User login
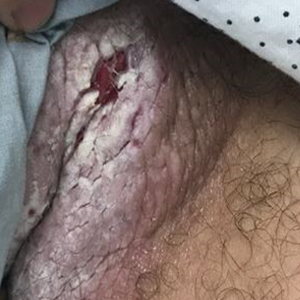
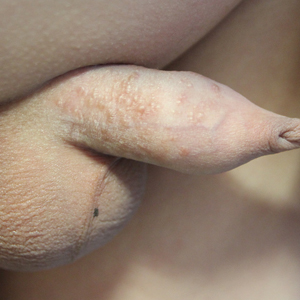
Multiple Yellow-Brown Papules on the Penis
The Diagnosis: Eruptive Syringoma
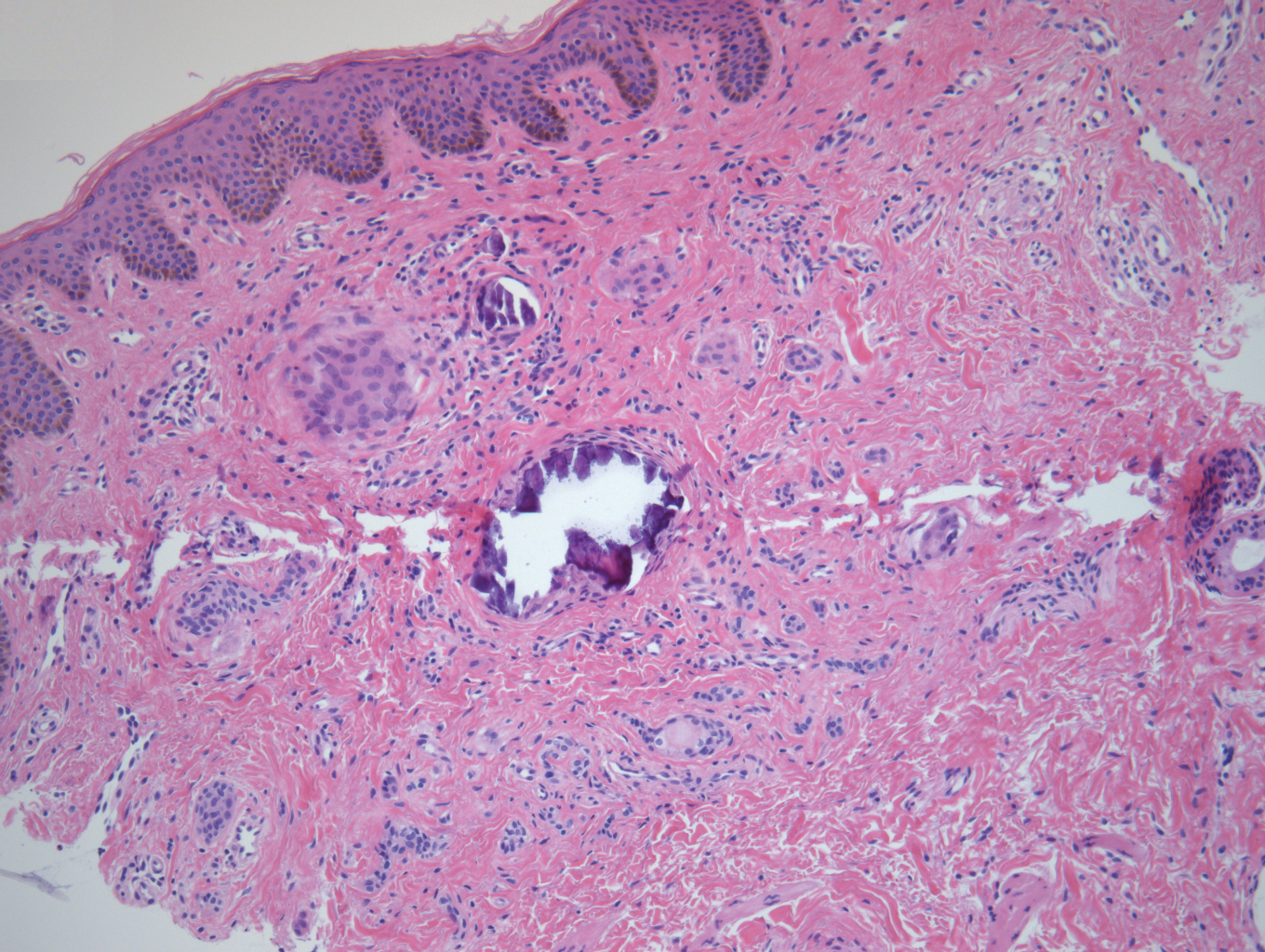
A punch biopsy of a lesion on the penis was performed. Histopathologic examination revealed many tadpole-shaped cords of epithelial cells and small ducts in the dermis (Figure). Based on clinical and histopathological findings, a diagnosis of eruptive syringoma was made. The patient declined treatment.
Syringomas are common benign eccrine neoplasms. They present clinically as small flesh-colored to brownish papules symmetrically distributed on the face, neck, trunk, pubic area, arms, and legs.1-3 Classic syringoma occurs more frequently in young adult women.1 Eruptive syringoma is a rare variant, and the age of onset ranges from 3 to 50 years.1-13 Eruptive syringoma is the term for multiple lesions that occur synchronously in any part of the body.1,4,13 The term eruptive is not the opposite of localized and refers to the time of onset of the lesion. There may be both a generalized eruptive syringoma or a localized eruptive syringoma depending on the distribution of the lesions.1 The most common site for syringoma occurrence is the eyelid; penile syringoma is extremely rare. Several cases of penile syringoma have been reported, but eruptive penile syringoma is rare.3,5-10,12,13
Histopathology is essential for the diagnosis of syringoma. Hematoxylin and eosin stain shows multiple small cystic ducts and epithelial cell nests in the dermis. Ductal structures sometimes appear tadpolelike or comma shaped depending on the section.1,2,7,12
The clinical differential diagnosis of syringoma includes sebaceous hyperplasia, verruca plana, molluscum contagiosum, bowenoid papulosis, condyloma acuminatum, lichen planus, lichen nitidus, milia, angiofibroma, epidermal cyst, calcinosis cutis, granuloma annulare, and sarcoidosis.3,8,12
Because syringoma is benign, treatment is not necessary unless there is a cosmetic problem.3,5,7,8,12 There is no satisfactory treatment of eruptive penile syringoma. Treatment options include topical tretinoin and adapalene, oral isotretinoin, cryotherapy, microelectrodesiccation with an epilating needle, dermabrasion, CO2 laser, and surgical excision.2,3,7,8,12
Adult patients with penile syringoma may be concerned about sexually transmitted diseases due to the appearance of the papules. If cosmesis is not an issue, clinicians should reassure the patient after a biopsy that the lesions are benign and self-limiting without recommending treatment.
- Ghanadan A, Khosravi M. Cutaneous syringoma: a clinicopathologic study of 34 new cases and review of the literature. Indian J Dermatol. 2013;58:326.
- Soler-Carrillo J, Estrach T, Mascaro JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246.
- Baek JO, Jee HJ, Kim TK, et al. Eruptive penile syringomas spreading to the pubic area and lower abdomen. Ann Dermatol. 2013;25:116-118.
- Pruzan DL, Esterly NB, Prose NS. Eruptive syringoma. Arch Dermatol. 1989;125:1119-1120.
- Olson JM, Robles DT, Argenyi ZB, et al. Multiple penile syringomas. J Am Acad Dermatol. 2008;59(2 suppl 1):S46-S47.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Huang C, Wang W, Wu B. Multiple brownish papules on the penile shaft. Indian J Dermatol Venereol Leprol. 2011;77:404.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:E21.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:e206-207.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:e21.
- Dhossche JM, Brodell RT, Al Hmada Y, et al. Skin-colored papules of the penis. Pediatr Dermatol. 2015;32:145-146.
- Todd PS, Gordon SC, Rovner RL, et al. Eruptive penile syringomas in an adolescent: novel approach with serial microexcisions and suture-adhesive repair. Pediatr Dermatol. 2016;33:E57-E60.
The Diagnosis: Eruptive Syringoma
A punch biopsy of a lesion on the penis was performed. Histopathologic examination revealed many tadpole-shaped cords of epithelial cells and small ducts in the dermis (Figure). Based on clinical and histopathological findings, a diagnosis of eruptive syringoma was made. The patient declined treatment.
Syringomas are common benign eccrine neoplasms. They present clinically as small flesh-colored to brownish papules symmetrically distributed on the face, neck, trunk, pubic area, arms, and legs.1-3 Classic syringoma occurs more frequently in young adult women.1 Eruptive syringoma is a rare variant, and the age of onset ranges from 3 to 50 years.1-13 Eruptive syringoma is the term for multiple lesions that occur synchronously in any part of the body.1,4,13 The term eruptive is not the opposite of localized and refers to the time of onset of the lesion. There may be both a generalized eruptive syringoma or a localized eruptive syringoma depending on the distribution of the lesions.1 The most common site for syringoma occurrence is the eyelid; penile syringoma is extremely rare. Several cases of penile syringoma have been reported, but eruptive penile syringoma is rare.3,5-10,12,13
Histopathology is essential for the diagnosis of syringoma. Hematoxylin and eosin stain shows multiple small cystic ducts and epithelial cell nests in the dermis. Ductal structures sometimes appear tadpolelike or comma shaped depending on the section.1,2,7,12
The clinical differential diagnosis of syringoma includes sebaceous hyperplasia, verruca plana, molluscum contagiosum, bowenoid papulosis, condyloma acuminatum, lichen planus, lichen nitidus, milia, angiofibroma, epidermal cyst, calcinosis cutis, granuloma annulare, and sarcoidosis.3,8,12
Because syringoma is benign, treatment is not necessary unless there is a cosmetic problem.3,5,7,8,12 There is no satisfactory treatment of eruptive penile syringoma. Treatment options include topical tretinoin and adapalene, oral isotretinoin, cryotherapy, microelectrodesiccation with an epilating needle, dermabrasion, CO2 laser, and surgical excision.2,3,7,8,12
Adult patients with penile syringoma may be concerned about sexually transmitted diseases due to the appearance of the papules. If cosmesis is not an issue, clinicians should reassure the patient after a biopsy that the lesions are benign and self-limiting without recommending treatment.
The Diagnosis: Eruptive Syringoma
A punch biopsy of a lesion on the penis was performed. Histopathologic examination revealed many tadpole-shaped cords of epithelial cells and small ducts in the dermis (Figure). Based on clinical and histopathological findings, a diagnosis of eruptive syringoma was made. The patient declined treatment.
Syringomas are common benign eccrine neoplasms. They present clinically as small flesh-colored to brownish papules symmetrically distributed on the face, neck, trunk, pubic area, arms, and legs.1-3 Classic syringoma occurs more frequently in young adult women.1 Eruptive syringoma is a rare variant, and the age of onset ranges from 3 to 50 years.1-13 Eruptive syringoma is the term for multiple lesions that occur synchronously in any part of the body.1,4,13 The term eruptive is not the opposite of localized and refers to the time of onset of the lesion. There may be both a generalized eruptive syringoma or a localized eruptive syringoma depending on the distribution of the lesions.1 The most common site for syringoma occurrence is the eyelid; penile syringoma is extremely rare. Several cases of penile syringoma have been reported, but eruptive penile syringoma is rare.3,5-10,12,13
Histopathology is essential for the diagnosis of syringoma. Hematoxylin and eosin stain shows multiple small cystic ducts and epithelial cell nests in the dermis. Ductal structures sometimes appear tadpolelike or comma shaped depending on the section.1,2,7,12
The clinical differential diagnosis of syringoma includes sebaceous hyperplasia, verruca plana, molluscum contagiosum, bowenoid papulosis, condyloma acuminatum, lichen planus, lichen nitidus, milia, angiofibroma, epidermal cyst, calcinosis cutis, granuloma annulare, and sarcoidosis.3,8,12
Because syringoma is benign, treatment is not necessary unless there is a cosmetic problem.3,5,7,8,12 There is no satisfactory treatment of eruptive penile syringoma. Treatment options include topical tretinoin and adapalene, oral isotretinoin, cryotherapy, microelectrodesiccation with an epilating needle, dermabrasion, CO2 laser, and surgical excision.2,3,7,8,12
Adult patients with penile syringoma may be concerned about sexually transmitted diseases due to the appearance of the papules. If cosmesis is not an issue, clinicians should reassure the patient after a biopsy that the lesions are benign and self-limiting without recommending treatment.
- Ghanadan A, Khosravi M. Cutaneous syringoma: a clinicopathologic study of 34 new cases and review of the literature. Indian J Dermatol. 2013;58:326.
- Soler-Carrillo J, Estrach T, Mascaro JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246.
- Baek JO, Jee HJ, Kim TK, et al. Eruptive penile syringomas spreading to the pubic area and lower abdomen. Ann Dermatol. 2013;25:116-118.
- Pruzan DL, Esterly NB, Prose NS. Eruptive syringoma. Arch Dermatol. 1989;125:1119-1120.
- Olson JM, Robles DT, Argenyi ZB, et al. Multiple penile syringomas. J Am Acad Dermatol. 2008;59(2 suppl 1):S46-S47.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Huang C, Wang W, Wu B. Multiple brownish papules on the penile shaft. Indian J Dermatol Venereol Leprol. 2011;77:404.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:E21.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:e206-207.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:e21.
- Dhossche JM, Brodell RT, Al Hmada Y, et al. Skin-colored papules of the penis. Pediatr Dermatol. 2015;32:145-146.
- Todd PS, Gordon SC, Rovner RL, et al. Eruptive penile syringomas in an adolescent: novel approach with serial microexcisions and suture-adhesive repair. Pediatr Dermatol. 2016;33:E57-E60.
- Ghanadan A, Khosravi M. Cutaneous syringoma: a clinicopathologic study of 34 new cases and review of the literature. Indian J Dermatol. 2013;58:326.
- Soler-Carrillo J, Estrach T, Mascaro JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246.
- Baek JO, Jee HJ, Kim TK, et al. Eruptive penile syringomas spreading to the pubic area and lower abdomen. Ann Dermatol. 2013;25:116-118.
- Pruzan DL, Esterly NB, Prose NS. Eruptive syringoma. Arch Dermatol. 1989;125:1119-1120.
- Olson JM, Robles DT, Argenyi ZB, et al. Multiple penile syringomas. J Am Acad Dermatol. 2008;59(2 suppl 1):S46-S47.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Huang C, Wang W, Wu B. Multiple brownish papules on the penile shaft. Indian J Dermatol Venereol Leprol. 2011;77:404.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:E21.
- Mitkov M, Balagula Y, Taube JM, et al. Plaque-like syringoma with involvement of deep reticular dermis. J Am Acad Dermatol. 2014;71:e206-207.
- Vaca EE, Mundinger GS, Zelken JA, et al. Surgical excision of multiple penile syringomas with scrotal flap reconstruction. Eplasty. 2014;14:e21.
- Dhossche JM, Brodell RT, Al Hmada Y, et al. Skin-colored papules of the penis. Pediatr Dermatol. 2015;32:145-146.
- Todd PS, Gordon SC, Rovner RL, et al. Eruptive penile syringomas in an adolescent: novel approach with serial microexcisions and suture-adhesive repair. Pediatr Dermatol. 2016;33:E57-E60.
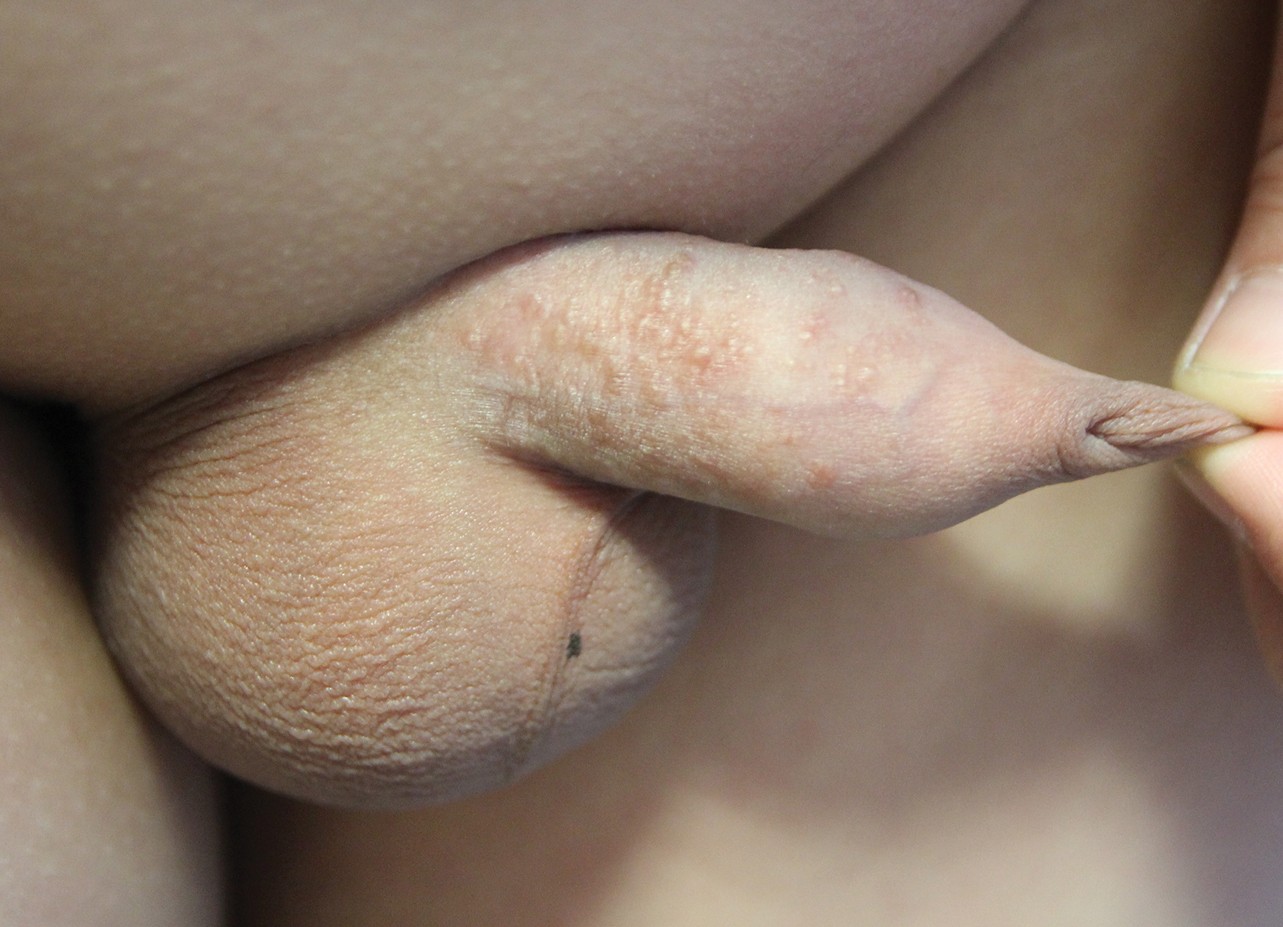
A 12-year-old boy presented with multiple asymptomatic, 0.1-cm, yellow-brown papules on the penile shaft of several years' duration. The lesions appeared suddenly. The patient had no history of trauma, injection, or an underlying disorder.
Erythematous Plaques on a Tattoo
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
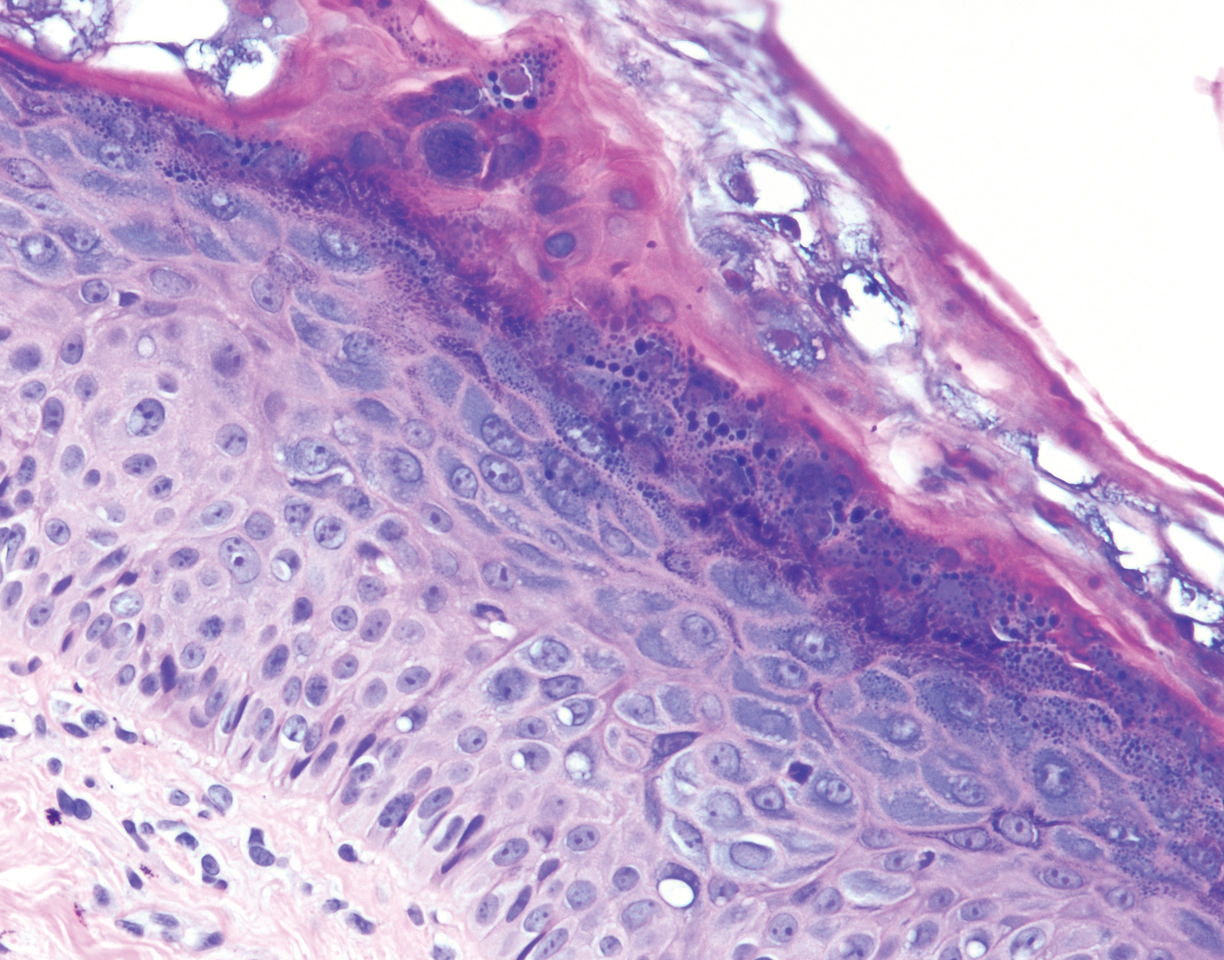
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
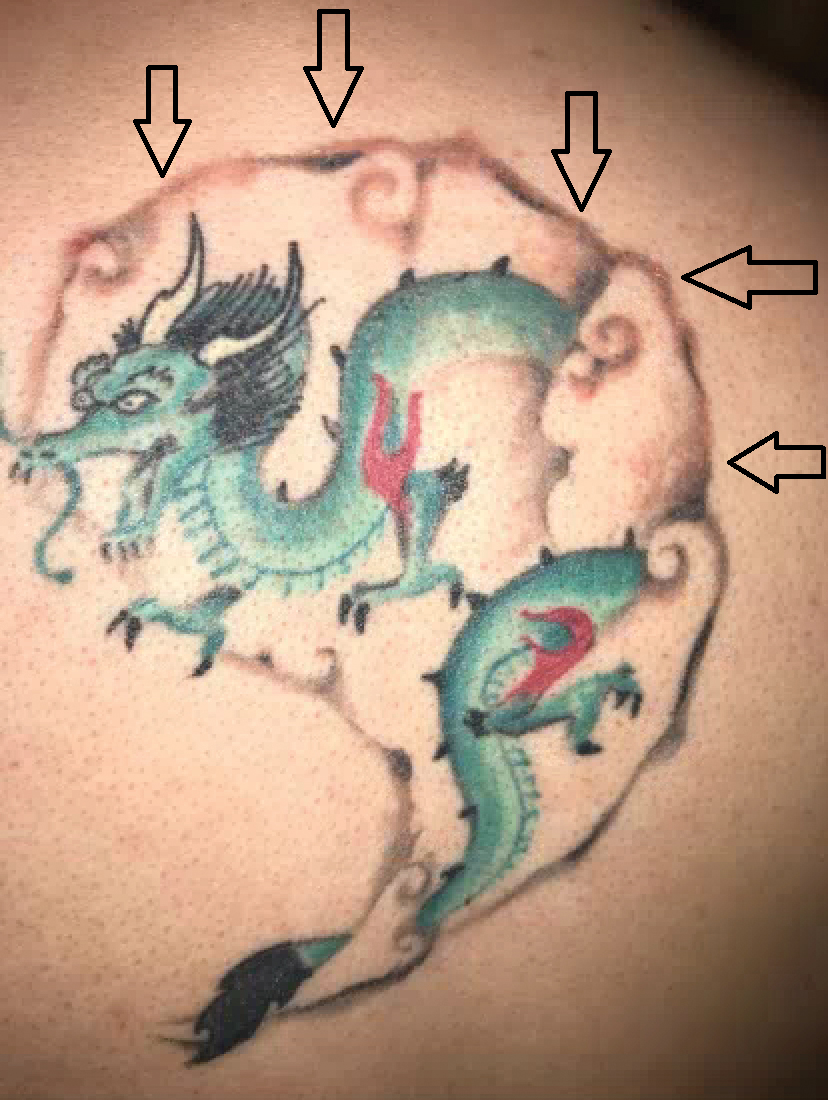
A 29-year-old man presented with increased redness, dryness, and pruritus at the periphery of a tattoo (arrows) on the upper back of 4 months' duration. He was diagnosed with human immunodeficiency virus 8 months prior to presentation and had a history of cystic fibrosis, eczema, and genital molluscum contagiosum. Laboratory analysis 1 month prior revealed a CD4 count of 42 cells/mm3 (reference range, 500-1200 cells/mm3), and the viral load was 2388 copies/mL (reference range, 20-10,000,000 copies/mL). Physical examination revealed multiple erythematous, eczematous, linear plaques along the dark gray lines of the tattoo. A 1.1.2 ×0.7.2 ×0.1-cm shave biopsy specimen was obtained. After the biopsy, tretinoin cream 0.1% and betamethasone dipropionate ointment 0.05% were prescribed to be alternately applied on the tattoo lesions until resolution.
Severe Gingival Swelling and Erythema
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
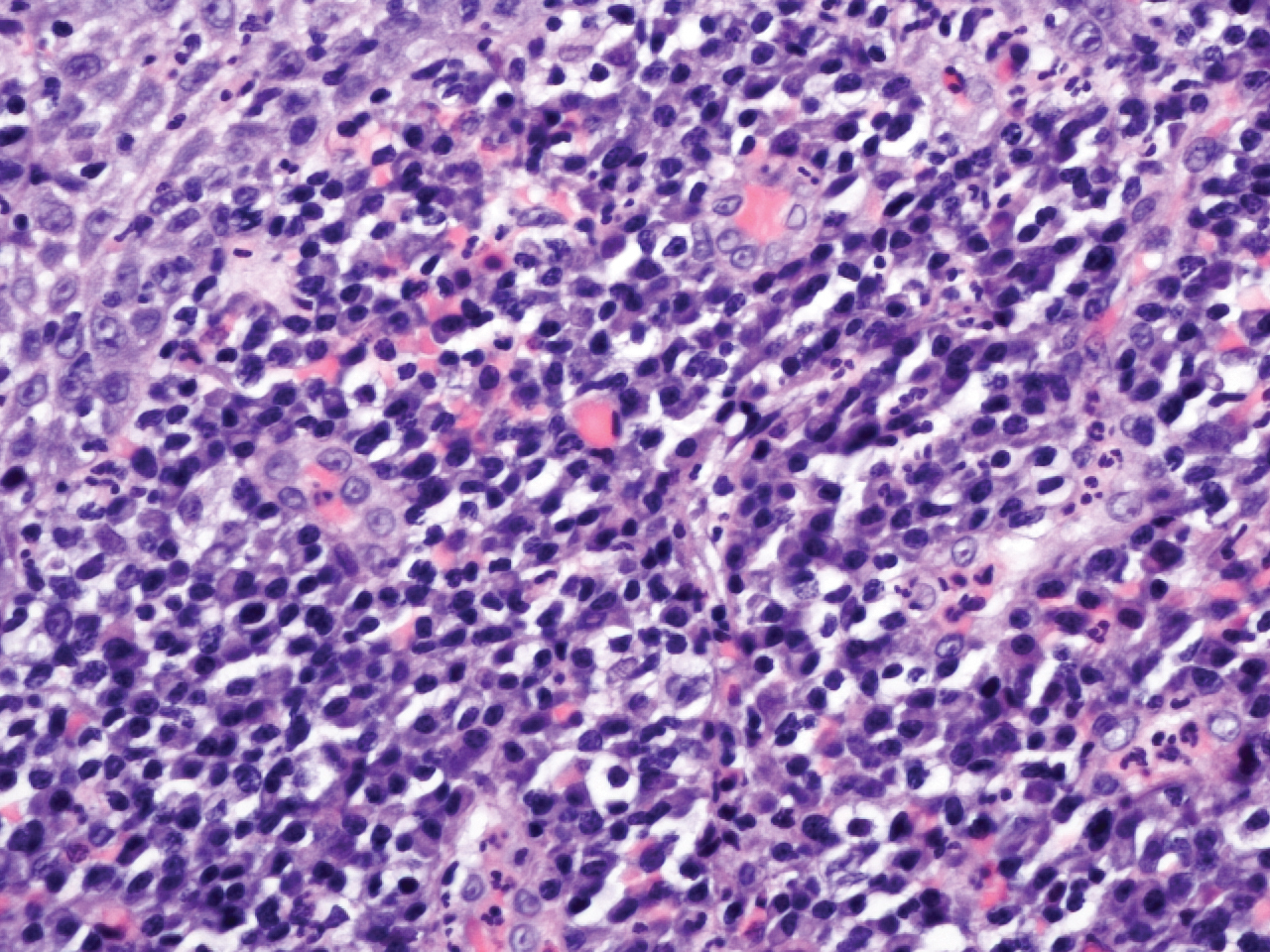
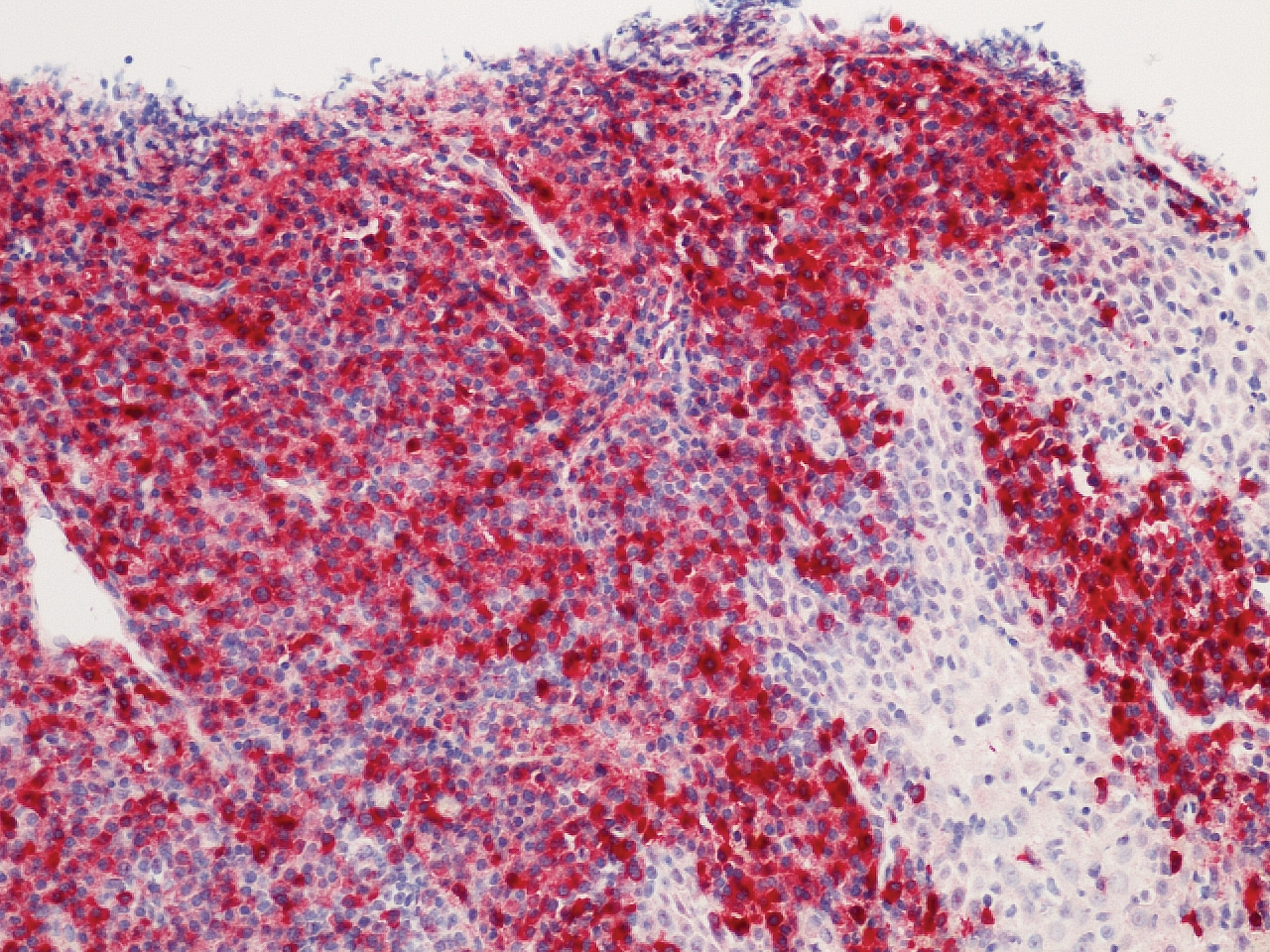
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
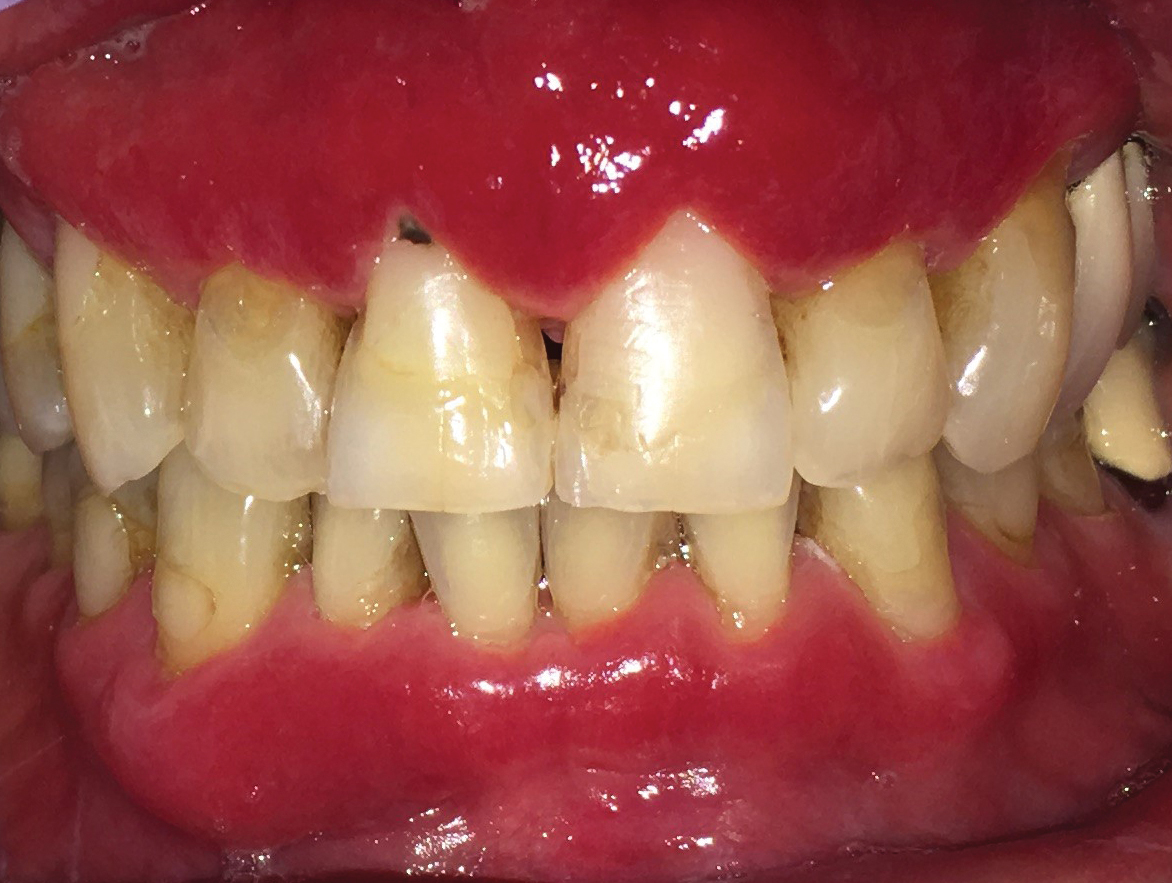
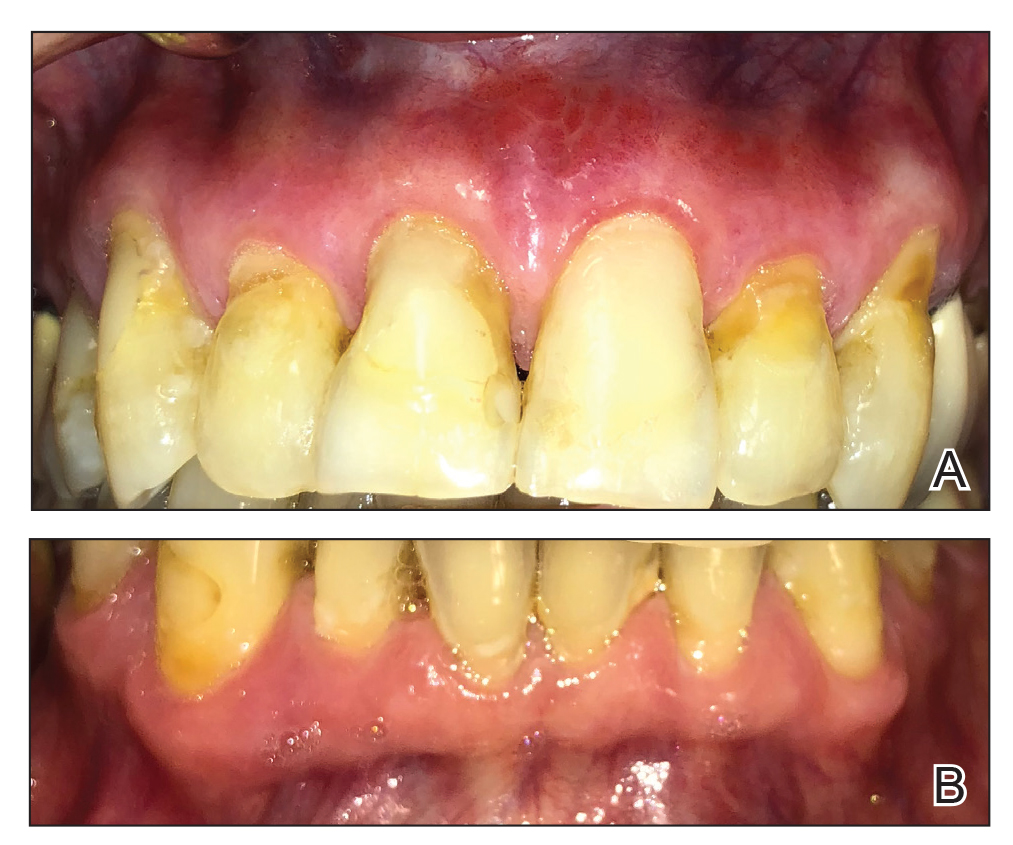
A 62-year-old man presented to an oral medicine specialist with gingival inflammation of at least 1 year's duration. He reported mild discomfort when consuming spicy foods and denied associated extraoral lesions. His medical history revealed hypertension, hypothyroidism, and psoriasis. Medications included lisinopril 10 mg and levothyroxine 100 µg daily. No known drug allergies were reported. His family and social history were noncontributory, and a detailed review of systems was unremarkable. Extraoral examination revealed no lymphadenopathy, salivary gland enlargement, or thyromegaly. Intraoral examination revealed diffuse enlargement of the maxillary and mandibular gingiva accompanied by severe erythema and bleeding on provocation. A 3-mm punch biopsy of the gingiva was performed for routine analysis and direct immunofluorescence.
Petechial Rash on the Thighs in an Immunosuppressed Patient
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
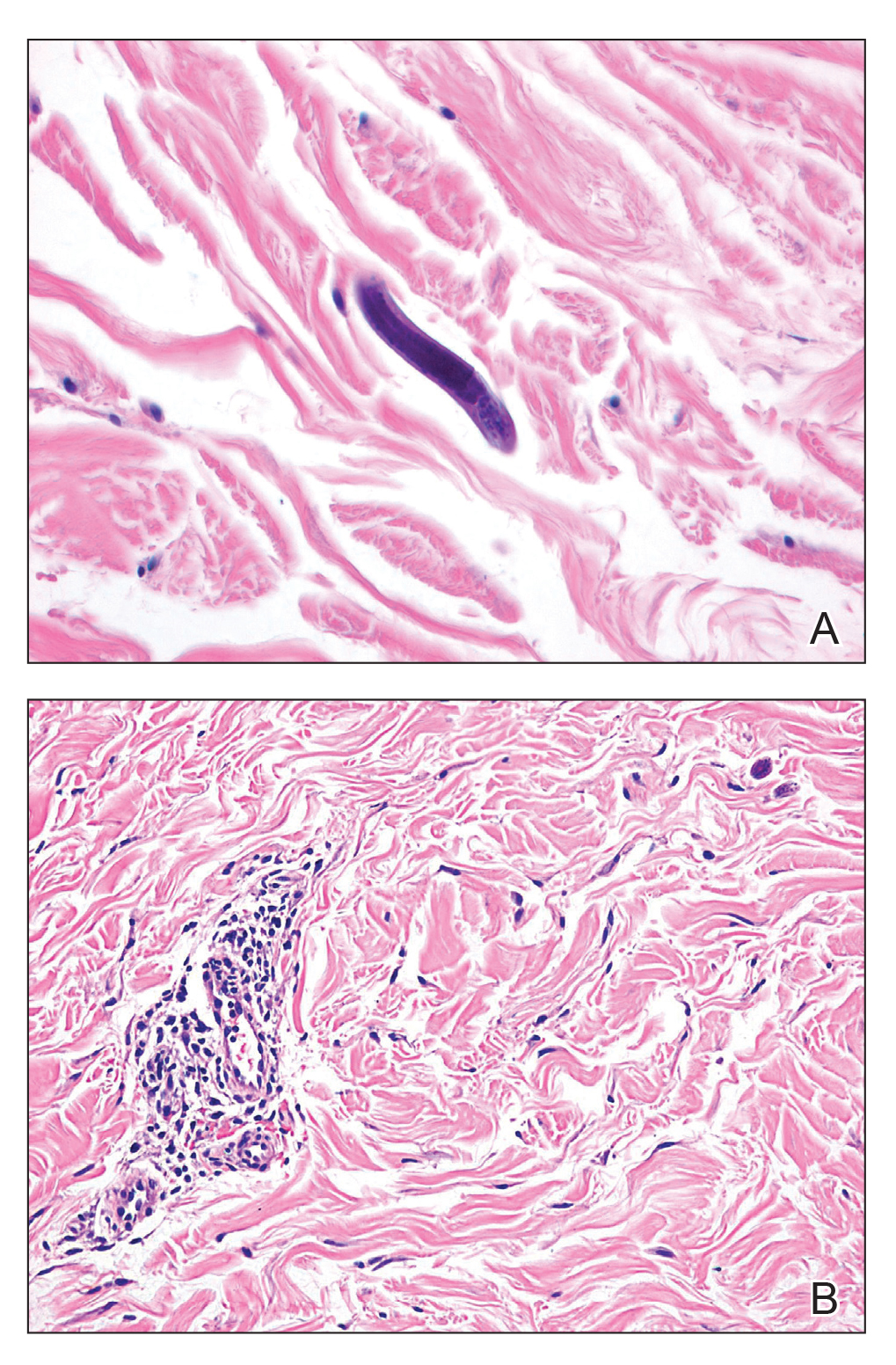
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
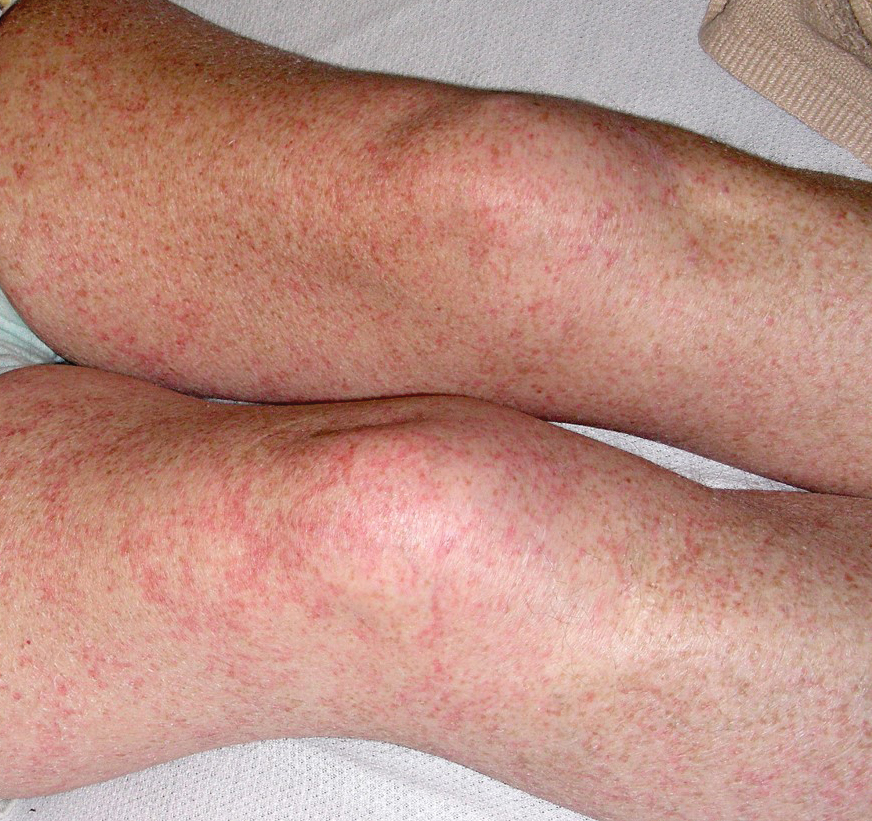
A 48-year-old woman from rural Virginia presented with centrifugally spreading, pruritic, blanchable macules over the lower abdomen and upper thighs noted 4 months after a pancreas transplant. After 3 weeks, the macules coalesced into reticulated nonblanching petechial patches. Fever, dyspnea, increasing xerosis, abdominal pain, and constipation were present. The patient had a medical history of type 1 diabetes mellitus requiring a pancreas transplant. Initial skin biopsy and fluorescence in situ hybridization to test for immune reaction to the XY-donor pancreas were negative. Mild transient eosinophilia was present at admission.
Asymptomatic Transient Lingual Hyperpigmentation
The Diagnosis: Pseudo-Black Hairy Tongue
Pseudo-black hairy tongue is a benign and painless disorder characterized by transient hyperpigmentation of the tongue with a substance that can be easily scraped off. In this case, the patient's lingual discoloration was secondary to the ingestion of bismuth salicylate. The phenomenon is thought to occur due to a reaction between bismuth and sulfur-containing compounds in the saliva, resulting in the characteristic black substance on the surface of the tongue that nestles between the lingual papillae.1 An associated feature may include black stools. Other etiologic factors involved in pseudo-black hairy tongue include food coloring, tobacco, and other drugs such antibiotics and antidepressants.2
The differential diagnosis of lingual hyperpigmentation includes lingua villosa nigra (also known as black hairy tongue), pigmented fungiform papillae of the tongue, acanthosis nigricans, and oral hairy leukoplakia. Lingua villosa nigra is a similar condition in which individuals present with a black tongue; however, the tongue also appears hairy. The tongue may appear as other colors such as brown, yellow, or green. Patients additionally may have symptoms of burning, dysgeusia, halitosis, or gagging. Poor oral hygiene, xerostomia, use of tobacco or alcohol, and different medications including antibiotics and antipsychotic medications increase the risk for developing lingua villosa nigra.2,3 This condition is distinguished from pseudo-black hairy tongue by proliferation and elongation of the filiform papillae.3 Pigmented fungiform papillae of the tongue is a normal variant of tongue morphology, is more common in individuals with darker skin types, and primarily affects the lateral aspect and apex of the tongue.4 Acanthosis nigricans can appear in the oral cavity as multiple pigmented papillary lesions on the dorsal and lateral regions of the tongue and frequently involves the lips; this condition may be associated with metabolic disorders or underlying malignancy.2,3 Oral hairy leukoplakia is caused by Epstein-Barr virus infection and typically presents as white plaques on the dorsal and ventral surfaces of the tongue; this condition largely is found in immunocompromised patients.5
In our patient there was an acute onset of tongue discoloration associated with ingestion of bismuth salicylate, no hypertrophy or lengthening of the lingual papillae, and no involvement of the patient's lips, which was consistent with the diagnosis of pseudo-black hairy tongue. Pseudo-black hairy tongue is transient and treated by discontinuation of offending agents and proper hygiene practices.
- Bradley B, Singleton M, Lin Wan Po A. Bismuth toxicity--a reassessment. J Clin Pharm Ther. 1989;14:423-441.
- Gurvits GE, Tan A. Black hairy tongue syndrome. World J Gastroenterol. 2014;20:10845-10850.
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569.
- Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
- Husak R, Garbe C, Orfanos CE. Oral hairy leukoplakia in 71 HIV-seropositive patients: clinical symptoms, relation to immunologic status, and prognostic significance. J Am Acad Dermatol. 1996;35:928-934.
The Diagnosis: Pseudo-Black Hairy Tongue
Pseudo-black hairy tongue is a benign and painless disorder characterized by transient hyperpigmentation of the tongue with a substance that can be easily scraped off. In this case, the patient's lingual discoloration was secondary to the ingestion of bismuth salicylate. The phenomenon is thought to occur due to a reaction between bismuth and sulfur-containing compounds in the saliva, resulting in the characteristic black substance on the surface of the tongue that nestles between the lingual papillae.1 An associated feature may include black stools. Other etiologic factors involved in pseudo-black hairy tongue include food coloring, tobacco, and other drugs such antibiotics and antidepressants.2
The differential diagnosis of lingual hyperpigmentation includes lingua villosa nigra (also known as black hairy tongue), pigmented fungiform papillae of the tongue, acanthosis nigricans, and oral hairy leukoplakia. Lingua villosa nigra is a similar condition in which individuals present with a black tongue; however, the tongue also appears hairy. The tongue may appear as other colors such as brown, yellow, or green. Patients additionally may have symptoms of burning, dysgeusia, halitosis, or gagging. Poor oral hygiene, xerostomia, use of tobacco or alcohol, and different medications including antibiotics and antipsychotic medications increase the risk for developing lingua villosa nigra.2,3 This condition is distinguished from pseudo-black hairy tongue by proliferation and elongation of the filiform papillae.3 Pigmented fungiform papillae of the tongue is a normal variant of tongue morphology, is more common in individuals with darker skin types, and primarily affects the lateral aspect and apex of the tongue.4 Acanthosis nigricans can appear in the oral cavity as multiple pigmented papillary lesions on the dorsal and lateral regions of the tongue and frequently involves the lips; this condition may be associated with metabolic disorders or underlying malignancy.2,3 Oral hairy leukoplakia is caused by Epstein-Barr virus infection and typically presents as white plaques on the dorsal and ventral surfaces of the tongue; this condition largely is found in immunocompromised patients.5
In our patient there was an acute onset of tongue discoloration associated with ingestion of bismuth salicylate, no hypertrophy or lengthening of the lingual papillae, and no involvement of the patient's lips, which was consistent with the diagnosis of pseudo-black hairy tongue. Pseudo-black hairy tongue is transient and treated by discontinuation of offending agents and proper hygiene practices.
The Diagnosis: Pseudo-Black Hairy Tongue
Pseudo-black hairy tongue is a benign and painless disorder characterized by transient hyperpigmentation of the tongue with a substance that can be easily scraped off. In this case, the patient's lingual discoloration was secondary to the ingestion of bismuth salicylate. The phenomenon is thought to occur due to a reaction between bismuth and sulfur-containing compounds in the saliva, resulting in the characteristic black substance on the surface of the tongue that nestles between the lingual papillae.1 An associated feature may include black stools. Other etiologic factors involved in pseudo-black hairy tongue include food coloring, tobacco, and other drugs such antibiotics and antidepressants.2
The differential diagnosis of lingual hyperpigmentation includes lingua villosa nigra (also known as black hairy tongue), pigmented fungiform papillae of the tongue, acanthosis nigricans, and oral hairy leukoplakia. Lingua villosa nigra is a similar condition in which individuals present with a black tongue; however, the tongue also appears hairy. The tongue may appear as other colors such as brown, yellow, or green. Patients additionally may have symptoms of burning, dysgeusia, halitosis, or gagging. Poor oral hygiene, xerostomia, use of tobacco or alcohol, and different medications including antibiotics and antipsychotic medications increase the risk for developing lingua villosa nigra.2,3 This condition is distinguished from pseudo-black hairy tongue by proliferation and elongation of the filiform papillae.3 Pigmented fungiform papillae of the tongue is a normal variant of tongue morphology, is more common in individuals with darker skin types, and primarily affects the lateral aspect and apex of the tongue.4 Acanthosis nigricans can appear in the oral cavity as multiple pigmented papillary lesions on the dorsal and lateral regions of the tongue and frequently involves the lips; this condition may be associated with metabolic disorders or underlying malignancy.2,3 Oral hairy leukoplakia is caused by Epstein-Barr virus infection and typically presents as white plaques on the dorsal and ventral surfaces of the tongue; this condition largely is found in immunocompromised patients.5
In our patient there was an acute onset of tongue discoloration associated with ingestion of bismuth salicylate, no hypertrophy or lengthening of the lingual papillae, and no involvement of the patient's lips, which was consistent with the diagnosis of pseudo-black hairy tongue. Pseudo-black hairy tongue is transient and treated by discontinuation of offending agents and proper hygiene practices.
- Bradley B, Singleton M, Lin Wan Po A. Bismuth toxicity--a reassessment. J Clin Pharm Ther. 1989;14:423-441.
- Gurvits GE, Tan A. Black hairy tongue syndrome. World J Gastroenterol. 2014;20:10845-10850.
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569.
- Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
- Husak R, Garbe C, Orfanos CE. Oral hairy leukoplakia in 71 HIV-seropositive patients: clinical symptoms, relation to immunologic status, and prognostic significance. J Am Acad Dermatol. 1996;35:928-934.
- Bradley B, Singleton M, Lin Wan Po A. Bismuth toxicity--a reassessment. J Clin Pharm Ther. 1989;14:423-441.
- Gurvits GE, Tan A. Black hairy tongue syndrome. World J Gastroenterol. 2014;20:10845-10850.
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569.
- Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
- Husak R, Garbe C, Orfanos CE. Oral hairy leukoplakia in 71 HIV-seropositive patients: clinical symptoms, relation to immunologic status, and prognostic significance. J Am Acad Dermatol. 1996;35:928-934.
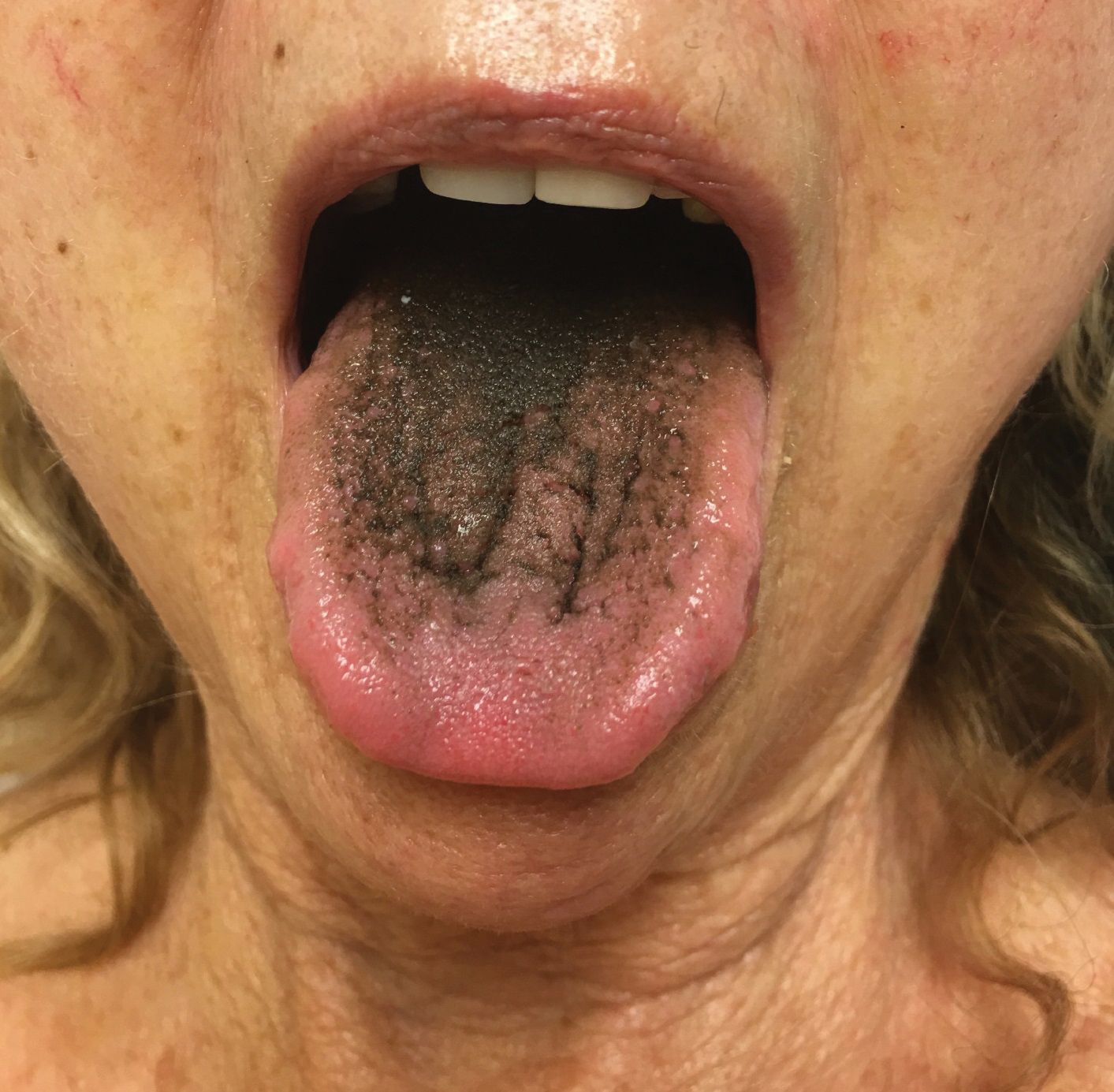
A 77-year-old woman incidentally was noted to have black discoloration of the tongue during a routine dermatologic examination. The patient was unaware of the tongue discoloration and reported that her tongue appeared normal the day prior. The tongue was asymptomatic. Clinical examination revealed black hyperpigmentation on the dorsal aspect of the tongue without appreciable hypertrophy or hyperkeratosis of the filiform papillae. The patient had a half-pack daily smoking habit for many years but had abstained from any smoking or tobacco use for the last 15 years. The patient endorsed good oral hygiene. Upon further questioning, the patient revealed that she had ingested 1 tablet of bismuth salicylate the prior night to relieve postprandial dyspepsia. A cotton-tipped applicator was rubbed gently against the affected area and removed some of the black pigment.
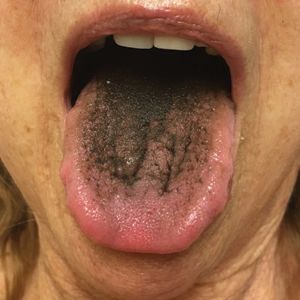
Painful Indurated Plaque on the Groin
The Diagnosis: Cutaneous Metastasis
Histopathology demonstrated ulceration of the epidermis with necrosis of the papillary dermis. There was a diffuse infiltration of pleomorphic and atypical epithelioid cells in the reticular dermis (Figure). Focally there was ductal and glandular differentiation. The stroma was sclerotic. At the deep aspect of the biopsy specimen, tumor cells intercalated between collagen bundles in linear strands. Atypical mitoses were common, and necrosis en masse was seen. An immunohistochemical panel also was performed. Tissue from the biopsy was strongly positive for CDX-2 and cytokeratin 20 and diffusely negative for cytokeratin 7, gross cystic disease fluid protein 15, and prostate-specific antigen. The other biopsy was sent for cultures and grew no organisms, which confirmed the diagnosis of cutaneous metastasis from the patient's primary colonic adenocarcinoma. Due to the poor prognosis and his overall poor health, our patient opted for palliative care.
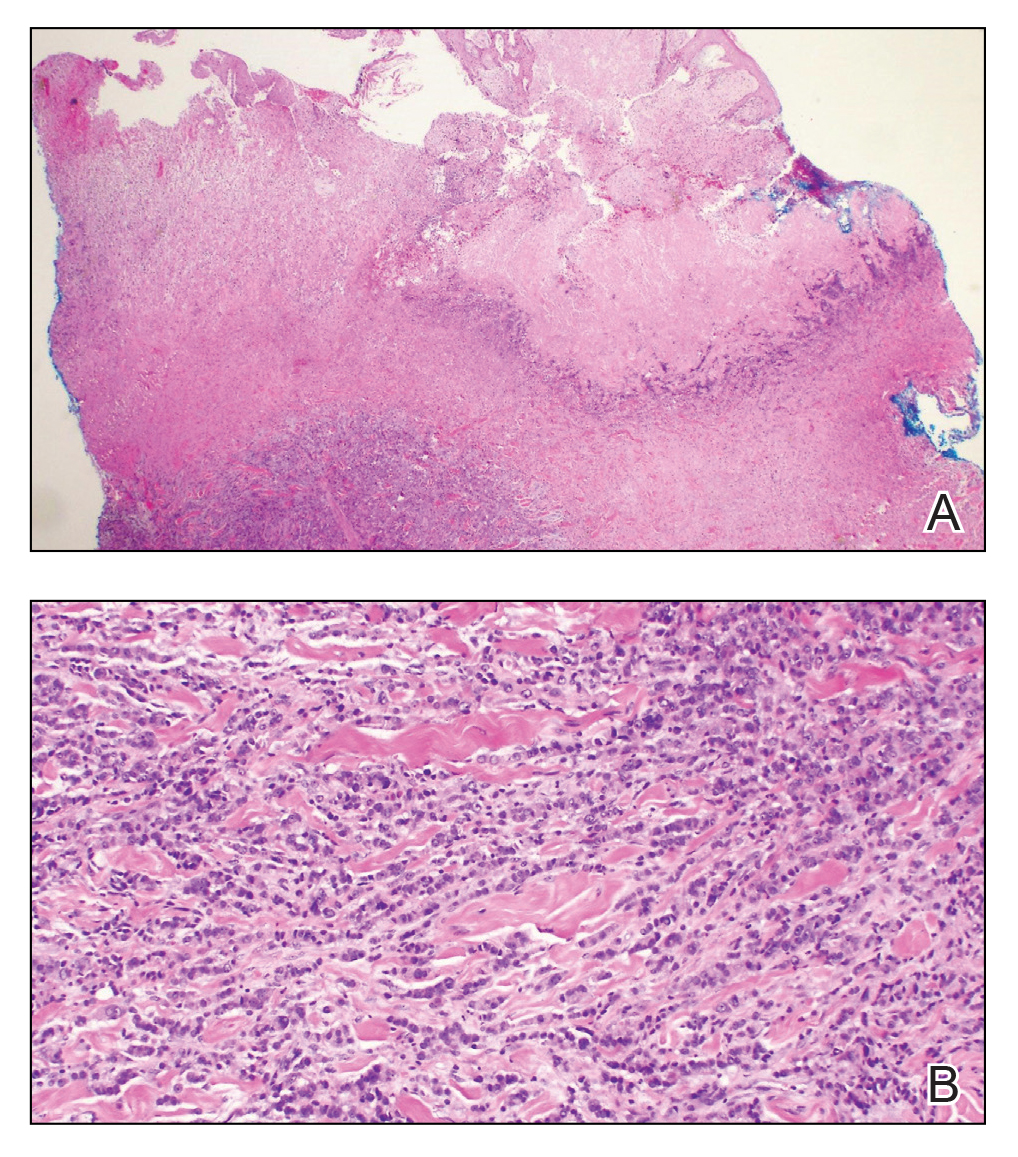
Based on large retrospective studies, the frequency of cutaneous metastasis for patients diagnosed with any malignancy is 0.7% to 9.0%.1-4 The third most common malignancy in both sexes is colorectal cancer, affecting approximately 5% of the US population.3 The frequency of cutaneous metastases from colorectal cancer is 0.81% to 3.9%.1,2,4,5 Generally, cutaneous metastases present within 2 to 3 years from diagnosis of primary malignancy.6,7 The most common sites for cutaneous metastases in a patient with colorectal cancer are the abdomen and pelvic region, often at surgical sites.1-4,6-9
The clinical presentation of cutaneous metastases varies greatly, and as a result, they commonly are misdiagnosed.6,7 Although treatment with many antibiotics and antifungals had failed in our patient, the examination still was concerning for a possible granulomatous infection vs malignancy. With the history of colon cancer, radiation treatment, and chemotherapy, the possible malignancy diagnoses included primary skin cancers, viral tumors, and cutaneous metastasis. The initial evaluations had focused on infectious causes and resulted in 6 weeks of misdiagnosis and inappropriate therapy. Despite cutaneous metastases being uncommon, there should be a high index of suspicion for lesions in patients who have a history of cancer, especially if the lesion does not respond to treatment.2,6,7
Physical examination in our patient showed a high tumor burden as well as evidence of carcinoma erysipeloides on the lower abdomen and thighs, in addition to carcinoma en cuirasse throughout the pubic region. Carcinoma erysipeloides was first described in 1893 in a patient with breast cancer: "The erythematous infiltration of the skin was very superficial, and was attended simply by redness with a slight degree of induration. Until touched by the finger the condition might easily have been taken for a slightly-marked form of erysipelas."10 The clinical findings are a result of lymphatic and vascular obstruction.3,9 The breast is the most common location to find carcinoma erysipeloides.3 It is an unusual occurrence to find it on the abdomen from colonic adenocarcinoma. The term cancer en cuirasse was coined in 1838 to describe the cutaneous manifestation of breast cancer that caused the skin to resemble the metal breastplate of a cuirasser.4 Similar to carcinoma erysipeloides, carcinoma en cuirasse most commonly is found as cutaneous metastasis from breast cancer, not from colonic adenocarcinoma.3
The histologic characteristics of cutaneous metastases in general are similar to the primary malignancy but can be more poorly differentiated.7 Generally, neoplastic cells are seen in the lymphatic and blood vessels, and a large portion of the tumor is confined to the deep dermis and in the subcutaneous fat.3,6 Histologic features of colonic adenocarcinoma metastases can demonstrate a well-differentiated, glandular architecture with mucin-secreting cells.3,8,9 There also is a histologic pattern of neoplastic cells arranging themselves between collagen bundles in linear strands; this finding more commonly is seen in adenocarcinoma of the breast but also was seen in our patient.3,9 With immunohistochemical staining, a truncated panel of cytokeratin 7, cytokeratin 20, and S-100 had a diagnostic accuracy of 100% for cutaneous metastases from colonic adenocarcinoma in one study. The pattern of all colonic adenocarcinomas was cytokeratin 20 positive and cytokeratin 7 and S-100 negative.6
Cutaneous metastases typically demonstrate widespread and rapidly progressive disease.3,9 Survival studies of cutaneous metastases showed that 48% to 66% of patients died within the first 6 months.3,6 Specifically, cutaneous metastases from colorectal cancers showed a median survival of 3 to 5 months.6,7 Currently there are no treatment guidelines for cutaneous metastases.
- Lookingbill DP, Spangler N, Helm K. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Gul U, Kilic A, Gonul M, et al. Spectrum of cutaneous metastases in 1287 cases of internal malignancies: a study from Turkey. Acta Derm Venereol. 2007;87:160-162.
- Hussein MR. Skin metastasis: a pathologist's perspective. J Cutan Pathol. 2010;37:E1-E20.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182; quiz 183-186.
- Hu S, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Saeed S, Keehn C, Morgan M. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Sariya D, Ruth K, Adams-McDonnell R. Clinicopathologic correlation of cutaneous metastases: experience of a cancer center. Arch Dermatol. 2007;143:613-620.
- Brownstein M, Helwig E. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
- McKee PH. Cutaneous metastases. J Cutan Pathol. 1985;12:239-250.
- Hutchinson J. Notes from congresses and continental hospitals: erythema-scirrhus of the skin in association with cancer of the breast. Arch Surg (London). 1893;4:220-222
The Diagnosis: Cutaneous Metastasis
Histopathology demonstrated ulceration of the epidermis with necrosis of the papillary dermis. There was a diffuse infiltration of pleomorphic and atypical epithelioid cells in the reticular dermis (Figure). Focally there was ductal and glandular differentiation. The stroma was sclerotic. At the deep aspect of the biopsy specimen, tumor cells intercalated between collagen bundles in linear strands. Atypical mitoses were common, and necrosis en masse was seen. An immunohistochemical panel also was performed. Tissue from the biopsy was strongly positive for CDX-2 and cytokeratin 20 and diffusely negative for cytokeratin 7, gross cystic disease fluid protein 15, and prostate-specific antigen. The other biopsy was sent for cultures and grew no organisms, which confirmed the diagnosis of cutaneous metastasis from the patient's primary colonic adenocarcinoma. Due to the poor prognosis and his overall poor health, our patient opted for palliative care.
Based on large retrospective studies, the frequency of cutaneous metastasis for patients diagnosed with any malignancy is 0.7% to 9.0%.1-4 The third most common malignancy in both sexes is colorectal cancer, affecting approximately 5% of the US population.3 The frequency of cutaneous metastases from colorectal cancer is 0.81% to 3.9%.1,2,4,5 Generally, cutaneous metastases present within 2 to 3 years from diagnosis of primary malignancy.6,7 The most common sites for cutaneous metastases in a patient with colorectal cancer are the abdomen and pelvic region, often at surgical sites.1-4,6-9
The clinical presentation of cutaneous metastases varies greatly, and as a result, they commonly are misdiagnosed.6,7 Although treatment with many antibiotics and antifungals had failed in our patient, the examination still was concerning for a possible granulomatous infection vs malignancy. With the history of colon cancer, radiation treatment, and chemotherapy, the possible malignancy diagnoses included primary skin cancers, viral tumors, and cutaneous metastasis. The initial evaluations had focused on infectious causes and resulted in 6 weeks of misdiagnosis and inappropriate therapy. Despite cutaneous metastases being uncommon, there should be a high index of suspicion for lesions in patients who have a history of cancer, especially if the lesion does not respond to treatment.2,6,7
Physical examination in our patient showed a high tumor burden as well as evidence of carcinoma erysipeloides on the lower abdomen and thighs, in addition to carcinoma en cuirasse throughout the pubic region. Carcinoma erysipeloides was first described in 1893 in a patient with breast cancer: "The erythematous infiltration of the skin was very superficial, and was attended simply by redness with a slight degree of induration. Until touched by the finger the condition might easily have been taken for a slightly-marked form of erysipelas."10 The clinical findings are a result of lymphatic and vascular obstruction.3,9 The breast is the most common location to find carcinoma erysipeloides.3 It is an unusual occurrence to find it on the abdomen from colonic adenocarcinoma. The term cancer en cuirasse was coined in 1838 to describe the cutaneous manifestation of breast cancer that caused the skin to resemble the metal breastplate of a cuirasser.4 Similar to carcinoma erysipeloides, carcinoma en cuirasse most commonly is found as cutaneous metastasis from breast cancer, not from colonic adenocarcinoma.3
The histologic characteristics of cutaneous metastases in general are similar to the primary malignancy but can be more poorly differentiated.7 Generally, neoplastic cells are seen in the lymphatic and blood vessels, and a large portion of the tumor is confined to the deep dermis and in the subcutaneous fat.3,6 Histologic features of colonic adenocarcinoma metastases can demonstrate a well-differentiated, glandular architecture with mucin-secreting cells.3,8,9 There also is a histologic pattern of neoplastic cells arranging themselves between collagen bundles in linear strands; this finding more commonly is seen in adenocarcinoma of the breast but also was seen in our patient.3,9 With immunohistochemical staining, a truncated panel of cytokeratin 7, cytokeratin 20, and S-100 had a diagnostic accuracy of 100% for cutaneous metastases from colonic adenocarcinoma in one study. The pattern of all colonic adenocarcinomas was cytokeratin 20 positive and cytokeratin 7 and S-100 negative.6
Cutaneous metastases typically demonstrate widespread and rapidly progressive disease.3,9 Survival studies of cutaneous metastases showed that 48% to 66% of patients died within the first 6 months.3,6 Specifically, cutaneous metastases from colorectal cancers showed a median survival of 3 to 5 months.6,7 Currently there are no treatment guidelines for cutaneous metastases.
The Diagnosis: Cutaneous Metastasis
Histopathology demonstrated ulceration of the epidermis with necrosis of the papillary dermis. There was a diffuse infiltration of pleomorphic and atypical epithelioid cells in the reticular dermis (Figure). Focally there was ductal and glandular differentiation. The stroma was sclerotic. At the deep aspect of the biopsy specimen, tumor cells intercalated between collagen bundles in linear strands. Atypical mitoses were common, and necrosis en masse was seen. An immunohistochemical panel also was performed. Tissue from the biopsy was strongly positive for CDX-2 and cytokeratin 20 and diffusely negative for cytokeratin 7, gross cystic disease fluid protein 15, and prostate-specific antigen. The other biopsy was sent for cultures and grew no organisms, which confirmed the diagnosis of cutaneous metastasis from the patient's primary colonic adenocarcinoma. Due to the poor prognosis and his overall poor health, our patient opted for palliative care.
Based on large retrospective studies, the frequency of cutaneous metastasis for patients diagnosed with any malignancy is 0.7% to 9.0%.1-4 The third most common malignancy in both sexes is colorectal cancer, affecting approximately 5% of the US population.3 The frequency of cutaneous metastases from colorectal cancer is 0.81% to 3.9%.1,2,4,5 Generally, cutaneous metastases present within 2 to 3 years from diagnosis of primary malignancy.6,7 The most common sites for cutaneous metastases in a patient with colorectal cancer are the abdomen and pelvic region, often at surgical sites.1-4,6-9
The clinical presentation of cutaneous metastases varies greatly, and as a result, they commonly are misdiagnosed.6,7 Although treatment with many antibiotics and antifungals had failed in our patient, the examination still was concerning for a possible granulomatous infection vs malignancy. With the history of colon cancer, radiation treatment, and chemotherapy, the possible malignancy diagnoses included primary skin cancers, viral tumors, and cutaneous metastasis. The initial evaluations had focused on infectious causes and resulted in 6 weeks of misdiagnosis and inappropriate therapy. Despite cutaneous metastases being uncommon, there should be a high index of suspicion for lesions in patients who have a history of cancer, especially if the lesion does not respond to treatment.2,6,7
Physical examination in our patient showed a high tumor burden as well as evidence of carcinoma erysipeloides on the lower abdomen and thighs, in addition to carcinoma en cuirasse throughout the pubic region. Carcinoma erysipeloides was first described in 1893 in a patient with breast cancer: "The erythematous infiltration of the skin was very superficial, and was attended simply by redness with a slight degree of induration. Until touched by the finger the condition might easily have been taken for a slightly-marked form of erysipelas."10 The clinical findings are a result of lymphatic and vascular obstruction.3,9 The breast is the most common location to find carcinoma erysipeloides.3 It is an unusual occurrence to find it on the abdomen from colonic adenocarcinoma. The term cancer en cuirasse was coined in 1838 to describe the cutaneous manifestation of breast cancer that caused the skin to resemble the metal breastplate of a cuirasser.4 Similar to carcinoma erysipeloides, carcinoma en cuirasse most commonly is found as cutaneous metastasis from breast cancer, not from colonic adenocarcinoma.3
The histologic characteristics of cutaneous metastases in general are similar to the primary malignancy but can be more poorly differentiated.7 Generally, neoplastic cells are seen in the lymphatic and blood vessels, and a large portion of the tumor is confined to the deep dermis and in the subcutaneous fat.3,6 Histologic features of colonic adenocarcinoma metastases can demonstrate a well-differentiated, glandular architecture with mucin-secreting cells.3,8,9 There also is a histologic pattern of neoplastic cells arranging themselves between collagen bundles in linear strands; this finding more commonly is seen in adenocarcinoma of the breast but also was seen in our patient.3,9 With immunohistochemical staining, a truncated panel of cytokeratin 7, cytokeratin 20, and S-100 had a diagnostic accuracy of 100% for cutaneous metastases from colonic adenocarcinoma in one study. The pattern of all colonic adenocarcinomas was cytokeratin 20 positive and cytokeratin 7 and S-100 negative.6
Cutaneous metastases typically demonstrate widespread and rapidly progressive disease.3,9 Survival studies of cutaneous metastases showed that 48% to 66% of patients died within the first 6 months.3,6 Specifically, cutaneous metastases from colorectal cancers showed a median survival of 3 to 5 months.6,7 Currently there are no treatment guidelines for cutaneous metastases.
- Lookingbill DP, Spangler N, Helm K. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Gul U, Kilic A, Gonul M, et al. Spectrum of cutaneous metastases in 1287 cases of internal malignancies: a study from Turkey. Acta Derm Venereol. 2007;87:160-162.
- Hussein MR. Skin metastasis: a pathologist's perspective. J Cutan Pathol. 2010;37:E1-E20.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182; quiz 183-186.
- Hu S, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Saeed S, Keehn C, Morgan M. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Sariya D, Ruth K, Adams-McDonnell R. Clinicopathologic correlation of cutaneous metastases: experience of a cancer center. Arch Dermatol. 2007;143:613-620.
- Brownstein M, Helwig E. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
- McKee PH. Cutaneous metastases. J Cutan Pathol. 1985;12:239-250.
- Hutchinson J. Notes from congresses and continental hospitals: erythema-scirrhus of the skin in association with cancer of the breast. Arch Surg (London). 1893;4:220-222
- Lookingbill DP, Spangler N, Helm K. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Gul U, Kilic A, Gonul M, et al. Spectrum of cutaneous metastases in 1287 cases of internal malignancies: a study from Turkey. Acta Derm Venereol. 2007;87:160-162.
- Hussein MR. Skin metastasis: a pathologist's perspective. J Cutan Pathol. 2010;37:E1-E20.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182; quiz 183-186.
- Hu S, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Saeed S, Keehn C, Morgan M. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Sariya D, Ruth K, Adams-McDonnell R. Clinicopathologic correlation of cutaneous metastases: experience of a cancer center. Arch Dermatol. 2007;143:613-620.
- Brownstein M, Helwig E. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
- McKee PH. Cutaneous metastases. J Cutan Pathol. 1985;12:239-250.
- Hutchinson J. Notes from congresses and continental hospitals: erythema-scirrhus of the skin in association with cancer of the breast. Arch Surg (London). 1893;4:220-222
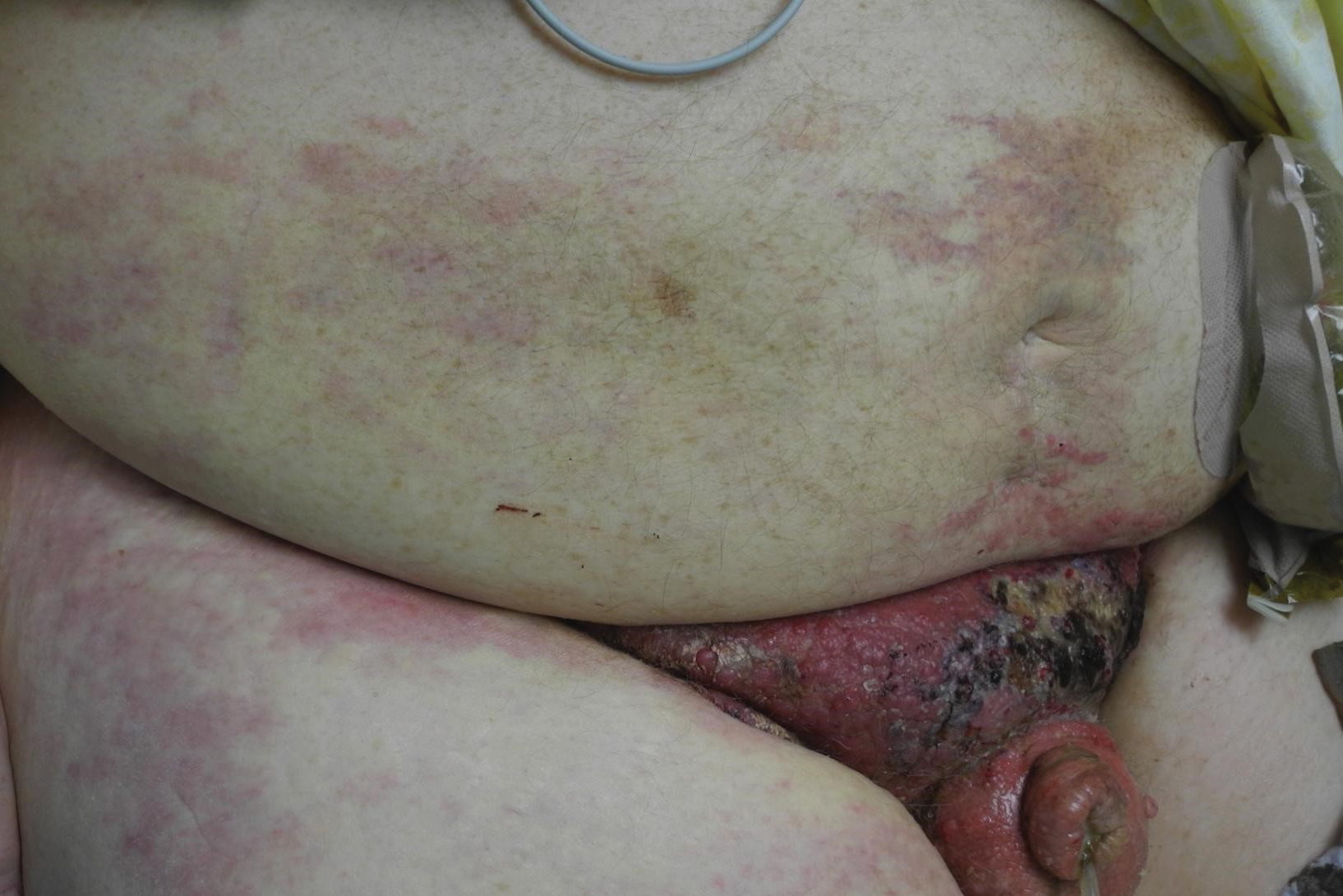
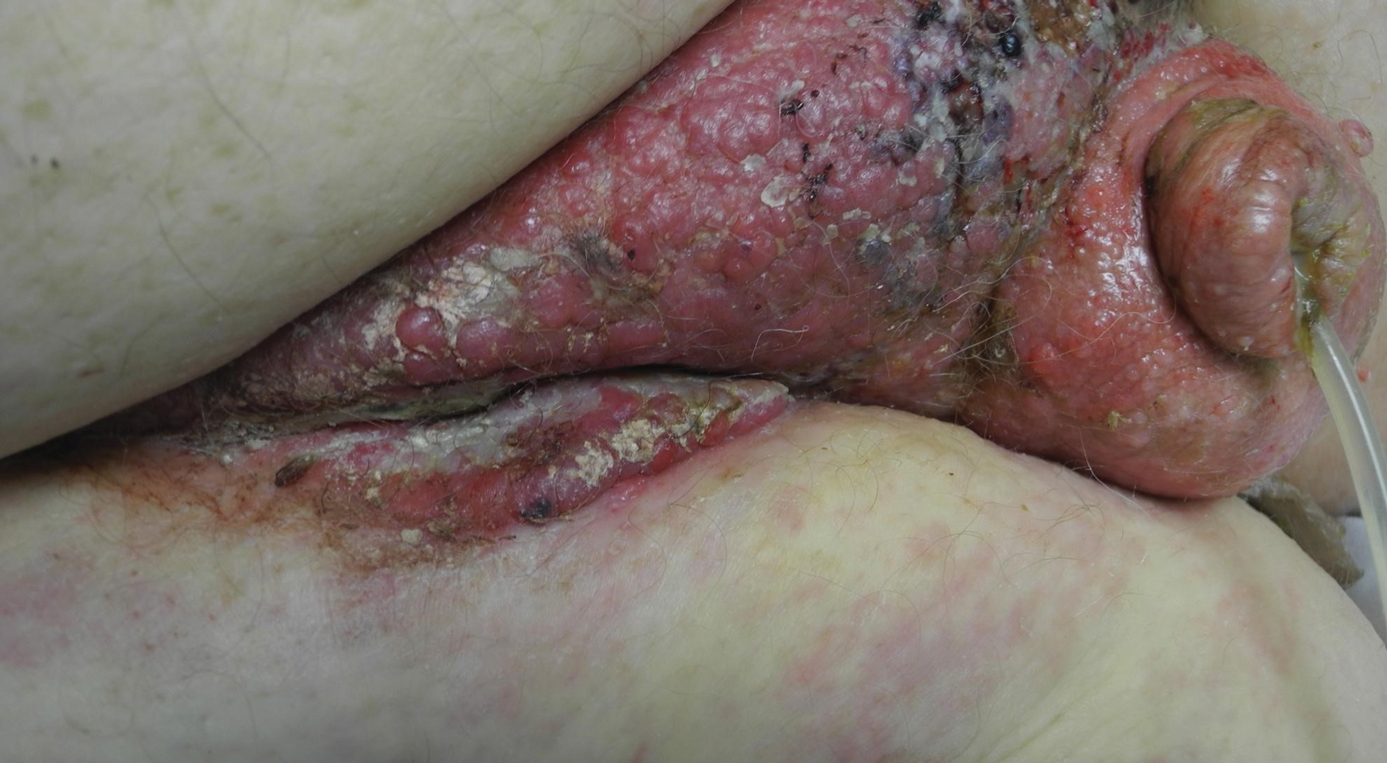
A 67-year-old man presented with a chronic lesion on the groin of 6 weeks' duration. The patient had a history of type 2 diabetes mellitus and colonic adenocarcinoma diagnosed 4 years prior that was treated with a colectomy, radiation therapy, and chemotherapy. Six weeks prior to the current presentation, the patient first sought treatment of swelling, redness, pain, and a bumpy texture on the groin. He was unsuccessfully managed by several physicians including at a long-term care facility where he was admitted and treated for presumed cellulitis. Attempted treatments included a topical antifungal, fluconazole, ciprofloxacin, metronidazole, cefepime, clindamycin, daptomycin, and vancomycin. The affected area continued to worsen along with the patient's overall health. He was transferred to the hospital for more advanced care and was evaluated by inpatient dermatology. Physical examination revealed firm, pink to red-brown, ulcerating papulonodules that coalesced into a large indurated plaque over the pubis, scrotum, penis, and inguinal folds (top). There also were red-violet, indurated plaques on the lower abdomen and bilateral proximal thighs (bottom). Punch biopsies were taken from the indurated area on the left side of the pubis--one for histopathologic evaluation and the other for bacterial, fungal, atypical mycobacterial, and Nocardia tissue cultures.
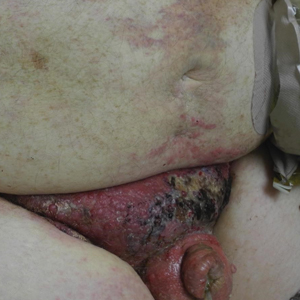
Solitary Papule on the Shoulder
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.

A 64-year-old man presented to dermatology for a full-body skin examination. He had no history of skin cancer. Physical examination revealed an asymptomatic, 4-mm, yellowish pink papule on the left posterior shoulder (top). Dermoscopy revealed yellow globules (bottom). The patient was unsure of the duration of the lesion and denied any prior trauma or medical procedure to the area. Subsequently, a shave biopsy was performed.

Petechiae and Ecchymoses on the Arm
The Diagnosis: Rumpel-Leede Phenomenon
Rumpel-Leede (R-L) phenomenon describes the rare benign occurrence of dermal capillaries acutely rupturing following the administration of a tourniquetlike force on an extremity,1 which manifests as asymptomatic petechiae and ecchymoses on a distal extremity, usually following noninvasive measurement of blood pressure.2 Rumpel-Leede phenomenon represents an underrecognized entity that either is excluded or minimally referenced in most dermatology textbooks. The R-L sign initially was described in the early 1900s after it was observed that tourniquets applied to the arms of patients with scarlet fever would lead to the development of petechiae in that extremity.3 It later was elucidated that underlying vascular disease predisposes to dermal capillary fragility, a risk factor for R-L phenomenon to occur upon application of a tourniquet. Rumpel-Leede phenomenon has been noted in patients with diabetes mellitus, acute or chronic hypertension, and thrombocytopenia.4 In addition to being hypertensive and diabetic, our patient had been taking amlodipine and diltiazem. Calcium channel blockers have been linked to R-L phenomenon in case reports as well as in a study of calcium channel blockers inducing capillary fragility in vivo.5 Rumpel-Leede phenomenon also has been noted in patients with tightly fitting garments and infants carried in baby carriers.1
The differential diagnosis for R-L phenomenon includes actinic purpura, small vessel vasculitis, disseminated intravascular coagulation (DIC), and deep vein thrombosis (DVT). Actinic purpura, also called solar purpura or senile purpura, represents the petechiae and ecchymoses that are associated with aging skin. It is thought to occur when DNA damage, UV-induced solar elastosis, and decreased lipids in the stratum corneum cause a weakened ability to contain red blood cell extravasation from capillaries.6 Due to the lack of history of trauma and clear association with the blood pressure cuff placement in our patient, a diagnosis of actinic purpura was unlikely. In small vessel vasculitis, patients classically present with nonblanching palpable purpura frequently distributed over the lower extremities. The isolation of the lesions to only the left arm lowered the suspicion for vasculitis. Cutaneous manifestations of DIC and other hypercoagulable states may include purpura, livedo reticularis, atrophie blanche, and in extreme cases purpura fulminans. Routine laboratory examination reveals thrombocytopenia, prolonged prothrombin time/partial thromboplastin time, and hemolytic anemia.7 Although our patient had the risk factors of recent infection and surgery, a hemoglobin level of 10.9 g/dL (reference range, 14.0-17.5 g/dL) and platelet count of 279,000/µL (reference range, 150,000-350,000/µL) excluded DIC as the probable diagnosis. Our patient was at an overall increased risk for DVT due to his prolonged hospital stay, increased age, and other factors. Despite these risk factors, the lack of pain or swelling made this diagnosis unlikely. Furthermore, our patient was heparinized throughout his hospital stay, and upper extremity DVT accounts for only 4% to 10% of the total DVT incidence.5
Although R-L phenomenon is a benign, self-limited condition, it may be necessary in some cases to rule out more serious underlying etiologies with investigative workup comprised of a complete blood cell count, coagulation profile, and basic metabolic panel. However, recognition of the R-L phenomenon in the right clinical context of localized petechiae or ecchymoses with a history of a tourniquetlike force may prevent an unnecessary and costly workup. Patients should be encouraged that R-L phenomenon will resolve over time with identification and correction of the tourniquetlike force. In this case, we recommended loosening of the sphygmomanometer cuff and alternating extremities to which the cuff was to be placed, which resulted in complete clearance of the petechiae and ecchymoses within 5 days.
- Nguyen T, Garcia D, Wang A, et al. Rumpel-Leede phenomenon associated with tourniquet-like forces of baby carriers in otherwise healthy infants. JAMA Dermatol. 2016;152:728-730.
- Chester M, Barwise J, Holzman M, et al. Acute dermal capillary rupture associated with noninvasive blood pressure monitoring. J Clin Anesth. 2007;19:473-475.
- Hartley A, Lim PB, Hayat SA. Rumpel-Leede phenomenon in a hypertensive patient due to mechanical trauma: a case report. J Med Case Rep. 2016;10:150.
- Varela D, Tran D, Ngamdu K, et al. Rumpel-Leede phenomenon presenting as a hypertensive urgency. Proc (Bayl Univ Med Cent). 2016;29:200-201.
- Cox NH, Walsh ML, Robson RH. Purpura and bleeding due to calcium-channel blockers: an underestimated problem? case reports and a pilot study. Clin Exp Dermatol. 2009;34:487-491.
- Ceilley RI. Treatment of actinic purpura. J Clin Aesthet Dermatol. 2017;10:44-50.
- Rajagopal R, Thachil J, Monagle P. Disseminated intravascular coagulation in paediatrics. Arch Dis Child. 2017;102:187-193.
- Kraaijpoel N, van Es N, Porreca E, et al. The diagnostic management of upper extremity deep venous thrombosis: a review of the literature. Thromb Res. 2017;156:54-59.
The Diagnosis: Rumpel-Leede Phenomenon
Rumpel-Leede (R-L) phenomenon describes the rare benign occurrence of dermal capillaries acutely rupturing following the administration of a tourniquetlike force on an extremity,1 which manifests as asymptomatic petechiae and ecchymoses on a distal extremity, usually following noninvasive measurement of blood pressure.2 Rumpel-Leede phenomenon represents an underrecognized entity that either is excluded or minimally referenced in most dermatology textbooks. The R-L sign initially was described in the early 1900s after it was observed that tourniquets applied to the arms of patients with scarlet fever would lead to the development of petechiae in that extremity.3 It later was elucidated that underlying vascular disease predisposes to dermal capillary fragility, a risk factor for R-L phenomenon to occur upon application of a tourniquet. Rumpel-Leede phenomenon has been noted in patients with diabetes mellitus, acute or chronic hypertension, and thrombocytopenia.4 In addition to being hypertensive and diabetic, our patient had been taking amlodipine and diltiazem. Calcium channel blockers have been linked to R-L phenomenon in case reports as well as in a study of calcium channel blockers inducing capillary fragility in vivo.5 Rumpel-Leede phenomenon also has been noted in patients with tightly fitting garments and infants carried in baby carriers.1
The differential diagnosis for R-L phenomenon includes actinic purpura, small vessel vasculitis, disseminated intravascular coagulation (DIC), and deep vein thrombosis (DVT). Actinic purpura, also called solar purpura or senile purpura, represents the petechiae and ecchymoses that are associated with aging skin. It is thought to occur when DNA damage, UV-induced solar elastosis, and decreased lipids in the stratum corneum cause a weakened ability to contain red blood cell extravasation from capillaries.6 Due to the lack of history of trauma and clear association with the blood pressure cuff placement in our patient, a diagnosis of actinic purpura was unlikely. In small vessel vasculitis, patients classically present with nonblanching palpable purpura frequently distributed over the lower extremities. The isolation of the lesions to only the left arm lowered the suspicion for vasculitis. Cutaneous manifestations of DIC and other hypercoagulable states may include purpura, livedo reticularis, atrophie blanche, and in extreme cases purpura fulminans. Routine laboratory examination reveals thrombocytopenia, prolonged prothrombin time/partial thromboplastin time, and hemolytic anemia.7 Although our patient had the risk factors of recent infection and surgery, a hemoglobin level of 10.9 g/dL (reference range, 14.0-17.5 g/dL) and platelet count of 279,000/µL (reference range, 150,000-350,000/µL) excluded DIC as the probable diagnosis. Our patient was at an overall increased risk for DVT due to his prolonged hospital stay, increased age, and other factors. Despite these risk factors, the lack of pain or swelling made this diagnosis unlikely. Furthermore, our patient was heparinized throughout his hospital stay, and upper extremity DVT accounts for only 4% to 10% of the total DVT incidence.5
Although R-L phenomenon is a benign, self-limited condition, it may be necessary in some cases to rule out more serious underlying etiologies with investigative workup comprised of a complete blood cell count, coagulation profile, and basic metabolic panel. However, recognition of the R-L phenomenon in the right clinical context of localized petechiae or ecchymoses with a history of a tourniquetlike force may prevent an unnecessary and costly workup. Patients should be encouraged that R-L phenomenon will resolve over time with identification and correction of the tourniquetlike force. In this case, we recommended loosening of the sphygmomanometer cuff and alternating extremities to which the cuff was to be placed, which resulted in complete clearance of the petechiae and ecchymoses within 5 days.
The Diagnosis: Rumpel-Leede Phenomenon
Rumpel-Leede (R-L) phenomenon describes the rare benign occurrence of dermal capillaries acutely rupturing following the administration of a tourniquetlike force on an extremity,1 which manifests as asymptomatic petechiae and ecchymoses on a distal extremity, usually following noninvasive measurement of blood pressure.2 Rumpel-Leede phenomenon represents an underrecognized entity that either is excluded or minimally referenced in most dermatology textbooks. The R-L sign initially was described in the early 1900s after it was observed that tourniquets applied to the arms of patients with scarlet fever would lead to the development of petechiae in that extremity.3 It later was elucidated that underlying vascular disease predisposes to dermal capillary fragility, a risk factor for R-L phenomenon to occur upon application of a tourniquet. Rumpel-Leede phenomenon has been noted in patients with diabetes mellitus, acute or chronic hypertension, and thrombocytopenia.4 In addition to being hypertensive and diabetic, our patient had been taking amlodipine and diltiazem. Calcium channel blockers have been linked to R-L phenomenon in case reports as well as in a study of calcium channel blockers inducing capillary fragility in vivo.5 Rumpel-Leede phenomenon also has been noted in patients with tightly fitting garments and infants carried in baby carriers.1
The differential diagnosis for R-L phenomenon includes actinic purpura, small vessel vasculitis, disseminated intravascular coagulation (DIC), and deep vein thrombosis (DVT). Actinic purpura, also called solar purpura or senile purpura, represents the petechiae and ecchymoses that are associated with aging skin. It is thought to occur when DNA damage, UV-induced solar elastosis, and decreased lipids in the stratum corneum cause a weakened ability to contain red blood cell extravasation from capillaries.6 Due to the lack of history of trauma and clear association with the blood pressure cuff placement in our patient, a diagnosis of actinic purpura was unlikely. In small vessel vasculitis, patients classically present with nonblanching palpable purpura frequently distributed over the lower extremities. The isolation of the lesions to only the left arm lowered the suspicion for vasculitis. Cutaneous manifestations of DIC and other hypercoagulable states may include purpura, livedo reticularis, atrophie blanche, and in extreme cases purpura fulminans. Routine laboratory examination reveals thrombocytopenia, prolonged prothrombin time/partial thromboplastin time, and hemolytic anemia.7 Although our patient had the risk factors of recent infection and surgery, a hemoglobin level of 10.9 g/dL (reference range, 14.0-17.5 g/dL) and platelet count of 279,000/µL (reference range, 150,000-350,000/µL) excluded DIC as the probable diagnosis. Our patient was at an overall increased risk for DVT due to his prolonged hospital stay, increased age, and other factors. Despite these risk factors, the lack of pain or swelling made this diagnosis unlikely. Furthermore, our patient was heparinized throughout his hospital stay, and upper extremity DVT accounts for only 4% to 10% of the total DVT incidence.5
Although R-L phenomenon is a benign, self-limited condition, it may be necessary in some cases to rule out more serious underlying etiologies with investigative workup comprised of a complete blood cell count, coagulation profile, and basic metabolic panel. However, recognition of the R-L phenomenon in the right clinical context of localized petechiae or ecchymoses with a history of a tourniquetlike force may prevent an unnecessary and costly workup. Patients should be encouraged that R-L phenomenon will resolve over time with identification and correction of the tourniquetlike force. In this case, we recommended loosening of the sphygmomanometer cuff and alternating extremities to which the cuff was to be placed, which resulted in complete clearance of the petechiae and ecchymoses within 5 days.
- Nguyen T, Garcia D, Wang A, et al. Rumpel-Leede phenomenon associated with tourniquet-like forces of baby carriers in otherwise healthy infants. JAMA Dermatol. 2016;152:728-730.
- Chester M, Barwise J, Holzman M, et al. Acute dermal capillary rupture associated with noninvasive blood pressure monitoring. J Clin Anesth. 2007;19:473-475.
- Hartley A, Lim PB, Hayat SA. Rumpel-Leede phenomenon in a hypertensive patient due to mechanical trauma: a case report. J Med Case Rep. 2016;10:150.
- Varela D, Tran D, Ngamdu K, et al. Rumpel-Leede phenomenon presenting as a hypertensive urgency. Proc (Bayl Univ Med Cent). 2016;29:200-201.
- Cox NH, Walsh ML, Robson RH. Purpura and bleeding due to calcium-channel blockers: an underestimated problem? case reports and a pilot study. Clin Exp Dermatol. 2009;34:487-491.
- Ceilley RI. Treatment of actinic purpura. J Clin Aesthet Dermatol. 2017;10:44-50.
- Rajagopal R, Thachil J, Monagle P. Disseminated intravascular coagulation in paediatrics. Arch Dis Child. 2017;102:187-193.
- Kraaijpoel N, van Es N, Porreca E, et al. The diagnostic management of upper extremity deep venous thrombosis: a review of the literature. Thromb Res. 2017;156:54-59.
- Nguyen T, Garcia D, Wang A, et al. Rumpel-Leede phenomenon associated with tourniquet-like forces of baby carriers in otherwise healthy infants. JAMA Dermatol. 2016;152:728-730.
- Chester M, Barwise J, Holzman M, et al. Acute dermal capillary rupture associated with noninvasive blood pressure monitoring. J Clin Anesth. 2007;19:473-475.
- Hartley A, Lim PB, Hayat SA. Rumpel-Leede phenomenon in a hypertensive patient due to mechanical trauma: a case report. J Med Case Rep. 2016;10:150.
- Varela D, Tran D, Ngamdu K, et al. Rumpel-Leede phenomenon presenting as a hypertensive urgency. Proc (Bayl Univ Med Cent). 2016;29:200-201.
- Cox NH, Walsh ML, Robson RH. Purpura and bleeding due to calcium-channel blockers: an underestimated problem? case reports and a pilot study. Clin Exp Dermatol. 2009;34:487-491.
- Ceilley RI. Treatment of actinic purpura. J Clin Aesthet Dermatol. 2017;10:44-50.
- Rajagopal R, Thachil J, Monagle P. Disseminated intravascular coagulation in paediatrics. Arch Dis Child. 2017;102:187-193.
- Kraaijpoel N, van Es N, Porreca E, et al. The diagnostic management of upper extremity deep venous thrombosis: a review of the literature. Thromb Res. 2017;156:54-59.
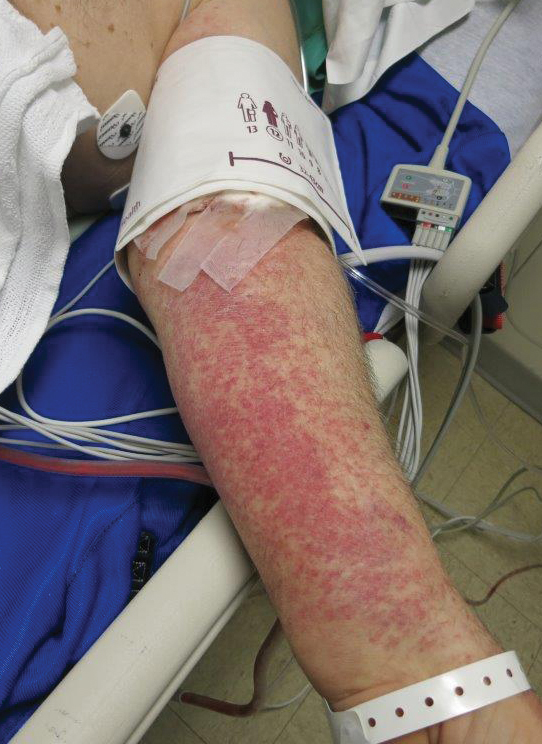
A 70-year-old man who had been admitted to the hospital a week prior for a right groin abscess overlying the site of a femoral graft developed a purpuric rash isolated to the left arm of 1 day's duration. The dermatology service was consulted by vascular surgery. The patient denied prior episodes of a similar rash, and there was no associated pruritus or pain. There was no history of trauma to the area. His medical history was remarkable for type 2 diabetes mellitus, hypertension, and peripheral vascular disease. His surgical history was notable for bilateral popliteal aneurysm repair and right femoral aneurysm repair. No pertinent family history was elicited. Cefepime, metronidazole, and vancomycin were administered for the groin abscess. Additional medications included amlodipine, atorvastatin, diltiazem, gabapentin, heparin, insulin, oxycodone, and acetaminophen.
Physical examination revealed broad ecchymoses on the left forearm with scattered petechiae as well as linear ecchymoses on the left upper arm distributed in the area where a sphygmomanometer cuff was applied. Full-body skin examination confirmed that the distribution of the petechiae and ecchymoses was limited to the left arm. The patient was normotensive at the time of examination. Laboratory evaluation revealed a hemoglobin level of 10.9 g/dL (reference range, 14.0-17.5 g/dL), a platelet count of 279,000/µL (reference range, 150,000-350,000/µL), and a glucose level of 121 mg/dL (reference range, 70-110 mg/dL).
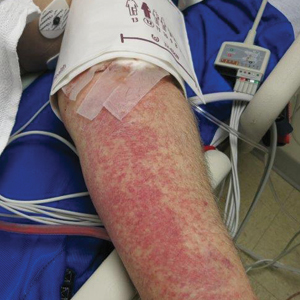
Minimally Hyperpigmented Plaque With Skin Thickening on the Neck
The Diagnosis: Fiddler's Neck
A thorough patient history revealed that the patient was retired and played violin regularly in the local orchestra. Fiddler's neck, or violin hickey, is an uncommon physical examination finding and often is considered a badge of honor by musicians who develop it. Fiddler's neck is a hobby-related callus seen in highly dedicated violin and viola players, and in some circles, it is known as a mark of greatness. In one instance, members of the public were asked to display a violin hickey before they were allowed to play a $3.5 million violin on public display in London, England.1 Fiddler's neck is a benign submandibular lesion caused by pressure and friction on the skin from extensive time spent playing the instrument. The primary cause is thought to be mechanical, but it is not fully understood why the lesion occurs in some musicians and not others, regardless of playing time.1 This submandibular fiddler's neck is distinct from a similarly named supraclavicular lesion, which represents an allergic contact dermatitis to the nickel bracket of the instrument's chin rest and presents with eczematous scale and/or vesicles.2,3 Submandibular fiddler's neck presents with some combination of erythema, edema, lichenification, and scarring just below the angle of the jaw. Occasionally, papules, pustules, and even cyst formation may be noted. Lesions are sometimes mistaken for malignancy or lymphedema. Therefore, a thorough history and clinical expertise are important, as surgical excision should be avoided.2
Depending on presentation, the differential diagnosis also may include malignant melanoma due to irregular pigmentation, branchial cleft cyst or sialolithiasis due to location and texture, or a tumor of the salivary gland.
Management of fiddler's neck may include topical steroids, neck or instrument padding, or decreased playing time. However, the lesion often is worn with pride, seen as a testament to the musician's dedication, and reassurance generally is most appropriate.1
- Roberts C. How to prevent or even cure a violin hickey. Strings. February 1, 2011. https://stringsmagazine.com/how-to-prevent-or-even-cure-a-violin-hickey/. Accessed January 31, 2020.
- Myint CW, Rutt AL, Sataloff RT. Fiddler's neck: a review. Ear Nose Throat J. 2017;96:76-79.
- Jue MS, Kim YS, Ro YS. Fiddler's neck accompanied by allergic contact dermatitis to nickel in a viola player. Ann Dermatol. 2010;22:88-90.
The Diagnosis: Fiddler's Neck
A thorough patient history revealed that the patient was retired and played violin regularly in the local orchestra. Fiddler's neck, or violin hickey, is an uncommon physical examination finding and often is considered a badge of honor by musicians who develop it. Fiddler's neck is a hobby-related callus seen in highly dedicated violin and viola players, and in some circles, it is known as a mark of greatness. In one instance, members of the public were asked to display a violin hickey before they were allowed to play a $3.5 million violin on public display in London, England.1 Fiddler's neck is a benign submandibular lesion caused by pressure and friction on the skin from extensive time spent playing the instrument. The primary cause is thought to be mechanical, but it is not fully understood why the lesion occurs in some musicians and not others, regardless of playing time.1 This submandibular fiddler's neck is distinct from a similarly named supraclavicular lesion, which represents an allergic contact dermatitis to the nickel bracket of the instrument's chin rest and presents with eczematous scale and/or vesicles.2,3 Submandibular fiddler's neck presents with some combination of erythema, edema, lichenification, and scarring just below the angle of the jaw. Occasionally, papules, pustules, and even cyst formation may be noted. Lesions are sometimes mistaken for malignancy or lymphedema. Therefore, a thorough history and clinical expertise are important, as surgical excision should be avoided.2
Depending on presentation, the differential diagnosis also may include malignant melanoma due to irregular pigmentation, branchial cleft cyst or sialolithiasis due to location and texture, or a tumor of the salivary gland.
Management of fiddler's neck may include topical steroids, neck or instrument padding, or decreased playing time. However, the lesion often is worn with pride, seen as a testament to the musician's dedication, and reassurance generally is most appropriate.1
The Diagnosis: Fiddler's Neck
A thorough patient history revealed that the patient was retired and played violin regularly in the local orchestra. Fiddler's neck, or violin hickey, is an uncommon physical examination finding and often is considered a badge of honor by musicians who develop it. Fiddler's neck is a hobby-related callus seen in highly dedicated violin and viola players, and in some circles, it is known as a mark of greatness. In one instance, members of the public were asked to display a violin hickey before they were allowed to play a $3.5 million violin on public display in London, England.1 Fiddler's neck is a benign submandibular lesion caused by pressure and friction on the skin from extensive time spent playing the instrument. The primary cause is thought to be mechanical, but it is not fully understood why the lesion occurs in some musicians and not others, regardless of playing time.1 This submandibular fiddler's neck is distinct from a similarly named supraclavicular lesion, which represents an allergic contact dermatitis to the nickel bracket of the instrument's chin rest and presents with eczematous scale and/or vesicles.2,3 Submandibular fiddler's neck presents with some combination of erythema, edema, lichenification, and scarring just below the angle of the jaw. Occasionally, papules, pustules, and even cyst formation may be noted. Lesions are sometimes mistaken for malignancy or lymphedema. Therefore, a thorough history and clinical expertise are important, as surgical excision should be avoided.2
Depending on presentation, the differential diagnosis also may include malignant melanoma due to irregular pigmentation, branchial cleft cyst or sialolithiasis due to location and texture, or a tumor of the salivary gland.
Management of fiddler's neck may include topical steroids, neck or instrument padding, or decreased playing time. However, the lesion often is worn with pride, seen as a testament to the musician's dedication, and reassurance generally is most appropriate.1
- Roberts C. How to prevent or even cure a violin hickey. Strings. February 1, 2011. https://stringsmagazine.com/how-to-prevent-or-even-cure-a-violin-hickey/. Accessed January 31, 2020.
- Myint CW, Rutt AL, Sataloff RT. Fiddler's neck: a review. Ear Nose Throat J. 2017;96:76-79.
- Jue MS, Kim YS, Ro YS. Fiddler's neck accompanied by allergic contact dermatitis to nickel in a viola player. Ann Dermatol. 2010;22:88-90.
- Roberts C. How to prevent or even cure a violin hickey. Strings. February 1, 2011. https://stringsmagazine.com/how-to-prevent-or-even-cure-a-violin-hickey/. Accessed January 31, 2020.
- Myint CW, Rutt AL, Sataloff RT. Fiddler's neck: a review. Ear Nose Throat J. 2017;96:76-79.
- Jue MS, Kim YS, Ro YS. Fiddler's neck accompanied by allergic contact dermatitis to nickel in a viola player. Ann Dermatol. 2010;22:88-90.
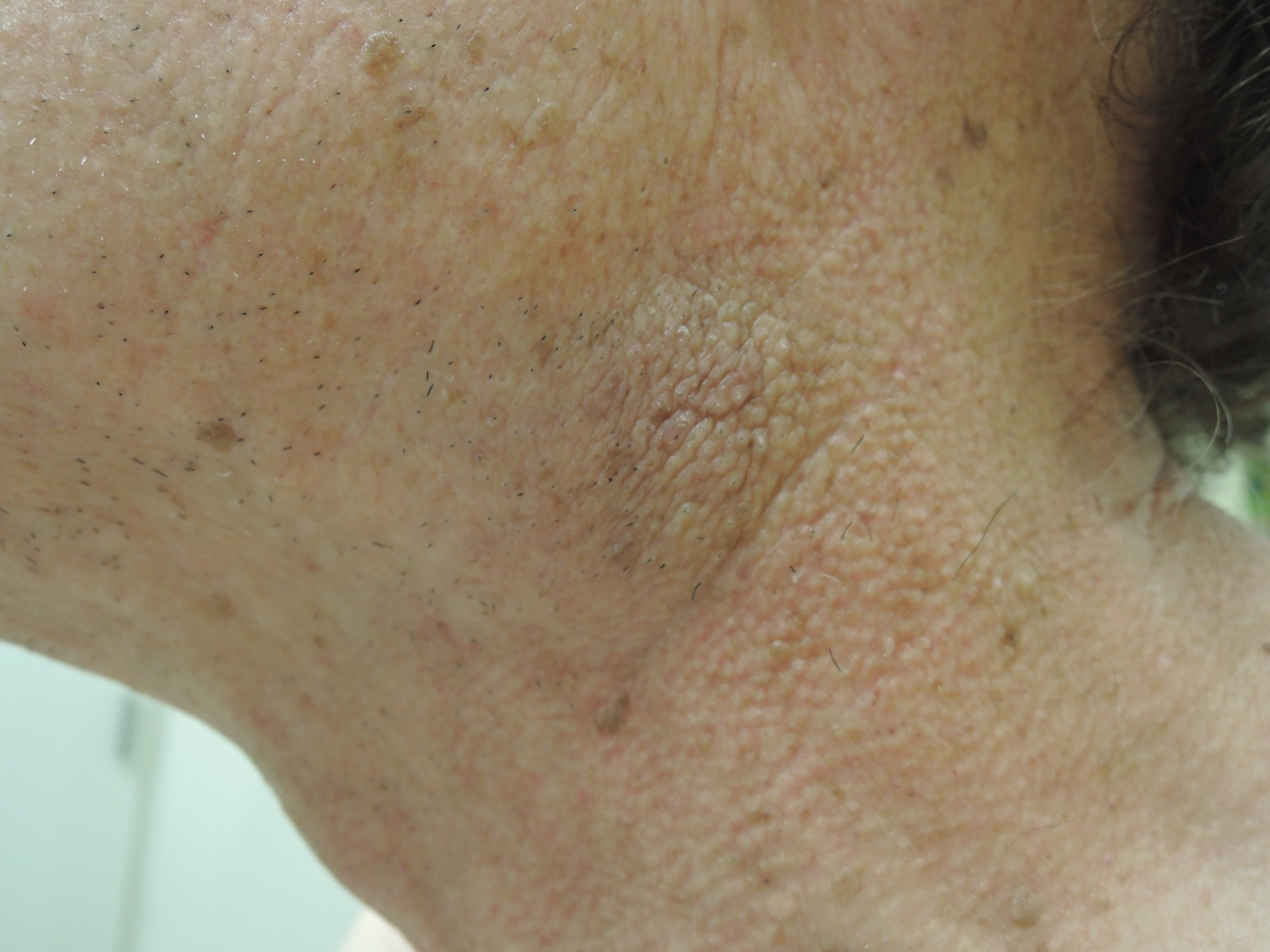
A 74-year-old man with a history of melanoma and basal cell carcinoma presented for an annual skin examination and displayed asymptomatic stable thickening of skin on the left side of the neck below the jawline of several years' duration. Physical examination revealed a 4×2-cm minimally hyperpigmented plaque with skin thickening and a pebbly appearing surface on the left lateral neck just inferior to the angle of the mandible.
Tender White Lesions on the Groin
The Diagnosis: Candidal Intertrigo
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
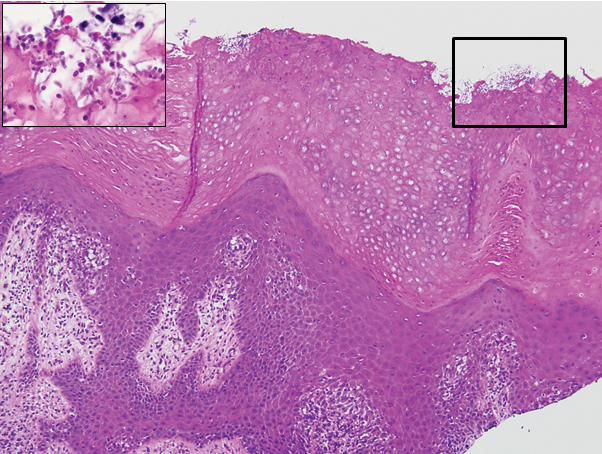
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6

The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
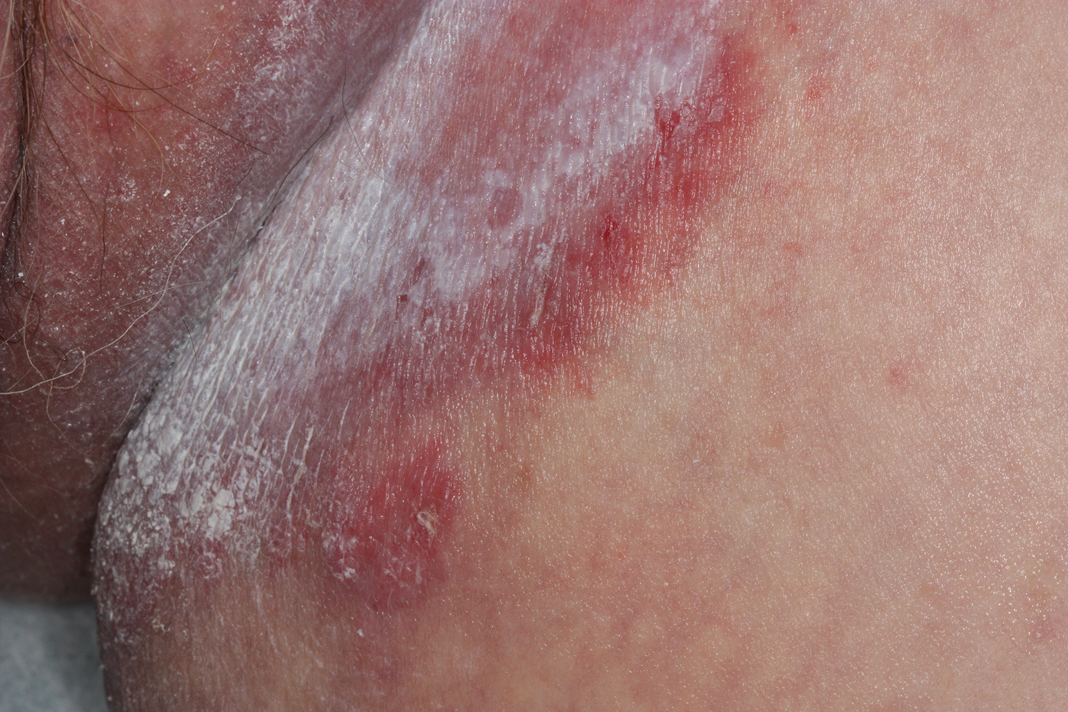
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
The Diagnosis: Candidal Intertrigo
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6
The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
The Diagnosis: Candidal Intertrigo
The biopsy confirmed a diagnosis of severe hyperkeratotic candidal intertrigo with no evidence of Hailey-Hailey disease. Hematoxylin and eosin- stained sections demonstrated irregular acanthosis and variable spongiosis. The stratum corneum was predominantly orthokeratotic with overlying psuedohyphae and yeast fungal elements (Figure 1).
Hyperimmunoglobulinemia E syndrome (HIES), also known as hyper-IgE syndrome or Job syndrome, is a rare immunodeficiency disorder characterized by an eczematous dermatitis-like rash, recurrent skin and lung abscesses, eosinophilia, and elevated serum IgE. Facial asymmetry, prominent forehead, deep-set eyes, broad nose, and roughened facial skin with large pores are characteristic of the sporadic and autosomal-recessive forms. Other common findings include retained primary teeth, hyperextensible joints, and recurrent mucocutaneous candidiasis.1
Although autosomal-dominant and autosomal-recessive inheritance patterns exist, sporadic mutations are the most common cause of HIES.2 Several genes have been implicated depending on the inheritance pattern. The majority of autosomal-dominant cases are associated with inactivating STAT3 (signal transducer and activator of transcription 3) mutations, whereas the majority of autosomal-recessive cases are associated with inactivating DOCK8 (dedicator of cytokinesis 8) mutations.1 Ultimately, all of these mutations lead to an impaired helper T cell (TH17) response, which is crucial for clearing fungal and extracellular bacterial infections.3
Skin eruptions typically are the first manifestation of HIES; they appear within the first week to month of life as papulopustular eruptions on the face and scalp and rapidly generalize to the rest of the body, favoring the shoulders, arms, chest, and buttocks. The pustules then coalesce into crusted plaques that resemble atopic dermatitis, frequently with superimposed Staphylococcus aureus infection. On microscopy, the pustules are folliculocentric and often contain eosinophils, whereas the plaques may contain intraepidermal collections of eosinophils.1
Mucocutaneous candidiasis is seen in approximately 60% of HIES cases and is closely linked to STAT3 inactivating mutations.3 Histologically, there is marked acanthosis with neutrophil exocytosis and abundant yeast and pseudohyphal forms within the stratum corneum (Figure 2).4 Cutaneous candidal infections typically require both oral and topical antifungal agents to clear the infection.3 Most cases of mucocutaneous candidiasis are caused by Candida albicans; however, other known culprits include Candida glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei.5,6 Of note, species identification and antifungal susceptibility studies may be useful in refractory cases, especially with C glabrata, which is known to acquire resistance to azoles, such as fluconazole, with emerging resistance to echinocandins.6
The differential diagnosis of this groin eruption included Hailey-Hailey disease; pemphigus vegetans, Hallopeau type; tinea cruris; and inverse psoriasis. Hailey-Hailey disease can be complicated by a superimposed candidal infection with similar clinical features, and biopsy may be required for definitive diagnosis. Hailey-Hailey disease typically presents with macerated fissured plaques that resemble macerated tissue paper with red fissures (Figure 3). Biopsy confirms full-thickness acantholysis resembling a dilapidated brick wall with minimal dyskeratosis.1 Pemphigus vegetans is a localized variant of pemphigus vulgaris with a predilection for flexural surfaces. The lesions progress to vegetating erosive plaques.4 The Hallopeau type often is studded with pustules and typically remains more localized than the Neumann type. Direct immunofluorescence demonstrates intercellular deposition of IgG and C3, and routine sections characteristically show pseudoepitheliomatous hyperplasia with intraepidermal eosinophilic microabscesses.1,4 Tinea cruris is characterized by erythematous annular lesions with raised scaly borders spreading down the inner thighs.7 The epidermis is variably spongiotic with parakeratosis, and neutrophils often present in a layered stratum corneum with basketweave keratin above a layer of more compact and eosinophilic keratin. Fungal stains, such as periodic acid-Schiff, will highlight the fungal hyphae within the stratum corneum. The inguinal folds are a typical location for inverse psoriasis, which generally appears as thin, sharply demarcated, shiny red plaques with less scale than plaque psoriasis.1 Psoriasiform hyperplasia with a diminished granular layer and tortuous papillary dermal vessels would be expected histologically.1
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
- James WD, Berger TG, Elston DM. Andrews' Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Schwartz RA, Tarlow MM. Dermatologic manifestations of Job syndrome. Medscape website. https://emedicine.medscape.com/article/1050852-overview. Updated April 22, 2019. Accessed March 28, 2020.
- Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int. 2012;61:191-196.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Pappas PG, Kauffman CA, Andes DR, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409-417.
- Center for Disease Control and Prevention. Antifungal resistance. https://www.cdc.gov/fungal/antifungal-resistance.html. Updated March 17, 2020. Accessed April 20, 2020.
- Tinea cruris. DermNet NZ website. https://www.dermnetnz.org/topics/tinea-cruris/. Published 2003. Accessed March 28, 2020.
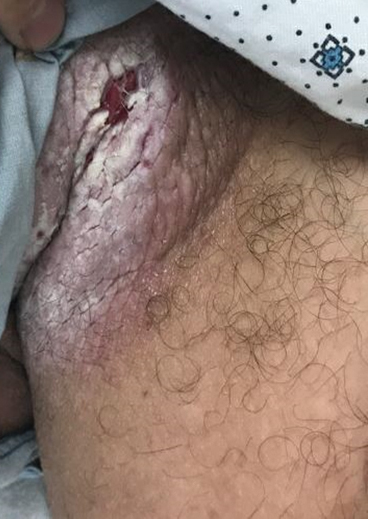
A 28-year-old man with a history of hyperimmunoglobulinemia E syndrome (previously known as Job syndrome), coarse facial features, and multiple skin and soft tissue infections presented to the university dermatology clinic with persistent white, macerated, fissured groin plaques that were present for months. The lesions were tender and pruritic with a burning sensation. Treatment with topical terbinafine and oral fluconazole was attempted without resolution of the eruption. A biopsy of the groin lesion was performed.