User login
Prescription cascade more likely after CCBs than other hypertension meds
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.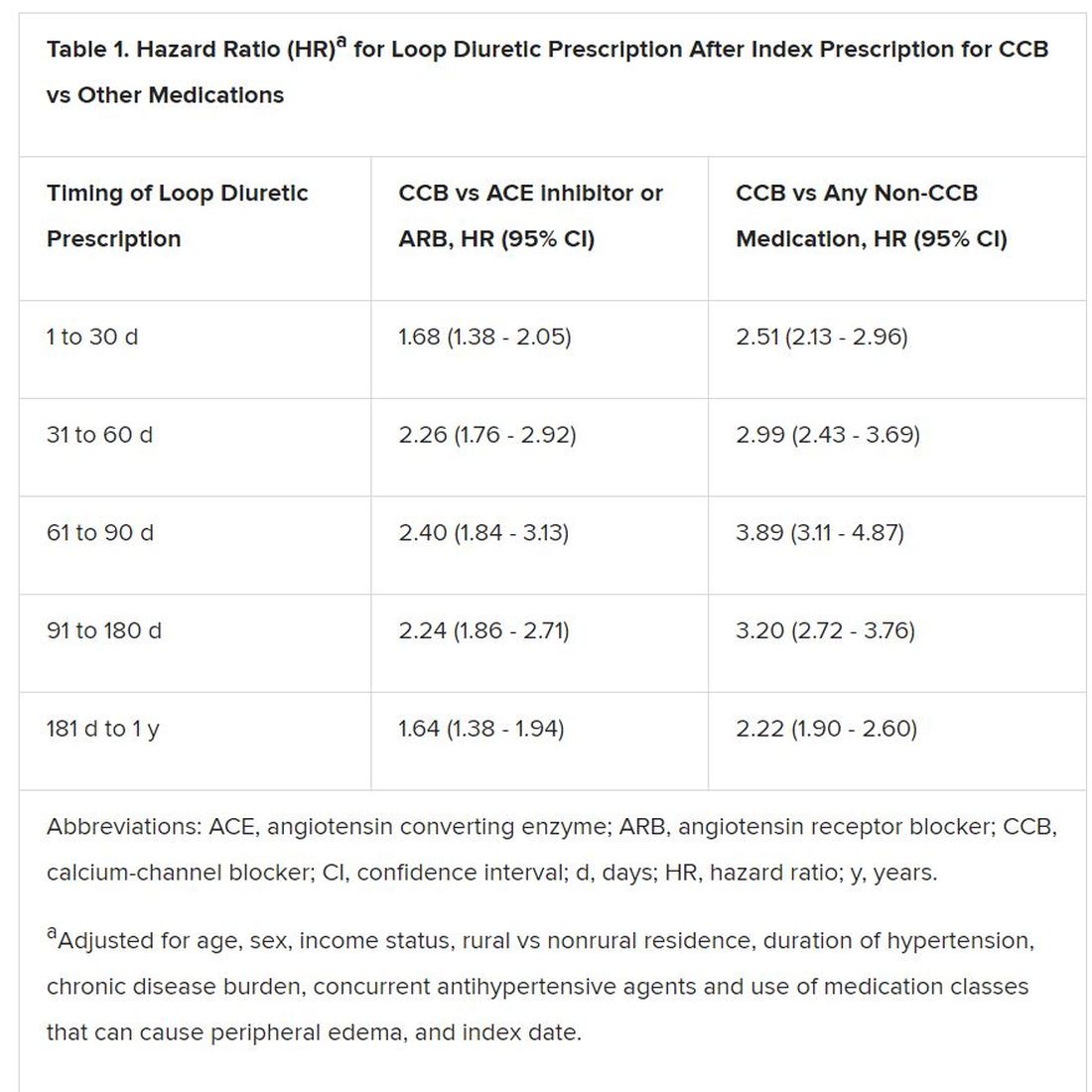
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Gene therapy appears effective in bladder cancer patients with few options
SAN FRANCISCO – (BCG), new research suggests.

The therapy, nadofaragene firadenovec, is a recombinant adenovirus that is instilled into the bladder and delivers the human interferon alpha-2b gene, leading to expression of the immune cytokine.
At 3 months, nadofaragene firadenovec had produced a complete response in 53.4% of patients with carcinoma in situ (CIS), and the rate of high-grade recurrence-free survival was 59.6% in all patients. Although most patients (70.1%) experienced adverse events in this trial, few had serious events (1.9%).
Stephen A. Boorjian, MD, of the Mayo Clinic in Rochester, Minn., presented these results at the 2020 Genitourinary Cancers Symposium, sponsored by the American Society for Clinical Oncology, ASTRO, and the Society of Urologic Oncology.
“The optimal management for patients with BCG-unresponsive non–muscle invasive bladder cancer remains to be established,” Dr. Boorjian said. “National and organizational guidelines recommend radical cystectomy in this setting, but we have to acknowledge that many of our patients will be either unwilling or unfit to undergo what is often a highly morbid operation.”
Dr. Boorjian and coinvestigators tested nadofaragene firadenovec in 107 patients with CIS, with or without high-grade Ta or T1 papillary disease, as well as 50 patients with high-grade Ta or T1 papillary disease only.
All patients received nadofaragene firadenovec once every 3 months for up four doses, with additional dosing at the investigator’s discretion. Although the trial was designated as phase 3, it did not have the typical randomized comparative design.
“The challenge in the BCG-unresponsive disease state is that, historically, there has been no validated good comparator to use here; there’s no standard of care for these patients,” Dr. Boorjian explained, noting that the design was chosen after discussion with the Food and Drug Administration.
Efficacy
The complete response rate among CIS patients at any time after instillation, the trial’s primary objective, was 53.4%. All of these responses occurred after a single dose of nadofaragene firadenovec. Responses were considered durable, as 45.5% of CIS patients who had a complete response at 3 months still had a complete response at 12 months.
The rate of high-grade recurrence-free survival at 3 months was 59.6% in the overall population, 53.4% in patients with CIS, and 72.9% in patients with papillary disease. At 12 months, the rate of high-grade recurrence-free survival was 30.5%, 24.3%, and 43.8%, respectively.
The rate of high-grade recurrence without muscle invasive bladder cancer was 71.8% in the CIS group and 52.1% in the papillary group. The rate of progression to muscle invasive bladder cancer or more advanced disease was 4.9% and 6.3%, respectively. The overall 2-year rate of cystectomy-free survival was 64%, and there were no bladder cancer deaths.
Safety
Nadofaragene firadenovec was considered safe and well tolerated. The most common study drug–related adverse events were irritative voiding symptoms, such as discharge, bladder spasm, micturition urgency, and hematuria.
Although 70.1% of patients experienced a local or systemic adverse event related to the study drug or procedure, few experienced serious adverse events (1.9%), grade 3 adverse events (3.8%), or treatment-emergent adverse events leading to discontinuation (1.9%). The serious adverse events were sepsis, syncope, and hematuria.
Next steps
“Nadofaragene firadenovec represents a promising option for patients with BCG-unresponsive [non–muscle invasive bladder cancer],” Dr. Boorjian said. “What we want to see going forward is durability of response, cystectomy-free survival, and metastasis-free and cancer-specific survival. Those are the harder endpoints that, as these data mature, we will want to see so that we are able to counsel our patients accordingly.”

The FDA recently approved pembrolizumab for this patient population, noted session cochair Guru Sonpavde, MD, of the Dana-Farber Cancer Institute in Boston.
“But nadofaragene firadenovec is much less toxic, it is given intravesically, and it’s only given once every 3 months,” he said. “The outcomes at the 1-year point look similar in terms of the durable complete response rate. So we hope that this therapy will be able to get to the clinic, but we have to wait for the FDA to decide.”
It remains to be seen how nadofaragene firadenovec compares with other emerging therapies for high-risk BCG-unresponsive non–muscle invasive bladder cancer, including pembrolizumab and potentially intravesical oportuzumab monatox (a targeted immunotoxin) and intravesical combination BCG and superagonist interleukin-15 (ALT-803), according to Dr. Boorjian.
“A unique feature of the trial I presented was that the results were validated by a mandatory 12-month study biopsy, which has not been the case in other trials,” he pointed out. “So this separates it a little bit when you are looking at pathologic response rates and high-grade recurrence–free rates. They are based on pathologic confirmation.”
Evaluators will ultimately compare these therapies on safety, efficacy, ease of administration, delivery schedule, and cost, Dr. Boorjian said. “At this stage, it’s very difficult to start to say which agent is going to be placed where in our management paradigm. We have to go step by step: Do the trial that we did, show our data, put it out for review, and then allow the community to engage in a discussion about which agent, and why, for which patient.”
This trial was funded by FKD Therapies Oy. Dr. Boorjian disclosed relationships with Ferring and Sanofi. Dr. Sonpavde disclosed relationships with many companies.
SOURCE: Boorjian SA et al. GUCS 2020, Abstract 442.
SAN FRANCISCO – (BCG), new research suggests.
The therapy, nadofaragene firadenovec, is a recombinant adenovirus that is instilled into the bladder and delivers the human interferon alpha-2b gene, leading to expression of the immune cytokine.
At 3 months, nadofaragene firadenovec had produced a complete response in 53.4% of patients with carcinoma in situ (CIS), and the rate of high-grade recurrence-free survival was 59.6% in all patients. Although most patients (70.1%) experienced adverse events in this trial, few had serious events (1.9%).
Stephen A. Boorjian, MD, of the Mayo Clinic in Rochester, Minn., presented these results at the 2020 Genitourinary Cancers Symposium, sponsored by the American Society for Clinical Oncology, ASTRO, and the Society of Urologic Oncology.
“The optimal management for patients with BCG-unresponsive non–muscle invasive bladder cancer remains to be established,” Dr. Boorjian said. “National and organizational guidelines recommend radical cystectomy in this setting, but we have to acknowledge that many of our patients will be either unwilling or unfit to undergo what is often a highly morbid operation.”
Dr. Boorjian and coinvestigators tested nadofaragene firadenovec in 107 patients with CIS, with or without high-grade Ta or T1 papillary disease, as well as 50 patients with high-grade Ta or T1 papillary disease only.
All patients received nadofaragene firadenovec once every 3 months for up four doses, with additional dosing at the investigator’s discretion. Although the trial was designated as phase 3, it did not have the typical randomized comparative design.
“The challenge in the BCG-unresponsive disease state is that, historically, there has been no validated good comparator to use here; there’s no standard of care for these patients,” Dr. Boorjian explained, noting that the design was chosen after discussion with the Food and Drug Administration.
Efficacy
The complete response rate among CIS patients at any time after instillation, the trial’s primary objective, was 53.4%. All of these responses occurred after a single dose of nadofaragene firadenovec. Responses were considered durable, as 45.5% of CIS patients who had a complete response at 3 months still had a complete response at 12 months.
The rate of high-grade recurrence-free survival at 3 months was 59.6% in the overall population, 53.4% in patients with CIS, and 72.9% in patients with papillary disease. At 12 months, the rate of high-grade recurrence-free survival was 30.5%, 24.3%, and 43.8%, respectively.
The rate of high-grade recurrence without muscle invasive bladder cancer was 71.8% in the CIS group and 52.1% in the papillary group. The rate of progression to muscle invasive bladder cancer or more advanced disease was 4.9% and 6.3%, respectively. The overall 2-year rate of cystectomy-free survival was 64%, and there were no bladder cancer deaths.
Safety
Nadofaragene firadenovec was considered safe and well tolerated. The most common study drug–related adverse events were irritative voiding symptoms, such as discharge, bladder spasm, micturition urgency, and hematuria.
Although 70.1% of patients experienced a local or systemic adverse event related to the study drug or procedure, few experienced serious adverse events (1.9%), grade 3 adverse events (3.8%), or treatment-emergent adverse events leading to discontinuation (1.9%). The serious adverse events were sepsis, syncope, and hematuria.
Next steps
“Nadofaragene firadenovec represents a promising option for patients with BCG-unresponsive [non–muscle invasive bladder cancer],” Dr. Boorjian said. “What we want to see going forward is durability of response, cystectomy-free survival, and metastasis-free and cancer-specific survival. Those are the harder endpoints that, as these data mature, we will want to see so that we are able to counsel our patients accordingly.”
The FDA recently approved pembrolizumab for this patient population, noted session cochair Guru Sonpavde, MD, of the Dana-Farber Cancer Institute in Boston.
“But nadofaragene firadenovec is much less toxic, it is given intravesically, and it’s only given once every 3 months,” he said. “The outcomes at the 1-year point look similar in terms of the durable complete response rate. So we hope that this therapy will be able to get to the clinic, but we have to wait for the FDA to decide.”
It remains to be seen how nadofaragene firadenovec compares with other emerging therapies for high-risk BCG-unresponsive non–muscle invasive bladder cancer, including pembrolizumab and potentially intravesical oportuzumab monatox (a targeted immunotoxin) and intravesical combination BCG and superagonist interleukin-15 (ALT-803), according to Dr. Boorjian.
“A unique feature of the trial I presented was that the results were validated by a mandatory 12-month study biopsy, which has not been the case in other trials,” he pointed out. “So this separates it a little bit when you are looking at pathologic response rates and high-grade recurrence–free rates. They are based on pathologic confirmation.”
Evaluators will ultimately compare these therapies on safety, efficacy, ease of administration, delivery schedule, and cost, Dr. Boorjian said. “At this stage, it’s very difficult to start to say which agent is going to be placed where in our management paradigm. We have to go step by step: Do the trial that we did, show our data, put it out for review, and then allow the community to engage in a discussion about which agent, and why, for which patient.”
This trial was funded by FKD Therapies Oy. Dr. Boorjian disclosed relationships with Ferring and Sanofi. Dr. Sonpavde disclosed relationships with many companies.
SOURCE: Boorjian SA et al. GUCS 2020, Abstract 442.
SAN FRANCISCO – (BCG), new research suggests.
The therapy, nadofaragene firadenovec, is a recombinant adenovirus that is instilled into the bladder and delivers the human interferon alpha-2b gene, leading to expression of the immune cytokine.
At 3 months, nadofaragene firadenovec had produced a complete response in 53.4% of patients with carcinoma in situ (CIS), and the rate of high-grade recurrence-free survival was 59.6% in all patients. Although most patients (70.1%) experienced adverse events in this trial, few had serious events (1.9%).
Stephen A. Boorjian, MD, of the Mayo Clinic in Rochester, Minn., presented these results at the 2020 Genitourinary Cancers Symposium, sponsored by the American Society for Clinical Oncology, ASTRO, and the Society of Urologic Oncology.
“The optimal management for patients with BCG-unresponsive non–muscle invasive bladder cancer remains to be established,” Dr. Boorjian said. “National and organizational guidelines recommend radical cystectomy in this setting, but we have to acknowledge that many of our patients will be either unwilling or unfit to undergo what is often a highly morbid operation.”
Dr. Boorjian and coinvestigators tested nadofaragene firadenovec in 107 patients with CIS, with or without high-grade Ta or T1 papillary disease, as well as 50 patients with high-grade Ta or T1 papillary disease only.
All patients received nadofaragene firadenovec once every 3 months for up four doses, with additional dosing at the investigator’s discretion. Although the trial was designated as phase 3, it did not have the typical randomized comparative design.
“The challenge in the BCG-unresponsive disease state is that, historically, there has been no validated good comparator to use here; there’s no standard of care for these patients,” Dr. Boorjian explained, noting that the design was chosen after discussion with the Food and Drug Administration.
Efficacy
The complete response rate among CIS patients at any time after instillation, the trial’s primary objective, was 53.4%. All of these responses occurred after a single dose of nadofaragene firadenovec. Responses were considered durable, as 45.5% of CIS patients who had a complete response at 3 months still had a complete response at 12 months.
The rate of high-grade recurrence-free survival at 3 months was 59.6% in the overall population, 53.4% in patients with CIS, and 72.9% in patients with papillary disease. At 12 months, the rate of high-grade recurrence-free survival was 30.5%, 24.3%, and 43.8%, respectively.
The rate of high-grade recurrence without muscle invasive bladder cancer was 71.8% in the CIS group and 52.1% in the papillary group. The rate of progression to muscle invasive bladder cancer or more advanced disease was 4.9% and 6.3%, respectively. The overall 2-year rate of cystectomy-free survival was 64%, and there were no bladder cancer deaths.
Safety
Nadofaragene firadenovec was considered safe and well tolerated. The most common study drug–related adverse events were irritative voiding symptoms, such as discharge, bladder spasm, micturition urgency, and hematuria.
Although 70.1% of patients experienced a local or systemic adverse event related to the study drug or procedure, few experienced serious adverse events (1.9%), grade 3 adverse events (3.8%), or treatment-emergent adverse events leading to discontinuation (1.9%). The serious adverse events were sepsis, syncope, and hematuria.
Next steps
“Nadofaragene firadenovec represents a promising option for patients with BCG-unresponsive [non–muscle invasive bladder cancer],” Dr. Boorjian said. “What we want to see going forward is durability of response, cystectomy-free survival, and metastasis-free and cancer-specific survival. Those are the harder endpoints that, as these data mature, we will want to see so that we are able to counsel our patients accordingly.”
The FDA recently approved pembrolizumab for this patient population, noted session cochair Guru Sonpavde, MD, of the Dana-Farber Cancer Institute in Boston.
“But nadofaragene firadenovec is much less toxic, it is given intravesically, and it’s only given once every 3 months,” he said. “The outcomes at the 1-year point look similar in terms of the durable complete response rate. So we hope that this therapy will be able to get to the clinic, but we have to wait for the FDA to decide.”
It remains to be seen how nadofaragene firadenovec compares with other emerging therapies for high-risk BCG-unresponsive non–muscle invasive bladder cancer, including pembrolizumab and potentially intravesical oportuzumab monatox (a targeted immunotoxin) and intravesical combination BCG and superagonist interleukin-15 (ALT-803), according to Dr. Boorjian.
“A unique feature of the trial I presented was that the results were validated by a mandatory 12-month study biopsy, which has not been the case in other trials,” he pointed out. “So this separates it a little bit when you are looking at pathologic response rates and high-grade recurrence–free rates. They are based on pathologic confirmation.”
Evaluators will ultimately compare these therapies on safety, efficacy, ease of administration, delivery schedule, and cost, Dr. Boorjian said. “At this stage, it’s very difficult to start to say which agent is going to be placed where in our management paradigm. We have to go step by step: Do the trial that we did, show our data, put it out for review, and then allow the community to engage in a discussion about which agent, and why, for which patient.”
This trial was funded by FKD Therapies Oy. Dr. Boorjian disclosed relationships with Ferring and Sanofi. Dr. Sonpavde disclosed relationships with many companies.
SOURCE: Boorjian SA et al. GUCS 2020, Abstract 442.
REPORTING FROM GUCS 2020
FDA, FTC uniting to promote biosimilars
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.

The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.

“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.
“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.
“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
Prescription osteoarthritis relief gets OTC approval
The Food and Drug Administration has approved formerly prescription-only Voltaren Arthritis Pain (diclofenac sodium topical gel, 1%) for nonprescription use via a process known as a prescription to over-the-counter (Rx-to-OTC) switch, according to a news release from the agency.

“As a result of the Rx-to-OTC switch process, many products sold over the counter today use ingredients or dosage strengths that were available only by prescription 30 years ago,” Karen Mahoney, MD, acting deputy director of the Office of Nonprescription Drugs in the FDA’s Center for Drug Evaluation and Research, said in the release.
This switch to nonprescription status is usually initiated by the manufacturer, who must provide data that demonstrates the drug in question is both safe and effective as self-medication in accordance with the proposed labeling and that consumers can use it safely and effectively without the supervision of a health care professional.
This particular therapy is a topical NSAID gel and was first approved by the FDA in 2007 with the indication for relief of osteoarthritis pain. It can take 7 days to have an effect, but if patients find it takes longer than that or they need to use it for more than 21 days, they should seek medical attention. The gel can cause severe allergic reactions, especially in people allergic to aspirin; patients who experience such reactions are advised to stop use and seek immediate medical care. Other concerns include potential for liver damage with extended use; the possibility of severe stomach bleeds; and risk of heart attack, heart failure, and stroke.
The gel will no longer be available in prescription form.
Full prescribing information can be found on the FDA website, as can the full news release regarding this approval.
The Food and Drug Administration has approved formerly prescription-only Voltaren Arthritis Pain (diclofenac sodium topical gel, 1%) for nonprescription use via a process known as a prescription to over-the-counter (Rx-to-OTC) switch, according to a news release from the agency.
“As a result of the Rx-to-OTC switch process, many products sold over the counter today use ingredients or dosage strengths that were available only by prescription 30 years ago,” Karen Mahoney, MD, acting deputy director of the Office of Nonprescription Drugs in the FDA’s Center for Drug Evaluation and Research, said in the release.
This switch to nonprescription status is usually initiated by the manufacturer, who must provide data that demonstrates the drug in question is both safe and effective as self-medication in accordance with the proposed labeling and that consumers can use it safely and effectively without the supervision of a health care professional.
This particular therapy is a topical NSAID gel and was first approved by the FDA in 2007 with the indication for relief of osteoarthritis pain. It can take 7 days to have an effect, but if patients find it takes longer than that or they need to use it for more than 21 days, they should seek medical attention. The gel can cause severe allergic reactions, especially in people allergic to aspirin; patients who experience such reactions are advised to stop use and seek immediate medical care. Other concerns include potential for liver damage with extended use; the possibility of severe stomach bleeds; and risk of heart attack, heart failure, and stroke.
The gel will no longer be available in prescription form.
Full prescribing information can be found on the FDA website, as can the full news release regarding this approval.
The Food and Drug Administration has approved formerly prescription-only Voltaren Arthritis Pain (diclofenac sodium topical gel, 1%) for nonprescription use via a process known as a prescription to over-the-counter (Rx-to-OTC) switch, according to a news release from the agency.
“As a result of the Rx-to-OTC switch process, many products sold over the counter today use ingredients or dosage strengths that were available only by prescription 30 years ago,” Karen Mahoney, MD, acting deputy director of the Office of Nonprescription Drugs in the FDA’s Center for Drug Evaluation and Research, said in the release.
This switch to nonprescription status is usually initiated by the manufacturer, who must provide data that demonstrates the drug in question is both safe and effective as self-medication in accordance with the proposed labeling and that consumers can use it safely and effectively without the supervision of a health care professional.
This particular therapy is a topical NSAID gel and was first approved by the FDA in 2007 with the indication for relief of osteoarthritis pain. It can take 7 days to have an effect, but if patients find it takes longer than that or they need to use it for more than 21 days, they should seek medical attention. The gel can cause severe allergic reactions, especially in people allergic to aspirin; patients who experience such reactions are advised to stop use and seek immediate medical care. Other concerns include potential for liver damage with extended use; the possibility of severe stomach bleeds; and risk of heart attack, heart failure, and stroke.
The gel will no longer be available in prescription form.
Full prescribing information can be found on the FDA website, as can the full news release regarding this approval.
Low-dose methotrexate trial pins down adverse event rates
A new study has found an elevated risk of some adverse events in patients treated with low-dose methotrexate, compared with patients treated with placebo.
“The data presented here provide an important source of new evidence to improve the monitoring guidelines and safe prescribing of LD-MTX [low-dose methotrexate],” wrote Daniel H. Solomon, MD, of Brigham and Women’s Hospital and Harvard Medical School in Boston, and coauthors. The study was published in Annals of Internal Medicine.

To determine the rates of adverse events (AEs) among LD-MTX users, along with assessing the risks of certain predefined AEs, the researchers enrolled 6,158 patients in the Cardiovascular Inflammation Reduction Trial (CIRT) and randomized 4,786 of those patients to two groups: those receiving LD-MTX (n = 2,391) and those receiving placebo (n = 2,395). The median dose was 15 mg per week, and median follow-up was 23 months. All participants in CIRT had a history of cardiovascular disease, along with diabetes or metabolic syndrome. Just over 81% of the participants were male, and nearly 85% were white. Their median age was nearly 66 years.
Of the participants in the LD-MTX group, 2,156 (90.2%) had an AE and 2,080 (87.0%) had an AE of interest, which included infectious, hematologic, pulmonary, hepatic, cancerous, and gastrointestinal AEs. Of the participants in the placebo group, 2,076 (86.7%) had an AE and 1,951 (81.5%) had an AE of interest. As such, the relative rate of an AE of interest was 17% higher in the LD-MTX group (hazard ratio, 1.17; 95% confidence interval, 1.10-1.25).
In regard to specific types of AEs, the rates of gastrointestinal (HR, 1.23; 95% CI, 1.03-1.47), pulmonary (HR, 1.42; 95% CI, 1.14-1.77), infectious (HR, 1.15; 95% CI, 1.01-1.30) and hematologic (HR, 1.22; 95% CI, 1.11-1.34) were higher for participants in the LD-MTX group. Five cases of cirrhosis were found in the LD-MTX group, compared with none in the placebo group; none of the patients with cirrhosis had severe liver test abnormalities before their diagnosis. While the risk of cancer overall was not elevated in the LD-MTX group, 53 participants in that group developed skin cancer, compared with 26 in the placebo group (HR, 2.04; 95% CI, 1.28-3.26). Renal AEs were among the few that decreased in LD-MTX users (HR, 0.85; 95% CI, 0.78-0.93).
“Methotrexate has become the standard of care for RA patients,” Dr. Solomon said in an interview, “and because it worked so well, we accepted it without large placebo-controlled trials and without a precise understanding of the risk factors for AEs. Until this study, our evidence basis for the side-effect profile was relatively weak.
“We had a limited data set but decades of experience,” he added. “Now we have better evidence, for example, that methotrexate is associated with elevations in liver function tests. We even found five cases of cirrhosis. And the people who developed cirrhosis didn’t have severe test abnormalities; just minor ones over many months. So now we have a better understanding of the potential impact of minor, yet chronic abnormalities.”
Dr. Solomon and coauthors acknowledged their study’s limitations, including CIRT not including patients with systemic rheumatic disease and the possibility that participants did not report AEs that occurred in between routine study visits. In addition, although the median follow-up of nearly 2 years was longer than in other LD-MTX trials, they noted that “it may still be too short to observe some AEs that require long-term exposure.”
Dr. Solomon and colleagues should be commended for undertaking a long-awaited randomized, placebo-controlled trial that adds much-needed insight into how and when to monitor patients being treated with MTX, Vivian P. Bykerk, MD, of the Hospital for Special Surgery and Weill Cornell Medical College in New York, wrote in an editorial (Ann Intern Med. 2020 Feb 17. doi: 10.7326/M20-0435).

Dr. Bykerk noted that although the results may not be applicable to patients with RA and other inflammatory arthritides who are treated with MTX – RA patients in particular are younger, more often female, have lower rates of diabetes, and usually receive higher doses than those used in CIRT — the risk estimates from the CIRT study are “largely congruent with those expected in MTX-treated patients with rheumatic diseases.”
Regardless, she emphasized that this is a step in a much-needed direction, reminding physicians that “MTX use has inherent risks” and that its AEs, although infrequent, are clinically serious.
The National Institutes of Health funded the study. Various authors reported receiving grants from the National Heart, Lung, and Blood Institute, along with grants, research support, and personal fees from numerous pharmaceutical companies before and during the study. Dr. Bykerk reported receiving personal fees, grants, and nonfinancial support from pharmaceutical companies, foundations, and the NIH.
SOURCE: Solomon DH et al. Ann Intern Med. 2020 Feb 17. doi: 10.7326/M19-3369.
A new study has found an elevated risk of some adverse events in patients treated with low-dose methotrexate, compared with patients treated with placebo.
“The data presented here provide an important source of new evidence to improve the monitoring guidelines and safe prescribing of LD-MTX [low-dose methotrexate],” wrote Daniel H. Solomon, MD, of Brigham and Women’s Hospital and Harvard Medical School in Boston, and coauthors. The study was published in Annals of Internal Medicine.
To determine the rates of adverse events (AEs) among LD-MTX users, along with assessing the risks of certain predefined AEs, the researchers enrolled 6,158 patients in the Cardiovascular Inflammation Reduction Trial (CIRT) and randomized 4,786 of those patients to two groups: those receiving LD-MTX (n = 2,391) and those receiving placebo (n = 2,395). The median dose was 15 mg per week, and median follow-up was 23 months. All participants in CIRT had a history of cardiovascular disease, along with diabetes or metabolic syndrome. Just over 81% of the participants were male, and nearly 85% were white. Their median age was nearly 66 years.
Of the participants in the LD-MTX group, 2,156 (90.2%) had an AE and 2,080 (87.0%) had an AE of interest, which included infectious, hematologic, pulmonary, hepatic, cancerous, and gastrointestinal AEs. Of the participants in the placebo group, 2,076 (86.7%) had an AE and 1,951 (81.5%) had an AE of interest. As such, the relative rate of an AE of interest was 17% higher in the LD-MTX group (hazard ratio, 1.17; 95% confidence interval, 1.10-1.25).
In regard to specific types of AEs, the rates of gastrointestinal (HR, 1.23; 95% CI, 1.03-1.47), pulmonary (HR, 1.42; 95% CI, 1.14-1.77), infectious (HR, 1.15; 95% CI, 1.01-1.30) and hematologic (HR, 1.22; 95% CI, 1.11-1.34) were higher for participants in the LD-MTX group. Five cases of cirrhosis were found in the LD-MTX group, compared with none in the placebo group; none of the patients with cirrhosis had severe liver test abnormalities before their diagnosis. While the risk of cancer overall was not elevated in the LD-MTX group, 53 participants in that group developed skin cancer, compared with 26 in the placebo group (HR, 2.04; 95% CI, 1.28-3.26). Renal AEs were among the few that decreased in LD-MTX users (HR, 0.85; 95% CI, 0.78-0.93).
“Methotrexate has become the standard of care for RA patients,” Dr. Solomon said in an interview, “and because it worked so well, we accepted it without large placebo-controlled trials and without a precise understanding of the risk factors for AEs. Until this study, our evidence basis for the side-effect profile was relatively weak.
“We had a limited data set but decades of experience,” he added. “Now we have better evidence, for example, that methotrexate is associated with elevations in liver function tests. We even found five cases of cirrhosis. And the people who developed cirrhosis didn’t have severe test abnormalities; just minor ones over many months. So now we have a better understanding of the potential impact of minor, yet chronic abnormalities.”
Dr. Solomon and coauthors acknowledged their study’s limitations, including CIRT not including patients with systemic rheumatic disease and the possibility that participants did not report AEs that occurred in between routine study visits. In addition, although the median follow-up of nearly 2 years was longer than in other LD-MTX trials, they noted that “it may still be too short to observe some AEs that require long-term exposure.”
Dr. Solomon and colleagues should be commended for undertaking a long-awaited randomized, placebo-controlled trial that adds much-needed insight into how and when to monitor patients being treated with MTX, Vivian P. Bykerk, MD, of the Hospital for Special Surgery and Weill Cornell Medical College in New York, wrote in an editorial (Ann Intern Med. 2020 Feb 17. doi: 10.7326/M20-0435).
Dr. Bykerk noted that although the results may not be applicable to patients with RA and other inflammatory arthritides who are treated with MTX – RA patients in particular are younger, more often female, have lower rates of diabetes, and usually receive higher doses than those used in CIRT — the risk estimates from the CIRT study are “largely congruent with those expected in MTX-treated patients with rheumatic diseases.”
Regardless, she emphasized that this is a step in a much-needed direction, reminding physicians that “MTX use has inherent risks” and that its AEs, although infrequent, are clinically serious.
The National Institutes of Health funded the study. Various authors reported receiving grants from the National Heart, Lung, and Blood Institute, along with grants, research support, and personal fees from numerous pharmaceutical companies before and during the study. Dr. Bykerk reported receiving personal fees, grants, and nonfinancial support from pharmaceutical companies, foundations, and the NIH.
SOURCE: Solomon DH et al. Ann Intern Med. 2020 Feb 17. doi: 10.7326/M19-3369.
A new study has found an elevated risk of some adverse events in patients treated with low-dose methotrexate, compared with patients treated with placebo.
“The data presented here provide an important source of new evidence to improve the monitoring guidelines and safe prescribing of LD-MTX [low-dose methotrexate],” wrote Daniel H. Solomon, MD, of Brigham and Women’s Hospital and Harvard Medical School in Boston, and coauthors. The study was published in Annals of Internal Medicine.
To determine the rates of adverse events (AEs) among LD-MTX users, along with assessing the risks of certain predefined AEs, the researchers enrolled 6,158 patients in the Cardiovascular Inflammation Reduction Trial (CIRT) and randomized 4,786 of those patients to two groups: those receiving LD-MTX (n = 2,391) and those receiving placebo (n = 2,395). The median dose was 15 mg per week, and median follow-up was 23 months. All participants in CIRT had a history of cardiovascular disease, along with diabetes or metabolic syndrome. Just over 81% of the participants were male, and nearly 85% were white. Their median age was nearly 66 years.
Of the participants in the LD-MTX group, 2,156 (90.2%) had an AE and 2,080 (87.0%) had an AE of interest, which included infectious, hematologic, pulmonary, hepatic, cancerous, and gastrointestinal AEs. Of the participants in the placebo group, 2,076 (86.7%) had an AE and 1,951 (81.5%) had an AE of interest. As such, the relative rate of an AE of interest was 17% higher in the LD-MTX group (hazard ratio, 1.17; 95% confidence interval, 1.10-1.25).
In regard to specific types of AEs, the rates of gastrointestinal (HR, 1.23; 95% CI, 1.03-1.47), pulmonary (HR, 1.42; 95% CI, 1.14-1.77), infectious (HR, 1.15; 95% CI, 1.01-1.30) and hematologic (HR, 1.22; 95% CI, 1.11-1.34) were higher for participants in the LD-MTX group. Five cases of cirrhosis were found in the LD-MTX group, compared with none in the placebo group; none of the patients with cirrhosis had severe liver test abnormalities before their diagnosis. While the risk of cancer overall was not elevated in the LD-MTX group, 53 participants in that group developed skin cancer, compared with 26 in the placebo group (HR, 2.04; 95% CI, 1.28-3.26). Renal AEs were among the few that decreased in LD-MTX users (HR, 0.85; 95% CI, 0.78-0.93).
“Methotrexate has become the standard of care for RA patients,” Dr. Solomon said in an interview, “and because it worked so well, we accepted it without large placebo-controlled trials and without a precise understanding of the risk factors for AEs. Until this study, our evidence basis for the side-effect profile was relatively weak.
“We had a limited data set but decades of experience,” he added. “Now we have better evidence, for example, that methotrexate is associated with elevations in liver function tests. We even found five cases of cirrhosis. And the people who developed cirrhosis didn’t have severe test abnormalities; just minor ones over many months. So now we have a better understanding of the potential impact of minor, yet chronic abnormalities.”
Dr. Solomon and coauthors acknowledged their study’s limitations, including CIRT not including patients with systemic rheumatic disease and the possibility that participants did not report AEs that occurred in between routine study visits. In addition, although the median follow-up of nearly 2 years was longer than in other LD-MTX trials, they noted that “it may still be too short to observe some AEs that require long-term exposure.”
Dr. Solomon and colleagues should be commended for undertaking a long-awaited randomized, placebo-controlled trial that adds much-needed insight into how and when to monitor patients being treated with MTX, Vivian P. Bykerk, MD, of the Hospital for Special Surgery and Weill Cornell Medical College in New York, wrote in an editorial (Ann Intern Med. 2020 Feb 17. doi: 10.7326/M20-0435).
Dr. Bykerk noted that although the results may not be applicable to patients with RA and other inflammatory arthritides who are treated with MTX – RA patients in particular are younger, more often female, have lower rates of diabetes, and usually receive higher doses than those used in CIRT — the risk estimates from the CIRT study are “largely congruent with those expected in MTX-treated patients with rheumatic diseases.”
Regardless, she emphasized that this is a step in a much-needed direction, reminding physicians that “MTX use has inherent risks” and that its AEs, although infrequent, are clinically serious.
The National Institutes of Health funded the study. Various authors reported receiving grants from the National Heart, Lung, and Blood Institute, along with grants, research support, and personal fees from numerous pharmaceutical companies before and during the study. Dr. Bykerk reported receiving personal fees, grants, and nonfinancial support from pharmaceutical companies, foundations, and the NIH.
SOURCE: Solomon DH et al. Ann Intern Med. 2020 Feb 17. doi: 10.7326/M19-3369.
FROM ANNALS OF INTERNAL MEDICINE
Another round of research shows ketamine may help alcoholism
More research suggests that a single infusion of ketamine combined with counseling may help alcohol-dependent patients curb their drinking.
In a pilot study of 40 participants, those who were randomly assigned to receive intravenous ketamine plus outpatient motivational enhancement therapy (MET) showed greater abstinence rates, longer time to relapse, and fewer heavy drinking days than did those who received MET plus midazolam.
The findings support a U.K. study published late last year showing that a single dose of intravenous ketamine plus therapy that focused on reactivating drinking-related “maladaptive reward memories” reduced drinking urges and alcohol intake more than just ketamine or a placebo infusion alone (Nat Commun. 2019 Nov 26;10[1]:5187).
of the New York State Psychiatric Institute, Columbia University, New York, said in an interview.
“It’s an important area of research to understand in order to make behavioral treatments more effective, and ketamine appears to have the properties to address those vulnerabilities,” Dr. Dakwar said.
The study was published in the American Journal of Psychiatry (2019 Dec 2. doi: 10.1176/appi.ajp.2019.19070684).
Real-world approach
Pathologic alcohol use is responsible for an estimated 3.8% of all deaths globally, yet current interventions for alcohol use disorder have limited efficacy, the researchers noted.
New treatments with innovative mechanisms would be valuable, they added.
Ketamine is a high-affinity N-methyl-d-aspartate receptor (NMDAR) antagonist.
Previously, research offered “promising results” with the use of ketamine for cocaine use disorder, including increased motivation to quit and decreased craving, Dr. Dakwar noted.
“Those results led us to think about how ketamine might be helpful for other substance use disorders, especially given the overlap in clinical vulnerabilities and epidemiology,” he said.
The study from the U.K. researchers was conducted in 90 patients with harmful drinking behavior but who had not been diagnosed with alcohol use disorder.
Dr. Dakwar noted that this was “a nontreatment study. None of the people there had alcohol use disorder; they were heavy drinkers. Also, the effects there were fairly modest.
“My interest was how to integrate ketamine into a clinical, real-world framework that could be helpful for people,” he added.
The study included 40 participants (52.5% women; 70.3% white; mean age, 53 years) with alcohol dependence whose average consumption was five drinks per day.
All entered a 5-week outpatient program of MET, which involved engaging in strategies to promote motivation and self-directed change.
During the program’s second week, the participants were randomly assigned to received a 52-minute IV infusion of ketamine 0.71 mg/kg (n = 17) or the benzodiazepine midazolam 0.025 mg/kg (n = 23).
This ketamine dose was selected “because it was the highest dose tolerated by participants in preliminary studies,” the researchers reported.
“Midazolam was chosen as the active control because it alters consciousness without any known persistent ... effect on alcohol dependence,” they added.
The “timeline follow back method” was used to assess alcohol use after treatment. Abstinence was confirmed by measuring urine ethyl glucuronide levels with urine toxicology tests.
Other measures included use of a visual analogue scale, the Clinical Institute Withdrawal Assessment, and the modified Perceived Stress Scale.
Primary outcome met
Results showed that 47.1% of the ketamine group and 59.1% of the midazolam group used alcohol during the 21 days after treatment infusion; 17.6% and 40.9%, respectively, had a heavy drinking day.
For the primary outcome measure of alcohol abstinence, the “quadratic effect of time was significant” (P = .004), as was time-by-treatment interaction (P less than .001).
Although the model-estimated proportions of alcohol abstinence remained stable for the ketamine group for 21 days post infusion, the proportions decreased significantly for the control group.
The odds of having a heavy drinking day did not change significantly after treatment for the ketamine group (odds ratio, 0.98; P = .74) but increased significantly with each postinfusion day for the midazolam group (OR, 1.19; P less than .001).
For the ketamine group, time to relapse was also significantly longer (P = .04).
No significant differences were found between the groups in rates of withdrawal, craving, or stress sensitivity.
A new direction?
The most common adverse events after treatment were sedation, seen in 12 members of the midazolam group and in 8 members of the ketamine group, and headache, seen in four and six members, respectively.
Although two ketamine-group members experienced mild agitation for up to 1 hour post infusion, no incidents of persistent psychoactive effects were reported in either group.
No participants who received ketamine dropped out during the study period; among those who received midazolam, six dropped out.
“These preliminary data suggest new directions in integrated pharmacotherapy-behavioral treatments for alcohol use disorder,” the investigators wrote.
However, a larger patient population will be needed in future research in order to “replicate these promising results,” they added.
Dr. Dakwar noted that the time to first drink after treatment was comparable between the groups.
“But what was different in the ketamine group was that they didn’t continue drinking after that first drink. They didn’t initiate heavy drinking, they didn’t relapse, they were able to bounce back and stay with the program,” he said.
“It was surprising but still consistent with the central hypothesis that ketamine provides this opportunity for setting the foundation for the requisite commitment so that, once things become difficult, they’re still able to maintain recovery,” Dr. Dakwar said.
‘Provocative findings’
In an accompanying editorial, Sanjay J. Mathew, MD, of the department of psychiatry and behavioral sciences at Baylor College of Medicine in Houston, and Rebecca B. Price, PhD, of the department of psychiatry at the University of Pittsburgh, noted that ketamine’s effects on abstinence “were robust” in this trial.
“It is also noteworthy that, in spite of recruiting from a population of patients with active and significant substance use history (a group that has routinely been excluded from ketamine trials in depression), no participant showed evidence of new drug-seeking behaviors,” Dr. Mathew and Dr. Price wrote.
“Overall, these findings are provocative and hypothesis generating but certainly not definitive because of the small sample size,” they add.
Other limitations cited include the short follow-up period and the fact that only half of the participants were available for a 6-month follow-up telephone interview. In addition, generalizability was limited because the population did not have additional medical or psychiatric illnesses or additional substance use disorders, the editorialists wrote.
Because of the limitations, the investigators “are appropriately circumspect about the immediate clinical implications of this small pilot study.”
Still, the results “affirm the potential of rational combinatorial approaches for a vexing medical and public health problem,” Dr. Mathew and Dr. Price concluded.
The study was funded by grants from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the New York State Psychiatric Institute. The study authors and Dr. Price reported no relevant financial relationships. Dr Mathew reported serving as a consultant to or having received research support from several companies, including Alkermes, Allergan, Clexio Biosciences, and Janssen. The original article includes a full list of his disclosures.
A version of this article first appeared on Medscape.com.
More research suggests that a single infusion of ketamine combined with counseling may help alcohol-dependent patients curb their drinking.
In a pilot study of 40 participants, those who were randomly assigned to receive intravenous ketamine plus outpatient motivational enhancement therapy (MET) showed greater abstinence rates, longer time to relapse, and fewer heavy drinking days than did those who received MET plus midazolam.
The findings support a U.K. study published late last year showing that a single dose of intravenous ketamine plus therapy that focused on reactivating drinking-related “maladaptive reward memories” reduced drinking urges and alcohol intake more than just ketamine or a placebo infusion alone (Nat Commun. 2019 Nov 26;10[1]:5187).
of the New York State Psychiatric Institute, Columbia University, New York, said in an interview.
“It’s an important area of research to understand in order to make behavioral treatments more effective, and ketamine appears to have the properties to address those vulnerabilities,” Dr. Dakwar said.
The study was published in the American Journal of Psychiatry (2019 Dec 2. doi: 10.1176/appi.ajp.2019.19070684).
Real-world approach
Pathologic alcohol use is responsible for an estimated 3.8% of all deaths globally, yet current interventions for alcohol use disorder have limited efficacy, the researchers noted.
New treatments with innovative mechanisms would be valuable, they added.
Ketamine is a high-affinity N-methyl-d-aspartate receptor (NMDAR) antagonist.
Previously, research offered “promising results” with the use of ketamine for cocaine use disorder, including increased motivation to quit and decreased craving, Dr. Dakwar noted.
“Those results led us to think about how ketamine might be helpful for other substance use disorders, especially given the overlap in clinical vulnerabilities and epidemiology,” he said.
The study from the U.K. researchers was conducted in 90 patients with harmful drinking behavior but who had not been diagnosed with alcohol use disorder.
Dr. Dakwar noted that this was “a nontreatment study. None of the people there had alcohol use disorder; they were heavy drinkers. Also, the effects there were fairly modest.
“My interest was how to integrate ketamine into a clinical, real-world framework that could be helpful for people,” he added.
The study included 40 participants (52.5% women; 70.3% white; mean age, 53 years) with alcohol dependence whose average consumption was five drinks per day.
All entered a 5-week outpatient program of MET, which involved engaging in strategies to promote motivation and self-directed change.
During the program’s second week, the participants were randomly assigned to received a 52-minute IV infusion of ketamine 0.71 mg/kg (n = 17) or the benzodiazepine midazolam 0.025 mg/kg (n = 23).
This ketamine dose was selected “because it was the highest dose tolerated by participants in preliminary studies,” the researchers reported.
“Midazolam was chosen as the active control because it alters consciousness without any known persistent ... effect on alcohol dependence,” they added.
The “timeline follow back method” was used to assess alcohol use after treatment. Abstinence was confirmed by measuring urine ethyl glucuronide levels with urine toxicology tests.
Other measures included use of a visual analogue scale, the Clinical Institute Withdrawal Assessment, and the modified Perceived Stress Scale.
Primary outcome met
Results showed that 47.1% of the ketamine group and 59.1% of the midazolam group used alcohol during the 21 days after treatment infusion; 17.6% and 40.9%, respectively, had a heavy drinking day.
For the primary outcome measure of alcohol abstinence, the “quadratic effect of time was significant” (P = .004), as was time-by-treatment interaction (P less than .001).
Although the model-estimated proportions of alcohol abstinence remained stable for the ketamine group for 21 days post infusion, the proportions decreased significantly for the control group.
The odds of having a heavy drinking day did not change significantly after treatment for the ketamine group (odds ratio, 0.98; P = .74) but increased significantly with each postinfusion day for the midazolam group (OR, 1.19; P less than .001).
For the ketamine group, time to relapse was also significantly longer (P = .04).
No significant differences were found between the groups in rates of withdrawal, craving, or stress sensitivity.
A new direction?
The most common adverse events after treatment were sedation, seen in 12 members of the midazolam group and in 8 members of the ketamine group, and headache, seen in four and six members, respectively.
Although two ketamine-group members experienced mild agitation for up to 1 hour post infusion, no incidents of persistent psychoactive effects were reported in either group.
No participants who received ketamine dropped out during the study period; among those who received midazolam, six dropped out.
“These preliminary data suggest new directions in integrated pharmacotherapy-behavioral treatments for alcohol use disorder,” the investigators wrote.
However, a larger patient population will be needed in future research in order to “replicate these promising results,” they added.
Dr. Dakwar noted that the time to first drink after treatment was comparable between the groups.
“But what was different in the ketamine group was that they didn’t continue drinking after that first drink. They didn’t initiate heavy drinking, they didn’t relapse, they were able to bounce back and stay with the program,” he said.
“It was surprising but still consistent with the central hypothesis that ketamine provides this opportunity for setting the foundation for the requisite commitment so that, once things become difficult, they’re still able to maintain recovery,” Dr. Dakwar said.
‘Provocative findings’
In an accompanying editorial, Sanjay J. Mathew, MD, of the department of psychiatry and behavioral sciences at Baylor College of Medicine in Houston, and Rebecca B. Price, PhD, of the department of psychiatry at the University of Pittsburgh, noted that ketamine’s effects on abstinence “were robust” in this trial.
“It is also noteworthy that, in spite of recruiting from a population of patients with active and significant substance use history (a group that has routinely been excluded from ketamine trials in depression), no participant showed evidence of new drug-seeking behaviors,” Dr. Mathew and Dr. Price wrote.
“Overall, these findings are provocative and hypothesis generating but certainly not definitive because of the small sample size,” they add.
Other limitations cited include the short follow-up period and the fact that only half of the participants were available for a 6-month follow-up telephone interview. In addition, generalizability was limited because the population did not have additional medical or psychiatric illnesses or additional substance use disorders, the editorialists wrote.
Because of the limitations, the investigators “are appropriately circumspect about the immediate clinical implications of this small pilot study.”
Still, the results “affirm the potential of rational combinatorial approaches for a vexing medical and public health problem,” Dr. Mathew and Dr. Price concluded.
The study was funded by grants from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the New York State Psychiatric Institute. The study authors and Dr. Price reported no relevant financial relationships. Dr Mathew reported serving as a consultant to or having received research support from several companies, including Alkermes, Allergan, Clexio Biosciences, and Janssen. The original article includes a full list of his disclosures.
A version of this article first appeared on Medscape.com.
More research suggests that a single infusion of ketamine combined with counseling may help alcohol-dependent patients curb their drinking.
In a pilot study of 40 participants, those who were randomly assigned to receive intravenous ketamine plus outpatient motivational enhancement therapy (MET) showed greater abstinence rates, longer time to relapse, and fewer heavy drinking days than did those who received MET plus midazolam.
The findings support a U.K. study published late last year showing that a single dose of intravenous ketamine plus therapy that focused on reactivating drinking-related “maladaptive reward memories” reduced drinking urges and alcohol intake more than just ketamine or a placebo infusion alone (Nat Commun. 2019 Nov 26;10[1]:5187).
of the New York State Psychiatric Institute, Columbia University, New York, said in an interview.
“It’s an important area of research to understand in order to make behavioral treatments more effective, and ketamine appears to have the properties to address those vulnerabilities,” Dr. Dakwar said.
The study was published in the American Journal of Psychiatry (2019 Dec 2. doi: 10.1176/appi.ajp.2019.19070684).
Real-world approach
Pathologic alcohol use is responsible for an estimated 3.8% of all deaths globally, yet current interventions for alcohol use disorder have limited efficacy, the researchers noted.
New treatments with innovative mechanisms would be valuable, they added.
Ketamine is a high-affinity N-methyl-d-aspartate receptor (NMDAR) antagonist.
Previously, research offered “promising results” with the use of ketamine for cocaine use disorder, including increased motivation to quit and decreased craving, Dr. Dakwar noted.
“Those results led us to think about how ketamine might be helpful for other substance use disorders, especially given the overlap in clinical vulnerabilities and epidemiology,” he said.
The study from the U.K. researchers was conducted in 90 patients with harmful drinking behavior but who had not been diagnosed with alcohol use disorder.
Dr. Dakwar noted that this was “a nontreatment study. None of the people there had alcohol use disorder; they were heavy drinkers. Also, the effects there were fairly modest.
“My interest was how to integrate ketamine into a clinical, real-world framework that could be helpful for people,” he added.
The study included 40 participants (52.5% women; 70.3% white; mean age, 53 years) with alcohol dependence whose average consumption was five drinks per day.
All entered a 5-week outpatient program of MET, which involved engaging in strategies to promote motivation and self-directed change.
During the program’s second week, the participants were randomly assigned to received a 52-minute IV infusion of ketamine 0.71 mg/kg (n = 17) or the benzodiazepine midazolam 0.025 mg/kg (n = 23).
This ketamine dose was selected “because it was the highest dose tolerated by participants in preliminary studies,” the researchers reported.
“Midazolam was chosen as the active control because it alters consciousness without any known persistent ... effect on alcohol dependence,” they added.
The “timeline follow back method” was used to assess alcohol use after treatment. Abstinence was confirmed by measuring urine ethyl glucuronide levels with urine toxicology tests.
Other measures included use of a visual analogue scale, the Clinical Institute Withdrawal Assessment, and the modified Perceived Stress Scale.
Primary outcome met
Results showed that 47.1% of the ketamine group and 59.1% of the midazolam group used alcohol during the 21 days after treatment infusion; 17.6% and 40.9%, respectively, had a heavy drinking day.
For the primary outcome measure of alcohol abstinence, the “quadratic effect of time was significant” (P = .004), as was time-by-treatment interaction (P less than .001).
Although the model-estimated proportions of alcohol abstinence remained stable for the ketamine group for 21 days post infusion, the proportions decreased significantly for the control group.
The odds of having a heavy drinking day did not change significantly after treatment for the ketamine group (odds ratio, 0.98; P = .74) but increased significantly with each postinfusion day for the midazolam group (OR, 1.19; P less than .001).
For the ketamine group, time to relapse was also significantly longer (P = .04).
No significant differences were found between the groups in rates of withdrawal, craving, or stress sensitivity.
A new direction?
The most common adverse events after treatment were sedation, seen in 12 members of the midazolam group and in 8 members of the ketamine group, and headache, seen in four and six members, respectively.
Although two ketamine-group members experienced mild agitation for up to 1 hour post infusion, no incidents of persistent psychoactive effects were reported in either group.
No participants who received ketamine dropped out during the study period; among those who received midazolam, six dropped out.
“These preliminary data suggest new directions in integrated pharmacotherapy-behavioral treatments for alcohol use disorder,” the investigators wrote.
However, a larger patient population will be needed in future research in order to “replicate these promising results,” they added.
Dr. Dakwar noted that the time to first drink after treatment was comparable between the groups.
“But what was different in the ketamine group was that they didn’t continue drinking after that first drink. They didn’t initiate heavy drinking, they didn’t relapse, they were able to bounce back and stay with the program,” he said.
“It was surprising but still consistent with the central hypothesis that ketamine provides this opportunity for setting the foundation for the requisite commitment so that, once things become difficult, they’re still able to maintain recovery,” Dr. Dakwar said.
‘Provocative findings’
In an accompanying editorial, Sanjay J. Mathew, MD, of the department of psychiatry and behavioral sciences at Baylor College of Medicine in Houston, and Rebecca B. Price, PhD, of the department of psychiatry at the University of Pittsburgh, noted that ketamine’s effects on abstinence “were robust” in this trial.
“It is also noteworthy that, in spite of recruiting from a population of patients with active and significant substance use history (a group that has routinely been excluded from ketamine trials in depression), no participant showed evidence of new drug-seeking behaviors,” Dr. Mathew and Dr. Price wrote.
“Overall, these findings are provocative and hypothesis generating but certainly not definitive because of the small sample size,” they add.
Other limitations cited include the short follow-up period and the fact that only half of the participants were available for a 6-month follow-up telephone interview. In addition, generalizability was limited because the population did not have additional medical or psychiatric illnesses or additional substance use disorders, the editorialists wrote.
Because of the limitations, the investigators “are appropriately circumspect about the immediate clinical implications of this small pilot study.”
Still, the results “affirm the potential of rational combinatorial approaches for a vexing medical and public health problem,” Dr. Mathew and Dr. Price concluded.
The study was funded by grants from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the New York State Psychiatric Institute. The study authors and Dr. Price reported no relevant financial relationships. Dr Mathew reported serving as a consultant to or having received research support from several companies, including Alkermes, Allergan, Clexio Biosciences, and Janssen. The original article includes a full list of his disclosures.
A version of this article first appeared on Medscape.com.
Tramadol use for noncancer pain linked with increased hip fracture risk
The risk of hip fracture was higher among patients treated with tramadol for chronic noncancer pain than among those treated with other commonly used NSAIDs in a large population-based cohort in the United Kingdom.
The incidence of hip fracture over a 12-month period among 293,912 propensity score-matched tramadol and codeine recipients in The Health Improvement Network (THIN) database during 2000-2017 was 3.7 vs. 2.9 per 1,000 person-years, respectively (hazard ratio for hip fracture, 1.28), Jie Wei, PhD, of Xiangya Hospital, Central South University, Changsha, China, and colleagues reported in the Journal of Bone and Mineral Research.
Hip fracture incidence per 1,000 person-years was also higher in propensity score–matched cohorts of patients receiving tramadol vs. naproxen (2.9 vs. 1.7; HR, 1.69), ibuprofen (3.4 vs. 2.0; HR, 1.65), celecoxib (3.4 vs. 1.8; HR, 1.85), or etoricoxib (2.9 vs. 1.5; HR, 1.96), the investigators found.
Tramadol is considered a weak opioid and is commonly used for the treatment of pain based on a lower perceived risk of serious cardiovascular and gastrointestinal effects versus NSAIDs, and of addiction and respiratory depression versus traditional opioids, they explained. Several professional organizations also have “strongly or conditionally recommended tramadol” as a first- or second-line treatment for conditions such as osteoarthritis, fibromyalgia, and chronic low back pain.
The potential mechanisms for the association between tramadol and hip fracture require further study, but “[c]onsidering the significant impact of hip fracture on morbidity, mortality, and health care costs, our results point to the need to consider tramadol’s associated risk of fracture in clinical practice and treatment guidelines,” they concluded.
This study was supported by the National Institutes of Health, the National Natural Science Foundation of China, and the Postdoctoral Science Foundation of Central South University. The authors reported having no conflicts of interest.
SOURCE: Wei J et al. J Bone Miner Res. 2019 Feb 5. doi: 10.1002/jbmr.3935.
The risk of hip fracture was higher among patients treated with tramadol for chronic noncancer pain than among those treated with other commonly used NSAIDs in a large population-based cohort in the United Kingdom.
The incidence of hip fracture over a 12-month period among 293,912 propensity score-matched tramadol and codeine recipients in The Health Improvement Network (THIN) database during 2000-2017 was 3.7 vs. 2.9 per 1,000 person-years, respectively (hazard ratio for hip fracture, 1.28), Jie Wei, PhD, of Xiangya Hospital, Central South University, Changsha, China, and colleagues reported in the Journal of Bone and Mineral Research.
Hip fracture incidence per 1,000 person-years was also higher in propensity score–matched cohorts of patients receiving tramadol vs. naproxen (2.9 vs. 1.7; HR, 1.69), ibuprofen (3.4 vs. 2.0; HR, 1.65), celecoxib (3.4 vs. 1.8; HR, 1.85), or etoricoxib (2.9 vs. 1.5; HR, 1.96), the investigators found.
Tramadol is considered a weak opioid and is commonly used for the treatment of pain based on a lower perceived risk of serious cardiovascular and gastrointestinal effects versus NSAIDs, and of addiction and respiratory depression versus traditional opioids, they explained. Several professional organizations also have “strongly or conditionally recommended tramadol” as a first- or second-line treatment for conditions such as osteoarthritis, fibromyalgia, and chronic low back pain.
The potential mechanisms for the association between tramadol and hip fracture require further study, but “[c]onsidering the significant impact of hip fracture on morbidity, mortality, and health care costs, our results point to the need to consider tramadol’s associated risk of fracture in clinical practice and treatment guidelines,” they concluded.
This study was supported by the National Institutes of Health, the National Natural Science Foundation of China, and the Postdoctoral Science Foundation of Central South University. The authors reported having no conflicts of interest.
SOURCE: Wei J et al. J Bone Miner Res. 2019 Feb 5. doi: 10.1002/jbmr.3935.
The risk of hip fracture was higher among patients treated with tramadol for chronic noncancer pain than among those treated with other commonly used NSAIDs in a large population-based cohort in the United Kingdom.
The incidence of hip fracture over a 12-month period among 293,912 propensity score-matched tramadol and codeine recipients in The Health Improvement Network (THIN) database during 2000-2017 was 3.7 vs. 2.9 per 1,000 person-years, respectively (hazard ratio for hip fracture, 1.28), Jie Wei, PhD, of Xiangya Hospital, Central South University, Changsha, China, and colleagues reported in the Journal of Bone and Mineral Research.
Hip fracture incidence per 1,000 person-years was also higher in propensity score–matched cohorts of patients receiving tramadol vs. naproxen (2.9 vs. 1.7; HR, 1.69), ibuprofen (3.4 vs. 2.0; HR, 1.65), celecoxib (3.4 vs. 1.8; HR, 1.85), or etoricoxib (2.9 vs. 1.5; HR, 1.96), the investigators found.
Tramadol is considered a weak opioid and is commonly used for the treatment of pain based on a lower perceived risk of serious cardiovascular and gastrointestinal effects versus NSAIDs, and of addiction and respiratory depression versus traditional opioids, they explained. Several professional organizations also have “strongly or conditionally recommended tramadol” as a first- or second-line treatment for conditions such as osteoarthritis, fibromyalgia, and chronic low back pain.
The potential mechanisms for the association between tramadol and hip fracture require further study, but “[c]onsidering the significant impact of hip fracture on morbidity, mortality, and health care costs, our results point to the need to consider tramadol’s associated risk of fracture in clinical practice and treatment guidelines,” they concluded.
This study was supported by the National Institutes of Health, the National Natural Science Foundation of China, and the Postdoctoral Science Foundation of Central South University. The authors reported having no conflicts of interest.
SOURCE: Wei J et al. J Bone Miner Res. 2019 Feb 5. doi: 10.1002/jbmr.3935.
FROM THE JOURNAL OF BONE AND MINERAL RESEARCH
FDA approves novel pandemic influenza vaccine
The Food and Drug Administration has approved the first and only adjuvanted, cell-based pandemic vaccine to provide active immunization against the influenza A virus H5N1 strain.
Influenza A (H5N1) monovalent vaccine, adjuvanted (Audenz, Seqirus) is for use in individuals aged 6 months and older. It’s designed to be rapidly deployed to help protect the U.S. population and can be stockpiled for first responders in the event of a pandemic.
The vaccine and formulated prefilled syringes used in the vaccine are produced in a state-of-the-art production facility built and supported through a multiyear public-private partnership between Seqirus and the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response at the U.S. Department of Health & Human Services.
“Pandemic influenza viruses can be deadly and spread rapidly, making production of safe, effective vaccines essential in saving lives,” BARDA Director Rick Bright, PhD, said in a company news release.
“With this licensure – the latest FDA-approved vaccine to prevent H5N1 influenza — we celebrate a decade-long partnership to achieve health security goals set by the National Strategy for Pandemic Influenza and the 2019 Executive Order to speed the availability of influenza vaccine. Ultimately, this latest licensure means we can protect more people in an influenza pandemic,” said Bright.
“The approval of Audenz represents a key advance in influenza prevention and pandemic preparedness, combining leading-edge, cell-based manufacturing and adjuvant technologies,” Russell Basser, MD, chief scientist and senior vice president of research and development at Seqirus, said in the news release. “This pandemic influenza vaccine exemplifies our commitment to developing innovative technologies that can help provide rapid response during a pandemic emergency.”
Audenz had FDA fast track designation, a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need.
This article first appeared on Medscape.com.
The Food and Drug Administration has approved the first and only adjuvanted, cell-based pandemic vaccine to provide active immunization against the influenza A virus H5N1 strain.
Influenza A (H5N1) monovalent vaccine, adjuvanted (Audenz, Seqirus) is for use in individuals aged 6 months and older. It’s designed to be rapidly deployed to help protect the U.S. population and can be stockpiled for first responders in the event of a pandemic.
The vaccine and formulated prefilled syringes used in the vaccine are produced in a state-of-the-art production facility built and supported through a multiyear public-private partnership between Seqirus and the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response at the U.S. Department of Health & Human Services.
“Pandemic influenza viruses can be deadly and spread rapidly, making production of safe, effective vaccines essential in saving lives,” BARDA Director Rick Bright, PhD, said in a company news release.
“With this licensure – the latest FDA-approved vaccine to prevent H5N1 influenza — we celebrate a decade-long partnership to achieve health security goals set by the National Strategy for Pandemic Influenza and the 2019 Executive Order to speed the availability of influenza vaccine. Ultimately, this latest licensure means we can protect more people in an influenza pandemic,” said Bright.
“The approval of Audenz represents a key advance in influenza prevention and pandemic preparedness, combining leading-edge, cell-based manufacturing and adjuvant technologies,” Russell Basser, MD, chief scientist and senior vice president of research and development at Seqirus, said in the news release. “This pandemic influenza vaccine exemplifies our commitment to developing innovative technologies that can help provide rapid response during a pandemic emergency.”
Audenz had FDA fast track designation, a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need.
This article first appeared on Medscape.com.
The Food and Drug Administration has approved the first and only adjuvanted, cell-based pandemic vaccine to provide active immunization against the influenza A virus H5N1 strain.
Influenza A (H5N1) monovalent vaccine, adjuvanted (Audenz, Seqirus) is for use in individuals aged 6 months and older. It’s designed to be rapidly deployed to help protect the U.S. population and can be stockpiled for first responders in the event of a pandemic.
The vaccine and formulated prefilled syringes used in the vaccine are produced in a state-of-the-art production facility built and supported through a multiyear public-private partnership between Seqirus and the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response at the U.S. Department of Health & Human Services.
“Pandemic influenza viruses can be deadly and spread rapidly, making production of safe, effective vaccines essential in saving lives,” BARDA Director Rick Bright, PhD, said in a company news release.
“With this licensure – the latest FDA-approved vaccine to prevent H5N1 influenza — we celebrate a decade-long partnership to achieve health security goals set by the National Strategy for Pandemic Influenza and the 2019 Executive Order to speed the availability of influenza vaccine. Ultimately, this latest licensure means we can protect more people in an influenza pandemic,” said Bright.
“The approval of Audenz represents a key advance in influenza prevention and pandemic preparedness, combining leading-edge, cell-based manufacturing and adjuvant technologies,” Russell Basser, MD, chief scientist and senior vice president of research and development at Seqirus, said in the news release. “This pandemic influenza vaccine exemplifies our commitment to developing innovative technologies that can help provide rapid response during a pandemic emergency.”
Audenz had FDA fast track designation, a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need.
This article first appeared on Medscape.com.
Stimulant Medication Prescribing Practices Within a VA Health Care System
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
Lenvatinib/pembrolizumab has good activity in advanced RCC, other solid tumors
A combination of the tyrosine kinase inhibitor lenvatinib (Lenvima) and the immune checkpoint inhibitor pembrolizumab (Keytruda) was safe and showed promising activity against advanced renal cell carcinoma and other solid tumors in a phase 1b/2 study.
Overall response rates (ORR) at 24 weeks ranged from 63% for patients with advanced renal cell carcinomas (RCC) to 25% for patients with urothelial cancers, reported Matthew H. Taylor, MD, of Knight Cancer Institute at Oregon Health & Science University in Portland, and colleagues.
The findings from this study sparked additional clinical trials for patients with gastric cancer, gastroesophageal cancer, and differentiated thyroid cancer, and set the stage for larger phase 3 trials in patients with advanced RCC, endometrial cancer, malignant melanoma, and non–small cell lung cancer (NSCLC).
“In the future, we also plan to study lenvatinib plus pembrolizumab in patients with RCC who have had disease progression after treatment with immune checkpoint inhibitors,” they wrote. The report was published in Journal of Clinical Oncology.
Lenvatinib is a multitargeted tyrosine kinase inhibitor (TKI) with action against vascular endothelial growth factor (VEGF) receptors 1-3, fibroblast growth factor (FGF) receptors 1-4, platelet-derived growth factor receptors alpha and the RET and KIT kinases.
“Preclinical and clinical studies suggest that modulation of VEGF-mediated immune suppression via angiogenesis inhibition could potentially augment the immunotherapeutic activity of immune checkpoint inhibitors,” the investigators wrote.
They reported results from the dose finding (1b) phase including 13 patients and initial phase 2 expansion cohorts with a total of 124 patients.
The maximum tolerated dose of lenvatinib in combination with pembrolizumab was established as 20 mg/day.
At 24 weeks of follow-up, the ORR for 30 patients with RCC was 63%; two additional patients had responses after week 24, for a total ORR at study cutoff in this cohort of 70%. The median duration of response for these patients was 20 months, and the median progression-free survival (PFS) was 19.8 months. At the time of data cutoff for this analysis, 9 of the 30 patients with RCC were still on treatment.
For 23 patients with endometrial cancer, the 24-week and overall ORR were 52%, with a median duration of response not reached, and a median PFS of 9.7 months. Seven patients were still on treatment at data cutoff.
For 21 patients with melanoma, the 24-week and overall ORR were 48%, median duration of response was 12.5 months, and median PFS was 5.5 months. Two of the patients were still on treatment at data cutoff.
For the 22 patients with squamous cell cancer of the head and neck, the 24-week ORR was 36%, with two patients having a response after week 24 for a total ORR at data cutoff of 46%. The median duration of response was 8.2 months and the median PFS was 4.7 months. Three patients remained on treatment at data cutoff.
For 21 patients with NSCLC, the 24-week and overall ORR were 33%, the median duration of response was 10.9 months, and median PFS was 5.9 months. Six of the patients were still receiving treatment at data cutoff.
For 20 patients with urothelial cancer, the 24-week and overall ORR were 25%, with a median duration of response not reached, and a median PFS of 5.4 months. Three patients were still receiving the combination at the time of data cutoff.
Treatment related adverse events (TRAEs) occurred in 133 of all 137 patients enrolled in the two study phases. The adverse events were similar across all cohorts, with any grade of events including fatigue in 58%, diarrhea in 52%, hypertension in 47%, hypothyroidism in 42%, and decreased appetite in 39%.
The most frequent grade 3 or 4 TRAEs were hypertension in 20%, fatigue in 12%, diarrhea in 9%, proteinuria in 8%, and increased lipase levels in 7%.
In all, 85% of patients had a TRAE leading to lenvatinib dose reduction and/or interruption, and 13% required lenvatinib discontinuation.
Events leading to pembrolizumab dose interruption occurred in 45% of patients, and pembrolizumab discontinuation in 15%.
The study was sponsored by Eisai with collaboration from Merck Sharp & Dohme. Dr. Taylor disclosed a consulting or advisory role for Bristol-Myers Squibb, Eisai, Array BioPharma, Loxo, Bayer, ArQule, Blueprint Medicines, Novartis, and Sanofi/Genzyme, and speakers bureau activities for BMS and Eisai.
SOURCE: Taylor MH et al. J Clin Oncol. 2020 Jan. 21 doi: 10.1200/JCO.19.01598.
A combination of the tyrosine kinase inhibitor lenvatinib (Lenvima) and the immune checkpoint inhibitor pembrolizumab (Keytruda) was safe and showed promising activity against advanced renal cell carcinoma and other solid tumors in a phase 1b/2 study.
Overall response rates (ORR) at 24 weeks ranged from 63% for patients with advanced renal cell carcinomas (RCC) to 25% for patients with urothelial cancers, reported Matthew H. Taylor, MD, of Knight Cancer Institute at Oregon Health & Science University in Portland, and colleagues.
The findings from this study sparked additional clinical trials for patients with gastric cancer, gastroesophageal cancer, and differentiated thyroid cancer, and set the stage for larger phase 3 trials in patients with advanced RCC, endometrial cancer, malignant melanoma, and non–small cell lung cancer (NSCLC).
“In the future, we also plan to study lenvatinib plus pembrolizumab in patients with RCC who have had disease progression after treatment with immune checkpoint inhibitors,” they wrote. The report was published in Journal of Clinical Oncology.
Lenvatinib is a multitargeted tyrosine kinase inhibitor (TKI) with action against vascular endothelial growth factor (VEGF) receptors 1-3, fibroblast growth factor (FGF) receptors 1-4, platelet-derived growth factor receptors alpha and the RET and KIT kinases.
“Preclinical and clinical studies suggest that modulation of VEGF-mediated immune suppression via angiogenesis inhibition could potentially augment the immunotherapeutic activity of immune checkpoint inhibitors,” the investigators wrote.
They reported results from the dose finding (1b) phase including 13 patients and initial phase 2 expansion cohorts with a total of 124 patients.
The maximum tolerated dose of lenvatinib in combination with pembrolizumab was established as 20 mg/day.
At 24 weeks of follow-up, the ORR for 30 patients with RCC was 63%; two additional patients had responses after week 24, for a total ORR at study cutoff in this cohort of 70%. The median duration of response for these patients was 20 months, and the median progression-free survival (PFS) was 19.8 months. At the time of data cutoff for this analysis, 9 of the 30 patients with RCC were still on treatment.
For 23 patients with endometrial cancer, the 24-week and overall ORR were 52%, with a median duration of response not reached, and a median PFS of 9.7 months. Seven patients were still on treatment at data cutoff.
For 21 patients with melanoma, the 24-week and overall ORR were 48%, median duration of response was 12.5 months, and median PFS was 5.5 months. Two of the patients were still on treatment at data cutoff.
For the 22 patients with squamous cell cancer of the head and neck, the 24-week ORR was 36%, with two patients having a response after week 24 for a total ORR at data cutoff of 46%. The median duration of response was 8.2 months and the median PFS was 4.7 months. Three patients remained on treatment at data cutoff.
For 21 patients with NSCLC, the 24-week and overall ORR were 33%, the median duration of response was 10.9 months, and median PFS was 5.9 months. Six of the patients were still receiving treatment at data cutoff.
For 20 patients with urothelial cancer, the 24-week and overall ORR were 25%, with a median duration of response not reached, and a median PFS of 5.4 months. Three patients were still receiving the combination at the time of data cutoff.
Treatment related adverse events (TRAEs) occurred in 133 of all 137 patients enrolled in the two study phases. The adverse events were similar across all cohorts, with any grade of events including fatigue in 58%, diarrhea in 52%, hypertension in 47%, hypothyroidism in 42%, and decreased appetite in 39%.
The most frequent grade 3 or 4 TRAEs were hypertension in 20%, fatigue in 12%, diarrhea in 9%, proteinuria in 8%, and increased lipase levels in 7%.
In all, 85% of patients had a TRAE leading to lenvatinib dose reduction and/or interruption, and 13% required lenvatinib discontinuation.
Events leading to pembrolizumab dose interruption occurred in 45% of patients, and pembrolizumab discontinuation in 15%.
The study was sponsored by Eisai with collaboration from Merck Sharp & Dohme. Dr. Taylor disclosed a consulting or advisory role for Bristol-Myers Squibb, Eisai, Array BioPharma, Loxo, Bayer, ArQule, Blueprint Medicines, Novartis, and Sanofi/Genzyme, and speakers bureau activities for BMS and Eisai.
SOURCE: Taylor MH et al. J Clin Oncol. 2020 Jan. 21 doi: 10.1200/JCO.19.01598.
A combination of the tyrosine kinase inhibitor lenvatinib (Lenvima) and the immune checkpoint inhibitor pembrolizumab (Keytruda) was safe and showed promising activity against advanced renal cell carcinoma and other solid tumors in a phase 1b/2 study.
Overall response rates (ORR) at 24 weeks ranged from 63% for patients with advanced renal cell carcinomas (RCC) to 25% for patients with urothelial cancers, reported Matthew H. Taylor, MD, of Knight Cancer Institute at Oregon Health & Science University in Portland, and colleagues.
The findings from this study sparked additional clinical trials for patients with gastric cancer, gastroesophageal cancer, and differentiated thyroid cancer, and set the stage for larger phase 3 trials in patients with advanced RCC, endometrial cancer, malignant melanoma, and non–small cell lung cancer (NSCLC).
“In the future, we also plan to study lenvatinib plus pembrolizumab in patients with RCC who have had disease progression after treatment with immune checkpoint inhibitors,” they wrote. The report was published in Journal of Clinical Oncology.
Lenvatinib is a multitargeted tyrosine kinase inhibitor (TKI) with action against vascular endothelial growth factor (VEGF) receptors 1-3, fibroblast growth factor (FGF) receptors 1-4, platelet-derived growth factor receptors alpha and the RET and KIT kinases.
“Preclinical and clinical studies suggest that modulation of VEGF-mediated immune suppression via angiogenesis inhibition could potentially augment the immunotherapeutic activity of immune checkpoint inhibitors,” the investigators wrote.
They reported results from the dose finding (1b) phase including 13 patients and initial phase 2 expansion cohorts with a total of 124 patients.
The maximum tolerated dose of lenvatinib in combination with pembrolizumab was established as 20 mg/day.
At 24 weeks of follow-up, the ORR for 30 patients with RCC was 63%; two additional patients had responses after week 24, for a total ORR at study cutoff in this cohort of 70%. The median duration of response for these patients was 20 months, and the median progression-free survival (PFS) was 19.8 months. At the time of data cutoff for this analysis, 9 of the 30 patients with RCC were still on treatment.
For 23 patients with endometrial cancer, the 24-week and overall ORR were 52%, with a median duration of response not reached, and a median PFS of 9.7 months. Seven patients were still on treatment at data cutoff.
For 21 patients with melanoma, the 24-week and overall ORR were 48%, median duration of response was 12.5 months, and median PFS was 5.5 months. Two of the patients were still on treatment at data cutoff.
For the 22 patients with squamous cell cancer of the head and neck, the 24-week ORR was 36%, with two patients having a response after week 24 for a total ORR at data cutoff of 46%. The median duration of response was 8.2 months and the median PFS was 4.7 months. Three patients remained on treatment at data cutoff.
For 21 patients with NSCLC, the 24-week and overall ORR were 33%, the median duration of response was 10.9 months, and median PFS was 5.9 months. Six of the patients were still receiving treatment at data cutoff.
For 20 patients with urothelial cancer, the 24-week and overall ORR were 25%, with a median duration of response not reached, and a median PFS of 5.4 months. Three patients were still receiving the combination at the time of data cutoff.
Treatment related adverse events (TRAEs) occurred in 133 of all 137 patients enrolled in the two study phases. The adverse events were similar across all cohorts, with any grade of events including fatigue in 58%, diarrhea in 52%, hypertension in 47%, hypothyroidism in 42%, and decreased appetite in 39%.
The most frequent grade 3 or 4 TRAEs were hypertension in 20%, fatigue in 12%, diarrhea in 9%, proteinuria in 8%, and increased lipase levels in 7%.
In all, 85% of patients had a TRAE leading to lenvatinib dose reduction and/or interruption, and 13% required lenvatinib discontinuation.
Events leading to pembrolizumab dose interruption occurred in 45% of patients, and pembrolizumab discontinuation in 15%.
The study was sponsored by Eisai with collaboration from Merck Sharp & Dohme. Dr. Taylor disclosed a consulting or advisory role for Bristol-Myers Squibb, Eisai, Array BioPharma, Loxo, Bayer, ArQule, Blueprint Medicines, Novartis, and Sanofi/Genzyme, and speakers bureau activities for BMS and Eisai.
SOURCE: Taylor MH et al. J Clin Oncol. 2020 Jan. 21 doi: 10.1200/JCO.19.01598.
FROM THE JOURNAL OF CLINICAL ONCOLOGY