subject to more medical scrutiny that might have been consider intrusive.”
The change “validates the important point that pregnancy potential is not exclusively defined by sociocultural constructs of gender and allow dermatologists to focus purely on what matters when prescribing isotretinoin – whether an individual is able to become pregnant or not, regardless of their gender identity,” Klint Peebles, MD, a dermatologist at Kaiser Permanente in Washington, D.C., and suburban Maryland, who has also advocated for the change, said in an interview.
FDA elaborates
The modification includes important changes for doctors, pharmacists, and patients alike, according to the FDA.
Health care providers must assign and confirm their currently enrolled patient’s risk category when they first log in to the IPLEDGE REMS website on or after Dec. 13, the effective date. They should be sure any patient whose prescription RMA (iPLEDGE authorization) expires on Dec. 11-12 is told to obtain their prescription before midnight, Eastern time, Dec. 10.
Pharmacists will be affected, too, since the iPLEDGE REMS changed to a new platform vendor and the current “switch” pharmacy management system will be removed as a method to verify authorization to dispense isotretinoin. With these changes, as of Dec. 13, pharmacists can’t use the switch system to obtain a predispense authorization, or RMA (risk management authorization). They will need to obtain an RMA online by accessing the iPLEDGE REMS website or via telephone to the PLEDGE REMS center, 866-495-0654, before dispensing the prescription.
Patients, beginning Dec. 13, will have the option of presenting a unique QR code at the pharmacy on their smartphone rather than providing the iPLEDGE identification number. The code can be accessed by logging into their account on the iPLEDGE REMS website.
Patients with an isotretinoin prescription RMA that expires Dec. 11-12, must obtain the prescription before 11:59 p.m. Eastern time on Dec. 10. If the RMA expires before the prescription is picked up, the patient must begin the authorization process all over again.
Dr. Safer, Dr. Yeung, and Dr. Peebles have no relevant disclosures.
More information on the update and the isotretinoin REMS program is available on the FDA website.
The Food and Drug Administration has approved a modification to the isotretinoin risk-mitigation program to make it more inclusive for transgender patients.
Beginning on Dec. 13, 2021, Previously, there were three risk categories: females of reproductive potential, females not of reproductive potential, and males.
In recent years, dermatologists and others have advocated for the change, hoping to make the process more inclusive and less intrusive for their transgender patients.
Isotretinoin (Accutane, Absorica, Amnesteem, Claravis, others) has a high risk of severe birth defects, and has been linked with other health issues, making it crucial for those with the ability to become pregnant to take contraceptive precautions while on the medication. Under the iPLEDGE program, physicians, patients, and pharmacies prescribing, using, or dispensing the drug must all be registered, with requirements that include the use of two forms of an effective contraceptive and regular pregnancy testing for patients who can become pregnant.
The FDA had given notification in June 2018 that the REMS modification and labeling change would be required, replacing the gender-specific language with gender-neutral language, according to an FDA spokesperson. The change was based on feedback that the gender-specific language can be a barrier to access for some patients. The FDA approved the modification on Oct. 8.
Expert reactions
“This is an exciting and welcome change from the FDA on iPLEDGE that many dermatologists, myself included, have advocated for quite a few years,” Howa Yeung, MD, MSc, assistant professor of dermatology at Emory University, Atlanta, said in an interview.
In a report on the dermatologic care for lesbian, gay, bisexual, and transgender persons published in the Journal of the American Academy of Dermatology, Dr. Yeung and his colleagues noted that more than 10 million lesbian, gay, bisexual and transgender people live in the United States and that improving their health is a public health priority.
“For cisgender patients, nothing has changed – patients will continue to receive appropriate educational material related to isotretinoin based on their pregnancy potential,” Dr. Yeung said. “For transgender and gender diverse patients, this is a huge step forward.”
Under the previous system, doctors were asked to register patients using gender binary categories, “which were confusing when they did not reflect reality” for these patients, Dr. Yeung said. The new system, Dr. Yeung added, “will make my job easier. I no longer have to struggle between respecting the patient’s gender identity and providing medically necessary care for patients with severe acne.”
“The new terminology is not just respectful, it also is simpler and makes more sense,” agreed Joshua D. Safer, MD, executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System and professor of medicine at the Icahn School of Medicine at Mount Sinai, New York. “As it stood, a transgender man with his uterus and ovaries in place might be missed in the pregnancy surveillance system because he could simply be labeled a man and not followed further. At the same time, both transgender women and cisgender women who were at no risk of pregnancy could be subject to more medical scrutiny that might have been consider intrusive.”
The change “validates the important point that pregnancy potential is not exclusively defined by sociocultural constructs of gender and allow dermatologists to focus purely on what matters when prescribing isotretinoin – whether an individual is able to become pregnant or not, regardless of their gender identity,” Klint Peebles, MD, a dermatologist at Kaiser Permanente in Washington, D.C., and suburban Maryland, who has also advocated for the change, said in an interview.
FDA elaborates
The modification includes important changes for doctors, pharmacists, and patients alike, according to the FDA.
Health care providers must assign and confirm their currently enrolled patient’s risk category when they first log in to the IPLEDGE REMS website on or after Dec. 13, the effective date. They should be sure any patient whose prescription RMA (iPLEDGE authorization) expires on Dec. 11-12 is told to obtain their prescription before midnight, Eastern time, Dec. 10.
Pharmacists will be affected, too, since the iPLEDGE REMS changed to a new platform vendor and the current “switch” pharmacy management system will be removed as a method to verify authorization to dispense isotretinoin. With these changes, as of Dec. 13, pharmacists can’t use the switch system to obtain a predispense authorization, or RMA (risk management authorization). They will need to obtain an RMA online by accessing the iPLEDGE REMS website or via telephone to the PLEDGE REMS center, 866-495-0654, before dispensing the prescription.
Patients, beginning Dec. 13, will have the option of presenting a unique QR code at the pharmacy on their smartphone rather than providing the iPLEDGE identification number. The code can be accessed by logging into their account on the iPLEDGE REMS website.
Patients with an isotretinoin prescription RMA that expires Dec. 11-12, must obtain the prescription before 11:59 p.m. Eastern time on Dec. 10. If the RMA expires before the prescription is picked up, the patient must begin the authorization process all over again.
Dr. Safer, Dr. Yeung, and Dr. Peebles have no relevant disclosures.
More information on the update and the isotretinoin REMS program is available on the FDA website.
9-step ladder may kids with allergies return to eggs
For many children in the process of outgrowing egg allergy, the step-wise reintroduction of foods that contain eggs can be achieved at home using a nine-rung laddered approach, according to updated guidelines from the British Society for Allergy and Clinical Immunology (BSACI).
Attempts to reintroduce egg into the child’s diet can start at the age of 12 months or 6 months from the last reaction, as long as past reactions have been mild to moderate and the child does not have asthma, according to guidelines from the BSACI, which represents allergists, pediatricians, and other health care practitioners.
According to the guidelines, the reintroduction needs to be guided by a specialist allergy service for children who have had severe reactions to egg or who have asthma.
Susan C. Leech, MB BChir, DCH, first author of the guidelines and a consultant in pediatric allergy with the Department of Child Health at Kings College Hospital, London, told this news organization that home reintroduction should begin slowly with small amounts of baked egg, starting with a pea-sized piece of cake, and should proceed gradually.
“Parents can be reassured that it’s a relatively safe thing to do as long as it’s done with caution,” said Dr. Leech.
The expanded guidelines include a new nine-step reintroduction ladder. It builds on a three-stage classification of egg-containing foods that was first introduced in BSACI guidelines in 2010.
On the bottom four rungs, children work their way through small but increasing amounts of fairy cakes (cupcakes), biscuits (cookies), and other foods containing baked eggs.
The next three rungs involve hard-boiled eggs, quiche, and other well-cooked egg products.
At the eighth rung, children can have small mouthfuls of runny scrambled eggs, mayonnaise, and other less-cooked or raw egg-containing products. At the top rung, children can have increasing amounts of those products as well as licks of cake batter.
The guidelines were published online September 29 in Clinical and Experimental Allergy along with a supplement that includes a series of examples showing how the guidelines apply to specific patient cases.
“These are examples only,” the guideline authors caution in the appendix. “Clinical judgment of severity is important as risk assessment is not always easy.”
Anna Nowak-Wegrzyn, MD, PhD, a professor of pediatrics at NYU Grossman School of Medicine and chief of pediatric allergy and immunology for Hassenfeld Children’s Hospital at NYU Langone, who was not involved in the BSACI guidelines, described the egg ladder as a “proactive” strategy that deserves further study and consideration.
“I think that this may be a valid approach,” said Dr. Nowak-Wegrzyn in an interview. “Eggs have good nutritional value, and they are present in a lot of foods, so avoidance creates logistical challenges.”
Using the egg ladder for home-based reintroduction may be especially suited in resource-poor areas where access to an allergist may be difficult, she said. It may also be suited for families that can’t visit the office because of pandemic-related restrictions.
“If the child had a severe reaction or if they have asthma, then it’s a no-go,” she added, “but if you have a patient who has a really mild reaction and you think that overall the risk of a significant reaction or bad symptoms is low, then it may be worth doing.”
Dr. Leech and Dr. Nowak-Wegrzyn have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For many children in the process of outgrowing egg allergy, the step-wise reintroduction of foods that contain eggs can be achieved at home using a nine-rung laddered approach, according to updated guidelines from the British Society for Allergy and Clinical Immunology (BSACI).
Attempts to reintroduce egg into the child’s diet can start at the age of 12 months or 6 months from the last reaction, as long as past reactions have been mild to moderate and the child does not have asthma, according to guidelines from the BSACI, which represents allergists, pediatricians, and other health care practitioners.
According to the guidelines, the reintroduction needs to be guided by a specialist allergy service for children who have had severe reactions to egg or who have asthma.
Susan C. Leech, MB BChir, DCH, first author of the guidelines and a consultant in pediatric allergy with the Department of Child Health at Kings College Hospital, London, told this news organization that home reintroduction should begin slowly with small amounts of baked egg, starting with a pea-sized piece of cake, and should proceed gradually.
“Parents can be reassured that it’s a relatively safe thing to do as long as it’s done with caution,” said Dr. Leech.
The expanded guidelines include a new nine-step reintroduction ladder. It builds on a three-stage classification of egg-containing foods that was first introduced in BSACI guidelines in 2010.
On the bottom four rungs, children work their way through small but increasing amounts of fairy cakes (cupcakes), biscuits (cookies), and other foods containing baked eggs.
The next three rungs involve hard-boiled eggs, quiche, and other well-cooked egg products.
At the eighth rung, children can have small mouthfuls of runny scrambled eggs, mayonnaise, and other less-cooked or raw egg-containing products. At the top rung, children can have increasing amounts of those products as well as licks of cake batter.
The guidelines were published online September 29 in Clinical and Experimental Allergy along with a supplement that includes a series of examples showing how the guidelines apply to specific patient cases.
“These are examples only,” the guideline authors caution in the appendix. “Clinical judgment of severity is important as risk assessment is not always easy.”
Anna Nowak-Wegrzyn, MD, PhD, a professor of pediatrics at NYU Grossman School of Medicine and chief of pediatric allergy and immunology for Hassenfeld Children’s Hospital at NYU Langone, who was not involved in the BSACI guidelines, described the egg ladder as a “proactive” strategy that deserves further study and consideration.
“I think that this may be a valid approach,” said Dr. Nowak-Wegrzyn in an interview. “Eggs have good nutritional value, and they are present in a lot of foods, so avoidance creates logistical challenges.”
Using the egg ladder for home-based reintroduction may be especially suited in resource-poor areas where access to an allergist may be difficult, she said. It may also be suited for families that can’t visit the office because of pandemic-related restrictions.
“If the child had a severe reaction or if they have asthma, then it’s a no-go,” she added, “but if you have a patient who has a really mild reaction and you think that overall the risk of a significant reaction or bad symptoms is low, then it may be worth doing.”
Dr. Leech and Dr. Nowak-Wegrzyn have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For many children in the process of outgrowing egg allergy, the step-wise reintroduction of foods that contain eggs can be achieved at home using a nine-rung laddered approach, according to updated guidelines from the British Society for Allergy and Clinical Immunology (BSACI).
Attempts to reintroduce egg into the child’s diet can start at the age of 12 months or 6 months from the last reaction, as long as past reactions have been mild to moderate and the child does not have asthma, according to guidelines from the BSACI, which represents allergists, pediatricians, and other health care practitioners.
According to the guidelines, the reintroduction needs to be guided by a specialist allergy service for children who have had severe reactions to egg or who have asthma.
Susan C. Leech, MB BChir, DCH, first author of the guidelines and a consultant in pediatric allergy with the Department of Child Health at Kings College Hospital, London, told this news organization that home reintroduction should begin slowly with small amounts of baked egg, starting with a pea-sized piece of cake, and should proceed gradually.
“Parents can be reassured that it’s a relatively safe thing to do as long as it’s done with caution,” said Dr. Leech.
The expanded guidelines include a new nine-step reintroduction ladder. It builds on a three-stage classification of egg-containing foods that was first introduced in BSACI guidelines in 2010.
On the bottom four rungs, children work their way through small but increasing amounts of fairy cakes (cupcakes), biscuits (cookies), and other foods containing baked eggs.
The next three rungs involve hard-boiled eggs, quiche, and other well-cooked egg products.
At the eighth rung, children can have small mouthfuls of runny scrambled eggs, mayonnaise, and other less-cooked or raw egg-containing products. At the top rung, children can have increasing amounts of those products as well as licks of cake batter.
The guidelines were published online September 29 in Clinical and Experimental Allergy along with a supplement that includes a series of examples showing how the guidelines apply to specific patient cases.
“These are examples only,” the guideline authors caution in the appendix. “Clinical judgment of severity is important as risk assessment is not always easy.”
Anna Nowak-Wegrzyn, MD, PhD, a professor of pediatrics at NYU Grossman School of Medicine and chief of pediatric allergy and immunology for Hassenfeld Children’s Hospital at NYU Langone, who was not involved in the BSACI guidelines, described the egg ladder as a “proactive” strategy that deserves further study and consideration.
“I think that this may be a valid approach,” said Dr. Nowak-Wegrzyn in an interview. “Eggs have good nutritional value, and they are present in a lot of foods, so avoidance creates logistical challenges.”
Using the egg ladder for home-based reintroduction may be especially suited in resource-poor areas where access to an allergist may be difficult, she said. It may also be suited for families that can’t visit the office because of pandemic-related restrictions.
“If the child had a severe reaction or if they have asthma, then it’s a no-go,” she added, “but if you have a patient who has a really mild reaction and you think that overall the risk of a significant reaction or bad symptoms is low, then it may be worth doing.”
Dr. Leech and Dr. Nowak-Wegrzyn have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.