User login
Full-dose anticoagulation reduces need for life support in COVID-19
Full-dose anticoagulation was superior to low, prophylactic doses in reducing the need for vital organ support such as ventilation in moderately ill patients hospitalized for COVID-19, according to a report released Jan. 22 by the National Institutes of Health (NIH).
“This is a major advance for patients hospitalized with COVID. Full dose of anticoagulation in these non-ICU patients improved outcomes and there’s a trend toward a reduction in mortality,” Judith Hochman, MD, director of the Cardiovascular Clinical Research Center at NYU Langone Medical Center, New York, said in an interview.
“We have treatments that are improving outcomes but not as many that reduce mortality, so we’re hopeful when the full dataset comes in that will be confirmed,” she said.
The observation of increased rates of blood clots and inflammation among COVID-19 patients, which can lead to complications such as lung failure, heart attack, and stroke, has given rise to various anticoagulant treatment protocols and a need for randomized data on routinely administering increased doses of anticoagulation to hospitalized patients.
Today’s top-line findings come from three linked clinical trials – REMAP-CAP, ACTIV-4, and ATTACC – examining the safety and efficacy of full-dose anticoagulation to treat moderately ill or critically ill adults hospitalized with COVID-19 compared with a lower dose typically used to prevent blood clots in hospitalized patients.
In December 2020, all three trials paused enrollment of the critically ill subgroup after results showed that full-dose anticoagulation started in the intensive care unit (ICU) was not beneficial and may have been harmful in some patients.
Moderately ill patients with COVID-19, defined as those who did not require ICU care or organ support, made up 80% of participants at enrollment in the three trials, Dr. Hochman said.
Among more than 1,000 moderately ill patients reviewed as of the data cut with the data safety monitoring board, full doses of low molecular weight or unfractionated heparin were superior to low prophylactic doses for the primary endpoint of need for ventilation or other organ supportive interventions at 21 days after randomization.
This met the predefined threshold for 99% probability of superiority and recruitment was stopped, Dr. Hochman reported. “Obviously safety figured into this decision. The risk/benefit ratio was very clear.”
The results do not pertain to patients with a previous indication for anticoagulation, who were excluded from the trials.
Data from an additional 1,000 patients will be reviewed and the data published sometime in the next 2-3 months, she said.
With large numbers of COVID-19 patients requiring hospitalization, the outcomes could help reduce the overload on intensive care units around the world, the NIH noted.
The results also highlight the critical role of timing in the course of COVID-19.
“We believe that full anticoagulation is effective early in the disease course,” Dr. Hochman said. “Based on the results so far from these three platform trials, those that were very, very sick at the time of enrollment really didn’t benefit and we needed to have caught them at an earlier stage.
“It’s possible that the people in the ICU are just different and the minute they get sick they need the ICU; so we haven’t clearly demonstrated this time course and when to intervene, but that’s the implication of the findings.”
The question of even earlier treatment is being examined in the partner ACTIV-4B trial, which is enrolling patients with COVID-19 illness not requiring hospitalization and randomizing them to the direct oral anticoagulant apixaban or aspirin or placebo.
“It’s a very important trial and we really want to get the message out that patients should volunteer for it,” said Dr. Hochman, principal investigator of the ACTIV-4 trial.
In the United States, the ACTIV-4 trial is being led by a collaborative effort involving a number of universities, including the University of Pittsburgh and New York University.
The REMAP-CAP, ACTIV-4, and ATTACC study platforms span five continents in more than 300 hospitals and are supported by multiple international funding organizations including the National Institutes of Health, Canadian Institutes of Health Research, the National Institute for Health Research (United Kingdom), the National Health and Medical Research Council (Australia), and the PREPARE and RECOVER consortia (European Union).
A version of this article first appeared on Medscape.com.
Full-dose anticoagulation was superior to low, prophylactic doses in reducing the need for vital organ support such as ventilation in moderately ill patients hospitalized for COVID-19, according to a report released Jan. 22 by the National Institutes of Health (NIH).
“This is a major advance for patients hospitalized with COVID. Full dose of anticoagulation in these non-ICU patients improved outcomes and there’s a trend toward a reduction in mortality,” Judith Hochman, MD, director of the Cardiovascular Clinical Research Center at NYU Langone Medical Center, New York, said in an interview.
“We have treatments that are improving outcomes but not as many that reduce mortality, so we’re hopeful when the full dataset comes in that will be confirmed,” she said.
The observation of increased rates of blood clots and inflammation among COVID-19 patients, which can lead to complications such as lung failure, heart attack, and stroke, has given rise to various anticoagulant treatment protocols and a need for randomized data on routinely administering increased doses of anticoagulation to hospitalized patients.
Today’s top-line findings come from three linked clinical trials – REMAP-CAP, ACTIV-4, and ATTACC – examining the safety and efficacy of full-dose anticoagulation to treat moderately ill or critically ill adults hospitalized with COVID-19 compared with a lower dose typically used to prevent blood clots in hospitalized patients.
In December 2020, all three trials paused enrollment of the critically ill subgroup after results showed that full-dose anticoagulation started in the intensive care unit (ICU) was not beneficial and may have been harmful in some patients.
Moderately ill patients with COVID-19, defined as those who did not require ICU care or organ support, made up 80% of participants at enrollment in the three trials, Dr. Hochman said.
Among more than 1,000 moderately ill patients reviewed as of the data cut with the data safety monitoring board, full doses of low molecular weight or unfractionated heparin were superior to low prophylactic doses for the primary endpoint of need for ventilation or other organ supportive interventions at 21 days after randomization.
This met the predefined threshold for 99% probability of superiority and recruitment was stopped, Dr. Hochman reported. “Obviously safety figured into this decision. The risk/benefit ratio was very clear.”
The results do not pertain to patients with a previous indication for anticoagulation, who were excluded from the trials.
Data from an additional 1,000 patients will be reviewed and the data published sometime in the next 2-3 months, she said.
With large numbers of COVID-19 patients requiring hospitalization, the outcomes could help reduce the overload on intensive care units around the world, the NIH noted.
The results also highlight the critical role of timing in the course of COVID-19.
“We believe that full anticoagulation is effective early in the disease course,” Dr. Hochman said. “Based on the results so far from these three platform trials, those that were very, very sick at the time of enrollment really didn’t benefit and we needed to have caught them at an earlier stage.
“It’s possible that the people in the ICU are just different and the minute they get sick they need the ICU; so we haven’t clearly demonstrated this time course and when to intervene, but that’s the implication of the findings.”
The question of even earlier treatment is being examined in the partner ACTIV-4B trial, which is enrolling patients with COVID-19 illness not requiring hospitalization and randomizing them to the direct oral anticoagulant apixaban or aspirin or placebo.
“It’s a very important trial and we really want to get the message out that patients should volunteer for it,” said Dr. Hochman, principal investigator of the ACTIV-4 trial.
In the United States, the ACTIV-4 trial is being led by a collaborative effort involving a number of universities, including the University of Pittsburgh and New York University.
The REMAP-CAP, ACTIV-4, and ATTACC study platforms span five continents in more than 300 hospitals and are supported by multiple international funding organizations including the National Institutes of Health, Canadian Institutes of Health Research, the National Institute for Health Research (United Kingdom), the National Health and Medical Research Council (Australia), and the PREPARE and RECOVER consortia (European Union).
A version of this article first appeared on Medscape.com.
Full-dose anticoagulation was superior to low, prophylactic doses in reducing the need for vital organ support such as ventilation in moderately ill patients hospitalized for COVID-19, according to a report released Jan. 22 by the National Institutes of Health (NIH).
“This is a major advance for patients hospitalized with COVID. Full dose of anticoagulation in these non-ICU patients improved outcomes and there’s a trend toward a reduction in mortality,” Judith Hochman, MD, director of the Cardiovascular Clinical Research Center at NYU Langone Medical Center, New York, said in an interview.
“We have treatments that are improving outcomes but not as many that reduce mortality, so we’re hopeful when the full dataset comes in that will be confirmed,” she said.
The observation of increased rates of blood clots and inflammation among COVID-19 patients, which can lead to complications such as lung failure, heart attack, and stroke, has given rise to various anticoagulant treatment protocols and a need for randomized data on routinely administering increased doses of anticoagulation to hospitalized patients.
Today’s top-line findings come from three linked clinical trials – REMAP-CAP, ACTIV-4, and ATTACC – examining the safety and efficacy of full-dose anticoagulation to treat moderately ill or critically ill adults hospitalized with COVID-19 compared with a lower dose typically used to prevent blood clots in hospitalized patients.
In December 2020, all three trials paused enrollment of the critically ill subgroup after results showed that full-dose anticoagulation started in the intensive care unit (ICU) was not beneficial and may have been harmful in some patients.
Moderately ill patients with COVID-19, defined as those who did not require ICU care or organ support, made up 80% of participants at enrollment in the three trials, Dr. Hochman said.
Among more than 1,000 moderately ill patients reviewed as of the data cut with the data safety monitoring board, full doses of low molecular weight or unfractionated heparin were superior to low prophylactic doses for the primary endpoint of need for ventilation or other organ supportive interventions at 21 days after randomization.
This met the predefined threshold for 99% probability of superiority and recruitment was stopped, Dr. Hochman reported. “Obviously safety figured into this decision. The risk/benefit ratio was very clear.”
The results do not pertain to patients with a previous indication for anticoagulation, who were excluded from the trials.
Data from an additional 1,000 patients will be reviewed and the data published sometime in the next 2-3 months, she said.
With large numbers of COVID-19 patients requiring hospitalization, the outcomes could help reduce the overload on intensive care units around the world, the NIH noted.
The results also highlight the critical role of timing in the course of COVID-19.
“We believe that full anticoagulation is effective early in the disease course,” Dr. Hochman said. “Based on the results so far from these three platform trials, those that were very, very sick at the time of enrollment really didn’t benefit and we needed to have caught them at an earlier stage.
“It’s possible that the people in the ICU are just different and the minute they get sick they need the ICU; so we haven’t clearly demonstrated this time course and when to intervene, but that’s the implication of the findings.”
The question of even earlier treatment is being examined in the partner ACTIV-4B trial, which is enrolling patients with COVID-19 illness not requiring hospitalization and randomizing them to the direct oral anticoagulant apixaban or aspirin or placebo.
“It’s a very important trial and we really want to get the message out that patients should volunteer for it,” said Dr. Hochman, principal investigator of the ACTIV-4 trial.
In the United States, the ACTIV-4 trial is being led by a collaborative effort involving a number of universities, including the University of Pittsburgh and New York University.
The REMAP-CAP, ACTIV-4, and ATTACC study platforms span five continents in more than 300 hospitals and are supported by multiple international funding organizations including the National Institutes of Health, Canadian Institutes of Health Research, the National Institute for Health Research (United Kingdom), the National Health and Medical Research Council (Australia), and the PREPARE and RECOVER consortia (European Union).
A version of this article first appeared on Medscape.com.
Controversy flares over ivermectin for COVID-19
The National Institutes of Health has dropped its recommendation against the inexpensive antiparasitic drug ivermectin for treatment of COVID-19, and the agency now advises it can’t recommend for or against its use, leaving the decision to physicians and their patients.
“Results from adequately powered, well-designed, and well-conducted clinical trials are needed to provide more specific, evidence-based guidance on the role of ivermectin for the treatment of COVID-19,” according to new NIH guidance released last week.
Passionate arguments have been waged for and against the drug’s use.
The NIH update disappointed members of the Front Line COVID-19 Critical Care Alliance (FLCCC), which outlined its case for endorsing ivermectin in a public statement Jan. 18. Point by point, the group of 10 physicians argued against each limitation that drove the NIH’s ruling.
The group’s members said that, although grateful the recommendation against the drug was dropped, a neutral approach is not acceptable as total U.S. deaths surpassed 400,000 since last spring – and currently approach 4,000 a day. Results from research are enough to support its use, and the drug will immediately save lives, they say.
“Patients do not have time to wait,” they write, “and we as health care providers in society do not have that time either.”
NIH, which in August had recommended against ivermectin’s use, invited the group to present evidence to its treatment guidance panel on Jan. 6 to detail the emerging science surrounding ivermectin. The group cited rapidly growing evidence of the drug’s effectiveness.
Pierre Kory, MD, president/cofounder of FLCCC and a pulmonary and critical care specialist at Aurora St. Luke’s Medical Center in Milwaukee, also spoke before a Senate panel on Dec. 8 in a widely shared impassioned video, touting ivermectin as a COVID-19 “miracle” drug, a term he said he doesn’t use lightly.
Dr. Kory pleaded with the NIH to consider the emerging data. “Please, I’m just asking that they review our manuscript,” he told the senators.
“We have immense amounts of data to show that ivermectin must be implemented and implemented now,” he said.
Some draw parallels to hydroxychloroquine
Critics have said there’s not enough data to institute a protocol, and some draw parallels to another repurposed drug – hydroxychloroquine (HCQ) – which was once considered a promising treatment for COVID-19, based on flawed and incomplete evidence, and now is not recommended.
Paul Sax, MD, a professor of medicine at Harvard and clinical director of the HIV program and division of infectious diseases at Brigham and Women’s Hospital in Boston, wrote in a blog post earlier this month in the New England Journal of Medicine Journal Watch that ivermectin has more robust evidence for it than HCQ ever did.
“But we’re not quite yet at the ‘practice changing’ level,” he writes. “Results from at least five randomized clinical trials are expected soon that might further inform the decision.”
He said the best argument for the drug is seen in this explanation of a meta-analysis of studies of between 100 and 500 patients by Andrew Hill, MD, with the department of pharmacology, University of Liverpool (England).
Dr. Sax advises against two biases in considering ivermectin. One is assuming that because HCQ failed, other antiparasitic drugs will too.
The second bias to avoid, he says, is discounting studies done in low- and middle-income countries because “they weren’t done in the right places.”
“That’s not just bias,” he says. “It’s also snobbery.”
Ivermectin has been approved by the U.S. Food and Drug Administration for treatment of onchocerciasis (river blindness) and strongyloidiasis, but is not FDA-approved for the treatment of any viral infection. It also is sometimes used to treat animals.
In dropping the recommendation against ivermectin, the NIH gave it the same neutral declaration as monoclonal antibodies and convalescent plasma.
Some physicians say they won’t prescribe it
Some physicians say they won’t be recommending it to their COVID-19 patients.
Amesh Adalja, MD, an infectious disease expert and senior scholar at the Johns Hopkins University Center for Health Security in Baltimore,said in an interview that the NIH update hasn’t changed his mind and he isn’t prescribing it for his patients.
He said although “there’s enough of a signal” that he would like to see more data, “we haven’t seen anything in terms of a really robust study.”
He noted that the Infectious Diseases Society of America has 15 recommendations for COVID-19 treatment “and not one of them has to do with ivermectin.”
He added, “It’s not enough to see if it works, but we need to see who it works in and when it works in them.”
He also acknowledged that “some prominent physicians” are recommending it.
Among them is Paul Marik, MD, endowed professor of medicine and chief of pulmonary and critical care medicine at Eastern Virginia Medical School in Norfolk. A cofounder of FLCCC, Dr. Marik has championed ivermectin and developed a protocol for its use to prevent and treat COVID-19.
The data surrounding ivermectin have met with hope, criticism, and warnings.
Australian researchers published a study ahead of print in Antiviral Research that found ivermectin inhibited the replication of SARS-CoV-2 in a laboratory setting.
The study concluded that the drug resulted post infection in a 5,000-fold reduction in viral RNA at 48 hours. After that study, however, the FDA in April warned consumers not to self-medicate with ivermectin products intended for animals.
The NIH acknowledged that several randomized trials and retrospective studies of ivermectin use in patients with COVID-19 have now been published in peer-reviewed journals or on preprint servers.
“Some clinical studies showed no benefits or worsening of disease after ivermectin use, whereas others reported shorter time to resolution of disease manifestations attributed to COVID-19, greater reduction in inflammatory markers, shorter time to viral clearance, or lower mortality rates in patients who received ivermectin than in patients who received comparator drugs or placebo,” the NIH guidance reads.
The NIH acknowledges limitations: the studies have been small; doses of ivermectin have varied; some patients were taking other medications at the same time (including doxycycline, hydroxychloroquine, azithromycin, zinc, and corticosteroids, which may be potential confounders); and patients’ severity of COVID was not always clearly described in the studies.
Nasia Safdar, MD, medical director of infection prevention at the University of Wisconsin Hospital in Madison, told this news organization she agrees more research is needed before ivermectin is recommended by regulatory bodies for COVID-19.
That said, Dr. Safdar added, “in individual circumstances if a physician is confronted with a patient in dire straits and you’re not sure what to do, might you consider it? I think after a discussion with the patient, perhaps, but the level of evidence certainly doesn’t rise to the level of a policy.”
A downside of recommending a treatment without conclusive data, even if harm isn’t the primary concern, she said, is that supplies could dwindle for its intended use in other diseases. Also, premature approval can limit the robust research needed to see not only whether it works better for prevention or treatment, but also if it’s effective depending on patient populations and the severity of COVID-19.
Dr. Adalja and Dr. Safdar have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The National Institutes of Health has dropped its recommendation against the inexpensive antiparasitic drug ivermectin for treatment of COVID-19, and the agency now advises it can’t recommend for or against its use, leaving the decision to physicians and their patients.
“Results from adequately powered, well-designed, and well-conducted clinical trials are needed to provide more specific, evidence-based guidance on the role of ivermectin for the treatment of COVID-19,” according to new NIH guidance released last week.
Passionate arguments have been waged for and against the drug’s use.
The NIH update disappointed members of the Front Line COVID-19 Critical Care Alliance (FLCCC), which outlined its case for endorsing ivermectin in a public statement Jan. 18. Point by point, the group of 10 physicians argued against each limitation that drove the NIH’s ruling.
The group’s members said that, although grateful the recommendation against the drug was dropped, a neutral approach is not acceptable as total U.S. deaths surpassed 400,000 since last spring – and currently approach 4,000 a day. Results from research are enough to support its use, and the drug will immediately save lives, they say.
“Patients do not have time to wait,” they write, “and we as health care providers in society do not have that time either.”
NIH, which in August had recommended against ivermectin’s use, invited the group to present evidence to its treatment guidance panel on Jan. 6 to detail the emerging science surrounding ivermectin. The group cited rapidly growing evidence of the drug’s effectiveness.
Pierre Kory, MD, president/cofounder of FLCCC and a pulmonary and critical care specialist at Aurora St. Luke’s Medical Center in Milwaukee, also spoke before a Senate panel on Dec. 8 in a widely shared impassioned video, touting ivermectin as a COVID-19 “miracle” drug, a term he said he doesn’t use lightly.
Dr. Kory pleaded with the NIH to consider the emerging data. “Please, I’m just asking that they review our manuscript,” he told the senators.
“We have immense amounts of data to show that ivermectin must be implemented and implemented now,” he said.
Some draw parallels to hydroxychloroquine
Critics have said there’s not enough data to institute a protocol, and some draw parallels to another repurposed drug – hydroxychloroquine (HCQ) – which was once considered a promising treatment for COVID-19, based on flawed and incomplete evidence, and now is not recommended.
Paul Sax, MD, a professor of medicine at Harvard and clinical director of the HIV program and division of infectious diseases at Brigham and Women’s Hospital in Boston, wrote in a blog post earlier this month in the New England Journal of Medicine Journal Watch that ivermectin has more robust evidence for it than HCQ ever did.
“But we’re not quite yet at the ‘practice changing’ level,” he writes. “Results from at least five randomized clinical trials are expected soon that might further inform the decision.”
He said the best argument for the drug is seen in this explanation of a meta-analysis of studies of between 100 and 500 patients by Andrew Hill, MD, with the department of pharmacology, University of Liverpool (England).
Dr. Sax advises against two biases in considering ivermectin. One is assuming that because HCQ failed, other antiparasitic drugs will too.
The second bias to avoid, he says, is discounting studies done in low- and middle-income countries because “they weren’t done in the right places.”
“That’s not just bias,” he says. “It’s also snobbery.”
Ivermectin has been approved by the U.S. Food and Drug Administration for treatment of onchocerciasis (river blindness) and strongyloidiasis, but is not FDA-approved for the treatment of any viral infection. It also is sometimes used to treat animals.
In dropping the recommendation against ivermectin, the NIH gave it the same neutral declaration as monoclonal antibodies and convalescent plasma.
Some physicians say they won’t prescribe it
Some physicians say they won’t be recommending it to their COVID-19 patients.
Amesh Adalja, MD, an infectious disease expert and senior scholar at the Johns Hopkins University Center for Health Security in Baltimore,said in an interview that the NIH update hasn’t changed his mind and he isn’t prescribing it for his patients.
He said although “there’s enough of a signal” that he would like to see more data, “we haven’t seen anything in terms of a really robust study.”
He noted that the Infectious Diseases Society of America has 15 recommendations for COVID-19 treatment “and not one of them has to do with ivermectin.”
He added, “It’s not enough to see if it works, but we need to see who it works in and when it works in them.”
He also acknowledged that “some prominent physicians” are recommending it.
Among them is Paul Marik, MD, endowed professor of medicine and chief of pulmonary and critical care medicine at Eastern Virginia Medical School in Norfolk. A cofounder of FLCCC, Dr. Marik has championed ivermectin and developed a protocol for its use to prevent and treat COVID-19.
The data surrounding ivermectin have met with hope, criticism, and warnings.
Australian researchers published a study ahead of print in Antiviral Research that found ivermectin inhibited the replication of SARS-CoV-2 in a laboratory setting.
The study concluded that the drug resulted post infection in a 5,000-fold reduction in viral RNA at 48 hours. After that study, however, the FDA in April warned consumers not to self-medicate with ivermectin products intended for animals.
The NIH acknowledged that several randomized trials and retrospective studies of ivermectin use in patients with COVID-19 have now been published in peer-reviewed journals or on preprint servers.
“Some clinical studies showed no benefits or worsening of disease after ivermectin use, whereas others reported shorter time to resolution of disease manifestations attributed to COVID-19, greater reduction in inflammatory markers, shorter time to viral clearance, or lower mortality rates in patients who received ivermectin than in patients who received comparator drugs or placebo,” the NIH guidance reads.
The NIH acknowledges limitations: the studies have been small; doses of ivermectin have varied; some patients were taking other medications at the same time (including doxycycline, hydroxychloroquine, azithromycin, zinc, and corticosteroids, which may be potential confounders); and patients’ severity of COVID was not always clearly described in the studies.
Nasia Safdar, MD, medical director of infection prevention at the University of Wisconsin Hospital in Madison, told this news organization she agrees more research is needed before ivermectin is recommended by regulatory bodies for COVID-19.
That said, Dr. Safdar added, “in individual circumstances if a physician is confronted with a patient in dire straits and you’re not sure what to do, might you consider it? I think after a discussion with the patient, perhaps, but the level of evidence certainly doesn’t rise to the level of a policy.”
A downside of recommending a treatment without conclusive data, even if harm isn’t the primary concern, she said, is that supplies could dwindle for its intended use in other diseases. Also, premature approval can limit the robust research needed to see not only whether it works better for prevention or treatment, but also if it’s effective depending on patient populations and the severity of COVID-19.
Dr. Adalja and Dr. Safdar have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The National Institutes of Health has dropped its recommendation against the inexpensive antiparasitic drug ivermectin for treatment of COVID-19, and the agency now advises it can’t recommend for or against its use, leaving the decision to physicians and their patients.
“Results from adequately powered, well-designed, and well-conducted clinical trials are needed to provide more specific, evidence-based guidance on the role of ivermectin for the treatment of COVID-19,” according to new NIH guidance released last week.
Passionate arguments have been waged for and against the drug’s use.
The NIH update disappointed members of the Front Line COVID-19 Critical Care Alliance (FLCCC), which outlined its case for endorsing ivermectin in a public statement Jan. 18. Point by point, the group of 10 physicians argued against each limitation that drove the NIH’s ruling.
The group’s members said that, although grateful the recommendation against the drug was dropped, a neutral approach is not acceptable as total U.S. deaths surpassed 400,000 since last spring – and currently approach 4,000 a day. Results from research are enough to support its use, and the drug will immediately save lives, they say.
“Patients do not have time to wait,” they write, “and we as health care providers in society do not have that time either.”
NIH, which in August had recommended against ivermectin’s use, invited the group to present evidence to its treatment guidance panel on Jan. 6 to detail the emerging science surrounding ivermectin. The group cited rapidly growing evidence of the drug’s effectiveness.
Pierre Kory, MD, president/cofounder of FLCCC and a pulmonary and critical care specialist at Aurora St. Luke’s Medical Center in Milwaukee, also spoke before a Senate panel on Dec. 8 in a widely shared impassioned video, touting ivermectin as a COVID-19 “miracle” drug, a term he said he doesn’t use lightly.
Dr. Kory pleaded with the NIH to consider the emerging data. “Please, I’m just asking that they review our manuscript,” he told the senators.
“We have immense amounts of data to show that ivermectin must be implemented and implemented now,” he said.
Some draw parallels to hydroxychloroquine
Critics have said there’s not enough data to institute a protocol, and some draw parallels to another repurposed drug – hydroxychloroquine (HCQ) – which was once considered a promising treatment for COVID-19, based on flawed and incomplete evidence, and now is not recommended.
Paul Sax, MD, a professor of medicine at Harvard and clinical director of the HIV program and division of infectious diseases at Brigham and Women’s Hospital in Boston, wrote in a blog post earlier this month in the New England Journal of Medicine Journal Watch that ivermectin has more robust evidence for it than HCQ ever did.
“But we’re not quite yet at the ‘practice changing’ level,” he writes. “Results from at least five randomized clinical trials are expected soon that might further inform the decision.”
He said the best argument for the drug is seen in this explanation of a meta-analysis of studies of between 100 and 500 patients by Andrew Hill, MD, with the department of pharmacology, University of Liverpool (England).
Dr. Sax advises against two biases in considering ivermectin. One is assuming that because HCQ failed, other antiparasitic drugs will too.
The second bias to avoid, he says, is discounting studies done in low- and middle-income countries because “they weren’t done in the right places.”
“That’s not just bias,” he says. “It’s also snobbery.”
Ivermectin has been approved by the U.S. Food and Drug Administration for treatment of onchocerciasis (river blindness) and strongyloidiasis, but is not FDA-approved for the treatment of any viral infection. It also is sometimes used to treat animals.
In dropping the recommendation against ivermectin, the NIH gave it the same neutral declaration as monoclonal antibodies and convalescent plasma.
Some physicians say they won’t prescribe it
Some physicians say they won’t be recommending it to their COVID-19 patients.
Amesh Adalja, MD, an infectious disease expert and senior scholar at the Johns Hopkins University Center for Health Security in Baltimore,said in an interview that the NIH update hasn’t changed his mind and he isn’t prescribing it for his patients.
He said although “there’s enough of a signal” that he would like to see more data, “we haven’t seen anything in terms of a really robust study.”
He noted that the Infectious Diseases Society of America has 15 recommendations for COVID-19 treatment “and not one of them has to do with ivermectin.”
He added, “It’s not enough to see if it works, but we need to see who it works in and when it works in them.”
He also acknowledged that “some prominent physicians” are recommending it.
Among them is Paul Marik, MD, endowed professor of medicine and chief of pulmonary and critical care medicine at Eastern Virginia Medical School in Norfolk. A cofounder of FLCCC, Dr. Marik has championed ivermectin and developed a protocol for its use to prevent and treat COVID-19.
The data surrounding ivermectin have met with hope, criticism, and warnings.
Australian researchers published a study ahead of print in Antiviral Research that found ivermectin inhibited the replication of SARS-CoV-2 in a laboratory setting.
The study concluded that the drug resulted post infection in a 5,000-fold reduction in viral RNA at 48 hours. After that study, however, the FDA in April warned consumers not to self-medicate with ivermectin products intended for animals.
The NIH acknowledged that several randomized trials and retrospective studies of ivermectin use in patients with COVID-19 have now been published in peer-reviewed journals or on preprint servers.
“Some clinical studies showed no benefits or worsening of disease after ivermectin use, whereas others reported shorter time to resolution of disease manifestations attributed to COVID-19, greater reduction in inflammatory markers, shorter time to viral clearance, or lower mortality rates in patients who received ivermectin than in patients who received comparator drugs or placebo,” the NIH guidance reads.
The NIH acknowledges limitations: the studies have been small; doses of ivermectin have varied; some patients were taking other medications at the same time (including doxycycline, hydroxychloroquine, azithromycin, zinc, and corticosteroids, which may be potential confounders); and patients’ severity of COVID was not always clearly described in the studies.
Nasia Safdar, MD, medical director of infection prevention at the University of Wisconsin Hospital in Madison, told this news organization she agrees more research is needed before ivermectin is recommended by regulatory bodies for COVID-19.
That said, Dr. Safdar added, “in individual circumstances if a physician is confronted with a patient in dire straits and you’re not sure what to do, might you consider it? I think after a discussion with the patient, perhaps, but the level of evidence certainly doesn’t rise to the level of a policy.”
A downside of recommending a treatment without conclusive data, even if harm isn’t the primary concern, she said, is that supplies could dwindle for its intended use in other diseases. Also, premature approval can limit the robust research needed to see not only whether it works better for prevention or treatment, but also if it’s effective depending on patient populations and the severity of COVID-19.
Dr. Adalja and Dr. Safdar have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Monoclonal antibody combo treatment reduces viral load in mild to moderate COVID-19
A combination treatment of neutralizing monoclonal antibodies bamlanivimab and etesevimab was associated with a statistically significant reduction in SARS-CoV-2 at day 11 compared with placebo among nonhospitalized patients who had mild to moderate COVID-19, new data indicate.
However, bamlanivimab alone in three different single-infusion doses showed no significant reduction in viral load, compared with placebo, according to the phase 2/3 study by Robert L. Gottlieb, MD, PhD, of the Baylor University Medical Center and the Baylor Scott & White Research Institute, both in Dallas, and colleagues.
Findings from the Blocking Viral Attachment and Cell Entry with SARS-CoV-2 Neutralizing Antibodies (BLAZE-1) study were published online Jan. 21 in JAMA. The results represent findings through Oct. 6, 2020.
BLAZE-1 was funded by Eli Lilly, which makes both of the antispike neutralizing antibodies. The trial was conducted at 49 U.S. centers and included 613 outpatients who tested positive for SARS-CoV-2 and had one or more mild to moderate symptoms.
Patients were randomized to one of five groups (four treatment groups and a placebo control), and researchers analyzed between-group differences.
All four treatment arms suggest a trend toward reduction in viral load, which was the primary endpoint of the trial, but only the combination showed a statistically significant reduction.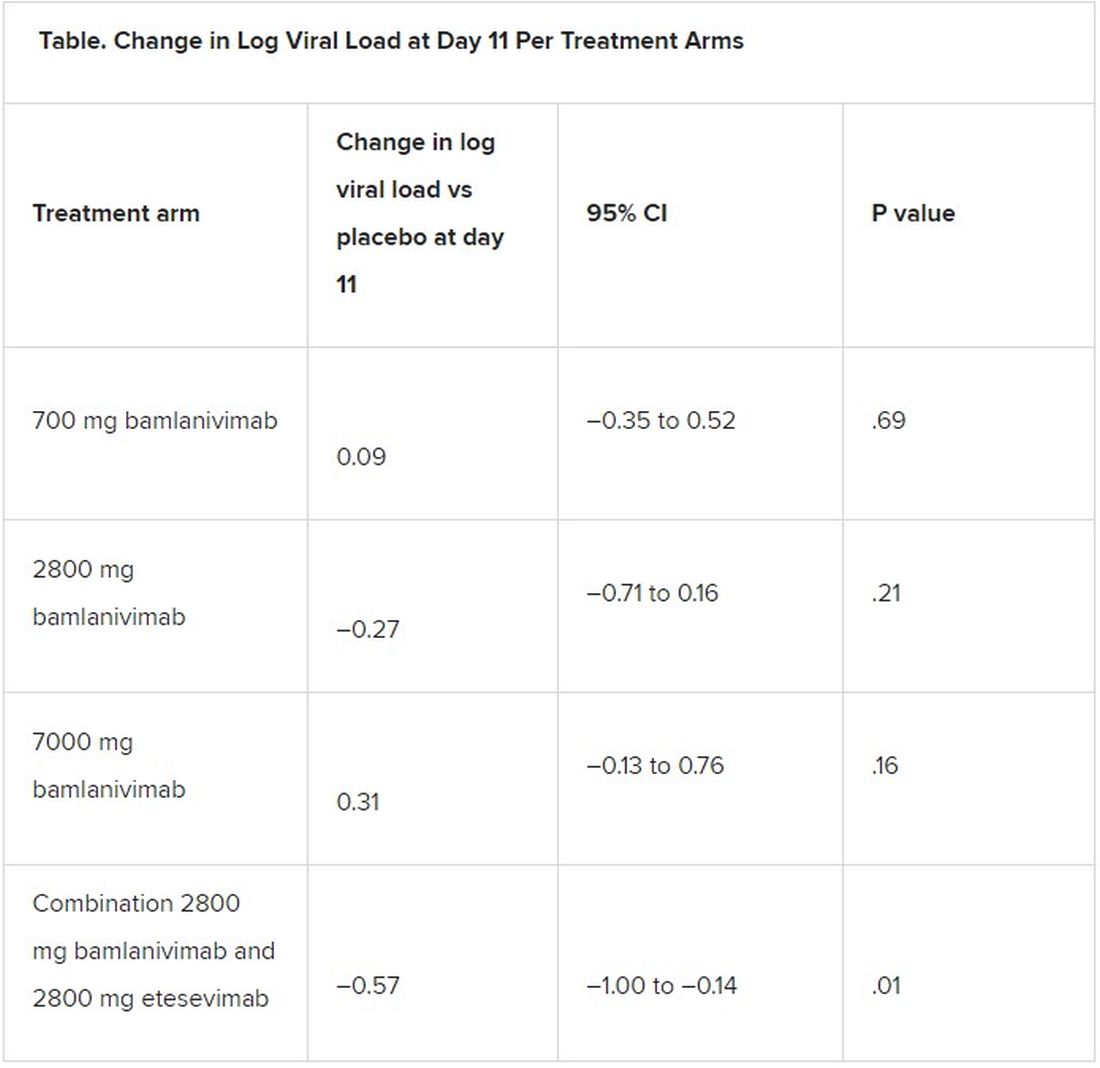
The average age of patients was 44.7 years, 54.6% were female, 42.5% were Hispanic, and 67.1% had at least one risk factor for severe COVID-19 (aged ≥55 years, body mass index of at least 30, or relevant comorbidity such as hypertension).
Among secondary outcomes, there were no consistent differences between the monotherapy groups or the combination group versus placebo for the other measures of viral load or clinical symptom scores.
The proportion of patients who had COVID-19–related hospitalizations or ED visits was 5.8% (nine events) for placebo; 1.0% (one event) for the 700-mg group; 1.9% (two events) for 2,800 mg; 2.0% (two events) for 7,000 mg; and 0.9% (one event) for combination treatment.
“Combining these two neutralizing monoclonal antibodies in clinical use may enhance viral load reduction and decrease treatment-emergent resistant variants,” the authors concluded.
Safety profile comparison
As for adverse events, immediate hypersensitivity reactions were reported in nine patients (six bamlanivimab, two combination treatment, and one placebo). No deaths occurred during the study.
Serious adverse events unrelated to SARS-CoV-2 infection or considered related to the study drug occurred in 0% (0/309) of patients in the bamlanivimab monotherapy groups; in 0.9% (1/112) of patients in the combination group; and in 0.6% (1/156) of patients in the placebo group.
The serious adverse event in the combination group was a urinary tract infection deemed unrelated to the study drug, the authors wrote.
The two most frequently reported side effects were nausea (3.0% for the 700-mg group; 3.7% for the 2,800-mg group; 5.0% for the 7,000-mg group; 3.6% for the combination group; and 3.8% for the placebo group) and diarrhea (1.0%, 1.9%, 5.9%, 0.9%, and 4.5%, respectively).
The authors included in the study’s limitations that the primary endpoint at day 11 may have been too late to best detect treatment effects.
“All patients, including those who received placebo, demonstrated substantial viral reduction by day 11,” they noted. “An earlier time point like day 3 or day 7 could possibly have been more appropriate to measure viral load.”
Currently, only remdesivir has been approved by the Food and Drug Administration for treating COVID-19, but convalescent plasma and neutralizing monoclonal antibodies have been granted emergency-use authorization.
In an accompanying editor’s note, Preeti N. Malani, MD, with the division of infectious diseases at the University of Michigan, Ann Arbor, and associate editor of JAMA, and Robert M. Golub, MD, deputy editor of JAMA, pointed out that these results differ from an earlier interim analysis of BLAZE-1 data.
A previous publication by Peter Chen, MD, with the department of medicine at Cedars Sinai Medical Center, Los Angeles, compared the three monotherapy groups (no combination group) with placebo, and in that study the 2,800-mg dose of bamlanivimab versus placebo achieved statistical significance for reduction in viral load from baseline at day 11, whereas the other two doses did not.
The editors explain that, in the study by Dr. Chen, “Follow-up for the placebo group was incomplete at the time of the database lock on Sept. 5, 2020. In the final analysis reported in the current article, the database was locked on Oct. 6, 2020, and the longer follow-up for the placebo group, which is now complete, resulted in changes in the primary outcome among that group.”
They concluded: “The comparison of the monotherapy groups against the final results for the placebo group led to changes in the effect sizes,” and the statistical significance of the 2,800-mg group was erased.
The editors pointed out that monoclonal antibodies are likely to benefit certain patients but definitive answers regarding which patients will benefit and under what circumstances will likely take more time than clinicians have to make decisions on treatment.
Meanwhile, as this news organization reported, the United States has spent $375 million on bamlanivimab and $450 million on Regeneron’s monoclonal antibody cocktail of casirivimab plus imdevimab, with the promise to spend billions more.
However, 80% of the 660,000 doses delivered by the two companies are still sitting on shelves, federal officials said in a press briefing last week, because of doubts about efficacy, lack of resources for infusion centers, and questions on reimbursement.
“While the world waits for widespread administration of effective vaccines and additional data on treatments, local efforts should work to improve testing access and turnaround time and reduce logistical barriers to ensure that monoclonal therapies can be provided to patients who are most likely to benefit,” Dr. Malani and Dr. Golub wrote.
This trial was sponsored and funded by Eli Lilly. Dr. Gottlieb disclosed personal fees and nonfinancial support (medication for another trial) from Gilead Sciences and serving on an advisory board for Sentinel. Several coauthors have financial ties to Eli Lilly. Dr. Malani reported serving on the National Institute of Allergy and Infectious Diseases COVID-19 Preventive Monoclonal Antibody data and safety monitoring board but was not compensated. Dr. Golub disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A combination treatment of neutralizing monoclonal antibodies bamlanivimab and etesevimab was associated with a statistically significant reduction in SARS-CoV-2 at day 11 compared with placebo among nonhospitalized patients who had mild to moderate COVID-19, new data indicate.
However, bamlanivimab alone in three different single-infusion doses showed no significant reduction in viral load, compared with placebo, according to the phase 2/3 study by Robert L. Gottlieb, MD, PhD, of the Baylor University Medical Center and the Baylor Scott & White Research Institute, both in Dallas, and colleagues.
Findings from the Blocking Viral Attachment and Cell Entry with SARS-CoV-2 Neutralizing Antibodies (BLAZE-1) study were published online Jan. 21 in JAMA. The results represent findings through Oct. 6, 2020.
BLAZE-1 was funded by Eli Lilly, which makes both of the antispike neutralizing antibodies. The trial was conducted at 49 U.S. centers and included 613 outpatients who tested positive for SARS-CoV-2 and had one or more mild to moderate symptoms.
Patients were randomized to one of five groups (four treatment groups and a placebo control), and researchers analyzed between-group differences.
All four treatment arms suggest a trend toward reduction in viral load, which was the primary endpoint of the trial, but only the combination showed a statistically significant reduction.
The average age of patients was 44.7 years, 54.6% were female, 42.5% were Hispanic, and 67.1% had at least one risk factor for severe COVID-19 (aged ≥55 years, body mass index of at least 30, or relevant comorbidity such as hypertension).
Among secondary outcomes, there were no consistent differences between the monotherapy groups or the combination group versus placebo for the other measures of viral load or clinical symptom scores.
The proportion of patients who had COVID-19–related hospitalizations or ED visits was 5.8% (nine events) for placebo; 1.0% (one event) for the 700-mg group; 1.9% (two events) for 2,800 mg; 2.0% (two events) for 7,000 mg; and 0.9% (one event) for combination treatment.
“Combining these two neutralizing monoclonal antibodies in clinical use may enhance viral load reduction and decrease treatment-emergent resistant variants,” the authors concluded.
Safety profile comparison
As for adverse events, immediate hypersensitivity reactions were reported in nine patients (six bamlanivimab, two combination treatment, and one placebo). No deaths occurred during the study.
Serious adverse events unrelated to SARS-CoV-2 infection or considered related to the study drug occurred in 0% (0/309) of patients in the bamlanivimab monotherapy groups; in 0.9% (1/112) of patients in the combination group; and in 0.6% (1/156) of patients in the placebo group.
The serious adverse event in the combination group was a urinary tract infection deemed unrelated to the study drug, the authors wrote.
The two most frequently reported side effects were nausea (3.0% for the 700-mg group; 3.7% for the 2,800-mg group; 5.0% for the 7,000-mg group; 3.6% for the combination group; and 3.8% for the placebo group) and diarrhea (1.0%, 1.9%, 5.9%, 0.9%, and 4.5%, respectively).
The authors included in the study’s limitations that the primary endpoint at day 11 may have been too late to best detect treatment effects.
“All patients, including those who received placebo, demonstrated substantial viral reduction by day 11,” they noted. “An earlier time point like day 3 or day 7 could possibly have been more appropriate to measure viral load.”
Currently, only remdesivir has been approved by the Food and Drug Administration for treating COVID-19, but convalescent plasma and neutralizing monoclonal antibodies have been granted emergency-use authorization.
In an accompanying editor’s note, Preeti N. Malani, MD, with the division of infectious diseases at the University of Michigan, Ann Arbor, and associate editor of JAMA, and Robert M. Golub, MD, deputy editor of JAMA, pointed out that these results differ from an earlier interim analysis of BLAZE-1 data.
A previous publication by Peter Chen, MD, with the department of medicine at Cedars Sinai Medical Center, Los Angeles, compared the three monotherapy groups (no combination group) with placebo, and in that study the 2,800-mg dose of bamlanivimab versus placebo achieved statistical significance for reduction in viral load from baseline at day 11, whereas the other two doses did not.
The editors explain that, in the study by Dr. Chen, “Follow-up for the placebo group was incomplete at the time of the database lock on Sept. 5, 2020. In the final analysis reported in the current article, the database was locked on Oct. 6, 2020, and the longer follow-up for the placebo group, which is now complete, resulted in changes in the primary outcome among that group.”
They concluded: “The comparison of the monotherapy groups against the final results for the placebo group led to changes in the effect sizes,” and the statistical significance of the 2,800-mg group was erased.
The editors pointed out that monoclonal antibodies are likely to benefit certain patients but definitive answers regarding which patients will benefit and under what circumstances will likely take more time than clinicians have to make decisions on treatment.
Meanwhile, as this news organization reported, the United States has spent $375 million on bamlanivimab and $450 million on Regeneron’s monoclonal antibody cocktail of casirivimab plus imdevimab, with the promise to spend billions more.
However, 80% of the 660,000 doses delivered by the two companies are still sitting on shelves, federal officials said in a press briefing last week, because of doubts about efficacy, lack of resources for infusion centers, and questions on reimbursement.
“While the world waits for widespread administration of effective vaccines and additional data on treatments, local efforts should work to improve testing access and turnaround time and reduce logistical barriers to ensure that monoclonal therapies can be provided to patients who are most likely to benefit,” Dr. Malani and Dr. Golub wrote.
This trial was sponsored and funded by Eli Lilly. Dr. Gottlieb disclosed personal fees and nonfinancial support (medication for another trial) from Gilead Sciences and serving on an advisory board for Sentinel. Several coauthors have financial ties to Eli Lilly. Dr. Malani reported serving on the National Institute of Allergy and Infectious Diseases COVID-19 Preventive Monoclonal Antibody data and safety monitoring board but was not compensated. Dr. Golub disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A combination treatment of neutralizing monoclonal antibodies bamlanivimab and etesevimab was associated with a statistically significant reduction in SARS-CoV-2 at day 11 compared with placebo among nonhospitalized patients who had mild to moderate COVID-19, new data indicate.
However, bamlanivimab alone in three different single-infusion doses showed no significant reduction in viral load, compared with placebo, according to the phase 2/3 study by Robert L. Gottlieb, MD, PhD, of the Baylor University Medical Center and the Baylor Scott & White Research Institute, both in Dallas, and colleagues.
Findings from the Blocking Viral Attachment and Cell Entry with SARS-CoV-2 Neutralizing Antibodies (BLAZE-1) study were published online Jan. 21 in JAMA. The results represent findings through Oct. 6, 2020.
BLAZE-1 was funded by Eli Lilly, which makes both of the antispike neutralizing antibodies. The trial was conducted at 49 U.S. centers and included 613 outpatients who tested positive for SARS-CoV-2 and had one or more mild to moderate symptoms.
Patients were randomized to one of five groups (four treatment groups and a placebo control), and researchers analyzed between-group differences.
All four treatment arms suggest a trend toward reduction in viral load, which was the primary endpoint of the trial, but only the combination showed a statistically significant reduction.
The average age of patients was 44.7 years, 54.6% were female, 42.5% were Hispanic, and 67.1% had at least one risk factor for severe COVID-19 (aged ≥55 years, body mass index of at least 30, or relevant comorbidity such as hypertension).
Among secondary outcomes, there were no consistent differences between the monotherapy groups or the combination group versus placebo for the other measures of viral load or clinical symptom scores.
The proportion of patients who had COVID-19–related hospitalizations or ED visits was 5.8% (nine events) for placebo; 1.0% (one event) for the 700-mg group; 1.9% (two events) for 2,800 mg; 2.0% (two events) for 7,000 mg; and 0.9% (one event) for combination treatment.
“Combining these two neutralizing monoclonal antibodies in clinical use may enhance viral load reduction and decrease treatment-emergent resistant variants,” the authors concluded.
Safety profile comparison
As for adverse events, immediate hypersensitivity reactions were reported in nine patients (six bamlanivimab, two combination treatment, and one placebo). No deaths occurred during the study.
Serious adverse events unrelated to SARS-CoV-2 infection or considered related to the study drug occurred in 0% (0/309) of patients in the bamlanivimab monotherapy groups; in 0.9% (1/112) of patients in the combination group; and in 0.6% (1/156) of patients in the placebo group.
The serious adverse event in the combination group was a urinary tract infection deemed unrelated to the study drug, the authors wrote.
The two most frequently reported side effects were nausea (3.0% for the 700-mg group; 3.7% for the 2,800-mg group; 5.0% for the 7,000-mg group; 3.6% for the combination group; and 3.8% for the placebo group) and diarrhea (1.0%, 1.9%, 5.9%, 0.9%, and 4.5%, respectively).
The authors included in the study’s limitations that the primary endpoint at day 11 may have been too late to best detect treatment effects.
“All patients, including those who received placebo, demonstrated substantial viral reduction by day 11,” they noted. “An earlier time point like day 3 or day 7 could possibly have been more appropriate to measure viral load.”
Currently, only remdesivir has been approved by the Food and Drug Administration for treating COVID-19, but convalescent plasma and neutralizing monoclonal antibodies have been granted emergency-use authorization.
In an accompanying editor’s note, Preeti N. Malani, MD, with the division of infectious diseases at the University of Michigan, Ann Arbor, and associate editor of JAMA, and Robert M. Golub, MD, deputy editor of JAMA, pointed out that these results differ from an earlier interim analysis of BLAZE-1 data.
A previous publication by Peter Chen, MD, with the department of medicine at Cedars Sinai Medical Center, Los Angeles, compared the three monotherapy groups (no combination group) with placebo, and in that study the 2,800-mg dose of bamlanivimab versus placebo achieved statistical significance for reduction in viral load from baseline at day 11, whereas the other two doses did not.
The editors explain that, in the study by Dr. Chen, “Follow-up for the placebo group was incomplete at the time of the database lock on Sept. 5, 2020. In the final analysis reported in the current article, the database was locked on Oct. 6, 2020, and the longer follow-up for the placebo group, which is now complete, resulted in changes in the primary outcome among that group.”
They concluded: “The comparison of the monotherapy groups against the final results for the placebo group led to changes in the effect sizes,” and the statistical significance of the 2,800-mg group was erased.
The editors pointed out that monoclonal antibodies are likely to benefit certain patients but definitive answers regarding which patients will benefit and under what circumstances will likely take more time than clinicians have to make decisions on treatment.
Meanwhile, as this news organization reported, the United States has spent $375 million on bamlanivimab and $450 million on Regeneron’s monoclonal antibody cocktail of casirivimab plus imdevimab, with the promise to spend billions more.
However, 80% of the 660,000 doses delivered by the two companies are still sitting on shelves, federal officials said in a press briefing last week, because of doubts about efficacy, lack of resources for infusion centers, and questions on reimbursement.
“While the world waits for widespread administration of effective vaccines and additional data on treatments, local efforts should work to improve testing access and turnaround time and reduce logistical barriers to ensure that monoclonal therapies can be provided to patients who are most likely to benefit,” Dr. Malani and Dr. Golub wrote.
This trial was sponsored and funded by Eli Lilly. Dr. Gottlieb disclosed personal fees and nonfinancial support (medication for another trial) from Gilead Sciences and serving on an advisory board for Sentinel. Several coauthors have financial ties to Eli Lilly. Dr. Malani reported serving on the National Institute of Allergy and Infectious Diseases COVID-19 Preventive Monoclonal Antibody data and safety monitoring board but was not compensated. Dr. Golub disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
ACEIs, ARBs safe to continue in COVID-19: Trial published
The BRACE-CORONA trial, the first randomized trial to address the question of whether patients with COVID-19 should continue to take ACE inhibitors (ACEIs) or angiotensin-receptor blockers (ARBs) – has now been published.
The study, which was conducted in patients hospitalized with COVID-19 who were taking ACEIs or ARBs before hospitalization, showed no significant difference in the mean number of days alive and out of the hospital for those assigned to discontinue versus those assigned to continue these medications.
There were, however, hints that continuing to take ACEIs or ARBs may be beneficial for patients with more severe COVID-19.
The study was first presented at last year’s European Society of Cardiology Congress and was reported by this news organization at that time. The study was published online in JAMA on Jan. 19, 2021.
“These findings do not support routinely discontinuing ACEIs or ARBs among patients hospitalized with mild to moderate COVID-19 if there is an indication for treatment,” the authors concluded.
Led by Renato D. Lopes, MD, Duke Clinical Research Institute, Durham, N.C., the researchers explained that there has been conflicting speculation about the effect of renin-angiotensin-aldosterone system (RAAS) inhibitors on the course of COVID-19.
On the one hand, observations from animal models suggest that ACEIs and ARBs up-regulate the expression of ACE2, a receptor involved in SARS-CoV-2 infection of host target cells. This led to suggestions that these medications may enhance viral binding and cell entry. Conversely, RAAS inhibitors could benefit patients with COVID-19 through effects on angiotensin II expression and subsequent increases in angiotensin 1-7 and 1-9, which have vasodilatory and anti-inflammatory effects that might attenuate lung injury.
The BRACE-CORONA trial included 659 patients hospitalized in Brazil with mild to moderate COVID-19 who were taking ACEIs or ARBs prior to hospitalization. The median age of the patients was 55 years. Of these patients, 57.1% were considered to have mild cases at hospital admission, and 42.9% were considered to have moderate cases.
Results showed no significant difference in the number of days alive and out of the hospital for patients in the discontinuation group (mean, 21.9 days) in comparison with patients in the continuation group (mean, 22.9 days). The mean ratio was 0.95 (95% confidence interval, 0.90-1.01).
There also was no statistically significant difference in deaths (2.7% of the discontinuation group vs. 2.8% for the continuation group); cardiovascular death (0.6% vs. 0.3%), or COVID-19 progression (38.3% vs. 32.3%).
The most common adverse events were respiratory failure requiring invasive mechanical ventilation (9.6% in the discontinuation group vs. 7.7% in the continuation group), shock requiring vasopressors (8.4% vs. 7.1%), acute MI (7.5% vs. 4.6%), new or worsening heart failure (4.2% vs. 4.9%), and acute kidney failure requiring hemodialysis (3.3% vs. 2.8%).
The authors note that hypertension is an important comorbidity in patients with COVID-19. Recent data suggest that immune dysfunction may contribute to poor outcomes among patients who have COVID-19 and hypertension.
It has been shown that, when use of long-term medications is discontinued during hospitalization, the use of those medications is often not resumed, owing to clinical inertia. Long-term outcomes worsen as a result, the authors reported. In the current study, all patients had hypertension, and more than 50% were obese; both of these comorbidities increase the risk for poor outcomes with COVID-19.
The investigators pointed out that a sensitivity analysis in which site was regarded as a random effect showed a statistically significant finding in favor of the group that continued ACEIs or ARBs. This finding was similar to that of the on-treatment analysis. There were also statistically significant interactions between treatment effect and some subgroups, such as patients with lower oxygen saturation and greater disease severity at hospital admission. For these patients, continuing ACEIs or ARBs may be beneficial.
“The primary analyses with the null results but wide 95% confidence intervals suggest that the study might have been underpowered to detect a statistically significant benefit of continuing ACEIs or ARBs,” they said.
Dr. Lopes has received grant support from Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi and consulting fees from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Medtronic, Merck, Pfizer, Portola, and Sanofi.
A version of this article first appeared on Medscape.com.
The BRACE-CORONA trial, the first randomized trial to address the question of whether patients with COVID-19 should continue to take ACE inhibitors (ACEIs) or angiotensin-receptor blockers (ARBs) – has now been published.
The study, which was conducted in patients hospitalized with COVID-19 who were taking ACEIs or ARBs before hospitalization, showed no significant difference in the mean number of days alive and out of the hospital for those assigned to discontinue versus those assigned to continue these medications.
There were, however, hints that continuing to take ACEIs or ARBs may be beneficial for patients with more severe COVID-19.
The study was first presented at last year’s European Society of Cardiology Congress and was reported by this news organization at that time. The study was published online in JAMA on Jan. 19, 2021.
“These findings do not support routinely discontinuing ACEIs or ARBs among patients hospitalized with mild to moderate COVID-19 if there is an indication for treatment,” the authors concluded.
Led by Renato D. Lopes, MD, Duke Clinical Research Institute, Durham, N.C., the researchers explained that there has been conflicting speculation about the effect of renin-angiotensin-aldosterone system (RAAS) inhibitors on the course of COVID-19.
On the one hand, observations from animal models suggest that ACEIs and ARBs up-regulate the expression of ACE2, a receptor involved in SARS-CoV-2 infection of host target cells. This led to suggestions that these medications may enhance viral binding and cell entry. Conversely, RAAS inhibitors could benefit patients with COVID-19 through effects on angiotensin II expression and subsequent increases in angiotensin 1-7 and 1-9, which have vasodilatory and anti-inflammatory effects that might attenuate lung injury.
The BRACE-CORONA trial included 659 patients hospitalized in Brazil with mild to moderate COVID-19 who were taking ACEIs or ARBs prior to hospitalization. The median age of the patients was 55 years. Of these patients, 57.1% were considered to have mild cases at hospital admission, and 42.9% were considered to have moderate cases.
Results showed no significant difference in the number of days alive and out of the hospital for patients in the discontinuation group (mean, 21.9 days) in comparison with patients in the continuation group (mean, 22.9 days). The mean ratio was 0.95 (95% confidence interval, 0.90-1.01).
There also was no statistically significant difference in deaths (2.7% of the discontinuation group vs. 2.8% for the continuation group); cardiovascular death (0.6% vs. 0.3%), or COVID-19 progression (38.3% vs. 32.3%).
The most common adverse events were respiratory failure requiring invasive mechanical ventilation (9.6% in the discontinuation group vs. 7.7% in the continuation group), shock requiring vasopressors (8.4% vs. 7.1%), acute MI (7.5% vs. 4.6%), new or worsening heart failure (4.2% vs. 4.9%), and acute kidney failure requiring hemodialysis (3.3% vs. 2.8%).
The authors note that hypertension is an important comorbidity in patients with COVID-19. Recent data suggest that immune dysfunction may contribute to poor outcomes among patients who have COVID-19 and hypertension.
It has been shown that, when use of long-term medications is discontinued during hospitalization, the use of those medications is often not resumed, owing to clinical inertia. Long-term outcomes worsen as a result, the authors reported. In the current study, all patients had hypertension, and more than 50% were obese; both of these comorbidities increase the risk for poor outcomes with COVID-19.
The investigators pointed out that a sensitivity analysis in which site was regarded as a random effect showed a statistically significant finding in favor of the group that continued ACEIs or ARBs. This finding was similar to that of the on-treatment analysis. There were also statistically significant interactions between treatment effect and some subgroups, such as patients with lower oxygen saturation and greater disease severity at hospital admission. For these patients, continuing ACEIs or ARBs may be beneficial.
“The primary analyses with the null results but wide 95% confidence intervals suggest that the study might have been underpowered to detect a statistically significant benefit of continuing ACEIs or ARBs,” they said.
Dr. Lopes has received grant support from Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi and consulting fees from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Medtronic, Merck, Pfizer, Portola, and Sanofi.
A version of this article first appeared on Medscape.com.
The BRACE-CORONA trial, the first randomized trial to address the question of whether patients with COVID-19 should continue to take ACE inhibitors (ACEIs) or angiotensin-receptor blockers (ARBs) – has now been published.
The study, which was conducted in patients hospitalized with COVID-19 who were taking ACEIs or ARBs before hospitalization, showed no significant difference in the mean number of days alive and out of the hospital for those assigned to discontinue versus those assigned to continue these medications.
There were, however, hints that continuing to take ACEIs or ARBs may be beneficial for patients with more severe COVID-19.
The study was first presented at last year’s European Society of Cardiology Congress and was reported by this news organization at that time. The study was published online in JAMA on Jan. 19, 2021.
“These findings do not support routinely discontinuing ACEIs or ARBs among patients hospitalized with mild to moderate COVID-19 if there is an indication for treatment,” the authors concluded.
Led by Renato D. Lopes, MD, Duke Clinical Research Institute, Durham, N.C., the researchers explained that there has been conflicting speculation about the effect of renin-angiotensin-aldosterone system (RAAS) inhibitors on the course of COVID-19.
On the one hand, observations from animal models suggest that ACEIs and ARBs up-regulate the expression of ACE2, a receptor involved in SARS-CoV-2 infection of host target cells. This led to suggestions that these medications may enhance viral binding and cell entry. Conversely, RAAS inhibitors could benefit patients with COVID-19 through effects on angiotensin II expression and subsequent increases in angiotensin 1-7 and 1-9, which have vasodilatory and anti-inflammatory effects that might attenuate lung injury.
The BRACE-CORONA trial included 659 patients hospitalized in Brazil with mild to moderate COVID-19 who were taking ACEIs or ARBs prior to hospitalization. The median age of the patients was 55 years. Of these patients, 57.1% were considered to have mild cases at hospital admission, and 42.9% were considered to have moderate cases.
Results showed no significant difference in the number of days alive and out of the hospital for patients in the discontinuation group (mean, 21.9 days) in comparison with patients in the continuation group (mean, 22.9 days). The mean ratio was 0.95 (95% confidence interval, 0.90-1.01).
There also was no statistically significant difference in deaths (2.7% of the discontinuation group vs. 2.8% for the continuation group); cardiovascular death (0.6% vs. 0.3%), or COVID-19 progression (38.3% vs. 32.3%).
The most common adverse events were respiratory failure requiring invasive mechanical ventilation (9.6% in the discontinuation group vs. 7.7% in the continuation group), shock requiring vasopressors (8.4% vs. 7.1%), acute MI (7.5% vs. 4.6%), new or worsening heart failure (4.2% vs. 4.9%), and acute kidney failure requiring hemodialysis (3.3% vs. 2.8%).
The authors note that hypertension is an important comorbidity in patients with COVID-19. Recent data suggest that immune dysfunction may contribute to poor outcomes among patients who have COVID-19 and hypertension.
It has been shown that, when use of long-term medications is discontinued during hospitalization, the use of those medications is often not resumed, owing to clinical inertia. Long-term outcomes worsen as a result, the authors reported. In the current study, all patients had hypertension, and more than 50% were obese; both of these comorbidities increase the risk for poor outcomes with COVID-19.
The investigators pointed out that a sensitivity analysis in which site was regarded as a random effect showed a statistically significant finding in favor of the group that continued ACEIs or ARBs. This finding was similar to that of the on-treatment analysis. There were also statistically significant interactions between treatment effect and some subgroups, such as patients with lower oxygen saturation and greater disease severity at hospital admission. For these patients, continuing ACEIs or ARBs may be beneficial.
“The primary analyses with the null results but wide 95% confidence intervals suggest that the study might have been underpowered to detect a statistically significant benefit of continuing ACEIs or ARBs,” they said.
Dr. Lopes has received grant support from Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi and consulting fees from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Medtronic, Merck, Pfizer, Portola, and Sanofi.
A version of this article first appeared on Medscape.com.
Repeated ketamine infusions linked to rapid relief of PTSD
Repeated intravenous infusions of ketamine provide rapid relief for patients with posttraumatic stress disorder, new research suggests.
In what investigators are calling the first randomized controlled trial of repeated ketamine administration for chronic PTSD, 30 patients received six infusions of ketamine or midazolam (used as a psychoactive placebo) over 2 consecutive weeks.
Between baseline and week 2, those receiving ketamine showed significantly greater improvement than those receiving midazolam. Total scores on the Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) for the first group were almost 12 points lower than the latter group at week 2, meeting the study’s primary outcome measure.
In addition, 67% vs. 20% of the patients, respectively, were considered to be treatment responders; time to loss of response for those in the ketamine group was 28 days.
Although the overall findings were as expected, “what was surprising was how robust the results were,” lead author Adriana Feder, MD, associate professor of psychiatry, Icahn School of Medicine, Mount Sinai, New York, told this news organization.
It was also a bit surprising that, in a study of just 30 participants, “we were able to show such a clear difference” between the two treatment groups, said Dr. Feder, who is also a coinventor on issued patents for the use of ketamine as therapy for PTSD, and codirector of the Ehrenkranz Lab for the Study of Human Resilience at Mount Sinai.
The findings were published online Jan. 5 in the American Journal of Psychiatry.
Unmet need
Ketamine is a glutamate N-methyl-D-aspartate (NMDA) receptor antagonist that was first approved by the U.S. Food and Drug Administration for anesthetic use in 1970. It has also been shown to be effective for treatment-resistant depression.
PTSD has a lifetime prevalence of about 6% in the United States. “While trauma-focused psychotherapies have the most empirical support, they are limited by significant rates of nonresponse, partial response, and treatment dropout,” the investigators write. Also, there are “few available pharmacotherapies for PTSD, and their efficacy is insufficient,” they add.
“There’s a real need for new treatment interventions that are effective for PTSD and also work rapidly, because it can take weeks to months for currently available treatments to work for PTSD,” Dr. Feder said.
The researchers previously conducted a “proof-of-concept” randomized controlled trial of single infusions of ketamine for chronic PTSD. Results published in 2014 in JAMA Psychiatry showed significant reduction in PTSD symptoms 24 hours after infusion.
For the current study, the investigative team wanted to assess whether ketamine was viable as a longer-term treatment.
“We were encouraged by our initial promising findings” of the earlier trial, Dr. Feder said. “We wanted to do the second study to see if ketamine really works for PTSD, to see if we could replicate the rapid improvement and also examine whether a course of six infusions over 2 weeks could maintain the improvement.”
Thirty patients (aged 18-70; mean age, 39 years) with chronic PTSD from civilian or military trauma were enrolled (mean PTSD duration, 15 years).
The most cited primary trauma was sexual assault or molestation (n = 13), followed by physical assault or abuse (n = 8), witnessing a violent assault or death (n = 4), witnessing the 9/11 attacks (n = 3), and combat exposure (n = 2).
During the 2-week treatment phase, half of the patients were randomly assigned to receive six infusions of ketamine hydrochloride at a dose of 0.5 mg/kg (86.7% women; mean CAPS-5 score, 42), while the other half received six infusions of midazolam at a dose of 0.045 mg/kg (66.7% women; mean CAPS-5 score, 40).
In addition to the primary outcome measure of 2-week changes on the CAPS-5, secondary outcomes included score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) and the Impact of Event Scale-Revised (IES-R).
Treatment response was defined as a 30% or more improvement in symptoms on the CAPS-5. A number of measures were also used to assess potential treatment-related adverse events (AEs).
Safe, effective
Results showed significantly lower total CAPS-5 scores for the ketamine group vs. the midazolam group at week 1 (score difference, 8.8 points; P = .03) and at week 2 (score difference, 11.88 points; P = .004).
Those receiving ketamine also showed improvements in three of the four PTSD symptom clusters on the CAPS-5: avoidance (P < .0001), negative mood and cognitions (P = .02), and intrusions (P = .03). The fourth symptom cluster – arousal and reactivity – did not show a significant improvement.
In addition, the ketamine group showed significantly greater improvement scores on the MADRS at both week 1 and week 2.
Treatment response at 2 weeks was achieved by 10 members of the ketamine group and by three members of the midazolam group (P = .03).
Secondary analyses showed rapid improvement in the treatment responders within the ketamine group, with a mean change of 26 points on the total IES-R score between baseline and 24 hours after their first infusion, and a mean change of 13.4 points on the MADRS total past-24-hour score, a 53% improvement on average.
“A response at 2 weeks is very rapid but they got better sometimes within the first day,” Dr. Feder noted.
There were no serious AEs reported. Although some dissociative symptoms occurred during ketamine infusions, with the highest levels reported at the end of the infusion, these symptoms had resolved by the next assessment, conducted 2 hours after infusion.
The most frequently reported AE in the ketamine group, compared with midazolam, after the start of infusions was blurred vision (53% vs. 0%), followed by dizziness (33% vs. 13%), fatigue (33% vs. 87%), headache (27% vs. 13%), and nausea or vomiting (20% vs. 7%).
‘Large-magnitude improvement’
The overall findings show that, in this patient population, “repeated intravenous ketamine infusions administered over 2 weeks were associated with a large-magnitude, clinically significant improvement in PTSD symptoms,” the investigators write.
The results “were very satisfying,” added Dr. Feder. “It was heartening also to hear what some of the participants would say. Some told us about how their symptoms and feelings had changed during the course of treatment with ketamine, where they felt stronger and better able to cope with their trauma and memories.”
She noted, however, that this was not a study designed to specifically assess ketamine in treatment-resistant PTSD. “Some patients had had multiple treatments before that hadn’t worked, while others had not received treatment before. Efficacy for treatment-resistant PTSD is an important question for future research,” Dr. Feder said.
Other areas worth future exploration include treatment efficacy in patients with different types of trauma and whether outcomes can last longer in patients receiving ketamine plus psychotherapy treatment, she noted.
“I don’t want to ignore the fact that currently available treatments work for a number of people with chronic PTSD. But because there are many more for whom [the treatments] don’t work, or they’re insufficiently helped by those treatments, this is certainly one potentially very promising approach that can be added” to a clinician’s toolbox, Dr. Feder said.
Speaks to clinical utility
Commenting for this news organization, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University, New Haven, Connecticut, called this a “very solid and well-designed” study.
“It definitely builds on what’s been found in the past, but it’s a critical piece of information speaking to the clinical utility of this treatment for PTSD,” said Dr. Sanacora, who is also director of the Yale Depression Research Program and was not involved with the current research.
He agreed with the investigators that PTSD has long been a condition that is difficult to treat.
“It’s an area that has a great unmet need for treatment options. Beyond that, as ketamine is becoming more widely used, there’s increasing demand for off-label uses. This [study] actually provides some evidence that there may be efficacy there,” Dr. Sanacora said.
Although he cautioned that this was a small study, and thus further research with a larger patient population will be needed, it provides a compelling foundation to build upon.
“This study provides clear evidence to support a larger study to really give a definitive statement on the efficacy and safety of its use for PTSD. I don’t think this is the study that provides that definitive evidence, but it is a very strong indication, and it very strongly supports the initiation of a large study to address that,” said Dr. Sanacora.
He noted that, although he’s used the term “cautious optimism” for studies in the past, he has “real optimism” that ketamine will be effective for PTSD based on the results of this current study.
“We still need some more data to really convince us of that before we can say with any clear statement that it is effective and safe, but I’m very optimistic,” Dr. Sanacora concluded.
The study was funded by the Brain and Behavior Research Foundation, Mount Sinai Innovation Partners and the Mount Sinai i3 Accelerator, Gerald and Glenda Greenwald, and the Ehrenkranz Laboratory for Human Resilience. Dr. Feder is a coinventor on issued patents for the use of ketamine as therapy for PTSD. A list of all disclosures for the other study authors are listed in the original article.
A version of this article first appeared on Medscape.com.
Repeated intravenous infusions of ketamine provide rapid relief for patients with posttraumatic stress disorder, new research suggests.
In what investigators are calling the first randomized controlled trial of repeated ketamine administration for chronic PTSD, 30 patients received six infusions of ketamine or midazolam (used as a psychoactive placebo) over 2 consecutive weeks.
Between baseline and week 2, those receiving ketamine showed significantly greater improvement than those receiving midazolam. Total scores on the Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) for the first group were almost 12 points lower than the latter group at week 2, meeting the study’s primary outcome measure.
In addition, 67% vs. 20% of the patients, respectively, were considered to be treatment responders; time to loss of response for those in the ketamine group was 28 days.
Although the overall findings were as expected, “what was surprising was how robust the results were,” lead author Adriana Feder, MD, associate professor of psychiatry, Icahn School of Medicine, Mount Sinai, New York, told this news organization.
It was also a bit surprising that, in a study of just 30 participants, “we were able to show such a clear difference” between the two treatment groups, said Dr. Feder, who is also a coinventor on issued patents for the use of ketamine as therapy for PTSD, and codirector of the Ehrenkranz Lab for the Study of Human Resilience at Mount Sinai.
The findings were published online Jan. 5 in the American Journal of Psychiatry.
Unmet need
Ketamine is a glutamate N-methyl-D-aspartate (NMDA) receptor antagonist that was first approved by the U.S. Food and Drug Administration for anesthetic use in 1970. It has also been shown to be effective for treatment-resistant depression.
PTSD has a lifetime prevalence of about 6% in the United States. “While trauma-focused psychotherapies have the most empirical support, they are limited by significant rates of nonresponse, partial response, and treatment dropout,” the investigators write. Also, there are “few available pharmacotherapies for PTSD, and their efficacy is insufficient,” they add.
“There’s a real need for new treatment interventions that are effective for PTSD and also work rapidly, because it can take weeks to months for currently available treatments to work for PTSD,” Dr. Feder said.
The researchers previously conducted a “proof-of-concept” randomized controlled trial of single infusions of ketamine for chronic PTSD. Results published in 2014 in JAMA Psychiatry showed significant reduction in PTSD symptoms 24 hours after infusion.
For the current study, the investigative team wanted to assess whether ketamine was viable as a longer-term treatment.
“We were encouraged by our initial promising findings” of the earlier trial, Dr. Feder said. “We wanted to do the second study to see if ketamine really works for PTSD, to see if we could replicate the rapid improvement and also examine whether a course of six infusions over 2 weeks could maintain the improvement.”
Thirty patients (aged 18-70; mean age, 39 years) with chronic PTSD from civilian or military trauma were enrolled (mean PTSD duration, 15 years).
The most cited primary trauma was sexual assault or molestation (n = 13), followed by physical assault or abuse (n = 8), witnessing a violent assault or death (n = 4), witnessing the 9/11 attacks (n = 3), and combat exposure (n = 2).
During the 2-week treatment phase, half of the patients were randomly assigned to receive six infusions of ketamine hydrochloride at a dose of 0.5 mg/kg (86.7% women; mean CAPS-5 score, 42), while the other half received six infusions of midazolam at a dose of 0.045 mg/kg (66.7% women; mean CAPS-5 score, 40).
In addition to the primary outcome measure of 2-week changes on the CAPS-5, secondary outcomes included score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) and the Impact of Event Scale-Revised (IES-R).
Treatment response was defined as a 30% or more improvement in symptoms on the CAPS-5. A number of measures were also used to assess potential treatment-related adverse events (AEs).
Safe, effective
Results showed significantly lower total CAPS-5 scores for the ketamine group vs. the midazolam group at week 1 (score difference, 8.8 points; P = .03) and at week 2 (score difference, 11.88 points; P = .004).
Those receiving ketamine also showed improvements in three of the four PTSD symptom clusters on the CAPS-5: avoidance (P < .0001), negative mood and cognitions (P = .02), and intrusions (P = .03). The fourth symptom cluster – arousal and reactivity – did not show a significant improvement.
In addition, the ketamine group showed significantly greater improvement scores on the MADRS at both week 1 and week 2.
Treatment response at 2 weeks was achieved by 10 members of the ketamine group and by three members of the midazolam group (P = .03).
Secondary analyses showed rapid improvement in the treatment responders within the ketamine group, with a mean change of 26 points on the total IES-R score between baseline and 24 hours after their first infusion, and a mean change of 13.4 points on the MADRS total past-24-hour score, a 53% improvement on average.
“A response at 2 weeks is very rapid but they got better sometimes within the first day,” Dr. Feder noted.
There were no serious AEs reported. Although some dissociative symptoms occurred during ketamine infusions, with the highest levels reported at the end of the infusion, these symptoms had resolved by the next assessment, conducted 2 hours after infusion.
The most frequently reported AE in the ketamine group, compared with midazolam, after the start of infusions was blurred vision (53% vs. 0%), followed by dizziness (33% vs. 13%), fatigue (33% vs. 87%), headache (27% vs. 13%), and nausea or vomiting (20% vs. 7%).
‘Large-magnitude improvement’
The overall findings show that, in this patient population, “repeated intravenous ketamine infusions administered over 2 weeks were associated with a large-magnitude, clinically significant improvement in PTSD symptoms,” the investigators write.
The results “were very satisfying,” added Dr. Feder. “It was heartening also to hear what some of the participants would say. Some told us about how their symptoms and feelings had changed during the course of treatment with ketamine, where they felt stronger and better able to cope with their trauma and memories.”
She noted, however, that this was not a study designed to specifically assess ketamine in treatment-resistant PTSD. “Some patients had had multiple treatments before that hadn’t worked, while others had not received treatment before. Efficacy for treatment-resistant PTSD is an important question for future research,” Dr. Feder said.
Other areas worth future exploration include treatment efficacy in patients with different types of trauma and whether outcomes can last longer in patients receiving ketamine plus psychotherapy treatment, she noted.
“I don’t want to ignore the fact that currently available treatments work for a number of people with chronic PTSD. But because there are many more for whom [the treatments] don’t work, or they’re insufficiently helped by those treatments, this is certainly one potentially very promising approach that can be added” to a clinician’s toolbox, Dr. Feder said.
Speaks to clinical utility
Commenting for this news organization, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University, New Haven, Connecticut, called this a “very solid and well-designed” study.
“It definitely builds on what’s been found in the past, but it’s a critical piece of information speaking to the clinical utility of this treatment for PTSD,” said Dr. Sanacora, who is also director of the Yale Depression Research Program and was not involved with the current research.
He agreed with the investigators that PTSD has long been a condition that is difficult to treat.
“It’s an area that has a great unmet need for treatment options. Beyond that, as ketamine is becoming more widely used, there’s increasing demand for off-label uses. This [study] actually provides some evidence that there may be efficacy there,” Dr. Sanacora said.
Although he cautioned that this was a small study, and thus further research with a larger patient population will be needed, it provides a compelling foundation to build upon.
“This study provides clear evidence to support a larger study to really give a definitive statement on the efficacy and safety of its use for PTSD. I don’t think this is the study that provides that definitive evidence, but it is a very strong indication, and it very strongly supports the initiation of a large study to address that,” said Dr. Sanacora.
He noted that, although he’s used the term “cautious optimism” for studies in the past, he has “real optimism” that ketamine will be effective for PTSD based on the results of this current study.
“We still need some more data to really convince us of that before we can say with any clear statement that it is effective and safe, but I’m very optimistic,” Dr. Sanacora concluded.
The study was funded by the Brain and Behavior Research Foundation, Mount Sinai Innovation Partners and the Mount Sinai i3 Accelerator, Gerald and Glenda Greenwald, and the Ehrenkranz Laboratory for Human Resilience. Dr. Feder is a coinventor on issued patents for the use of ketamine as therapy for PTSD. A list of all disclosures for the other study authors are listed in the original article.
A version of this article first appeared on Medscape.com.
Repeated intravenous infusions of ketamine provide rapid relief for patients with posttraumatic stress disorder, new research suggests.
In what investigators are calling the first randomized controlled trial of repeated ketamine administration for chronic PTSD, 30 patients received six infusions of ketamine or midazolam (used as a psychoactive placebo) over 2 consecutive weeks.
Between baseline and week 2, those receiving ketamine showed significantly greater improvement than those receiving midazolam. Total scores on the Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) for the first group were almost 12 points lower than the latter group at week 2, meeting the study’s primary outcome measure.
In addition, 67% vs. 20% of the patients, respectively, were considered to be treatment responders; time to loss of response for those in the ketamine group was 28 days.
Although the overall findings were as expected, “what was surprising was how robust the results were,” lead author Adriana Feder, MD, associate professor of psychiatry, Icahn School of Medicine, Mount Sinai, New York, told this news organization.
It was also a bit surprising that, in a study of just 30 participants, “we were able to show such a clear difference” between the two treatment groups, said Dr. Feder, who is also a coinventor on issued patents for the use of ketamine as therapy for PTSD, and codirector of the Ehrenkranz Lab for the Study of Human Resilience at Mount Sinai.
The findings were published online Jan. 5 in the American Journal of Psychiatry.
Unmet need
Ketamine is a glutamate N-methyl-D-aspartate (NMDA) receptor antagonist that was first approved by the U.S. Food and Drug Administration for anesthetic use in 1970. It has also been shown to be effective for treatment-resistant depression.
PTSD has a lifetime prevalence of about 6% in the United States. “While trauma-focused psychotherapies have the most empirical support, they are limited by significant rates of nonresponse, partial response, and treatment dropout,” the investigators write. Also, there are “few available pharmacotherapies for PTSD, and their efficacy is insufficient,” they add.
“There’s a real need for new treatment interventions that are effective for PTSD and also work rapidly, because it can take weeks to months for currently available treatments to work for PTSD,” Dr. Feder said.
The researchers previously conducted a “proof-of-concept” randomized controlled trial of single infusions of ketamine for chronic PTSD. Results published in 2014 in JAMA Psychiatry showed significant reduction in PTSD symptoms 24 hours after infusion.
For the current study, the investigative team wanted to assess whether ketamine was viable as a longer-term treatment.
“We were encouraged by our initial promising findings” of the earlier trial, Dr. Feder said. “We wanted to do the second study to see if ketamine really works for PTSD, to see if we could replicate the rapid improvement and also examine whether a course of six infusions over 2 weeks could maintain the improvement.”
Thirty patients (aged 18-70; mean age, 39 years) with chronic PTSD from civilian or military trauma were enrolled (mean PTSD duration, 15 years).
The most cited primary trauma was sexual assault or molestation (n = 13), followed by physical assault or abuse (n = 8), witnessing a violent assault or death (n = 4), witnessing the 9/11 attacks (n = 3), and combat exposure (n = 2).
During the 2-week treatment phase, half of the patients were randomly assigned to receive six infusions of ketamine hydrochloride at a dose of 0.5 mg/kg (86.7% women; mean CAPS-5 score, 42), while the other half received six infusions of midazolam at a dose of 0.045 mg/kg (66.7% women; mean CAPS-5 score, 40).
In addition to the primary outcome measure of 2-week changes on the CAPS-5, secondary outcomes included score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) and the Impact of Event Scale-Revised (IES-R).
Treatment response was defined as a 30% or more improvement in symptoms on the CAPS-5. A number of measures were also used to assess potential treatment-related adverse events (AEs).
Safe, effective
Results showed significantly lower total CAPS-5 scores for the ketamine group vs. the midazolam group at week 1 (score difference, 8.8 points; P = .03) and at week 2 (score difference, 11.88 points; P = .004).
Those receiving ketamine also showed improvements in three of the four PTSD symptom clusters on the CAPS-5: avoidance (P < .0001), negative mood and cognitions (P = .02), and intrusions (P = .03). The fourth symptom cluster – arousal and reactivity – did not show a significant improvement.
In addition, the ketamine group showed significantly greater improvement scores on the MADRS at both week 1 and week 2.
Treatment response at 2 weeks was achieved by 10 members of the ketamine group and by three members of the midazolam group (P = .03).
Secondary analyses showed rapid improvement in the treatment responders within the ketamine group, with a mean change of 26 points on the total IES-R score between baseline and 24 hours after their first infusion, and a mean change of 13.4 points on the MADRS total past-24-hour score, a 53% improvement on average.
“A response at 2 weeks is very rapid but they got better sometimes within the first day,” Dr. Feder noted.
There were no serious AEs reported. Although some dissociative symptoms occurred during ketamine infusions, with the highest levels reported at the end of the infusion, these symptoms had resolved by the next assessment, conducted 2 hours after infusion.
The most frequently reported AE in the ketamine group, compared with midazolam, after the start of infusions was blurred vision (53% vs. 0%), followed by dizziness (33% vs. 13%), fatigue (33% vs. 87%), headache (27% vs. 13%), and nausea or vomiting (20% vs. 7%).
‘Large-magnitude improvement’
The overall findings show that, in this patient population, “repeated intravenous ketamine infusions administered over 2 weeks were associated with a large-magnitude, clinically significant improvement in PTSD symptoms,” the investigators write.
The results “were very satisfying,” added Dr. Feder. “It was heartening also to hear what some of the participants would say. Some told us about how their symptoms and feelings had changed during the course of treatment with ketamine, where they felt stronger and better able to cope with their trauma and memories.”
She noted, however, that this was not a study designed to specifically assess ketamine in treatment-resistant PTSD. “Some patients had had multiple treatments before that hadn’t worked, while others had not received treatment before. Efficacy for treatment-resistant PTSD is an important question for future research,” Dr. Feder said.
Other areas worth future exploration include treatment efficacy in patients with different types of trauma and whether outcomes can last longer in patients receiving ketamine plus psychotherapy treatment, she noted.
“I don’t want to ignore the fact that currently available treatments work for a number of people with chronic PTSD. But because there are many more for whom [the treatments] don’t work, or they’re insufficiently helped by those treatments, this is certainly one potentially very promising approach that can be added” to a clinician’s toolbox, Dr. Feder said.
Speaks to clinical utility
Commenting for this news organization, Gerard Sanacora, MD, PhD, professor of psychiatry at Yale University, New Haven, Connecticut, called this a “very solid and well-designed” study.
“It definitely builds on what’s been found in the past, but it’s a critical piece of information speaking to the clinical utility of this treatment for PTSD,” said Dr. Sanacora, who is also director of the Yale Depression Research Program and was not involved with the current research.
He agreed with the investigators that PTSD has long been a condition that is difficult to treat.
“It’s an area that has a great unmet need for treatment options. Beyond that, as ketamine is becoming more widely used, there’s increasing demand for off-label uses. This [study] actually provides some evidence that there may be efficacy there,” Dr. Sanacora said.
Although he cautioned that this was a small study, and thus further research with a larger patient population will be needed, it provides a compelling foundation to build upon.
“This study provides clear evidence to support a larger study to really give a definitive statement on the efficacy and safety of its use for PTSD. I don’t think this is the study that provides that definitive evidence, but it is a very strong indication, and it very strongly supports the initiation of a large study to address that,” said Dr. Sanacora.
He noted that, although he’s used the term “cautious optimism” for studies in the past, he has “real optimism” that ketamine will be effective for PTSD based on the results of this current study.
“We still need some more data to really convince us of that before we can say with any clear statement that it is effective and safe, but I’m very optimistic,” Dr. Sanacora concluded.
The study was funded by the Brain and Behavior Research Foundation, Mount Sinai Innovation Partners and the Mount Sinai i3 Accelerator, Gerald and Glenda Greenwald, and the Ehrenkranz Laboratory for Human Resilience. Dr. Feder is a coinventor on issued patents for the use of ketamine as therapy for PTSD. A list of all disclosures for the other study authors are listed in the original article.
A version of this article first appeared on Medscape.com.
Further warning on SGLT2 inhibitor use and DKA risk in COVID-19
a new case series suggests.
Five patients with type 2 diabetes who were taking SGLT2 inhibitors presented in DKA despite having glucose levels below 300 mg/dL. The report was published online last month in AACE Clinical Case Reports by Rebecca J. Vitale, MD, and colleagues at Brigham and Women’s Hospital, Boston.
“A cluster of euglycemic DKA cases at our hospital during the first wave of the pandemic suggests that patients with diabetes taking SGLT2 inhibitors may be at enhanced risk for euDKA when they contract COVID-19,” senior author Naomi D.L. Fisher, MD, said in an interview.
Dr. Fisher, an endocrinologist, added: “This complication is preventable with the simple measure of holding the drug. We are hopeful that widespread patient and physician education will prevent future cases of euDKA as COVID-19 infections continue to surge.”
These cases underscore recommendations published early in the COVID-19 pandemic by an international panel, she noted.
“Patients who are acutely ill with nausea, vomiting, abdominal pain, or diarrhea, or who are experiencing loss of appetite with reduced food and fluid intake, should be advised to hold their SGLT2 inhibitor. This medication should not be resumed until patients are feeling better and eating and drinking normally.”
On the other hand, “If patients with asymptomatic or mild COVID-19 infection are otherwise well, and are eating and drinking normally, there is no evidence that SGLT2 inhibitors need to be stopped. These patients should monitor [themselves] closely for worsening symptoms, especially resulting in poor hydration and nutrition, which would be reason to discontinue their medication.”
Pay special attention to the elderly, those with complications
However, special consideration should be given to elderly patients and those with medical conditions known to increase the likelihood of severe infection, like heart failure and chronic obstructive pulmonary disease, Dr. Fisher added.
The SGLT2 inhibitor class of drugs causes significant urinary glucose excretion, and they are also diuretics. A decrease in available glucose and volume depletion are probably both important contributors to euDKA, she explained.
With COVID-19 infection the euDKA risk is compounded by several mechanisms. Most cases of euDKA are associated with an underlying state of starvation that can be triggered by vomiting, diarrhea, loss of appetite, and poor oral intake.
In addition – although not yet known for certain – SARS-CoV-2 may also be toxic to pancreatic beta cells and thus reduce insulin secretion. The maladaptive inflammatory response seen with COVID-19 may also contribute, she said.
The patients in the current case series were three men and two women seen between March and May 2020. They ranged in age from 52 to 79 years.
None had a prior history of DKA or any known diabetes complications. In all of them, antihyperglycemic medications, including SGLT2 inhibitors, were stopped on hospital admission. The patients were initially treated with intravenous insulin, and then subcutaneous insulin after the DKA diagnosis.
Three of the patients were discharged to rehabilitation facilities on hospital days 28-47 and one (age 53 years) was discharged home on day 11. The other patient also had hypertension and nonalcoholic steatohepatitis.
A version of this article first appeared on Medscape.com.
a new case series suggests.
Five patients with type 2 diabetes who were taking SGLT2 inhibitors presented in DKA despite having glucose levels below 300 mg/dL. The report was published online last month in AACE Clinical Case Reports by Rebecca J. Vitale, MD, and colleagues at Brigham and Women’s Hospital, Boston.
“A cluster of euglycemic DKA cases at our hospital during the first wave of the pandemic suggests that patients with diabetes taking SGLT2 inhibitors may be at enhanced risk for euDKA when they contract COVID-19,” senior author Naomi D.L. Fisher, MD, said in an interview.
Dr. Fisher, an endocrinologist, added: “This complication is preventable with the simple measure of holding the drug. We are hopeful that widespread patient and physician education will prevent future cases of euDKA as COVID-19 infections continue to surge.”
These cases underscore recommendations published early in the COVID-19 pandemic by an international panel, she noted.
“Patients who are acutely ill with nausea, vomiting, abdominal pain, or diarrhea, or who are experiencing loss of appetite with reduced food and fluid intake, should be advised to hold their SGLT2 inhibitor. This medication should not be resumed until patients are feeling better and eating and drinking normally.”
On the other hand, “If patients with asymptomatic or mild COVID-19 infection are otherwise well, and are eating and drinking normally, there is no evidence that SGLT2 inhibitors need to be stopped. These patients should monitor [themselves] closely for worsening symptoms, especially resulting in poor hydration and nutrition, which would be reason to discontinue their medication.”
Pay special attention to the elderly, those with complications
However, special consideration should be given to elderly patients and those with medical conditions known to increase the likelihood of severe infection, like heart failure and chronic obstructive pulmonary disease, Dr. Fisher added.
The SGLT2 inhibitor class of drugs causes significant urinary glucose excretion, and they are also diuretics. A decrease in available glucose and volume depletion are probably both important contributors to euDKA, she explained.
With COVID-19 infection the euDKA risk is compounded by several mechanisms. Most cases of euDKA are associated with an underlying state of starvation that can be triggered by vomiting, diarrhea, loss of appetite, and poor oral intake.
In addition – although not yet known for certain – SARS-CoV-2 may also be toxic to pancreatic beta cells and thus reduce insulin secretion. The maladaptive inflammatory response seen with COVID-19 may also contribute, she said.
The patients in the current case series were three men and two women seen between March and May 2020. They ranged in age from 52 to 79 years.
None had a prior history of DKA or any known diabetes complications. In all of them, antihyperglycemic medications, including SGLT2 inhibitors, were stopped on hospital admission. The patients were initially treated with intravenous insulin, and then subcutaneous insulin after the DKA diagnosis.
Three of the patients were discharged to rehabilitation facilities on hospital days 28-47 and one (age 53 years) was discharged home on day 11. The other patient also had hypertension and nonalcoholic steatohepatitis.
A version of this article first appeared on Medscape.com.
a new case series suggests.
Five patients with type 2 diabetes who were taking SGLT2 inhibitors presented in DKA despite having glucose levels below 300 mg/dL. The report was published online last month in AACE Clinical Case Reports by Rebecca J. Vitale, MD, and colleagues at Brigham and Women’s Hospital, Boston.
“A cluster of euglycemic DKA cases at our hospital during the first wave of the pandemic suggests that patients with diabetes taking SGLT2 inhibitors may be at enhanced risk for euDKA when they contract COVID-19,” senior author Naomi D.L. Fisher, MD, said in an interview.
Dr. Fisher, an endocrinologist, added: “This complication is preventable with the simple measure of holding the drug. We are hopeful that widespread patient and physician education will prevent future cases of euDKA as COVID-19 infections continue to surge.”
These cases underscore recommendations published early in the COVID-19 pandemic by an international panel, she noted.
“Patients who are acutely ill with nausea, vomiting, abdominal pain, or diarrhea, or who are experiencing loss of appetite with reduced food and fluid intake, should be advised to hold their SGLT2 inhibitor. This medication should not be resumed until patients are feeling better and eating and drinking normally.”
On the other hand, “If patients with asymptomatic or mild COVID-19 infection are otherwise well, and are eating and drinking normally, there is no evidence that SGLT2 inhibitors need to be stopped. These patients should monitor [themselves] closely for worsening symptoms, especially resulting in poor hydration and nutrition, which would be reason to discontinue their medication.”
Pay special attention to the elderly, those with complications
However, special consideration should be given to elderly patients and those with medical conditions known to increase the likelihood of severe infection, like heart failure and chronic obstructive pulmonary disease, Dr. Fisher added.
The SGLT2 inhibitor class of drugs causes significant urinary glucose excretion, and they are also diuretics. A decrease in available glucose and volume depletion are probably both important contributors to euDKA, she explained.
With COVID-19 infection the euDKA risk is compounded by several mechanisms. Most cases of euDKA are associated with an underlying state of starvation that can be triggered by vomiting, diarrhea, loss of appetite, and poor oral intake.
In addition – although not yet known for certain – SARS-CoV-2 may also be toxic to pancreatic beta cells and thus reduce insulin secretion. The maladaptive inflammatory response seen with COVID-19 may also contribute, she said.
The patients in the current case series were three men and two women seen between March and May 2020. They ranged in age from 52 to 79 years.
None had a prior history of DKA or any known diabetes complications. In all of them, antihyperglycemic medications, including SGLT2 inhibitors, were stopped on hospital admission. The patients were initially treated with intravenous insulin, and then subcutaneous insulin after the DKA diagnosis.
Three of the patients were discharged to rehabilitation facilities on hospital days 28-47 and one (age 53 years) was discharged home on day 11. The other patient also had hypertension and nonalcoholic steatohepatitis.
A version of this article first appeared on Medscape.com.
Gut microbiome may predict nivolumab efficacy in gastric cancer
Researchers have demonstrated bacterial invasion of the epithelial cell pathway in the gut microbiome and suggest that this could potentially become a novel biomarker.
“In addition, we found gastric cancer–specific gut microbiome predictive of responses to immune checkpoint inhibitors,” said study author Yu Sunakawa, MD, PhD, an associate professor in the department of clinical oncology at St. Marianna University, Kawasaki, Japan.
Dr. Sunakawa presented the study’s results at the 2021 Gastrointestinal Cancers Symposium.
The gut microbiome holds great interest as a potential biomarker for response. Previous studies suggested that it may hold the key to immunotherapy responses. The concept has been demonstrated in several studies involving patients with melanoma, but this is the first study in patients with gastric cancer.
Nivolumab monotherapy has been shown to provide a survival benefit with a manageable safety profile in previously treated patients with gastric cancer or gastroesophageal junction (GEJ) cancer, Dr. Sunakawa noted. However, fewer than half of patients responded to therapy.
“The disease control rate was about 40%, and many patients did not experience any tumor degradation,” he said. “About 60% of the patients did not respond to nivolumab as a late-line therapy.”
In the observational/translational DELIVER trial, investigators enrolled 501 patients with recurrent or metastatic adenocarcinoma of the stomach or GEJ. The patients were recruited from 50 sites in Japan.
The primary endpoint was the relationship between the genomic pathway in the gut microbiome and efficacy of nivolumab and whether there was progressive disease or not at the first evaluation, as determined in accordance with Response Evaluation Criteria in Solid Tumors criteria.
Genomic data were measured by genome shotgun sequence at a central laboratory. Biomarkers were analyzed by Wilcoxon rank sum test in the first 200 patients, who constituted the training cohort. The top 30 biomarker candidates were validated in the last 300 patients (the validation cohort) using the Bonferroni method.
Clinical and genomic data were available for 437 patients (87%). Of this group, 180 constituted the training cohort, and 257, the validation cohort.
The phylogenetic composition of common bacterial taxa was similar for both cohorts.
In the training cohort, 62.2% of patients had progressive disease, as did 53.2% in the validation cohort. The microbiome was more diverse among the patients who did not have progressive disease than among those who did have progressive disease.
The authors noted that, although there was no statistically significant pathway to be validated for a primary endpoint using the Bonferroni method, bacterial invasion of epithelial cells in the KEGG pathway was associated with clinical outcomes in both the training cohort (P = .057) and the validation cohort (P = .014). However, these pathways were not significantly associated with progressive disease after Bonferroni correction, a conservative test that adjusts for multiple comparisons.
An exploratory analysis of genus showed that Odoribacter and Veillonella species were associated with tumor response to nivolumab in both cohorts.
Dr. Sunakawa noted that biomarker analyses are ongoing. The researchers are investigating the relationships between microbiome and survival times, as well as other endpoints.
Still some gaps
In a discussion of the study, Jonathan Yeung, MD, PhD, of Princess Margaret Cancer Center, Toronto, congratulated the investigators on their study, noting that “the logistical hurdles must have been tremendous to obtain these data.”
However, Dr. Yeung pointed out some limitations and gaps in the data that were presented. For example, he found that the ratio of the training set to the validation set was unusual. “The training set is usually larger and usually an 80/20 ratio,” he said. “In their design, the validation set is larger, and I’m quite curious about their rationale.
“The conclusion of the study is that a more diverse microbiome was observed in patients with a tumor response than in those without a response,” he continued, “but they don’t actually show the statistical test used to make this conclusion. There is considerable overlap between the groups, and more compelling data are needed to make that conclusion.”
Another limitation was the marked imbalance in the number of patients whose condition responded to nivolumab in comparison with those whose condition did not (20 vs. 417 patients). This could have affected the statistical power of the study.
But overall, Dr. Yeung congratulated the authors for presenting a very impressive dataset. “The preliminary data are very interesting, and I look forward to the final results,” he said.
The study was funded by Ono Pharmaceutical and Bristol-Myers Squibb, which markets nivolumab. Dr. Sunakawa has received honoraria from Bayer Yakuhin, Bristol-Myers Squibb Japan, Chugai, Kyowa Hakko Kirin, Lilly Japan, Nippon Kayaku, Sanofi, Taiho, Takeda, and Yakult Honsha. He has held a consulting or advisory role for Bristol-Myers Squibb Japan, Daiichi Sankyo, and Takeda and has received research funding from Chugai Pharma, Daiichi Sankyo, Lilly Japan, Sanofi, Taiho Pharmaceutical, and Takeda. The Gastrointestinal Cancers Symposium is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology.
A version of this article first appeared on Medscape.com.
Researchers have demonstrated bacterial invasion of the epithelial cell pathway in the gut microbiome and suggest that this could potentially become a novel biomarker.
“In addition, we found gastric cancer–specific gut microbiome predictive of responses to immune checkpoint inhibitors,” said study author Yu Sunakawa, MD, PhD, an associate professor in the department of clinical oncology at St. Marianna University, Kawasaki, Japan.
Dr. Sunakawa presented the study’s results at the 2021 Gastrointestinal Cancers Symposium.
The gut microbiome holds great interest as a potential biomarker for response. Previous studies suggested that it may hold the key to immunotherapy responses. The concept has been demonstrated in several studies involving patients with melanoma, but this is the first study in patients with gastric cancer.
Nivolumab monotherapy has been shown to provide a survival benefit with a manageable safety profile in previously treated patients with gastric cancer or gastroesophageal junction (GEJ) cancer, Dr. Sunakawa noted. However, fewer than half of patients responded to therapy.
“The disease control rate was about 40%, and many patients did not experience any tumor degradation,” he said. “About 60% of the patients did not respond to nivolumab as a late-line therapy.”
In the observational/translational DELIVER trial, investigators enrolled 501 patients with recurrent or metastatic adenocarcinoma of the stomach or GEJ. The patients were recruited from 50 sites in Japan.
The primary endpoint was the relationship between the genomic pathway in the gut microbiome and efficacy of nivolumab and whether there was progressive disease or not at the first evaluation, as determined in accordance with Response Evaluation Criteria in Solid Tumors criteria.
Genomic data were measured by genome shotgun sequence at a central laboratory. Biomarkers were analyzed by Wilcoxon rank sum test in the first 200 patients, who constituted the training cohort. The top 30 biomarker candidates were validated in the last 300 patients (the validation cohort) using the Bonferroni method.
Clinical and genomic data were available for 437 patients (87%). Of this group, 180 constituted the training cohort, and 257, the validation cohort.
The phylogenetic composition of common bacterial taxa was similar for both cohorts.
In the training cohort, 62.2% of patients had progressive disease, as did 53.2% in the validation cohort. The microbiome was more diverse among the patients who did not have progressive disease than among those who did have progressive disease.
The authors noted that, although there was no statistically significant pathway to be validated for a primary endpoint using the Bonferroni method, bacterial invasion of epithelial cells in the KEGG pathway was associated with clinical outcomes in both the training cohort (P = .057) and the validation cohort (P = .014). However, these pathways were not significantly associated with progressive disease after Bonferroni correction, a conservative test that adjusts for multiple comparisons.
An exploratory analysis of genus showed that Odoribacter and Veillonella species were associated with tumor response to nivolumab in both cohorts.
Dr. Sunakawa noted that biomarker analyses are ongoing. The researchers are investigating the relationships between microbiome and survival times, as well as other endpoints.
Still some gaps
In a discussion of the study, Jonathan Yeung, MD, PhD, of Princess Margaret Cancer Center, Toronto, congratulated the investigators on their study, noting that “the logistical hurdles must have been tremendous to obtain these data.”
However, Dr. Yeung pointed out some limitations and gaps in the data that were presented. For example, he found that the ratio of the training set to the validation set was unusual. “The training set is usually larger and usually an 80/20 ratio,” he said. “In their design, the validation set is larger, and I’m quite curious about their rationale.
“The conclusion of the study is that a more diverse microbiome was observed in patients with a tumor response than in those without a response,” he continued, “but they don’t actually show the statistical test used to make this conclusion. There is considerable overlap between the groups, and more compelling data are needed to make that conclusion.”
Another limitation was the marked imbalance in the number of patients whose condition responded to nivolumab in comparison with those whose condition did not (20 vs. 417 patients). This could have affected the statistical power of the study.
But overall, Dr. Yeung congratulated the authors for presenting a very impressive dataset. “The preliminary data are very interesting, and I look forward to the final results,” he said.
The study was funded by Ono Pharmaceutical and Bristol-Myers Squibb, which markets nivolumab. Dr. Sunakawa has received honoraria from Bayer Yakuhin, Bristol-Myers Squibb Japan, Chugai, Kyowa Hakko Kirin, Lilly Japan, Nippon Kayaku, Sanofi, Taiho, Takeda, and Yakult Honsha. He has held a consulting or advisory role for Bristol-Myers Squibb Japan, Daiichi Sankyo, and Takeda and has received research funding from Chugai Pharma, Daiichi Sankyo, Lilly Japan, Sanofi, Taiho Pharmaceutical, and Takeda. The Gastrointestinal Cancers Symposium is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology.
A version of this article first appeared on Medscape.com.
Researchers have demonstrated bacterial invasion of the epithelial cell pathway in the gut microbiome and suggest that this could potentially become a novel biomarker.
“In addition, we found gastric cancer–specific gut microbiome predictive of responses to immune checkpoint inhibitors,” said study author Yu Sunakawa, MD, PhD, an associate professor in the department of clinical oncology at St. Marianna University, Kawasaki, Japan.
Dr. Sunakawa presented the study’s results at the 2021 Gastrointestinal Cancers Symposium.
The gut microbiome holds great interest as a potential biomarker for response. Previous studies suggested that it may hold the key to immunotherapy responses. The concept has been demonstrated in several studies involving patients with melanoma, but this is the first study in patients with gastric cancer.
Nivolumab monotherapy has been shown to provide a survival benefit with a manageable safety profile in previously treated patients with gastric cancer or gastroesophageal junction (GEJ) cancer, Dr. Sunakawa noted. However, fewer than half of patients responded to therapy.
“The disease control rate was about 40%, and many patients did not experience any tumor degradation,” he said. “About 60% of the patients did not respond to nivolumab as a late-line therapy.”
In the observational/translational DELIVER trial, investigators enrolled 501 patients with recurrent or metastatic adenocarcinoma of the stomach or GEJ. The patients were recruited from 50 sites in Japan.
The primary endpoint was the relationship between the genomic pathway in the gut microbiome and efficacy of nivolumab and whether there was progressive disease or not at the first evaluation, as determined in accordance with Response Evaluation Criteria in Solid Tumors criteria.
Genomic data were measured by genome shotgun sequence at a central laboratory. Biomarkers were analyzed by Wilcoxon rank sum test in the first 200 patients, who constituted the training cohort. The top 30 biomarker candidates were validated in the last 300 patients (the validation cohort) using the Bonferroni method.
Clinical and genomic data were available for 437 patients (87%). Of this group, 180 constituted the training cohort, and 257, the validation cohort.
The phylogenetic composition of common bacterial taxa was similar for both cohorts.
In the training cohort, 62.2% of patients had progressive disease, as did 53.2% in the validation cohort. The microbiome was more diverse among the patients who did not have progressive disease than among those who did have progressive disease.
The authors noted that, although there was no statistically significant pathway to be validated for a primary endpoint using the Bonferroni method, bacterial invasion of epithelial cells in the KEGG pathway was associated with clinical outcomes in both the training cohort (P = .057) and the validation cohort (P = .014). However, these pathways were not significantly associated with progressive disease after Bonferroni correction, a conservative test that adjusts for multiple comparisons.
An exploratory analysis of genus showed that Odoribacter and Veillonella species were associated with tumor response to nivolumab in both cohorts.
Dr. Sunakawa noted that biomarker analyses are ongoing. The researchers are investigating the relationships between microbiome and survival times, as well as other endpoints.
Still some gaps
In a discussion of the study, Jonathan Yeung, MD, PhD, of Princess Margaret Cancer Center, Toronto, congratulated the investigators on their study, noting that “the logistical hurdles must have been tremendous to obtain these data.”
However, Dr. Yeung pointed out some limitations and gaps in the data that were presented. For example, he found that the ratio of the training set to the validation set was unusual. “The training set is usually larger and usually an 80/20 ratio,” he said. “In their design, the validation set is larger, and I’m quite curious about their rationale.
“The conclusion of the study is that a more diverse microbiome was observed in patients with a tumor response than in those without a response,” he continued, “but they don’t actually show the statistical test used to make this conclusion. There is considerable overlap between the groups, and more compelling data are needed to make that conclusion.”
Another limitation was the marked imbalance in the number of patients whose condition responded to nivolumab in comparison with those whose condition did not (20 vs. 417 patients). This could have affected the statistical power of the study.
But overall, Dr. Yeung congratulated the authors for presenting a very impressive dataset. “The preliminary data are very interesting, and I look forward to the final results,” he said.
The study was funded by Ono Pharmaceutical and Bristol-Myers Squibb, which markets nivolumab. Dr. Sunakawa has received honoraria from Bayer Yakuhin, Bristol-Myers Squibb Japan, Chugai, Kyowa Hakko Kirin, Lilly Japan, Nippon Kayaku, Sanofi, Taiho, Takeda, and Yakult Honsha. He has held a consulting or advisory role for Bristol-Myers Squibb Japan, Daiichi Sankyo, and Takeda and has received research funding from Chugai Pharma, Daiichi Sankyo, Lilly Japan, Sanofi, Taiho Pharmaceutical, and Takeda. The Gastrointestinal Cancers Symposium is sponsored by the American Gastroenterological Association, the American Society for Clinical Oncology, the American Society for Radiation Oncology, and the Society of Surgical Oncology.
A version of this article first appeared on Medscape.com.
Biomarker HF risk score envisioned as SGLT2 inhibitor lodestar in diabetes
A scoring system that predicts risk for new heart failure over 5 years that is based solely on a few familiar, readily available biomarkers could potentially help steer patients with diabetes or even prediabetes toward HF-preventive therapies, researchers proposed based on a new study.
They foresee the risk-stratification tool, based on data pooled from three major community-based cohort studies but not independently validated, as a way to select patients with diabetes and prediabetes for treatment with SGLT2 inhibitors.
Several members of that drug class, conceived as antidiabetic agents, have been shown to help in prevention or treatment of HF in patients with diabetes and those without diabetes but at increased cardiovascular (CV) risk. Yet their uptake in practice has been lagging, the group noted.
Most HF benefits in the SGLT2 inhibitor trials “were seen in patients who have established cardiovascular disease – basically a history of heart attack or stroke,” Ambarish Pandey, MD, MSCS, University of Texas Southwestern Medical Center, Dallas, said in an interview.
“So we wanted to see how we can identify high-risk patients without a history of cardiovascular disease using these biomarkers, as an approach to targeting SGLT2 inhibitors, which are fairly expensive therapies,” he said. Without such risk stratification, “you end up treating so many more patients to get very modest returns.”
The group developed a scoring system based on four biomarkers that are “easily measured with inexpensive tests,” Dr. Pandey said: high-sensitivity-assay cardiac troponin T (hs-cTnT) and C-reactive protein (hs-CRP) levels, N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) levels, and electrocardiography for evidence of left-ventricular hypertrophy (ECG-LVH).
The derivation cohort consisted of participants in the Atherosclerosis Risk in Communities RIC, Dallas Heart Study, and Multi-Ethnic Study of Atherosclerosis Multi-Ethnic Study of Atherosclerosis epidemiologic studies who were free of coronary heart disease, stroke, or HF for whom there were sufficient data on CV risk factors and the four biomarkers. None were taking SGLT2 inhibitors at enrollment in their respective studies, the researchers noted.
Members of the pooled cohorts who had diabetes or prediabetes were assigned 1 point for each abnormal biomarker. The 5-year risk for incident HF went up continuously along with the score in people with diabetes and in those with prediabetes, the latter defined as a fasting plasma glucose level from 100 mg/dL to less than 126 mg/dL.
For those with a score of 1, compared with 0, for example, the risk for HF went up 82% with diabetes and 40% with prediabetes. But for those with a score of 3 or 4, the risk went up more than four and a half times with diabetes and more than three and a half times for those with prediabetes. Risk increases were independent of other likely HF risk factors and consistently significant.
The analysis was published Jan. 6 in JACC: Heart Failure.
The biomarker score should be especially useful in patients considered at low to intermediate risk, based on clinical characteristics, as a means to identify residual HF risk and, potentially, select candidates for SGLT2-inhibitor therapy, Dr. Pandey said.
“The other purpose of the study was to broaden the scope of heart failure prevention in dysglycemia by looking also at prediabetes, not just diabetes,” he said. There isn’t much high-quality evidence supporting SGLT2-inhibitor therapy in prediabetes, but it follows that the drugs may be helpful in prediabetes because they are protective in patients with and without diabetes.
“Our work suggests that prediabetes patients who have elevated biomarkers are at a higher risk of heart failure,” Dr. Pandey said, suggesting that the HF risk score could potentially help select their drug therapy as well.
The current study seems “to provide a proof of concept that one can use circulating biomarkers to more precisely identify patients in whom therapies might be expected to exert greatest benefit,” which is especially important for potentially expensive agents like the SGLT2 inhibitors, James L. Januzzi, MD, Massachusetts General Hospital, Boston, said in an interview.
Importantly in the analysis, a greater number of biomarker abnormalities not only corresponded to rising levels of risk, the risk increases were “dramatic,” and therefore so was the supposed potential benefit of SGLT2-inhibitor therapy, said Dr. Januzzi, who isn’t a coauthor but was an editor for its publication in JACC: Heart Failure.
The uptake of SGLT2 inhibitors for heart failure in practice has been less rapid than hoped, he observed, so if “this hypothetical construct holds up” for the drug class, “it might actually help kick-start focusing on who might optimally receive the drugs.”
Elevated levels of hs-cTnT, hs-CRP, and NT-proBNP, as well as presence of ECG-LVH, were each independently associated with a significantly increased 5-year risk for HF in unadjusted and adjusted analyses of the 6,799 people in the pooled cohort, 33.2% of whom had diabetes and 66.8% of whom had prediabetes, the group writes.
The scoring system would require validation in other cohorts before it could be used, Dr. Pandey observed; once there is “robust validation,” it might be applied first to patients with dysglycemia at intermediate CV risk by standard clinical measures.
Certainly the HF risk-stratification scoring system requires validation in other studies, Dr. Januzzi agreed. But it is intuitively appealing, and the study’s results are consistent with “data that we’re submitting for publication imminently” based on the CANVAS CV-outcomes trial of the SGLT2 inhibitor canagliflozin (Invokana) in patients with diabetes.
Dr. Pandey disclosed receiving support from the Gilead Sciences Research Scholar Program and serving on an advisory board of Roche Diagnostics. Dr. Januzzi disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on end-point committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda.
A version of this article first appeared on Medscape.com.
A scoring system that predicts risk for new heart failure over 5 years that is based solely on a few familiar, readily available biomarkers could potentially help steer patients with diabetes or even prediabetes toward HF-preventive therapies, researchers proposed based on a new study.
They foresee the risk-stratification tool, based on data pooled from three major community-based cohort studies but not independently validated, as a way to select patients with diabetes and prediabetes for treatment with SGLT2 inhibitors.
Several members of that drug class, conceived as antidiabetic agents, have been shown to help in prevention or treatment of HF in patients with diabetes and those without diabetes but at increased cardiovascular (CV) risk. Yet their uptake in practice has been lagging, the group noted.
Most HF benefits in the SGLT2 inhibitor trials “were seen in patients who have established cardiovascular disease – basically a history of heart attack or stroke,” Ambarish Pandey, MD, MSCS, University of Texas Southwestern Medical Center, Dallas, said in an interview.
“So we wanted to see how we can identify high-risk patients without a history of cardiovascular disease using these biomarkers, as an approach to targeting SGLT2 inhibitors, which are fairly expensive therapies,” he said. Without such risk stratification, “you end up treating so many more patients to get very modest returns.”
The group developed a scoring system based on four biomarkers that are “easily measured with inexpensive tests,” Dr. Pandey said: high-sensitivity-assay cardiac troponin T (hs-cTnT) and C-reactive protein (hs-CRP) levels, N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) levels, and electrocardiography for evidence of left-ventricular hypertrophy (ECG-LVH).
The derivation cohort consisted of participants in the Atherosclerosis Risk in Communities RIC, Dallas Heart Study, and Multi-Ethnic Study of Atherosclerosis Multi-Ethnic Study of Atherosclerosis epidemiologic studies who were free of coronary heart disease, stroke, or HF for whom there were sufficient data on CV risk factors and the four biomarkers. None were taking SGLT2 inhibitors at enrollment in their respective studies, the researchers noted.
Members of the pooled cohorts who had diabetes or prediabetes were assigned 1 point for each abnormal biomarker. The 5-year risk for incident HF went up continuously along with the score in people with diabetes and in those with prediabetes, the latter defined as a fasting plasma glucose level from 100 mg/dL to less than 126 mg/dL.
For those with a score of 1, compared with 0, for example, the risk for HF went up 82% with diabetes and 40% with prediabetes. But for those with a score of 3 or 4, the risk went up more than four and a half times with diabetes and more than three and a half times for those with prediabetes. Risk increases were independent of other likely HF risk factors and consistently significant.
The analysis was published Jan. 6 in JACC: Heart Failure.
The biomarker score should be especially useful in patients considered at low to intermediate risk, based on clinical characteristics, as a means to identify residual HF risk and, potentially, select candidates for SGLT2-inhibitor therapy, Dr. Pandey said.
“The other purpose of the study was to broaden the scope of heart failure prevention in dysglycemia by looking also at prediabetes, not just diabetes,” he said. There isn’t much high-quality evidence supporting SGLT2-inhibitor therapy in prediabetes, but it follows that the drugs may be helpful in prediabetes because they are protective in patients with and without diabetes.
“Our work suggests that prediabetes patients who have elevated biomarkers are at a higher risk of heart failure,” Dr. Pandey said, suggesting that the HF risk score could potentially help select their drug therapy as well.
The current study seems “to provide a proof of concept that one can use circulating biomarkers to more precisely identify patients in whom therapies might be expected to exert greatest benefit,” which is especially important for potentially expensive agents like the SGLT2 inhibitors, James L. Januzzi, MD, Massachusetts General Hospital, Boston, said in an interview.
Importantly in the analysis, a greater number of biomarker abnormalities not only corresponded to rising levels of risk, the risk increases were “dramatic,” and therefore so was the supposed potential benefit of SGLT2-inhibitor therapy, said Dr. Januzzi, who isn’t a coauthor but was an editor for its publication in JACC: Heart Failure.
The uptake of SGLT2 inhibitors for heart failure in practice has been less rapid than hoped, he observed, so if “this hypothetical construct holds up” for the drug class, “it might actually help kick-start focusing on who might optimally receive the drugs.”
Elevated levels of hs-cTnT, hs-CRP, and NT-proBNP, as well as presence of ECG-LVH, were each independently associated with a significantly increased 5-year risk for HF in unadjusted and adjusted analyses of the 6,799 people in the pooled cohort, 33.2% of whom had diabetes and 66.8% of whom had prediabetes, the group writes.
The scoring system would require validation in other cohorts before it could be used, Dr. Pandey observed; once there is “robust validation,” it might be applied first to patients with dysglycemia at intermediate CV risk by standard clinical measures.
Certainly the HF risk-stratification scoring system requires validation in other studies, Dr. Januzzi agreed. But it is intuitively appealing, and the study’s results are consistent with “data that we’re submitting for publication imminently” based on the CANVAS CV-outcomes trial of the SGLT2 inhibitor canagliflozin (Invokana) in patients with diabetes.
Dr. Pandey disclosed receiving support from the Gilead Sciences Research Scholar Program and serving on an advisory board of Roche Diagnostics. Dr. Januzzi disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on end-point committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda.
A version of this article first appeared on Medscape.com.
A scoring system that predicts risk for new heart failure over 5 years that is based solely on a few familiar, readily available biomarkers could potentially help steer patients with diabetes or even prediabetes toward HF-preventive therapies, researchers proposed based on a new study.
They foresee the risk-stratification tool, based on data pooled from three major community-based cohort studies but not independently validated, as a way to select patients with diabetes and prediabetes for treatment with SGLT2 inhibitors.
Several members of that drug class, conceived as antidiabetic agents, have been shown to help in prevention or treatment of HF in patients with diabetes and those without diabetes but at increased cardiovascular (CV) risk. Yet their uptake in practice has been lagging, the group noted.
Most HF benefits in the SGLT2 inhibitor trials “were seen in patients who have established cardiovascular disease – basically a history of heart attack or stroke,” Ambarish Pandey, MD, MSCS, University of Texas Southwestern Medical Center, Dallas, said in an interview.
“So we wanted to see how we can identify high-risk patients without a history of cardiovascular disease using these biomarkers, as an approach to targeting SGLT2 inhibitors, which are fairly expensive therapies,” he said. Without such risk stratification, “you end up treating so many more patients to get very modest returns.”
The group developed a scoring system based on four biomarkers that are “easily measured with inexpensive tests,” Dr. Pandey said: high-sensitivity-assay cardiac troponin T (hs-cTnT) and C-reactive protein (hs-CRP) levels, N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) levels, and electrocardiography for evidence of left-ventricular hypertrophy (ECG-LVH).
The derivation cohort consisted of participants in the Atherosclerosis Risk in Communities RIC, Dallas Heart Study, and Multi-Ethnic Study of Atherosclerosis Multi-Ethnic Study of Atherosclerosis epidemiologic studies who were free of coronary heart disease, stroke, or HF for whom there were sufficient data on CV risk factors and the four biomarkers. None were taking SGLT2 inhibitors at enrollment in their respective studies, the researchers noted.
Members of the pooled cohorts who had diabetes or prediabetes were assigned 1 point for each abnormal biomarker. The 5-year risk for incident HF went up continuously along with the score in people with diabetes and in those with prediabetes, the latter defined as a fasting plasma glucose level from 100 mg/dL to less than 126 mg/dL.
For those with a score of 1, compared with 0, for example, the risk for HF went up 82% with diabetes and 40% with prediabetes. But for those with a score of 3 or 4, the risk went up more than four and a half times with diabetes and more than three and a half times for those with prediabetes. Risk increases were independent of other likely HF risk factors and consistently significant.
The analysis was published Jan. 6 in JACC: Heart Failure.
The biomarker score should be especially useful in patients considered at low to intermediate risk, based on clinical characteristics, as a means to identify residual HF risk and, potentially, select candidates for SGLT2-inhibitor therapy, Dr. Pandey said.
“The other purpose of the study was to broaden the scope of heart failure prevention in dysglycemia by looking also at prediabetes, not just diabetes,” he said. There isn’t much high-quality evidence supporting SGLT2-inhibitor therapy in prediabetes, but it follows that the drugs may be helpful in prediabetes because they are protective in patients with and without diabetes.
“Our work suggests that prediabetes patients who have elevated biomarkers are at a higher risk of heart failure,” Dr. Pandey said, suggesting that the HF risk score could potentially help select their drug therapy as well.
The current study seems “to provide a proof of concept that one can use circulating biomarkers to more precisely identify patients in whom therapies might be expected to exert greatest benefit,” which is especially important for potentially expensive agents like the SGLT2 inhibitors, James L. Januzzi, MD, Massachusetts General Hospital, Boston, said in an interview.
Importantly in the analysis, a greater number of biomarker abnormalities not only corresponded to rising levels of risk, the risk increases were “dramatic,” and therefore so was the supposed potential benefit of SGLT2-inhibitor therapy, said Dr. Januzzi, who isn’t a coauthor but was an editor for its publication in JACC: Heart Failure.
The uptake of SGLT2 inhibitors for heart failure in practice has been less rapid than hoped, he observed, so if “this hypothetical construct holds up” for the drug class, “it might actually help kick-start focusing on who might optimally receive the drugs.”
Elevated levels of hs-cTnT, hs-CRP, and NT-proBNP, as well as presence of ECG-LVH, were each independently associated with a significantly increased 5-year risk for HF in unadjusted and adjusted analyses of the 6,799 people in the pooled cohort, 33.2% of whom had diabetes and 66.8% of whom had prediabetes, the group writes.
The scoring system would require validation in other cohorts before it could be used, Dr. Pandey observed; once there is “robust validation,” it might be applied first to patients with dysglycemia at intermediate CV risk by standard clinical measures.
Certainly the HF risk-stratification scoring system requires validation in other studies, Dr. Januzzi agreed. But it is intuitively appealing, and the study’s results are consistent with “data that we’re submitting for publication imminently” based on the CANVAS CV-outcomes trial of the SGLT2 inhibitor canagliflozin (Invokana) in patients with diabetes.
Dr. Pandey disclosed receiving support from the Gilead Sciences Research Scholar Program and serving on an advisory board of Roche Diagnostics. Dr. Januzzi disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on end-point committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda.
A version of this article first appeared on Medscape.com.
Arthritis drugs ‘impressive’ for severe COVID but not ‘magic cure’
New findings suggest that monoclonal antibodies used to treat RA could improve severe COVID-19 outcomes, including risk for death.
Given within 24 hours of critical illness, tocilizumab (Actemra) was associated with a median of 10 days free of respiratory and cardiovascular support up to day 21, the primary outcome. Similarly, sarilumab (Kevzara) was linked to a median of 11 days. In contrast, the usual care control group experienced zero such days in the hospital.
However, the Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community-Acquired Pneumonia (REMAP-CAP) trial comes with a caveat. The preprint findings have not yet been peer reviewed and “should not be used to guide clinical practice,” the authors stated.
The results were published online Jan. 7 in MedRxiv.
Nevertheless, the trial also revealed a mortality benefit associated with the two interleukin-6 antagonists. The hospital mortality rate was 22% with sarilumab, 28% with tocilizumab, and almost 36% with usual care.
“That’s a big change in survival. They are both lifesaving drugs,” lead coinvestigator Anthony Gordon, an Imperial College London professor of anesthesia and critical care, commented in a recent story by Reuters.
Consider the big picture
“What I think is important is ... this is one of many trials,” Paul Auwaerter, MD, MBA, said in an interview. Many other studies looking at monoclonal antibody therapy for people with COVID-19 were halted because they did not show improvement.

One exception is the EMPACTA trial, which suggested that tocilizumab was effective if given before a person becomes ill enough to be placed on a ventilator, said Dr. Auwaerter, clinical director of the division of infectious diseases at Johns Hopkins Medicine and a contributor to this news organization. “It appeared to reduce the need for mechanical ventilation or death.”
“These two trials are the first randomized, prospective trials that show a benefit on a background of others which have not,” Dr. Auwaerter added.
Interim findings
The REMAP-CAP investigators randomly assigned adults within 24 hours of critical care for COVID-19 to 8 mg/kg tocilizumab, 400 mg sarilumab, or usual care at 113 sites in six countries. There were 353 participants in the tocilizumab arm, 48 in the sarilumab group, and 402 in the control group.
Compared with the control group, the 10 days free of organ support in the tocilizumab cohort was associated with an adjusted odds ratio of 1.64 (95% confidence interval, 1.25-2.14). The 11 days free of organ support in the sarilumab cohort was likewise superior to control (adjusted odds ratio, 1.76; 95% CI, 1.17-2.91).
“All secondary outcomes and analyses supported efficacy of these IL-6 receptor antagonists,” the authors note. These endpoints included 90-day survival, time to intensive care unit discharge, and hospital discharge.
Cautious optimism?
“The results were quite impressive – having 10 or 11 fewer days in the ICU, compared to standard of care,” Deepa Gotur, MD, said in an interview. “Choosing the right patient population and providing the anti-IL-6 treatment at the right time would be the key here.”

In addition to not yet receiving peer review, an open-label design, a relatively short follow-up of 21 days, and steroids becoming standard of care about halfway through the trial are potential limitations, said Dr. Gotur, an intensivist at Houston Methodist Hospital and associate professor of clinical medicine at Weill Cornell Medicine, New York.
“This is an interesting study,” Carl J. Fichtenbaum, MD, professor of clinical medicine at the University of Cincinnati, said in a comment.
Additional detail on how many participants in each group received steroids is warranted, Dr. Fichtenbaum said. “The analysis did not carefully adjust for the use of steroids that might have influenced outcomes.”

Dr. Fichtenbaum said it’s important to look at what is distinctive about REMAP-CAP because “there are several other studies showing opposite results.”
Dr. Gotur was an investigator on a previous study evaluating tocilizumab for patients already on mechanical ventilation. “One of the key differences between this and other studies is that they included more of the ICU population,” she said. “They also included patients within 24 hours of requiring organ support, cardiac, as well as respiratory support.” Some other research included less-acute patients, including all comers into the ED who required oxygen and received tocilizumab.
The prior studies also evaluated cytokine or inflammatory markers. In contrast, REMAP-CAP researchers “looked at organ failure itself ... which I think makes sense,” Dr. Gotur said.
Cytokine release syndrome can cause organ damage or organ failure, she added, “but these markers are all over the place. I’ve seen patients who are very, very sick despite having a low [C-reactive protein] or IL-6 level.”
Backing from the British
Citing the combined 24% decrease in the risk for death associated with these agents in the REMAP-CAP trial, the U.K. government announced Jan. 7 it will work to make tocilizumab and sarilumab available to citizens with severe COVID-19.
Experts in the United Kingdom shared their perspectives on the REMAP-CAP interim findings through the U.K. Science Media Centre.
“There are few treatments for severe COVID-19,” said Robin Ferner, MD, honorary professor of clinical pharmacology at the University of Birmingham (England) and honorary consultant physician at City Hospital Birmingham. “If the published data from REMAP-CAP are supported by further studies, this suggests that two IL-6 receptor antagonists can reduce the death rate in the most severely ill patients.”
Dr. Ferner added that the findings are not a “magic cure,” however. He pointed out that of 401 patients given the drugs, 109 died, and with standard treatment, 144 out of 402 died.
Peter Horby, MD, PhD, was more optimistic. “It is great to see a positive result at a time that we really need good news and more tools to fight COVID. This is great achievement for REMAP-CAP,” he said.
“We hope to soon have results from RECOVERY on the effect of tocilizumab in less severely ill patients in the hospital,” said Dr. Horby, cochief investigator of the RECOVERY trial and professor of emerging infectious diseases at the Centre for Tropical Medicine and Global Health at the University of Oxford (England).
Stephen Evans, BA, MSc, FRCP, professor of pharmacoepidemiology at the London School of Hygiene & Tropical Medicine, said, “This is a high-quality trial, and although published as a preprint, is of much higher quality than many non–peer-reviewed papers.”
Dr. Evans also noted the addition of steroid therapy for many participants. “Partway through the trial, the RECOVERY trial findings showed that the corticosteroid drug dexamethasone had notable mortality benefits. Consequently, quite a number of the patients in this trial had also received a corticosteroid.”
“It does look as though these drugs give some additional benefit beyond that given by dexamethasone,” he added.
Awaiting peer review
“We need to wait for the final results and ensure it was adequately powered with enough observations to make us confident in the results,” Dr. Fichtenbaum said.
“We in the United States have to step back and look at the entire set of studies and also, for this particular one, REMAP-CAP, to be in a peer-reviewed publication,” Dr. Auwaerter said. Preprints are often released “in the setting of the pandemic, where there may be important findings, especially if they impact mortality or severity of illness.”
“We need to make sure these findings, as outlined, hold up,” he said.
In the meantime, Dr. Auwaerter added, “Exactly how this will fit in is unclear. But it’s important to me as another potential drug that can help our critically ill patients.”
The REMAP-CAP study is ongoing and updated results will be provided online.
Dr. Auwaerter disclosed that he is a consultant for EMD Serono and a member of the data monitoring safety board for Humanigen. Dr. Gotur, Dr. Fichtenbaum, Dr. Ferner, and Dr. Evans disclosed no relevant financial relationships. Dr. Horby reported that Oxford University receives funding for the RECOVERY trial from U.K. Research and Innovation and the National Institute for Health Research. Roche Products and Sanofi supported REMAP-CAP through provision of tocilizumab and sarilumab in the United Kingdom.
A version of this article first appeared on Medscape.com.
New findings suggest that monoclonal antibodies used to treat RA could improve severe COVID-19 outcomes, including risk for death.
Given within 24 hours of critical illness, tocilizumab (Actemra) was associated with a median of 10 days free of respiratory and cardiovascular support up to day 21, the primary outcome. Similarly, sarilumab (Kevzara) was linked to a median of 11 days. In contrast, the usual care control group experienced zero such days in the hospital.
However, the Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community-Acquired Pneumonia (REMAP-CAP) trial comes with a caveat. The preprint findings have not yet been peer reviewed and “should not be used to guide clinical practice,” the authors stated.
The results were published online Jan. 7 in MedRxiv.
Nevertheless, the trial also revealed a mortality benefit associated with the two interleukin-6 antagonists. The hospital mortality rate was 22% with sarilumab, 28% with tocilizumab, and almost 36% with usual care.
“That’s a big change in survival. They are both lifesaving drugs,” lead coinvestigator Anthony Gordon, an Imperial College London professor of anesthesia and critical care, commented in a recent story by Reuters.
Consider the big picture
“What I think is important is ... this is one of many trials,” Paul Auwaerter, MD, MBA, said in an interview. Many other studies looking at monoclonal antibody therapy for people with COVID-19 were halted because they did not show improvement.
One exception is the EMPACTA trial, which suggested that tocilizumab was effective if given before a person becomes ill enough to be placed on a ventilator, said Dr. Auwaerter, clinical director of the division of infectious diseases at Johns Hopkins Medicine and a contributor to this news organization. “It appeared to reduce the need for mechanical ventilation or death.”
“These two trials are the first randomized, prospective trials that show a benefit on a background of others which have not,” Dr. Auwaerter added.
Interim findings
The REMAP-CAP investigators randomly assigned adults within 24 hours of critical care for COVID-19 to 8 mg/kg tocilizumab, 400 mg sarilumab, or usual care at 113 sites in six countries. There were 353 participants in the tocilizumab arm, 48 in the sarilumab group, and 402 in the control group.
Compared with the control group, the 10 days free of organ support in the tocilizumab cohort was associated with an adjusted odds ratio of 1.64 (95% confidence interval, 1.25-2.14). The 11 days free of organ support in the sarilumab cohort was likewise superior to control (adjusted odds ratio, 1.76; 95% CI, 1.17-2.91).
“All secondary outcomes and analyses supported efficacy of these IL-6 receptor antagonists,” the authors note. These endpoints included 90-day survival, time to intensive care unit discharge, and hospital discharge.
Cautious optimism?
“The results were quite impressive – having 10 or 11 fewer days in the ICU, compared to standard of care,” Deepa Gotur, MD, said in an interview. “Choosing the right patient population and providing the anti-IL-6 treatment at the right time would be the key here.”
In addition to not yet receiving peer review, an open-label design, a relatively short follow-up of 21 days, and steroids becoming standard of care about halfway through the trial are potential limitations, said Dr. Gotur, an intensivist at Houston Methodist Hospital and associate professor of clinical medicine at Weill Cornell Medicine, New York.
“This is an interesting study,” Carl J. Fichtenbaum, MD, professor of clinical medicine at the University of Cincinnati, said in a comment.
Additional detail on how many participants in each group received steroids is warranted, Dr. Fichtenbaum said. “The analysis did not carefully adjust for the use of steroids that might have influenced outcomes.”
Dr. Fichtenbaum said it’s important to look at what is distinctive about REMAP-CAP because “there are several other studies showing opposite results.”
Dr. Gotur was an investigator on a previous study evaluating tocilizumab for patients already on mechanical ventilation. “One of the key differences between this and other studies is that they included more of the ICU population,” she said. “They also included patients within 24 hours of requiring organ support, cardiac, as well as respiratory support.” Some other research included less-acute patients, including all comers into the ED who required oxygen and received tocilizumab.
The prior studies also evaluated cytokine or inflammatory markers. In contrast, REMAP-CAP researchers “looked at organ failure itself ... which I think makes sense,” Dr. Gotur said.
Cytokine release syndrome can cause organ damage or organ failure, she added, “but these markers are all over the place. I’ve seen patients who are very, very sick despite having a low [C-reactive protein] or IL-6 level.”
Backing from the British
Citing the combined 24% decrease in the risk for death associated with these agents in the REMAP-CAP trial, the U.K. government announced Jan. 7 it will work to make tocilizumab and sarilumab available to citizens with severe COVID-19.
Experts in the United Kingdom shared their perspectives on the REMAP-CAP interim findings through the U.K. Science Media Centre.
“There are few treatments for severe COVID-19,” said Robin Ferner, MD, honorary professor of clinical pharmacology at the University of Birmingham (England) and honorary consultant physician at City Hospital Birmingham. “If the published data from REMAP-CAP are supported by further studies, this suggests that two IL-6 receptor antagonists can reduce the death rate in the most severely ill patients.”
Dr. Ferner added that the findings are not a “magic cure,” however. He pointed out that of 401 patients given the drugs, 109 died, and with standard treatment, 144 out of 402 died.
Peter Horby, MD, PhD, was more optimistic. “It is great to see a positive result at a time that we really need good news and more tools to fight COVID. This is great achievement for REMAP-CAP,” he said.
“We hope to soon have results from RECOVERY on the effect of tocilizumab in less severely ill patients in the hospital,” said Dr. Horby, cochief investigator of the RECOVERY trial and professor of emerging infectious diseases at the Centre for Tropical Medicine and Global Health at the University of Oxford (England).
Stephen Evans, BA, MSc, FRCP, professor of pharmacoepidemiology at the London School of Hygiene & Tropical Medicine, said, “This is a high-quality trial, and although published as a preprint, is of much higher quality than many non–peer-reviewed papers.”
Dr. Evans also noted the addition of steroid therapy for many participants. “Partway through the trial, the RECOVERY trial findings showed that the corticosteroid drug dexamethasone had notable mortality benefits. Consequently, quite a number of the patients in this trial had also received a corticosteroid.”
“It does look as though these drugs give some additional benefit beyond that given by dexamethasone,” he added.
Awaiting peer review
“We need to wait for the final results and ensure it was adequately powered with enough observations to make us confident in the results,” Dr. Fichtenbaum said.
“We in the United States have to step back and look at the entire set of studies and also, for this particular one, REMAP-CAP, to be in a peer-reviewed publication,” Dr. Auwaerter said. Preprints are often released “in the setting of the pandemic, where there may be important findings, especially if they impact mortality or severity of illness.”
“We need to make sure these findings, as outlined, hold up,” he said.
In the meantime, Dr. Auwaerter added, “Exactly how this will fit in is unclear. But it’s important to me as another potential drug that can help our critically ill patients.”
The REMAP-CAP study is ongoing and updated results will be provided online.
Dr. Auwaerter disclosed that he is a consultant for EMD Serono and a member of the data monitoring safety board for Humanigen. Dr. Gotur, Dr. Fichtenbaum, Dr. Ferner, and Dr. Evans disclosed no relevant financial relationships. Dr. Horby reported that Oxford University receives funding for the RECOVERY trial from U.K. Research and Innovation and the National Institute for Health Research. Roche Products and Sanofi supported REMAP-CAP through provision of tocilizumab and sarilumab in the United Kingdom.
A version of this article first appeared on Medscape.com.
New findings suggest that monoclonal antibodies used to treat RA could improve severe COVID-19 outcomes, including risk for death.
Given within 24 hours of critical illness, tocilizumab (Actemra) was associated with a median of 10 days free of respiratory and cardiovascular support up to day 21, the primary outcome. Similarly, sarilumab (Kevzara) was linked to a median of 11 days. In contrast, the usual care control group experienced zero such days in the hospital.
However, the Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community-Acquired Pneumonia (REMAP-CAP) trial comes with a caveat. The preprint findings have not yet been peer reviewed and “should not be used to guide clinical practice,” the authors stated.
The results were published online Jan. 7 in MedRxiv.
Nevertheless, the trial also revealed a mortality benefit associated with the two interleukin-6 antagonists. The hospital mortality rate was 22% with sarilumab, 28% with tocilizumab, and almost 36% with usual care.
“That’s a big change in survival. They are both lifesaving drugs,” lead coinvestigator Anthony Gordon, an Imperial College London professor of anesthesia and critical care, commented in a recent story by Reuters.
Consider the big picture
“What I think is important is ... this is one of many trials,” Paul Auwaerter, MD, MBA, said in an interview. Many other studies looking at monoclonal antibody therapy for people with COVID-19 were halted because they did not show improvement.
One exception is the EMPACTA trial, which suggested that tocilizumab was effective if given before a person becomes ill enough to be placed on a ventilator, said Dr. Auwaerter, clinical director of the division of infectious diseases at Johns Hopkins Medicine and a contributor to this news organization. “It appeared to reduce the need for mechanical ventilation or death.”
“These two trials are the first randomized, prospective trials that show a benefit on a background of others which have not,” Dr. Auwaerter added.
Interim findings
The REMAP-CAP investigators randomly assigned adults within 24 hours of critical care for COVID-19 to 8 mg/kg tocilizumab, 400 mg sarilumab, or usual care at 113 sites in six countries. There were 353 participants in the tocilizumab arm, 48 in the sarilumab group, and 402 in the control group.
Compared with the control group, the 10 days free of organ support in the tocilizumab cohort was associated with an adjusted odds ratio of 1.64 (95% confidence interval, 1.25-2.14). The 11 days free of organ support in the sarilumab cohort was likewise superior to control (adjusted odds ratio, 1.76; 95% CI, 1.17-2.91).
“All secondary outcomes and analyses supported efficacy of these IL-6 receptor antagonists,” the authors note. These endpoints included 90-day survival, time to intensive care unit discharge, and hospital discharge.
Cautious optimism?
“The results were quite impressive – having 10 or 11 fewer days in the ICU, compared to standard of care,” Deepa Gotur, MD, said in an interview. “Choosing the right patient population and providing the anti-IL-6 treatment at the right time would be the key here.”
In addition to not yet receiving peer review, an open-label design, a relatively short follow-up of 21 days, and steroids becoming standard of care about halfway through the trial are potential limitations, said Dr. Gotur, an intensivist at Houston Methodist Hospital and associate professor of clinical medicine at Weill Cornell Medicine, New York.
“This is an interesting study,” Carl J. Fichtenbaum, MD, professor of clinical medicine at the University of Cincinnati, said in a comment.
Additional detail on how many participants in each group received steroids is warranted, Dr. Fichtenbaum said. “The analysis did not carefully adjust for the use of steroids that might have influenced outcomes.”
Dr. Fichtenbaum said it’s important to look at what is distinctive about REMAP-CAP because “there are several other studies showing opposite results.”
Dr. Gotur was an investigator on a previous study evaluating tocilizumab for patients already on mechanical ventilation. “One of the key differences between this and other studies is that they included more of the ICU population,” she said. “They also included patients within 24 hours of requiring organ support, cardiac, as well as respiratory support.” Some other research included less-acute patients, including all comers into the ED who required oxygen and received tocilizumab.
The prior studies also evaluated cytokine or inflammatory markers. In contrast, REMAP-CAP researchers “looked at organ failure itself ... which I think makes sense,” Dr. Gotur said.
Cytokine release syndrome can cause organ damage or organ failure, she added, “but these markers are all over the place. I’ve seen patients who are very, very sick despite having a low [C-reactive protein] or IL-6 level.”
Backing from the British
Citing the combined 24% decrease in the risk for death associated with these agents in the REMAP-CAP trial, the U.K. government announced Jan. 7 it will work to make tocilizumab and sarilumab available to citizens with severe COVID-19.
Experts in the United Kingdom shared their perspectives on the REMAP-CAP interim findings through the U.K. Science Media Centre.
“There are few treatments for severe COVID-19,” said Robin Ferner, MD, honorary professor of clinical pharmacology at the University of Birmingham (England) and honorary consultant physician at City Hospital Birmingham. “If the published data from REMAP-CAP are supported by further studies, this suggests that two IL-6 receptor antagonists can reduce the death rate in the most severely ill patients.”
Dr. Ferner added that the findings are not a “magic cure,” however. He pointed out that of 401 patients given the drugs, 109 died, and with standard treatment, 144 out of 402 died.
Peter Horby, MD, PhD, was more optimistic. “It is great to see a positive result at a time that we really need good news and more tools to fight COVID. This is great achievement for REMAP-CAP,” he said.
“We hope to soon have results from RECOVERY on the effect of tocilizumab in less severely ill patients in the hospital,” said Dr. Horby, cochief investigator of the RECOVERY trial and professor of emerging infectious diseases at the Centre for Tropical Medicine and Global Health at the University of Oxford (England).
Stephen Evans, BA, MSc, FRCP, professor of pharmacoepidemiology at the London School of Hygiene & Tropical Medicine, said, “This is a high-quality trial, and although published as a preprint, is of much higher quality than many non–peer-reviewed papers.”
Dr. Evans also noted the addition of steroid therapy for many participants. “Partway through the trial, the RECOVERY trial findings showed that the corticosteroid drug dexamethasone had notable mortality benefits. Consequently, quite a number of the patients in this trial had also received a corticosteroid.”
“It does look as though these drugs give some additional benefit beyond that given by dexamethasone,” he added.
Awaiting peer review
“We need to wait for the final results and ensure it was adequately powered with enough observations to make us confident in the results,” Dr. Fichtenbaum said.
“We in the United States have to step back and look at the entire set of studies and also, for this particular one, REMAP-CAP, to be in a peer-reviewed publication,” Dr. Auwaerter said. Preprints are often released “in the setting of the pandemic, where there may be important findings, especially if they impact mortality or severity of illness.”
“We need to make sure these findings, as outlined, hold up,” he said.
In the meantime, Dr. Auwaerter added, “Exactly how this will fit in is unclear. But it’s important to me as another potential drug that can help our critically ill patients.”
The REMAP-CAP study is ongoing and updated results will be provided online.
Dr. Auwaerter disclosed that he is a consultant for EMD Serono and a member of the data monitoring safety board for Humanigen. Dr. Gotur, Dr. Fichtenbaum, Dr. Ferner, and Dr. Evans disclosed no relevant financial relationships. Dr. Horby reported that Oxford University receives funding for the RECOVERY trial from U.K. Research and Innovation and the National Institute for Health Research. Roche Products and Sanofi supported REMAP-CAP through provision of tocilizumab and sarilumab in the United Kingdom.
A version of this article first appeared on Medscape.com.
Could an osteoporosis drug reduce need for hip revision surgery?
A single injection of denosumab (Prolia, Amgen), frequently used to treat osteoporosis, may reduce the need for revision surgery in patients with symptomatic osteolysis following total hip arthroplasty, a new proof-of-concept study suggests.
Aseptic loosening is the result of wear-induced osteolysis caused by the prosthetic hip and is a major contributor to the need for revision surgery in many parts of the world.
“The only established treatment for prosthesis-related osteolysis after joint replacement is revision surgery, which carries substantially greater morbidity and mortality than primary joint replacement,” Mohit M. Mahatma, MRes, of the University of Sheffield, England, and colleagues wrote in their article, published online Jan. 11 in The Lancet Rheumatology.
As well as an increased risk of infection and other complications, revision surgery is much more costly than a first-time operation, they added.
“The results of this proof-of-concept clinical trial indicate that denosumab is effective at reducing bone resorption activity within osteolytic lesion tissue and is well tolerated within the limitations of the single dose used here,” they concluded.
Commenting on the findings, Antonia Chen, MD, associate professor of orthopedic surgery, Harvard Medical School, Boston, emphasized that further studies are needed to assess the effectiveness of this strategy to reduce the need for hip revision surgery.
Nevertheless, “osteolysis is still unfortunately a problem we do have to deal with and we do not have any other way to prevent it,” she said in an interview. “So it’s a good start ... although further studies are definitely needed,” Dr. Chen added.
In an accompanying editorial, Hannu Aro, MD, Turku University Hospital in Finland, agreed: “Without a doubt, the trial is a breakthrough, but it represents only the first step in the development of pharmacological therapy aiming to slow, prevent, or even reverse the process of wear-induced periprosthetic osteolysis.”
Small single-center study
The phase 2, single-center, randomized, controlled trial involved 22 patients who had previously undergone hip replacement surgery at Sheffield Teaching Hospitals and were scheduled for revision surgery due to symptomatic osteolysis. They were randomized to a single subcutaneous injection of denosumab at a dose of 60 mg, or placebo, on their second hospital visit.
“The primary outcome was the between-group difference in the number of osteoclasts per mm of osteolytic membrane at the osteolytic membrane-bone interface at week 8,” the authors noted.
At this time point, there were 83% fewer osteoclasts at the interface in the denosumab group compared with placebo, at a median of 0.05 per mm in the treatment group compared with 0.30 per mm in the placebo group (P = .011).
Secondary histological outcomes were also significantly improved in favor of the denosumab group compared with placebo.
Potential to prevent half of all hip revision surgeries?
Patients who received denosumab also demonstrated an acute fall in serum and urinary markers of bone resorption following administration of the drug, reaching a nadir at week 4, which was maintained until revision surgery at week 8.
In contrast, “no change in these markers was observed in the placebo group [P < .0003 for all biomarkers],” the investigators noted. Rates of adverse events were comparable in both treatment groups.
As the authors explained, osteolysis occurs following joint replacement surgery when particles of plastic wear off from the prosthesis, triggering an immune reaction that attacks the bone around the implant, causing the joint to loosen.
“It is very clear from our bone biopsies and bone imaging that the [denosumab] injection stops the bone absorbing the microplastic particles from the replacement joint and therefore could prevent the bone from being eaten away and the need for revision surgery,” senior author Mark Wilkinson, MBChB, PhD, honorary consultant orthopedic surgeon, Sheffield Teaching Hospitals, said in a press release from his institution.
“This study is a significant breakthrough as we’ve demonstrated that there is a drug, already available and successful in the treatment of osteoporosis, that has the potential to prevent up to half of all revised replacement surgeries which are caused by osteolysis,” he added.
Dr. Wilkinson and coauthors said their results justify the need for future trials targeting earlier-stage disease to further test the use of denosumab to prevent or reduce the need for revision surgery.
In 2018, aseptic loosening accounted for over half of all revision procedures, as reported to the National Joint Registry in England and Wales.
Older polyethylene prostheses are the main culprit
Commenting further on the study, Dr. Chen noted that osteolysis still plagues orthopedic surgeons because the original polyethylene prostheses were not very good. A better prosthesis developed at Massachusetts General Hospital is made up of highly crossed-link polyethylene and still wears over time but to a much lesser extent than the older polyethylene prostheses.
Metal and ceramic prostheses also can induce osteolysis, but again to a much lesser extent than the older polyethylene implants.
“Any particle can technically cause osteolysis but plastic produces the most particles,” Dr. Chen explained. Although hip revision rates in the United States are low to begin with, aseptic loosening is still one of the main reasons that patients need to undergo revision surgery, she observed.
“A lot of patients are still living with the old plastic [implants] so there is still a need for something like this,” she stressed.
However, many questions about this potential new strategy remain to be answered, including when best to initiate treatment and how to manage patients at risk for osteolysis 20-30 years after they have received their original implant.
In his editorial, Dr. Aro said that serious adverse consequences often become evident 10-20 years after patients have undergone the original hip replacement procedures, when they are potentially less physically fit than they were at the time of the operation and thus less able to withstand the rigors of a difficult revision surgery.
“In this context, the concept of nonsurgical pharmacological treatment of periprosthetic osteolysis ... brings a new hope for the ever-increasing population of patients with total hip arthroplasty to avoid revision surgery,” Dr. Aro suggested.
However, Dr. Aro cautioned that reduction of bone turnover by antiresorptive agents such as denosumab has been associated with the development of atypical femoral fractures.
The study was funded by Amgen. Dr. Wilkinson has reported receiving a grant from Amgen. Dr. Chen has reported serving as a consultant for Striker and b-One Ortho. Dr. Aro has reported receiving a grant to his institution from Amgen Finland and the Academy of Finland. He has also served as a member of an advisory scientific board for Amgen Finland.
A version of this article first appeared on Medscape.com.
A single injection of denosumab (Prolia, Amgen), frequently used to treat osteoporosis, may reduce the need for revision surgery in patients with symptomatic osteolysis following total hip arthroplasty, a new proof-of-concept study suggests.
Aseptic loosening is the result of wear-induced osteolysis caused by the prosthetic hip and is a major contributor to the need for revision surgery in many parts of the world.
“The only established treatment for prosthesis-related osteolysis after joint replacement is revision surgery, which carries substantially greater morbidity and mortality than primary joint replacement,” Mohit M. Mahatma, MRes, of the University of Sheffield, England, and colleagues wrote in their article, published online Jan. 11 in The Lancet Rheumatology.
As well as an increased risk of infection and other complications, revision surgery is much more costly than a first-time operation, they added.
“The results of this proof-of-concept clinical trial indicate that denosumab is effective at reducing bone resorption activity within osteolytic lesion tissue and is well tolerated within the limitations of the single dose used here,” they concluded.
Commenting on the findings, Antonia Chen, MD, associate professor of orthopedic surgery, Harvard Medical School, Boston, emphasized that further studies are needed to assess the effectiveness of this strategy to reduce the need for hip revision surgery.
Nevertheless, “osteolysis is still unfortunately a problem we do have to deal with and we do not have any other way to prevent it,” she said in an interview. “So it’s a good start ... although further studies are definitely needed,” Dr. Chen added.
In an accompanying editorial, Hannu Aro, MD, Turku University Hospital in Finland, agreed: “Without a doubt, the trial is a breakthrough, but it represents only the first step in the development of pharmacological therapy aiming to slow, prevent, or even reverse the process of wear-induced periprosthetic osteolysis.”
Small single-center study
The phase 2, single-center, randomized, controlled trial involved 22 patients who had previously undergone hip replacement surgery at Sheffield Teaching Hospitals and were scheduled for revision surgery due to symptomatic osteolysis. They were randomized to a single subcutaneous injection of denosumab at a dose of 60 mg, or placebo, on their second hospital visit.
“The primary outcome was the between-group difference in the number of osteoclasts per mm of osteolytic membrane at the osteolytic membrane-bone interface at week 8,” the authors noted.
At this time point, there were 83% fewer osteoclasts at the interface in the denosumab group compared with placebo, at a median of 0.05 per mm in the treatment group compared with 0.30 per mm in the placebo group (P = .011).
Secondary histological outcomes were also significantly improved in favor of the denosumab group compared with placebo.
Potential to prevent half of all hip revision surgeries?
Patients who received denosumab also demonstrated an acute fall in serum and urinary markers of bone resorption following administration of the drug, reaching a nadir at week 4, which was maintained until revision surgery at week 8.
In contrast, “no change in these markers was observed in the placebo group [P < .0003 for all biomarkers],” the investigators noted. Rates of adverse events were comparable in both treatment groups.
As the authors explained, osteolysis occurs following joint replacement surgery when particles of plastic wear off from the prosthesis, triggering an immune reaction that attacks the bone around the implant, causing the joint to loosen.
“It is very clear from our bone biopsies and bone imaging that the [denosumab] injection stops the bone absorbing the microplastic particles from the replacement joint and therefore could prevent the bone from being eaten away and the need for revision surgery,” senior author Mark Wilkinson, MBChB, PhD, honorary consultant orthopedic surgeon, Sheffield Teaching Hospitals, said in a press release from his institution.
“This study is a significant breakthrough as we’ve demonstrated that there is a drug, already available and successful in the treatment of osteoporosis, that has the potential to prevent up to half of all revised replacement surgeries which are caused by osteolysis,” he added.
Dr. Wilkinson and coauthors said their results justify the need for future trials targeting earlier-stage disease to further test the use of denosumab to prevent or reduce the need for revision surgery.
In 2018, aseptic loosening accounted for over half of all revision procedures, as reported to the National Joint Registry in England and Wales.
Older polyethylene prostheses are the main culprit
Commenting further on the study, Dr. Chen noted that osteolysis still plagues orthopedic surgeons because the original polyethylene prostheses were not very good. A better prosthesis developed at Massachusetts General Hospital is made up of highly crossed-link polyethylene and still wears over time but to a much lesser extent than the older polyethylene prostheses.
Metal and ceramic prostheses also can induce osteolysis, but again to a much lesser extent than the older polyethylene implants.
“Any particle can technically cause osteolysis but plastic produces the most particles,” Dr. Chen explained. Although hip revision rates in the United States are low to begin with, aseptic loosening is still one of the main reasons that patients need to undergo revision surgery, she observed.
“A lot of patients are still living with the old plastic [implants] so there is still a need for something like this,” she stressed.
However, many questions about this potential new strategy remain to be answered, including when best to initiate treatment and how to manage patients at risk for osteolysis 20-30 years after they have received their original implant.
In his editorial, Dr. Aro said that serious adverse consequences often become evident 10-20 years after patients have undergone the original hip replacement procedures, when they are potentially less physically fit than they were at the time of the operation and thus less able to withstand the rigors of a difficult revision surgery.
“In this context, the concept of nonsurgical pharmacological treatment of periprosthetic osteolysis ... brings a new hope for the ever-increasing population of patients with total hip arthroplasty to avoid revision surgery,” Dr. Aro suggested.
However, Dr. Aro cautioned that reduction of bone turnover by antiresorptive agents such as denosumab has been associated with the development of atypical femoral fractures.
The study was funded by Amgen. Dr. Wilkinson has reported receiving a grant from Amgen. Dr. Chen has reported serving as a consultant for Striker and b-One Ortho. Dr. Aro has reported receiving a grant to his institution from Amgen Finland and the Academy of Finland. He has also served as a member of an advisory scientific board for Amgen Finland.
A version of this article first appeared on Medscape.com.
A single injection of denosumab (Prolia, Amgen), frequently used to treat osteoporosis, may reduce the need for revision surgery in patients with symptomatic osteolysis following total hip arthroplasty, a new proof-of-concept study suggests.
Aseptic loosening is the result of wear-induced osteolysis caused by the prosthetic hip and is a major contributor to the need for revision surgery in many parts of the world.
“The only established treatment for prosthesis-related osteolysis after joint replacement is revision surgery, which carries substantially greater morbidity and mortality than primary joint replacement,” Mohit M. Mahatma, MRes, of the University of Sheffield, England, and colleagues wrote in their article, published online Jan. 11 in The Lancet Rheumatology.
As well as an increased risk of infection and other complications, revision surgery is much more costly than a first-time operation, they added.
“The results of this proof-of-concept clinical trial indicate that denosumab is effective at reducing bone resorption activity within osteolytic lesion tissue and is well tolerated within the limitations of the single dose used here,” they concluded.
Commenting on the findings, Antonia Chen, MD, associate professor of orthopedic surgery, Harvard Medical School, Boston, emphasized that further studies are needed to assess the effectiveness of this strategy to reduce the need for hip revision surgery.
Nevertheless, “osteolysis is still unfortunately a problem we do have to deal with and we do not have any other way to prevent it,” she said in an interview. “So it’s a good start ... although further studies are definitely needed,” Dr. Chen added.
In an accompanying editorial, Hannu Aro, MD, Turku University Hospital in Finland, agreed: “Without a doubt, the trial is a breakthrough, but it represents only the first step in the development of pharmacological therapy aiming to slow, prevent, or even reverse the process of wear-induced periprosthetic osteolysis.”
Small single-center study
The phase 2, single-center, randomized, controlled trial involved 22 patients who had previously undergone hip replacement surgery at Sheffield Teaching Hospitals and were scheduled for revision surgery due to symptomatic osteolysis. They were randomized to a single subcutaneous injection of denosumab at a dose of 60 mg, or placebo, on their second hospital visit.
“The primary outcome was the between-group difference in the number of osteoclasts per mm of osteolytic membrane at the osteolytic membrane-bone interface at week 8,” the authors noted.
At this time point, there were 83% fewer osteoclasts at the interface in the denosumab group compared with placebo, at a median of 0.05 per mm in the treatment group compared with 0.30 per mm in the placebo group (P = .011).
Secondary histological outcomes were also significantly improved in favor of the denosumab group compared with placebo.
Potential to prevent half of all hip revision surgeries?
Patients who received denosumab also demonstrated an acute fall in serum and urinary markers of bone resorption following administration of the drug, reaching a nadir at week 4, which was maintained until revision surgery at week 8.
In contrast, “no change in these markers was observed in the placebo group [P < .0003 for all biomarkers],” the investigators noted. Rates of adverse events were comparable in both treatment groups.
As the authors explained, osteolysis occurs following joint replacement surgery when particles of plastic wear off from the prosthesis, triggering an immune reaction that attacks the bone around the implant, causing the joint to loosen.
“It is very clear from our bone biopsies and bone imaging that the [denosumab] injection stops the bone absorbing the microplastic particles from the replacement joint and therefore could prevent the bone from being eaten away and the need for revision surgery,” senior author Mark Wilkinson, MBChB, PhD, honorary consultant orthopedic surgeon, Sheffield Teaching Hospitals, said in a press release from his institution.
“This study is a significant breakthrough as we’ve demonstrated that there is a drug, already available and successful in the treatment of osteoporosis, that has the potential to prevent up to half of all revised replacement surgeries which are caused by osteolysis,” he added.
Dr. Wilkinson and coauthors said their results justify the need for future trials targeting earlier-stage disease to further test the use of denosumab to prevent or reduce the need for revision surgery.
In 2018, aseptic loosening accounted for over half of all revision procedures, as reported to the National Joint Registry in England and Wales.
Older polyethylene prostheses are the main culprit
Commenting further on the study, Dr. Chen noted that osteolysis still plagues orthopedic surgeons because the original polyethylene prostheses were not very good. A better prosthesis developed at Massachusetts General Hospital is made up of highly crossed-link polyethylene and still wears over time but to a much lesser extent than the older polyethylene prostheses.
Metal and ceramic prostheses also can induce osteolysis, but again to a much lesser extent than the older polyethylene implants.
“Any particle can technically cause osteolysis but plastic produces the most particles,” Dr. Chen explained. Although hip revision rates in the United States are low to begin with, aseptic loosening is still one of the main reasons that patients need to undergo revision surgery, she observed.
“A lot of patients are still living with the old plastic [implants] so there is still a need for something like this,” she stressed.
However, many questions about this potential new strategy remain to be answered, including when best to initiate treatment and how to manage patients at risk for osteolysis 20-30 years after they have received their original implant.
In his editorial, Dr. Aro said that serious adverse consequences often become evident 10-20 years after patients have undergone the original hip replacement procedures, when they are potentially less physically fit than they were at the time of the operation and thus less able to withstand the rigors of a difficult revision surgery.
“In this context, the concept of nonsurgical pharmacological treatment of periprosthetic osteolysis ... brings a new hope for the ever-increasing population of patients with total hip arthroplasty to avoid revision surgery,” Dr. Aro suggested.
However, Dr. Aro cautioned that reduction of bone turnover by antiresorptive agents such as denosumab has been associated with the development of atypical femoral fractures.
The study was funded by Amgen. Dr. Wilkinson has reported receiving a grant from Amgen. Dr. Chen has reported serving as a consultant for Striker and b-One Ortho. Dr. Aro has reported receiving a grant to his institution from Amgen Finland and the Academy of Finland. He has also served as a member of an advisory scientific board for Amgen Finland.
A version of this article first appeared on Medscape.com.