User login
Unilateral Vesicular Eruption in a Neonate
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.

A 4-day-old female neonate presented to the dermatology clinic with a vesicular eruption on the left leg of 1 day's duration. The eruption was asymptomatic without any extracutaneous findings. This term infant was born without complication, and the mother denied any symptoms consistent with herpes simplex virus infection. Physical examination revealed yellow-red vesicles on an erythematous base in a blaschkoid distribution on the left leg. The rest of the examination was unremarkable. Herpes simplex virus polymerase chain reaction testing was negative.
Bleeding Hand Mass in an Older Man
The Diagnosis: Epithelioid Angiosarcoma
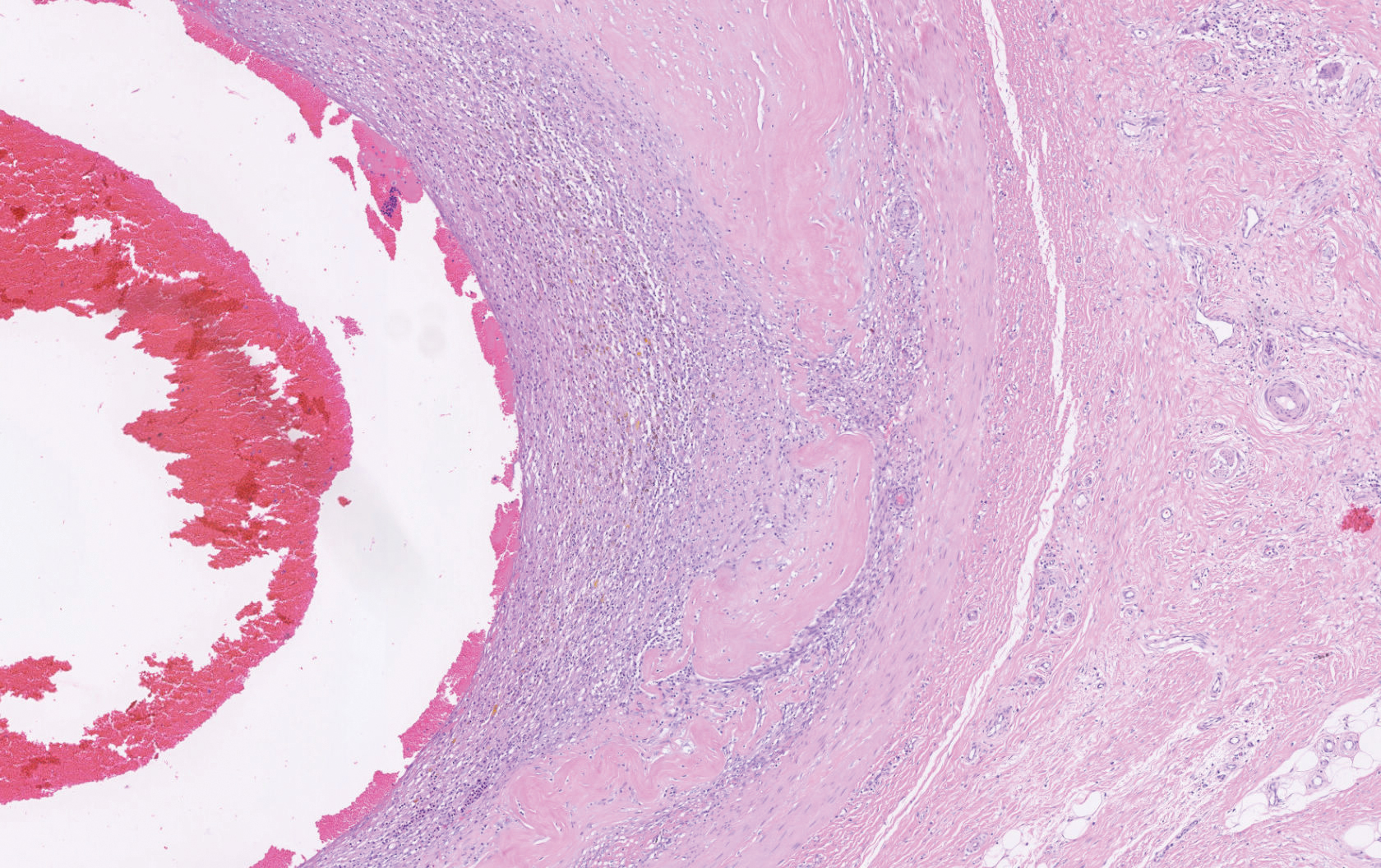
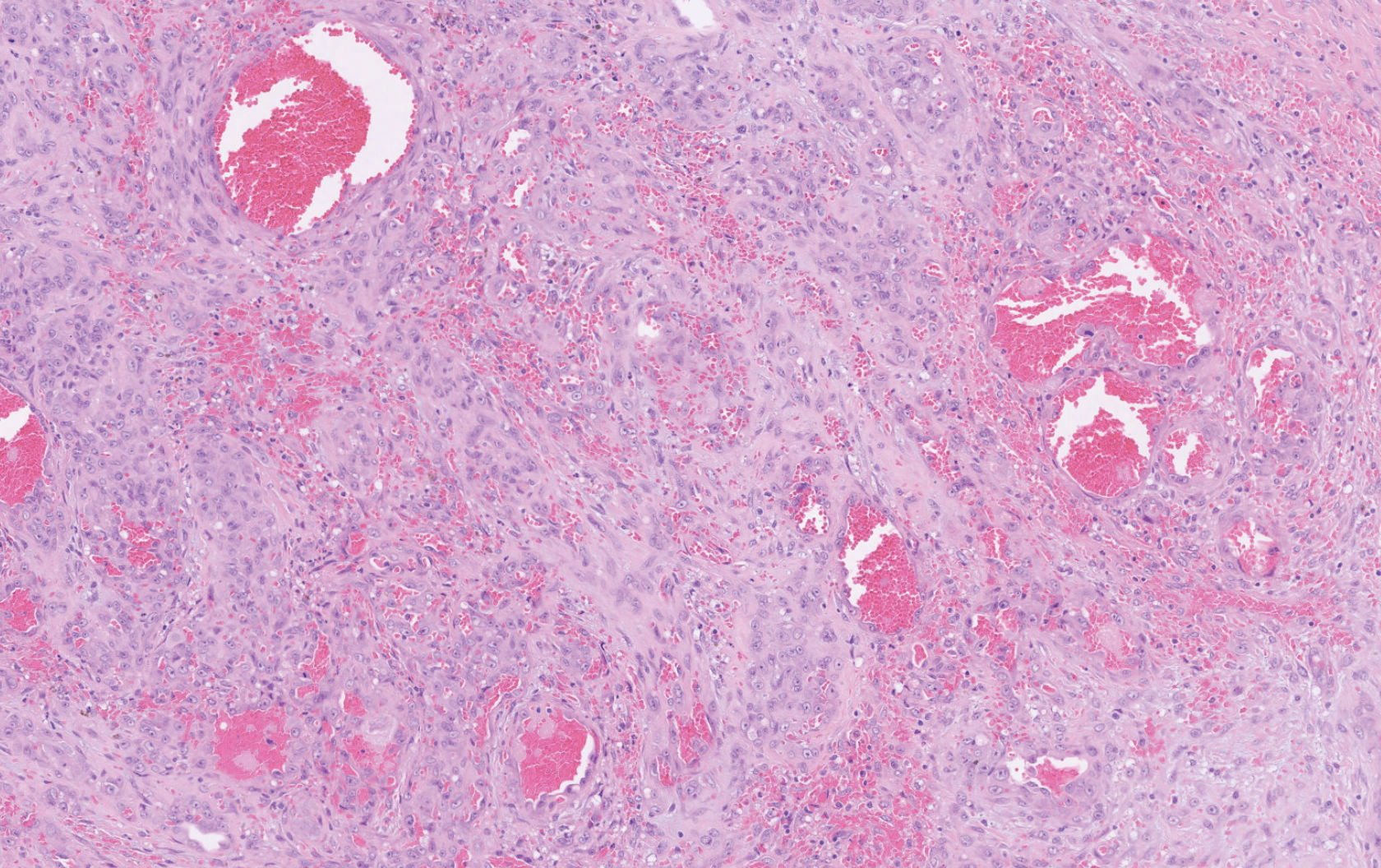
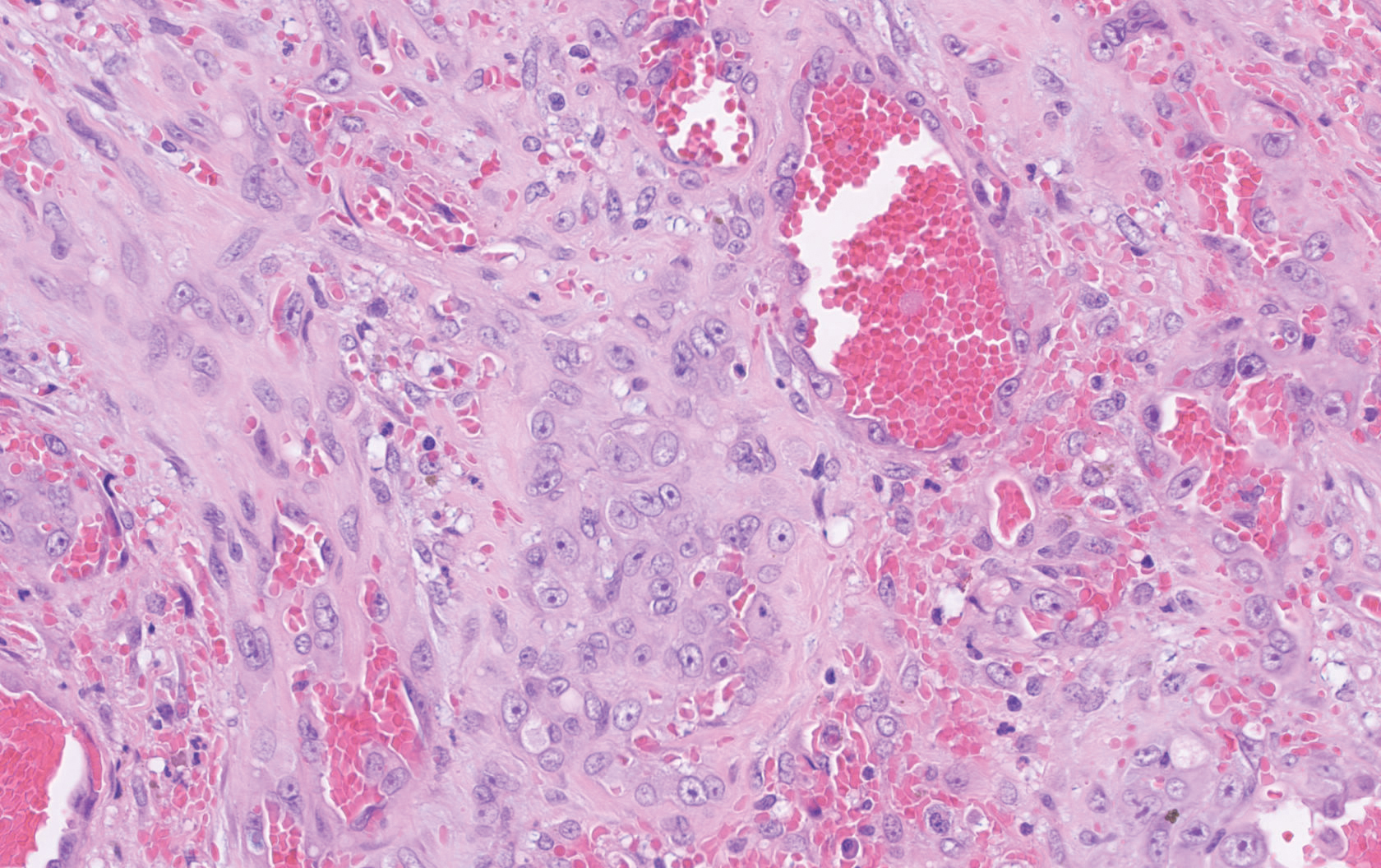
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.

Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
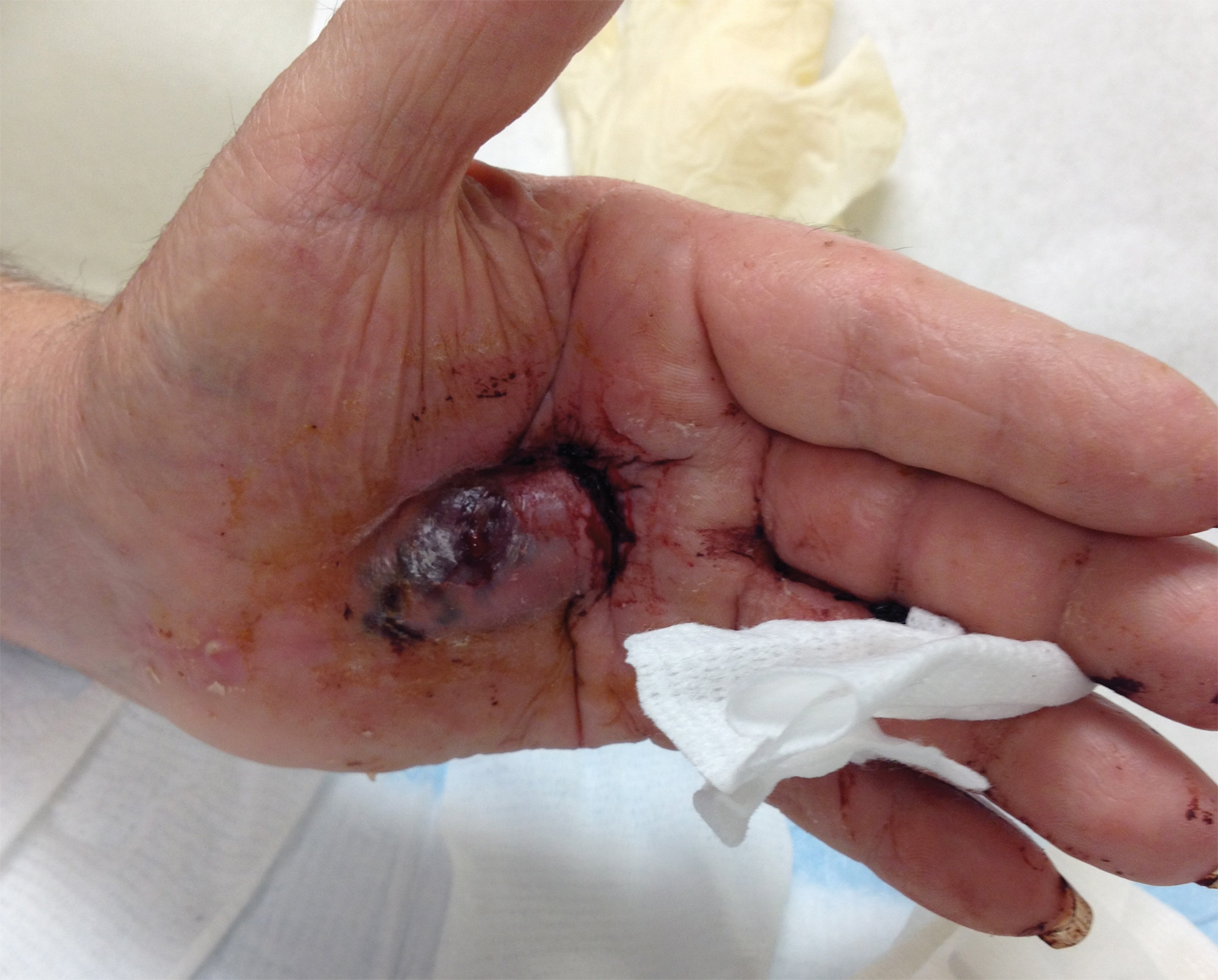
A 72-year-old man presented for evaluation of a mass on the left hand that continued to grow over the last few months and eventually bled. The patient first noticed a small firm lump on the palm approximately 1 year prior to presentation, and it was originally diagnosed as a Dupuytren contracture by his primary care physician. Months later, the lesion grew and began to bleed. Magnetic resonance imaging showed large hematomas of the hand with areas of nodular enhancement. The mass was located between the third and fourth proximal phalanges and abutted the extensor tendon. Complete excision yielded a definitive diagnosis.
Broadly Distributed Vascular Macules in a Pediatric Patient
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
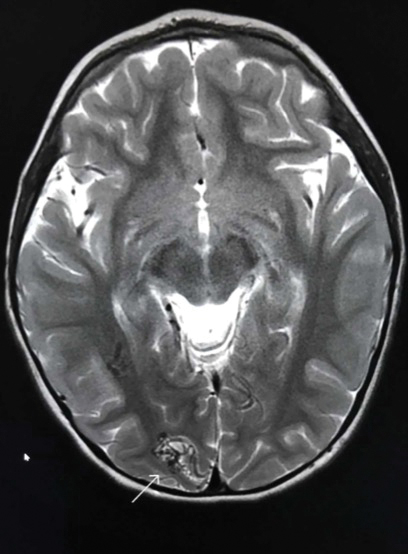
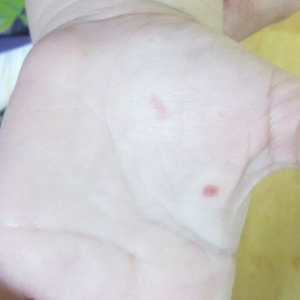
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
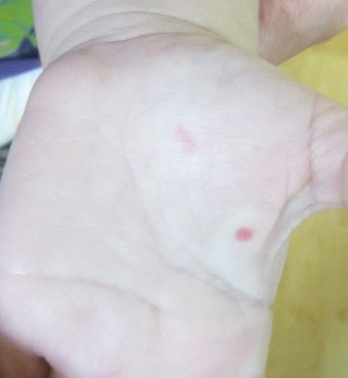
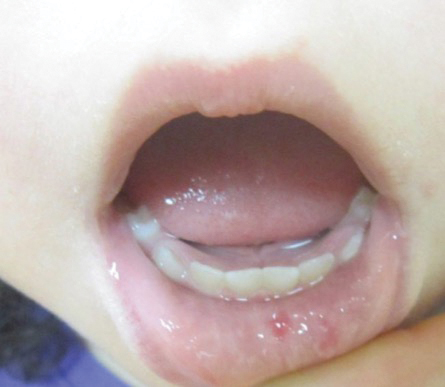
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
A 2-year-old girl presented with an erythematous macule on the left nasal sidewall that had been present since birth as well as other similar-appearing macules that had slowly evolved over the last 2 years. The patient was born via normal spontaneous vaginal delivery to healthy parents. She had 2 healthy siblings. Her parents reported that she was otherwise growing and developing normally. The patient had no history of epistaxis, and there was no family history of vascular anomalies. Physical examination revealed 2- to 6-mm vascular macules that blanched with pressure and filled quickly thereafter on the left nasal sidewall, upper (top) and lower extremities, and trunk. Some macules were surrounded by blanched halos. Several 1-mm red macules also were noted on the exterior and interior of the mucosal lower lip (bottom).
Smooth Papules on the Left Hand
The Diagnosis: Adult Colloid Milium
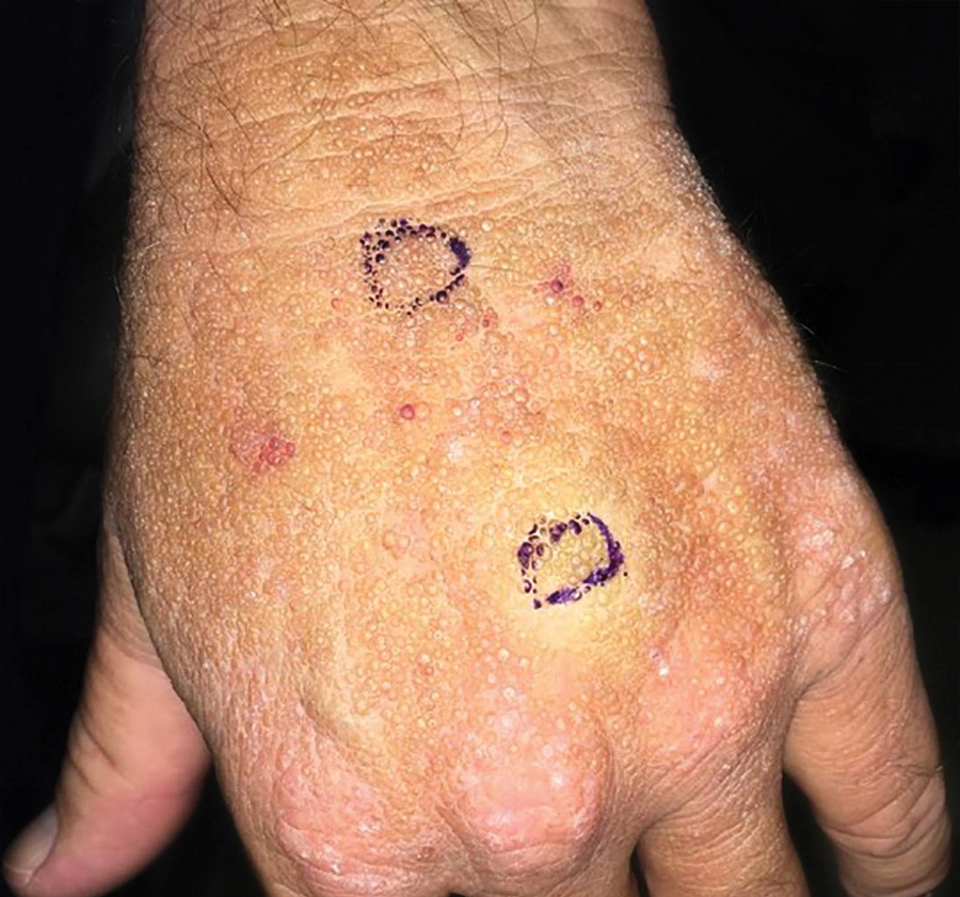
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).

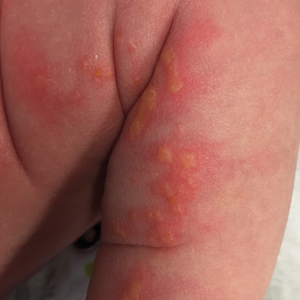
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
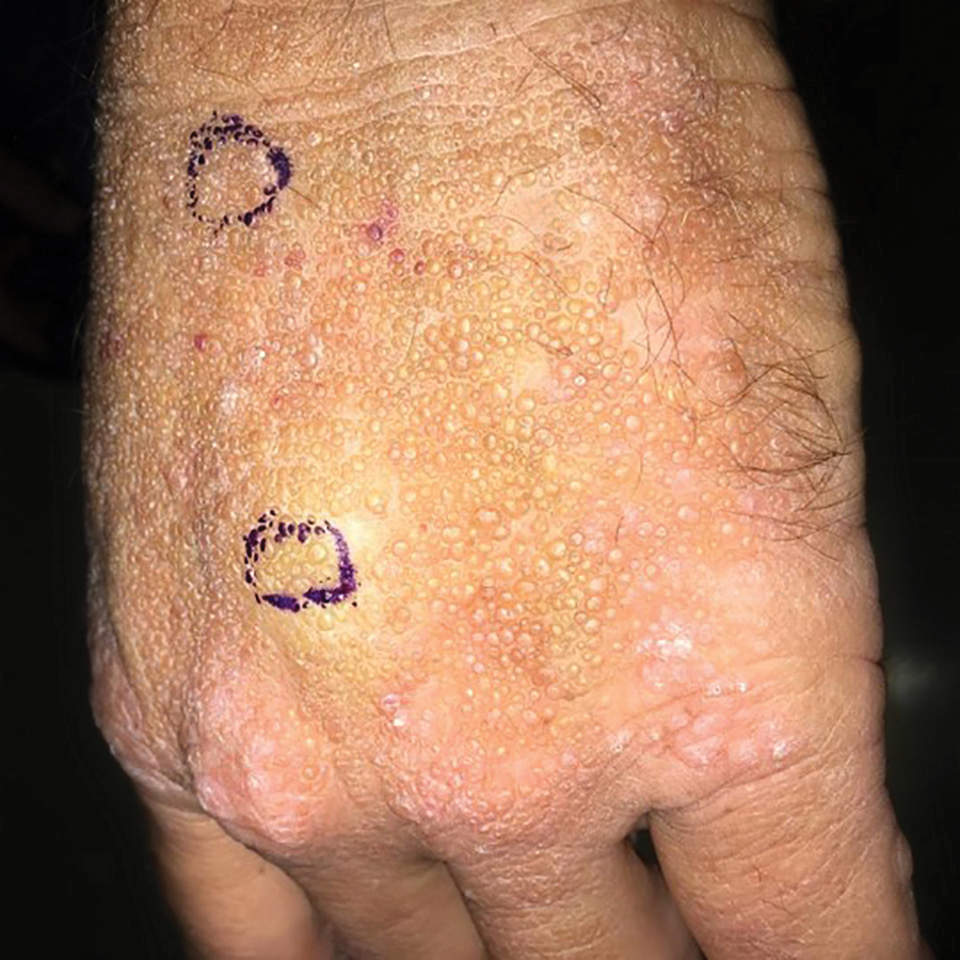
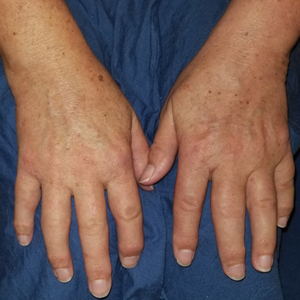
A 41-year-old man presented to the outpatient dermatology clinic with multiple smooth papules on the left hand of 7 years' duration. The papules had been steadily increasing in number, and the patient reported that they were frequently symptomatic with a burning itching sensation. Physical examination revealed multiple 1- to 3-mm, dome-shaped, translucent to flesh-colored papules on the left hand with a few scattered bright red papules. No similar lesions were present on the right hand or elsewhere on the body. He had a history of hypertension but was otherwise healthy with no other chronic medical conditions.
Periorbital Swelling and Rash Following Trauma
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
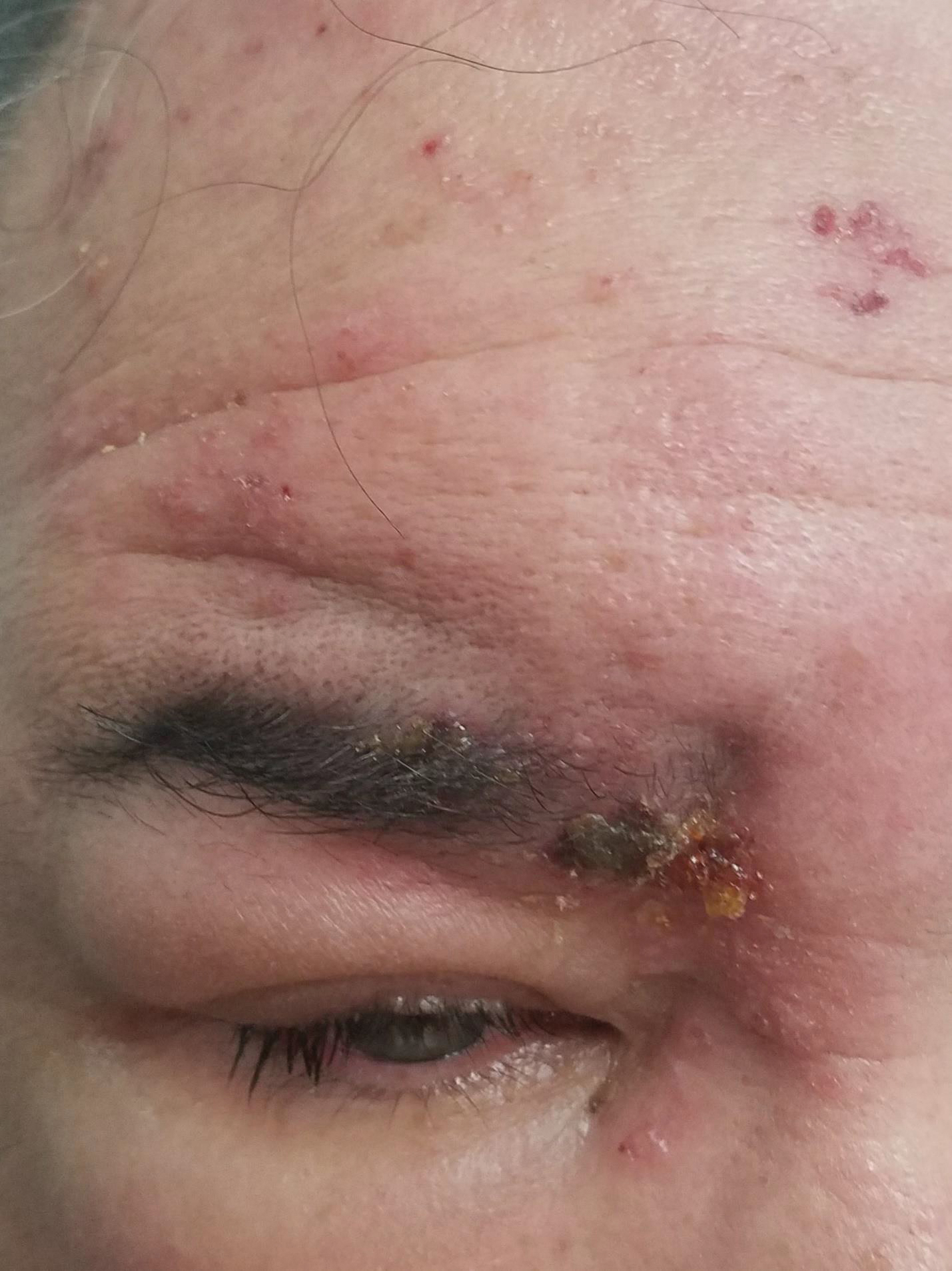
A 56-year-old man presented to an urgent care clinic with right periorbital swelling. He reported hitting his head on the door to a storage unit 2 days prior but did not lose consciousness. The swelling presented 2 days later. He reported mild headache and swelling around the right eye that coincided with an uncomfortable rash on the face and scalp. He also reported visual disruption due to the swelling but denied any eye pain, discharge from the eye, or painful eye movements. He had no lesions on the lips or inside the mouth. He denied any history of skin conditions. He further denied fever, joint pain, or any other systemic symptoms. His chronic medical conditions included diabetes mellitus, hypertension, and hyperlipidemia that were stable on metformin, carvedilol, amlodipine, enalapril, and simvastatin, which he had taken for several years. He had not started any new medications, and there were no recent changes in the dosing of his medications.
Friable Scalp Nodule
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
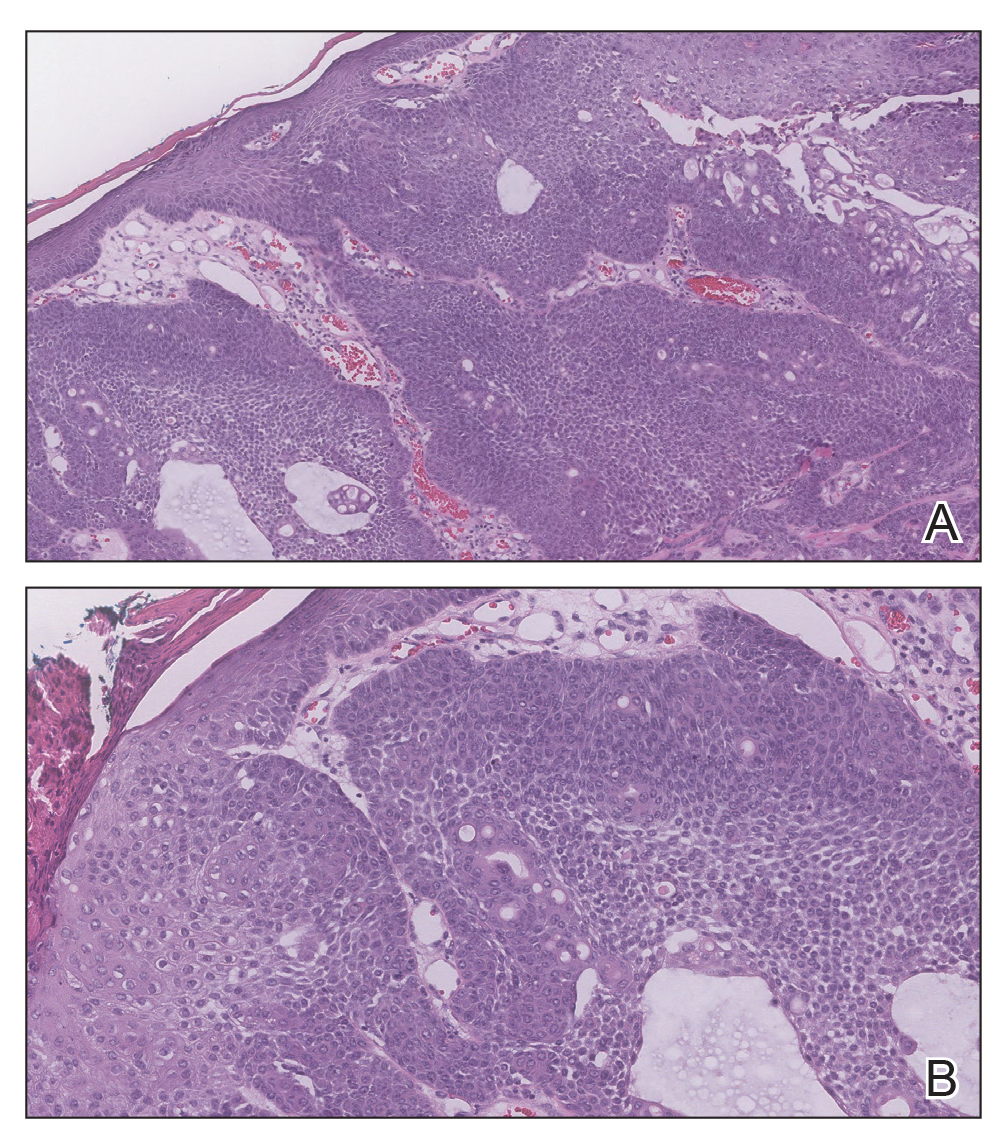
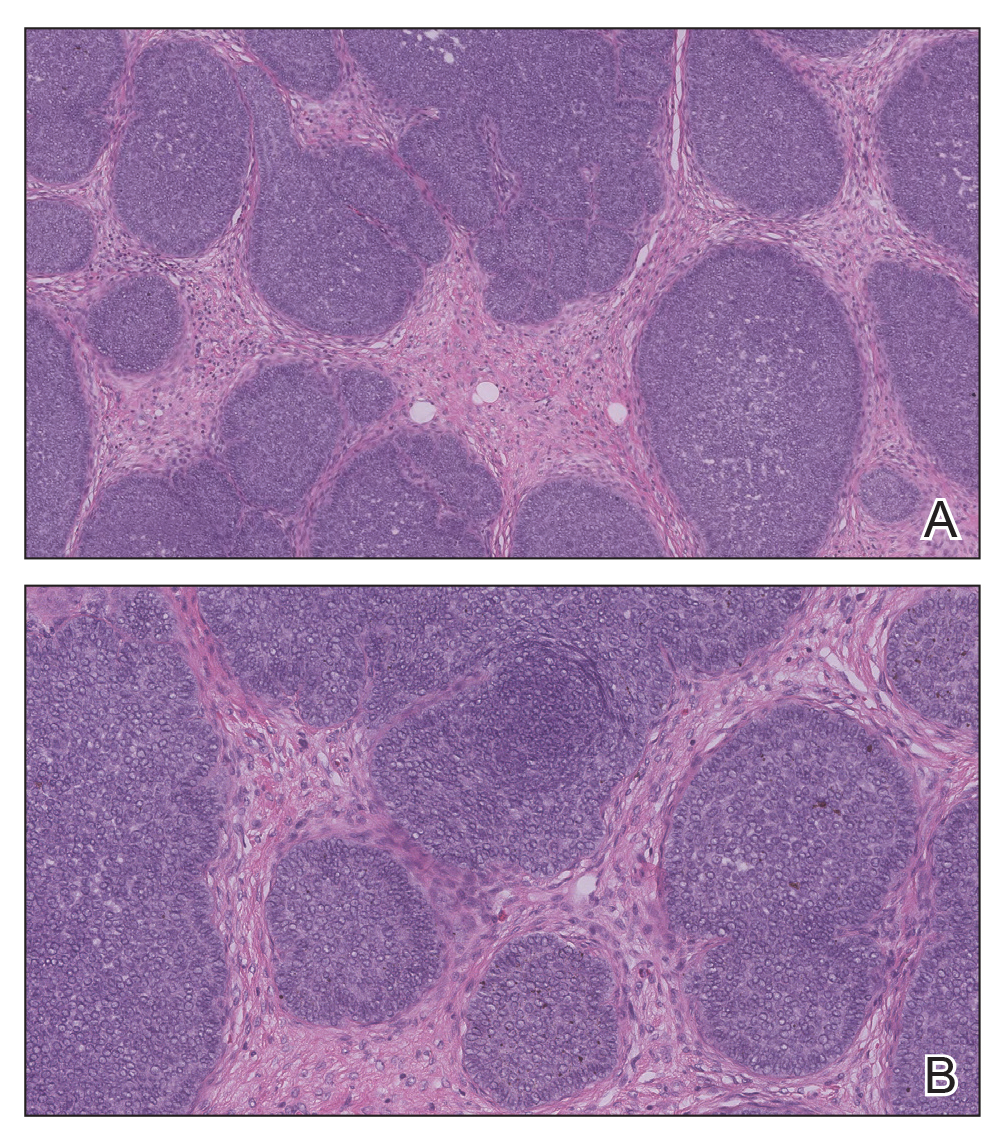
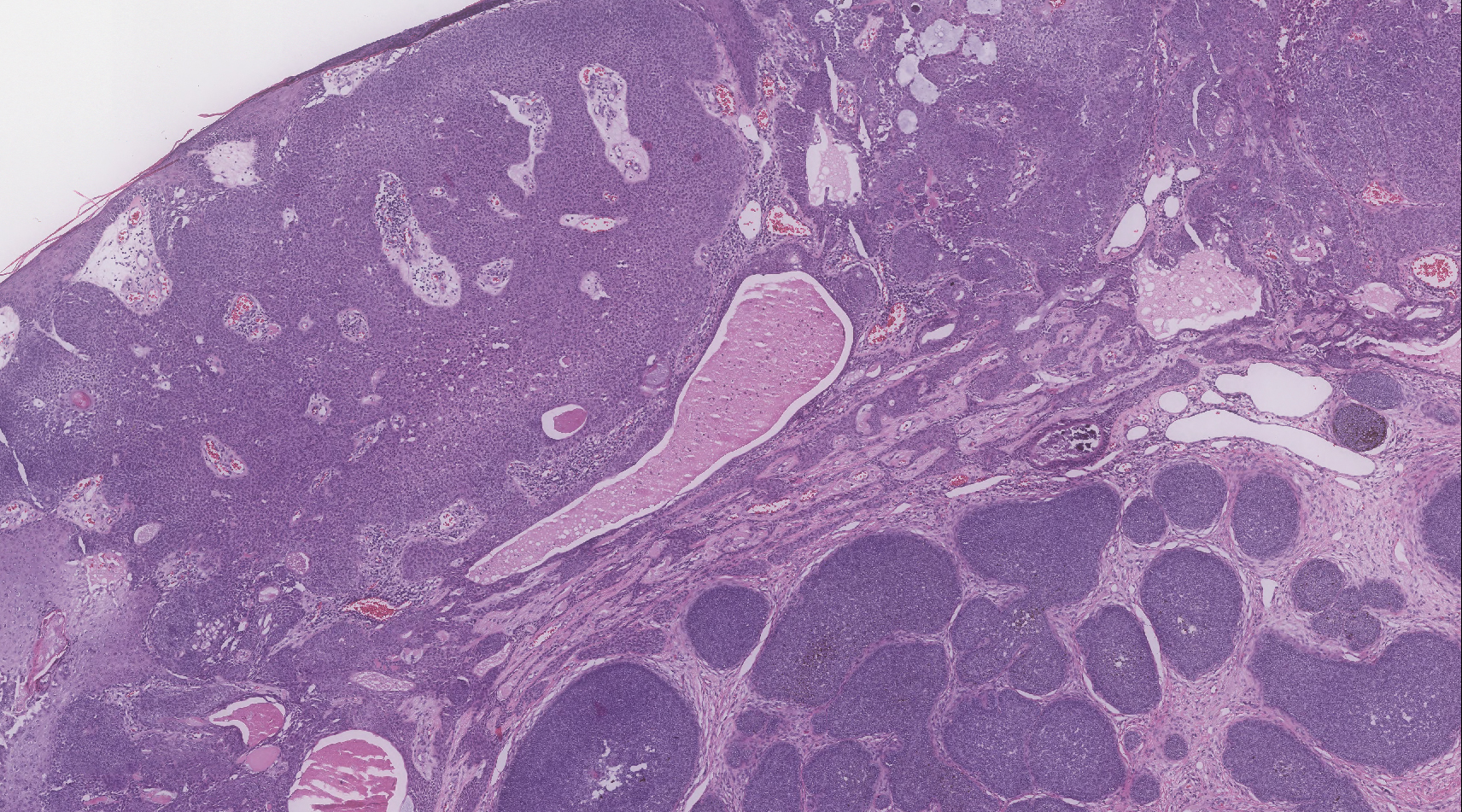
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
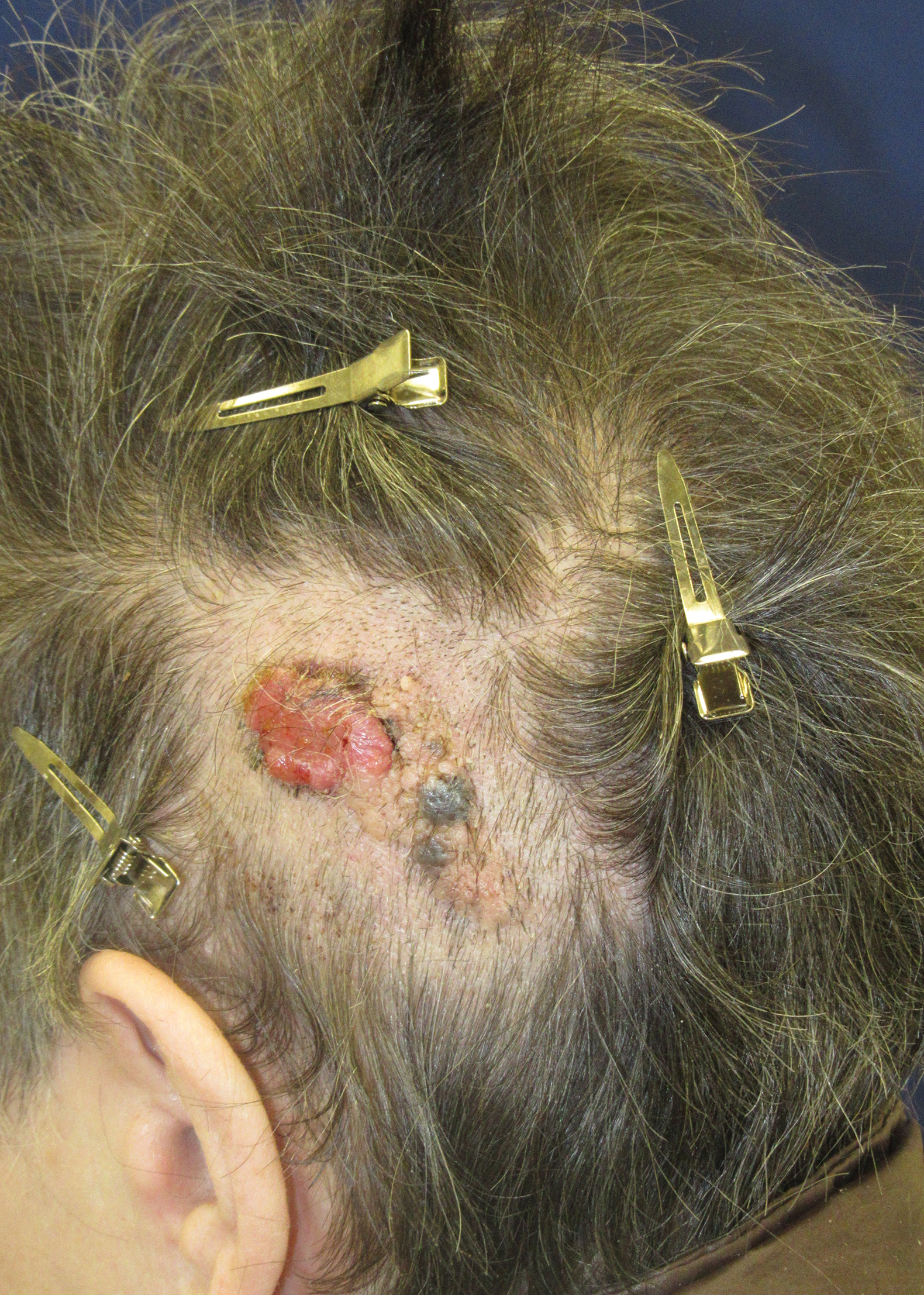
A 75-year-old woman presented with an enlarging plaque on the scalp of 5 years' duration. Physical examination revealed a 5.6.2 ×2.9-cm, tan-colored, verrucous plaque with an overlying pink friable nodule on the left occipital scalp. The lesion was not painful or pruritic, and the patient did not have any constitutional symptoms such as fever, night sweats, or weight loss. The patient denied prior tanning bed use and reported intermittent sun exposure over her lifetime. She denied any prior surgical intervention. There was no family history of similar lesions.
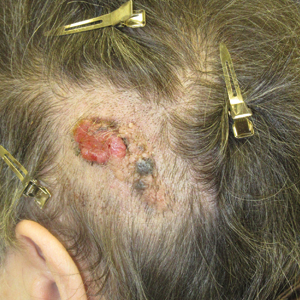
Brown-Black Punctate Macule on the Left Palm
The Diagnosis: Talon Noir
Paring of the stratum corneum overlying the lesion revealed coagulated blood, leading to a diagnosis of talon noir. Talon noir, also known as calcaneal petechiae, is a benign lesion that is typically found on the heel of the foot or palm of the hand.1 To the naked eye, talon noir appears as a brown-black asymmetric macule often with an overlying callus. Dermatoscopic visualization reveals grouped, reddish-colored globules composed of intracorneal hemorrhages, often in a linear pattern without any disruption of the normal skin surface architecture.1,2
Talon noir is the product of shear stress and often is seen in individuals who participate in sports such as baseball, hockey, soccer, and football.1,3 Lateral shearing forces cause tearing of blood vessels within the papillary dermis, which leads to punctate papillary dermal hemorrhages and extravasation of blood into the epidermis, resulting in intracorneal hemorrhage.1,4 Talon noir lesions are completely asymptomatic and typically resolve without intervention within 2 to 3 weeks of discontinuation of the precipitating sport or trauma.4
Recognizing talon noir is important, as it can occasionally be mistaken for acral lentiginous melanoma junctional nevus, tinea nigra, or verruca vulgaris. Paring of a lesion that is suspicious for talon noir is a simple and important step for ruling out a more ominous diagnosis (ie, acral lentiginous melanoma). If paring reveals coagulated blood, then junctional nevus, acral lentiginous melanoma, and tinea nigra can be excluded from the differential diagnosis. To rule out verruca vulgaris, one must prove that there is no disruption of the normal skin architecture, which can be confirmed with dermatoscopic visualization. Verrucae characteristically cause disruption of normal skin architecture, and junctional nevi would reveal a pigment pattern on dermatoscopy. This case illustrates how simple bedside procedures—dermoscopy and paring—can reassure patients and caregivers of the benign nature of talon noir.
1. Googe AB, Schulmeier JS, Jackson AR, et al. Talon noir: paring can eliminate the need for biopsy. Postgrad Med J. 2014;90:730-731.
2. Lao M, Weissler A, Siegfried E. Talon noir. J Pediatr. 2013;163:919.
3. Ayres S, Mihan R. Calcaneal petechiae. Arch Dermatol. 1972;106:262.
4. Bender TW. Cutaneous manifestations of disease in athletes. Skinmed. 2003;2:34-40
The Diagnosis: Talon Noir
Paring of the stratum corneum overlying the lesion revealed coagulated blood, leading to a diagnosis of talon noir. Talon noir, also known as calcaneal petechiae, is a benign lesion that is typically found on the heel of the foot or palm of the hand.1 To the naked eye, talon noir appears as a brown-black asymmetric macule often with an overlying callus. Dermatoscopic visualization reveals grouped, reddish-colored globules composed of intracorneal hemorrhages, often in a linear pattern without any disruption of the normal skin surface architecture.1,2
Talon noir is the product of shear stress and often is seen in individuals who participate in sports such as baseball, hockey, soccer, and football.1,3 Lateral shearing forces cause tearing of blood vessels within the papillary dermis, which leads to punctate papillary dermal hemorrhages and extravasation of blood into the epidermis, resulting in intracorneal hemorrhage.1,4 Talon noir lesions are completely asymptomatic and typically resolve without intervention within 2 to 3 weeks of discontinuation of the precipitating sport or trauma.4
Recognizing talon noir is important, as it can occasionally be mistaken for acral lentiginous melanoma junctional nevus, tinea nigra, or verruca vulgaris. Paring of a lesion that is suspicious for talon noir is a simple and important step for ruling out a more ominous diagnosis (ie, acral lentiginous melanoma). If paring reveals coagulated blood, then junctional nevus, acral lentiginous melanoma, and tinea nigra can be excluded from the differential diagnosis. To rule out verruca vulgaris, one must prove that there is no disruption of the normal skin architecture, which can be confirmed with dermatoscopic visualization. Verrucae characteristically cause disruption of normal skin architecture, and junctional nevi would reveal a pigment pattern on dermatoscopy. This case illustrates how simple bedside procedures—dermoscopy and paring—can reassure patients and caregivers of the benign nature of talon noir.
The Diagnosis: Talon Noir
Paring of the stratum corneum overlying the lesion revealed coagulated blood, leading to a diagnosis of talon noir. Talon noir, also known as calcaneal petechiae, is a benign lesion that is typically found on the heel of the foot or palm of the hand.1 To the naked eye, talon noir appears as a brown-black asymmetric macule often with an overlying callus. Dermatoscopic visualization reveals grouped, reddish-colored globules composed of intracorneal hemorrhages, often in a linear pattern without any disruption of the normal skin surface architecture.1,2
Talon noir is the product of shear stress and often is seen in individuals who participate in sports such as baseball, hockey, soccer, and football.1,3 Lateral shearing forces cause tearing of blood vessels within the papillary dermis, which leads to punctate papillary dermal hemorrhages and extravasation of blood into the epidermis, resulting in intracorneal hemorrhage.1,4 Talon noir lesions are completely asymptomatic and typically resolve without intervention within 2 to 3 weeks of discontinuation of the precipitating sport or trauma.4
Recognizing talon noir is important, as it can occasionally be mistaken for acral lentiginous melanoma junctional nevus, tinea nigra, or verruca vulgaris. Paring of a lesion that is suspicious for talon noir is a simple and important step for ruling out a more ominous diagnosis (ie, acral lentiginous melanoma). If paring reveals coagulated blood, then junctional nevus, acral lentiginous melanoma, and tinea nigra can be excluded from the differential diagnosis. To rule out verruca vulgaris, one must prove that there is no disruption of the normal skin architecture, which can be confirmed with dermatoscopic visualization. Verrucae characteristically cause disruption of normal skin architecture, and junctional nevi would reveal a pigment pattern on dermatoscopy. This case illustrates how simple bedside procedures—dermoscopy and paring—can reassure patients and caregivers of the benign nature of talon noir.
1. Googe AB, Schulmeier JS, Jackson AR, et al. Talon noir: paring can eliminate the need for biopsy. Postgrad Med J. 2014;90:730-731.
2. Lao M, Weissler A, Siegfried E. Talon noir. J Pediatr. 2013;163:919.
3. Ayres S, Mihan R. Calcaneal petechiae. Arch Dermatol. 1972;106:262.
4. Bender TW. Cutaneous manifestations of disease in athletes. Skinmed. 2003;2:34-40
1. Googe AB, Schulmeier JS, Jackson AR, et al. Talon noir: paring can eliminate the need for biopsy. Postgrad Med J. 2014;90:730-731.
2. Lao M, Weissler A, Siegfried E. Talon noir. J Pediatr. 2013;163:919.
3. Ayres S, Mihan R. Calcaneal petechiae. Arch Dermatol. 1972;106:262.
4. Bender TW. Cutaneous manifestations of disease in athletes. Skinmed. 2003;2:34-40
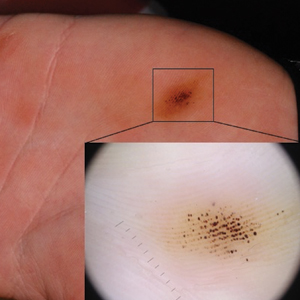
A 16-year-old adolescent boy presented to our clinic with a “new brown mole” on the left palm that had appeared within the last few months. The patient did not recall if it had changed in size, shape, or color, and there was no associated pain or itching. He denied any trauma to the hand, but he actively played both hockey and baseball. Physical examination revealed calloused palms bilaterally. One of the calluses was present over the hypothenar eminence, and centrally there were grouped brown-black punctate macules, some that coalesced into larger macules. Dermatoscopic examination (inset) revealed punctate rustcolored macules in a parallel ridge pattern. There was no disruption of the normal skin architecture.
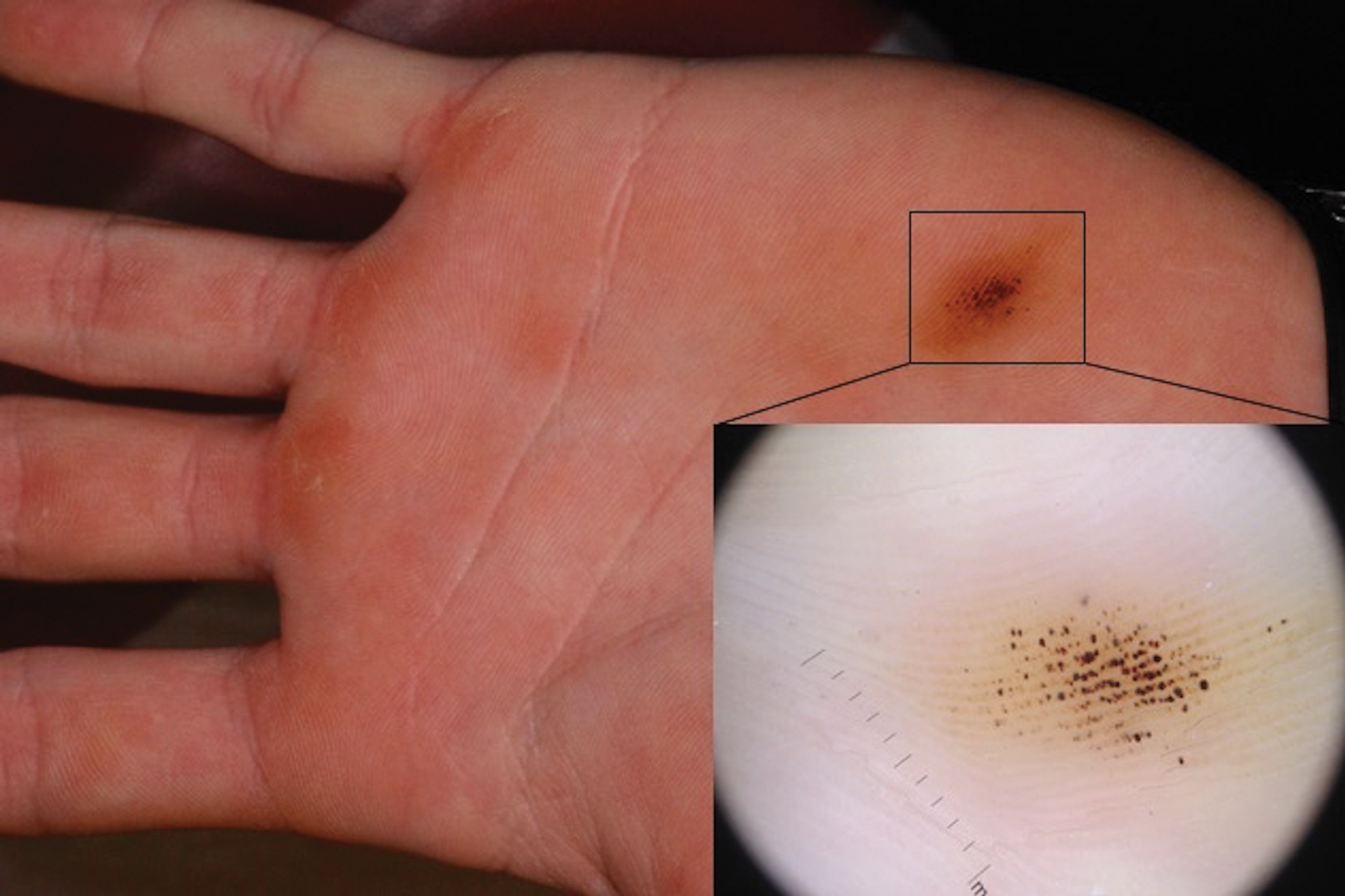
Firm Tumor Encasing the Left Second Toe of an Infant
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF), or recurring digital fibrous tumor of childhood, is a benign juvenile myofibromatosis that presents as a firm, flesh-colored or slightly erythematous, dome-shaped papule or nodule on the dorsolateral aspects of the digits, usually sparing the thumbs and great toes.1 The tumor appears most commonly at birth and in infants younger than 1 year. It grows slowly over the first month, then rapidly over the next 10 to 14 months.1,2
Although lesions usually regress spontaneously within a few years, excision may be necessary when functional impairment and joint deformity occur. Tumors, however, may recur locally.1,2
Histologically, IDFs are composed of spindled myofibroblasts with characteristic round eosinophilic intracytoplasmic inclusion bodies, which represent actin and vimentin filaments.1 In our case, histopathologic evaluation showed a proliferation of fibrous spindle cells with pathognomonic eosinophilic intracytoplasmic inclusions consistent with IDF (Figure).
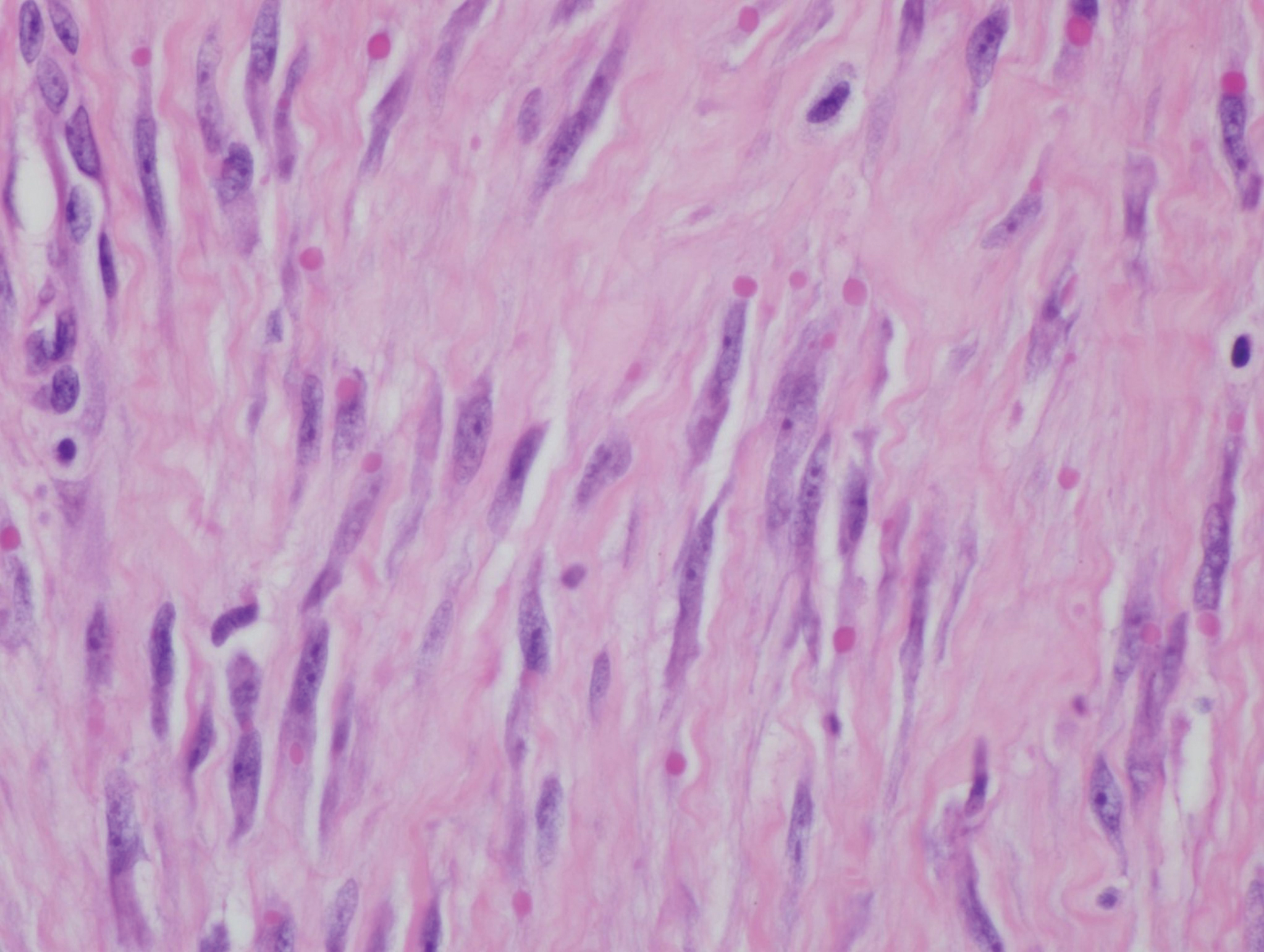
Fibrosarcomas are high-grade and low-grade soft-tissue neoplasms comprised of atypical spindle cells in a herringbone pattern with mitotic figures on pathology.3 They typically present as a slowly growing subcutaneous tumor on the lower extremities of young to middle-aged adults that may progress to become a palpable tender nodule. Infantile hemangiomas, the most common benign soft-tissue tumors of childhood, are vascular proliferations more commonly seen in low-birth-weight female white infants of multiple gestation pregnancies. Superficial hemangiomas present as bright red and lobular nodules or plaques. Deep hemangiomas present as ill-defined, blue-violaceous nodules with no overlying skin changes. Mixed hemangiomas present with features of both superficial and deep hemangiomas. Infantile hemangiomas experience a proliferative phase until 9 to 12 months of age, followed by a gradual involutional phase ending with possible residual telangiectases or fibrofatty change. Unlike IDFs, infantile hemangiomas favor the head and neck over other areas of the body. Keloids are firm, smooth, variably colored papules or plaques of haphazardly arranged thick dermal collagen bundles. They usually develop within a year of skin injury and extend beyond the original injury margin into normal tissue. Supernumerary digits present as small fleshy papules or larger nodules with a vestigial nail, most commonly on the ulnar side of the fifth finger or the radial side of the thumb. Histologically, they are composed of fascicles of nerve fibers.3
Treatment in our patient included partial amputation of the left second toe and excision with local tissue rearrangement by a plastic surgeon. His postoperative course was complicated by a minor wound infection, which resolved with a 7-day course of cephalexin. Since then, the patient has healed well, with gradual toe tissue and nail growth. No recurrence was reported 11 months after surgery.
- Heymann WR. Infantile digital fibromatosis. J Am Acad Dermatol. 2008;59:122-123.
- Niamba P, Léauté-Labrèze C, Boralevi F, et al. Further documentation of spontaneous regression of infantile digital fibromatosis. Pediatr Dermatol. 2007;24:280-284.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Philadelphia, PA: Elsevier Limited; 2018.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF), or recurring digital fibrous tumor of childhood, is a benign juvenile myofibromatosis that presents as a firm, flesh-colored or slightly erythematous, dome-shaped papule or nodule on the dorsolateral aspects of the digits, usually sparing the thumbs and great toes.1 The tumor appears most commonly at birth and in infants younger than 1 year. It grows slowly over the first month, then rapidly over the next 10 to 14 months.1,2
Although lesions usually regress spontaneously within a few years, excision may be necessary when functional impairment and joint deformity occur. Tumors, however, may recur locally.1,2
Histologically, IDFs are composed of spindled myofibroblasts with characteristic round eosinophilic intracytoplasmic inclusion bodies, which represent actin and vimentin filaments.1 In our case, histopathologic evaluation showed a proliferation of fibrous spindle cells with pathognomonic eosinophilic intracytoplasmic inclusions consistent with IDF (Figure).
Fibrosarcomas are high-grade and low-grade soft-tissue neoplasms comprised of atypical spindle cells in a herringbone pattern with mitotic figures on pathology.3 They typically present as a slowly growing subcutaneous tumor on the lower extremities of young to middle-aged adults that may progress to become a palpable tender nodule. Infantile hemangiomas, the most common benign soft-tissue tumors of childhood, are vascular proliferations more commonly seen in low-birth-weight female white infants of multiple gestation pregnancies. Superficial hemangiomas present as bright red and lobular nodules or plaques. Deep hemangiomas present as ill-defined, blue-violaceous nodules with no overlying skin changes. Mixed hemangiomas present with features of both superficial and deep hemangiomas. Infantile hemangiomas experience a proliferative phase until 9 to 12 months of age, followed by a gradual involutional phase ending with possible residual telangiectases or fibrofatty change. Unlike IDFs, infantile hemangiomas favor the head and neck over other areas of the body. Keloids are firm, smooth, variably colored papules or plaques of haphazardly arranged thick dermal collagen bundles. They usually develop within a year of skin injury and extend beyond the original injury margin into normal tissue. Supernumerary digits present as small fleshy papules or larger nodules with a vestigial nail, most commonly on the ulnar side of the fifth finger or the radial side of the thumb. Histologically, they are composed of fascicles of nerve fibers.3
Treatment in our patient included partial amputation of the left second toe and excision with local tissue rearrangement by a plastic surgeon. His postoperative course was complicated by a minor wound infection, which resolved with a 7-day course of cephalexin. Since then, the patient has healed well, with gradual toe tissue and nail growth. No recurrence was reported 11 months after surgery.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF), or recurring digital fibrous tumor of childhood, is a benign juvenile myofibromatosis that presents as a firm, flesh-colored or slightly erythematous, dome-shaped papule or nodule on the dorsolateral aspects of the digits, usually sparing the thumbs and great toes.1 The tumor appears most commonly at birth and in infants younger than 1 year. It grows slowly over the first month, then rapidly over the next 10 to 14 months.1,2
Although lesions usually regress spontaneously within a few years, excision may be necessary when functional impairment and joint deformity occur. Tumors, however, may recur locally.1,2
Histologically, IDFs are composed of spindled myofibroblasts with characteristic round eosinophilic intracytoplasmic inclusion bodies, which represent actin and vimentin filaments.1 In our case, histopathologic evaluation showed a proliferation of fibrous spindle cells with pathognomonic eosinophilic intracytoplasmic inclusions consistent with IDF (Figure).
Fibrosarcomas are high-grade and low-grade soft-tissue neoplasms comprised of atypical spindle cells in a herringbone pattern with mitotic figures on pathology.3 They typically present as a slowly growing subcutaneous tumor on the lower extremities of young to middle-aged adults that may progress to become a palpable tender nodule. Infantile hemangiomas, the most common benign soft-tissue tumors of childhood, are vascular proliferations more commonly seen in low-birth-weight female white infants of multiple gestation pregnancies. Superficial hemangiomas present as bright red and lobular nodules or plaques. Deep hemangiomas present as ill-defined, blue-violaceous nodules with no overlying skin changes. Mixed hemangiomas present with features of both superficial and deep hemangiomas. Infantile hemangiomas experience a proliferative phase until 9 to 12 months of age, followed by a gradual involutional phase ending with possible residual telangiectases or fibrofatty change. Unlike IDFs, infantile hemangiomas favor the head and neck over other areas of the body. Keloids are firm, smooth, variably colored papules or plaques of haphazardly arranged thick dermal collagen bundles. They usually develop within a year of skin injury and extend beyond the original injury margin into normal tissue. Supernumerary digits present as small fleshy papules or larger nodules with a vestigial nail, most commonly on the ulnar side of the fifth finger or the radial side of the thumb. Histologically, they are composed of fascicles of nerve fibers.3
Treatment in our patient included partial amputation of the left second toe and excision with local tissue rearrangement by a plastic surgeon. His postoperative course was complicated by a minor wound infection, which resolved with a 7-day course of cephalexin. Since then, the patient has healed well, with gradual toe tissue and nail growth. No recurrence was reported 11 months after surgery.
- Heymann WR. Infantile digital fibromatosis. J Am Acad Dermatol. 2008;59:122-123.
- Niamba P, Léauté-Labrèze C, Boralevi F, et al. Further documentation of spontaneous regression of infantile digital fibromatosis. Pediatr Dermatol. 2007;24:280-284.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Philadelphia, PA: Elsevier Limited; 2018.
- Heymann WR. Infantile digital fibromatosis. J Am Acad Dermatol. 2008;59:122-123.
- Niamba P, Léauté-Labrèze C, Boralevi F, et al. Further documentation of spontaneous regression of infantile digital fibromatosis. Pediatr Dermatol. 2007;24:280-284.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Philadelphia, PA: Elsevier Limited; 2018.
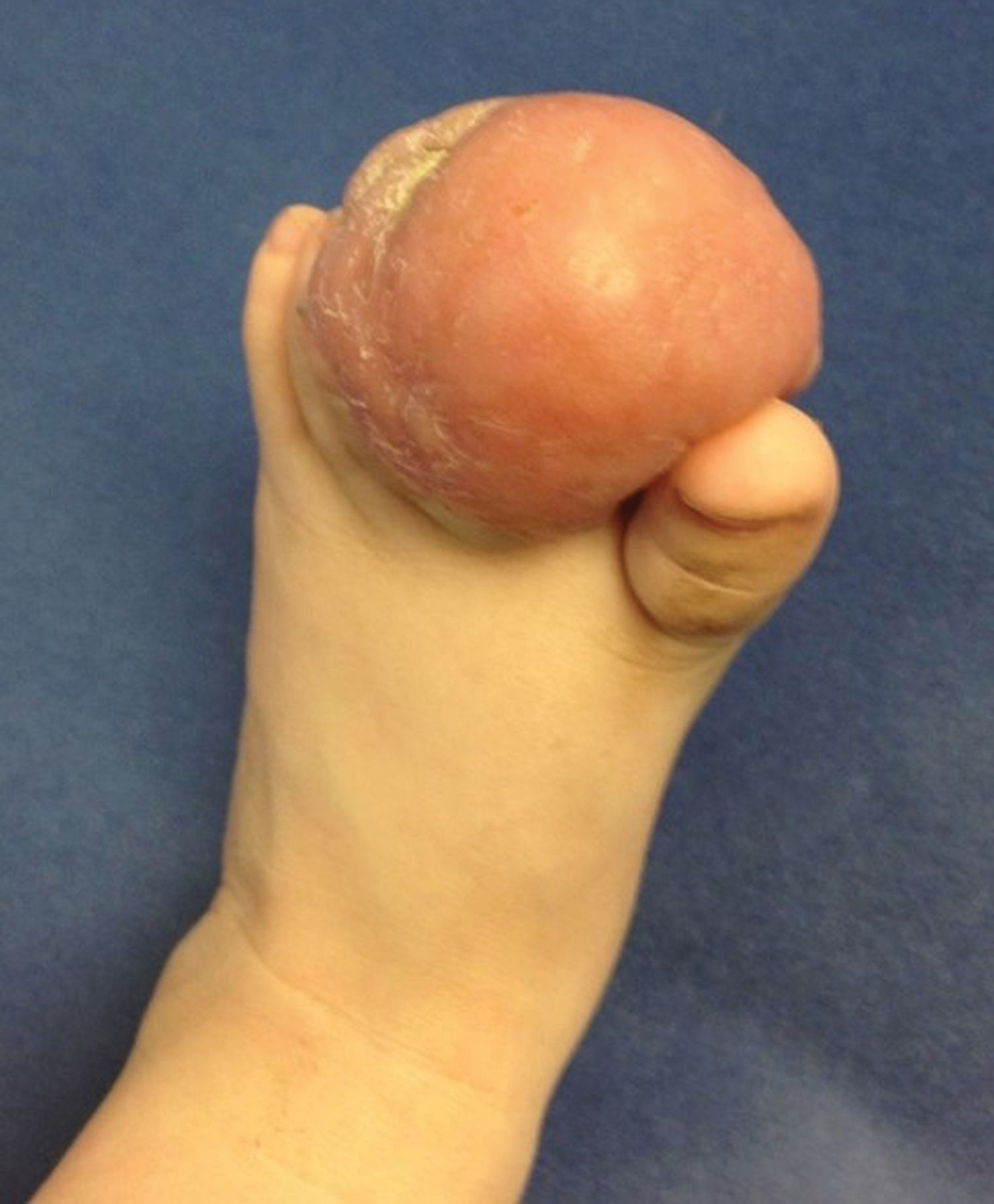
A 10-month-old infant boy presented to the dermatology clinic with a firm, nonpainful, 5.5.×5.6-cm tumor encasing the left second toe, with associated skin breakdown, gait impairment, and lateral displacement of the third toe. The lesion began as a small bump under the toenail at 2 months of age; it then grew rapidly without bleeding or ulceration. It was diagnosed as a hemangioma at 4 months of age, and oral propranolol was initiated for 3 months, without suppression of tumor growth. The patient was referred to the pediatric dermatology department where a clinical diagnosis was made, and the patient was subsequently referred to plastic surgery for excision.
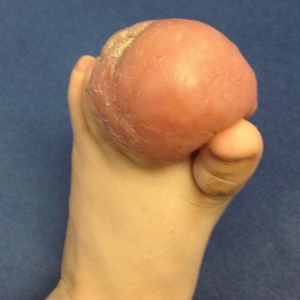
Numerous Flesh-Colored Nodules on the Trunk
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
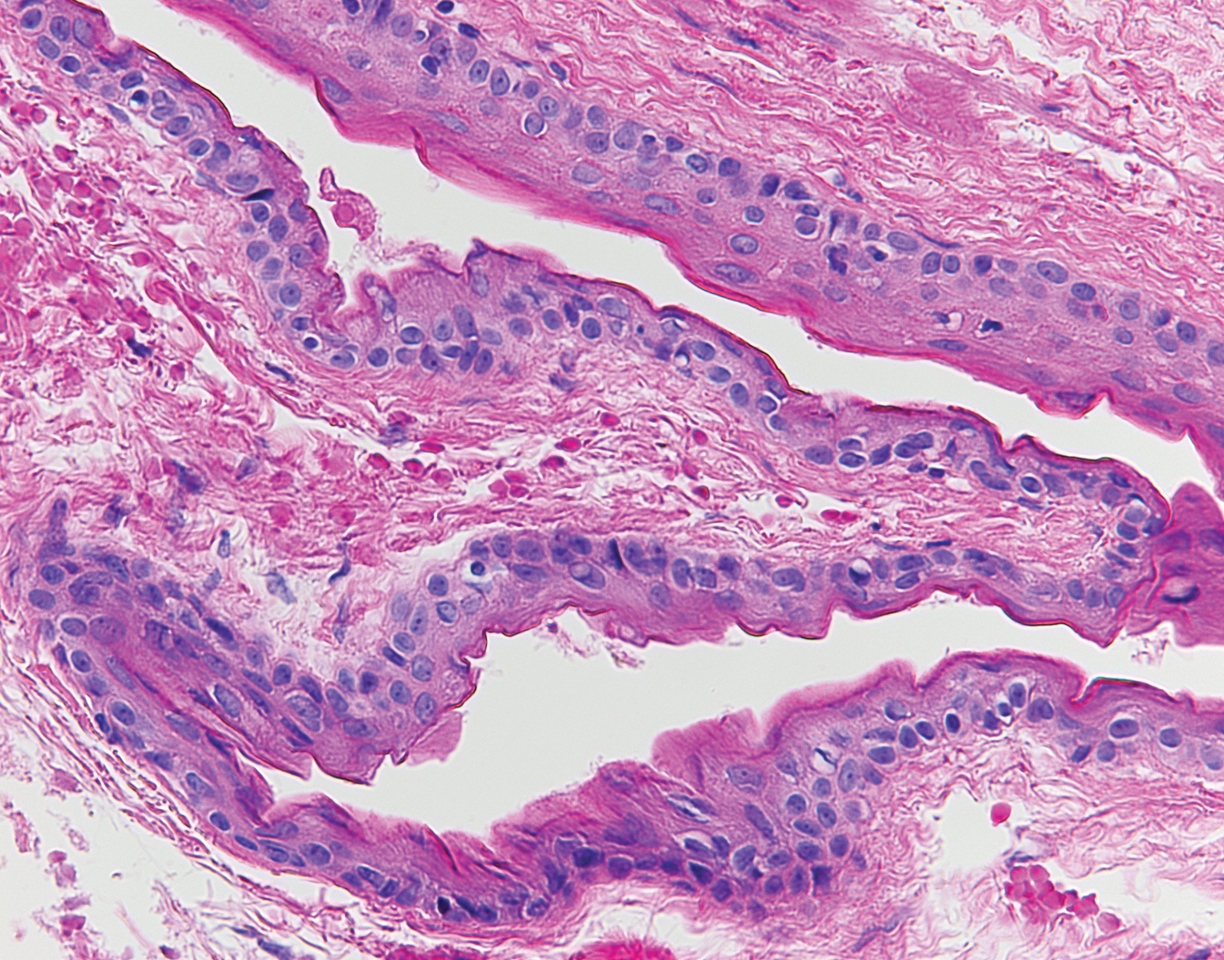
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
The Diagnosis: Steatocystoma Multiplex
The punch biopsy of an abdominal lesion demonstrated a folded cyst wall with a wavy eosinophilic cuticle (Figure), characteristics consistent with steatocystoma multiplex (SM).
Also known as eruptive steatocystoma, SM consists of numerous flesh-colored, dome-shaped papules and nodules that most commonly arise during adolescence, with a median age of onset of 26 years.1 These hamartomatous nevoid malformations arise in areas with well-developed pilosebaceous units, such as the upper extremities, neck, axillae, and trunk.1,2 They occur less commonly on the scalp, face, and acral surfaces.2-5 The lesions range in size from 2 to 30 mm6 and usually are asymptomatic.1 Occasionally, steatocystomas become tender or can rupture.7
Steatocystoma multiplex may arise sporadically or may be inherited in an autosomal-dominant fashion. Mutations in exon 1 of the keratin 17 gene, KRT17, have been identified in autosomal-dominant SM.6,8 KRT17 mutations also are responsible for pachyonychia congenita type 2, which is associated with SM.9 Some patients with pachyonychia congenita type 2 who have prominent SM and mild nail findings may be misdiagnosed as having pure SM.2
The histopathologic features of SM were described in a study by Cho and colleagues1 of 64 patients. Steatocystomas have cyst walls that may be either intricately folded or round/oval, comprised of an average of 4.9 epithelial cell layers. In most cases, the cyst wall contains sebaceous lobules. In all cases, an acellular eosinophilic cuticle was present, and no granular layer was seen. Few vellus hairs may be observed in the cystic cavity.1
The differential diagnosis of SM includes eruptive vellus hair cysts, lipomas, Muir-Torre syndrome, and Gardner syndrome. Some have suggested that eruptive vellus hair cysts and SM exist on a disease spectrum because of their similar clinical presentation.10 In contrast to SM, however, eruptive vellus hair cysts originate in the infundibulum of the hair shaft rather than the sebaceous duct, and more numerous vellus hair shafts are seen on histopathology.1
Various treatment modalities have been described, including isotretinoin for inflamed lesions,11 cryotherapy for noninflamed lesions,11 aspiration of lesions smallerthan 1 cm,12 and electrocautery combined with topical retinoids.13 Laser treatment has been described, with a 1450-nm diode laser used to target the abnormal sebaceous glands and a 1550-nm fractionated erbium-doped fiber laser used to target the dermal cysts.14 Carbon dioxide lasers also may be used to open the cyst for drainage.15 Surgical excision or mini-incision also may be performed.16,17
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his assistance with interpretation of the dermatopathology in this case.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Rollins T, Levin RM, Heymann WR. Acral steatocystoma multiplex.J Am Acad Dermatol. 2000;43(2, pt 2):396-399.
- Setoyama M, Mizoguchi S, Usuki K, et al. Steatocystoma multiplex: a case with unusual clinical and histological manifestation. Am J Dermatopathol. 1997;19:89-92.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273-278.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sato K, Shibuya K, Taguchi H, et al. Aspiration therapy in steatocystoma multiplex. Arch Dermatol. 1993;129:35-37.
- Papakonstantinou E, Franke I, Gollnick H. Facial steatocystoma multiplex combined with eruptive vellus hair cysts: a hybrid? J Eur Acad Dermatol Venereol. 2015;29:2051-2053.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7, pt 1):1104-1106.
- Rossi R, Cappugi P, Battini M, et al. CO2 laser therapy in a case of steatocystoma multiplex with prominent nodules on the face and neck. Int J Dermatol. 2003;42:302-304.
- Schmook T, Burg G, Hafner J. Surgical pearl: mini-incisions for the extraction of steatocystoma multiplex. J Am Acad Dermatol. 2001;44:1041-1042.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.

A 33-year-old woman presented with numerous firm, noncompressible, flesh-colored nodules that measured 3 to 4 mm and were distributed across the abdomen, chest, back, and neck. The lesions had been present for approximately 10 years. The patient denied any lesion-associated pain, itching, or bleeding, and there was no family history of similar lesions. A punch biopsy of a lesion on the central abdomen was obtained.
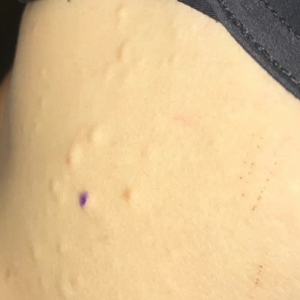
Widespread Skin Thickening
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
![]()
A 62-year-old woman presented with widespread skin thickening and tightness that progressed over 2 months. Physical examination showed generalized, nonpruritic, nonpainful, flesh-colored papules along the fingers, bilateral arms and legs, chest, neck, forehead, chin, and ears. She reported mild acid reflux on review of systems. She denied a history of Raynaud phenomenon and had normal-appearing nail beds on capillaroscopy. Laboratory studies including autoimmune serologies were normal, aside from a mildly elevated monoclonal IgG spike.