User login
Recall widens for diabetes drug metformin
The recall of extended-release metformin continues this month as 76 more lots have been flagged for a possible cancer-causing ingredient.
The Food and Drug Administration announced the latest recall, involving Marksans Pharma Limited and Sun Pharmaceutical Industries products, on Oct. 5. It involves the 500-mg and 700-mg tablets. More than 175 different drug combinations have been recalled since late May.
Consumers can see all the recalled metformin products at this FDA website. The agency says that immediate-release metformin does not appear to have the same contamination problem.
The FDA has been investigating the presence of nitrosamines, known to be possible carcinogens, in the popular diabetes medications since December, when they were first discovered in drugs in other countries. The agency said this month they still do not know the source of nitrosamines in the medications.
The investigation and subsequent recalls follow similar ones for contamination of popular heartburn and blood pressure drugs also for nitrosamines, such as N-Nitrosodimethylamine (NDMA).
The FDA says patients taking metformin products that have been recalled should continue taking the medication until a doctor or pharmacist gives them a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking the medication without first talking to their doctor.
The agency has asked drug manufacturers to test products before batches are released into the market. The companies must tell the FDA if any product shows levels of nitrosamines above the acceptable limit.
The risk from nitrosamines is not clear. The FDA says they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
This article first appeared on WebMD.com.
The recall of extended-release metformin continues this month as 76 more lots have been flagged for a possible cancer-causing ingredient.
The Food and Drug Administration announced the latest recall, involving Marksans Pharma Limited and Sun Pharmaceutical Industries products, on Oct. 5. It involves the 500-mg and 700-mg tablets. More than 175 different drug combinations have been recalled since late May.
Consumers can see all the recalled metformin products at this FDA website. The agency says that immediate-release metformin does not appear to have the same contamination problem.
The FDA has been investigating the presence of nitrosamines, known to be possible carcinogens, in the popular diabetes medications since December, when they were first discovered in drugs in other countries. The agency said this month they still do not know the source of nitrosamines in the medications.
The investigation and subsequent recalls follow similar ones for contamination of popular heartburn and blood pressure drugs also for nitrosamines, such as N-Nitrosodimethylamine (NDMA).
The FDA says patients taking metformin products that have been recalled should continue taking the medication until a doctor or pharmacist gives them a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking the medication without first talking to their doctor.
The agency has asked drug manufacturers to test products before batches are released into the market. The companies must tell the FDA if any product shows levels of nitrosamines above the acceptable limit.
The risk from nitrosamines is not clear. The FDA says they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
This article first appeared on WebMD.com.
The recall of extended-release metformin continues this month as 76 more lots have been flagged for a possible cancer-causing ingredient.
The Food and Drug Administration announced the latest recall, involving Marksans Pharma Limited and Sun Pharmaceutical Industries products, on Oct. 5. It involves the 500-mg and 700-mg tablets. More than 175 different drug combinations have been recalled since late May.
Consumers can see all the recalled metformin products at this FDA website. The agency says that immediate-release metformin does not appear to have the same contamination problem.
The FDA has been investigating the presence of nitrosamines, known to be possible carcinogens, in the popular diabetes medications since December, when they were first discovered in drugs in other countries. The agency said this month they still do not know the source of nitrosamines in the medications.
The investigation and subsequent recalls follow similar ones for contamination of popular heartburn and blood pressure drugs also for nitrosamines, such as N-Nitrosodimethylamine (NDMA).
The FDA says patients taking metformin products that have been recalled should continue taking the medication until a doctor or pharmacist gives them a replacement or a different treatment option. It could be dangerous for patients with type 2 diabetes to stop taking the medication without first talking to their doctor.
The agency has asked drug manufacturers to test products before batches are released into the market. The companies must tell the FDA if any product shows levels of nitrosamines above the acceptable limit.
The risk from nitrosamines is not clear. The FDA says they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
This article first appeared on WebMD.com.
FDA issues new NSAIDs warning for second half of pregnancy
The U.S. Food and Drug Administration released new warnings Oct. 15 that most nonsteroidal anti-inflammatory agents (NSAIDs) carry an elevated risk for kidney complications in unborn children when taken around weeks 20 or later in pregnancy.
Citing newly available research, the agency states the risk of low amniotic fluid (known as oligohydramnios) can occur, which in turn can cause rare but serious kidney problems in the offspring. Pregnancy complications also can result.
The FDA action expands on earlier warnings about agents in this drug class, which the FDA previously cautioned about taking after week 30 of pregnancy because of heart-related risks.
Manufacturers of both over-the-counter and prescription NSAIDs – including ibuprofen, naproxen, diclofenac, and celecoxib – will be required to update their labeling with the new warning.
Low-dose (81-mg) aspirin is excluded from this warning.
“Low-dose aspirin may be an important treatment for some women during pregnancy and should be taken under the direction of a healthcare professional,” the agency stated in a news release.
“It is important that women understand the benefits and risks of the medications they may take over the course of their pregnancy,” Patrizia Cavazzoni, MD, acting director of FDA’s Center for Drug Evaluation and Research, states in the release. “To this end, the agency is using its regulatory authority to inform women and their healthcare providers about the risks if NSAIDs are used after around 20 weeks of pregnancy and beyond.”
Oligohydramnios can arise quickly – in as little as 2 days – or weeks after starting regular NSAID use in this patient population. The condition usually resolves if a pregnant woman stops taking the NSAID, the agency notes.
If a health care provider believes NSAIDs are necessary between about 20 and 30 weeks of pregnancy, use should be limited to the lowest effective dose and shortest duration possible, the Drug Safety Communication notes.
As a reminder, health care professionals and patients should report side effects from NSAIDs to the FDA’s MedWatch program.
A version of this article originally appeared on Medscape.com.
The U.S. Food and Drug Administration released new warnings Oct. 15 that most nonsteroidal anti-inflammatory agents (NSAIDs) carry an elevated risk for kidney complications in unborn children when taken around weeks 20 or later in pregnancy.
Citing newly available research, the agency states the risk of low amniotic fluid (known as oligohydramnios) can occur, which in turn can cause rare but serious kidney problems in the offspring. Pregnancy complications also can result.
The FDA action expands on earlier warnings about agents in this drug class, which the FDA previously cautioned about taking after week 30 of pregnancy because of heart-related risks.
Manufacturers of both over-the-counter and prescription NSAIDs – including ibuprofen, naproxen, diclofenac, and celecoxib – will be required to update their labeling with the new warning.
Low-dose (81-mg) aspirin is excluded from this warning.
“Low-dose aspirin may be an important treatment for some women during pregnancy and should be taken under the direction of a healthcare professional,” the agency stated in a news release.
“It is important that women understand the benefits and risks of the medications they may take over the course of their pregnancy,” Patrizia Cavazzoni, MD, acting director of FDA’s Center for Drug Evaluation and Research, states in the release. “To this end, the agency is using its regulatory authority to inform women and their healthcare providers about the risks if NSAIDs are used after around 20 weeks of pregnancy and beyond.”
Oligohydramnios can arise quickly – in as little as 2 days – or weeks after starting regular NSAID use in this patient population. The condition usually resolves if a pregnant woman stops taking the NSAID, the agency notes.
If a health care provider believes NSAIDs are necessary between about 20 and 30 weeks of pregnancy, use should be limited to the lowest effective dose and shortest duration possible, the Drug Safety Communication notes.
As a reminder, health care professionals and patients should report side effects from NSAIDs to the FDA’s MedWatch program.
A version of this article originally appeared on Medscape.com.
The U.S. Food and Drug Administration released new warnings Oct. 15 that most nonsteroidal anti-inflammatory agents (NSAIDs) carry an elevated risk for kidney complications in unborn children when taken around weeks 20 or later in pregnancy.
Citing newly available research, the agency states the risk of low amniotic fluid (known as oligohydramnios) can occur, which in turn can cause rare but serious kidney problems in the offspring. Pregnancy complications also can result.
The FDA action expands on earlier warnings about agents in this drug class, which the FDA previously cautioned about taking after week 30 of pregnancy because of heart-related risks.
Manufacturers of both over-the-counter and prescription NSAIDs – including ibuprofen, naproxen, diclofenac, and celecoxib – will be required to update their labeling with the new warning.
Low-dose (81-mg) aspirin is excluded from this warning.
“Low-dose aspirin may be an important treatment for some women during pregnancy and should be taken under the direction of a healthcare professional,” the agency stated in a news release.
“It is important that women understand the benefits and risks of the medications they may take over the course of their pregnancy,” Patrizia Cavazzoni, MD, acting director of FDA’s Center for Drug Evaluation and Research, states in the release. “To this end, the agency is using its regulatory authority to inform women and their healthcare providers about the risks if NSAIDs are used after around 20 weeks of pregnancy and beyond.”
Oligohydramnios can arise quickly – in as little as 2 days – or weeks after starting regular NSAID use in this patient population. The condition usually resolves if a pregnant woman stops taking the NSAID, the agency notes.
If a health care provider believes NSAIDs are necessary between about 20 and 30 weeks of pregnancy, use should be limited to the lowest effective dose and shortest duration possible, the Drug Safety Communication notes.
As a reminder, health care professionals and patients should report side effects from NSAIDs to the FDA’s MedWatch program.
A version of this article originally appeared on Medscape.com.
Upadacitinib more effective, less safe than abatacept for RA
Upadacitinib (Rinvoq) proved superior to abatacept in both disease activity and remission in rheumatoid arthritis patients yet led to more adverse events, according to a new study that compared the two drugs.
“Additional data from longer and larger trials are needed to better understand long-term outcomes and safety of upadacitinib as compared with other drugs for the treatment of rheumatoid arthritis,” wrote Andrea Rubbert-Roth, MD, of the Cantonal Clinic St. Gallen in St. Gallen, Switzerland, and her colleagues. The study was published in the New England Journal of Medicine.
The Food and Drug Administration approved upadacitinib for the treatment of rheumatoid arthritis in August 2019.
To compare the Janus kinase (JAK) inhibitor upadacitinib and the biologic disease-modifying antirheumatic drug (DMARD) abatacept as safe and effective treatments for RA, the researchers launched a randomized, double-blind, phase 3 clinical trial dubbed SELECT-CHOICE at 120 sites in 28 countries. All patients had moderate to severe active disease after previously having inadequate responses to at least one biologic DMARD. Slightly more than 82% of the participants were female, with a mean age of 55 years and mean RA duration of 12 years.
Patients were assigned either 15 mg of oral upadacitinib daily (n = 303) or intravenous abatacept at day 1 and weeks 2, 4, 8, 12, 16 and 20 (n = 309) with dosage tied to body weight, each in combination with stable synthetic DMARDs. Disease activity was measured after 12 weeks via the Disease Activity Score for 28 joints using C-reactive protein (DAS28-CRP). A DAS28-CRP of more than 5.1 was categorized as high disease activity, while 3.2-5.1 meant moderate disease activity, 2.6-3.2 meant low disease activity, and less than 2.6 indicated remission.
The mean DAS28-CRP at baseline was 5.70 in the upadacitinib group and 5.88 in the abatacept group. After 12 weeks, the mean change from baseline was –2.52 points and –2.00 points, respectively (difference, –0.52 points; 95% confidence interval, –0.69 to –0.35; P < .001 for noninferiority; P < .001 for superiority). In patients with a DAS28-CRP of less than 2.6, the percentage of those having remission was 30% with upadacitinib and 13.3% with abatacept (difference, 16.8 percentage points; 95% CI, 10.4 to 23.2; P < .001 for superiority).
Over the 24-week trial period, the incidence of all adverse events (209 vs. 189) and serious adverse events (10 vs. 5) was higher in the upadacitinib group than in the abatacept group. There were 23 cases of hepatic disorder with upadacitinib, compared with 5 with abatacept; 2 thromboembolic events with upadacitinib, compared with 0 with abatacept; and 2 deaths with upadacitinib, compared with 1 with abatacept.

“The thing that bothers me, actually, is the adverse events,” Daniel E. Furst, MD, professor of medicine (emeritus) and rheumatology at the University of California, Los Angeles, said in an interview. “There were a fair number of them, all of which were a little higher in upadacitinib. They certainly made very little of those.”
He noted several other concerns about the study, including a potential geographic effect stemming from 60% of the study’s centers being in South and Central America and Eastern Europe. “Those patients don’t always have very good medical care,” he said. “They have an inherent, underlying placebo response that can be much different than Western Europe and North America.”
He also questioned their choice of primary endpoint metric.
“I think a much more legitimate way at looking at remission is the CDAI [Clinical Disease Activity Index] rather than the DAS28,” he said. “The DAS28, even at its best, is low disease activity, not true remission.”
“Bottom line,” he added, “this is a legitimate study that supports previous findings. One more important thing that is overlooked, though, is an economic analysis. A true economic analysis would be very important to place this in the armamentarium.”
Study affirms upadacitinib’s place in the RA treatment pecking order

By showing that upadacitinib was not only noninferior but superior to abatacept in decreasing disease activity, Rubbert-Roth and colleagues have positioned the JAK inhibitor at “the forefront of treatment for rheumatoid arthritis,” wrote Guro L. Goll, MD, PhD, and Tore K. Kvien, MD, PhD, of Diakonhjemmet Hospital in Oslo, in an accompanying editorial.
Though the authors noted that the 24-week trial was likely too short to make meaningful assumptions about long-term outcomes, they recognized the notably improved treatment outcomes over the study period and stated the importance of “head-to-head trials ... to inform evidence-based clinical decisions.” Similar to Dr. Furst, however, they stated an interest in “detailed data on changes in the CDAI score as a continuous measure.”

They also acknowledged the significant increase in adverse events among patients in the upadacitinib group, underlining the need to learn more in forthcoming, lengthier trials. “Rheumatologists will be looking hard at future data,” they wrote, “to assess whether improved treatment outcomes justify an increased risk of adverse events.”
The study was supported by AbbVie. The authors acknowledged numerous potential conflicts of interest, including receiving research grants and fees from various pharmaceutical companies for consulting, lectures, and being on advisory boards.
SOURCE: Rubbert-Roth A et al. N Engl J Med. 2020 Oct 14. doi: 10.1056/NEJMoa2008250.
Upadacitinib (Rinvoq) proved superior to abatacept in both disease activity and remission in rheumatoid arthritis patients yet led to more adverse events, according to a new study that compared the two drugs.
“Additional data from longer and larger trials are needed to better understand long-term outcomes and safety of upadacitinib as compared with other drugs for the treatment of rheumatoid arthritis,” wrote Andrea Rubbert-Roth, MD, of the Cantonal Clinic St. Gallen in St. Gallen, Switzerland, and her colleagues. The study was published in the New England Journal of Medicine.
The Food and Drug Administration approved upadacitinib for the treatment of rheumatoid arthritis in August 2019.
To compare the Janus kinase (JAK) inhibitor upadacitinib and the biologic disease-modifying antirheumatic drug (DMARD) abatacept as safe and effective treatments for RA, the researchers launched a randomized, double-blind, phase 3 clinical trial dubbed SELECT-CHOICE at 120 sites in 28 countries. All patients had moderate to severe active disease after previously having inadequate responses to at least one biologic DMARD. Slightly more than 82% of the participants were female, with a mean age of 55 years and mean RA duration of 12 years.
Patients were assigned either 15 mg of oral upadacitinib daily (n = 303) or intravenous abatacept at day 1 and weeks 2, 4, 8, 12, 16 and 20 (n = 309) with dosage tied to body weight, each in combination with stable synthetic DMARDs. Disease activity was measured after 12 weeks via the Disease Activity Score for 28 joints using C-reactive protein (DAS28-CRP). A DAS28-CRP of more than 5.1 was categorized as high disease activity, while 3.2-5.1 meant moderate disease activity, 2.6-3.2 meant low disease activity, and less than 2.6 indicated remission.
The mean DAS28-CRP at baseline was 5.70 in the upadacitinib group and 5.88 in the abatacept group. After 12 weeks, the mean change from baseline was –2.52 points and –2.00 points, respectively (difference, –0.52 points; 95% confidence interval, –0.69 to –0.35; P < .001 for noninferiority; P < .001 for superiority). In patients with a DAS28-CRP of less than 2.6, the percentage of those having remission was 30% with upadacitinib and 13.3% with abatacept (difference, 16.8 percentage points; 95% CI, 10.4 to 23.2; P < .001 for superiority).
Over the 24-week trial period, the incidence of all adverse events (209 vs. 189) and serious adverse events (10 vs. 5) was higher in the upadacitinib group than in the abatacept group. There were 23 cases of hepatic disorder with upadacitinib, compared with 5 with abatacept; 2 thromboembolic events with upadacitinib, compared with 0 with abatacept; and 2 deaths with upadacitinib, compared with 1 with abatacept.
“The thing that bothers me, actually, is the adverse events,” Daniel E. Furst, MD, professor of medicine (emeritus) and rheumatology at the University of California, Los Angeles, said in an interview. “There were a fair number of them, all of which were a little higher in upadacitinib. They certainly made very little of those.”
He noted several other concerns about the study, including a potential geographic effect stemming from 60% of the study’s centers being in South and Central America and Eastern Europe. “Those patients don’t always have very good medical care,” he said. “They have an inherent, underlying placebo response that can be much different than Western Europe and North America.”
He also questioned their choice of primary endpoint metric.
“I think a much more legitimate way at looking at remission is the CDAI [Clinical Disease Activity Index] rather than the DAS28,” he said. “The DAS28, even at its best, is low disease activity, not true remission.”
“Bottom line,” he added, “this is a legitimate study that supports previous findings. One more important thing that is overlooked, though, is an economic analysis. A true economic analysis would be very important to place this in the armamentarium.”
Study affirms upadacitinib’s place in the RA treatment pecking order
By showing that upadacitinib was not only noninferior but superior to abatacept in decreasing disease activity, Rubbert-Roth and colleagues have positioned the JAK inhibitor at “the forefront of treatment for rheumatoid arthritis,” wrote Guro L. Goll, MD, PhD, and Tore K. Kvien, MD, PhD, of Diakonhjemmet Hospital in Oslo, in an accompanying editorial.
Though the authors noted that the 24-week trial was likely too short to make meaningful assumptions about long-term outcomes, they recognized the notably improved treatment outcomes over the study period and stated the importance of “head-to-head trials ... to inform evidence-based clinical decisions.” Similar to Dr. Furst, however, they stated an interest in “detailed data on changes in the CDAI score as a continuous measure.”
They also acknowledged the significant increase in adverse events among patients in the upadacitinib group, underlining the need to learn more in forthcoming, lengthier trials. “Rheumatologists will be looking hard at future data,” they wrote, “to assess whether improved treatment outcomes justify an increased risk of adverse events.”
The study was supported by AbbVie. The authors acknowledged numerous potential conflicts of interest, including receiving research grants and fees from various pharmaceutical companies for consulting, lectures, and being on advisory boards.
SOURCE: Rubbert-Roth A et al. N Engl J Med. 2020 Oct 14. doi: 10.1056/NEJMoa2008250.
Upadacitinib (Rinvoq) proved superior to abatacept in both disease activity and remission in rheumatoid arthritis patients yet led to more adverse events, according to a new study that compared the two drugs.
“Additional data from longer and larger trials are needed to better understand long-term outcomes and safety of upadacitinib as compared with other drugs for the treatment of rheumatoid arthritis,” wrote Andrea Rubbert-Roth, MD, of the Cantonal Clinic St. Gallen in St. Gallen, Switzerland, and her colleagues. The study was published in the New England Journal of Medicine.
The Food and Drug Administration approved upadacitinib for the treatment of rheumatoid arthritis in August 2019.
To compare the Janus kinase (JAK) inhibitor upadacitinib and the biologic disease-modifying antirheumatic drug (DMARD) abatacept as safe and effective treatments for RA, the researchers launched a randomized, double-blind, phase 3 clinical trial dubbed SELECT-CHOICE at 120 sites in 28 countries. All patients had moderate to severe active disease after previously having inadequate responses to at least one biologic DMARD. Slightly more than 82% of the participants were female, with a mean age of 55 years and mean RA duration of 12 years.
Patients were assigned either 15 mg of oral upadacitinib daily (n = 303) or intravenous abatacept at day 1 and weeks 2, 4, 8, 12, 16 and 20 (n = 309) with dosage tied to body weight, each in combination with stable synthetic DMARDs. Disease activity was measured after 12 weeks via the Disease Activity Score for 28 joints using C-reactive protein (DAS28-CRP). A DAS28-CRP of more than 5.1 was categorized as high disease activity, while 3.2-5.1 meant moderate disease activity, 2.6-3.2 meant low disease activity, and less than 2.6 indicated remission.
The mean DAS28-CRP at baseline was 5.70 in the upadacitinib group and 5.88 in the abatacept group. After 12 weeks, the mean change from baseline was –2.52 points and –2.00 points, respectively (difference, –0.52 points; 95% confidence interval, –0.69 to –0.35; P < .001 for noninferiority; P < .001 for superiority). In patients with a DAS28-CRP of less than 2.6, the percentage of those having remission was 30% with upadacitinib and 13.3% with abatacept (difference, 16.8 percentage points; 95% CI, 10.4 to 23.2; P < .001 for superiority).
Over the 24-week trial period, the incidence of all adverse events (209 vs. 189) and serious adverse events (10 vs. 5) was higher in the upadacitinib group than in the abatacept group. There were 23 cases of hepatic disorder with upadacitinib, compared with 5 with abatacept; 2 thromboembolic events with upadacitinib, compared with 0 with abatacept; and 2 deaths with upadacitinib, compared with 1 with abatacept.
“The thing that bothers me, actually, is the adverse events,” Daniel E. Furst, MD, professor of medicine (emeritus) and rheumatology at the University of California, Los Angeles, said in an interview. “There were a fair number of them, all of which were a little higher in upadacitinib. They certainly made very little of those.”
He noted several other concerns about the study, including a potential geographic effect stemming from 60% of the study’s centers being in South and Central America and Eastern Europe. “Those patients don’t always have very good medical care,” he said. “They have an inherent, underlying placebo response that can be much different than Western Europe and North America.”
He also questioned their choice of primary endpoint metric.
“I think a much more legitimate way at looking at remission is the CDAI [Clinical Disease Activity Index] rather than the DAS28,” he said. “The DAS28, even at its best, is low disease activity, not true remission.”
“Bottom line,” he added, “this is a legitimate study that supports previous findings. One more important thing that is overlooked, though, is an economic analysis. A true economic analysis would be very important to place this in the armamentarium.”
Study affirms upadacitinib’s place in the RA treatment pecking order
By showing that upadacitinib was not only noninferior but superior to abatacept in decreasing disease activity, Rubbert-Roth and colleagues have positioned the JAK inhibitor at “the forefront of treatment for rheumatoid arthritis,” wrote Guro L. Goll, MD, PhD, and Tore K. Kvien, MD, PhD, of Diakonhjemmet Hospital in Oslo, in an accompanying editorial.
Though the authors noted that the 24-week trial was likely too short to make meaningful assumptions about long-term outcomes, they recognized the notably improved treatment outcomes over the study period and stated the importance of “head-to-head trials ... to inform evidence-based clinical decisions.” Similar to Dr. Furst, however, they stated an interest in “detailed data on changes in the CDAI score as a continuous measure.”
They also acknowledged the significant increase in adverse events among patients in the upadacitinib group, underlining the need to learn more in forthcoming, lengthier trials. “Rheumatologists will be looking hard at future data,” they wrote, “to assess whether improved treatment outcomes justify an increased risk of adverse events.”
The study was supported by AbbVie. The authors acknowledged numerous potential conflicts of interest, including receiving research grants and fees from various pharmaceutical companies for consulting, lectures, and being on advisory boards.
SOURCE: Rubbert-Roth A et al. N Engl J Med. 2020 Oct 14. doi: 10.1056/NEJMoa2008250.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Upadacitinib decreased disease activity but was associated with more serious adverse events, compared with abatacept, over a 24-week trial period.
Major finding: After 12 weeks, the mean change from baseline in the DAS28-CRP was –2.52 points with upadacitinib and –2.00 points with abatacept (difference, –0.52 points; 95% CI, –0.69 to –0.35; P < .001).
Study details: A randomized, double-blind, phase 3 clinical trial of RA patients who had previous inadequate responses to at least one biologic DMARD.
Disclosures: The study was supported by AbbVie. The authors acknowledged numerous potential conflicts of interest, including receiving research grants and fees from various pharmaceutical companies for consulting, lectures, and being on advisory boards.
Source: Rubbert-Roth A et al. N Engl J Med. 2020 Oct 14. doi: 10.1056/NEJMoa2008250
Dual therapy serves as well as triple for most HIV patients
based on a meta-analysis including data from more than 5,000 patients.
Although triple therapy remains the standard of care, the availability of more potent drugs has revived interest in dual and mono therapies, wrote Pisaturo Mariantonietta, MD, of the University of Campania Luigi Vanvitelli, Naples, Italy, and colleagues.
In a study published in Clinical Microbiology and Infection, the researchers identified 14 articles including 5,205 treatment-naive HIV adults. The studies were published between 2008 and 2020; 13 were randomized, controlled trials.
The dual therapies used in the studies included atazanavir/r plus maraviroc; lopinavir/r plus lamivudine; raltegravir plus darunavir/r; lopinavir/r plus tenofovir, raltegravir, efavirenz, or maraviroc; atazanavir/r plus raltegravir and darunavir/r plus maraviroc; and dolutegravir plus lamivudine.
Overall, no significant differences occurred in the primary endpoint of treatment failure across 10 studies between dual therapy and triple therapy patients based on data at 48 weeks (relative risk 1.20). “The rate of treatment failure did not differ among the two groups when stratifying the patients according to the drug used in the dual regimen,” the researchers said.
Low viral load’s link to treatment failure
Among 2,398 patients with a low HIV viral load (less than 100,000 copies/mL), dual therapy patients were significantly more likely to experience treatment failure than were triple therapy patients (RR, 1.47, P = .007). No differences were noted between dual and triple therapy failure among patients with high HIV viral loads at baseline. Patterns were similar at 96 weeks, but only three studies included 96-week data, the researchers said.
The rate of discontinuation because of adverse events was not significantly different between the groups at 48 weeks.
The study findings were limited by several factors, including the use of different regimens in the dual strategies, some of which are no longer in use, as well as there being insufficient data to fully compare outcomes at 96 weeks, and lack of information on cerebrospinal fluid viral load, the researchers noted.
However, the results suggest that dual therapy might be considered for HIV-naive patients with a low viral load, they said.
“Further RCTs that will evaluate the efficacy of antiretroviral regimens in use today among difficult-to-treat populations, such as patients with high viral load, including both intention-to-treat and per-protocol analysis, are needed to address this topic,” they concluded.
Consider range of patient factors when choosing therapies
Conducting the study at this time was important because of the expanding options for treating HIV patients, Donna E. Sweet, MD, an HIV specialist and professor of medicine at the University of Kansas, Wichita, said in an interview.
“We now have two single tablet formulations that are dual rather than triple therapy, and as treaters we are all trying to know when to use them,” she explained.
Dr. Sweet said she was not surprised by the study findings, given that well-conducted, randomized, controlled trials allowed the combination therapies to be approved.
Some of the key challenges to identifying the optimal treatment for HIV patients include factoring in the use of concomitant medications that could lead to drug-drug interactions, noted Dr. Sweet, who serves an editorial advisory board member of Internal Medicine News.
The take-home message for clinicians, in her opinion, is that “less drugs may mean less toxicity, but we don’t want to sacrifice efficacy,” she said. “There may be patients who are better suited than others for two vs. three drugs,” Dr. Sweet emphasized.
The next steps for research on the value of dual vs. triple therapy should include longer term efficacy studies, especially in those with lower CD4 counts and higher viral loads, said Dr. Sweet. In addition to factors such as CD4 counts and viral load, the food requirements of certain ART regimens could affect adherence and therefore a clinician decision to use two drugs rather than three, she noted.
Dr. Sweet disclosed past relationships with ViiV, Gilead, Merck, and Janssen on their speakers bureaus, and current advisory roles with Gilead and ViiV.
The study received no outside funding. Lead author Dr. Mariantonietta and several coauthors disclosed relationships with companies including ViiV Healthcare, AbbVie, Janssen-Cilag and Gilead Science, and Merck Sharp & Dohme, but no conflicts in connection with this study.
SOURCE: Mariantonietta P et al. Clin Microbiol Infect. 2020 Oct 5. doi: 10.1016/j.cmi.2020.09.048.
based on a meta-analysis including data from more than 5,000 patients.
Although triple therapy remains the standard of care, the availability of more potent drugs has revived interest in dual and mono therapies, wrote Pisaturo Mariantonietta, MD, of the University of Campania Luigi Vanvitelli, Naples, Italy, and colleagues.
In a study published in Clinical Microbiology and Infection, the researchers identified 14 articles including 5,205 treatment-naive HIV adults. The studies were published between 2008 and 2020; 13 were randomized, controlled trials.
The dual therapies used in the studies included atazanavir/r plus maraviroc; lopinavir/r plus lamivudine; raltegravir plus darunavir/r; lopinavir/r plus tenofovir, raltegravir, efavirenz, or maraviroc; atazanavir/r plus raltegravir and darunavir/r plus maraviroc; and dolutegravir plus lamivudine.
Overall, no significant differences occurred in the primary endpoint of treatment failure across 10 studies between dual therapy and triple therapy patients based on data at 48 weeks (relative risk 1.20). “The rate of treatment failure did not differ among the two groups when stratifying the patients according to the drug used in the dual regimen,” the researchers said.
Low viral load’s link to treatment failure
Among 2,398 patients with a low HIV viral load (less than 100,000 copies/mL), dual therapy patients were significantly more likely to experience treatment failure than were triple therapy patients (RR, 1.47, P = .007). No differences were noted between dual and triple therapy failure among patients with high HIV viral loads at baseline. Patterns were similar at 96 weeks, but only three studies included 96-week data, the researchers said.
The rate of discontinuation because of adverse events was not significantly different between the groups at 48 weeks.
The study findings were limited by several factors, including the use of different regimens in the dual strategies, some of which are no longer in use, as well as there being insufficient data to fully compare outcomes at 96 weeks, and lack of information on cerebrospinal fluid viral load, the researchers noted.
However, the results suggest that dual therapy might be considered for HIV-naive patients with a low viral load, they said.
“Further RCTs that will evaluate the efficacy of antiretroviral regimens in use today among difficult-to-treat populations, such as patients with high viral load, including both intention-to-treat and per-protocol analysis, are needed to address this topic,” they concluded.
Consider range of patient factors when choosing therapies
Conducting the study at this time was important because of the expanding options for treating HIV patients, Donna E. Sweet, MD, an HIV specialist and professor of medicine at the University of Kansas, Wichita, said in an interview.
“We now have two single tablet formulations that are dual rather than triple therapy, and as treaters we are all trying to know when to use them,” she explained.
Dr. Sweet said she was not surprised by the study findings, given that well-conducted, randomized, controlled trials allowed the combination therapies to be approved.
Some of the key challenges to identifying the optimal treatment for HIV patients include factoring in the use of concomitant medications that could lead to drug-drug interactions, noted Dr. Sweet, who serves an editorial advisory board member of Internal Medicine News.
The take-home message for clinicians, in her opinion, is that “less drugs may mean less toxicity, but we don’t want to sacrifice efficacy,” she said. “There may be patients who are better suited than others for two vs. three drugs,” Dr. Sweet emphasized.
The next steps for research on the value of dual vs. triple therapy should include longer term efficacy studies, especially in those with lower CD4 counts and higher viral loads, said Dr. Sweet. In addition to factors such as CD4 counts and viral load, the food requirements of certain ART regimens could affect adherence and therefore a clinician decision to use two drugs rather than three, she noted.
Dr. Sweet disclosed past relationships with ViiV, Gilead, Merck, and Janssen on their speakers bureaus, and current advisory roles with Gilead and ViiV.
The study received no outside funding. Lead author Dr. Mariantonietta and several coauthors disclosed relationships with companies including ViiV Healthcare, AbbVie, Janssen-Cilag and Gilead Science, and Merck Sharp & Dohme, but no conflicts in connection with this study.
SOURCE: Mariantonietta P et al. Clin Microbiol Infect. 2020 Oct 5. doi: 10.1016/j.cmi.2020.09.048.
based on a meta-analysis including data from more than 5,000 patients.
Although triple therapy remains the standard of care, the availability of more potent drugs has revived interest in dual and mono therapies, wrote Pisaturo Mariantonietta, MD, of the University of Campania Luigi Vanvitelli, Naples, Italy, and colleagues.
In a study published in Clinical Microbiology and Infection, the researchers identified 14 articles including 5,205 treatment-naive HIV adults. The studies were published between 2008 and 2020; 13 were randomized, controlled trials.
The dual therapies used in the studies included atazanavir/r plus maraviroc; lopinavir/r plus lamivudine; raltegravir plus darunavir/r; lopinavir/r plus tenofovir, raltegravir, efavirenz, or maraviroc; atazanavir/r plus raltegravir and darunavir/r plus maraviroc; and dolutegravir plus lamivudine.
Overall, no significant differences occurred in the primary endpoint of treatment failure across 10 studies between dual therapy and triple therapy patients based on data at 48 weeks (relative risk 1.20). “The rate of treatment failure did not differ among the two groups when stratifying the patients according to the drug used in the dual regimen,” the researchers said.
Low viral load’s link to treatment failure
Among 2,398 patients with a low HIV viral load (less than 100,000 copies/mL), dual therapy patients were significantly more likely to experience treatment failure than were triple therapy patients (RR, 1.47, P = .007). No differences were noted between dual and triple therapy failure among patients with high HIV viral loads at baseline. Patterns were similar at 96 weeks, but only three studies included 96-week data, the researchers said.
The rate of discontinuation because of adverse events was not significantly different between the groups at 48 weeks.
The study findings were limited by several factors, including the use of different regimens in the dual strategies, some of which are no longer in use, as well as there being insufficient data to fully compare outcomes at 96 weeks, and lack of information on cerebrospinal fluid viral load, the researchers noted.
However, the results suggest that dual therapy might be considered for HIV-naive patients with a low viral load, they said.
“Further RCTs that will evaluate the efficacy of antiretroviral regimens in use today among difficult-to-treat populations, such as patients with high viral load, including both intention-to-treat and per-protocol analysis, are needed to address this topic,” they concluded.
Consider range of patient factors when choosing therapies
Conducting the study at this time was important because of the expanding options for treating HIV patients, Donna E. Sweet, MD, an HIV specialist and professor of medicine at the University of Kansas, Wichita, said in an interview.
“We now have two single tablet formulations that are dual rather than triple therapy, and as treaters we are all trying to know when to use them,” she explained.
Dr. Sweet said she was not surprised by the study findings, given that well-conducted, randomized, controlled trials allowed the combination therapies to be approved.
Some of the key challenges to identifying the optimal treatment for HIV patients include factoring in the use of concomitant medications that could lead to drug-drug interactions, noted Dr. Sweet, who serves an editorial advisory board member of Internal Medicine News.
The take-home message for clinicians, in her opinion, is that “less drugs may mean less toxicity, but we don’t want to sacrifice efficacy,” she said. “There may be patients who are better suited than others for two vs. three drugs,” Dr. Sweet emphasized.
The next steps for research on the value of dual vs. triple therapy should include longer term efficacy studies, especially in those with lower CD4 counts and higher viral loads, said Dr. Sweet. In addition to factors such as CD4 counts and viral load, the food requirements of certain ART regimens could affect adherence and therefore a clinician decision to use two drugs rather than three, she noted.
Dr. Sweet disclosed past relationships with ViiV, Gilead, Merck, and Janssen on their speakers bureaus, and current advisory roles with Gilead and ViiV.
The study received no outside funding. Lead author Dr. Mariantonietta and several coauthors disclosed relationships with companies including ViiV Healthcare, AbbVie, Janssen-Cilag and Gilead Science, and Merck Sharp & Dohme, but no conflicts in connection with this study.
SOURCE: Mariantonietta P et al. Clin Microbiol Infect. 2020 Oct 5. doi: 10.1016/j.cmi.2020.09.048.
FROM CLINICAL MICROBIOLOGY AND INFECTION
T2D treatments create tension between glycemic and cardiovascular goals
It was no surprise that updated guidelines recently published by the European Society of Cardiology for managing cardiovascular disease in patients with diabetes highlighted optimized treatment from a cardiovascular disease perspective, while a nearly concurrent update from two major diabetes societies saw the same issue from a more glycemic point of view.
This difference led to divergent approaches to managing hyperglycemia in patients with type 2 diabetes (T2D). The two diabetes societies that wrote one set of recommendations, the American Diabetes Association and the European Association for the Study of Diabetes, put metformin at the pinnacle of their drug hierarchy. Patients with T2D and established atherosclerotic cardiovascular disease (CVD), chronic kidney disease, or heart failure should all receive metformin first unless contraindicated or not tolerated, their updated consensus report said.
Once metformin is on board, a clinician can then add a second diabetes agent from among the two drug classes recently proven to also reduce cardiovascular and renal events, either the SGLT2 (sodium-glucose transporter 2) inhibitors, or GLP-1 (glucagonlike peptide–1) receptor agonists, they advised.
Cardiovascular disease focus represents a ‘major paradigm shift’
In contrast, the ESC guidelines called for upfront, systematic assessment of CVD risk in patients with T2D before treatment starts, and for patients in high- or very high–risk strata, the guidelines recommended starting the patient first on an SGLT2 inhibitor or a GLP-1 receptor agonist, and only adding metformin in patients who need additional glycemic control.
The guidelines also recommended starting treatment-naive patients with moderate CVD risk on metformin. For patients already on metformin, the new ESC guidelines called for adding an agent from at least one of these two drug classes with proven CVD benefits for those at high or very high CVD risk. The guidelines also note that the CVD benefits of the two newer drug classes differ and hence require further individualization depending on the risks faced by each patient, such as the risk for heart failure hospitalizations.
It’s an approach “driven by data from the cardiovascular outcome trials,” that showed several drugs from both the SGLT2 inhibitor and GLP-1 receptor agonist classes have substantial benefit for preventing cardiovascular events, renal events, hospitalizations for heart failure, and in some studies all-cause mortality, said Francesco Cosentino, MD, during a discussion of the guideline differences at the virtual annual meeting of the European Association for the Study of Diabetes.
The ESC approach also represents “a major paradigm shift,” a “change from a glucose-centric approach to an approach driven by cardiovascular disease events,” summed up Dr. Cosentino, professor of cardiology at the Karolinska Institute in Stockholm and chair of the task force that wrote the ESC’s 2019 updated guidelines. The ESC approach advocates initiating drugs for treating patients with T2D “based on cardiovascular disease risk classification,” he highlighted. Results from some SGLT2 inhibitor cardiovascular outcome trials showed that the CVD benefit was similar regardless of whether or not patients also received metformin.
ADA, EASD call for ‘a different emphasis’
“There is a different emphasis” in the statement issued by the diabetologists of the ADA and EASD, admitted Peter J. Grant, MD, a professor of diabetes and endocrinology at the University of Leeds (England) and cochair of the ESC guidelines task force. Dr. Grant represented the EASD on the task force, and the Association collaborated with the ESC in producing its guidelines.

“The ADA and EASD recommendations “look primarily at glucose control, with cardiovascular disease management as secondary.” In contrast, the ESC guidelines “are primarily cardiovascular disease risk guidelines, with a glucose interest,” Dr. Grant declared.
Despite his involvement in writing the ESC guidelines, Dr. Grant tilted toward the ADA/EASD statement as more globally relevant.
“There is much more to vasculopathy in diabetes than just macrovascular disease. Many patients with type 2 diabetes without macrovascular complications have microvascular disease,” including the potential for retinopathy, nephropathy, and neuropathy, he said. These complications can also have a strong impact on psychological well being and treatment satisfaction.
“It’s important that we’re not glucocentric any more, but it’s equally important that we treat glucose because it has such a benefit for microvascular disease.” Dr. Grant also cited metformin’s long history of safety and good tolerance, clinician comfort prescribing it, and its low price. Heavier reliance on SGLT2 inhibitors and GLP-1 receptor agonists will be expensive for the short term while the cost of these drugs remains high, which places a higher burden on “knowing we’re doing it right,” said Dr. Grant.
Dr. Cosentino pointed out that the higher cost of the drugs in the two classes shown to exert important cardiovascular and renal effects needs to be considered in a cost-effectiveness context, not just by cost alone.
‘Clinical inertia’ could be a danger
Dr. Cosentino played down a major disagreement between the two guidelines, suggesting that “focusing on the differences leads to clinical inertia” by the practicing community when they are unsure how to reconcile the two positions.
Dr. Grant agreed that adding a second drug to metformin right away made sense in at least selected patients. “Look at each patient and decide whether they need glycemic control. If so, and if they also have cardiovascular disease, use both drugs,” metformin, plus one agent from one of the two newer classes.
Something both experts agreed on is that it’s time to generally steer clear of sulfonylurea drugs. “We have evidence for harmful effects from sulfonylureas,” Dr. Cosentino said.
“I’d dump sulfonylureas,” was Dr. Grant’s assessment, but he added that they still have a role for patients who need additional glycemic control but can’t afford the newer drugs.
Dr. Cosentino has had financial relationships with Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Merck, Mundipharma, Novo Nordisk, and Pfizer, Dr. Grant has lectured on behalf of AstraZeneca, GlaxoSmithKline, Merck, Novo Nordisk, the Medicines Company, and Takeda, and he has been an adviser to Amgen, AstraZeneca, Novartis, Novo Nordisk, and Synexus.
It was no surprise that updated guidelines recently published by the European Society of Cardiology for managing cardiovascular disease in patients with diabetes highlighted optimized treatment from a cardiovascular disease perspective, while a nearly concurrent update from two major diabetes societies saw the same issue from a more glycemic point of view.
This difference led to divergent approaches to managing hyperglycemia in patients with type 2 diabetes (T2D). The two diabetes societies that wrote one set of recommendations, the American Diabetes Association and the European Association for the Study of Diabetes, put metformin at the pinnacle of their drug hierarchy. Patients with T2D and established atherosclerotic cardiovascular disease (CVD), chronic kidney disease, or heart failure should all receive metformin first unless contraindicated or not tolerated, their updated consensus report said.
Once metformin is on board, a clinician can then add a second diabetes agent from among the two drug classes recently proven to also reduce cardiovascular and renal events, either the SGLT2 (sodium-glucose transporter 2) inhibitors, or GLP-1 (glucagonlike peptide–1) receptor agonists, they advised.
Cardiovascular disease focus represents a ‘major paradigm shift’
In contrast, the ESC guidelines called for upfront, systematic assessment of CVD risk in patients with T2D before treatment starts, and for patients in high- or very high–risk strata, the guidelines recommended starting the patient first on an SGLT2 inhibitor or a GLP-1 receptor agonist, and only adding metformin in patients who need additional glycemic control.
The guidelines also recommended starting treatment-naive patients with moderate CVD risk on metformin. For patients already on metformin, the new ESC guidelines called for adding an agent from at least one of these two drug classes with proven CVD benefits for those at high or very high CVD risk. The guidelines also note that the CVD benefits of the two newer drug classes differ and hence require further individualization depending on the risks faced by each patient, such as the risk for heart failure hospitalizations.
It’s an approach “driven by data from the cardiovascular outcome trials,” that showed several drugs from both the SGLT2 inhibitor and GLP-1 receptor agonist classes have substantial benefit for preventing cardiovascular events, renal events, hospitalizations for heart failure, and in some studies all-cause mortality, said Francesco Cosentino, MD, during a discussion of the guideline differences at the virtual annual meeting of the European Association for the Study of Diabetes.
The ESC approach also represents “a major paradigm shift,” a “change from a glucose-centric approach to an approach driven by cardiovascular disease events,” summed up Dr. Cosentino, professor of cardiology at the Karolinska Institute in Stockholm and chair of the task force that wrote the ESC’s 2019 updated guidelines. The ESC approach advocates initiating drugs for treating patients with T2D “based on cardiovascular disease risk classification,” he highlighted. Results from some SGLT2 inhibitor cardiovascular outcome trials showed that the CVD benefit was similar regardless of whether or not patients also received metformin.
ADA, EASD call for ‘a different emphasis’
“There is a different emphasis” in the statement issued by the diabetologists of the ADA and EASD, admitted Peter J. Grant, MD, a professor of diabetes and endocrinology at the University of Leeds (England) and cochair of the ESC guidelines task force. Dr. Grant represented the EASD on the task force, and the Association collaborated with the ESC in producing its guidelines.
“The ADA and EASD recommendations “look primarily at glucose control, with cardiovascular disease management as secondary.” In contrast, the ESC guidelines “are primarily cardiovascular disease risk guidelines, with a glucose interest,” Dr. Grant declared.
Despite his involvement in writing the ESC guidelines, Dr. Grant tilted toward the ADA/EASD statement as more globally relevant.
“There is much more to vasculopathy in diabetes than just macrovascular disease. Many patients with type 2 diabetes without macrovascular complications have microvascular disease,” including the potential for retinopathy, nephropathy, and neuropathy, he said. These complications can also have a strong impact on psychological well being and treatment satisfaction.
“It’s important that we’re not glucocentric any more, but it’s equally important that we treat glucose because it has such a benefit for microvascular disease.” Dr. Grant also cited metformin’s long history of safety and good tolerance, clinician comfort prescribing it, and its low price. Heavier reliance on SGLT2 inhibitors and GLP-1 receptor agonists will be expensive for the short term while the cost of these drugs remains high, which places a higher burden on “knowing we’re doing it right,” said Dr. Grant.
Dr. Cosentino pointed out that the higher cost of the drugs in the two classes shown to exert important cardiovascular and renal effects needs to be considered in a cost-effectiveness context, not just by cost alone.
‘Clinical inertia’ could be a danger
Dr. Cosentino played down a major disagreement between the two guidelines, suggesting that “focusing on the differences leads to clinical inertia” by the practicing community when they are unsure how to reconcile the two positions.
Dr. Grant agreed that adding a second drug to metformin right away made sense in at least selected patients. “Look at each patient and decide whether they need glycemic control. If so, and if they also have cardiovascular disease, use both drugs,” metformin, plus one agent from one of the two newer classes.
Something both experts agreed on is that it’s time to generally steer clear of sulfonylurea drugs. “We have evidence for harmful effects from sulfonylureas,” Dr. Cosentino said.
“I’d dump sulfonylureas,” was Dr. Grant’s assessment, but he added that they still have a role for patients who need additional glycemic control but can’t afford the newer drugs.
Dr. Cosentino has had financial relationships with Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Merck, Mundipharma, Novo Nordisk, and Pfizer, Dr. Grant has lectured on behalf of AstraZeneca, GlaxoSmithKline, Merck, Novo Nordisk, the Medicines Company, and Takeda, and he has been an adviser to Amgen, AstraZeneca, Novartis, Novo Nordisk, and Synexus.
It was no surprise that updated guidelines recently published by the European Society of Cardiology for managing cardiovascular disease in patients with diabetes highlighted optimized treatment from a cardiovascular disease perspective, while a nearly concurrent update from two major diabetes societies saw the same issue from a more glycemic point of view.
This difference led to divergent approaches to managing hyperglycemia in patients with type 2 diabetes (T2D). The two diabetes societies that wrote one set of recommendations, the American Diabetes Association and the European Association for the Study of Diabetes, put metformin at the pinnacle of their drug hierarchy. Patients with T2D and established atherosclerotic cardiovascular disease (CVD), chronic kidney disease, or heart failure should all receive metformin first unless contraindicated or not tolerated, their updated consensus report said.
Once metformin is on board, a clinician can then add a second diabetes agent from among the two drug classes recently proven to also reduce cardiovascular and renal events, either the SGLT2 (sodium-glucose transporter 2) inhibitors, or GLP-1 (glucagonlike peptide–1) receptor agonists, they advised.
Cardiovascular disease focus represents a ‘major paradigm shift’
In contrast, the ESC guidelines called for upfront, systematic assessment of CVD risk in patients with T2D before treatment starts, and for patients in high- or very high–risk strata, the guidelines recommended starting the patient first on an SGLT2 inhibitor or a GLP-1 receptor agonist, and only adding metformin in patients who need additional glycemic control.
The guidelines also recommended starting treatment-naive patients with moderate CVD risk on metformin. For patients already on metformin, the new ESC guidelines called for adding an agent from at least one of these two drug classes with proven CVD benefits for those at high or very high CVD risk. The guidelines also note that the CVD benefits of the two newer drug classes differ and hence require further individualization depending on the risks faced by each patient, such as the risk for heart failure hospitalizations.
It’s an approach “driven by data from the cardiovascular outcome trials,” that showed several drugs from both the SGLT2 inhibitor and GLP-1 receptor agonist classes have substantial benefit for preventing cardiovascular events, renal events, hospitalizations for heart failure, and in some studies all-cause mortality, said Francesco Cosentino, MD, during a discussion of the guideline differences at the virtual annual meeting of the European Association for the Study of Diabetes.
The ESC approach also represents “a major paradigm shift,” a “change from a glucose-centric approach to an approach driven by cardiovascular disease events,” summed up Dr. Cosentino, professor of cardiology at the Karolinska Institute in Stockholm and chair of the task force that wrote the ESC’s 2019 updated guidelines. The ESC approach advocates initiating drugs for treating patients with T2D “based on cardiovascular disease risk classification,” he highlighted. Results from some SGLT2 inhibitor cardiovascular outcome trials showed that the CVD benefit was similar regardless of whether or not patients also received metformin.
ADA, EASD call for ‘a different emphasis’
“There is a different emphasis” in the statement issued by the diabetologists of the ADA and EASD, admitted Peter J. Grant, MD, a professor of diabetes and endocrinology at the University of Leeds (England) and cochair of the ESC guidelines task force. Dr. Grant represented the EASD on the task force, and the Association collaborated with the ESC in producing its guidelines.
“The ADA and EASD recommendations “look primarily at glucose control, with cardiovascular disease management as secondary.” In contrast, the ESC guidelines “are primarily cardiovascular disease risk guidelines, with a glucose interest,” Dr. Grant declared.
Despite his involvement in writing the ESC guidelines, Dr. Grant tilted toward the ADA/EASD statement as more globally relevant.
“There is much more to vasculopathy in diabetes than just macrovascular disease. Many patients with type 2 diabetes without macrovascular complications have microvascular disease,” including the potential for retinopathy, nephropathy, and neuropathy, he said. These complications can also have a strong impact on psychological well being and treatment satisfaction.
“It’s important that we’re not glucocentric any more, but it’s equally important that we treat glucose because it has such a benefit for microvascular disease.” Dr. Grant also cited metformin’s long history of safety and good tolerance, clinician comfort prescribing it, and its low price. Heavier reliance on SGLT2 inhibitors and GLP-1 receptor agonists will be expensive for the short term while the cost of these drugs remains high, which places a higher burden on “knowing we’re doing it right,” said Dr. Grant.
Dr. Cosentino pointed out that the higher cost of the drugs in the two classes shown to exert important cardiovascular and renal effects needs to be considered in a cost-effectiveness context, not just by cost alone.
‘Clinical inertia’ could be a danger
Dr. Cosentino played down a major disagreement between the two guidelines, suggesting that “focusing on the differences leads to clinical inertia” by the practicing community when they are unsure how to reconcile the two positions.
Dr. Grant agreed that adding a second drug to metformin right away made sense in at least selected patients. “Look at each patient and decide whether they need glycemic control. If so, and if they also have cardiovascular disease, use both drugs,” metformin, plus one agent from one of the two newer classes.
Something both experts agreed on is that it’s time to generally steer clear of sulfonylurea drugs. “We have evidence for harmful effects from sulfonylureas,” Dr. Cosentino said.
“I’d dump sulfonylureas,” was Dr. Grant’s assessment, but he added that they still have a role for patients who need additional glycemic control but can’t afford the newer drugs.
Dr. Cosentino has had financial relationships with Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Merck, Mundipharma, Novo Nordisk, and Pfizer, Dr. Grant has lectured on behalf of AstraZeneca, GlaxoSmithKline, Merck, Novo Nordisk, the Medicines Company, and Takeda, and he has been an adviser to Amgen, AstraZeneca, Novartis, Novo Nordisk, and Synexus.
FROM EASD 2020
Remdesivir effective, well-tolerated in final trial report
Drug beats placebo across multiple endpoints in COVID-19 patients
In May 2020, remdesivir received Food and Drug Administration approval for emergency treatment of severe COVID-19 on the basis of a preliminary report on this trial. In August 2020, the FDA expanded the indication to include all hospitalized adult and pediatric patients with suspected or laboratory-confirmed COVID-19 infection irrespective of severity.
“Our findings were consistent with the findings of the preliminary report: a 10-day course of remdesivir was superior to placebo in the treatment of hospitalized patients with COVID-19,” reported a team of investigators led by John H. Beigel, MD, of the Division of Microbiology and Infectious Diseases at the National Institute of Allergy and Infectious Diseases, in the New England Journal of Medicine.
The drug’s broadened indication was not based on the ACTT-1 trial, according to Dr. Beigel. “Other data have demonstrated that remdesivir shortens recovery in patients with lower acuity. In our study, evidence of pneumonia was an enrollment requirement,” he explained in an interview.
In the newly published final ACTT-1 data, the median time to recovery was 10 days for those on active therapy versus 15 days for those randomized to placebo. With a rate ratio of 1.29 (P less than .001), this translated to a recovery that was about one third faster.
In this final report, remdesivir’s significant advantage over placebo regarding the trial’s primary endpoint was reinforced by efficacy on multiple secondary endpoints.
This benefits on multiple secondary endpoints included a 50% greater odds ratio (OR, 1.5; 95% CI, 1.2-1.9) of significant clinical improvement by day 15 after adjustment for baseline severity, a shorter initial length of hospital stay (12 vs. 17 days) and fewer days on oxygen supplementation (13 vs. 21 days) for the subgroup of patients on oxygen at enrollment.
Although the numerically lower mortality in the remdesivir arm (6.75 vs. 11.9%) did not reach statistical significance, Dr. Beigel said, “mortality was moving in the same direction as the other key endpoints.”
According to the study investigators, the types of rates of adverse events on remdesivir, which inhibits viral replication, “were generally similar in the remdesivir and placebo groups.”
In ACTT-1, 1,062 patients were randomized to remdesivir (200 mg loading dose followed by 100 mg daily for up to 9 days) or placebo. Patients were enrolled at study sites in North America, Europe, and Asia.
The data of ACTT-1 confirm a benefit from remdesivir in hospitalized COVID-19 patients with severe disease, but Dr. Beigel said he agrees with the current FDA indication that supports treatment in any hospitalized COVID-19 patient.
“We saw bigger benefits in patients with more severe infections. The benefits are not as large in patients with mild disease, but I think remdesivir should be considered in any hospitalized patient,” Dr. Beigel said.
This point of view is shared.
“I would give this drug to anyone in the hospital infected with COVID-19 assuming there was an ample supply and no need for rationing,” said Donna E. Sweet, MD, professor of internal medicine, University of Kansas, Wichita. She noted that this study has implications for hospital and hospital staff, as well as for patients.
“This type of reduction in recovery time means a reduction in potential exposures to hospital staff, a reduced need for PPE [personal protective equipment], and it will free up beds in the ICU [intensive care unit],” said Dr. Sweet, who also serves as an editorial advisory board member for Internal Medicine News.
An infectious disease specialist at the University of Minnesota also considers remdesivir to have an important role for conserving resources that deserves emphasis.
The reduction in time to recovery “is of benefit to the health system by maintaining hospital bed capacity,” said David R. Boulware, MD, professor of medicine at the University of Minnesota, Minneapolis.
According to his reading of the available data, including those from ACTT-1, the benefit appears to be greatest in those with a moderate degree of illness, which he defined as “sick enough to be hospitalized and require oxygen, yet not severely sick [and] requiring a ventilator or [extracorporeal membrane oxygenation].”
This does not preclude a benefit in those with more severe or milder disease, but patients with mild disease “are likely to recover regardless – or despite – whatever therapy they receive,” he said.
Dr. Beigel, the principal investigator of this trial, reports no potential conflicts of interest.
SOURCE: Beigel JH et al. N Engl J Med. 2020 Oct 8. doi: 10.1056/NEJMoa2007764.
Drug beats placebo across multiple endpoints in COVID-19 patients
Drug beats placebo across multiple endpoints in COVID-19 patients
In May 2020, remdesivir received Food and Drug Administration approval for emergency treatment of severe COVID-19 on the basis of a preliminary report on this trial. In August 2020, the FDA expanded the indication to include all hospitalized adult and pediatric patients with suspected or laboratory-confirmed COVID-19 infection irrespective of severity.
“Our findings were consistent with the findings of the preliminary report: a 10-day course of remdesivir was superior to placebo in the treatment of hospitalized patients with COVID-19,” reported a team of investigators led by John H. Beigel, MD, of the Division of Microbiology and Infectious Diseases at the National Institute of Allergy and Infectious Diseases, in the New England Journal of Medicine.
The drug’s broadened indication was not based on the ACTT-1 trial, according to Dr. Beigel. “Other data have demonstrated that remdesivir shortens recovery in patients with lower acuity. In our study, evidence of pneumonia was an enrollment requirement,” he explained in an interview.
In the newly published final ACTT-1 data, the median time to recovery was 10 days for those on active therapy versus 15 days for those randomized to placebo. With a rate ratio of 1.29 (P less than .001), this translated to a recovery that was about one third faster.
In this final report, remdesivir’s significant advantage over placebo regarding the trial’s primary endpoint was reinforced by efficacy on multiple secondary endpoints.
This benefits on multiple secondary endpoints included a 50% greater odds ratio (OR, 1.5; 95% CI, 1.2-1.9) of significant clinical improvement by day 15 after adjustment for baseline severity, a shorter initial length of hospital stay (12 vs. 17 days) and fewer days on oxygen supplementation (13 vs. 21 days) for the subgroup of patients on oxygen at enrollment.
Although the numerically lower mortality in the remdesivir arm (6.75 vs. 11.9%) did not reach statistical significance, Dr. Beigel said, “mortality was moving in the same direction as the other key endpoints.”
According to the study investigators, the types of rates of adverse events on remdesivir, which inhibits viral replication, “were generally similar in the remdesivir and placebo groups.”
In ACTT-1, 1,062 patients were randomized to remdesivir (200 mg loading dose followed by 100 mg daily for up to 9 days) or placebo. Patients were enrolled at study sites in North America, Europe, and Asia.
The data of ACTT-1 confirm a benefit from remdesivir in hospitalized COVID-19 patients with severe disease, but Dr. Beigel said he agrees with the current FDA indication that supports treatment in any hospitalized COVID-19 patient.
“We saw bigger benefits in patients with more severe infections. The benefits are not as large in patients with mild disease, but I think remdesivir should be considered in any hospitalized patient,” Dr. Beigel said.
This point of view is shared.
“I would give this drug to anyone in the hospital infected with COVID-19 assuming there was an ample supply and no need for rationing,” said Donna E. Sweet, MD, professor of internal medicine, University of Kansas, Wichita. She noted that this study has implications for hospital and hospital staff, as well as for patients.
“This type of reduction in recovery time means a reduction in potential exposures to hospital staff, a reduced need for PPE [personal protective equipment], and it will free up beds in the ICU [intensive care unit],” said Dr. Sweet, who also serves as an editorial advisory board member for Internal Medicine News.
An infectious disease specialist at the University of Minnesota also considers remdesivir to have an important role for conserving resources that deserves emphasis.
The reduction in time to recovery “is of benefit to the health system by maintaining hospital bed capacity,” said David R. Boulware, MD, professor of medicine at the University of Minnesota, Minneapolis.
According to his reading of the available data, including those from ACTT-1, the benefit appears to be greatest in those with a moderate degree of illness, which he defined as “sick enough to be hospitalized and require oxygen, yet not severely sick [and] requiring a ventilator or [extracorporeal membrane oxygenation].”
This does not preclude a benefit in those with more severe or milder disease, but patients with mild disease “are likely to recover regardless – or despite – whatever therapy they receive,” he said.
Dr. Beigel, the principal investigator of this trial, reports no potential conflicts of interest.
SOURCE: Beigel JH et al. N Engl J Med. 2020 Oct 8. doi: 10.1056/NEJMoa2007764.
In May 2020, remdesivir received Food and Drug Administration approval for emergency treatment of severe COVID-19 on the basis of a preliminary report on this trial. In August 2020, the FDA expanded the indication to include all hospitalized adult and pediatric patients with suspected or laboratory-confirmed COVID-19 infection irrespective of severity.
“Our findings were consistent with the findings of the preliminary report: a 10-day course of remdesivir was superior to placebo in the treatment of hospitalized patients with COVID-19,” reported a team of investigators led by John H. Beigel, MD, of the Division of Microbiology and Infectious Diseases at the National Institute of Allergy and Infectious Diseases, in the New England Journal of Medicine.
The drug’s broadened indication was not based on the ACTT-1 trial, according to Dr. Beigel. “Other data have demonstrated that remdesivir shortens recovery in patients with lower acuity. In our study, evidence of pneumonia was an enrollment requirement,” he explained in an interview.
In the newly published final ACTT-1 data, the median time to recovery was 10 days for those on active therapy versus 15 days for those randomized to placebo. With a rate ratio of 1.29 (P less than .001), this translated to a recovery that was about one third faster.
In this final report, remdesivir’s significant advantage over placebo regarding the trial’s primary endpoint was reinforced by efficacy on multiple secondary endpoints.
This benefits on multiple secondary endpoints included a 50% greater odds ratio (OR, 1.5; 95% CI, 1.2-1.9) of significant clinical improvement by day 15 after adjustment for baseline severity, a shorter initial length of hospital stay (12 vs. 17 days) and fewer days on oxygen supplementation (13 vs. 21 days) for the subgroup of patients on oxygen at enrollment.
Although the numerically lower mortality in the remdesivir arm (6.75 vs. 11.9%) did not reach statistical significance, Dr. Beigel said, “mortality was moving in the same direction as the other key endpoints.”
According to the study investigators, the types of rates of adverse events on remdesivir, which inhibits viral replication, “were generally similar in the remdesivir and placebo groups.”
In ACTT-1, 1,062 patients were randomized to remdesivir (200 mg loading dose followed by 100 mg daily for up to 9 days) or placebo. Patients were enrolled at study sites in North America, Europe, and Asia.
The data of ACTT-1 confirm a benefit from remdesivir in hospitalized COVID-19 patients with severe disease, but Dr. Beigel said he agrees with the current FDA indication that supports treatment in any hospitalized COVID-19 patient.
“We saw bigger benefits in patients with more severe infections. The benefits are not as large in patients with mild disease, but I think remdesivir should be considered in any hospitalized patient,” Dr. Beigel said.
This point of view is shared.
“I would give this drug to anyone in the hospital infected with COVID-19 assuming there was an ample supply and no need for rationing,” said Donna E. Sweet, MD, professor of internal medicine, University of Kansas, Wichita. She noted that this study has implications for hospital and hospital staff, as well as for patients.
“This type of reduction in recovery time means a reduction in potential exposures to hospital staff, a reduced need for PPE [personal protective equipment], and it will free up beds in the ICU [intensive care unit],” said Dr. Sweet, who also serves as an editorial advisory board member for Internal Medicine News.
An infectious disease specialist at the University of Minnesota also considers remdesivir to have an important role for conserving resources that deserves emphasis.
The reduction in time to recovery “is of benefit to the health system by maintaining hospital bed capacity,” said David R. Boulware, MD, professor of medicine at the University of Minnesota, Minneapolis.
According to his reading of the available data, including those from ACTT-1, the benefit appears to be greatest in those with a moderate degree of illness, which he defined as “sick enough to be hospitalized and require oxygen, yet not severely sick [and] requiring a ventilator or [extracorporeal membrane oxygenation].”
This does not preclude a benefit in those with more severe or milder disease, but patients with mild disease “are likely to recover regardless – or despite – whatever therapy they receive,” he said.
Dr. Beigel, the principal investigator of this trial, reports no potential conflicts of interest.
SOURCE: Beigel JH et al. N Engl J Med. 2020 Oct 8. doi: 10.1056/NEJMoa2007764.
More data on impact of corticosteroids on COVID-19 mortality in patients with COPD
, a study of almost 1 million individuals in the United Kingdom has shown.
Patients with chronic obstructive pulmonary disease or asthma who used ICS on a regular basis were more likely to die from COVID-19 than COPD or asthma patients who were prescribed non-ICS therapies, reported co-lead author Anna Schultze, PhD, of London School of Hygiene & Tropical Medicine and colleagues.

Of note, the increased risk of death among ICS users likely stemmed from greater severity of preexisting chronic respiratory conditions, instead of directly from ICS usage, which has little apparent impact on COVID-19 mortality, the investigators wrote in Lancet Respiratory Medicine.
These findings conflict with a hypothesis proposed early in the pandemic: that ICS may protect individuals from SARS-CoV-2 infection and poor outcomes with COVID-19.
According to Megan Conroy, MD, of the department of internal medicine at the Ohio State University Wexner Medical Center, Columbus, this hypothesis was based on some unexpected epidemiological findings.
“In general, we tend to think people with underlying lung disease – like COPD or asthma – to be at higher risk for severe forms of lower respiratory tract infections,” Dr. Conroy said. “Somewhat surprisingly, early data in the pandemic showed patients with COPD and asthma [were] underrepresented [among patients with COVID] when compared to the prevalence of these diseases in the population.”
This raised the possibility of an incidental protective effect from regular ICS therapy, which “had some strong theoretic pathophysiologic basis,” Dr. Conroy said, referring to research that demonstrated ICS-mediated downregulation of SARS-CoV-2 entry receptors ACE2 and TMPRSS2.
Dr. Schultze and colleagues noted that investigators for two ongoing randomized controlled trials (NCT04331054, NCT04330586) are studying ICS as an intervention for COVID-19; but neither trial includes individuals already taking ICS for chronic respiratory disease.
The present observational study therefore aimed to assess mortality risk within this population. Data were drawn from electronic health records and a U.K. national mortality database, with follow-up ranging from March 1 to May 6, 2020. Eligibility required a relevant prescription within 4 months of first follow-up. In the COPD group, patients were prescribed a long-acting beta agonist plus a long-acting muscarinic antagonist (LABA–LAMA), LABA alone, LABA plus ICS, LABA–LAMA plus ICS, or ICS alone (if prescribed LABA within 4 months).
In the asthma group, patients received low/medium-dose ICS, high-dose ICS, or a short-acting beta agonist (SABA) alone. Patients with COPD were at least 35 years of age, while those with asthma were 18 years or older. Hazard ratios were adjusted for a variety of covariates, including respiratory disease–exacerbation history, age, sex, body mass index, hypertension, diabetes, and others.
These eligibility criteria returned 148,557 patients with COPD and 818,490 with asthma.
Patients with COPD who were prescribed ICS plus LABA-LAMA or ICS plus LABA had an increased risk of COVID-19-related death, compared with those who did not receive ICS (adjusted hazard ratio, 1.39; 95% confidence interval, 1.10-1.76). Separate analyses of patients who received a triple combination (LABA–LAMA plus ICS) versus those who took a dual combination (LABA plus ICS) showed that triple-combination therapy was significantly associated with increased COVID-19-related mortality (aHR, 1.43; 95% CI, 1.12-1.83), while dual-combination therapy was less so (aHR, 1.29; 95% CI, 0.96-1.74). Non–COVID-19–related mortality was significantly increased for all COPD patients who were prescribed ICS, with or without adjustment for covariates.
Asthma patients prescribed high-dose ICS instead of SABA alone had a slightly greater risk of COVID-19–related death, based on an adjusted hazard ratio of 1.55 (95% CI, 1.10-2.18). Those with asthma who received low/medium–dose ICS demonstrated a slight trend toward increased mortality risk, but this was not significant (aHR, 1.14; 95% CI, 0.85-1.54). ICS usage in the asthma group was not linked with a significant increase in non–COVID-19–related death.
“In summary, we found no evidence of a beneficial effect of regular ICS use among people with COPD and asthma on COVID-19–related mortality,” the investigators concluded.
In agreement with the investigators, Dr. Conroy said that the increased mortality rate among ICS users should not be misconstrued as a medication-related risk.
“While the study found that those with COPD or asthma taking ICS and high-dose ICS were at an increased risk of death, this could easily be explained by the likelihood that those are the patients who are more likely to have more severe underlying lung disease,” Dr. Conroy said. “While this observational study did attempt to control for exacerbation history, the ability to do so by electronic health records data is certainly imperfect.”
With this in mind, patients with chronic respiratory disease should be encouraged to adhere to their usual treatment regimen, Dr. Conroy added.
“There isn’t evidence to increase or decrease medications just because of the pandemic,” she said. “A patient with asthma or COPD should continue to take the medications that are needed to achieve good control of their lung disease.”
The study was funded by the U.K. Medical Research Council. The investigators reported additional relationships with the Wellcome Trust, the Good Thinking Foundation, the Laura and John Arnold Foundation, and others. Dr. Conroy reported no conflicts of interest.
SOURCE: Schultze A et al. Lancet Respir Med. 2020 Sep 24. doi: 10.1016/ S2213-2600(20)30415-X.
, a study of almost 1 million individuals in the United Kingdom has shown.
Patients with chronic obstructive pulmonary disease or asthma who used ICS on a regular basis were more likely to die from COVID-19 than COPD or asthma patients who were prescribed non-ICS therapies, reported co-lead author Anna Schultze, PhD, of London School of Hygiene & Tropical Medicine and colleagues.
Of note, the increased risk of death among ICS users likely stemmed from greater severity of preexisting chronic respiratory conditions, instead of directly from ICS usage, which has little apparent impact on COVID-19 mortality, the investigators wrote in Lancet Respiratory Medicine.
These findings conflict with a hypothesis proposed early in the pandemic: that ICS may protect individuals from SARS-CoV-2 infection and poor outcomes with COVID-19.
According to Megan Conroy, MD, of the department of internal medicine at the Ohio State University Wexner Medical Center, Columbus, this hypothesis was based on some unexpected epidemiological findings.
“In general, we tend to think people with underlying lung disease – like COPD or asthma – to be at higher risk for severe forms of lower respiratory tract infections,” Dr. Conroy said. “Somewhat surprisingly, early data in the pandemic showed patients with COPD and asthma [were] underrepresented [among patients with COVID] when compared to the prevalence of these diseases in the population.”
This raised the possibility of an incidental protective effect from regular ICS therapy, which “had some strong theoretic pathophysiologic basis,” Dr. Conroy said, referring to research that demonstrated ICS-mediated downregulation of SARS-CoV-2 entry receptors ACE2 and TMPRSS2.
Dr. Schultze and colleagues noted that investigators for two ongoing randomized controlled trials (NCT04331054, NCT04330586) are studying ICS as an intervention for COVID-19; but neither trial includes individuals already taking ICS for chronic respiratory disease.
The present observational study therefore aimed to assess mortality risk within this population. Data were drawn from electronic health records and a U.K. national mortality database, with follow-up ranging from March 1 to May 6, 2020. Eligibility required a relevant prescription within 4 months of first follow-up. In the COPD group, patients were prescribed a long-acting beta agonist plus a long-acting muscarinic antagonist (LABA–LAMA), LABA alone, LABA plus ICS, LABA–LAMA plus ICS, or ICS alone (if prescribed LABA within 4 months).
In the asthma group, patients received low/medium-dose ICS, high-dose ICS, or a short-acting beta agonist (SABA) alone. Patients with COPD were at least 35 years of age, while those with asthma were 18 years or older. Hazard ratios were adjusted for a variety of covariates, including respiratory disease–exacerbation history, age, sex, body mass index, hypertension, diabetes, and others.
These eligibility criteria returned 148,557 patients with COPD and 818,490 with asthma.
Patients with COPD who were prescribed ICS plus LABA-LAMA or ICS plus LABA had an increased risk of COVID-19-related death, compared with those who did not receive ICS (adjusted hazard ratio, 1.39; 95% confidence interval, 1.10-1.76). Separate analyses of patients who received a triple combination (LABA–LAMA plus ICS) versus those who took a dual combination (LABA plus ICS) showed that triple-combination therapy was significantly associated with increased COVID-19-related mortality (aHR, 1.43; 95% CI, 1.12-1.83), while dual-combination therapy was less so (aHR, 1.29; 95% CI, 0.96-1.74). Non–COVID-19–related mortality was significantly increased for all COPD patients who were prescribed ICS, with or without adjustment for covariates.
Asthma patients prescribed high-dose ICS instead of SABA alone had a slightly greater risk of COVID-19–related death, based on an adjusted hazard ratio of 1.55 (95% CI, 1.10-2.18). Those with asthma who received low/medium–dose ICS demonstrated a slight trend toward increased mortality risk, but this was not significant (aHR, 1.14; 95% CI, 0.85-1.54). ICS usage in the asthma group was not linked with a significant increase in non–COVID-19–related death.
“In summary, we found no evidence of a beneficial effect of regular ICS use among people with COPD and asthma on COVID-19–related mortality,” the investigators concluded.
In agreement with the investigators, Dr. Conroy said that the increased mortality rate among ICS users should not be misconstrued as a medication-related risk.
“While the study found that those with COPD or asthma taking ICS and high-dose ICS were at an increased risk of death, this could easily be explained by the likelihood that those are the patients who are more likely to have more severe underlying lung disease,” Dr. Conroy said. “While this observational study did attempt to control for exacerbation history, the ability to do so by electronic health records data is certainly imperfect.”
With this in mind, patients with chronic respiratory disease should be encouraged to adhere to their usual treatment regimen, Dr. Conroy added.
“There isn’t evidence to increase or decrease medications just because of the pandemic,” she said. “A patient with asthma or COPD should continue to take the medications that are needed to achieve good control of their lung disease.”
The study was funded by the U.K. Medical Research Council. The investigators reported additional relationships with the Wellcome Trust, the Good Thinking Foundation, the Laura and John Arnold Foundation, and others. Dr. Conroy reported no conflicts of interest.
SOURCE: Schultze A et al. Lancet Respir Med. 2020 Sep 24. doi: 10.1016/ S2213-2600(20)30415-X.
, a study of almost 1 million individuals in the United Kingdom has shown.
Patients with chronic obstructive pulmonary disease or asthma who used ICS on a regular basis were more likely to die from COVID-19 than COPD or asthma patients who were prescribed non-ICS therapies, reported co-lead author Anna Schultze, PhD, of London School of Hygiene & Tropical Medicine and colleagues.
Of note, the increased risk of death among ICS users likely stemmed from greater severity of preexisting chronic respiratory conditions, instead of directly from ICS usage, which has little apparent impact on COVID-19 mortality, the investigators wrote in Lancet Respiratory Medicine.
These findings conflict with a hypothesis proposed early in the pandemic: that ICS may protect individuals from SARS-CoV-2 infection and poor outcomes with COVID-19.
According to Megan Conroy, MD, of the department of internal medicine at the Ohio State University Wexner Medical Center, Columbus, this hypothesis was based on some unexpected epidemiological findings.
“In general, we tend to think people with underlying lung disease – like COPD or asthma – to be at higher risk for severe forms of lower respiratory tract infections,” Dr. Conroy said. “Somewhat surprisingly, early data in the pandemic showed patients with COPD and asthma [were] underrepresented [among patients with COVID] when compared to the prevalence of these diseases in the population.”
This raised the possibility of an incidental protective effect from regular ICS therapy, which “had some strong theoretic pathophysiologic basis,” Dr. Conroy said, referring to research that demonstrated ICS-mediated downregulation of SARS-CoV-2 entry receptors ACE2 and TMPRSS2.
Dr. Schultze and colleagues noted that investigators for two ongoing randomized controlled trials (NCT04331054, NCT04330586) are studying ICS as an intervention for COVID-19; but neither trial includes individuals already taking ICS for chronic respiratory disease.
The present observational study therefore aimed to assess mortality risk within this population. Data were drawn from electronic health records and a U.K. national mortality database, with follow-up ranging from March 1 to May 6, 2020. Eligibility required a relevant prescription within 4 months of first follow-up. In the COPD group, patients were prescribed a long-acting beta agonist plus a long-acting muscarinic antagonist (LABA–LAMA), LABA alone, LABA plus ICS, LABA–LAMA plus ICS, or ICS alone (if prescribed LABA within 4 months).
In the asthma group, patients received low/medium-dose ICS, high-dose ICS, or a short-acting beta agonist (SABA) alone. Patients with COPD were at least 35 years of age, while those with asthma were 18 years or older. Hazard ratios were adjusted for a variety of covariates, including respiratory disease–exacerbation history, age, sex, body mass index, hypertension, diabetes, and others.
These eligibility criteria returned 148,557 patients with COPD and 818,490 with asthma.
Patients with COPD who were prescribed ICS plus LABA-LAMA or ICS plus LABA had an increased risk of COVID-19-related death, compared with those who did not receive ICS (adjusted hazard ratio, 1.39; 95% confidence interval, 1.10-1.76). Separate analyses of patients who received a triple combination (LABA–LAMA plus ICS) versus those who took a dual combination (LABA plus ICS) showed that triple-combination therapy was significantly associated with increased COVID-19-related mortality (aHR, 1.43; 95% CI, 1.12-1.83), while dual-combination therapy was less so (aHR, 1.29; 95% CI, 0.96-1.74). Non–COVID-19–related mortality was significantly increased for all COPD patients who were prescribed ICS, with or without adjustment for covariates.
Asthma patients prescribed high-dose ICS instead of SABA alone had a slightly greater risk of COVID-19–related death, based on an adjusted hazard ratio of 1.55 (95% CI, 1.10-2.18). Those with asthma who received low/medium–dose ICS demonstrated a slight trend toward increased mortality risk, but this was not significant (aHR, 1.14; 95% CI, 0.85-1.54). ICS usage in the asthma group was not linked with a significant increase in non–COVID-19–related death.
“In summary, we found no evidence of a beneficial effect of regular ICS use among people with COPD and asthma on COVID-19–related mortality,” the investigators concluded.
In agreement with the investigators, Dr. Conroy said that the increased mortality rate among ICS users should not be misconstrued as a medication-related risk.
“While the study found that those with COPD or asthma taking ICS and high-dose ICS were at an increased risk of death, this could easily be explained by the likelihood that those are the patients who are more likely to have more severe underlying lung disease,” Dr. Conroy said. “While this observational study did attempt to control for exacerbation history, the ability to do so by electronic health records data is certainly imperfect.”
With this in mind, patients with chronic respiratory disease should be encouraged to adhere to their usual treatment regimen, Dr. Conroy added.
“There isn’t evidence to increase or decrease medications just because of the pandemic,” she said. “A patient with asthma or COPD should continue to take the medications that are needed to achieve good control of their lung disease.”
The study was funded by the U.K. Medical Research Council. The investigators reported additional relationships with the Wellcome Trust, the Good Thinking Foundation, the Laura and John Arnold Foundation, and others. Dr. Conroy reported no conflicts of interest.
SOURCE: Schultze A et al. Lancet Respir Med. 2020 Sep 24. doi: 10.1016/ S2213-2600(20)30415-X.
FROM LANCET RESPIRATORY MEDICINE
Retrospective Review on the Safety and Efficacy of Direct Oral Anticoagulants Compared With Warfarin in Patients With Cirrhosis
Coagulation in patients with cirrhosis is a complicated area of evolving research. Patients with cirrhosis were originally thought to be naturally anticoagulated due to the decreased production of clotting factors and platelets, combined with an increased international normalized ratio (INR).1 New data have shown that patients with cirrhosis are at a concomitant risk of bleeding and thrombosis due to increased platelet aggregation, decreased fibrinolysis, and decreased production of natural anticoagulants such as protein C and antithrombin.1 Traditionally, patients with cirrhosis needing anticoagulation therapy for comorbid conditions, such as nonvalvular atrial fibrillation (NVAF) or venous thromboembolism (VTE) were placed on warfarin therapy. Managing warfarin in patients with cirrhosis poses a challenge to clinicians due to the many food and drug interactions, narrow therapeutic index, and complications with maintaining a therapeutic INR.1
Direct oral anticoagulants (DOACs) have several benefits over warfarin therapy, including convenience, decreased monitoring, decreased drug and dietary restrictions, and faster onset of action.2 Conversely, DOACs undergo extensive hepatic metabolism giving rise to concerns about supratherapeutic drug levels and increased bleeding rates in patients with liver dysfunction.1 Consequently, patients with cirrhosis were excluded from the pivotal trials establishing DOACs for NVAF and VTE treatment. Exclusion of these patients in major clinical trials alongside the challenges of managing warfarin warrant an evaluation of the efficacy and safety of DOACs in patients with cirrhosis.
Recent retrospective studies have examined the use of DOACs in patients with cirrhosis and found favorable results. A retrospective chart review by Intagliata and colleagues consisting of 39 patients with cirrhosis using either a DOAC or warfarin found similar rates of all-cause bleeding and major bleeding between the 2 groups.3 A retrospective cohort study by Hum and colleagues consisting of 45 patients with cirrhosis compared the use of DOACs with warfarin or low-molecular weight heparin (LMWH).4 Hum and colleagues found patients prescribed a DOAC had significantly fewer major bleeding events than did patients using warfarin or LMWH.4 The largest retrospective cohort study consisted of 233 patients with chronic liver disease and found no differences among all-cause bleeding and major bleeding rates between patients using DOACs compared with those of patients using warfarin.5
The purpose of this research is to evaluate the safety and efficacy of DOACs in veteran patients with cirrhosis compared with patients using warfarin.
Methods
A retrospective single-center chart review was conducted at the Michael E. DeBakey Veterans Affairs Medical Center (MEDVAMC) in Houston, Texas, between October 31, 2014 and October 31, 2018. Patients included in the study were adults aged ≥ 18 years with a diagnosis of cirrhosis and prescribed any of the following oral anticoagulants: apixaban, dabigatran, edoxaban, rivaroxaban, or warfarin. Patients prescribed apixaban, dabigatran, edoxaban, or rivaroxaban were collectively grouped into the DOAC group, while patients prescribed warfarin were classified as the standard of care comparator group.
A diagnosis of cirrhosis was confirmed using a combination of the codes from the ninth and tenth editions of the International Classification of Diseases (ICD) for cirrhosis, documentation of diagnostic confirmation by clinicians from the gastroenterology or hepatology services, and positive liver biopsy result. Liver function tests, liver ultrasound results, and FibroSure biomarker assays were used to aid in confirming the diagnosis of cirrhosis but were not considered definitive. Patients were excluded from the trial if they had indications for anticoagulation other than NVAF and VTE and/or were prescribed triple antithrombotic therapy (dual antiplatelet therapy plus an anticoagulant). Patients who switched anticoagulant therapy during the trial period (ie, switched from warfarin to a DOAC) were also excluded from the analysis.
Patient demographic characteristics that were collected included weight; body mass index (BMI); etiology of cirrhosis; Child-Turcotte-Pugh, Model for End-Stage Liver Disease (MELD), and CHA2DS2-VASc score; concomitant antiplatelet, nonsteroidal anti-inflammatory drug (NSAID), proton pump inhibitor (PPI), and histamine-2 receptor antagonist
Two patient lists were used to identify patients for inclusion in the warfarin arm. The first patient list was generated using the US Department of Veterans Affairs (VA) Cirrhosis Tracker, which identified patients with an ICD-9/10 code for cirrhosis and an INR laboratory value. Patients generated from the VA Cirrhosis Tracker with an INR > 1.5 were screened for a warfarin prescription and then evaluated for full study inclusion. The second patient list was generated using the VA Advanced Liver Disease Dashboard which identified patients with ICD-9/10 codes for advanced liver disease and an active warfarin prescription. Patients with an active warfarin prescription were then evaluated for full study inclusion. A single patient list was generated to identify patients for inclusion in the DOAC arm. This patient list was generated using the VA DOAC dashboard, which identified patients with an active DOAC prescription and an ICD-9/10 code for cirrhosis. Patients with an ICD-9/10 code for cirrhosis and prescribed a DOAC were screened for full study inclusion. Patient data were collected from the MEDVAMC Computerized Patient Record System (CPRS) electronic health record (EHR). The research study was approved by the Baylor College of Medicine Institutional Review Board and the VA Office of Research and Development.
Outcomes
The primary endpoint for the study was all-cause bleeding. The secondary endpoints for the study were major bleeding and failed efficacy. Major bleeding was defined using the International Society on Thrombosis and Haemostasis (ISTH) 2005 definition: fatal bleeding, symptomatic bleeding in a critical organ area (ie, intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, or intramuscular with compartment syndrome), or bleeding causing a fall in hemoglobin level of > 2 g/dL or leading to the transfusion of ≥ 2 units of red cells.6 Failed efficacy was a combination endpoint that included development of VTE, stroke, myocardial infarction (MI), and/or death. A prespecified subgroup analysis was conducted at the end of the study period to analyze trends in the DOAC and warfarin groups with respect to all-cause bleeding. All-cause bleeding risk was stratified by weight, BMI, Child-Turcotte-Pugh score, MELD score, presence of gastric and/or esophageal varices, active malignancies, percentage of time within therapeutic INR range in the warfarin group, indications for anticoagulation, and antiplatelet, NSAID, PPI, and H2RA therapy.
Statistical Analysis
Data were analyzed using descriptive and inferential statistics. Continuous data were analyzed using the Student t test, and categorical data were analyzed using the Fisher exact test. Previous studies determined an all-cause bleeding rate of 10 to 17% for warfarin compared with 5% for DOACs.7,8 To detect a 12% difference in the all-cause bleeding rate between DOACs and warfarin, 212 patients would be needed to achieve 80% power at an α level of 0.05.
Results
A total of 170 patients were screened, and after applying inclusion and exclusion criteria, 79 patients were enrolled in the study (Figure). The DOAC group included 42 patients, and the warfarin group included 37 patients. In the DOAC group, 69.1% (n = 29) of patients were taking apixaban, 21.4% (n = 9) rivaroxaban, and 9.5% (n = 4) dabigatran. There were no patients prescribed edoxaban during the study period.
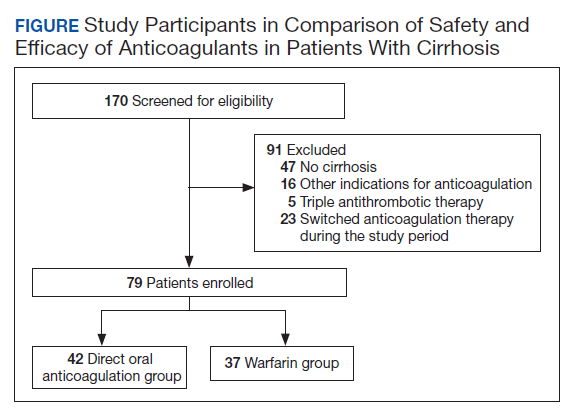
Baseline characteristics were similar between the 2 groups except for Child-Turcotte-Pugh score, MELD score, mean INR, and number of days on anticoagulation therapy (Table 1). Most of the patients were male (98.7%), and the mean age was 71 years. The most common causes of cirrhosis were viral (29.1%), nonalcoholic fatty liver disease (NAFLD) (24.1%), multiple causes (22.8%), and alcohol (21.5%). Sixty-two patients (78.5%) had a NVAF indication for anticoagulation. The average CHA2DS2-VASc score was 3.7. Aspirin was prescribed in 51.9% (n = 41) of patients, and PPIs were prescribed in 48.1% (n = 38) of patients. At inclusion, esophageal varices were present in 13 patients and active malignancies were present in 6 patients.
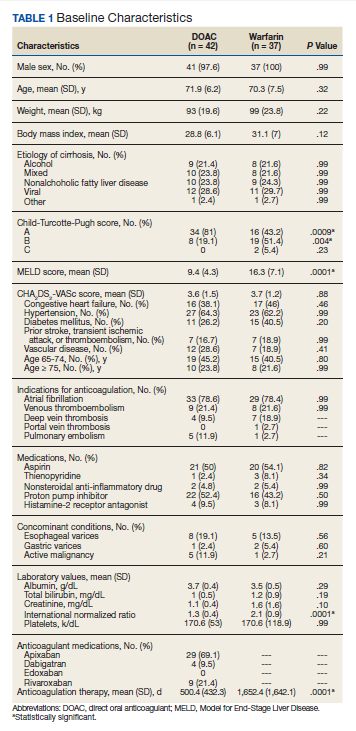
Statistically significant differences in baseline characteristics were found between mean INR, Child-Turcotte-Pugh scores, MELD scores, and number of days on anticoagulant therapy. The mean INR was 1.3 in the DOAC group compared with 2.1 in the warfarin group (P = .0001). Eighty-one percent (n = 34) of patients in the DOAC group had a Child-Turcotte-Pugh score of A compared with 43.2% (n = 16) of patients in the warfarin group (P = .0009). Eight patients in the DOAC group had a Child-Turcotte-Pugh score of B compared with 19 patients in the warfarin group (P = .004). The mean MELD score was 9.4 in the DOAC group compared with 16.3 in the warfarin group (P = .0001). The mean days on anticoagulant therapy was 500.4 days for the DOAC group compared with 1,652.4 days for the warfarin group (P = .0001).
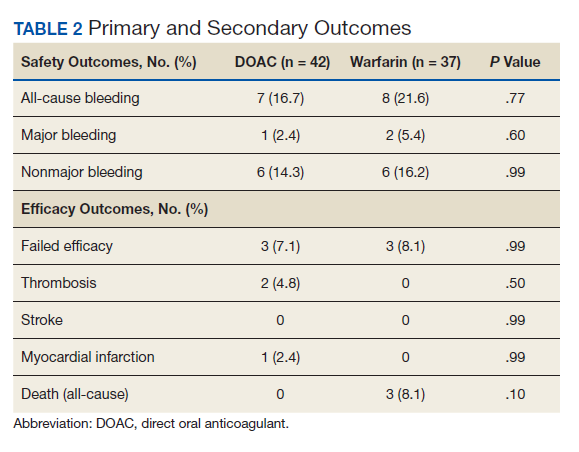
Safety Outcome
The primary outcome comparing all-cause bleeding rates between patients on DOACs compared with warfarin are listed in Table 2. With respect to the primary outcome, 7 (16.7%) patients on DOACs experienced a bleeding event compared with 8 (21.6%) patients on warfarin (P = .77). No statistically significant differences were detected between the DOAC and warfarin groups with respect to all-cause bleeding. Seven bleeding events occurred in the DOAC group; 1 met the qualification for major bleeding with a suspected gastrointestinal (GI) bleed.6 The other 6 bleeding episodes in the DOAC group consisted of hematoma, epistaxis, hematuria, and hematochezia. Eight bleeding events occurred in the warfarin group; 2 met the qualification for major bleeding with an intracranial hemorrhage and upper GI bleed.6 The other 6 bleeding episodes in the warfarin group consisted of epistaxis, bleeding gums, hematuria, and hematochezia. There were no statistically significant differences between the rates of major bleeding and nonmajor bleeding between the DOAC and warfarin groups.
Efficacy Outcomes
There were 3 events in the DOAC group and 3 events in the warfarin group (P = .99). In the DOAC group, 2 patients experienced a pulmonary embolism, and 1 patient experienced a MI. In the warfarin group, 3 patients died (end-stage heart failure, unknown cause due to death at an outside hospital, and sepsis/organ failure). There were no statistically significant differences between the composite endpoint of failed efficacy or the individual endpoints of VTE, stroke, MI, and death.
Subgroup Analysis
A prespecified subgroup analysis was conducted to determine risk factors for all-cause bleeding within each treatment group (Table 3). No significant trends were observed in the following risk factors: Child-Turcotte-Pugh score, indication for anticoagulation, use of NSAIDs, PPIs or H2RAs, presence of gastric or esophageal varices, active malignancies, and time within therapeutic INR range in the warfarin group. Patients with bleeding events had slightly increased weight and BMI vs patients without bleeding events. Within the warfarin group, patients with bleeding events had slightly elevated MELD scores compared to patients without bleeding events. There was an equal balance of patients prescribed aspirin therapy between the groups with and without bleeding events. Overall, no significant risk factors were identified for all-cause bleeding.
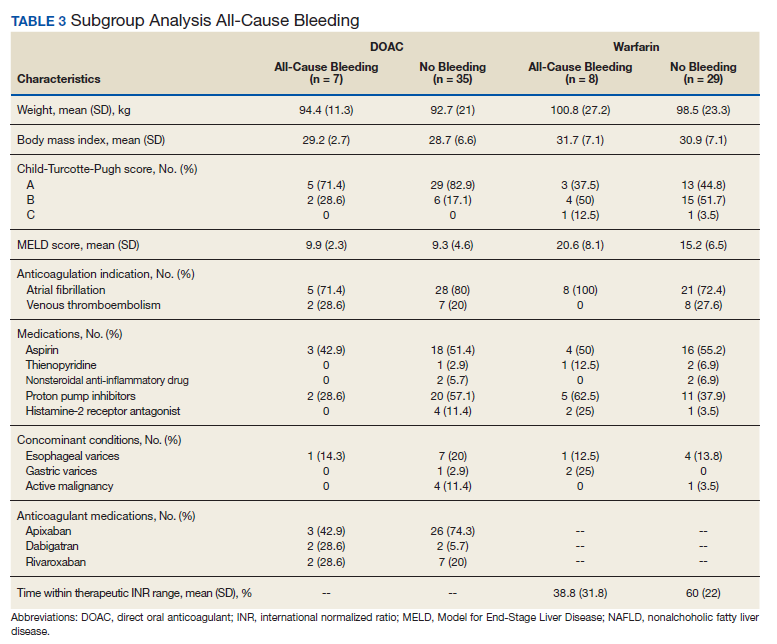
Discussion
Initially, patients with cirrhosis were excluded from DOAC trials due to concerns for increased bleeding risk with hepatically eliminated medications. New retrospective research has concluded that in patients with cirrhosis, DOACs have similar or lower bleeding rates when compared directly to warfarin.9,10
In this study, no statistically significant differences were detected between the primary and secondary outcomes of all-cause bleeding, major bleeding, or failed efficacy. Subgroup analysis did not identify any significant risk factors with respect to all-cause bleeding among patients in the DOAC and warfarin groups. To meet 80% power, 212 patients needed to be enrolled in the study; however, only 79 patients were enrolled, and power was not met. The results of this study should be interpreted cautiously as hypothesis-generating due to the small sample size. Strengths of this study include similar baseline characteristics between the DOAC and warfarin groups, 4-year length of retrospective data review, and availability of both inpatient and outpatient EHR limiting the amount of missing data points.
Baseline characteristics were similar between the groups except for mean INR, Child-Turcotte-Pugh score, MELD score, and number of days on anticoagulation therapy. The difference in mean INR between groups is expected as patients in the warfarin group have a goal INR of 2 to 3 to maintain therapeutic efficacy and safety. INR is not used as a marker of efficacy or safety with DOACs; therefore, a consistent elevation in INR is not expected. Child- Turcotte-Pugh scores are calculated using INR levels.11 When calculating the score, patients with an INR < 1.7 receive 1 point; patients with an INR between 1.7 and 2.3 receive 2 points.11 Therefore, patients in the warfarin group will have artificially inflated Child-Turcotte-Pugh scores as this group has goal INR levels of 2 to 3. This makes Child-Turcotte-Pugh scores unreliable markers of disease severity in patients using warfarin therapy. When the INR scores for patients prescribed warfarin were replaced with values < 1.7, the statistical difference disappeared between the warfarin and DOAC groups. The same effect is seen on MELD scores for patients prescribed warfarin therapy. The MELD score is calculated using INR levels.12 MELD scores also will be artificially elevated in patients prescribed warfarin therapy due to the INR elevation to between 2 and 3. When MELD scores for patients prescribed warfarin were replaced with values similar to those in the DOAC group, the statistical difference disappeared between the warfarin and DOAC groups.
The last statistically significant difference was found in number of days on anticoagulant therapy. This difference was expected as warfarin is the standard of care for anticoagulation treatment in patients with cirrhosis. The first DOAC, dabigatran, was not approved by the US Food and Drug Administration until 2010.13 DOACs have only recently been used in patients with cirrhosis accounting for the statistically significant difference in days on anticoagulation therapy between the warfarin and DOAC groups.
Limitations
The inability to meet power or evaluate adherence and appropriate renal dose adjustments for DOACs limited this study. This study was conducted at a single center in a predominantly male veteran population and therefore may not be generalizable to other populations. A majority of patients in the DOAC group were prescribed apixaban (69.1%), which may have affected the overall rate of major bleeding in the DOAC group. Pivotal trials of apixaban have shown a consistent decreased risk of major bleeding in patients with NVAF or VTE when compared with warfarin.14,15 Therefore, the results of this study may not be generalizable to all DOACs.
An inherent limitation of this study was the inability to collect data verifying adherence in the DOAC group. However, in the warfarin group, percentage of time within the therapeutic INR range of 2 to 3 was collected. While not a direct marker of adherence, this does allow for limited evaluation of therapeutic efficacy and safety within the warfarin group. Last, proper dosing of DOACs in patients with and without adequate renal function was not evaluated in this study.
Conclusions
The results of this study are consistent with other retrospective research and literature reviews. There were no statistically significant differences identified between the rates of all-cause bleeding, major bleeding, and failed efficacy between the DOAC and warfarin groups. DOACs may be a safe alternative to warfarin in patients with cirrhosis requiring anticoagulation for NVAF or VTE, but large randomized trials are required to confirm these results.
1. Qamar A, Vaduganathan M, Greenberger NJ, Giugliano RP. Oral anticoagulation in patients with liver disease. J Am Coll Cardiol. 2018;71(19):2162-2175. doi:10.1016/j.jacc.2018.03.023
2. Priyanka P, Kupec JT, Krafft M, Shah NA, Reynolds GJ. Newer oral anticoagulants in the treatment of acute portal vein thrombosis in patients with and without cirrhosis. Int J Hepatol. 2018;2018:8432781. Published 2018 Jun 5. doi:10.1155/2018/8432781
3. Intagliata NM, Henry ZH, Maitland H, et al. Direct oral anticoagulants in cirrhosis patients pose similar risks of bleeding when compared to traditional anticoagulation. Dig Dis Sci. 2016;61(6):1721-1727. doi:10.1007/s10620-015-4012-2
4. Hum J, Shatzel JJ, Jou JH, Deloughery TG. The efficacy and safety of direct oral anticoagulants vs traditional anticoagulants in cirrhosis. Eur J Haematol. 2017;98(4):393-397. doi:10.1111/ejh.12844
5. Goriacko P, Veltri KT. Safety of direct oral anticoagulants vs warfarin in patients with chronic liver disease and atrial fibrillation. Eur J Haematol. 2018;100(5):488-493. doi:10.1111/ejh.13045
6. Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692-694. doi:10.1111/j.1538-7836.2005.01204.x
7. Rubboli A, Becattini C, Verheugt FW. Incidence, clinical impact and risk of bleeding during oral anticoagulation therapy. World J Cardiol. 2011;3(11):351-358. doi:10.4330/wjc.v3.i11.351
8. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
9. Hoolwerf EW, Kraaijpoel N, Büller HR, van Es N. Direct oral anticoagulants in patients with liver cirrhosis: A systematic review. Thromb Res. 2018;170:102-108. doi:10.1016/j.thromres.2018.08.011
10. Steuber TD, Howard ML, Nisly SA. Direct oral anticoagulants in chronic liver disease. Ann Pharmacother. 2019;53(10):1042-1049. doi:10.1177/1060028019841582
11. Janevska D, Chaloska-Ivanova V, Janevski V. Hepatocellular carcinoma: risk factors, diagnosis and treatment. Open Access Maced J Med Sci. 2015;3(4):732-736. doi:10.3889/oamjms.2015.111
12. Singal AK, Kamath PS. Model for End-Stage Liver Disease. J Clin Exp Hepatol. 2013;3(1):50-60. doi:10.1016/j.jceh.2012.11.002
13. Joppa SA, Salciccioli J, Adamski J, et al. A practical review of the emerging direct anticoagulants, laboratory monitoring, and reversal agents. J Clin Med. 2018;7(2):29. Published 2018 Feb 11. doi:10.3390/jcm7020029
14. Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. doi:10.1056/NEJMoa1107039
15. Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799-808. doi:10.1056/NEJMoa1302507
Coagulation in patients with cirrhosis is a complicated area of evolving research. Patients with cirrhosis were originally thought to be naturally anticoagulated due to the decreased production of clotting factors and platelets, combined with an increased international normalized ratio (INR).1 New data have shown that patients with cirrhosis are at a concomitant risk of bleeding and thrombosis due to increased platelet aggregation, decreased fibrinolysis, and decreased production of natural anticoagulants such as protein C and antithrombin.1 Traditionally, patients with cirrhosis needing anticoagulation therapy for comorbid conditions, such as nonvalvular atrial fibrillation (NVAF) or venous thromboembolism (VTE) were placed on warfarin therapy. Managing warfarin in patients with cirrhosis poses a challenge to clinicians due to the many food and drug interactions, narrow therapeutic index, and complications with maintaining a therapeutic INR.1
Direct oral anticoagulants (DOACs) have several benefits over warfarin therapy, including convenience, decreased monitoring, decreased drug and dietary restrictions, and faster onset of action.2 Conversely, DOACs undergo extensive hepatic metabolism giving rise to concerns about supratherapeutic drug levels and increased bleeding rates in patients with liver dysfunction.1 Consequently, patients with cirrhosis were excluded from the pivotal trials establishing DOACs for NVAF and VTE treatment. Exclusion of these patients in major clinical trials alongside the challenges of managing warfarin warrant an evaluation of the efficacy and safety of DOACs in patients with cirrhosis.
Recent retrospective studies have examined the use of DOACs in patients with cirrhosis and found favorable results. A retrospective chart review by Intagliata and colleagues consisting of 39 patients with cirrhosis using either a DOAC or warfarin found similar rates of all-cause bleeding and major bleeding between the 2 groups.3 A retrospective cohort study by Hum and colleagues consisting of 45 patients with cirrhosis compared the use of DOACs with warfarin or low-molecular weight heparin (LMWH).4 Hum and colleagues found patients prescribed a DOAC had significantly fewer major bleeding events than did patients using warfarin or LMWH.4 The largest retrospective cohort study consisted of 233 patients with chronic liver disease and found no differences among all-cause bleeding and major bleeding rates between patients using DOACs compared with those of patients using warfarin.5
The purpose of this research is to evaluate the safety and efficacy of DOACs in veteran patients with cirrhosis compared with patients using warfarin.
Methods
A retrospective single-center chart review was conducted at the Michael E. DeBakey Veterans Affairs Medical Center (MEDVAMC) in Houston, Texas, between October 31, 2014 and October 31, 2018. Patients included in the study were adults aged ≥ 18 years with a diagnosis of cirrhosis and prescribed any of the following oral anticoagulants: apixaban, dabigatran, edoxaban, rivaroxaban, or warfarin. Patients prescribed apixaban, dabigatran, edoxaban, or rivaroxaban were collectively grouped into the DOAC group, while patients prescribed warfarin were classified as the standard of care comparator group.
A diagnosis of cirrhosis was confirmed using a combination of the codes from the ninth and tenth editions of the International Classification of Diseases (ICD) for cirrhosis, documentation of diagnostic confirmation by clinicians from the gastroenterology or hepatology services, and positive liver biopsy result. Liver function tests, liver ultrasound results, and FibroSure biomarker assays were used to aid in confirming the diagnosis of cirrhosis but were not considered definitive. Patients were excluded from the trial if they had indications for anticoagulation other than NVAF and VTE and/or were prescribed triple antithrombotic therapy (dual antiplatelet therapy plus an anticoagulant). Patients who switched anticoagulant therapy during the trial period (ie, switched from warfarin to a DOAC) were also excluded from the analysis.
Patient demographic characteristics that were collected included weight; body mass index (BMI); etiology of cirrhosis; Child-Turcotte-Pugh, Model for End-Stage Liver Disease (MELD), and CHA2DS2-VASc score; concomitant antiplatelet, nonsteroidal anti-inflammatory drug (NSAID), proton pump inhibitor (PPI), and histamine-2 receptor antagonist
Two patient lists were used to identify patients for inclusion in the warfarin arm. The first patient list was generated using the US Department of Veterans Affairs (VA) Cirrhosis Tracker, which identified patients with an ICD-9/10 code for cirrhosis and an INR laboratory value. Patients generated from the VA Cirrhosis Tracker with an INR > 1.5 were screened for a warfarin prescription and then evaluated for full study inclusion. The second patient list was generated using the VA Advanced Liver Disease Dashboard which identified patients with ICD-9/10 codes for advanced liver disease and an active warfarin prescription. Patients with an active warfarin prescription were then evaluated for full study inclusion. A single patient list was generated to identify patients for inclusion in the DOAC arm. This patient list was generated using the VA DOAC dashboard, which identified patients with an active DOAC prescription and an ICD-9/10 code for cirrhosis. Patients with an ICD-9/10 code for cirrhosis and prescribed a DOAC were screened for full study inclusion. Patient data were collected from the MEDVAMC Computerized Patient Record System (CPRS) electronic health record (EHR). The research study was approved by the Baylor College of Medicine Institutional Review Board and the VA Office of Research and Development.
Outcomes
The primary endpoint for the study was all-cause bleeding. The secondary endpoints for the study were major bleeding and failed efficacy. Major bleeding was defined using the International Society on Thrombosis and Haemostasis (ISTH) 2005 definition: fatal bleeding, symptomatic bleeding in a critical organ area (ie, intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, or intramuscular with compartment syndrome), or bleeding causing a fall in hemoglobin level of > 2 g/dL or leading to the transfusion of ≥ 2 units of red cells.6 Failed efficacy was a combination endpoint that included development of VTE, stroke, myocardial infarction (MI), and/or death. A prespecified subgroup analysis was conducted at the end of the study period to analyze trends in the DOAC and warfarin groups with respect to all-cause bleeding. All-cause bleeding risk was stratified by weight, BMI, Child-Turcotte-Pugh score, MELD score, presence of gastric and/or esophageal varices, active malignancies, percentage of time within therapeutic INR range in the warfarin group, indications for anticoagulation, and antiplatelet, NSAID, PPI, and H2RA therapy.
Statistical Analysis
Data were analyzed using descriptive and inferential statistics. Continuous data were analyzed using the Student t test, and categorical data were analyzed using the Fisher exact test. Previous studies determined an all-cause bleeding rate of 10 to 17% for warfarin compared with 5% for DOACs.7,8 To detect a 12% difference in the all-cause bleeding rate between DOACs and warfarin, 212 patients would be needed to achieve 80% power at an α level of 0.05.
Results
A total of 170 patients were screened, and after applying inclusion and exclusion criteria, 79 patients were enrolled in the study (Figure). The DOAC group included 42 patients, and the warfarin group included 37 patients. In the DOAC group, 69.1% (n = 29) of patients were taking apixaban, 21.4% (n = 9) rivaroxaban, and 9.5% (n = 4) dabigatran. There were no patients prescribed edoxaban during the study period.
Baseline characteristics were similar between the 2 groups except for Child-Turcotte-Pugh score, MELD score, mean INR, and number of days on anticoagulation therapy (Table 1). Most of the patients were male (98.7%), and the mean age was 71 years. The most common causes of cirrhosis were viral (29.1%), nonalcoholic fatty liver disease (NAFLD) (24.1%), multiple causes (22.8%), and alcohol (21.5%). Sixty-two patients (78.5%) had a NVAF indication for anticoagulation. The average CHA2DS2-VASc score was 3.7. Aspirin was prescribed in 51.9% (n = 41) of patients, and PPIs were prescribed in 48.1% (n = 38) of patients. At inclusion, esophageal varices were present in 13 patients and active malignancies were present in 6 patients.
Statistically significant differences in baseline characteristics were found between mean INR, Child-Turcotte-Pugh scores, MELD scores, and number of days on anticoagulant therapy. The mean INR was 1.3 in the DOAC group compared with 2.1 in the warfarin group (P = .0001). Eighty-one percent (n = 34) of patients in the DOAC group had a Child-Turcotte-Pugh score of A compared with 43.2% (n = 16) of patients in the warfarin group (P = .0009). Eight patients in the DOAC group had a Child-Turcotte-Pugh score of B compared with 19 patients in the warfarin group (P = .004). The mean MELD score was 9.4 in the DOAC group compared with 16.3 in the warfarin group (P = .0001). The mean days on anticoagulant therapy was 500.4 days for the DOAC group compared with 1,652.4 days for the warfarin group (P = .0001).
Safety Outcome
The primary outcome comparing all-cause bleeding rates between patients on DOACs compared with warfarin are listed in Table 2. With respect to the primary outcome, 7 (16.7%) patients on DOACs experienced a bleeding event compared with 8 (21.6%) patients on warfarin (P = .77). No statistically significant differences were detected between the DOAC and warfarin groups with respect to all-cause bleeding. Seven bleeding events occurred in the DOAC group; 1 met the qualification for major bleeding with a suspected gastrointestinal (GI) bleed.6 The other 6 bleeding episodes in the DOAC group consisted of hematoma, epistaxis, hematuria, and hematochezia. Eight bleeding events occurred in the warfarin group; 2 met the qualification for major bleeding with an intracranial hemorrhage and upper GI bleed.6 The other 6 bleeding episodes in the warfarin group consisted of epistaxis, bleeding gums, hematuria, and hematochezia. There were no statistically significant differences between the rates of major bleeding and nonmajor bleeding between the DOAC and warfarin groups.
Efficacy Outcomes
There were 3 events in the DOAC group and 3 events in the warfarin group (P = .99). In the DOAC group, 2 patients experienced a pulmonary embolism, and 1 patient experienced a MI. In the warfarin group, 3 patients died (end-stage heart failure, unknown cause due to death at an outside hospital, and sepsis/organ failure). There were no statistically significant differences between the composite endpoint of failed efficacy or the individual endpoints of VTE, stroke, MI, and death.
Subgroup Analysis
A prespecified subgroup analysis was conducted to determine risk factors for all-cause bleeding within each treatment group (Table 3). No significant trends were observed in the following risk factors: Child-Turcotte-Pugh score, indication for anticoagulation, use of NSAIDs, PPIs or H2RAs, presence of gastric or esophageal varices, active malignancies, and time within therapeutic INR range in the warfarin group. Patients with bleeding events had slightly increased weight and BMI vs patients without bleeding events. Within the warfarin group, patients with bleeding events had slightly elevated MELD scores compared to patients without bleeding events. There was an equal balance of patients prescribed aspirin therapy between the groups with and without bleeding events. Overall, no significant risk factors were identified for all-cause bleeding.
Discussion
Initially, patients with cirrhosis were excluded from DOAC trials due to concerns for increased bleeding risk with hepatically eliminated medications. New retrospective research has concluded that in patients with cirrhosis, DOACs have similar or lower bleeding rates when compared directly to warfarin.9,10
In this study, no statistically significant differences were detected between the primary and secondary outcomes of all-cause bleeding, major bleeding, or failed efficacy. Subgroup analysis did not identify any significant risk factors with respect to all-cause bleeding among patients in the DOAC and warfarin groups. To meet 80% power, 212 patients needed to be enrolled in the study; however, only 79 patients were enrolled, and power was not met. The results of this study should be interpreted cautiously as hypothesis-generating due to the small sample size. Strengths of this study include similar baseline characteristics between the DOAC and warfarin groups, 4-year length of retrospective data review, and availability of both inpatient and outpatient EHR limiting the amount of missing data points.
Baseline characteristics were similar between the groups except for mean INR, Child-Turcotte-Pugh score, MELD score, and number of days on anticoagulation therapy. The difference in mean INR between groups is expected as patients in the warfarin group have a goal INR of 2 to 3 to maintain therapeutic efficacy and safety. INR is not used as a marker of efficacy or safety with DOACs; therefore, a consistent elevation in INR is not expected. Child- Turcotte-Pugh scores are calculated using INR levels.11 When calculating the score, patients with an INR < 1.7 receive 1 point; patients with an INR between 1.7 and 2.3 receive 2 points.11 Therefore, patients in the warfarin group will have artificially inflated Child-Turcotte-Pugh scores as this group has goal INR levels of 2 to 3. This makes Child-Turcotte-Pugh scores unreliable markers of disease severity in patients using warfarin therapy. When the INR scores for patients prescribed warfarin were replaced with values < 1.7, the statistical difference disappeared between the warfarin and DOAC groups. The same effect is seen on MELD scores for patients prescribed warfarin therapy. The MELD score is calculated using INR levels.12 MELD scores also will be artificially elevated in patients prescribed warfarin therapy due to the INR elevation to between 2 and 3. When MELD scores for patients prescribed warfarin were replaced with values similar to those in the DOAC group, the statistical difference disappeared between the warfarin and DOAC groups.
The last statistically significant difference was found in number of days on anticoagulant therapy. This difference was expected as warfarin is the standard of care for anticoagulation treatment in patients with cirrhosis. The first DOAC, dabigatran, was not approved by the US Food and Drug Administration until 2010.13 DOACs have only recently been used in patients with cirrhosis accounting for the statistically significant difference in days on anticoagulation therapy between the warfarin and DOAC groups.
Limitations
The inability to meet power or evaluate adherence and appropriate renal dose adjustments for DOACs limited this study. This study was conducted at a single center in a predominantly male veteran population and therefore may not be generalizable to other populations. A majority of patients in the DOAC group were prescribed apixaban (69.1%), which may have affected the overall rate of major bleeding in the DOAC group. Pivotal trials of apixaban have shown a consistent decreased risk of major bleeding in patients with NVAF or VTE when compared with warfarin.14,15 Therefore, the results of this study may not be generalizable to all DOACs.
An inherent limitation of this study was the inability to collect data verifying adherence in the DOAC group. However, in the warfarin group, percentage of time within the therapeutic INR range of 2 to 3 was collected. While not a direct marker of adherence, this does allow for limited evaluation of therapeutic efficacy and safety within the warfarin group. Last, proper dosing of DOACs in patients with and without adequate renal function was not evaluated in this study.
Conclusions
The results of this study are consistent with other retrospective research and literature reviews. There were no statistically significant differences identified between the rates of all-cause bleeding, major bleeding, and failed efficacy between the DOAC and warfarin groups. DOACs may be a safe alternative to warfarin in patients with cirrhosis requiring anticoagulation for NVAF or VTE, but large randomized trials are required to confirm these results.
Coagulation in patients with cirrhosis is a complicated area of evolving research. Patients with cirrhosis were originally thought to be naturally anticoagulated due to the decreased production of clotting factors and platelets, combined with an increased international normalized ratio (INR).1 New data have shown that patients with cirrhosis are at a concomitant risk of bleeding and thrombosis due to increased platelet aggregation, decreased fibrinolysis, and decreased production of natural anticoagulants such as protein C and antithrombin.1 Traditionally, patients with cirrhosis needing anticoagulation therapy for comorbid conditions, such as nonvalvular atrial fibrillation (NVAF) or venous thromboembolism (VTE) were placed on warfarin therapy. Managing warfarin in patients with cirrhosis poses a challenge to clinicians due to the many food and drug interactions, narrow therapeutic index, and complications with maintaining a therapeutic INR.1
Direct oral anticoagulants (DOACs) have several benefits over warfarin therapy, including convenience, decreased monitoring, decreased drug and dietary restrictions, and faster onset of action.2 Conversely, DOACs undergo extensive hepatic metabolism giving rise to concerns about supratherapeutic drug levels and increased bleeding rates in patients with liver dysfunction.1 Consequently, patients with cirrhosis were excluded from the pivotal trials establishing DOACs for NVAF and VTE treatment. Exclusion of these patients in major clinical trials alongside the challenges of managing warfarin warrant an evaluation of the efficacy and safety of DOACs in patients with cirrhosis.
Recent retrospective studies have examined the use of DOACs in patients with cirrhosis and found favorable results. A retrospective chart review by Intagliata and colleagues consisting of 39 patients with cirrhosis using either a DOAC or warfarin found similar rates of all-cause bleeding and major bleeding between the 2 groups.3 A retrospective cohort study by Hum and colleagues consisting of 45 patients with cirrhosis compared the use of DOACs with warfarin or low-molecular weight heparin (LMWH).4 Hum and colleagues found patients prescribed a DOAC had significantly fewer major bleeding events than did patients using warfarin or LMWH.4 The largest retrospective cohort study consisted of 233 patients with chronic liver disease and found no differences among all-cause bleeding and major bleeding rates between patients using DOACs compared with those of patients using warfarin.5
The purpose of this research is to evaluate the safety and efficacy of DOACs in veteran patients with cirrhosis compared with patients using warfarin.
Methods
A retrospective single-center chart review was conducted at the Michael E. DeBakey Veterans Affairs Medical Center (MEDVAMC) in Houston, Texas, between October 31, 2014 and October 31, 2018. Patients included in the study were adults aged ≥ 18 years with a diagnosis of cirrhosis and prescribed any of the following oral anticoagulants: apixaban, dabigatran, edoxaban, rivaroxaban, or warfarin. Patients prescribed apixaban, dabigatran, edoxaban, or rivaroxaban were collectively grouped into the DOAC group, while patients prescribed warfarin were classified as the standard of care comparator group.
A diagnosis of cirrhosis was confirmed using a combination of the codes from the ninth and tenth editions of the International Classification of Diseases (ICD) for cirrhosis, documentation of diagnostic confirmation by clinicians from the gastroenterology or hepatology services, and positive liver biopsy result. Liver function tests, liver ultrasound results, and FibroSure biomarker assays were used to aid in confirming the diagnosis of cirrhosis but were not considered definitive. Patients were excluded from the trial if they had indications for anticoagulation other than NVAF and VTE and/or were prescribed triple antithrombotic therapy (dual antiplatelet therapy plus an anticoagulant). Patients who switched anticoagulant therapy during the trial period (ie, switched from warfarin to a DOAC) were also excluded from the analysis.
Patient demographic characteristics that were collected included weight; body mass index (BMI); etiology of cirrhosis; Child-Turcotte-Pugh, Model for End-Stage Liver Disease (MELD), and CHA2DS2-VASc score; concomitant antiplatelet, nonsteroidal anti-inflammatory drug (NSAID), proton pump inhibitor (PPI), and histamine-2 receptor antagonist
Two patient lists were used to identify patients for inclusion in the warfarin arm. The first patient list was generated using the US Department of Veterans Affairs (VA) Cirrhosis Tracker, which identified patients with an ICD-9/10 code for cirrhosis and an INR laboratory value. Patients generated from the VA Cirrhosis Tracker with an INR > 1.5 were screened for a warfarin prescription and then evaluated for full study inclusion. The second patient list was generated using the VA Advanced Liver Disease Dashboard which identified patients with ICD-9/10 codes for advanced liver disease and an active warfarin prescription. Patients with an active warfarin prescription were then evaluated for full study inclusion. A single patient list was generated to identify patients for inclusion in the DOAC arm. This patient list was generated using the VA DOAC dashboard, which identified patients with an active DOAC prescription and an ICD-9/10 code for cirrhosis. Patients with an ICD-9/10 code for cirrhosis and prescribed a DOAC were screened for full study inclusion. Patient data were collected from the MEDVAMC Computerized Patient Record System (CPRS) electronic health record (EHR). The research study was approved by the Baylor College of Medicine Institutional Review Board and the VA Office of Research and Development.
Outcomes
The primary endpoint for the study was all-cause bleeding. The secondary endpoints for the study were major bleeding and failed efficacy. Major bleeding was defined using the International Society on Thrombosis and Haemostasis (ISTH) 2005 definition: fatal bleeding, symptomatic bleeding in a critical organ area (ie, intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, or intramuscular with compartment syndrome), or bleeding causing a fall in hemoglobin level of > 2 g/dL or leading to the transfusion of ≥ 2 units of red cells.6 Failed efficacy was a combination endpoint that included development of VTE, stroke, myocardial infarction (MI), and/or death. A prespecified subgroup analysis was conducted at the end of the study period to analyze trends in the DOAC and warfarin groups with respect to all-cause bleeding. All-cause bleeding risk was stratified by weight, BMI, Child-Turcotte-Pugh score, MELD score, presence of gastric and/or esophageal varices, active malignancies, percentage of time within therapeutic INR range in the warfarin group, indications for anticoagulation, and antiplatelet, NSAID, PPI, and H2RA therapy.
Statistical Analysis
Data were analyzed using descriptive and inferential statistics. Continuous data were analyzed using the Student t test, and categorical data were analyzed using the Fisher exact test. Previous studies determined an all-cause bleeding rate of 10 to 17% for warfarin compared with 5% for DOACs.7,8 To detect a 12% difference in the all-cause bleeding rate between DOACs and warfarin, 212 patients would be needed to achieve 80% power at an α level of 0.05.
Results
A total of 170 patients were screened, and after applying inclusion and exclusion criteria, 79 patients were enrolled in the study (Figure). The DOAC group included 42 patients, and the warfarin group included 37 patients. In the DOAC group, 69.1% (n = 29) of patients were taking apixaban, 21.4% (n = 9) rivaroxaban, and 9.5% (n = 4) dabigatran. There were no patients prescribed edoxaban during the study period.
Baseline characteristics were similar between the 2 groups except for Child-Turcotte-Pugh score, MELD score, mean INR, and number of days on anticoagulation therapy (Table 1). Most of the patients were male (98.7%), and the mean age was 71 years. The most common causes of cirrhosis were viral (29.1%), nonalcoholic fatty liver disease (NAFLD) (24.1%), multiple causes (22.8%), and alcohol (21.5%). Sixty-two patients (78.5%) had a NVAF indication for anticoagulation. The average CHA2DS2-VASc score was 3.7. Aspirin was prescribed in 51.9% (n = 41) of patients, and PPIs were prescribed in 48.1% (n = 38) of patients. At inclusion, esophageal varices were present in 13 patients and active malignancies were present in 6 patients.
Statistically significant differences in baseline characteristics were found between mean INR, Child-Turcotte-Pugh scores, MELD scores, and number of days on anticoagulant therapy. The mean INR was 1.3 in the DOAC group compared with 2.1 in the warfarin group (P = .0001). Eighty-one percent (n = 34) of patients in the DOAC group had a Child-Turcotte-Pugh score of A compared with 43.2% (n = 16) of patients in the warfarin group (P = .0009). Eight patients in the DOAC group had a Child-Turcotte-Pugh score of B compared with 19 patients in the warfarin group (P = .004). The mean MELD score was 9.4 in the DOAC group compared with 16.3 in the warfarin group (P = .0001). The mean days on anticoagulant therapy was 500.4 days for the DOAC group compared with 1,652.4 days for the warfarin group (P = .0001).
Safety Outcome
The primary outcome comparing all-cause bleeding rates between patients on DOACs compared with warfarin are listed in Table 2. With respect to the primary outcome, 7 (16.7%) patients on DOACs experienced a bleeding event compared with 8 (21.6%) patients on warfarin (P = .77). No statistically significant differences were detected between the DOAC and warfarin groups with respect to all-cause bleeding. Seven bleeding events occurred in the DOAC group; 1 met the qualification for major bleeding with a suspected gastrointestinal (GI) bleed.6 The other 6 bleeding episodes in the DOAC group consisted of hematoma, epistaxis, hematuria, and hematochezia. Eight bleeding events occurred in the warfarin group; 2 met the qualification for major bleeding with an intracranial hemorrhage and upper GI bleed.6 The other 6 bleeding episodes in the warfarin group consisted of epistaxis, bleeding gums, hematuria, and hematochezia. There were no statistically significant differences between the rates of major bleeding and nonmajor bleeding between the DOAC and warfarin groups.
Efficacy Outcomes
There were 3 events in the DOAC group and 3 events in the warfarin group (P = .99). In the DOAC group, 2 patients experienced a pulmonary embolism, and 1 patient experienced a MI. In the warfarin group, 3 patients died (end-stage heart failure, unknown cause due to death at an outside hospital, and sepsis/organ failure). There were no statistically significant differences between the composite endpoint of failed efficacy or the individual endpoints of VTE, stroke, MI, and death.
Subgroup Analysis
A prespecified subgroup analysis was conducted to determine risk factors for all-cause bleeding within each treatment group (Table 3). No significant trends were observed in the following risk factors: Child-Turcotte-Pugh score, indication for anticoagulation, use of NSAIDs, PPIs or H2RAs, presence of gastric or esophageal varices, active malignancies, and time within therapeutic INR range in the warfarin group. Patients with bleeding events had slightly increased weight and BMI vs patients without bleeding events. Within the warfarin group, patients with bleeding events had slightly elevated MELD scores compared to patients without bleeding events. There was an equal balance of patients prescribed aspirin therapy between the groups with and without bleeding events. Overall, no significant risk factors were identified for all-cause bleeding.
Discussion
Initially, patients with cirrhosis were excluded from DOAC trials due to concerns for increased bleeding risk with hepatically eliminated medications. New retrospective research has concluded that in patients with cirrhosis, DOACs have similar or lower bleeding rates when compared directly to warfarin.9,10
In this study, no statistically significant differences were detected between the primary and secondary outcomes of all-cause bleeding, major bleeding, or failed efficacy. Subgroup analysis did not identify any significant risk factors with respect to all-cause bleeding among patients in the DOAC and warfarin groups. To meet 80% power, 212 patients needed to be enrolled in the study; however, only 79 patients were enrolled, and power was not met. The results of this study should be interpreted cautiously as hypothesis-generating due to the small sample size. Strengths of this study include similar baseline characteristics between the DOAC and warfarin groups, 4-year length of retrospective data review, and availability of both inpatient and outpatient EHR limiting the amount of missing data points.
Baseline characteristics were similar between the groups except for mean INR, Child-Turcotte-Pugh score, MELD score, and number of days on anticoagulation therapy. The difference in mean INR between groups is expected as patients in the warfarin group have a goal INR of 2 to 3 to maintain therapeutic efficacy and safety. INR is not used as a marker of efficacy or safety with DOACs; therefore, a consistent elevation in INR is not expected. Child- Turcotte-Pugh scores are calculated using INR levels.11 When calculating the score, patients with an INR < 1.7 receive 1 point; patients with an INR between 1.7 and 2.3 receive 2 points.11 Therefore, patients in the warfarin group will have artificially inflated Child-Turcotte-Pugh scores as this group has goal INR levels of 2 to 3. This makes Child-Turcotte-Pugh scores unreliable markers of disease severity in patients using warfarin therapy. When the INR scores for patients prescribed warfarin were replaced with values < 1.7, the statistical difference disappeared between the warfarin and DOAC groups. The same effect is seen on MELD scores for patients prescribed warfarin therapy. The MELD score is calculated using INR levels.12 MELD scores also will be artificially elevated in patients prescribed warfarin therapy due to the INR elevation to between 2 and 3. When MELD scores for patients prescribed warfarin were replaced with values similar to those in the DOAC group, the statistical difference disappeared between the warfarin and DOAC groups.
The last statistically significant difference was found in number of days on anticoagulant therapy. This difference was expected as warfarin is the standard of care for anticoagulation treatment in patients with cirrhosis. The first DOAC, dabigatran, was not approved by the US Food and Drug Administration until 2010.13 DOACs have only recently been used in patients with cirrhosis accounting for the statistically significant difference in days on anticoagulation therapy between the warfarin and DOAC groups.
Limitations
The inability to meet power or evaluate adherence and appropriate renal dose adjustments for DOACs limited this study. This study was conducted at a single center in a predominantly male veteran population and therefore may not be generalizable to other populations. A majority of patients in the DOAC group were prescribed apixaban (69.1%), which may have affected the overall rate of major bleeding in the DOAC group. Pivotal trials of apixaban have shown a consistent decreased risk of major bleeding in patients with NVAF or VTE when compared with warfarin.14,15 Therefore, the results of this study may not be generalizable to all DOACs.
An inherent limitation of this study was the inability to collect data verifying adherence in the DOAC group. However, in the warfarin group, percentage of time within the therapeutic INR range of 2 to 3 was collected. While not a direct marker of adherence, this does allow for limited evaluation of therapeutic efficacy and safety within the warfarin group. Last, proper dosing of DOACs in patients with and without adequate renal function was not evaluated in this study.
Conclusions
The results of this study are consistent with other retrospective research and literature reviews. There were no statistically significant differences identified between the rates of all-cause bleeding, major bleeding, and failed efficacy between the DOAC and warfarin groups. DOACs may be a safe alternative to warfarin in patients with cirrhosis requiring anticoagulation for NVAF or VTE, but large randomized trials are required to confirm these results.
1. Qamar A, Vaduganathan M, Greenberger NJ, Giugliano RP. Oral anticoagulation in patients with liver disease. J Am Coll Cardiol. 2018;71(19):2162-2175. doi:10.1016/j.jacc.2018.03.023
2. Priyanka P, Kupec JT, Krafft M, Shah NA, Reynolds GJ. Newer oral anticoagulants in the treatment of acute portal vein thrombosis in patients with and without cirrhosis. Int J Hepatol. 2018;2018:8432781. Published 2018 Jun 5. doi:10.1155/2018/8432781
3. Intagliata NM, Henry ZH, Maitland H, et al. Direct oral anticoagulants in cirrhosis patients pose similar risks of bleeding when compared to traditional anticoagulation. Dig Dis Sci. 2016;61(6):1721-1727. doi:10.1007/s10620-015-4012-2
4. Hum J, Shatzel JJ, Jou JH, Deloughery TG. The efficacy and safety of direct oral anticoagulants vs traditional anticoagulants in cirrhosis. Eur J Haematol. 2017;98(4):393-397. doi:10.1111/ejh.12844
5. Goriacko P, Veltri KT. Safety of direct oral anticoagulants vs warfarin in patients with chronic liver disease and atrial fibrillation. Eur J Haematol. 2018;100(5):488-493. doi:10.1111/ejh.13045
6. Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692-694. doi:10.1111/j.1538-7836.2005.01204.x
7. Rubboli A, Becattini C, Verheugt FW. Incidence, clinical impact and risk of bleeding during oral anticoagulation therapy. World J Cardiol. 2011;3(11):351-358. doi:10.4330/wjc.v3.i11.351
8. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
9. Hoolwerf EW, Kraaijpoel N, Büller HR, van Es N. Direct oral anticoagulants in patients with liver cirrhosis: A systematic review. Thromb Res. 2018;170:102-108. doi:10.1016/j.thromres.2018.08.011
10. Steuber TD, Howard ML, Nisly SA. Direct oral anticoagulants in chronic liver disease. Ann Pharmacother. 2019;53(10):1042-1049. doi:10.1177/1060028019841582
11. Janevska D, Chaloska-Ivanova V, Janevski V. Hepatocellular carcinoma: risk factors, diagnosis and treatment. Open Access Maced J Med Sci. 2015;3(4):732-736. doi:10.3889/oamjms.2015.111
12. Singal AK, Kamath PS. Model for End-Stage Liver Disease. J Clin Exp Hepatol. 2013;3(1):50-60. doi:10.1016/j.jceh.2012.11.002
13. Joppa SA, Salciccioli J, Adamski J, et al. A practical review of the emerging direct anticoagulants, laboratory monitoring, and reversal agents. J Clin Med. 2018;7(2):29. Published 2018 Feb 11. doi:10.3390/jcm7020029
14. Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. doi:10.1056/NEJMoa1107039
15. Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799-808. doi:10.1056/NEJMoa1302507
1. Qamar A, Vaduganathan M, Greenberger NJ, Giugliano RP. Oral anticoagulation in patients with liver disease. J Am Coll Cardiol. 2018;71(19):2162-2175. doi:10.1016/j.jacc.2018.03.023
2. Priyanka P, Kupec JT, Krafft M, Shah NA, Reynolds GJ. Newer oral anticoagulants in the treatment of acute portal vein thrombosis in patients with and without cirrhosis. Int J Hepatol. 2018;2018:8432781. Published 2018 Jun 5. doi:10.1155/2018/8432781
3. Intagliata NM, Henry ZH, Maitland H, et al. Direct oral anticoagulants in cirrhosis patients pose similar risks of bleeding when compared to traditional anticoagulation. Dig Dis Sci. 2016;61(6):1721-1727. doi:10.1007/s10620-015-4012-2
4. Hum J, Shatzel JJ, Jou JH, Deloughery TG. The efficacy and safety of direct oral anticoagulants vs traditional anticoagulants in cirrhosis. Eur J Haematol. 2017;98(4):393-397. doi:10.1111/ejh.12844
5. Goriacko P, Veltri KT. Safety of direct oral anticoagulants vs warfarin in patients with chronic liver disease and atrial fibrillation. Eur J Haematol. 2018;100(5):488-493. doi:10.1111/ejh.13045
6. Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692-694. doi:10.1111/j.1538-7836.2005.01204.x
7. Rubboli A, Becattini C, Verheugt FW. Incidence, clinical impact and risk of bleeding during oral anticoagulation therapy. World J Cardiol. 2011;3(11):351-358. doi:10.4330/wjc.v3.i11.351
8. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
9. Hoolwerf EW, Kraaijpoel N, Büller HR, van Es N. Direct oral anticoagulants in patients with liver cirrhosis: A systematic review. Thromb Res. 2018;170:102-108. doi:10.1016/j.thromres.2018.08.011
10. Steuber TD, Howard ML, Nisly SA. Direct oral anticoagulants in chronic liver disease. Ann Pharmacother. 2019;53(10):1042-1049. doi:10.1177/1060028019841582
11. Janevska D, Chaloska-Ivanova V, Janevski V. Hepatocellular carcinoma: risk factors, diagnosis and treatment. Open Access Maced J Med Sci. 2015;3(4):732-736. doi:10.3889/oamjms.2015.111
12. Singal AK, Kamath PS. Model for End-Stage Liver Disease. J Clin Exp Hepatol. 2013;3(1):50-60. doi:10.1016/j.jceh.2012.11.002
13. Joppa SA, Salciccioli J, Adamski J, et al. A practical review of the emerging direct anticoagulants, laboratory monitoring, and reversal agents. J Clin Med. 2018;7(2):29. Published 2018 Feb 11. doi:10.3390/jcm7020029
14. Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. doi:10.1056/NEJMoa1107039
15. Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799-808. doi:10.1056/NEJMoa1302507
Multidisciplinary Transitional Pain Service for the Veteran Population
Despite advancements in techniques, postsurgical pain continues to be a prominent part of the patient experience. Often this experience can lead to developing chronic postsurgical pain that interferes with quality of life after the expected time to recovery.1-3 As many as 14% of patients who undergo surgery without any history of opioid use develop chronic opioid use that persists after recovery from their operation.4-8 For patients with existing chronic opioid use or a history of substance use disorder (SUD), surgeons, primary care providers, or addiction providers often do not provide sufficient presurgical planning or postsurgical coordination of care. This lack of pain care coordination can increase the risk of inadequate pain control, opioid use escalation, or SUD relapse after surgery.
Convincing arguments have been made that a perioperative surgical home can improve significantly the quality of perioperative care.9-14 This report describes our experience implementing a perioperative surgical home at the US Department of Veterans Affairs (VA) Salt Lake City VA Medical Center (SLCVAMC), focusing on pain management extending from the preoperative period until 6 months or more after surgery. This type of Transitional Pain Service (TPS) has been described previously.15-17 Our service differs from those described previously by enrolling all patients before surgery rather than select postsurgical enrollment of only patients with a history of opioid use or SUD or patients who struggle with persistent postsurgical pain.
Methods
In January 2018, we developed and implemented a new TPS at the SLCVAMC. The transitional pain team consisted of an anesthesiologist with specialization in acute pain management, a nurse practitioner (NP) with experience in both acute and chronic pain management, 2 nurse care coordinators, and a psychologist (Figure 1). Before implementation, a needs assessment took place with these key stakeholders and others at SLCVAMC to identify the following specific goals of the TPS: (1) reduce pain through pharmacologic and nonpharmacologic interventions; (2) eliminate new chronic opioid use in previously nonopioid user (NOU) patients; (3) address chronic opioid use in previous chronic opioid users (COUs) by providing support for opioid taper and alternative analgesic therapies for their chronic pain conditions; and (4) improve continuity of care by close coordination with the surgical team, primary care providers (PCPs), and mental health or chronic pain providers as needed.
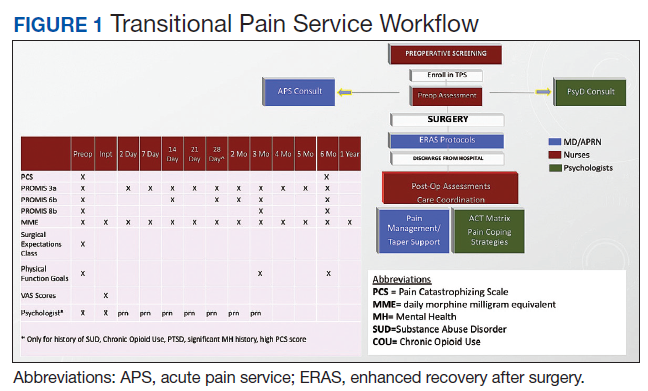
Once these TPS goals were defined, the Consolidated Framework for Implementation Research (CFIR) guided the implementation. CFIR is a theory-based implementation framework consisting of 5 domains: intervention characteristics, inner setting, outer setting, characteristics of individuals, and process. These domains were used to identify barriers and facilitators during the early implementation process and helped refine TPS as it was put into clinical practice.
Patient Selection
During the initial implementation of TPS, enrollment was limited to patients scheduled for elective primary or revision knee, hip, or shoulder replacement as well as rotator cuff repair surgery. But as the TPS workflow became established after iterative refinement, we expanded the program to enroll patients with established risk factors for OUD having other types of surgery (Table 1). The diagnosis of risk factors, such as history of SUD, chronic opioid use, or significant mental health disorders (ie, history of suicidal ideation or attempt, posttraumatic stress disorder, and inpatient psychiatric care) were confirmed through both in-person interviews and electronic health record (EHR) documentation. The overall goal was to identify all at-risk patients as soon as they were indicated for surgery, to allow time for evaluation, education, developing an individualized pain plan, and opioid taper prior to surgery if indicated.
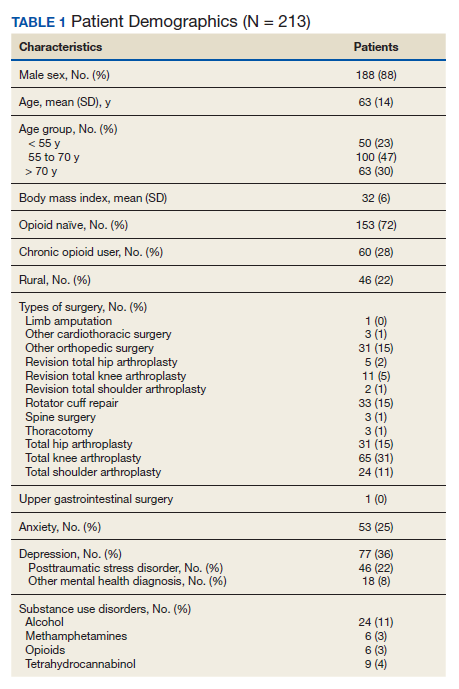
Preoperative Procedures
Once identified, patients were contacted by a TPS team member and invited to attend a onetime 90-minute presurgical expectations class held at SLCVAMC. The education curriculum was developed by the whole team, and classes were taught primarily by the TPS psychologist. The class included education about expectations for postoperative pain, available analgesic therapies, opioid education, appropriate use of opioids, and the effect of psychological factors on pain. Pain coping strategies were introduced using a mindfulness-based intervention (MBI) and the Acceptance and Commitment Therapy (ACT) matrix. Classes were offered multiple times a week to help maximize convenience for patients and were separate from the anesthesia preoperative evaluation. Patients attended class only once. High-risk patients (patients with chronic opioid therapy, recent history of or current SUDs, significant comorbid mental health issues) were encouraged to attend this class one-on-one with the TPS psychologist rather than in the group setting, so individual attention to mental health and SUD issues could be addressed directly.
Baseline history, morphine equivalent daily dose (MEDD), and patient-reported outcomes using measures from the Patient-Reported Outcome Measurement System (PROMIS) for pain intensity (PROMIS 3a), pain interference (PROMIS 6b), and physical function (PROMIS 8b), and a pain-catastrophizing scale (PCS) score were obtained on all patients.18 PROMIS measures are validated questionnaires developed with the National Institutes of Health to standardize and quantify patient-reported outcomes in many domains.19 Patients with a history of SUD or COU met with the anesthesiologist and/or NP, and a personalized pain plan was developed that included preoperative opioid taper, buprenorphine use strategy, or opioid-free strategies.
Hospital Procedures
On the day of surgery, the TPS team met with the patient preoperatively and implemented an individualized pain plan that included multimodal analgesic techniques with nonsteroidal anti-inflammatory drugs, acetaminophen, gabapentinoids, and regional anesthesia, where appropriate (Table 2). Enhanced recovery after surgery protocols were developed in conjunction with the surgeons to include local infiltration analgesia by the surgeon, postoperative multimodal analgesic strategies, and intensive physical therapy starting the day of surgery for inpatient procedures.
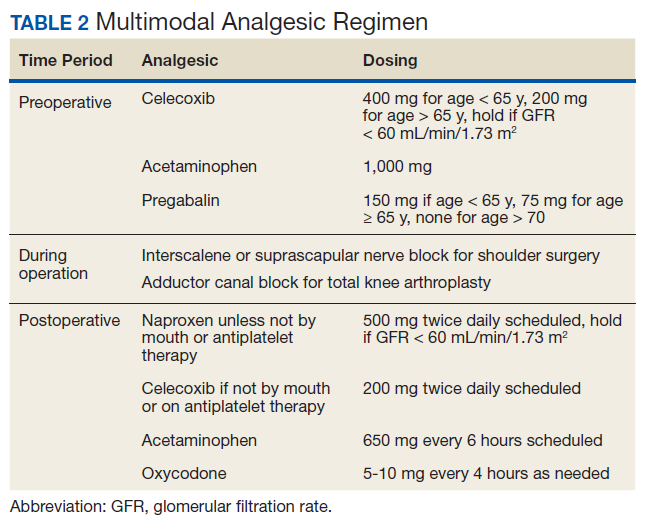
After surgery, the TPS team followed up with patients daily and provided recommendations for analgesic therapies. Patients were offered daily sessions with the psychologist to reinforce and practice nonpharmacologic pain-coping strategies, such as meditation and relaxation. Prior to patient discharge, the TPS team provided recommendations for discharge medications and an opioid taper plan. For some patients taking buprenorphine before surgery who had stopped this therapy prior to or during their hospital stay, TPS providers transitioned them back to buprenorphine before discharge.
Postoperative Procedures
Patients were called by the nurse care coordinators at postdischarge days 2, 7, 10, 14, 21, 28, and then monthly for ≥ 6 months. For patients who had not stopped opioid use or returned to their preoperative baseline opioid dose, weekly calls were made until opioid taper goals were achieved. At each call, nurses collected PROMIS scores for the previous 24 hours, the most recent 24-hour MEDD, the date of last opioid use, and the number of remaining opioid tablets after opioid cessation. In addition, nurses provided active listening and supportive care and encouragement as well as care coordination for issues related to rehabilitation facilities, physical therapy, transportation, medication questions, and wound questions. Nurses notified the anesthesiologist or NP when patients were unable to taper opioid use or had poor pain control as indicated by their PROMIS scores, opioid use, or directly expressed by the patient.
The TPS team prescribed alternative analgesic therapies, opioid taper plans, and communicated with surgeons and primary care providers if limited continued opioid therapy was recommended. Individual sessions with the psychologist were available to patients after discharge with a focus on ACT-matrix therapy and consultation with long-term mental health and/or substance abuse providers as indicated. Frequent communication and care coordination were maintained with the surgical team, the PCP, and other providers on the mental health or chronic pain services. This care coordination often included postsurgical joint clinic appointments in which TPS providers and nurses would be present with the surgeon or the PCP.
For patients with inadequately treated chronic pain conditions or who required long-term opioid tapers, we developed a combined clinic with the TPS and Anesthesia Chronic Pain group. This clinic allows patients to be seen by both services in the same setting, allowing a warm handoff by TPS to the chronic pain team.
Heath and Decision Support Tools
An electronic dashboard registry of surgical episodes managed by TPS was developed to achieve clinical, administrative, and quality improvement goals. The dashboard registry consists of surgical episode data, opioid doses, patient-reported outcomes, and clinical decision-making processes. Custom-built note templates capture pertinent data through embedded data labels, called health factors. Data are captured as part of routine clinical care, recorded in Computerized Patient Record System as health factors. They are available in the VA Corporate Data Warehouse as structured data. Workflows are executed daily to keep the dashboard registry current, clean, and able to process new data. Information displays direct daily clinical workflow and support point-of-care clinical decision making (Figures 2, 3, and 4). Data are aggregated across patient-care encounters and allow nurse care coordinators to concisely review pertinent patient data prior to delivering care. These data include surgical history, comorbidities, timeline of opioid use, and PROMIS scores during their course of recovery. This system allows TPS to optimize care delivery by providing longitudinal data across the surgical episode, thereby reducing the time needed to review records. Secondary purposes of captured data include measuring clinic performance and quality improvement to improve care delivery.
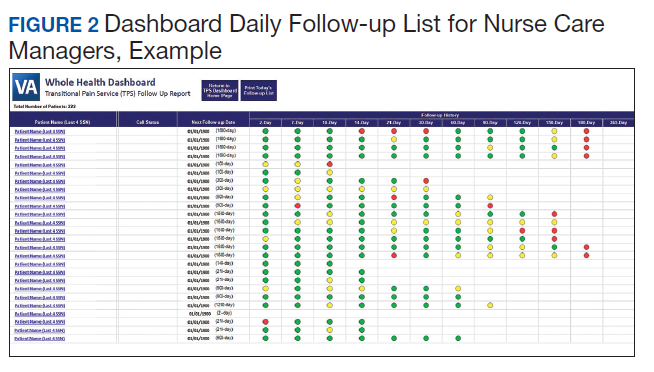
Results
The TPS intervention was implemented January 1, 2018. Two-hundred thirteen patients were enrolled between January and December 2018, which included 60 (28%) patients with a history of chronic opioid use and 153 (72%) patients who were considered opioid naïve. A total of 99% of patients had ≥ 1 successful follow-up within 14 days after discharge, 96% had ≥ 1 follow-up between 14 and 30 days after surgery, and 72% had completed personal follow-up 90 days after discharge (Table 3). For patients who TPS was unable to contact in person or by phone, 90-day MEDD was obtained using prescription and Controlled Substance Database reviews. The protocol for this retrospective analysis was approved by the University of Utah Institutional Review Board and the VA Research Review Committee.
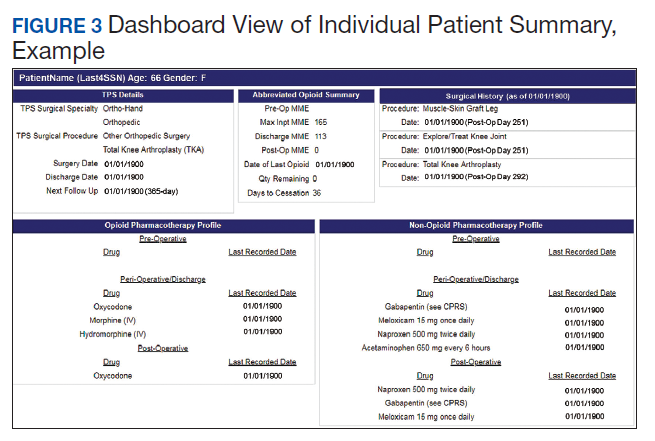
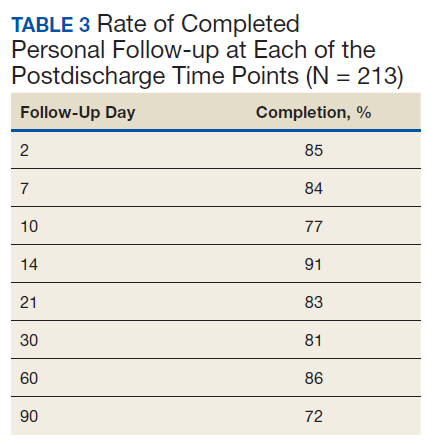
By 90 days after surgery, 26 (43.3%) COUs were off opioids completely, 17 (28.3%) had decreased their opioid dose from their preoperative baseline MEDD (120 [SD, 108] vs 55 [SD, 45]), 14 (23.3%) returned to their baseline dose, and 3 (5%) increased from their baseline dose. Of the 153 patients who were NOUs before surgery, only 1 (0.7%) was taking opioids after 90 days. TPS continued to work closely with the patient and their PCP and that patient was finally able to stop opioid use 262 days after discharge. Ten patients had an additional surgery within 90 days of the initial surgery. Of these, 6 were COU, of whom 3 stopped all opioids by 90 days from their original surgery, 2 had no change in MEDD at 90 days, and 1 had a lower MEDD at 90 days. Of the 4 NOU who had additional surgery, all were off opioids by 90 days from the original surgery.
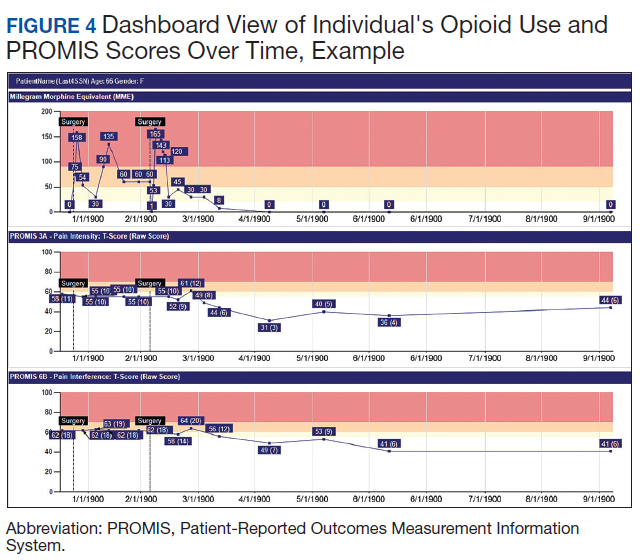
Although difficult to quantify, a meaningful outcome of TPS has been to improve satisfaction substantially among health care providers caring for complex patients at risk for chronic opioid abuse. This group includes the many members of the surgical team, PCPs, and addiction specialists who appreciate the close care coordination and assistance in caring for patients with difficult issues, especially with opioid tapers or SUDs. We also have noticed changes in prescribing practices among surgeons and PCPs for their patients who are not part of TPS.
Discussion
With any new clinical service, there are obstacles and challenges. TPS requires a considerable investment in personnel, and currently no mechanism is in place for obtaining payment for many of the provided services. We were fortunate the VA Whole Health Initiative, the VA Office of Rural Health, and the VA Centers of Innovation provided support for the development, implementation, and pilot evaluation of TPS. After we presented our initial results to hospital leadership, we also received hospital support to expand TPS service to include a total of 4 nurse care coordinators and 2 psychologists. We are currently performing a cost analysis of the service but recognize that this model may be difficult to reproduce at other institutions without a change in reimbursement standards.
Developing a working relationship with the surgical and primary care services required a concerted effort from the TPS team and a number of months to become effective. As most veterans receive primary care, mental health care, and surgical care within the VA system, this model lends itself to close care coordination. Initially there was skepticism about TPS recommendations to reduce opioid use, especially from PCPs who had cared for complex patients over many years. But this uncertainty went away as we showed evidence of close patient follow-up and detailed communication. TPS soon became the designated service for both primary care and surgical providers who were otherwise uncomfortable with how to approach opioid tapers and nonopioid pain strategies. In fact, a substantial portion of our referrals now come directly from the PCP who is referring a high-risk patient for evaluation for surgery rather than from the surgeons, and joint visits with TPS and primary care have become commonplace.
Challenges abound when working with patients with substance abuse history, opioid use history, high anxiety, significant pain catastrophizing, and those who have had previous negative experiences with surgery. We have found that the most important facet of our service comes from the amount of time and effort team members, especially the nurses, spend helping patients. Much of the nurses' work focuses on nonpain-related issues, such as assisting patients with finding transportation, housing issues, questions about medications, help scheduling appointments, etc. Through this concerted effort, patients gain trust in TPS providers and are willing to listen to and experiment with our recommendations. Many patients who were initially extremely unreceptive to the presurgery education asked for our support weeks after surgery to help with postsurgery pain.
Another challenge we continue to experience comes from the success of the program.
Conclusions
The multidisciplinary TPS supports greater preoperative to postoperative longitudinal care for surgical patients. This endeavor has resulted in better patient preparation before surgery and improved care coordination after surgery, with specific improvements in appropriate use of opioid medications and smooth transitions of care for patients with ongoing and complex needs. Development of sophisticated note templates and customized health information technology allows for accurate follow-through and data gathering for quality improvement, facilitating data-driven improvements and proving value to the facility.
Given that TPS is a multidisciplinary program with multiple interventions, it is difficult to pinpoint which specific aspects of TPS are most effective in achieving success. For example, although we have little doubt that the work our psychologists do with our patients is beneficial and even essential for the success we have had with some of our most difficult patients, it is less clear whether it matters if they use mindfulness, ACT matrix, or cognitive behavioral therapy. We think that an important part of TPS is the frequent human interaction with a caring individual. Therefore, as TPS continues to grow, maintaining the ability to provide frequent personal interaction is a priority.
The role of opioids in acute pain deserves further scrutiny. In 2018, with TPS use of opioids after orthopedic surgery decreased by > 40% from the previous year. Despite this more restricted use of opioids, pain interference and physical function scores indicated that surgical patients do not seem to experience increased pain or reduced physical function. In addition, stopping opioid use for COUs did not seem to affect the quality of recovery, pain, or physical function. Future prospective controlled studies of TPS are needed to confirm these findings and identify which aspects of TPS are most effective in improving functional recovery of patients. Also, more evidence is needed to determine the appropriateness or need for opioids in acute postsurgical pain.
TPS has expanded to include all surgical specialties. Given the high burden and limited resources, we have chosen to focus on patients at higher risk for chronic postsurgical pain by type of surgery (eg, thoracotomy, open abdominal, limb amputation, major joint surgery) and/or history of substance abuse or chronic opioid use. To better direct scarce resources where it would be of most benefit, we are now enrolling only NOUs without other risk factors postoperatively if they request a refill of opioids or are otherwise struggling with pain control after surgery. Whether this approach affects the success we had in the first year in preventing new COUs after surgery remains to be seen.
It is unlikely that any single model of a perioperative surgical home will fit the needs of the many different types of medical systems that exist. The TPS model fits well in large hospital systems, like the VA, where patients receive most of their care within the same system. However, it seems to us that the optimal TPS program in any health system will provide education, support, and care coordination beginning preoperatively to prepare the patient for surgery and then to facilitate care coordination to transition patients back to their PCPs or on to specialized chronic care.
Acknowledgments
We would like to acknowledge the contributions of Candice Harmon, RN; David Merrill, RN; Amy Beckstead, RN, who have provided invaluable assistance with establishing the TPS program at the VA Salt Lake City and helping with the evaluation process.
Funding for the implementation and evaluation of the TPS was received from the VA Whole Health Initiative, the VA Center of Innovation, the VA Office of Rural Health, and National Institutes of Health Grant UL1TR002538.
1. Ilfeld BM, Madison SJ, Suresh PJ. Persistent postmastectomy pain and pain-related physical and emotional functioning with and without a continuous paravertebral nerve block: a prospective 1-year follow-up assessment of a randomized, triple-masked, placebo-controlled study. Ann Surg Oncol. 2015;22(6):2017-2025. doi:10.1245/s10434-014-4248-7
2. Richebé P, Capdevila X, Rivat C. Persistent postsurgical pain. Anesthesiology. 2018;129(3):590-607. doi:10.1097/aln.0000000000002238
3. Glare P, Aubrey KR, Myles PS. Transition from acute to chronic pain after surgery. Lancet. 2019;393(10180):1537-1546. doi:10.1016/s0140-6736(19)30352-6
4. Brummett CM, Waljee JF, Goesling J, et al. New persistent opioid use after minor and major surgical procedures in US adults. JAMA Surgery. 2017;152(6):e170504-e170504. doi:10.1001/jamasurg.2017.0504
5. Swenson CW, Kamdar NS, Seiler K, Morgan DM, Lin P, As-Sanie S. Definition development and prevalence of new persistent opioid use following hysterectomy. Am J Obstet Gynecol. 2018;219(5):486.e1-486.e7. doi:10.1016/j.ajog.2018.06.010
6. Bartels K, Fernandez-Bustamante A, McWilliams SK, Hopfer CJ, Mikulich-Gilbertson SK. Long-term opioid use after inpatient surgery - a retrospective cohort study. Drug Alcohol Depend. 2018;187:61-65. doi:10.1016/j.drugalcdep.2018.02.013
7. Bedard N, DeMik D, Dowdle S, Callaghan J. Trends and risk factors for prolonged opioid use after unicompartmental knee arthroplasty. Bone Joint J. 2018;100-B(1)(suppl A):62-67. doi:10.1302/0301-620x.100b1.bjj-2017-0547.r1
8. Politzer CS, Kildow BJ, Goltz DE, Green CL, Bolognesi MP, Seyler T. Trends in opioid utilization before and after total knee arthroplasty. J Arthroplasty. 2018;33(7S):S147-S153.e1. doi:10.1016/j.arth.2017.10.060
9. Mariano ER, Walters TL, Kim ET, Kain ZN. Why the perioperative surgical home makes sense for Veterans Affairs health care. Anesth Analg. 2015;120(5):1163-1166. doi:10.1213/ane.0000000000000712
10. Walters TL, Howard SK, Kou A, et al. Design and implementation of a perioperative surgical home at a Veterans Affairs hospital. Semin Cardiothorac Vasc Anesth. 2016;20(2):133-140. doi:10.1177/1089253215607066
11. Walters TL, Mariano ER, Clark DJ. Perioperative surgical home and the integral role of pain medicine. Pain Med. 2015;16(9):1666-1672. doi:10.1111/pme.12796
12. Vetter TR, Kain ZN. Role of the perioperative surgical home in optimizing the perioperative use of opioids. Anesth Analg. 2017;125(5):1653-1657. doi:10.1213/ane.0000000000002280
13. Shafer SL. Anesthesia & Analgesia’s 2015 collection on the perioperative surgical home. Anesth Analg. 2015;120(5):966-967. doi:10.1213/ane.0000000000000696
14. Wenzel JT, Schwenk ES, Baratta JL, Viscusi ER. Managing opioid-tolerant patients in the perioperative surgical home. Anesthesiol Clin. 2016;34(2):287-301. doi:10.1016/j.anclin.2016.01.005
15. Katz J, Weinrib A, Fashler SR, et al. The Toronto General Hospital Transitional Pain Service: development and implementation of a multidisciplinary program to prevent chronic postsurgical pain. J Pain Res. 2015;8:695-702. doi:10.2147/jpr.s91924
16. Tiippana E, Hamunen K, Heiskanen T, Nieminen T, Kalso E, Kontinen VK. New approach for treatment of prolonged postoperative pain: APS Out-Patient Clinic. Scand J Pain. 2016;12(1):19-24. doi:10.1016/j.sjpain.2016.02.008
17. Katz J, Weinrib AZ, Clarke H. Chronic postsurgical pain: from risk factor identification to multidisciplinary management at the Toronto General Hospital Transitional Pain Service. Can J Pain. 2019;3(2):49-58. doi:10.1080/24740527.2019.1574537
18. Sullivan MJ, Bishop SR, Pivik J. The Pain Catastrophizing Scale: development and validation. Psychol Assess. 1995;7(4):524-532. doi:10.1037/1040-3590.7.4.524
19. HealthMeasures. Intro to PROMIS. https://www.healthmeasures.net/explore-measurement-systems/promis. Accessed September 28, 2020.
Despite advancements in techniques, postsurgical pain continues to be a prominent part of the patient experience. Often this experience can lead to developing chronic postsurgical pain that interferes with quality of life after the expected time to recovery.1-3 As many as 14% of patients who undergo surgery without any history of opioid use develop chronic opioid use that persists after recovery from their operation.4-8 For patients with existing chronic opioid use or a history of substance use disorder (SUD), surgeons, primary care providers, or addiction providers often do not provide sufficient presurgical planning or postsurgical coordination of care. This lack of pain care coordination can increase the risk of inadequate pain control, opioid use escalation, or SUD relapse after surgery.
Convincing arguments have been made that a perioperative surgical home can improve significantly the quality of perioperative care.9-14 This report describes our experience implementing a perioperative surgical home at the US Department of Veterans Affairs (VA) Salt Lake City VA Medical Center (SLCVAMC), focusing on pain management extending from the preoperative period until 6 months or more after surgery. This type of Transitional Pain Service (TPS) has been described previously.15-17 Our service differs from those described previously by enrolling all patients before surgery rather than select postsurgical enrollment of only patients with a history of opioid use or SUD or patients who struggle with persistent postsurgical pain.
Methods
In January 2018, we developed and implemented a new TPS at the SLCVAMC. The transitional pain team consisted of an anesthesiologist with specialization in acute pain management, a nurse practitioner (NP) with experience in both acute and chronic pain management, 2 nurse care coordinators, and a psychologist (Figure 1). Before implementation, a needs assessment took place with these key stakeholders and others at SLCVAMC to identify the following specific goals of the TPS: (1) reduce pain through pharmacologic and nonpharmacologic interventions; (2) eliminate new chronic opioid use in previously nonopioid user (NOU) patients; (3) address chronic opioid use in previous chronic opioid users (COUs) by providing support for opioid taper and alternative analgesic therapies for their chronic pain conditions; and (4) improve continuity of care by close coordination with the surgical team, primary care providers (PCPs), and mental health or chronic pain providers as needed.
Once these TPS goals were defined, the Consolidated Framework for Implementation Research (CFIR) guided the implementation. CFIR is a theory-based implementation framework consisting of 5 domains: intervention characteristics, inner setting, outer setting, characteristics of individuals, and process. These domains were used to identify barriers and facilitators during the early implementation process and helped refine TPS as it was put into clinical practice.
Patient Selection
During the initial implementation of TPS, enrollment was limited to patients scheduled for elective primary or revision knee, hip, or shoulder replacement as well as rotator cuff repair surgery. But as the TPS workflow became established after iterative refinement, we expanded the program to enroll patients with established risk factors for OUD having other types of surgery (Table 1). The diagnosis of risk factors, such as history of SUD, chronic opioid use, or significant mental health disorders (ie, history of suicidal ideation or attempt, posttraumatic stress disorder, and inpatient psychiatric care) were confirmed through both in-person interviews and electronic health record (EHR) documentation. The overall goal was to identify all at-risk patients as soon as they were indicated for surgery, to allow time for evaluation, education, developing an individualized pain plan, and opioid taper prior to surgery if indicated.
Preoperative Procedures
Once identified, patients were contacted by a TPS team member and invited to attend a onetime 90-minute presurgical expectations class held at SLCVAMC. The education curriculum was developed by the whole team, and classes were taught primarily by the TPS psychologist. The class included education about expectations for postoperative pain, available analgesic therapies, opioid education, appropriate use of opioids, and the effect of psychological factors on pain. Pain coping strategies were introduced using a mindfulness-based intervention (MBI) and the Acceptance and Commitment Therapy (ACT) matrix. Classes were offered multiple times a week to help maximize convenience for patients and were separate from the anesthesia preoperative evaluation. Patients attended class only once. High-risk patients (patients with chronic opioid therapy, recent history of or current SUDs, significant comorbid mental health issues) were encouraged to attend this class one-on-one with the TPS psychologist rather than in the group setting, so individual attention to mental health and SUD issues could be addressed directly.
Baseline history, morphine equivalent daily dose (MEDD), and patient-reported outcomes using measures from the Patient-Reported Outcome Measurement System (PROMIS) for pain intensity (PROMIS 3a), pain interference (PROMIS 6b), and physical function (PROMIS 8b), and a pain-catastrophizing scale (PCS) score were obtained on all patients.18 PROMIS measures are validated questionnaires developed with the National Institutes of Health to standardize and quantify patient-reported outcomes in many domains.19 Patients with a history of SUD or COU met with the anesthesiologist and/or NP, and a personalized pain plan was developed that included preoperative opioid taper, buprenorphine use strategy, or opioid-free strategies.
Hospital Procedures
On the day of surgery, the TPS team met with the patient preoperatively and implemented an individualized pain plan that included multimodal analgesic techniques with nonsteroidal anti-inflammatory drugs, acetaminophen, gabapentinoids, and regional anesthesia, where appropriate (Table 2). Enhanced recovery after surgery protocols were developed in conjunction with the surgeons to include local infiltration analgesia by the surgeon, postoperative multimodal analgesic strategies, and intensive physical therapy starting the day of surgery for inpatient procedures.
After surgery, the TPS team followed up with patients daily and provided recommendations for analgesic therapies. Patients were offered daily sessions with the psychologist to reinforce and practice nonpharmacologic pain-coping strategies, such as meditation and relaxation. Prior to patient discharge, the TPS team provided recommendations for discharge medications and an opioid taper plan. For some patients taking buprenorphine before surgery who had stopped this therapy prior to or during their hospital stay, TPS providers transitioned them back to buprenorphine before discharge.
Postoperative Procedures
Patients were called by the nurse care coordinators at postdischarge days 2, 7, 10, 14, 21, 28, and then monthly for ≥ 6 months. For patients who had not stopped opioid use or returned to their preoperative baseline opioid dose, weekly calls were made until opioid taper goals were achieved. At each call, nurses collected PROMIS scores for the previous 24 hours, the most recent 24-hour MEDD, the date of last opioid use, and the number of remaining opioid tablets after opioid cessation. In addition, nurses provided active listening and supportive care and encouragement as well as care coordination for issues related to rehabilitation facilities, physical therapy, transportation, medication questions, and wound questions. Nurses notified the anesthesiologist or NP when patients were unable to taper opioid use or had poor pain control as indicated by their PROMIS scores, opioid use, or directly expressed by the patient.
The TPS team prescribed alternative analgesic therapies, opioid taper plans, and communicated with surgeons and primary care providers if limited continued opioid therapy was recommended. Individual sessions with the psychologist were available to patients after discharge with a focus on ACT-matrix therapy and consultation with long-term mental health and/or substance abuse providers as indicated. Frequent communication and care coordination were maintained with the surgical team, the PCP, and other providers on the mental health or chronic pain services. This care coordination often included postsurgical joint clinic appointments in which TPS providers and nurses would be present with the surgeon or the PCP.
For patients with inadequately treated chronic pain conditions or who required long-term opioid tapers, we developed a combined clinic with the TPS and Anesthesia Chronic Pain group. This clinic allows patients to be seen by both services in the same setting, allowing a warm handoff by TPS to the chronic pain team.
Heath and Decision Support Tools
An electronic dashboard registry of surgical episodes managed by TPS was developed to achieve clinical, administrative, and quality improvement goals. The dashboard registry consists of surgical episode data, opioid doses, patient-reported outcomes, and clinical decision-making processes. Custom-built note templates capture pertinent data through embedded data labels, called health factors. Data are captured as part of routine clinical care, recorded in Computerized Patient Record System as health factors. They are available in the VA Corporate Data Warehouse as structured data. Workflows are executed daily to keep the dashboard registry current, clean, and able to process new data. Information displays direct daily clinical workflow and support point-of-care clinical decision making (Figures 2, 3, and 4). Data are aggregated across patient-care encounters and allow nurse care coordinators to concisely review pertinent patient data prior to delivering care. These data include surgical history, comorbidities, timeline of opioid use, and PROMIS scores during their course of recovery. This system allows TPS to optimize care delivery by providing longitudinal data across the surgical episode, thereby reducing the time needed to review records. Secondary purposes of captured data include measuring clinic performance and quality improvement to improve care delivery.
Results
The TPS intervention was implemented January 1, 2018. Two-hundred thirteen patients were enrolled between January and December 2018, which included 60 (28%) patients with a history of chronic opioid use and 153 (72%) patients who were considered opioid naïve. A total of 99% of patients had ≥ 1 successful follow-up within 14 days after discharge, 96% had ≥ 1 follow-up between 14 and 30 days after surgery, and 72% had completed personal follow-up 90 days after discharge (Table 3). For patients who TPS was unable to contact in person or by phone, 90-day MEDD was obtained using prescription and Controlled Substance Database reviews. The protocol for this retrospective analysis was approved by the University of Utah Institutional Review Board and the VA Research Review Committee.
By 90 days after surgery, 26 (43.3%) COUs were off opioids completely, 17 (28.3%) had decreased their opioid dose from their preoperative baseline MEDD (120 [SD, 108] vs 55 [SD, 45]), 14 (23.3%) returned to their baseline dose, and 3 (5%) increased from their baseline dose. Of the 153 patients who were NOUs before surgery, only 1 (0.7%) was taking opioids after 90 days. TPS continued to work closely with the patient and their PCP and that patient was finally able to stop opioid use 262 days after discharge. Ten patients had an additional surgery within 90 days of the initial surgery. Of these, 6 were COU, of whom 3 stopped all opioids by 90 days from their original surgery, 2 had no change in MEDD at 90 days, and 1 had a lower MEDD at 90 days. Of the 4 NOU who had additional surgery, all were off opioids by 90 days from the original surgery.
Although difficult to quantify, a meaningful outcome of TPS has been to improve satisfaction substantially among health care providers caring for complex patients at risk for chronic opioid abuse. This group includes the many members of the surgical team, PCPs, and addiction specialists who appreciate the close care coordination and assistance in caring for patients with difficult issues, especially with opioid tapers or SUDs. We also have noticed changes in prescribing practices among surgeons and PCPs for their patients who are not part of TPS.
Discussion
With any new clinical service, there are obstacles and challenges. TPS requires a considerable investment in personnel, and currently no mechanism is in place for obtaining payment for many of the provided services. We were fortunate the VA Whole Health Initiative, the VA Office of Rural Health, and the VA Centers of Innovation provided support for the development, implementation, and pilot evaluation of TPS. After we presented our initial results to hospital leadership, we also received hospital support to expand TPS service to include a total of 4 nurse care coordinators and 2 psychologists. We are currently performing a cost analysis of the service but recognize that this model may be difficult to reproduce at other institutions without a change in reimbursement standards.
Developing a working relationship with the surgical and primary care services required a concerted effort from the TPS team and a number of months to become effective. As most veterans receive primary care, mental health care, and surgical care within the VA system, this model lends itself to close care coordination. Initially there was skepticism about TPS recommendations to reduce opioid use, especially from PCPs who had cared for complex patients over many years. But this uncertainty went away as we showed evidence of close patient follow-up and detailed communication. TPS soon became the designated service for both primary care and surgical providers who were otherwise uncomfortable with how to approach opioid tapers and nonopioid pain strategies. In fact, a substantial portion of our referrals now come directly from the PCP who is referring a high-risk patient for evaluation for surgery rather than from the surgeons, and joint visits with TPS and primary care have become commonplace.
Challenges abound when working with patients with substance abuse history, opioid use history, high anxiety, significant pain catastrophizing, and those who have had previous negative experiences with surgery. We have found that the most important facet of our service comes from the amount of time and effort team members, especially the nurses, spend helping patients. Much of the nurses' work focuses on nonpain-related issues, such as assisting patients with finding transportation, housing issues, questions about medications, help scheduling appointments, etc. Through this concerted effort, patients gain trust in TPS providers and are willing to listen to and experiment with our recommendations. Many patients who were initially extremely unreceptive to the presurgery education asked for our support weeks after surgery to help with postsurgery pain.
Another challenge we continue to experience comes from the success of the program.
Conclusions
The multidisciplinary TPS supports greater preoperative to postoperative longitudinal care for surgical patients. This endeavor has resulted in better patient preparation before surgery and improved care coordination after surgery, with specific improvements in appropriate use of opioid medications and smooth transitions of care for patients with ongoing and complex needs. Development of sophisticated note templates and customized health information technology allows for accurate follow-through and data gathering for quality improvement, facilitating data-driven improvements and proving value to the facility.
Given that TPS is a multidisciplinary program with multiple interventions, it is difficult to pinpoint which specific aspects of TPS are most effective in achieving success. For example, although we have little doubt that the work our psychologists do with our patients is beneficial and even essential for the success we have had with some of our most difficult patients, it is less clear whether it matters if they use mindfulness, ACT matrix, or cognitive behavioral therapy. We think that an important part of TPS is the frequent human interaction with a caring individual. Therefore, as TPS continues to grow, maintaining the ability to provide frequent personal interaction is a priority.
The role of opioids in acute pain deserves further scrutiny. In 2018, with TPS use of opioids after orthopedic surgery decreased by > 40% from the previous year. Despite this more restricted use of opioids, pain interference and physical function scores indicated that surgical patients do not seem to experience increased pain or reduced physical function. In addition, stopping opioid use for COUs did not seem to affect the quality of recovery, pain, or physical function. Future prospective controlled studies of TPS are needed to confirm these findings and identify which aspects of TPS are most effective in improving functional recovery of patients. Also, more evidence is needed to determine the appropriateness or need for opioids in acute postsurgical pain.
TPS has expanded to include all surgical specialties. Given the high burden and limited resources, we have chosen to focus on patients at higher risk for chronic postsurgical pain by type of surgery (eg, thoracotomy, open abdominal, limb amputation, major joint surgery) and/or history of substance abuse or chronic opioid use. To better direct scarce resources where it would be of most benefit, we are now enrolling only NOUs without other risk factors postoperatively if they request a refill of opioids or are otherwise struggling with pain control after surgery. Whether this approach affects the success we had in the first year in preventing new COUs after surgery remains to be seen.
It is unlikely that any single model of a perioperative surgical home will fit the needs of the many different types of medical systems that exist. The TPS model fits well in large hospital systems, like the VA, where patients receive most of their care within the same system. However, it seems to us that the optimal TPS program in any health system will provide education, support, and care coordination beginning preoperatively to prepare the patient for surgery and then to facilitate care coordination to transition patients back to their PCPs or on to specialized chronic care.
Acknowledgments
We would like to acknowledge the contributions of Candice Harmon, RN; David Merrill, RN; Amy Beckstead, RN, who have provided invaluable assistance with establishing the TPS program at the VA Salt Lake City and helping with the evaluation process.
Funding for the implementation and evaluation of the TPS was received from the VA Whole Health Initiative, the VA Center of Innovation, the VA Office of Rural Health, and National Institutes of Health Grant UL1TR002538.
Despite advancements in techniques, postsurgical pain continues to be a prominent part of the patient experience. Often this experience can lead to developing chronic postsurgical pain that interferes with quality of life after the expected time to recovery.1-3 As many as 14% of patients who undergo surgery without any history of opioid use develop chronic opioid use that persists after recovery from their operation.4-8 For patients with existing chronic opioid use or a history of substance use disorder (SUD), surgeons, primary care providers, or addiction providers often do not provide sufficient presurgical planning or postsurgical coordination of care. This lack of pain care coordination can increase the risk of inadequate pain control, opioid use escalation, or SUD relapse after surgery.
Convincing arguments have been made that a perioperative surgical home can improve significantly the quality of perioperative care.9-14 This report describes our experience implementing a perioperative surgical home at the US Department of Veterans Affairs (VA) Salt Lake City VA Medical Center (SLCVAMC), focusing on pain management extending from the preoperative period until 6 months or more after surgery. This type of Transitional Pain Service (TPS) has been described previously.15-17 Our service differs from those described previously by enrolling all patients before surgery rather than select postsurgical enrollment of only patients with a history of opioid use or SUD or patients who struggle with persistent postsurgical pain.
Methods
In January 2018, we developed and implemented a new TPS at the SLCVAMC. The transitional pain team consisted of an anesthesiologist with specialization in acute pain management, a nurse practitioner (NP) with experience in both acute and chronic pain management, 2 nurse care coordinators, and a psychologist (Figure 1). Before implementation, a needs assessment took place with these key stakeholders and others at SLCVAMC to identify the following specific goals of the TPS: (1) reduce pain through pharmacologic and nonpharmacologic interventions; (2) eliminate new chronic opioid use in previously nonopioid user (NOU) patients; (3) address chronic opioid use in previous chronic opioid users (COUs) by providing support for opioid taper and alternative analgesic therapies for their chronic pain conditions; and (4) improve continuity of care by close coordination with the surgical team, primary care providers (PCPs), and mental health or chronic pain providers as needed.
Once these TPS goals were defined, the Consolidated Framework for Implementation Research (CFIR) guided the implementation. CFIR is a theory-based implementation framework consisting of 5 domains: intervention characteristics, inner setting, outer setting, characteristics of individuals, and process. These domains were used to identify barriers and facilitators during the early implementation process and helped refine TPS as it was put into clinical practice.
Patient Selection
During the initial implementation of TPS, enrollment was limited to patients scheduled for elective primary or revision knee, hip, or shoulder replacement as well as rotator cuff repair surgery. But as the TPS workflow became established after iterative refinement, we expanded the program to enroll patients with established risk factors for OUD having other types of surgery (Table 1). The diagnosis of risk factors, such as history of SUD, chronic opioid use, or significant mental health disorders (ie, history of suicidal ideation or attempt, posttraumatic stress disorder, and inpatient psychiatric care) were confirmed through both in-person interviews and electronic health record (EHR) documentation. The overall goal was to identify all at-risk patients as soon as they were indicated for surgery, to allow time for evaluation, education, developing an individualized pain plan, and opioid taper prior to surgery if indicated.
Preoperative Procedures
Once identified, patients were contacted by a TPS team member and invited to attend a onetime 90-minute presurgical expectations class held at SLCVAMC. The education curriculum was developed by the whole team, and classes were taught primarily by the TPS psychologist. The class included education about expectations for postoperative pain, available analgesic therapies, opioid education, appropriate use of opioids, and the effect of psychological factors on pain. Pain coping strategies were introduced using a mindfulness-based intervention (MBI) and the Acceptance and Commitment Therapy (ACT) matrix. Classes were offered multiple times a week to help maximize convenience for patients and were separate from the anesthesia preoperative evaluation. Patients attended class only once. High-risk patients (patients with chronic opioid therapy, recent history of or current SUDs, significant comorbid mental health issues) were encouraged to attend this class one-on-one with the TPS psychologist rather than in the group setting, so individual attention to mental health and SUD issues could be addressed directly.
Baseline history, morphine equivalent daily dose (MEDD), and patient-reported outcomes using measures from the Patient-Reported Outcome Measurement System (PROMIS) for pain intensity (PROMIS 3a), pain interference (PROMIS 6b), and physical function (PROMIS 8b), and a pain-catastrophizing scale (PCS) score were obtained on all patients.18 PROMIS measures are validated questionnaires developed with the National Institutes of Health to standardize and quantify patient-reported outcomes in many domains.19 Patients with a history of SUD or COU met with the anesthesiologist and/or NP, and a personalized pain plan was developed that included preoperative opioid taper, buprenorphine use strategy, or opioid-free strategies.
Hospital Procedures
On the day of surgery, the TPS team met with the patient preoperatively and implemented an individualized pain plan that included multimodal analgesic techniques with nonsteroidal anti-inflammatory drugs, acetaminophen, gabapentinoids, and regional anesthesia, where appropriate (Table 2). Enhanced recovery after surgery protocols were developed in conjunction with the surgeons to include local infiltration analgesia by the surgeon, postoperative multimodal analgesic strategies, and intensive physical therapy starting the day of surgery for inpatient procedures.
After surgery, the TPS team followed up with patients daily and provided recommendations for analgesic therapies. Patients were offered daily sessions with the psychologist to reinforce and practice nonpharmacologic pain-coping strategies, such as meditation and relaxation. Prior to patient discharge, the TPS team provided recommendations for discharge medications and an opioid taper plan. For some patients taking buprenorphine before surgery who had stopped this therapy prior to or during their hospital stay, TPS providers transitioned them back to buprenorphine before discharge.
Postoperative Procedures
Patients were called by the nurse care coordinators at postdischarge days 2, 7, 10, 14, 21, 28, and then monthly for ≥ 6 months. For patients who had not stopped opioid use or returned to their preoperative baseline opioid dose, weekly calls were made until opioid taper goals were achieved. At each call, nurses collected PROMIS scores for the previous 24 hours, the most recent 24-hour MEDD, the date of last opioid use, and the number of remaining opioid tablets after opioid cessation. In addition, nurses provided active listening and supportive care and encouragement as well as care coordination for issues related to rehabilitation facilities, physical therapy, transportation, medication questions, and wound questions. Nurses notified the anesthesiologist or NP when patients were unable to taper opioid use or had poor pain control as indicated by their PROMIS scores, opioid use, or directly expressed by the patient.
The TPS team prescribed alternative analgesic therapies, opioid taper plans, and communicated with surgeons and primary care providers if limited continued opioid therapy was recommended. Individual sessions with the psychologist were available to patients after discharge with a focus on ACT-matrix therapy and consultation with long-term mental health and/or substance abuse providers as indicated. Frequent communication and care coordination were maintained with the surgical team, the PCP, and other providers on the mental health or chronic pain services. This care coordination often included postsurgical joint clinic appointments in which TPS providers and nurses would be present with the surgeon or the PCP.
For patients with inadequately treated chronic pain conditions or who required long-term opioid tapers, we developed a combined clinic with the TPS and Anesthesia Chronic Pain group. This clinic allows patients to be seen by both services in the same setting, allowing a warm handoff by TPS to the chronic pain team.
Heath and Decision Support Tools
An electronic dashboard registry of surgical episodes managed by TPS was developed to achieve clinical, administrative, and quality improvement goals. The dashboard registry consists of surgical episode data, opioid doses, patient-reported outcomes, and clinical decision-making processes. Custom-built note templates capture pertinent data through embedded data labels, called health factors. Data are captured as part of routine clinical care, recorded in Computerized Patient Record System as health factors. They are available in the VA Corporate Data Warehouse as structured data. Workflows are executed daily to keep the dashboard registry current, clean, and able to process new data. Information displays direct daily clinical workflow and support point-of-care clinical decision making (Figures 2, 3, and 4). Data are aggregated across patient-care encounters and allow nurse care coordinators to concisely review pertinent patient data prior to delivering care. These data include surgical history, comorbidities, timeline of opioid use, and PROMIS scores during their course of recovery. This system allows TPS to optimize care delivery by providing longitudinal data across the surgical episode, thereby reducing the time needed to review records. Secondary purposes of captured data include measuring clinic performance and quality improvement to improve care delivery.
Results
The TPS intervention was implemented January 1, 2018. Two-hundred thirteen patients were enrolled between January and December 2018, which included 60 (28%) patients with a history of chronic opioid use and 153 (72%) patients who were considered opioid naïve. A total of 99% of patients had ≥ 1 successful follow-up within 14 days after discharge, 96% had ≥ 1 follow-up between 14 and 30 days after surgery, and 72% had completed personal follow-up 90 days after discharge (Table 3). For patients who TPS was unable to contact in person or by phone, 90-day MEDD was obtained using prescription and Controlled Substance Database reviews. The protocol for this retrospective analysis was approved by the University of Utah Institutional Review Board and the VA Research Review Committee.
By 90 days after surgery, 26 (43.3%) COUs were off opioids completely, 17 (28.3%) had decreased their opioid dose from their preoperative baseline MEDD (120 [SD, 108] vs 55 [SD, 45]), 14 (23.3%) returned to their baseline dose, and 3 (5%) increased from their baseline dose. Of the 153 patients who were NOUs before surgery, only 1 (0.7%) was taking opioids after 90 days. TPS continued to work closely with the patient and their PCP and that patient was finally able to stop opioid use 262 days after discharge. Ten patients had an additional surgery within 90 days of the initial surgery. Of these, 6 were COU, of whom 3 stopped all opioids by 90 days from their original surgery, 2 had no change in MEDD at 90 days, and 1 had a lower MEDD at 90 days. Of the 4 NOU who had additional surgery, all were off opioids by 90 days from the original surgery.
Although difficult to quantify, a meaningful outcome of TPS has been to improve satisfaction substantially among health care providers caring for complex patients at risk for chronic opioid abuse. This group includes the many members of the surgical team, PCPs, and addiction specialists who appreciate the close care coordination and assistance in caring for patients with difficult issues, especially with opioid tapers or SUDs. We also have noticed changes in prescribing practices among surgeons and PCPs for their patients who are not part of TPS.
Discussion
With any new clinical service, there are obstacles and challenges. TPS requires a considerable investment in personnel, and currently no mechanism is in place for obtaining payment for many of the provided services. We were fortunate the VA Whole Health Initiative, the VA Office of Rural Health, and the VA Centers of Innovation provided support for the development, implementation, and pilot evaluation of TPS. After we presented our initial results to hospital leadership, we also received hospital support to expand TPS service to include a total of 4 nurse care coordinators and 2 psychologists. We are currently performing a cost analysis of the service but recognize that this model may be difficult to reproduce at other institutions without a change in reimbursement standards.
Developing a working relationship with the surgical and primary care services required a concerted effort from the TPS team and a number of months to become effective. As most veterans receive primary care, mental health care, and surgical care within the VA system, this model lends itself to close care coordination. Initially there was skepticism about TPS recommendations to reduce opioid use, especially from PCPs who had cared for complex patients over many years. But this uncertainty went away as we showed evidence of close patient follow-up and detailed communication. TPS soon became the designated service for both primary care and surgical providers who were otherwise uncomfortable with how to approach opioid tapers and nonopioid pain strategies. In fact, a substantial portion of our referrals now come directly from the PCP who is referring a high-risk patient for evaluation for surgery rather than from the surgeons, and joint visits with TPS and primary care have become commonplace.
Challenges abound when working with patients with substance abuse history, opioid use history, high anxiety, significant pain catastrophizing, and those who have had previous negative experiences with surgery. We have found that the most important facet of our service comes from the amount of time and effort team members, especially the nurses, spend helping patients. Much of the nurses' work focuses on nonpain-related issues, such as assisting patients with finding transportation, housing issues, questions about medications, help scheduling appointments, etc. Through this concerted effort, patients gain trust in TPS providers and are willing to listen to and experiment with our recommendations. Many patients who were initially extremely unreceptive to the presurgery education asked for our support weeks after surgery to help with postsurgery pain.
Another challenge we continue to experience comes from the success of the program.
Conclusions
The multidisciplinary TPS supports greater preoperative to postoperative longitudinal care for surgical patients. This endeavor has resulted in better patient preparation before surgery and improved care coordination after surgery, with specific improvements in appropriate use of opioid medications and smooth transitions of care for patients with ongoing and complex needs. Development of sophisticated note templates and customized health information technology allows for accurate follow-through and data gathering for quality improvement, facilitating data-driven improvements and proving value to the facility.
Given that TPS is a multidisciplinary program with multiple interventions, it is difficult to pinpoint which specific aspects of TPS are most effective in achieving success. For example, although we have little doubt that the work our psychologists do with our patients is beneficial and even essential for the success we have had with some of our most difficult patients, it is less clear whether it matters if they use mindfulness, ACT matrix, or cognitive behavioral therapy. We think that an important part of TPS is the frequent human interaction with a caring individual. Therefore, as TPS continues to grow, maintaining the ability to provide frequent personal interaction is a priority.
The role of opioids in acute pain deserves further scrutiny. In 2018, with TPS use of opioids after orthopedic surgery decreased by > 40% from the previous year. Despite this more restricted use of opioids, pain interference and physical function scores indicated that surgical patients do not seem to experience increased pain or reduced physical function. In addition, stopping opioid use for COUs did not seem to affect the quality of recovery, pain, or physical function. Future prospective controlled studies of TPS are needed to confirm these findings and identify which aspects of TPS are most effective in improving functional recovery of patients. Also, more evidence is needed to determine the appropriateness or need for opioids in acute postsurgical pain.
TPS has expanded to include all surgical specialties. Given the high burden and limited resources, we have chosen to focus on patients at higher risk for chronic postsurgical pain by type of surgery (eg, thoracotomy, open abdominal, limb amputation, major joint surgery) and/or history of substance abuse or chronic opioid use. To better direct scarce resources where it would be of most benefit, we are now enrolling only NOUs without other risk factors postoperatively if they request a refill of opioids or are otherwise struggling with pain control after surgery. Whether this approach affects the success we had in the first year in preventing new COUs after surgery remains to be seen.
It is unlikely that any single model of a perioperative surgical home will fit the needs of the many different types of medical systems that exist. The TPS model fits well in large hospital systems, like the VA, where patients receive most of their care within the same system. However, it seems to us that the optimal TPS program in any health system will provide education, support, and care coordination beginning preoperatively to prepare the patient for surgery and then to facilitate care coordination to transition patients back to their PCPs or on to specialized chronic care.
Acknowledgments
We would like to acknowledge the contributions of Candice Harmon, RN; David Merrill, RN; Amy Beckstead, RN, who have provided invaluable assistance with establishing the TPS program at the VA Salt Lake City and helping with the evaluation process.
Funding for the implementation and evaluation of the TPS was received from the VA Whole Health Initiative, the VA Center of Innovation, the VA Office of Rural Health, and National Institutes of Health Grant UL1TR002538.
1. Ilfeld BM, Madison SJ, Suresh PJ. Persistent postmastectomy pain and pain-related physical and emotional functioning with and without a continuous paravertebral nerve block: a prospective 1-year follow-up assessment of a randomized, triple-masked, placebo-controlled study. Ann Surg Oncol. 2015;22(6):2017-2025. doi:10.1245/s10434-014-4248-7
2. Richebé P, Capdevila X, Rivat C. Persistent postsurgical pain. Anesthesiology. 2018;129(3):590-607. doi:10.1097/aln.0000000000002238
3. Glare P, Aubrey KR, Myles PS. Transition from acute to chronic pain after surgery. Lancet. 2019;393(10180):1537-1546. doi:10.1016/s0140-6736(19)30352-6
4. Brummett CM, Waljee JF, Goesling J, et al. New persistent opioid use after minor and major surgical procedures in US adults. JAMA Surgery. 2017;152(6):e170504-e170504. doi:10.1001/jamasurg.2017.0504
5. Swenson CW, Kamdar NS, Seiler K, Morgan DM, Lin P, As-Sanie S. Definition development and prevalence of new persistent opioid use following hysterectomy. Am J Obstet Gynecol. 2018;219(5):486.e1-486.e7. doi:10.1016/j.ajog.2018.06.010
6. Bartels K, Fernandez-Bustamante A, McWilliams SK, Hopfer CJ, Mikulich-Gilbertson SK. Long-term opioid use after inpatient surgery - a retrospective cohort study. Drug Alcohol Depend. 2018;187:61-65. doi:10.1016/j.drugalcdep.2018.02.013
7. Bedard N, DeMik D, Dowdle S, Callaghan J. Trends and risk factors for prolonged opioid use after unicompartmental knee arthroplasty. Bone Joint J. 2018;100-B(1)(suppl A):62-67. doi:10.1302/0301-620x.100b1.bjj-2017-0547.r1
8. Politzer CS, Kildow BJ, Goltz DE, Green CL, Bolognesi MP, Seyler T. Trends in opioid utilization before and after total knee arthroplasty. J Arthroplasty. 2018;33(7S):S147-S153.e1. doi:10.1016/j.arth.2017.10.060
9. Mariano ER, Walters TL, Kim ET, Kain ZN. Why the perioperative surgical home makes sense for Veterans Affairs health care. Anesth Analg. 2015;120(5):1163-1166. doi:10.1213/ane.0000000000000712
10. Walters TL, Howard SK, Kou A, et al. Design and implementation of a perioperative surgical home at a Veterans Affairs hospital. Semin Cardiothorac Vasc Anesth. 2016;20(2):133-140. doi:10.1177/1089253215607066
11. Walters TL, Mariano ER, Clark DJ. Perioperative surgical home and the integral role of pain medicine. Pain Med. 2015;16(9):1666-1672. doi:10.1111/pme.12796
12. Vetter TR, Kain ZN. Role of the perioperative surgical home in optimizing the perioperative use of opioids. Anesth Analg. 2017;125(5):1653-1657. doi:10.1213/ane.0000000000002280
13. Shafer SL. Anesthesia & Analgesia’s 2015 collection on the perioperative surgical home. Anesth Analg. 2015;120(5):966-967. doi:10.1213/ane.0000000000000696
14. Wenzel JT, Schwenk ES, Baratta JL, Viscusi ER. Managing opioid-tolerant patients in the perioperative surgical home. Anesthesiol Clin. 2016;34(2):287-301. doi:10.1016/j.anclin.2016.01.005
15. Katz J, Weinrib A, Fashler SR, et al. The Toronto General Hospital Transitional Pain Service: development and implementation of a multidisciplinary program to prevent chronic postsurgical pain. J Pain Res. 2015;8:695-702. doi:10.2147/jpr.s91924
16. Tiippana E, Hamunen K, Heiskanen T, Nieminen T, Kalso E, Kontinen VK. New approach for treatment of prolonged postoperative pain: APS Out-Patient Clinic. Scand J Pain. 2016;12(1):19-24. doi:10.1016/j.sjpain.2016.02.008
17. Katz J, Weinrib AZ, Clarke H. Chronic postsurgical pain: from risk factor identification to multidisciplinary management at the Toronto General Hospital Transitional Pain Service. Can J Pain. 2019;3(2):49-58. doi:10.1080/24740527.2019.1574537
18. Sullivan MJ, Bishop SR, Pivik J. The Pain Catastrophizing Scale: development and validation. Psychol Assess. 1995;7(4):524-532. doi:10.1037/1040-3590.7.4.524
19. HealthMeasures. Intro to PROMIS. https://www.healthmeasures.net/explore-measurement-systems/promis. Accessed September 28, 2020.
1. Ilfeld BM, Madison SJ, Suresh PJ. Persistent postmastectomy pain and pain-related physical and emotional functioning with and without a continuous paravertebral nerve block: a prospective 1-year follow-up assessment of a randomized, triple-masked, placebo-controlled study. Ann Surg Oncol. 2015;22(6):2017-2025. doi:10.1245/s10434-014-4248-7
2. Richebé P, Capdevila X, Rivat C. Persistent postsurgical pain. Anesthesiology. 2018;129(3):590-607. doi:10.1097/aln.0000000000002238
3. Glare P, Aubrey KR, Myles PS. Transition from acute to chronic pain after surgery. Lancet. 2019;393(10180):1537-1546. doi:10.1016/s0140-6736(19)30352-6
4. Brummett CM, Waljee JF, Goesling J, et al. New persistent opioid use after minor and major surgical procedures in US adults. JAMA Surgery. 2017;152(6):e170504-e170504. doi:10.1001/jamasurg.2017.0504
5. Swenson CW, Kamdar NS, Seiler K, Morgan DM, Lin P, As-Sanie S. Definition development and prevalence of new persistent opioid use following hysterectomy. Am J Obstet Gynecol. 2018;219(5):486.e1-486.e7. doi:10.1016/j.ajog.2018.06.010
6. Bartels K, Fernandez-Bustamante A, McWilliams SK, Hopfer CJ, Mikulich-Gilbertson SK. Long-term opioid use after inpatient surgery - a retrospective cohort study. Drug Alcohol Depend. 2018;187:61-65. doi:10.1016/j.drugalcdep.2018.02.013
7. Bedard N, DeMik D, Dowdle S, Callaghan J. Trends and risk factors for prolonged opioid use after unicompartmental knee arthroplasty. Bone Joint J. 2018;100-B(1)(suppl A):62-67. doi:10.1302/0301-620x.100b1.bjj-2017-0547.r1
8. Politzer CS, Kildow BJ, Goltz DE, Green CL, Bolognesi MP, Seyler T. Trends in opioid utilization before and after total knee arthroplasty. J Arthroplasty. 2018;33(7S):S147-S153.e1. doi:10.1016/j.arth.2017.10.060
9. Mariano ER, Walters TL, Kim ET, Kain ZN. Why the perioperative surgical home makes sense for Veterans Affairs health care. Anesth Analg. 2015;120(5):1163-1166. doi:10.1213/ane.0000000000000712
10. Walters TL, Howard SK, Kou A, et al. Design and implementation of a perioperative surgical home at a Veterans Affairs hospital. Semin Cardiothorac Vasc Anesth. 2016;20(2):133-140. doi:10.1177/1089253215607066
11. Walters TL, Mariano ER, Clark DJ. Perioperative surgical home and the integral role of pain medicine. Pain Med. 2015;16(9):1666-1672. doi:10.1111/pme.12796
12. Vetter TR, Kain ZN. Role of the perioperative surgical home in optimizing the perioperative use of opioids. Anesth Analg. 2017;125(5):1653-1657. doi:10.1213/ane.0000000000002280
13. Shafer SL. Anesthesia & Analgesia’s 2015 collection on the perioperative surgical home. Anesth Analg. 2015;120(5):966-967. doi:10.1213/ane.0000000000000696
14. Wenzel JT, Schwenk ES, Baratta JL, Viscusi ER. Managing opioid-tolerant patients in the perioperative surgical home. Anesthesiol Clin. 2016;34(2):287-301. doi:10.1016/j.anclin.2016.01.005
15. Katz J, Weinrib A, Fashler SR, et al. The Toronto General Hospital Transitional Pain Service: development and implementation of a multidisciplinary program to prevent chronic postsurgical pain. J Pain Res. 2015;8:695-702. doi:10.2147/jpr.s91924
16. Tiippana E, Hamunen K, Heiskanen T, Nieminen T, Kalso E, Kontinen VK. New approach for treatment of prolonged postoperative pain: APS Out-Patient Clinic. Scand J Pain. 2016;12(1):19-24. doi:10.1016/j.sjpain.2016.02.008
17. Katz J, Weinrib AZ, Clarke H. Chronic postsurgical pain: from risk factor identification to multidisciplinary management at the Toronto General Hospital Transitional Pain Service. Can J Pain. 2019;3(2):49-58. doi:10.1080/24740527.2019.1574537
18. Sullivan MJ, Bishop SR, Pivik J. The Pain Catastrophizing Scale: development and validation. Psychol Assess. 1995;7(4):524-532. doi:10.1037/1040-3590.7.4.524
19. HealthMeasures. Intro to PROMIS. https://www.healthmeasures.net/explore-measurement-systems/promis. Accessed September 28, 2020.
Experts assess infection risks for patients on biologics
In a new review, a group of infectious disease experts have summarized and made recommendations about recent findings regarding infections that can occur during treatment with an evolving set of targeted and biologic therapies for rheumatoid arthritis and psoriatic arthritis.
“We claim for the need for multicenter registries and multidisciplinary approaches, for new vaccines trials in RA and PsA, and for better defining when and how biologics can be restarted after severe infections,” lead author Olivier Lortholary, MD, of the Institut Pasteur in Paris, and his coauthors wrote in Annals of the Rheumatic Diseases.

“The take-home message is that different DMARDs [disease-modifying antirheumatic drugs], in many ways, are very similar,” said coauthor Kevin L. Winthrop, MD, MPH, professor of public health and ophthalmology at Oregon Health & Science University, Portland, in an interview. “They all have fairly similar risks when it comes to ‘classical’ or routine bacterial infections. But when you talk about opportunistic infections, you start seeing the differences between these drugs.”
The experts began by addressing the current view of the infectious risk of biologic therapies, citing a recent meta-analysis in which standard (odds ratio, 1.31; 95% confidence interval, 1.09-1.58) and high (OR, 1.90; 95% CI, 1.50-2.39) doses of biologics were associated with increased risk of serious infection. They also noted that the ‘healthy drug survivor effect’ tends to confound long-term extensions of randomized clinical trials involving biologics.
“That is largely because people who are more likely to do well or have proven themselves to do well with that infection, they tend to stay in [trials] and stay on drugs,” Dr. Winthrop said. “The ones who develop infections are more likely to drop out. You see this survival of the fittest-type situation, where healthy users dominate a cohort over time. That’s why you see incidence rates decreasing.”
In response, Arthur Kavanaugh, MD, professor of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego, and the director of the Center for Innovative Therapy there, backed the idea of a general ‘depletion of the susceptibles’ but warned doctors to evaluate each patient and situation accordingly. “Providers need to be vigilant throughout for common infections, rarer infections, and infections at greatest risk for the individual patient based on factors like comorbidities and concomitant medications,” he said in an interview.
When considering restarting a biologic in a patient who recently suffered a serious infection, the experts prescribed no general rule and noted that it will “depend on the type of infection, on the mechanism of action of the drug, on the other available drugs for the considered disease and, of course, on the willingness of the patients to restart a drug possibly having [given] him/her a side effect.”
Assessing infection risk related to various inhibitors
Regarding infections caused by TNF-alpha inhibitors (TNFIs), the experts acknowledged a broad increase in risk for mycobacterial and fungal infections, especially tuberculosis and histoplasmosis. They added that patients on TNFIs are more prone to developing pneumonia and soft tissue infections, while smaller studies have indicated a higher risk of listeriosis, legionellosis, herpes zoster (HZ), and reactivation of chronic hepatitis B virus infection.
As for recommendations, they endorsed discontinuing TNFIs when a serious infection occurs and not restarting until after treatment and clinical response. Patients should be screened for latent tuberculosis infection (LTBI) before starting the drug, and anti-TB drugs should be presented to patients with LTBI so they do not progress to active TB.
Regarding other biologics, they cited several studies indicating that IL-6 inhibitors can increase infection risks in RA patients at a rate similar to TNFIs. Among the most common infections were pneumonia and cellulitis. In addition, although PsA patients on IL-17 inhibitors have a dose-dependent risk of mild to moderate mucocutaneous candidiasis, there was no increased risk of serious opportunistic infections like TB.
In assessing JAK inhibitors, they cited a pooled analysis that indicated pneumonia and skin and soft-tissue infections as the most common and noted the high incidence of HZ, compared with other infections. They added that abatacept (Orencia) did not appear to increase risk of infections in RA patients, such as HZ, dermatomycosis, candidiasis, or endemic mycoses. Those same patients did not see an increased overall infection risk after treatment with rituximab (Rituxan), and clinical trials containing treatment with apremilast (Otezla) reported a rare occurrence of serious infections.
Recommendation-wise, they endorsed screening for LTBI before starting IL-6 inhibitors and antiviral prophylaxis with acyclovir in particularly at-risk patients on JAK inhibitors. Age-appropriate influenza vaccinations were also recommended for rituximab, because of the development of rituximab-induced hypogammaglobulinemia.
Prediction and prevention
When it comes to predicting infections in patients on biologics, the experts wrote that it “remains a challenge.” The potential effects of pretreatment underlying disease, the lack of validated biomarkers, and the relatively low rate of infections all combine to stymie prediction. That said, they acknowledged ongoing efforts in monitoring lymphocyte subpopulation counts and immunoglobin levels, as well as a clinical score called the RABBIT Risk Score for Infections, which was validated in two separate cohorts.

“As Yogi Berra said, predictions are hard, especially about the future,” Dr. Kavanaugh said. “Discussions with your patient are always important.”
In regard to overall prevention, they acknowledged that most of their recommendations are of low evidence, except for antiviral prophylaxis for hepatitis B patients on rituximab and the aforementioned LTBI therapy in patients on TNFIs. Broadly, they advocated for all RA and PsA patients to receive a full infectious disease evaluation before the start of targeted and biologic therapies.
They also addressed vaccinations, recommending an evaluation of the patient’s immunization history and potentially planning a catch-up schedule for those in need of the influenza vaccine, a diphtheria-tetanus-pertussis booster, or the pneumococcal vaccine. More broadly, they stated that “a better response is expected if [non-live] vaccination is performed before the introduction of immunosuppressive drugs.” They added that live vaccines should be administered as soon as possible.
What rheumatologists can do
“So how do you mitigate risk?” Dr. Winthrop asked. “You have to be able to predict the risk, see what’s modifiable, and try to act on it. A lot of the risk of infection has more to do with the patient than the therapy.
“You try to minimize what you’re doing to the patient, particularly around steroids,” he said. “And then you think about screening and vaccinations. Rheumatologists need to be involved in those conversations because they’re the ones who know how these drugs interact with vaccines. A lot of the drugs might dumb down vaccine responses. Be sure to consider that and give the vaccines at times that will optimize their immunogenicity and likely efficacy.”
“Thankfully, infections are not that common,” Dr. Kavanaugh said. “Rheumatologists depend on data from trials, but more safety data comes from registry data and personal and shared experience.”
The authors declared no potential conflicts of interest.
SOURCE: Lortholary O et al. Ann Rheum Dis. 2020 Sep 22. doi: 10.1136/annrheumdis-2020-217092.
In a new review, a group of infectious disease experts have summarized and made recommendations about recent findings regarding infections that can occur during treatment with an evolving set of targeted and biologic therapies for rheumatoid arthritis and psoriatic arthritis.
“We claim for the need for multicenter registries and multidisciplinary approaches, for new vaccines trials in RA and PsA, and for better defining when and how biologics can be restarted after severe infections,” lead author Olivier Lortholary, MD, of the Institut Pasteur in Paris, and his coauthors wrote in Annals of the Rheumatic Diseases.
“The take-home message is that different DMARDs [disease-modifying antirheumatic drugs], in many ways, are very similar,” said coauthor Kevin L. Winthrop, MD, MPH, professor of public health and ophthalmology at Oregon Health & Science University, Portland, in an interview. “They all have fairly similar risks when it comes to ‘classical’ or routine bacterial infections. But when you talk about opportunistic infections, you start seeing the differences between these drugs.”
The experts began by addressing the current view of the infectious risk of biologic therapies, citing a recent meta-analysis in which standard (odds ratio, 1.31; 95% confidence interval, 1.09-1.58) and high (OR, 1.90; 95% CI, 1.50-2.39) doses of biologics were associated with increased risk of serious infection. They also noted that the ‘healthy drug survivor effect’ tends to confound long-term extensions of randomized clinical trials involving biologics.
“That is largely because people who are more likely to do well or have proven themselves to do well with that infection, they tend to stay in [trials] and stay on drugs,” Dr. Winthrop said. “The ones who develop infections are more likely to drop out. You see this survival of the fittest-type situation, where healthy users dominate a cohort over time. That’s why you see incidence rates decreasing.”
In response, Arthur Kavanaugh, MD, professor of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego, and the director of the Center for Innovative Therapy there, backed the idea of a general ‘depletion of the susceptibles’ but warned doctors to evaluate each patient and situation accordingly. “Providers need to be vigilant throughout for common infections, rarer infections, and infections at greatest risk for the individual patient based on factors like comorbidities and concomitant medications,” he said in an interview.
When considering restarting a biologic in a patient who recently suffered a serious infection, the experts prescribed no general rule and noted that it will “depend on the type of infection, on the mechanism of action of the drug, on the other available drugs for the considered disease and, of course, on the willingness of the patients to restart a drug possibly having [given] him/her a side effect.”
Assessing infection risk related to various inhibitors
Regarding infections caused by TNF-alpha inhibitors (TNFIs), the experts acknowledged a broad increase in risk for mycobacterial and fungal infections, especially tuberculosis and histoplasmosis. They added that patients on TNFIs are more prone to developing pneumonia and soft tissue infections, while smaller studies have indicated a higher risk of listeriosis, legionellosis, herpes zoster (HZ), and reactivation of chronic hepatitis B virus infection.
As for recommendations, they endorsed discontinuing TNFIs when a serious infection occurs and not restarting until after treatment and clinical response. Patients should be screened for latent tuberculosis infection (LTBI) before starting the drug, and anti-TB drugs should be presented to patients with LTBI so they do not progress to active TB.
Regarding other biologics, they cited several studies indicating that IL-6 inhibitors can increase infection risks in RA patients at a rate similar to TNFIs. Among the most common infections were pneumonia and cellulitis. In addition, although PsA patients on IL-17 inhibitors have a dose-dependent risk of mild to moderate mucocutaneous candidiasis, there was no increased risk of serious opportunistic infections like TB.
In assessing JAK inhibitors, they cited a pooled analysis that indicated pneumonia and skin and soft-tissue infections as the most common and noted the high incidence of HZ, compared with other infections. They added that abatacept (Orencia) did not appear to increase risk of infections in RA patients, such as HZ, dermatomycosis, candidiasis, or endemic mycoses. Those same patients did not see an increased overall infection risk after treatment with rituximab (Rituxan), and clinical trials containing treatment with apremilast (Otezla) reported a rare occurrence of serious infections.
Recommendation-wise, they endorsed screening for LTBI before starting IL-6 inhibitors and antiviral prophylaxis with acyclovir in particularly at-risk patients on JAK inhibitors. Age-appropriate influenza vaccinations were also recommended for rituximab, because of the development of rituximab-induced hypogammaglobulinemia.
Prediction and prevention
When it comes to predicting infections in patients on biologics, the experts wrote that it “remains a challenge.” The potential effects of pretreatment underlying disease, the lack of validated biomarkers, and the relatively low rate of infections all combine to stymie prediction. That said, they acknowledged ongoing efforts in monitoring lymphocyte subpopulation counts and immunoglobin levels, as well as a clinical score called the RABBIT Risk Score for Infections, which was validated in two separate cohorts.
“As Yogi Berra said, predictions are hard, especially about the future,” Dr. Kavanaugh said. “Discussions with your patient are always important.”
In regard to overall prevention, they acknowledged that most of their recommendations are of low evidence, except for antiviral prophylaxis for hepatitis B patients on rituximab and the aforementioned LTBI therapy in patients on TNFIs. Broadly, they advocated for all RA and PsA patients to receive a full infectious disease evaluation before the start of targeted and biologic therapies.
They also addressed vaccinations, recommending an evaluation of the patient’s immunization history and potentially planning a catch-up schedule for those in need of the influenza vaccine, a diphtheria-tetanus-pertussis booster, or the pneumococcal vaccine. More broadly, they stated that “a better response is expected if [non-live] vaccination is performed before the introduction of immunosuppressive drugs.” They added that live vaccines should be administered as soon as possible.
What rheumatologists can do
“So how do you mitigate risk?” Dr. Winthrop asked. “You have to be able to predict the risk, see what’s modifiable, and try to act on it. A lot of the risk of infection has more to do with the patient than the therapy.
“You try to minimize what you’re doing to the patient, particularly around steroids,” he said. “And then you think about screening and vaccinations. Rheumatologists need to be involved in those conversations because they’re the ones who know how these drugs interact with vaccines. A lot of the drugs might dumb down vaccine responses. Be sure to consider that and give the vaccines at times that will optimize their immunogenicity and likely efficacy.”
“Thankfully, infections are not that common,” Dr. Kavanaugh said. “Rheumatologists depend on data from trials, but more safety data comes from registry data and personal and shared experience.”
The authors declared no potential conflicts of interest.
SOURCE: Lortholary O et al. Ann Rheum Dis. 2020 Sep 22. doi: 10.1136/annrheumdis-2020-217092.
In a new review, a group of infectious disease experts have summarized and made recommendations about recent findings regarding infections that can occur during treatment with an evolving set of targeted and biologic therapies for rheumatoid arthritis and psoriatic arthritis.
“We claim for the need for multicenter registries and multidisciplinary approaches, for new vaccines trials in RA and PsA, and for better defining when and how biologics can be restarted after severe infections,” lead author Olivier Lortholary, MD, of the Institut Pasteur in Paris, and his coauthors wrote in Annals of the Rheumatic Diseases.
“The take-home message is that different DMARDs [disease-modifying antirheumatic drugs], in many ways, are very similar,” said coauthor Kevin L. Winthrop, MD, MPH, professor of public health and ophthalmology at Oregon Health & Science University, Portland, in an interview. “They all have fairly similar risks when it comes to ‘classical’ or routine bacterial infections. But when you talk about opportunistic infections, you start seeing the differences between these drugs.”
The experts began by addressing the current view of the infectious risk of biologic therapies, citing a recent meta-analysis in which standard (odds ratio, 1.31; 95% confidence interval, 1.09-1.58) and high (OR, 1.90; 95% CI, 1.50-2.39) doses of biologics were associated with increased risk of serious infection. They also noted that the ‘healthy drug survivor effect’ tends to confound long-term extensions of randomized clinical trials involving biologics.
“That is largely because people who are more likely to do well or have proven themselves to do well with that infection, they tend to stay in [trials] and stay on drugs,” Dr. Winthrop said. “The ones who develop infections are more likely to drop out. You see this survival of the fittest-type situation, where healthy users dominate a cohort over time. That’s why you see incidence rates decreasing.”
In response, Arthur Kavanaugh, MD, professor of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego, and the director of the Center for Innovative Therapy there, backed the idea of a general ‘depletion of the susceptibles’ but warned doctors to evaluate each patient and situation accordingly. “Providers need to be vigilant throughout for common infections, rarer infections, and infections at greatest risk for the individual patient based on factors like comorbidities and concomitant medications,” he said in an interview.
When considering restarting a biologic in a patient who recently suffered a serious infection, the experts prescribed no general rule and noted that it will “depend on the type of infection, on the mechanism of action of the drug, on the other available drugs for the considered disease and, of course, on the willingness of the patients to restart a drug possibly having [given] him/her a side effect.”
Assessing infection risk related to various inhibitors
Regarding infections caused by TNF-alpha inhibitors (TNFIs), the experts acknowledged a broad increase in risk for mycobacterial and fungal infections, especially tuberculosis and histoplasmosis. They added that patients on TNFIs are more prone to developing pneumonia and soft tissue infections, while smaller studies have indicated a higher risk of listeriosis, legionellosis, herpes zoster (HZ), and reactivation of chronic hepatitis B virus infection.
As for recommendations, they endorsed discontinuing TNFIs when a serious infection occurs and not restarting until after treatment and clinical response. Patients should be screened for latent tuberculosis infection (LTBI) before starting the drug, and anti-TB drugs should be presented to patients with LTBI so they do not progress to active TB.
Regarding other biologics, they cited several studies indicating that IL-6 inhibitors can increase infection risks in RA patients at a rate similar to TNFIs. Among the most common infections were pneumonia and cellulitis. In addition, although PsA patients on IL-17 inhibitors have a dose-dependent risk of mild to moderate mucocutaneous candidiasis, there was no increased risk of serious opportunistic infections like TB.
In assessing JAK inhibitors, they cited a pooled analysis that indicated pneumonia and skin and soft-tissue infections as the most common and noted the high incidence of HZ, compared with other infections. They added that abatacept (Orencia) did not appear to increase risk of infections in RA patients, such as HZ, dermatomycosis, candidiasis, or endemic mycoses. Those same patients did not see an increased overall infection risk after treatment with rituximab (Rituxan), and clinical trials containing treatment with apremilast (Otezla) reported a rare occurrence of serious infections.
Recommendation-wise, they endorsed screening for LTBI before starting IL-6 inhibitors and antiviral prophylaxis with acyclovir in particularly at-risk patients on JAK inhibitors. Age-appropriate influenza vaccinations were also recommended for rituximab, because of the development of rituximab-induced hypogammaglobulinemia.
Prediction and prevention
When it comes to predicting infections in patients on biologics, the experts wrote that it “remains a challenge.” The potential effects of pretreatment underlying disease, the lack of validated biomarkers, and the relatively low rate of infections all combine to stymie prediction. That said, they acknowledged ongoing efforts in monitoring lymphocyte subpopulation counts and immunoglobin levels, as well as a clinical score called the RABBIT Risk Score for Infections, which was validated in two separate cohorts.
“As Yogi Berra said, predictions are hard, especially about the future,” Dr. Kavanaugh said. “Discussions with your patient are always important.”
In regard to overall prevention, they acknowledged that most of their recommendations are of low evidence, except for antiviral prophylaxis for hepatitis B patients on rituximab and the aforementioned LTBI therapy in patients on TNFIs. Broadly, they advocated for all RA and PsA patients to receive a full infectious disease evaluation before the start of targeted and biologic therapies.
They also addressed vaccinations, recommending an evaluation of the patient’s immunization history and potentially planning a catch-up schedule for those in need of the influenza vaccine, a diphtheria-tetanus-pertussis booster, or the pneumococcal vaccine. More broadly, they stated that “a better response is expected if [non-live] vaccination is performed before the introduction of immunosuppressive drugs.” They added that live vaccines should be administered as soon as possible.
What rheumatologists can do
“So how do you mitigate risk?” Dr. Winthrop asked. “You have to be able to predict the risk, see what’s modifiable, and try to act on it. A lot of the risk of infection has more to do with the patient than the therapy.
“You try to minimize what you’re doing to the patient, particularly around steroids,” he said. “And then you think about screening and vaccinations. Rheumatologists need to be involved in those conversations because they’re the ones who know how these drugs interact with vaccines. A lot of the drugs might dumb down vaccine responses. Be sure to consider that and give the vaccines at times that will optimize their immunogenicity and likely efficacy.”
“Thankfully, infections are not that common,” Dr. Kavanaugh said. “Rheumatologists depend on data from trials, but more safety data comes from registry data and personal and shared experience.”
The authors declared no potential conflicts of interest.
SOURCE: Lortholary O et al. Ann Rheum Dis. 2020 Sep 22. doi: 10.1136/annrheumdis-2020-217092.
FROM ANNALS OF THE RHEUMATIC DISEASES