User login
Survey finds Mohs surgeons favor nicotinamide for chemoprevention
, in a survey of members of the American College of Mohs Surgeons.
Although nicotinamide, a vitamin B3 derivative, has been shown to reduce keratinocyte carcinoma (KC) in high-risk patients, it is not approved by the Food and Drug Administration for chemoprevention, and no safe upper limit has been established in clinical trials to date, wrote Sheena Desai of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
The investigators emailed an anonymous 12-question survey to 1,500 members of the American College of Mohs Surgeons. Of the 170 who responded, 10 were excluded for discordant responses, leaving 160 participants whose replies were included in a multiple logistic regression analysis. The respondents were mainly U.S. board-certified dermatologists and Mohs surgeons (99.4% for both); 86.9% were in clinical practice, including 78.8% in private practice, according to the report of the results, published in Dermatologic Surgery.
Overall, 76.9% of the respondents said they recommended nicotinamide for preventing KC, and 20% said they had recommended nicotinamide to more than 100 patients in the past year. In addition, 45% of respondents reported patients who had been taking nicotinamide for 2 years or more. Overall, 63.8% of the respondents expressed no concerns about long-term safety of nicotinamide, compared with 28.1% who said they were uncertain about long-term safety. Those who expressed concern or uncertainty about long-term safety were significantly less likely to recommend nicotinamide for KC prevention in the past year (odds ratio, 0.30; 95% confidence interval [CI] 0.13-0.71). Clinicians with more than 10 years in practice were significantly less likely to recommend nicotinamide for chemoprevention (OR, 0.20; 95% CI 0.05-0.82).
The study findings were limited by several factors, including the low number of responses and the potential lack of generalizability to clinicians other than Mohs surgeons, the researchers noted. “Additional studies on nicotinamide safety and use patterns, including cost-effectiveness analyses, are needed given the widespread use identified in this study,” they concluded.
Limited safety data highlight research gaps
The study is particularly important at this time because nicotinamide has been increasingly used for KC chemoprevention since a randomized, controlled trial published in 2015 in the New England Journal of Medicine showed benefits, corresponding author Rebecca I. Hartman, MD, of the department of dermatology, Brigham and Women’s Hospital and Harvard University, Boston, said in an interview. That study of high-risk patients found that nicotinamide, 500 mg twice a day, was safe and effective in lowering the rates of new nonmelanoma skin cancers and AKs after 12 months .

“However, because this is not a prescription medication, but rather an OTC vitamin supplement, data on its use are not available,” she said.
Dr. Hartman said she was not surprised that nicotinamide is being used frequently by a majority of the survey respondents. “Most are using this if someone has two KCs over 2 years, which is a quite common occurrence,” she noted. However, “I was a bit surprised that nearly two-thirds had no safety concerns with long-term use, even though this has not been well-studied,” she added.
“Like anything we recommend, we must consider the risks and benefits,” Dr. Hartman said of nicotinamide. “Unfortunately, we don’t know the risks well, since this hasn’t been well-characterized with regular long-term use in these doses,” and more research is needed, she said. “The risks are likely low, as this is a vitamin that has been used for years in various OTC supplements,” she added. “However, there are some data showing slightly increased all-cause mortality with similar doses of a related medicine, niacin, in cardiovascular patients. For this reason, I recommend the medication when a patient’s KCs are really becoming burdensome – several KCs in a year or two – or when they are high-risk due to immunosuppression,” she explained.
“We also must consider the individual patient. For a healthy younger patient who has a public-facing job and as a result is very averse to developing any KCs on his or her face and very motivated to try prevention, it may make sense to try nicotinamide,” Dr. Hartman said. But for an older patient with cardiovascular comorbidities who is not bothered by a KC on his or her back or extremities, “this medication may not have a favorable risk-benefit profile.”
To address safety concerns, “researchers need to examine whether there are any harms in long-term regular nicotinamide use for KC prevention,” Dr. Hartman said. “This is something we hope to do in our patients; however, it is challenging to study in a retrospective way since the harm is likely small and there are so many other features that influence mortality as an outcome,” she noted.
The study received no outside funding. The researchers had no financial conflicts to disclose.
, in a survey of members of the American College of Mohs Surgeons.
Although nicotinamide, a vitamin B3 derivative, has been shown to reduce keratinocyte carcinoma (KC) in high-risk patients, it is not approved by the Food and Drug Administration for chemoprevention, and no safe upper limit has been established in clinical trials to date, wrote Sheena Desai of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
The investigators emailed an anonymous 12-question survey to 1,500 members of the American College of Mohs Surgeons. Of the 170 who responded, 10 were excluded for discordant responses, leaving 160 participants whose replies were included in a multiple logistic regression analysis. The respondents were mainly U.S. board-certified dermatologists and Mohs surgeons (99.4% for both); 86.9% were in clinical practice, including 78.8% in private practice, according to the report of the results, published in Dermatologic Surgery.
Overall, 76.9% of the respondents said they recommended nicotinamide for preventing KC, and 20% said they had recommended nicotinamide to more than 100 patients in the past year. In addition, 45% of respondents reported patients who had been taking nicotinamide for 2 years or more. Overall, 63.8% of the respondents expressed no concerns about long-term safety of nicotinamide, compared with 28.1% who said they were uncertain about long-term safety. Those who expressed concern or uncertainty about long-term safety were significantly less likely to recommend nicotinamide for KC prevention in the past year (odds ratio, 0.30; 95% confidence interval [CI] 0.13-0.71). Clinicians with more than 10 years in practice were significantly less likely to recommend nicotinamide for chemoprevention (OR, 0.20; 95% CI 0.05-0.82).
The study findings were limited by several factors, including the low number of responses and the potential lack of generalizability to clinicians other than Mohs surgeons, the researchers noted. “Additional studies on nicotinamide safety and use patterns, including cost-effectiveness analyses, are needed given the widespread use identified in this study,” they concluded.
Limited safety data highlight research gaps
The study is particularly important at this time because nicotinamide has been increasingly used for KC chemoprevention since a randomized, controlled trial published in 2015 in the New England Journal of Medicine showed benefits, corresponding author Rebecca I. Hartman, MD, of the department of dermatology, Brigham and Women’s Hospital and Harvard University, Boston, said in an interview. That study of high-risk patients found that nicotinamide, 500 mg twice a day, was safe and effective in lowering the rates of new nonmelanoma skin cancers and AKs after 12 months .
“However, because this is not a prescription medication, but rather an OTC vitamin supplement, data on its use are not available,” she said.
Dr. Hartman said she was not surprised that nicotinamide is being used frequently by a majority of the survey respondents. “Most are using this if someone has two KCs over 2 years, which is a quite common occurrence,” she noted. However, “I was a bit surprised that nearly two-thirds had no safety concerns with long-term use, even though this has not been well-studied,” she added.
“Like anything we recommend, we must consider the risks and benefits,” Dr. Hartman said of nicotinamide. “Unfortunately, we don’t know the risks well, since this hasn’t been well-characterized with regular long-term use in these doses,” and more research is needed, she said. “The risks are likely low, as this is a vitamin that has been used for years in various OTC supplements,” she added. “However, there are some data showing slightly increased all-cause mortality with similar doses of a related medicine, niacin, in cardiovascular patients. For this reason, I recommend the medication when a patient’s KCs are really becoming burdensome – several KCs in a year or two – or when they are high-risk due to immunosuppression,” she explained.
“We also must consider the individual patient. For a healthy younger patient who has a public-facing job and as a result is very averse to developing any KCs on his or her face and very motivated to try prevention, it may make sense to try nicotinamide,” Dr. Hartman said. But for an older patient with cardiovascular comorbidities who is not bothered by a KC on his or her back or extremities, “this medication may not have a favorable risk-benefit profile.”
To address safety concerns, “researchers need to examine whether there are any harms in long-term regular nicotinamide use for KC prevention,” Dr. Hartman said. “This is something we hope to do in our patients; however, it is challenging to study in a retrospective way since the harm is likely small and there are so many other features that influence mortality as an outcome,” she noted.
The study received no outside funding. The researchers had no financial conflicts to disclose.
, in a survey of members of the American College of Mohs Surgeons.
Although nicotinamide, a vitamin B3 derivative, has been shown to reduce keratinocyte carcinoma (KC) in high-risk patients, it is not approved by the Food and Drug Administration for chemoprevention, and no safe upper limit has been established in clinical trials to date, wrote Sheena Desai of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
The investigators emailed an anonymous 12-question survey to 1,500 members of the American College of Mohs Surgeons. Of the 170 who responded, 10 were excluded for discordant responses, leaving 160 participants whose replies were included in a multiple logistic regression analysis. The respondents were mainly U.S. board-certified dermatologists and Mohs surgeons (99.4% for both); 86.9% were in clinical practice, including 78.8% in private practice, according to the report of the results, published in Dermatologic Surgery.
Overall, 76.9% of the respondents said they recommended nicotinamide for preventing KC, and 20% said they had recommended nicotinamide to more than 100 patients in the past year. In addition, 45% of respondents reported patients who had been taking nicotinamide for 2 years or more. Overall, 63.8% of the respondents expressed no concerns about long-term safety of nicotinamide, compared with 28.1% who said they were uncertain about long-term safety. Those who expressed concern or uncertainty about long-term safety were significantly less likely to recommend nicotinamide for KC prevention in the past year (odds ratio, 0.30; 95% confidence interval [CI] 0.13-0.71). Clinicians with more than 10 years in practice were significantly less likely to recommend nicotinamide for chemoprevention (OR, 0.20; 95% CI 0.05-0.82).
The study findings were limited by several factors, including the low number of responses and the potential lack of generalizability to clinicians other than Mohs surgeons, the researchers noted. “Additional studies on nicotinamide safety and use patterns, including cost-effectiveness analyses, are needed given the widespread use identified in this study,” they concluded.
Limited safety data highlight research gaps
The study is particularly important at this time because nicotinamide has been increasingly used for KC chemoprevention since a randomized, controlled trial published in 2015 in the New England Journal of Medicine showed benefits, corresponding author Rebecca I. Hartman, MD, of the department of dermatology, Brigham and Women’s Hospital and Harvard University, Boston, said in an interview. That study of high-risk patients found that nicotinamide, 500 mg twice a day, was safe and effective in lowering the rates of new nonmelanoma skin cancers and AKs after 12 months .
“However, because this is not a prescription medication, but rather an OTC vitamin supplement, data on its use are not available,” she said.
Dr. Hartman said she was not surprised that nicotinamide is being used frequently by a majority of the survey respondents. “Most are using this if someone has two KCs over 2 years, which is a quite common occurrence,” she noted. However, “I was a bit surprised that nearly two-thirds had no safety concerns with long-term use, even though this has not been well-studied,” she added.
“Like anything we recommend, we must consider the risks and benefits,” Dr. Hartman said of nicotinamide. “Unfortunately, we don’t know the risks well, since this hasn’t been well-characterized with regular long-term use in these doses,” and more research is needed, she said. “The risks are likely low, as this is a vitamin that has been used for years in various OTC supplements,” she added. “However, there are some data showing slightly increased all-cause mortality with similar doses of a related medicine, niacin, in cardiovascular patients. For this reason, I recommend the medication when a patient’s KCs are really becoming burdensome – several KCs in a year or two – or when they are high-risk due to immunosuppression,” she explained.
“We also must consider the individual patient. For a healthy younger patient who has a public-facing job and as a result is very averse to developing any KCs on his or her face and very motivated to try prevention, it may make sense to try nicotinamide,” Dr. Hartman said. But for an older patient with cardiovascular comorbidities who is not bothered by a KC on his or her back or extremities, “this medication may not have a favorable risk-benefit profile.”
To address safety concerns, “researchers need to examine whether there are any harms in long-term regular nicotinamide use for KC prevention,” Dr. Hartman said. “This is something we hope to do in our patients; however, it is challenging to study in a retrospective way since the harm is likely small and there are so many other features that influence mortality as an outcome,” she noted.
The study received no outside funding. The researchers had no financial conflicts to disclose.
FROM DERMATOLOGIC SURGERY
FDA approves frontline immunotherapy for gastric cancers

This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
How some COVID-19 vaccines could cause rare blood clots
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
Adjunctive MDMA safe, effective for severe PTSD
Adding 3,4-methylenedioxymethamphetamine (MDMA) to integrative psychotherapy may significantly improve symptoms and well-being for patients with severe posttraumatic stress disorder, including those with the dissociative subtype, new research suggests.
MAPP1 is the first phase 3 randomized controlled trial of MDMA-assisted therapy in this population. Participants who received the active treatment showed greater improvement in PTSD symptoms, mood, and empathy in comparison with participants who received placebo.
MDMA was “extremely effective, particularly for a subpopulation that ordinarily does not respond well to conventional treatment,” study coinvestigator Bessel van der Kolk, MD, professor of psychiatry at Boston University School of Medicine, told delegates attending the virtual European Psychiatric Association (EPA) 2021 Congress.
Growing interest
, particularly because failure rates with most available evidence-based treatments have been relatively high.
As previously reported by this news organization, in 2017, the U.S. Food and Drug Administration approved the trial design of Dr. van der Kolk’s and colleagues’ MAPP1 study after granting MDMA breakthrough designation.
The MAPP1 investigators assessed 90 patients with PTSD (mean age, 41 years; 77% White; 66% women) from 50 sites. For the majority of patients (84%), trauma history was developmental. “In other words, trauma [occurred] very early in life, usually at the hands of their own caregivers,” Dr. van der Kolk noted.
In addition, 18% of the patients were veterans, and 12% had combat exposure. The average duration of PTSD before enrollment was 18 years. All patients underwent screening and three preparatory psychotherapy sessions at enrollment.
Participants were randomly assigned to receive MDMA 80 mg or 120 mg (n = 46) or placebo (n = 44) followed by three integrative psychotherapy sessions lasting a total of 8 hours. A supplemental dose of 40 or 60 mg of MDMA could be administered from 1.5 to 2 hours after the first dose.
The patients stayed in the laboratory on the evening of the treatment session and attended a debriefing the next morning. The session was repeated a month later and again a month after that. In between, patients had telephone contact with the raters, who were blinded to the treatment received.
Follow-up assessments were conducted 2 months after the third treatment session and again at 12 months. The primary outcome measure was change in Clinician Administered PTSD Scale for DSM 5 (CAPS-5) score from baseline.
‘Dramatic improvement’
Results showed that both the MDMA and placebo groups experienced a statistically significant improvement in PTSD symptoms, “but MDMA had a dramatically significant improvement, with an effect size of over 0.9,” Dr. van der Kolk said.
The MDMA group also reported enhanced mood and well-being, increased responsiveness to emotional and sensory stimuli, a greater sense of closeness to other people, and a greater feeling of empathy.
Patients also reported having heightened openness, “and clearly the issue of empathy for themselves and others was a very large part of the process,” said Dr. van der Kolk.
“But for me, the most interesting part of the study is that the Adverse Childhood Experiences scale had no effect,” he noted. In other words, “the amount of childhood adverse experiences did not predict outcomes, which was very surprising because usually those patients are very treatment resistant.”
Dr. van der Kolk added that the dissociative subtype of PTSD was first described in the DSM-5 and that patients are “notoriously unresponsive to most unconventional treatments.”
In the current study, 13 patients met the criteria for the subtype, and investigators found they “did better than people with classical PTSD,” Dr. van der Kolk said. He added that this is a “very, very important finding.”
Carefully controlled
Overall, 82% of patients reported a significant improvement by the end of the study; 56% reported that they no longer had PTSD.
In addition, 67% of patients no longer met diagnostic criteria for PTSD. These included patients who had crossed over to active treatment from the placebo group.
Eleven patients (12%) experienced relapse by 12 months; in nine of the cases, this was due to the presence of additional stressors.
There were “very few adverse side effects” during the study, Dr. van der Kolk noted. In addition, “there were really no serious mental side effects,” despite the patients’ “opening up so much very painful material,” he added.
The most common adverse events among the MDMA group were muscle tightness (63%), decreased appetite (52%), nausea (30%), hyperhidrosis (20%), and feeling cold (20%). These effects were “quite small [and] the sort of side effects you would expect in response to an amphetamine substance like MDMA,” said Dr. van der Kolk.
“An important reason why we think the side effect profile is so good is because the study was extremely carefully done, very carefully controlled,” he added. “There was a great deal of support, [and] we paid an enormous amount of attention to creating a very safe context in which this drug was being used.”
However, he expressed concern that “as people see the very good results, they may skimp a little bit on the creation of the context and not have as careful a psychotherapy protocol as we had here.”
‘On the right track’
Commenting on the findings for this news organization, David Nutt, MD, PhD, Edmond J. Safra Professor of Neuropsychopharmacology, Imperial College London, said the results are proof that the investigators’ “earlier smaller trials of MDMA were on the right track.”

“This larger and multicenter trial shows that MDMA therapy can be broadened into newer research groups, which augurs well for the much larger rollout that will be required once it gets a license,” said Dr. Nutt, who was not involved with the research.
He added, “the prior evidence of the safety of MDMA has [now] been confirmed.”
The study represents an “important step in the path to the clinical use of MDMA for PTSD,” Dr. Nutt said.
The study was sponsored by the Multidisciplinary Association for Psychedelic Studies. The investigators and Dr. Nutt have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Adding 3,4-methylenedioxymethamphetamine (MDMA) to integrative psychotherapy may significantly improve symptoms and well-being for patients with severe posttraumatic stress disorder, including those with the dissociative subtype, new research suggests.
MAPP1 is the first phase 3 randomized controlled trial of MDMA-assisted therapy in this population. Participants who received the active treatment showed greater improvement in PTSD symptoms, mood, and empathy in comparison with participants who received placebo.
MDMA was “extremely effective, particularly for a subpopulation that ordinarily does not respond well to conventional treatment,” study coinvestigator Bessel van der Kolk, MD, professor of psychiatry at Boston University School of Medicine, told delegates attending the virtual European Psychiatric Association (EPA) 2021 Congress.
Growing interest
, particularly because failure rates with most available evidence-based treatments have been relatively high.
As previously reported by this news organization, in 2017, the U.S. Food and Drug Administration approved the trial design of Dr. van der Kolk’s and colleagues’ MAPP1 study after granting MDMA breakthrough designation.
The MAPP1 investigators assessed 90 patients with PTSD (mean age, 41 years; 77% White; 66% women) from 50 sites. For the majority of patients (84%), trauma history was developmental. “In other words, trauma [occurred] very early in life, usually at the hands of their own caregivers,” Dr. van der Kolk noted.
In addition, 18% of the patients were veterans, and 12% had combat exposure. The average duration of PTSD before enrollment was 18 years. All patients underwent screening and three preparatory psychotherapy sessions at enrollment.
Participants were randomly assigned to receive MDMA 80 mg or 120 mg (n = 46) or placebo (n = 44) followed by three integrative psychotherapy sessions lasting a total of 8 hours. A supplemental dose of 40 or 60 mg of MDMA could be administered from 1.5 to 2 hours after the first dose.
The patients stayed in the laboratory on the evening of the treatment session and attended a debriefing the next morning. The session was repeated a month later and again a month after that. In between, patients had telephone contact with the raters, who were blinded to the treatment received.
Follow-up assessments were conducted 2 months after the third treatment session and again at 12 months. The primary outcome measure was change in Clinician Administered PTSD Scale for DSM 5 (CAPS-5) score from baseline.
‘Dramatic improvement’
Results showed that both the MDMA and placebo groups experienced a statistically significant improvement in PTSD symptoms, “but MDMA had a dramatically significant improvement, with an effect size of over 0.9,” Dr. van der Kolk said.
The MDMA group also reported enhanced mood and well-being, increased responsiveness to emotional and sensory stimuli, a greater sense of closeness to other people, and a greater feeling of empathy.
Patients also reported having heightened openness, “and clearly the issue of empathy for themselves and others was a very large part of the process,” said Dr. van der Kolk.
“But for me, the most interesting part of the study is that the Adverse Childhood Experiences scale had no effect,” he noted. In other words, “the amount of childhood adverse experiences did not predict outcomes, which was very surprising because usually those patients are very treatment resistant.”
Dr. van der Kolk added that the dissociative subtype of PTSD was first described in the DSM-5 and that patients are “notoriously unresponsive to most unconventional treatments.”
In the current study, 13 patients met the criteria for the subtype, and investigators found they “did better than people with classical PTSD,” Dr. van der Kolk said. He added that this is a “very, very important finding.”
Carefully controlled
Overall, 82% of patients reported a significant improvement by the end of the study; 56% reported that they no longer had PTSD.
In addition, 67% of patients no longer met diagnostic criteria for PTSD. These included patients who had crossed over to active treatment from the placebo group.
Eleven patients (12%) experienced relapse by 12 months; in nine of the cases, this was due to the presence of additional stressors.
There were “very few adverse side effects” during the study, Dr. van der Kolk noted. In addition, “there were really no serious mental side effects,” despite the patients’ “opening up so much very painful material,” he added.
The most common adverse events among the MDMA group were muscle tightness (63%), decreased appetite (52%), nausea (30%), hyperhidrosis (20%), and feeling cold (20%). These effects were “quite small [and] the sort of side effects you would expect in response to an amphetamine substance like MDMA,” said Dr. van der Kolk.
“An important reason why we think the side effect profile is so good is because the study was extremely carefully done, very carefully controlled,” he added. “There was a great deal of support, [and] we paid an enormous amount of attention to creating a very safe context in which this drug was being used.”
However, he expressed concern that “as people see the very good results, they may skimp a little bit on the creation of the context and not have as careful a psychotherapy protocol as we had here.”
‘On the right track’
Commenting on the findings for this news organization, David Nutt, MD, PhD, Edmond J. Safra Professor of Neuropsychopharmacology, Imperial College London, said the results are proof that the investigators’ “earlier smaller trials of MDMA were on the right track.”
“This larger and multicenter trial shows that MDMA therapy can be broadened into newer research groups, which augurs well for the much larger rollout that will be required once it gets a license,” said Dr. Nutt, who was not involved with the research.
He added, “the prior evidence of the safety of MDMA has [now] been confirmed.”
The study represents an “important step in the path to the clinical use of MDMA for PTSD,” Dr. Nutt said.
The study was sponsored by the Multidisciplinary Association for Psychedelic Studies. The investigators and Dr. Nutt have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Adding 3,4-methylenedioxymethamphetamine (MDMA) to integrative psychotherapy may significantly improve symptoms and well-being for patients with severe posttraumatic stress disorder, including those with the dissociative subtype, new research suggests.
MAPP1 is the first phase 3 randomized controlled trial of MDMA-assisted therapy in this population. Participants who received the active treatment showed greater improvement in PTSD symptoms, mood, and empathy in comparison with participants who received placebo.
MDMA was “extremely effective, particularly for a subpopulation that ordinarily does not respond well to conventional treatment,” study coinvestigator Bessel van der Kolk, MD, professor of psychiatry at Boston University School of Medicine, told delegates attending the virtual European Psychiatric Association (EPA) 2021 Congress.
Growing interest
, particularly because failure rates with most available evidence-based treatments have been relatively high.
As previously reported by this news organization, in 2017, the U.S. Food and Drug Administration approved the trial design of Dr. van der Kolk’s and colleagues’ MAPP1 study after granting MDMA breakthrough designation.
The MAPP1 investigators assessed 90 patients with PTSD (mean age, 41 years; 77% White; 66% women) from 50 sites. For the majority of patients (84%), trauma history was developmental. “In other words, trauma [occurred] very early in life, usually at the hands of their own caregivers,” Dr. van der Kolk noted.
In addition, 18% of the patients were veterans, and 12% had combat exposure. The average duration of PTSD before enrollment was 18 years. All patients underwent screening and three preparatory psychotherapy sessions at enrollment.
Participants were randomly assigned to receive MDMA 80 mg or 120 mg (n = 46) or placebo (n = 44) followed by three integrative psychotherapy sessions lasting a total of 8 hours. A supplemental dose of 40 or 60 mg of MDMA could be administered from 1.5 to 2 hours after the first dose.
The patients stayed in the laboratory on the evening of the treatment session and attended a debriefing the next morning. The session was repeated a month later and again a month after that. In between, patients had telephone contact with the raters, who were blinded to the treatment received.
Follow-up assessments were conducted 2 months after the third treatment session and again at 12 months. The primary outcome measure was change in Clinician Administered PTSD Scale for DSM 5 (CAPS-5) score from baseline.
‘Dramatic improvement’
Results showed that both the MDMA and placebo groups experienced a statistically significant improvement in PTSD symptoms, “but MDMA had a dramatically significant improvement, with an effect size of over 0.9,” Dr. van der Kolk said.
The MDMA group also reported enhanced mood and well-being, increased responsiveness to emotional and sensory stimuli, a greater sense of closeness to other people, and a greater feeling of empathy.
Patients also reported having heightened openness, “and clearly the issue of empathy for themselves and others was a very large part of the process,” said Dr. van der Kolk.
“But for me, the most interesting part of the study is that the Adverse Childhood Experiences scale had no effect,” he noted. In other words, “the amount of childhood adverse experiences did not predict outcomes, which was very surprising because usually those patients are very treatment resistant.”
Dr. van der Kolk added that the dissociative subtype of PTSD was first described in the DSM-5 and that patients are “notoriously unresponsive to most unconventional treatments.”
In the current study, 13 patients met the criteria for the subtype, and investigators found they “did better than people with classical PTSD,” Dr. van der Kolk said. He added that this is a “very, very important finding.”
Carefully controlled
Overall, 82% of patients reported a significant improvement by the end of the study; 56% reported that they no longer had PTSD.
In addition, 67% of patients no longer met diagnostic criteria for PTSD. These included patients who had crossed over to active treatment from the placebo group.
Eleven patients (12%) experienced relapse by 12 months; in nine of the cases, this was due to the presence of additional stressors.
There were “very few adverse side effects” during the study, Dr. van der Kolk noted. In addition, “there were really no serious mental side effects,” despite the patients’ “opening up so much very painful material,” he added.
The most common adverse events among the MDMA group were muscle tightness (63%), decreased appetite (52%), nausea (30%), hyperhidrosis (20%), and feeling cold (20%). These effects were “quite small [and] the sort of side effects you would expect in response to an amphetamine substance like MDMA,” said Dr. van der Kolk.
“An important reason why we think the side effect profile is so good is because the study was extremely carefully done, very carefully controlled,” he added. “There was a great deal of support, [and] we paid an enormous amount of attention to creating a very safe context in which this drug was being used.”
However, he expressed concern that “as people see the very good results, they may skimp a little bit on the creation of the context and not have as careful a psychotherapy protocol as we had here.”
‘On the right track’
Commenting on the findings for this news organization, David Nutt, MD, PhD, Edmond J. Safra Professor of Neuropsychopharmacology, Imperial College London, said the results are proof that the investigators’ “earlier smaller trials of MDMA were on the right track.”
“This larger and multicenter trial shows that MDMA therapy can be broadened into newer research groups, which augurs well for the much larger rollout that will be required once it gets a license,” said Dr. Nutt, who was not involved with the research.
He added, “the prior evidence of the safety of MDMA has [now] been confirmed.”
The study represents an “important step in the path to the clinical use of MDMA for PTSD,” Dr. Nutt said.
The study was sponsored by the Multidisciplinary Association for Psychedelic Studies. The investigators and Dr. Nutt have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
New guidelines on antibiotic prescribing focus on shorter courses
An antibiotic course of 5 days is usually just as effective as longer courses but with fewer side effects and decreased overall antibiotic exposure for a number of common bacterial conditions, according to new clinical guidelines published by the American College of Physicians.
The guidelines focus on treatment of uncomplicated cases involving pneumonia, urinary tract infections (UTIs), cellulitis, chronic obstructive pulmonary disease (COPD) exacerbations, and acute bronchitis. The goal of the guidelines is to continue improving antibiotic stewardship given the increasing threat of antibiotic resistance and the adverse effects of antibiotics.
“Any use of antibiotics (including necessary use) has downstream effects outside of treating infection,” Dawn Nolt, MD, MPH, a professor of pediatric infection disease at Oregon Health & Science University, Portland, said in an interview. Dr. Nolt was not involved in developing these guidelines. “Undesirable outcomes include allergic reactions, diarrhea, and antibiotic-resistant bacteria. When we reduce unnecessary antibiotic, we reduce undesirable outcomes,” she said.
According to background information in the paper, 1 in 10 patients receives an antibiotic prescription during visits, yet nearly a third of these (30%) are unnecessary and last too long, especially for sinusitis and bronchitis. Meanwhile, overuse of antibiotics, particularly broad-spectrum ones, leads to resistance and adverse effects in up to 20% of patients.
“Prescribing practices can vary based on the type of provider, the setting where the antibiotic is being prescribed, what geographic area you are looking at, the medical reason for which the antibiotic is being prescribed, the actual germ being targeted, and the type of patient,” Dr. Nolt said. “But this variability can be reduced when prescribing providers are aware and follow best practice standards as through this article.”
The new ACP guidelines are a distillation of recommendations from preexisting infectious disease organizations, Dr. Nolt said, but aimed specifically at those practicing internal medicine.
“We define appropriate antibiotic use as prescribing the right antibiotic at the right dose for the right duration for a specific condition,” Rachael A. Lee, MD, MSPH, of the University of Alabama at Birmingham, and colleagues wrote in the article detailing the new guidelines. “Despite evidence and guidelines supporting shorter durations of antibiotic use, many physicians do not prescribe short-course therapy, frequently defaulting to 10-day courses regardless of the condition.”
The reasons for this default response vary. Though some clinicians prescribe longer courses specifically to prevent antibiotic resistance, no evidence shows that continuing to take antibiotics after symptoms have resolved actually reduces likelihood of resistance, the authors noted.
“In fact, resistance is a documented side effect of prolonged antibiotic use due to natural selection pressure,” they wrote.
Another common reason is habit.
“This was the ‘conventional wisdom’ for so long, just trying to make sure all bacteria causing the infection were completely eradicated, with no stragglers that had been exposed to the antibiotic but were not gone and now could evolve into resistant organisms,” Jacqueline W. Fincher, MD, a primary care physician and president of the ACP, said in an interview. “While antibiotic stewardship has been very important for over a decade, we now have more recent head-to-head studies/data showing that, in these four conditions, shorter courses of treatment are just as efficacious with less side effects and adverse events.”
The researchers reviewed all existing clinical guidelines related to bronchitis with COPD exacerbations, community-acquired pneumonia, UTIs, and cellulitis, as well as any other relevant studies in the literature. Although they did not conduct a formal systematic review, they compiled the guidelines specifically for all internists, family physicians and other clinicians caring for patients with these conditions.
“Although most patients with these infections will be seen in the outpatient setting, these best-practice advice statements also apply to patients who present in the inpatient setting,” the authors wrote. They also note the importance of ensuring the patient has the correct diagnosis and appropriate corresponding antibiotic prescription. “If a patient is not improving with appropriate antibiotics, it is important for the clinician to reassess for other causes of symptoms rather than defaulting to a longer duration of antibiotic therapy,” they wrote, calling a longer course “the exception and not the rule.”
Acute bronchitis with COPD exacerbations
Antibiotic treatment for COPD exacerbations and acute uncomplicated bronchitis with signs of a bacterial infection should last no longer than 5 days. The authors define this condition as an acute respiratory infection with a normal chest x-ray, most often caused by a virus. Although patients with bronchitis do not automatically need antibiotics if there’s no evidence of pneumonia, the authors did advise antibiotics in cases involving COPD and a high likelihood of bacterial infection. Clinicians should base their choice of antibiotics on the most common bacterial etiology: Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis. Ideal candidates for therapy may include aminopenicillin with clavulanic acid, a macrolide, or a tetracycline.
Community-acquired pneumonia
The initial course of antibiotics should be at least 5 days for pneumonia and only extended after considering validated evidence of the patient’s clinical stability, such as resuming normal vital signs, mental activity, and the ability to eat. Multiple randomized, controlled trials have shown no improved benefit from longer courses, though longer courses are linked to increased adverse events and mortality.
Again, antibiotics used should “cover common pathogens, such as S. pneumoniae, H. influenzae, Mycoplasma pneumoniae, and Staphylococcus aureus, and atypical pathogens, such as Legionella species,” the authors wrote. Options include “amoxicillin, doxycycline, or a macrolide for healthy adults or a beta-lactam with a macrolide or a respiratory fluoroquinolone in patients with comorbidities.”
UTIs: Uncomplicated cystitis and pyelonephritis
For women’s bacterial cystitis – 75% of which is caused by Escherichia coli – the guidelines recommend nitrofurantoin for 5 days, trimethoprim-sulfamethoxazole for 3 days, or fosfomycin as a single dose. For uncomplicated pyelonephritis in both men and women, clinicians can consider fluoroquinolones for 5-7 days or trimethoprim-sulfamethoxazole for 14 days, depending on antibiotic susceptibility.
This recommendation does not include UTIs in women who are pregnant or UTIs with other functional abnormalities present, such as obstruction. The authors also intentionally left out acute bacterial prostatitis because of its complexity and how long it can take to treat.
Cellulitis
MRSA, which has been increasing in prevalence, is a leading cause of skin and soft-tissue infections, such as necrotizing infections, cellulitis, and erysipelas. Unless the patient has penetrating trauma, evidence of MRSA infection elsewhere, injection drug use, nasal colonization of MRSA, or systemic inflammatory response syndrome, the guidelines recommend a 5- to 6-day course of cephalosporin, penicillin, or clindamycin, extended only if the infection has not improved in 5 days. Further research can narrow down the most appropriate treatment course.
This guidance does not apply to purulent cellulitis, such as conditions with abscesses, furuncles, or carbuncles that typically require incision and drainage.
Continuing to get the message out
Dr. Fincher emphasized the importance of continuing to disseminate messaging for clinicians about reducing unnecessary antibiotic use.
“In medicine we are constantly bombarded with new information. It is those patients and disease states that we see and treat every day that are especially important for us as physicians and other clinicians to keep our skills and knowledge base up to date when it comes to use of antibiotics,” Dr. Fincher said in an interview. “We just need to continue to educate and push out the data, guidelines, and recommendations.”
Dr. Nolt added that it’s important to emphasize how to translate these national recommendations into local practices since local guidance can also raise awareness and encourage local compliance.
Other strategies for reducing overuse of antibiotics “include restriction on antibiotics available at health care systems (formulary restriction), not allowing use of antibiotics unless there is discussion about the patient’s case (preauthorization), and reviewing cases of patients on antibiotics and advising on next steps (prospective audit and feedback),” she said.
The research was funded by the ACP. Dr. Lee has received personal fees from this news organization and Prime Education. Dr. Fincher owns stock in Johnson & Johnson and Procter and Gamble. Dr. Nolt and the article’s coauthors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
An antibiotic course of 5 days is usually just as effective as longer courses but with fewer side effects and decreased overall antibiotic exposure for a number of common bacterial conditions, according to new clinical guidelines published by the American College of Physicians.
The guidelines focus on treatment of uncomplicated cases involving pneumonia, urinary tract infections (UTIs), cellulitis, chronic obstructive pulmonary disease (COPD) exacerbations, and acute bronchitis. The goal of the guidelines is to continue improving antibiotic stewardship given the increasing threat of antibiotic resistance and the adverse effects of antibiotics.
“Any use of antibiotics (including necessary use) has downstream effects outside of treating infection,” Dawn Nolt, MD, MPH, a professor of pediatric infection disease at Oregon Health & Science University, Portland, said in an interview. Dr. Nolt was not involved in developing these guidelines. “Undesirable outcomes include allergic reactions, diarrhea, and antibiotic-resistant bacteria. When we reduce unnecessary antibiotic, we reduce undesirable outcomes,” she said.
According to background information in the paper, 1 in 10 patients receives an antibiotic prescription during visits, yet nearly a third of these (30%) are unnecessary and last too long, especially for sinusitis and bronchitis. Meanwhile, overuse of antibiotics, particularly broad-spectrum ones, leads to resistance and adverse effects in up to 20% of patients.
“Prescribing practices can vary based on the type of provider, the setting where the antibiotic is being prescribed, what geographic area you are looking at, the medical reason for which the antibiotic is being prescribed, the actual germ being targeted, and the type of patient,” Dr. Nolt said. “But this variability can be reduced when prescribing providers are aware and follow best practice standards as through this article.”
The new ACP guidelines are a distillation of recommendations from preexisting infectious disease organizations, Dr. Nolt said, but aimed specifically at those practicing internal medicine.
“We define appropriate antibiotic use as prescribing the right antibiotic at the right dose for the right duration for a specific condition,” Rachael A. Lee, MD, MSPH, of the University of Alabama at Birmingham, and colleagues wrote in the article detailing the new guidelines. “Despite evidence and guidelines supporting shorter durations of antibiotic use, many physicians do not prescribe short-course therapy, frequently defaulting to 10-day courses regardless of the condition.”
The reasons for this default response vary. Though some clinicians prescribe longer courses specifically to prevent antibiotic resistance, no evidence shows that continuing to take antibiotics after symptoms have resolved actually reduces likelihood of resistance, the authors noted.
“In fact, resistance is a documented side effect of prolonged antibiotic use due to natural selection pressure,” they wrote.
Another common reason is habit.
“This was the ‘conventional wisdom’ for so long, just trying to make sure all bacteria causing the infection were completely eradicated, with no stragglers that had been exposed to the antibiotic but were not gone and now could evolve into resistant organisms,” Jacqueline W. Fincher, MD, a primary care physician and president of the ACP, said in an interview. “While antibiotic stewardship has been very important for over a decade, we now have more recent head-to-head studies/data showing that, in these four conditions, shorter courses of treatment are just as efficacious with less side effects and adverse events.”
The researchers reviewed all existing clinical guidelines related to bronchitis with COPD exacerbations, community-acquired pneumonia, UTIs, and cellulitis, as well as any other relevant studies in the literature. Although they did not conduct a formal systematic review, they compiled the guidelines specifically for all internists, family physicians and other clinicians caring for patients with these conditions.
“Although most patients with these infections will be seen in the outpatient setting, these best-practice advice statements also apply to patients who present in the inpatient setting,” the authors wrote. They also note the importance of ensuring the patient has the correct diagnosis and appropriate corresponding antibiotic prescription. “If a patient is not improving with appropriate antibiotics, it is important for the clinician to reassess for other causes of symptoms rather than defaulting to a longer duration of antibiotic therapy,” they wrote, calling a longer course “the exception and not the rule.”
Acute bronchitis with COPD exacerbations
Antibiotic treatment for COPD exacerbations and acute uncomplicated bronchitis with signs of a bacterial infection should last no longer than 5 days. The authors define this condition as an acute respiratory infection with a normal chest x-ray, most often caused by a virus. Although patients with bronchitis do not automatically need antibiotics if there’s no evidence of pneumonia, the authors did advise antibiotics in cases involving COPD and a high likelihood of bacterial infection. Clinicians should base their choice of antibiotics on the most common bacterial etiology: Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis. Ideal candidates for therapy may include aminopenicillin with clavulanic acid, a macrolide, or a tetracycline.
Community-acquired pneumonia
The initial course of antibiotics should be at least 5 days for pneumonia and only extended after considering validated evidence of the patient’s clinical stability, such as resuming normal vital signs, mental activity, and the ability to eat. Multiple randomized, controlled trials have shown no improved benefit from longer courses, though longer courses are linked to increased adverse events and mortality.
Again, antibiotics used should “cover common pathogens, such as S. pneumoniae, H. influenzae, Mycoplasma pneumoniae, and Staphylococcus aureus, and atypical pathogens, such as Legionella species,” the authors wrote. Options include “amoxicillin, doxycycline, or a macrolide for healthy adults or a beta-lactam with a macrolide or a respiratory fluoroquinolone in patients with comorbidities.”
UTIs: Uncomplicated cystitis and pyelonephritis
For women’s bacterial cystitis – 75% of which is caused by Escherichia coli – the guidelines recommend nitrofurantoin for 5 days, trimethoprim-sulfamethoxazole for 3 days, or fosfomycin as a single dose. For uncomplicated pyelonephritis in both men and women, clinicians can consider fluoroquinolones for 5-7 days or trimethoprim-sulfamethoxazole for 14 days, depending on antibiotic susceptibility.
This recommendation does not include UTIs in women who are pregnant or UTIs with other functional abnormalities present, such as obstruction. The authors also intentionally left out acute bacterial prostatitis because of its complexity and how long it can take to treat.
Cellulitis
MRSA, which has been increasing in prevalence, is a leading cause of skin and soft-tissue infections, such as necrotizing infections, cellulitis, and erysipelas. Unless the patient has penetrating trauma, evidence of MRSA infection elsewhere, injection drug use, nasal colonization of MRSA, or systemic inflammatory response syndrome, the guidelines recommend a 5- to 6-day course of cephalosporin, penicillin, or clindamycin, extended only if the infection has not improved in 5 days. Further research can narrow down the most appropriate treatment course.
This guidance does not apply to purulent cellulitis, such as conditions with abscesses, furuncles, or carbuncles that typically require incision and drainage.
Continuing to get the message out
Dr. Fincher emphasized the importance of continuing to disseminate messaging for clinicians about reducing unnecessary antibiotic use.
“In medicine we are constantly bombarded with new information. It is those patients and disease states that we see and treat every day that are especially important for us as physicians and other clinicians to keep our skills and knowledge base up to date when it comes to use of antibiotics,” Dr. Fincher said in an interview. “We just need to continue to educate and push out the data, guidelines, and recommendations.”
Dr. Nolt added that it’s important to emphasize how to translate these national recommendations into local practices since local guidance can also raise awareness and encourage local compliance.
Other strategies for reducing overuse of antibiotics “include restriction on antibiotics available at health care systems (formulary restriction), not allowing use of antibiotics unless there is discussion about the patient’s case (preauthorization), and reviewing cases of patients on antibiotics and advising on next steps (prospective audit and feedback),” she said.
The research was funded by the ACP. Dr. Lee has received personal fees from this news organization and Prime Education. Dr. Fincher owns stock in Johnson & Johnson and Procter and Gamble. Dr. Nolt and the article’s coauthors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
An antibiotic course of 5 days is usually just as effective as longer courses but with fewer side effects and decreased overall antibiotic exposure for a number of common bacterial conditions, according to new clinical guidelines published by the American College of Physicians.
The guidelines focus on treatment of uncomplicated cases involving pneumonia, urinary tract infections (UTIs), cellulitis, chronic obstructive pulmonary disease (COPD) exacerbations, and acute bronchitis. The goal of the guidelines is to continue improving antibiotic stewardship given the increasing threat of antibiotic resistance and the adverse effects of antibiotics.
“Any use of antibiotics (including necessary use) has downstream effects outside of treating infection,” Dawn Nolt, MD, MPH, a professor of pediatric infection disease at Oregon Health & Science University, Portland, said in an interview. Dr. Nolt was not involved in developing these guidelines. “Undesirable outcomes include allergic reactions, diarrhea, and antibiotic-resistant bacteria. When we reduce unnecessary antibiotic, we reduce undesirable outcomes,” she said.
According to background information in the paper, 1 in 10 patients receives an antibiotic prescription during visits, yet nearly a third of these (30%) are unnecessary and last too long, especially for sinusitis and bronchitis. Meanwhile, overuse of antibiotics, particularly broad-spectrum ones, leads to resistance and adverse effects in up to 20% of patients.
“Prescribing practices can vary based on the type of provider, the setting where the antibiotic is being prescribed, what geographic area you are looking at, the medical reason for which the antibiotic is being prescribed, the actual germ being targeted, and the type of patient,” Dr. Nolt said. “But this variability can be reduced when prescribing providers are aware and follow best practice standards as through this article.”
The new ACP guidelines are a distillation of recommendations from preexisting infectious disease organizations, Dr. Nolt said, but aimed specifically at those practicing internal medicine.
“We define appropriate antibiotic use as prescribing the right antibiotic at the right dose for the right duration for a specific condition,” Rachael A. Lee, MD, MSPH, of the University of Alabama at Birmingham, and colleagues wrote in the article detailing the new guidelines. “Despite evidence and guidelines supporting shorter durations of antibiotic use, many physicians do not prescribe short-course therapy, frequently defaulting to 10-day courses regardless of the condition.”
The reasons for this default response vary. Though some clinicians prescribe longer courses specifically to prevent antibiotic resistance, no evidence shows that continuing to take antibiotics after symptoms have resolved actually reduces likelihood of resistance, the authors noted.
“In fact, resistance is a documented side effect of prolonged antibiotic use due to natural selection pressure,” they wrote.
Another common reason is habit.
“This was the ‘conventional wisdom’ for so long, just trying to make sure all bacteria causing the infection were completely eradicated, with no stragglers that had been exposed to the antibiotic but were not gone and now could evolve into resistant organisms,” Jacqueline W. Fincher, MD, a primary care physician and president of the ACP, said in an interview. “While antibiotic stewardship has been very important for over a decade, we now have more recent head-to-head studies/data showing that, in these four conditions, shorter courses of treatment are just as efficacious with less side effects and adverse events.”
The researchers reviewed all existing clinical guidelines related to bronchitis with COPD exacerbations, community-acquired pneumonia, UTIs, and cellulitis, as well as any other relevant studies in the literature. Although they did not conduct a formal systematic review, they compiled the guidelines specifically for all internists, family physicians and other clinicians caring for patients with these conditions.
“Although most patients with these infections will be seen in the outpatient setting, these best-practice advice statements also apply to patients who present in the inpatient setting,” the authors wrote. They also note the importance of ensuring the patient has the correct diagnosis and appropriate corresponding antibiotic prescription. “If a patient is not improving with appropriate antibiotics, it is important for the clinician to reassess for other causes of symptoms rather than defaulting to a longer duration of antibiotic therapy,” they wrote, calling a longer course “the exception and not the rule.”
Acute bronchitis with COPD exacerbations
Antibiotic treatment for COPD exacerbations and acute uncomplicated bronchitis with signs of a bacterial infection should last no longer than 5 days. The authors define this condition as an acute respiratory infection with a normal chest x-ray, most often caused by a virus. Although patients with bronchitis do not automatically need antibiotics if there’s no evidence of pneumonia, the authors did advise antibiotics in cases involving COPD and a high likelihood of bacterial infection. Clinicians should base their choice of antibiotics on the most common bacterial etiology: Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis. Ideal candidates for therapy may include aminopenicillin with clavulanic acid, a macrolide, or a tetracycline.
Community-acquired pneumonia
The initial course of antibiotics should be at least 5 days for pneumonia and only extended after considering validated evidence of the patient’s clinical stability, such as resuming normal vital signs, mental activity, and the ability to eat. Multiple randomized, controlled trials have shown no improved benefit from longer courses, though longer courses are linked to increased adverse events and mortality.
Again, antibiotics used should “cover common pathogens, such as S. pneumoniae, H. influenzae, Mycoplasma pneumoniae, and Staphylococcus aureus, and atypical pathogens, such as Legionella species,” the authors wrote. Options include “amoxicillin, doxycycline, or a macrolide for healthy adults or a beta-lactam with a macrolide or a respiratory fluoroquinolone in patients with comorbidities.”
UTIs: Uncomplicated cystitis and pyelonephritis
For women’s bacterial cystitis – 75% of which is caused by Escherichia coli – the guidelines recommend nitrofurantoin for 5 days, trimethoprim-sulfamethoxazole for 3 days, or fosfomycin as a single dose. For uncomplicated pyelonephritis in both men and women, clinicians can consider fluoroquinolones for 5-7 days or trimethoprim-sulfamethoxazole for 14 days, depending on antibiotic susceptibility.
This recommendation does not include UTIs in women who are pregnant or UTIs with other functional abnormalities present, such as obstruction. The authors also intentionally left out acute bacterial prostatitis because of its complexity and how long it can take to treat.
Cellulitis
MRSA, which has been increasing in prevalence, is a leading cause of skin and soft-tissue infections, such as necrotizing infections, cellulitis, and erysipelas. Unless the patient has penetrating trauma, evidence of MRSA infection elsewhere, injection drug use, nasal colonization of MRSA, or systemic inflammatory response syndrome, the guidelines recommend a 5- to 6-day course of cephalosporin, penicillin, or clindamycin, extended only if the infection has not improved in 5 days. Further research can narrow down the most appropriate treatment course.
This guidance does not apply to purulent cellulitis, such as conditions with abscesses, furuncles, or carbuncles that typically require incision and drainage.
Continuing to get the message out
Dr. Fincher emphasized the importance of continuing to disseminate messaging for clinicians about reducing unnecessary antibiotic use.
“In medicine we are constantly bombarded with new information. It is those patients and disease states that we see and treat every day that are especially important for us as physicians and other clinicians to keep our skills and knowledge base up to date when it comes to use of antibiotics,” Dr. Fincher said in an interview. “We just need to continue to educate and push out the data, guidelines, and recommendations.”
Dr. Nolt added that it’s important to emphasize how to translate these national recommendations into local practices since local guidance can also raise awareness and encourage local compliance.
Other strategies for reducing overuse of antibiotics “include restriction on antibiotics available at health care systems (formulary restriction), not allowing use of antibiotics unless there is discussion about the patient’s case (preauthorization), and reviewing cases of patients on antibiotics and advising on next steps (prospective audit and feedback),” she said.
The research was funded by the ACP. Dr. Lee has received personal fees from this news organization and Prime Education. Dr. Fincher owns stock in Johnson & Johnson and Procter and Gamble. Dr. Nolt and the article’s coauthors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers stress importance of second COVID-19 vaccine dose for infliximab users
Patients being treated with infliximab had weakened immune responses to the first dose of the ChAdOx1 nCoV-19 (Oxford/AstraZeneca) and BNT162b2 (Pfizer/BioNTech) vaccines, compared with patients on vedolizumab (Entyvio), although a very significant number of patients from both groups seroconverted after their second dose, according to a new U.K. study of patients with inflammatory bowel disease (IBD).
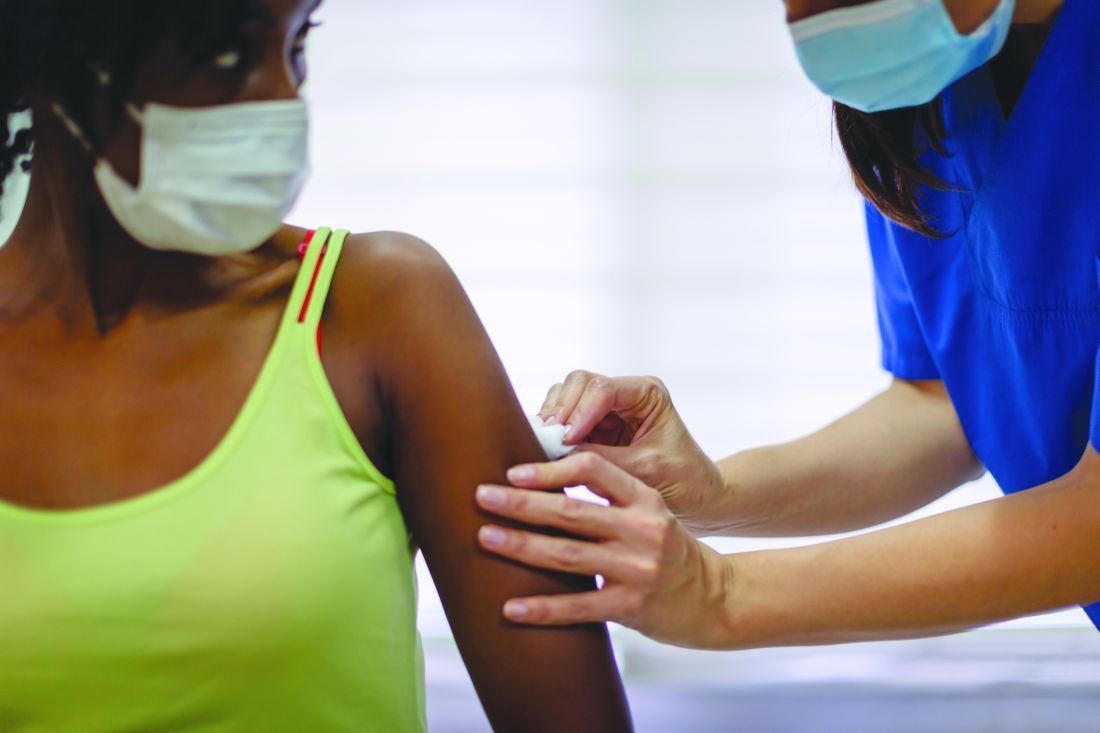
“Antibody testing and adapted vaccine schedules should be considered to protect these at-risk patients,” Nicholas A. Kennedy, PhD, MBBS, of the University of Exeter (England) and colleagues wrote in a preprint published March 29 on MedRxiv.
Infliximab is an anti–tumor necrosis factor (anti-TNF) monoclonal antibody that’s approved to treat adult and pediatric Crohn’s disease and ulcerative colitis, as well as rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis, whereas vedolizumab, a gut selective anti-integrin alpha4beta7 monoclonal antibody that is not associated with impaired systemic immune responses, is approved to treat Crohn’s disease and ulcerative colitis in adults.
A previous study from Kennedy and colleagues revealed that IBD patients on infliximab showed a weakened COVID-19 antibody response compared with patients on vedolizumab. To determine if treatment with anti-TNF drugs impacted the efficacy of the first shot of these two-dose COVID-19 vaccines, the researchers used data from the CLARITY IBD study to assess 865 infliximab- and 428 vedolizumab-treated participants without evidence of prior SARS-CoV-2 infection who had received uninterrupted biologic therapy since being recruited between Sept. 22 and Dec. 23, 2020.
In the 3-10 weeks after initial vaccination, geometric mean concentrations for SARS-CoV-2 anti-spike protein receptor-binding protein antibodies were lower in patients on infliximab, compared with patients on vedolizumab for both the Pfizer (6.0 U/mL [5.9] versus 28.8 U/mL [5.4], P < .0001) and AstraZeneca (4.7 U/mL [4.9] versus 13.8 U/mL [5.9]; P < .0001) vaccines. The researchers’ multivariable models reinforced those findings, with antibody concentrations lower in infliximab-treated patients for both the Pfizer (fold change, 0.29; 95% confidence interval, 0.21-0.40; P < .0001) and AstraZeneca (FC, 0.39; 95% CI, 0.30-0.51; P < .0001) vaccines.
After second doses of the two-dose Pfizer vaccine, 85% of patients on infliximab and 86% of patients on vedolizumab seroconverted (P = .68); similarly high seroconversion rates were seen in patients who had been infected with SARS-CoV-2 prior to receiving either vaccine. Several patient characteristics were associated with lower antibody concentrations regardless of vaccine type: being 60 years or older, use of immunomodulators, having Crohn’s disease, and being a smoker. Alternatively, non-White ethnicity was associated with higher antibody concentrations.
Evidence has ‘unclear clinical significance’
“These data, which require peer review, do not change my opinion on the safety and efficacy of COVID-19 vaccines in patients taking TNF inhibitors such as infliximab as monotherapy for the treatment of psoriatic disease,” Joel M. Gelfand MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, said in an interview.

“First, two peer-reviewed studies found good antibody response in patients on TNF inhibitors receiving COVID-19 vaccines (doi: 10.1136/annrheumdis-2021-220289; 10.1136/annrheumdis-2021-220272). Second, antibody responses were robust in the small cohort that received the second dose of a COVID-19 vaccine. We already know that, for the two messenger RNA-based vaccines available under emergency use authorization in the U.S., a second dose is required for optimal efficacy. Thus, evidence of a reduced antibody response after just one dose is of unclear clinical significance. Third, antibody responses are only a surrogate marker, and a low antibody response doesn’t necessarily mean the patient will not be protected by the vaccine.”
Focus on the second dose of a two-dose regimen
“Tell me about the response in people who got both doses of a vaccine that you’re supposed to get both doses of,” Jeffrey Curtis, MD, professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama at Birmingham, said in an interview. “The number of patients in that subset was small [n = 27] but in my opinion that’s the most clinically relevant analysis and the one that patients and clinicians want answered.”

He also emphasized the uncertainty around what ‘protection’ means in these early days of studying COVID-19 vaccine responses. “You can define seroprotection or seroconversion as some absolute level of an antibody response, but if you want to say ‘Mrs. Smith, your antibody level was X,’ on whatever arbitrary scale with whoever’s arbitrary lab test, nobody actually knows that Mrs. Smith is now protected from SARS-CoV-2, or how protected,” he said.
“What is not terribly controversial is: If you can’t detect antibodies, the vaccine didn’t ‘take,’ if you will. But if I tell you that the mean antibody level was X with one drug and then 2X with another drug, does that mean that you’re twice as protected? We don’t know that. I’m fearful that people are looking at these studies and thinking that more is better. It might be, but we don’t know that to be true.”
Debating the cause of weakened immune responses
“The biological plausibility of being on an anti-TNF affecting your immune reaction to a messenger RNA or even a replication-deficient viral vector vaccine doesn’t make sense,” David T. Rubin, MD, professor of medicine at the University of Chicago and chair of the National Scientific Advisory Committee of the Crohn’s and Colitis Foundation, said in an interview.

“I’m sure immunologists may differ with me on this, but given what we have come to appreciate about these vaccine mechanisms, this finding doesn’t make intuitive sense. So we need to make sure that, when this happens, we look to the next studies and try to understand, was there any other confounder that may have resulted in these findings that was not adequately adjusted for or addressed in some other way?
“When you have a study of this size, you argue, ‘Because it’s so large, any effect that was seen must be real,’ ” he added. “Alternatively, to have a study of this size, by its very nature you are limited in being able to control for certain other factors or differences between the groups.”
That said, he commended the authors for their study and acknowledged the potential questions it raises about the single-shot Johnson & Johnson vaccine. “If you only get one and you’re on infliximab, this study implies that maybe that’s not enough,” he said. “Despite the fact that Johnson & Johnson was approved as a single dose, it may be necessary to think about it as the first of two, or maybe it’s not the preferred vaccine in this group of patients.”
The study was supported by the Royal Devon and Exeter and Hull University Hospital Foundation NHS Trusts and unrestricted educational grants from Biogen (Switzerland), Celltrion Healthcare (South Korea), Galapagos NV (Belgium), and F. Hoffmann-La Roche (Switzerland). The authors acknowledged numerous potential conflicts of interest, including receiving grants, personal fees, and nonfinancial support from various pharmaceutical companies.
Patients being treated with infliximab had weakened immune responses to the first dose of the ChAdOx1 nCoV-19 (Oxford/AstraZeneca) and BNT162b2 (Pfizer/BioNTech) vaccines, compared with patients on vedolizumab (Entyvio), although a very significant number of patients from both groups seroconverted after their second dose, according to a new U.K. study of patients with inflammatory bowel disease (IBD).
“Antibody testing and adapted vaccine schedules should be considered to protect these at-risk patients,” Nicholas A. Kennedy, PhD, MBBS, of the University of Exeter (England) and colleagues wrote in a preprint published March 29 on MedRxiv.
Infliximab is an anti–tumor necrosis factor (anti-TNF) monoclonal antibody that’s approved to treat adult and pediatric Crohn’s disease and ulcerative colitis, as well as rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis, whereas vedolizumab, a gut selective anti-integrin alpha4beta7 monoclonal antibody that is not associated with impaired systemic immune responses, is approved to treat Crohn’s disease and ulcerative colitis in adults.
A previous study from Kennedy and colleagues revealed that IBD patients on infliximab showed a weakened COVID-19 antibody response compared with patients on vedolizumab. To determine if treatment with anti-TNF drugs impacted the efficacy of the first shot of these two-dose COVID-19 vaccines, the researchers used data from the CLARITY IBD study to assess 865 infliximab- and 428 vedolizumab-treated participants without evidence of prior SARS-CoV-2 infection who had received uninterrupted biologic therapy since being recruited between Sept. 22 and Dec. 23, 2020.
In the 3-10 weeks after initial vaccination, geometric mean concentrations for SARS-CoV-2 anti-spike protein receptor-binding protein antibodies were lower in patients on infliximab, compared with patients on vedolizumab for both the Pfizer (6.0 U/mL [5.9] versus 28.8 U/mL [5.4], P < .0001) and AstraZeneca (4.7 U/mL [4.9] versus 13.8 U/mL [5.9]; P < .0001) vaccines. The researchers’ multivariable models reinforced those findings, with antibody concentrations lower in infliximab-treated patients for both the Pfizer (fold change, 0.29; 95% confidence interval, 0.21-0.40; P < .0001) and AstraZeneca (FC, 0.39; 95% CI, 0.30-0.51; P < .0001) vaccines.
After second doses of the two-dose Pfizer vaccine, 85% of patients on infliximab and 86% of patients on vedolizumab seroconverted (P = .68); similarly high seroconversion rates were seen in patients who had been infected with SARS-CoV-2 prior to receiving either vaccine. Several patient characteristics were associated with lower antibody concentrations regardless of vaccine type: being 60 years or older, use of immunomodulators, having Crohn’s disease, and being a smoker. Alternatively, non-White ethnicity was associated with higher antibody concentrations.
Evidence has ‘unclear clinical significance’
“These data, which require peer review, do not change my opinion on the safety and efficacy of COVID-19 vaccines in patients taking TNF inhibitors such as infliximab as monotherapy for the treatment of psoriatic disease,” Joel M. Gelfand MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, said in an interview.
“First, two peer-reviewed studies found good antibody response in patients on TNF inhibitors receiving COVID-19 vaccines (doi: 10.1136/annrheumdis-2021-220289; 10.1136/annrheumdis-2021-220272). Second, antibody responses were robust in the small cohort that received the second dose of a COVID-19 vaccine. We already know that, for the two messenger RNA-based vaccines available under emergency use authorization in the U.S., a second dose is required for optimal efficacy. Thus, evidence of a reduced antibody response after just one dose is of unclear clinical significance. Third, antibody responses are only a surrogate marker, and a low antibody response doesn’t necessarily mean the patient will not be protected by the vaccine.”
Focus on the second dose of a two-dose regimen
“Tell me about the response in people who got both doses of a vaccine that you’re supposed to get both doses of,” Jeffrey Curtis, MD, professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama at Birmingham, said in an interview. “The number of patients in that subset was small [n = 27] but in my opinion that’s the most clinically relevant analysis and the one that patients and clinicians want answered.”
He also emphasized the uncertainty around what ‘protection’ means in these early days of studying COVID-19 vaccine responses. “You can define seroprotection or seroconversion as some absolute level of an antibody response, but if you want to say ‘Mrs. Smith, your antibody level was X,’ on whatever arbitrary scale with whoever’s arbitrary lab test, nobody actually knows that Mrs. Smith is now protected from SARS-CoV-2, or how protected,” he said.
“What is not terribly controversial is: If you can’t detect antibodies, the vaccine didn’t ‘take,’ if you will. But if I tell you that the mean antibody level was X with one drug and then 2X with another drug, does that mean that you’re twice as protected? We don’t know that. I’m fearful that people are looking at these studies and thinking that more is better. It might be, but we don’t know that to be true.”
Debating the cause of weakened immune responses
“The biological plausibility of being on an anti-TNF affecting your immune reaction to a messenger RNA or even a replication-deficient viral vector vaccine doesn’t make sense,” David T. Rubin, MD, professor of medicine at the University of Chicago and chair of the National Scientific Advisory Committee of the Crohn’s and Colitis Foundation, said in an interview.
“I’m sure immunologists may differ with me on this, but given what we have come to appreciate about these vaccine mechanisms, this finding doesn’t make intuitive sense. So we need to make sure that, when this happens, we look to the next studies and try to understand, was there any other confounder that may have resulted in these findings that was not adequately adjusted for or addressed in some other way?
“When you have a study of this size, you argue, ‘Because it’s so large, any effect that was seen must be real,’ ” he added. “Alternatively, to have a study of this size, by its very nature you are limited in being able to control for certain other factors or differences between the groups.”
That said, he commended the authors for their study and acknowledged the potential questions it raises about the single-shot Johnson & Johnson vaccine. “If you only get one and you’re on infliximab, this study implies that maybe that’s not enough,” he said. “Despite the fact that Johnson & Johnson was approved as a single dose, it may be necessary to think about it as the first of two, or maybe it’s not the preferred vaccine in this group of patients.”
The study was supported by the Royal Devon and Exeter and Hull University Hospital Foundation NHS Trusts and unrestricted educational grants from Biogen (Switzerland), Celltrion Healthcare (South Korea), Galapagos NV (Belgium), and F. Hoffmann-La Roche (Switzerland). The authors acknowledged numerous potential conflicts of interest, including receiving grants, personal fees, and nonfinancial support from various pharmaceutical companies.
Patients being treated with infliximab had weakened immune responses to the first dose of the ChAdOx1 nCoV-19 (Oxford/AstraZeneca) and BNT162b2 (Pfizer/BioNTech) vaccines, compared with patients on vedolizumab (Entyvio), although a very significant number of patients from both groups seroconverted after their second dose, according to a new U.K. study of patients with inflammatory bowel disease (IBD).
“Antibody testing and adapted vaccine schedules should be considered to protect these at-risk patients,” Nicholas A. Kennedy, PhD, MBBS, of the University of Exeter (England) and colleagues wrote in a preprint published March 29 on MedRxiv.
Infliximab is an anti–tumor necrosis factor (anti-TNF) monoclonal antibody that’s approved to treat adult and pediatric Crohn’s disease and ulcerative colitis, as well as rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis, whereas vedolizumab, a gut selective anti-integrin alpha4beta7 monoclonal antibody that is not associated with impaired systemic immune responses, is approved to treat Crohn’s disease and ulcerative colitis in adults.
A previous study from Kennedy and colleagues revealed that IBD patients on infliximab showed a weakened COVID-19 antibody response compared with patients on vedolizumab. To determine if treatment with anti-TNF drugs impacted the efficacy of the first shot of these two-dose COVID-19 vaccines, the researchers used data from the CLARITY IBD study to assess 865 infliximab- and 428 vedolizumab-treated participants without evidence of prior SARS-CoV-2 infection who had received uninterrupted biologic therapy since being recruited between Sept. 22 and Dec. 23, 2020.
In the 3-10 weeks after initial vaccination, geometric mean concentrations for SARS-CoV-2 anti-spike protein receptor-binding protein antibodies were lower in patients on infliximab, compared with patients on vedolizumab for both the Pfizer (6.0 U/mL [5.9] versus 28.8 U/mL [5.4], P < .0001) and AstraZeneca (4.7 U/mL [4.9] versus 13.8 U/mL [5.9]; P < .0001) vaccines. The researchers’ multivariable models reinforced those findings, with antibody concentrations lower in infliximab-treated patients for both the Pfizer (fold change, 0.29; 95% confidence interval, 0.21-0.40; P < .0001) and AstraZeneca (FC, 0.39; 95% CI, 0.30-0.51; P < .0001) vaccines.
After second doses of the two-dose Pfizer vaccine, 85% of patients on infliximab and 86% of patients on vedolizumab seroconverted (P = .68); similarly high seroconversion rates were seen in patients who had been infected with SARS-CoV-2 prior to receiving either vaccine. Several patient characteristics were associated with lower antibody concentrations regardless of vaccine type: being 60 years or older, use of immunomodulators, having Crohn’s disease, and being a smoker. Alternatively, non-White ethnicity was associated with higher antibody concentrations.
Evidence has ‘unclear clinical significance’
“These data, which require peer review, do not change my opinion on the safety and efficacy of COVID-19 vaccines in patients taking TNF inhibitors such as infliximab as monotherapy for the treatment of psoriatic disease,” Joel M. Gelfand MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, said in an interview.
“First, two peer-reviewed studies found good antibody response in patients on TNF inhibitors receiving COVID-19 vaccines (doi: 10.1136/annrheumdis-2021-220289; 10.1136/annrheumdis-2021-220272). Second, antibody responses were robust in the small cohort that received the second dose of a COVID-19 vaccine. We already know that, for the two messenger RNA-based vaccines available under emergency use authorization in the U.S., a second dose is required for optimal efficacy. Thus, evidence of a reduced antibody response after just one dose is of unclear clinical significance. Third, antibody responses are only a surrogate marker, and a low antibody response doesn’t necessarily mean the patient will not be protected by the vaccine.”
Focus on the second dose of a two-dose regimen
“Tell me about the response in people who got both doses of a vaccine that you’re supposed to get both doses of,” Jeffrey Curtis, MD, professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama at Birmingham, said in an interview. “The number of patients in that subset was small [n = 27] but in my opinion that’s the most clinically relevant analysis and the one that patients and clinicians want answered.”
He also emphasized the uncertainty around what ‘protection’ means in these early days of studying COVID-19 vaccine responses. “You can define seroprotection or seroconversion as some absolute level of an antibody response, but if you want to say ‘Mrs. Smith, your antibody level was X,’ on whatever arbitrary scale with whoever’s arbitrary lab test, nobody actually knows that Mrs. Smith is now protected from SARS-CoV-2, or how protected,” he said.
“What is not terribly controversial is: If you can’t detect antibodies, the vaccine didn’t ‘take,’ if you will. But if I tell you that the mean antibody level was X with one drug and then 2X with another drug, does that mean that you’re twice as protected? We don’t know that. I’m fearful that people are looking at these studies and thinking that more is better. It might be, but we don’t know that to be true.”
Debating the cause of weakened immune responses
“The biological plausibility of being on an anti-TNF affecting your immune reaction to a messenger RNA or even a replication-deficient viral vector vaccine doesn’t make sense,” David T. Rubin, MD, professor of medicine at the University of Chicago and chair of the National Scientific Advisory Committee of the Crohn’s and Colitis Foundation, said in an interview.
“I’m sure immunologists may differ with me on this, but given what we have come to appreciate about these vaccine mechanisms, this finding doesn’t make intuitive sense. So we need to make sure that, when this happens, we look to the next studies and try to understand, was there any other confounder that may have resulted in these findings that was not adequately adjusted for or addressed in some other way?
“When you have a study of this size, you argue, ‘Because it’s so large, any effect that was seen must be real,’ ” he added. “Alternatively, to have a study of this size, by its very nature you are limited in being able to control for certain other factors or differences between the groups.”
That said, he commended the authors for their study and acknowledged the potential questions it raises about the single-shot Johnson & Johnson vaccine. “If you only get one and you’re on infliximab, this study implies that maybe that’s not enough,” he said. “Despite the fact that Johnson & Johnson was approved as a single dose, it may be necessary to think about it as the first of two, or maybe it’s not the preferred vaccine in this group of patients.”
The study was supported by the Royal Devon and Exeter and Hull University Hospital Foundation NHS Trusts and unrestricted educational grants from Biogen (Switzerland), Celltrion Healthcare (South Korea), Galapagos NV (Belgium), and F. Hoffmann-La Roche (Switzerland). The authors acknowledged numerous potential conflicts of interest, including receiving grants, personal fees, and nonfinancial support from various pharmaceutical companies.
FROM MEDRXIV
FDA approves new ready-to-inject glucagon product
The Food and Drug Administration has approved dasiglucagon (Zegalogue 0.6 mg/0.6 mL, Zealand Pharma) autoinjector and prefilled syringe for the treatment of severe hypoglycemia in people with diabetes aged 6 years and older.

The product has a shelf-life of 36 months at refrigerated temperatures and is stable for up to 12 months at room temperature.
“This approval will help enable appropriate children and adults with diabetes to be able to address sudden and severe hypoglycemia, which can quickly progress from a mild event to an emergency,” Jeremy Pettus, MD, assistant professor of medicine at the University of California, San Diego, said in a company statement.
The approval marks the latest step in the development of newer glucagon formulations that are easier to use in hypoglycemic emergencies than the traditional formulation that requires several steps for reconstitution.
The first intranasal glucagon (Baqsimi, Eli Lilly) was approved in the United States in July 2019 for people with diabetes age 4 years and older.
In September 2019, the FDA approved another prefilled glucagon rescue pen (Gvoke HypoPen, Xeris Pharmaceuticals) for the treatment of severe hypoglycemia in adult and pediatric patients age 2 years and older with diabetes.
Dasiglucagon is currently in phase 3 trials as a subcutaneous infusion for treating congenital hyperinsulinemia, and in phase 2 trials as part of a bihormonal artificial pancreas pump system.
The FDA approval was based on results from three randomized, double-blind, placebo-controlled, phase 3 studies of dasiglucagon in children age 6-17 years and adults with type 1 diabetes.
The primary endpoint was time to achieving an increase in blood glucose of 20 mg/dL or greater from time of administration without additional intervention within 45 minutes. That endpoint was achieved in all three studies, with a median time to blood glucose recovery of 10 minutes overall, with 99% of adults recovering within 15 minutes.
The most common adverse events reported in 2% or more of study participants were nausea, vomiting, headache, and injection-site pain in both children and adults. Diarrhea was also reported in adults.
Full launch is expected in late June 2021.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved dasiglucagon (Zegalogue 0.6 mg/0.6 mL, Zealand Pharma) autoinjector and prefilled syringe for the treatment of severe hypoglycemia in people with diabetes aged 6 years and older.
The product has a shelf-life of 36 months at refrigerated temperatures and is stable for up to 12 months at room temperature.
“This approval will help enable appropriate children and adults with diabetes to be able to address sudden and severe hypoglycemia, which can quickly progress from a mild event to an emergency,” Jeremy Pettus, MD, assistant professor of medicine at the University of California, San Diego, said in a company statement.
The approval marks the latest step in the development of newer glucagon formulations that are easier to use in hypoglycemic emergencies than the traditional formulation that requires several steps for reconstitution.
The first intranasal glucagon (Baqsimi, Eli Lilly) was approved in the United States in July 2019 for people with diabetes age 4 years and older.
In September 2019, the FDA approved another prefilled glucagon rescue pen (Gvoke HypoPen, Xeris Pharmaceuticals) for the treatment of severe hypoglycemia in adult and pediatric patients age 2 years and older with diabetes.
Dasiglucagon is currently in phase 3 trials as a subcutaneous infusion for treating congenital hyperinsulinemia, and in phase 2 trials as part of a bihormonal artificial pancreas pump system.
The FDA approval was based on results from three randomized, double-blind, placebo-controlled, phase 3 studies of dasiglucagon in children age 6-17 years and adults with type 1 diabetes.
The primary endpoint was time to achieving an increase in blood glucose of 20 mg/dL or greater from time of administration without additional intervention within 45 minutes. That endpoint was achieved in all three studies, with a median time to blood glucose recovery of 10 minutes overall, with 99% of adults recovering within 15 minutes.
The most common adverse events reported in 2% or more of study participants were nausea, vomiting, headache, and injection-site pain in both children and adults. Diarrhea was also reported in adults.
Full launch is expected in late June 2021.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved dasiglucagon (Zegalogue 0.6 mg/0.6 mL, Zealand Pharma) autoinjector and prefilled syringe for the treatment of severe hypoglycemia in people with diabetes aged 6 years and older.
The product has a shelf-life of 36 months at refrigerated temperatures and is stable for up to 12 months at room temperature.
“This approval will help enable appropriate children and adults with diabetes to be able to address sudden and severe hypoglycemia, which can quickly progress from a mild event to an emergency,” Jeremy Pettus, MD, assistant professor of medicine at the University of California, San Diego, said in a company statement.
The approval marks the latest step in the development of newer glucagon formulations that are easier to use in hypoglycemic emergencies than the traditional formulation that requires several steps for reconstitution.
The first intranasal glucagon (Baqsimi, Eli Lilly) was approved in the United States in July 2019 for people with diabetes age 4 years and older.
In September 2019, the FDA approved another prefilled glucagon rescue pen (Gvoke HypoPen, Xeris Pharmaceuticals) for the treatment of severe hypoglycemia in adult and pediatric patients age 2 years and older with diabetes.
Dasiglucagon is currently in phase 3 trials as a subcutaneous infusion for treating congenital hyperinsulinemia, and in phase 2 trials as part of a bihormonal artificial pancreas pump system.
The FDA approval was based on results from three randomized, double-blind, placebo-controlled, phase 3 studies of dasiglucagon in children age 6-17 years and adults with type 1 diabetes.
The primary endpoint was time to achieving an increase in blood glucose of 20 mg/dL or greater from time of administration without additional intervention within 45 minutes. That endpoint was achieved in all three studies, with a median time to blood glucose recovery of 10 minutes overall, with 99% of adults recovering within 15 minutes.
The most common adverse events reported in 2% or more of study participants were nausea, vomiting, headache, and injection-site pain in both children and adults. Diarrhea was also reported in adults.
Full launch is expected in late June 2021.
A version of this article first appeared on Medscape.com.
Servier and French drug safety agency found guilty on diet drug
More than 10 years after the withdrawal of the weight-loss drug Mediator (benfluorex) from the market in France, the Paris Court issued its judgment on March 29, 2021, against Servier Laboratories and the French National Agency for the Safety of Medicines and Health Products (ANSM).
Servier Laboratories was convicted of “aggravated deception” and fined 2.7 million euros (approximately $3.2 million) but were found not guilty of fraud. ANSM will also have to pay a fine.
Mediator was brought to the market in 1976 for the treatment of hyperlipidemia and for overweight patients with type 2 diabetes but was used off label as an appetite suppressant. It was taken by 5 million people and was only removed from the market in France in 2009 because of its toxic effects.
Mediator was taken off the market in Spain 6 years earlier, and in Switzerland 12 years earlier, and more than 30 years before in Belgium. It was never marketed in the United States.
The number of deaths because of heart valve damage related to the drug in France has been estimated at 220-300 in the short term (2.5 years) and 1,300-1,800 in the long term. In addition, the drug has been responsible for 3,100-4,200 hospital admissions for valvular insufficiency and pulmonary arterial hypertension.
“Despite knowing the risks for very many years ... [Servier Laboratories] never took the necessary measures and thus deceived” consumers of Mediator, declared the president of the criminal court, Sylvie Daunois.
Servier has “weakened confidence in the health system,” she added.
“I am very happy that ‘aggravated deception,’ the heart of the case, has been recognized and condemned,” Irène Frachon, MD, a pulmonologist at Brest (France) University Hospital and whistleblower on the Mediator scandal, said in an interview.
However, Dr. Frachon continued: “The major problem, putting a toxic agent on the market for years, is a given. But the weakness of the sentences gives a mixed message.
“The judgment is too cautious in its punishments,” she added, pointing out that, “in the case of contaminated blood, there were prison sentences.”
Servier deceived doctors and patients
The French trial in September 2019 was extraordinary, with about 100 witnesses, nearly 400 lawyers, and 5,000 victims.
On June 23, 2020, the prosecutor, Aude Le Guilcher, requested at the end of her indictment that the six companies of the Servier group be fined, notably for “deception, homicide, involuntary injuries, and fraud,” to the tune of 20.3 million euros (approximately $23.8 million).
Against the former No. 2 of Servier, Jean-Philippe Seta, Ms. Le Guilcher requested 5 years in prison, with 2 years suspended, and a 200,000 euro (approximately $235,000) fine.
The same sum was requested against ANSM for homicide and unintentional injuries.
In the end, Mr. Seta, the former right hand of Jacques Servier, who died in 2004, was sentenced to 4 years in prison, suspended. For their part, ANSM was fined 303,000 euros(approximately $350,000).
It is now clearly established that Servier Laboratories knowingly concealed the similarity of Mediator to the fenfluramine family of compounds, which was banned in 1990 because of adverse effects.
The group also deceived doctors who prescribed the drug and patients who took it by hiding its toxicity.
Mediator should never have been authorized for use
In terms of the fraud charges, the prosecutor estimated that the losses incurred by the primary health insurance industry were in the region of several hundred million euros.
She argued that Mediator should never have been reimbursed, as “it should never have benefited from market authorization, which it received solely due to the fraudulent actions of the company.”
But because of the statute of limitations, this argument was not heard, explained Dr. Frachon, “and the same is true of conflicts of interest, where limitations led to them being discharged.
“We understand the legal difficulties, but it’s a shame in terms of the signal sent.”
“I hope the medical world will learn the lesson and not continue with ‘business as usual’ with people who are delinquents. I think it will be essential to restore public confidence,” concluded Dr. Frachon.
No conflicts of interest or funding were declared.
A version of this article first appeared on Medscape.com.
More than 10 years after the withdrawal of the weight-loss drug Mediator (benfluorex) from the market in France, the Paris Court issued its judgment on March 29, 2021, against Servier Laboratories and the French National Agency for the Safety of Medicines and Health Products (ANSM).
Servier Laboratories was convicted of “aggravated deception” and fined 2.7 million euros (approximately $3.2 million) but were found not guilty of fraud. ANSM will also have to pay a fine.
Mediator was brought to the market in 1976 for the treatment of hyperlipidemia and for overweight patients with type 2 diabetes but was used off label as an appetite suppressant. It was taken by 5 million people and was only removed from the market in France in 2009 because of its toxic effects.
Mediator was taken off the market in Spain 6 years earlier, and in Switzerland 12 years earlier, and more than 30 years before in Belgium. It was never marketed in the United States.
The number of deaths because of heart valve damage related to the drug in France has been estimated at 220-300 in the short term (2.5 years) and 1,300-1,800 in the long term. In addition, the drug has been responsible for 3,100-4,200 hospital admissions for valvular insufficiency and pulmonary arterial hypertension.
“Despite knowing the risks for very many years ... [Servier Laboratories] never took the necessary measures and thus deceived” consumers of Mediator, declared the president of the criminal court, Sylvie Daunois.
Servier has “weakened confidence in the health system,” she added.
“I am very happy that ‘aggravated deception,’ the heart of the case, has been recognized and condemned,” Irène Frachon, MD, a pulmonologist at Brest (France) University Hospital and whistleblower on the Mediator scandal, said in an interview.
However, Dr. Frachon continued: “The major problem, putting a toxic agent on the market for years, is a given. But the weakness of the sentences gives a mixed message.
“The judgment is too cautious in its punishments,” she added, pointing out that, “in the case of contaminated blood, there were prison sentences.”
Servier deceived doctors and patients
The French trial in September 2019 was extraordinary, with about 100 witnesses, nearly 400 lawyers, and 5,000 victims.
On June 23, 2020, the prosecutor, Aude Le Guilcher, requested at the end of her indictment that the six companies of the Servier group be fined, notably for “deception, homicide, involuntary injuries, and fraud,” to the tune of 20.3 million euros (approximately $23.8 million).
Against the former No. 2 of Servier, Jean-Philippe Seta, Ms. Le Guilcher requested 5 years in prison, with 2 years suspended, and a 200,000 euro (approximately $235,000) fine.
The same sum was requested against ANSM for homicide and unintentional injuries.
In the end, Mr. Seta, the former right hand of Jacques Servier, who died in 2004, was sentenced to 4 years in prison, suspended. For their part, ANSM was fined 303,000 euros(approximately $350,000).
It is now clearly established that Servier Laboratories knowingly concealed the similarity of Mediator to the fenfluramine family of compounds, which was banned in 1990 because of adverse effects.
The group also deceived doctors who prescribed the drug and patients who took it by hiding its toxicity.
Mediator should never have been authorized for use
In terms of the fraud charges, the prosecutor estimated that the losses incurred by the primary health insurance industry were in the region of several hundred million euros.
She argued that Mediator should never have been reimbursed, as “it should never have benefited from market authorization, which it received solely due to the fraudulent actions of the company.”
But because of the statute of limitations, this argument was not heard, explained Dr. Frachon, “and the same is true of conflicts of interest, where limitations led to them being discharged.
“We understand the legal difficulties, but it’s a shame in terms of the signal sent.”
“I hope the medical world will learn the lesson and not continue with ‘business as usual’ with people who are delinquents. I think it will be essential to restore public confidence,” concluded Dr. Frachon.
No conflicts of interest or funding were declared.
A version of this article first appeared on Medscape.com.
More than 10 years after the withdrawal of the weight-loss drug Mediator (benfluorex) from the market in France, the Paris Court issued its judgment on March 29, 2021, against Servier Laboratories and the French National Agency for the Safety of Medicines and Health Products (ANSM).
Servier Laboratories was convicted of “aggravated deception” and fined 2.7 million euros (approximately $3.2 million) but were found not guilty of fraud. ANSM will also have to pay a fine.
Mediator was brought to the market in 1976 for the treatment of hyperlipidemia and for overweight patients with type 2 diabetes but was used off label as an appetite suppressant. It was taken by 5 million people and was only removed from the market in France in 2009 because of its toxic effects.
Mediator was taken off the market in Spain 6 years earlier, and in Switzerland 12 years earlier, and more than 30 years before in Belgium. It was never marketed in the United States.
The number of deaths because of heart valve damage related to the drug in France has been estimated at 220-300 in the short term (2.5 years) and 1,300-1,800 in the long term. In addition, the drug has been responsible for 3,100-4,200 hospital admissions for valvular insufficiency and pulmonary arterial hypertension.
“Despite knowing the risks for very many years ... [Servier Laboratories] never took the necessary measures and thus deceived” consumers of Mediator, declared the president of the criminal court, Sylvie Daunois.
Servier has “weakened confidence in the health system,” she added.
“I am very happy that ‘aggravated deception,’ the heart of the case, has been recognized and condemned,” Irène Frachon, MD, a pulmonologist at Brest (France) University Hospital and whistleblower on the Mediator scandal, said in an interview.
However, Dr. Frachon continued: “The major problem, putting a toxic agent on the market for years, is a given. But the weakness of the sentences gives a mixed message.
“The judgment is too cautious in its punishments,” she added, pointing out that, “in the case of contaminated blood, there were prison sentences.”
Servier deceived doctors and patients
The French trial in September 2019 was extraordinary, with about 100 witnesses, nearly 400 lawyers, and 5,000 victims.
On June 23, 2020, the prosecutor, Aude Le Guilcher, requested at the end of her indictment that the six companies of the Servier group be fined, notably for “deception, homicide, involuntary injuries, and fraud,” to the tune of 20.3 million euros (approximately $23.8 million).
Against the former No. 2 of Servier, Jean-Philippe Seta, Ms. Le Guilcher requested 5 years in prison, with 2 years suspended, and a 200,000 euro (approximately $235,000) fine.
The same sum was requested against ANSM for homicide and unintentional injuries.
In the end, Mr. Seta, the former right hand of Jacques Servier, who died in 2004, was sentenced to 4 years in prison, suspended. For their part, ANSM was fined 303,000 euros(approximately $350,000).
It is now clearly established that Servier Laboratories knowingly concealed the similarity of Mediator to the fenfluramine family of compounds, which was banned in 1990 because of adverse effects.
The group also deceived doctors who prescribed the drug and patients who took it by hiding its toxicity.
Mediator should never have been authorized for use
In terms of the fraud charges, the prosecutor estimated that the losses incurred by the primary health insurance industry were in the region of several hundred million euros.
She argued that Mediator should never have been reimbursed, as “it should never have benefited from market authorization, which it received solely due to the fraudulent actions of the company.”
But because of the statute of limitations, this argument was not heard, explained Dr. Frachon, “and the same is true of conflicts of interest, where limitations led to them being discharged.
“We understand the legal difficulties, but it’s a shame in terms of the signal sent.”
“I hope the medical world will learn the lesson and not continue with ‘business as usual’ with people who are delinquents. I think it will be essential to restore public confidence,” concluded Dr. Frachon.
No conflicts of interest or funding were declared.
A version of this article first appeared on Medscape.com.
Steroids can be stopped in some older multiple myeloma patients
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
National Psoriasis Foundation recommends some stop methotrexate for 2 weeks after J&J vaccine
The , Joel M. Gelfand, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
The new guidance states: “Patients 60 or older who have at least one comorbidity associated with an increased risk for poor COVID-19 outcomes, and who are taking methotrexate with well-controlled psoriatic disease, may, in consultation with their prescriber, consider holding it for 2 weeks after receiving the Ad26.COV2.S [Johnson & Johnson] vaccine in order to potentially improve vaccine response.”
The key word here is “potentially.” There is no hard evidence that a 2-week hold on methotrexate after receiving the killed adenovirus vaccine will actually provide a clinically meaningful benefit. But it’s a hypothetical possibility. The rationale stems from a small randomized trial conducted in South Korea several years ago in which patients with rheumatoid arthritis were assigned to hold or continue their methotrexate for the first 2 weeks after receiving an inactivated-virus influenza vaccine. The antibody response to the vaccine was better in those who temporarily halted their methotrexate, explained Dr. Gelfand, cochair of the NPF COVID-19 Task Force and professor of dermatology and of epidemiology at the University of Pennsylvania, Philadelphia.
“If you have a patient on methotrexate who’s 60 or older and whose psoriasis is completely controlled and quiescent and the patient is concerned about how well the vaccine is going to work, this is a reasonable thing to consider in someone who’s at higher risk for poor outcomes if they get infected,” he said.
If the informed patient wants to continue on methotrexate without interruption, that’s fine, too, in light of the lack of compelling evidence on this issue, the dermatologist added at the conference, sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The NPF task force does not extend the recommendation to consider holding methotrexate in recipients of the mRNA-based Moderna and Pfizer vaccines because of their very different mechanisms of action. Nor is it recommended to hold biologic agents after receiving any of the available COVID-19 vaccines. Studies have shown no altered immunologic response to influenza or pneumococcal vaccines in patients who continued on tumor necrosis factor inhibitors or interleukin-17 inhibitors. The interleukin-23 inhibitors haven’t been studied in this regard.
The task force recommends that most psoriasis patients should continue on treatment throughout the pandemic, and newly diagnosed patients should commence appropriate therapy as if there was no pandemic.
“We’ve learned that many patients who stopped their treatment for psoriatic disease early in the pandemic came to regret that decision because their psoriasis flared and got worse and required reinstitution of therapy,” Dr. Gelfand said. “The current data is largely reassuring that if there is an effect of our therapies on the risk of COVID, it must be rather small and therefore unlikely to be clinically meaningful for our patients.”
Dr. Gelfand reported serving as a consultant to and recipient of institutional research grants from Pfizer and numerous other pharmaceutical companies.
MedscapeLIVE and this news organization are owned by the same parent company.
The , Joel M. Gelfand, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
The new guidance states: “Patients 60 or older who have at least one comorbidity associated with an increased risk for poor COVID-19 outcomes, and who are taking methotrexate with well-controlled psoriatic disease, may, in consultation with their prescriber, consider holding it for 2 weeks after receiving the Ad26.COV2.S [Johnson & Johnson] vaccine in order to potentially improve vaccine response.”
The key word here is “potentially.” There is no hard evidence that a 2-week hold on methotrexate after receiving the killed adenovirus vaccine will actually provide a clinically meaningful benefit. But it’s a hypothetical possibility. The rationale stems from a small randomized trial conducted in South Korea several years ago in which patients with rheumatoid arthritis were assigned to hold or continue their methotrexate for the first 2 weeks after receiving an inactivated-virus influenza vaccine. The antibody response to the vaccine was better in those who temporarily halted their methotrexate, explained Dr. Gelfand, cochair of the NPF COVID-19 Task Force and professor of dermatology and of epidemiology at the University of Pennsylvania, Philadelphia.
“If you have a patient on methotrexate who’s 60 or older and whose psoriasis is completely controlled and quiescent and the patient is concerned about how well the vaccine is going to work, this is a reasonable thing to consider in someone who’s at higher risk for poor outcomes if they get infected,” he said.
If the informed patient wants to continue on methotrexate without interruption, that’s fine, too, in light of the lack of compelling evidence on this issue, the dermatologist added at the conference, sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The NPF task force does not extend the recommendation to consider holding methotrexate in recipients of the mRNA-based Moderna and Pfizer vaccines because of their very different mechanisms of action. Nor is it recommended to hold biologic agents after receiving any of the available COVID-19 vaccines. Studies have shown no altered immunologic response to influenza or pneumococcal vaccines in patients who continued on tumor necrosis factor inhibitors or interleukin-17 inhibitors. The interleukin-23 inhibitors haven’t been studied in this regard.
The task force recommends that most psoriasis patients should continue on treatment throughout the pandemic, and newly diagnosed patients should commence appropriate therapy as if there was no pandemic.
“We’ve learned that many patients who stopped their treatment for psoriatic disease early in the pandemic came to regret that decision because their psoriasis flared and got worse and required reinstitution of therapy,” Dr. Gelfand said. “The current data is largely reassuring that if there is an effect of our therapies on the risk of COVID, it must be rather small and therefore unlikely to be clinically meaningful for our patients.”
Dr. Gelfand reported serving as a consultant to and recipient of institutional research grants from Pfizer and numerous other pharmaceutical companies.
MedscapeLIVE and this news organization are owned by the same parent company.
The , Joel M. Gelfand, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
The new guidance states: “Patients 60 or older who have at least one comorbidity associated with an increased risk for poor COVID-19 outcomes, and who are taking methotrexate with well-controlled psoriatic disease, may, in consultation with their prescriber, consider holding it for 2 weeks after receiving the Ad26.COV2.S [Johnson & Johnson] vaccine in order to potentially improve vaccine response.”
The key word here is “potentially.” There is no hard evidence that a 2-week hold on methotrexate after receiving the killed adenovirus vaccine will actually provide a clinically meaningful benefit. But it’s a hypothetical possibility. The rationale stems from a small randomized trial conducted in South Korea several years ago in which patients with rheumatoid arthritis were assigned to hold or continue their methotrexate for the first 2 weeks after receiving an inactivated-virus influenza vaccine. The antibody response to the vaccine was better in those who temporarily halted their methotrexate, explained Dr. Gelfand, cochair of the NPF COVID-19 Task Force and professor of dermatology and of epidemiology at the University of Pennsylvania, Philadelphia.
“If you have a patient on methotrexate who’s 60 or older and whose psoriasis is completely controlled and quiescent and the patient is concerned about how well the vaccine is going to work, this is a reasonable thing to consider in someone who’s at higher risk for poor outcomes if they get infected,” he said.
If the informed patient wants to continue on methotrexate without interruption, that’s fine, too, in light of the lack of compelling evidence on this issue, the dermatologist added at the conference, sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The NPF task force does not extend the recommendation to consider holding methotrexate in recipients of the mRNA-based Moderna and Pfizer vaccines because of their very different mechanisms of action. Nor is it recommended to hold biologic agents after receiving any of the available COVID-19 vaccines. Studies have shown no altered immunologic response to influenza or pneumococcal vaccines in patients who continued on tumor necrosis factor inhibitors or interleukin-17 inhibitors. The interleukin-23 inhibitors haven’t been studied in this regard.
The task force recommends that most psoriasis patients should continue on treatment throughout the pandemic, and newly diagnosed patients should commence appropriate therapy as if there was no pandemic.
“We’ve learned that many patients who stopped their treatment for psoriatic disease early in the pandemic came to regret that decision because their psoriasis flared and got worse and required reinstitution of therapy,” Dr. Gelfand said. “The current data is largely reassuring that if there is an effect of our therapies on the risk of COVID, it must be rather small and therefore unlikely to be clinically meaningful for our patients.”
Dr. Gelfand reported serving as a consultant to and recipient of institutional research grants from Pfizer and numerous other pharmaceutical companies.
MedscapeLIVE and this news organization are owned by the same parent company.
FROM INNOVATIONS IN DERMATOLOGY