User login
Many institutions exceed recommended radiation doses during lung cancer screening
according to a study published in JAMA Internal Medicine.
Various institutional characteristics, such as allowing any radiologist to establish CT scan protocols, are associated with a greater likelihood of using higher radiation doses. “Dose optimization practices may benefit from being tailored to specific practice types, as well as different organizational structures, to have a higher likelihood of meeting dose guidelines,” wrote Joshua Demb, PhD, MPH, a cancer epidemiologist at the University of California, San Diego, and colleagues.
Lung cancer screening benefits patients when low-dose CT is used, but not when high-dose CT is used, because radiation from higher doses may cause as many cancers as are detected by screening. The Centers for Medicare & Medicaid Services require institutions to use low-dose techniques and participate in a dose registry to be reimbursed for lung cancer screening. The American College of Radiology recommends that lung cancer screening scans have a volume CT dose index (CTDIvol) of 3 mGy or lower and an effective dose (ED) of 1 millisieverts (mSv) or lower.
A prospective study of registry data
Dr. Demb and colleagues conducted a study to describe CT radiation doses for lung cancer screening in current practice and to identify the factors that explain variation in doses between institutions. They prospectively collected lung cancer screening examination dose metrics from 2016 to 2017 at U.S. institutions participating in the University of California, San Francisco, International Dose Registry. Eligible institutions performed a minimum of 24 lung cancer screening scans during the study period. At baseline, the investigators surveyed institutions about their characteristics (for example, how they perform and oversee CT). Dr. Demb and colleagues estimated mixed-effects linear and logistic regression models using forward variable selection. They conducted their analysis between 2018 and 2019.
The researchers chose four outcome measures. The first was mean CTDIvol, reflecting the average radiation dose per slice. The second was mean ED, reflecting the total dose received and estimated future cancer risk. The third was the proportion of CT scans using radiation doses above ACR benchmarks. The fourth was the proportion of CT scans using radiation doses above the 75th percentile of registry doses (CTDIvol greater than 2.7 mGy and ED greater than 1.4 mSv).
Institutional characteristics associated with radiation dose
Dr. Demb and colleagues collected data from 72 institutions about 12,529 patients undergoing CT scans for lung cancer screening. Approximately 58% of patients were men, and the patients’ median age was 65 years. The mean CTDIvol, adjusted for patient size, was 2.4 mGy. The mean ED for lung cancer screening, adjusted for chest diameter, was 1.2 mSv.
A total of 15 institutions (21%) had a median adjusted CTDIvol value higher than the ACR guideline, and 47 (65%) had a median adjusted ED higher than the ACR guideline. Approximately 18% of CT scans had a CTDIvol higher than guidelines, and 50% had an ED higher than ACR guidelines.
Institutions that permitted any radiologist to establish CT protocols had 44% higher mean CTDIvol and 27% higher mean ED, compared with institutions that restricted who could establish protocols. Institutions that permitted any radiologist to establish protocols also had higher odds of conducting examinations that exceeded ACR CTDIvol guidelines (odds ratio, 12.0) and of being in the 75th percentile of the registry CTDIvol (OR, 19.0) or ED (OR, 8.5) values.
In contrast, having lead radiologists establish CT protocols resulted in lower odds of using doses that exceeded ACR ED guidelines (OR, 0.01). Employing external, rather than internal, medical physicists was associated with increased odds of exceeding ACR CTDIvol guidelines (OR, 6.1). Having medical physicists establish protocols was associated with decreased odds of exceeding the 75th percentile of the registry CTDIvol (OR, 0.09) values. Institutions that updated protocols as needed, rather than annually, had 27% higher mean CTDIvol.
“Although we cannot establish causality in this observational study, our results suggest that considering these factors (for example, allowing only lead radiologists to establish protocols) could have a meaningful impact on dose, and could be important areas to develop interventions to optimize doses of CT protocols” the investigators wrote.
The Patient Centered Outcomes Research Institute and the National Institutes of Health supported this research. The authors reported no conflicts of interest.
SOURCE: Demb J et al. JAMA Intern Med. 2019 Sep 23. doi: 10.1001/jamainternmed.2019.3893.
according to a study published in JAMA Internal Medicine.
Various institutional characteristics, such as allowing any radiologist to establish CT scan protocols, are associated with a greater likelihood of using higher radiation doses. “Dose optimization practices may benefit from being tailored to specific practice types, as well as different organizational structures, to have a higher likelihood of meeting dose guidelines,” wrote Joshua Demb, PhD, MPH, a cancer epidemiologist at the University of California, San Diego, and colleagues.
Lung cancer screening benefits patients when low-dose CT is used, but not when high-dose CT is used, because radiation from higher doses may cause as many cancers as are detected by screening. The Centers for Medicare & Medicaid Services require institutions to use low-dose techniques and participate in a dose registry to be reimbursed for lung cancer screening. The American College of Radiology recommends that lung cancer screening scans have a volume CT dose index (CTDIvol) of 3 mGy or lower and an effective dose (ED) of 1 millisieverts (mSv) or lower.
A prospective study of registry data
Dr. Demb and colleagues conducted a study to describe CT radiation doses for lung cancer screening in current practice and to identify the factors that explain variation in doses between institutions. They prospectively collected lung cancer screening examination dose metrics from 2016 to 2017 at U.S. institutions participating in the University of California, San Francisco, International Dose Registry. Eligible institutions performed a minimum of 24 lung cancer screening scans during the study period. At baseline, the investigators surveyed institutions about their characteristics (for example, how they perform and oversee CT). Dr. Demb and colleagues estimated mixed-effects linear and logistic regression models using forward variable selection. They conducted their analysis between 2018 and 2019.
The researchers chose four outcome measures. The first was mean CTDIvol, reflecting the average radiation dose per slice. The second was mean ED, reflecting the total dose received and estimated future cancer risk. The third was the proportion of CT scans using radiation doses above ACR benchmarks. The fourth was the proportion of CT scans using radiation doses above the 75th percentile of registry doses (CTDIvol greater than 2.7 mGy and ED greater than 1.4 mSv).
Institutional characteristics associated with radiation dose
Dr. Demb and colleagues collected data from 72 institutions about 12,529 patients undergoing CT scans for lung cancer screening. Approximately 58% of patients were men, and the patients’ median age was 65 years. The mean CTDIvol, adjusted for patient size, was 2.4 mGy. The mean ED for lung cancer screening, adjusted for chest diameter, was 1.2 mSv.
A total of 15 institutions (21%) had a median adjusted CTDIvol value higher than the ACR guideline, and 47 (65%) had a median adjusted ED higher than the ACR guideline. Approximately 18% of CT scans had a CTDIvol higher than guidelines, and 50% had an ED higher than ACR guidelines.
Institutions that permitted any radiologist to establish CT protocols had 44% higher mean CTDIvol and 27% higher mean ED, compared with institutions that restricted who could establish protocols. Institutions that permitted any radiologist to establish protocols also had higher odds of conducting examinations that exceeded ACR CTDIvol guidelines (odds ratio, 12.0) and of being in the 75th percentile of the registry CTDIvol (OR, 19.0) or ED (OR, 8.5) values.
In contrast, having lead radiologists establish CT protocols resulted in lower odds of using doses that exceeded ACR ED guidelines (OR, 0.01). Employing external, rather than internal, medical physicists was associated with increased odds of exceeding ACR CTDIvol guidelines (OR, 6.1). Having medical physicists establish protocols was associated with decreased odds of exceeding the 75th percentile of the registry CTDIvol (OR, 0.09) values. Institutions that updated protocols as needed, rather than annually, had 27% higher mean CTDIvol.
“Although we cannot establish causality in this observational study, our results suggest that considering these factors (for example, allowing only lead radiologists to establish protocols) could have a meaningful impact on dose, and could be important areas to develop interventions to optimize doses of CT protocols” the investigators wrote.
The Patient Centered Outcomes Research Institute and the National Institutes of Health supported this research. The authors reported no conflicts of interest.
SOURCE: Demb J et al. JAMA Intern Med. 2019 Sep 23. doi: 10.1001/jamainternmed.2019.3893.
according to a study published in JAMA Internal Medicine.
Various institutional characteristics, such as allowing any radiologist to establish CT scan protocols, are associated with a greater likelihood of using higher radiation doses. “Dose optimization practices may benefit from being tailored to specific practice types, as well as different organizational structures, to have a higher likelihood of meeting dose guidelines,” wrote Joshua Demb, PhD, MPH, a cancer epidemiologist at the University of California, San Diego, and colleagues.
Lung cancer screening benefits patients when low-dose CT is used, but not when high-dose CT is used, because radiation from higher doses may cause as many cancers as are detected by screening. The Centers for Medicare & Medicaid Services require institutions to use low-dose techniques and participate in a dose registry to be reimbursed for lung cancer screening. The American College of Radiology recommends that lung cancer screening scans have a volume CT dose index (CTDIvol) of 3 mGy or lower and an effective dose (ED) of 1 millisieverts (mSv) or lower.
A prospective study of registry data
Dr. Demb and colleagues conducted a study to describe CT radiation doses for lung cancer screening in current practice and to identify the factors that explain variation in doses between institutions. They prospectively collected lung cancer screening examination dose metrics from 2016 to 2017 at U.S. institutions participating in the University of California, San Francisco, International Dose Registry. Eligible institutions performed a minimum of 24 lung cancer screening scans during the study period. At baseline, the investigators surveyed institutions about their characteristics (for example, how they perform and oversee CT). Dr. Demb and colleagues estimated mixed-effects linear and logistic regression models using forward variable selection. They conducted their analysis between 2018 and 2019.
The researchers chose four outcome measures. The first was mean CTDIvol, reflecting the average radiation dose per slice. The second was mean ED, reflecting the total dose received and estimated future cancer risk. The third was the proportion of CT scans using radiation doses above ACR benchmarks. The fourth was the proportion of CT scans using radiation doses above the 75th percentile of registry doses (CTDIvol greater than 2.7 mGy and ED greater than 1.4 mSv).
Institutional characteristics associated with radiation dose
Dr. Demb and colleagues collected data from 72 institutions about 12,529 patients undergoing CT scans for lung cancer screening. Approximately 58% of patients were men, and the patients’ median age was 65 years. The mean CTDIvol, adjusted for patient size, was 2.4 mGy. The mean ED for lung cancer screening, adjusted for chest diameter, was 1.2 mSv.
A total of 15 institutions (21%) had a median adjusted CTDIvol value higher than the ACR guideline, and 47 (65%) had a median adjusted ED higher than the ACR guideline. Approximately 18% of CT scans had a CTDIvol higher than guidelines, and 50% had an ED higher than ACR guidelines.
Institutions that permitted any radiologist to establish CT protocols had 44% higher mean CTDIvol and 27% higher mean ED, compared with institutions that restricted who could establish protocols. Institutions that permitted any radiologist to establish protocols also had higher odds of conducting examinations that exceeded ACR CTDIvol guidelines (odds ratio, 12.0) and of being in the 75th percentile of the registry CTDIvol (OR, 19.0) or ED (OR, 8.5) values.
In contrast, having lead radiologists establish CT protocols resulted in lower odds of using doses that exceeded ACR ED guidelines (OR, 0.01). Employing external, rather than internal, medical physicists was associated with increased odds of exceeding ACR CTDIvol guidelines (OR, 6.1). Having medical physicists establish protocols was associated with decreased odds of exceeding the 75th percentile of the registry CTDIvol (OR, 0.09) values. Institutions that updated protocols as needed, rather than annually, had 27% higher mean CTDIvol.
“Although we cannot establish causality in this observational study, our results suggest that considering these factors (for example, allowing only lead radiologists to establish protocols) could have a meaningful impact on dose, and could be important areas to develop interventions to optimize doses of CT protocols” the investigators wrote.
The Patient Centered Outcomes Research Institute and the National Institutes of Health supported this research. The authors reported no conflicts of interest.
SOURCE: Demb J et al. JAMA Intern Med. 2019 Sep 23. doi: 10.1001/jamainternmed.2019.3893.
FROM JAMA INTERNAL MEDICINE
Key clinical point: A significant proportion of institutions exceed guideline-recommended dose levels for CT screening for lung cancer.
Major finding: About 21% of institutions have median volume CT dose index above American College of Radiology guidelines, and 65% have median effective dose above ACR guidelines.
Study details: A prospective study of data for 12,529 patients undergoing screening at 72 institutions.
Disclosures: The Patient Centered Outcomes Research Institute and the National Institutes of Health supported this research. The authors reported no conflicts of interest.
Source: Demb J et al. JAMA Intern Med. 2019 Sep 23. doi: 10.1001/jamainternmed.2019.3893.
New guideline conditionally recommends long-term home NIV for COPD patients
from a European Respiratory Society task force.
“Our recommendations, based on the best available evidence, can guide the management of chronic hypercapnic respiratory failure in COPD patients aimed at improving patient outcomes,” wrote Begum Ergan, MD, of Dokuz Eylul University, Izmir, Turkey, and coauthors. The guideline was published in the European Respiratory Journal.
To provide insight into the clinical application of LTH-NIV, the European Respiratory Society convened a task force of 20 clinicians, methodologists, and experts. Their four recommendations were developed based on the GRADE (Grading, Recommendation, Assessment, Development and Evaluation) methodology.
The first recommendation was to use LTH-NIV for patients with chronic stable hypercapnic COPD. Though an analysis of randomized, controlled trials showed little effect on mortality or hospitalizations, pooled analyses showed that NIV may decrease dyspnea scores (standardized mean difference, –0.51; 95% confidence interval, –0.06 to –0.95) and increase health-related quality of life (SMD, 0.49; 95% CI, –0.01 to 0.98).
The second was to use LTH-NIV in patients with COPD following a life-threatening episode of acute hypercapnic respiratory failure requiring acute NIV, if hypercapnia persists. Though it was not associated with a reduction in mortality (risk ratio, 0.92; 95% CI, 0.67-1.25), it was found to potentially reduce exacerbations (SMD, 0.19; 95% CI, –0.40 to 0.01) and hospitalizations (RR, 0.61; 95% CI, 0.30-1.24).
The third was to titrate LTH-NIV to normalize or reduce PaCO2 levels in patients with COPD. While this recommendation was issued with a very low certainty of evidence, it was driven by the “minimal potential harms of targeted PaCO2 reduction.”
The fourth was to use fixed pressure support mode as first-choice ventilator mode in patients with COPD using LTH-NIV. The six trials on this subject did not provide insight into long-term outcomes, nor were there significant improvements seen in health-related quality of life, sleep quality, or exercise tolerance. As such, it was also issued with a very low certainty of evidence.
The authors acknowledged all four recommendations as weak and conditional, “due to limitations in the certainty of the available evidence.” As such, they noted that their recommendations “require consideration of individual preferences, resource considerations, technical expertise, and clinical circumstances prior to implementation in clinical practice.”
The authors reported numerous disclosures, including receiving grants and personal fees from various medical supply companies.
SOURCE: Ergan B et al. Eur Respir J. 2019 Aug 29. doi: 10.1183/13993003.01003-2019.
from a European Respiratory Society task force.
“Our recommendations, based on the best available evidence, can guide the management of chronic hypercapnic respiratory failure in COPD patients aimed at improving patient outcomes,” wrote Begum Ergan, MD, of Dokuz Eylul University, Izmir, Turkey, and coauthors. The guideline was published in the European Respiratory Journal.
To provide insight into the clinical application of LTH-NIV, the European Respiratory Society convened a task force of 20 clinicians, methodologists, and experts. Their four recommendations were developed based on the GRADE (Grading, Recommendation, Assessment, Development and Evaluation) methodology.
The first recommendation was to use LTH-NIV for patients with chronic stable hypercapnic COPD. Though an analysis of randomized, controlled trials showed little effect on mortality or hospitalizations, pooled analyses showed that NIV may decrease dyspnea scores (standardized mean difference, –0.51; 95% confidence interval, –0.06 to –0.95) and increase health-related quality of life (SMD, 0.49; 95% CI, –0.01 to 0.98).
The second was to use LTH-NIV in patients with COPD following a life-threatening episode of acute hypercapnic respiratory failure requiring acute NIV, if hypercapnia persists. Though it was not associated with a reduction in mortality (risk ratio, 0.92; 95% CI, 0.67-1.25), it was found to potentially reduce exacerbations (SMD, 0.19; 95% CI, –0.40 to 0.01) and hospitalizations (RR, 0.61; 95% CI, 0.30-1.24).
The third was to titrate LTH-NIV to normalize or reduce PaCO2 levels in patients with COPD. While this recommendation was issued with a very low certainty of evidence, it was driven by the “minimal potential harms of targeted PaCO2 reduction.”
The fourth was to use fixed pressure support mode as first-choice ventilator mode in patients with COPD using LTH-NIV. The six trials on this subject did not provide insight into long-term outcomes, nor were there significant improvements seen in health-related quality of life, sleep quality, or exercise tolerance. As such, it was also issued with a very low certainty of evidence.
The authors acknowledged all four recommendations as weak and conditional, “due to limitations in the certainty of the available evidence.” As such, they noted that their recommendations “require consideration of individual preferences, resource considerations, technical expertise, and clinical circumstances prior to implementation in clinical practice.”
The authors reported numerous disclosures, including receiving grants and personal fees from various medical supply companies.
SOURCE: Ergan B et al. Eur Respir J. 2019 Aug 29. doi: 10.1183/13993003.01003-2019.
from a European Respiratory Society task force.
“Our recommendations, based on the best available evidence, can guide the management of chronic hypercapnic respiratory failure in COPD patients aimed at improving patient outcomes,” wrote Begum Ergan, MD, of Dokuz Eylul University, Izmir, Turkey, and coauthors. The guideline was published in the European Respiratory Journal.
To provide insight into the clinical application of LTH-NIV, the European Respiratory Society convened a task force of 20 clinicians, methodologists, and experts. Their four recommendations were developed based on the GRADE (Grading, Recommendation, Assessment, Development and Evaluation) methodology.
The first recommendation was to use LTH-NIV for patients with chronic stable hypercapnic COPD. Though an analysis of randomized, controlled trials showed little effect on mortality or hospitalizations, pooled analyses showed that NIV may decrease dyspnea scores (standardized mean difference, –0.51; 95% confidence interval, –0.06 to –0.95) and increase health-related quality of life (SMD, 0.49; 95% CI, –0.01 to 0.98).
The second was to use LTH-NIV in patients with COPD following a life-threatening episode of acute hypercapnic respiratory failure requiring acute NIV, if hypercapnia persists. Though it was not associated with a reduction in mortality (risk ratio, 0.92; 95% CI, 0.67-1.25), it was found to potentially reduce exacerbations (SMD, 0.19; 95% CI, –0.40 to 0.01) and hospitalizations (RR, 0.61; 95% CI, 0.30-1.24).
The third was to titrate LTH-NIV to normalize or reduce PaCO2 levels in patients with COPD. While this recommendation was issued with a very low certainty of evidence, it was driven by the “minimal potential harms of targeted PaCO2 reduction.”
The fourth was to use fixed pressure support mode as first-choice ventilator mode in patients with COPD using LTH-NIV. The six trials on this subject did not provide insight into long-term outcomes, nor were there significant improvements seen in health-related quality of life, sleep quality, or exercise tolerance. As such, it was also issued with a very low certainty of evidence.
The authors acknowledged all four recommendations as weak and conditional, “due to limitations in the certainty of the available evidence.” As such, they noted that their recommendations “require consideration of individual preferences, resource considerations, technical expertise, and clinical circumstances prior to implementation in clinical practice.”
The authors reported numerous disclosures, including receiving grants and personal fees from various medical supply companies.
SOURCE: Ergan B et al. Eur Respir J. 2019 Aug 29. doi: 10.1183/13993003.01003-2019.
FROM THE EUROPEAN RESPIRATORY JOURNAL
Growing vaping habit may lead to nicotine addiction in adolescents
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
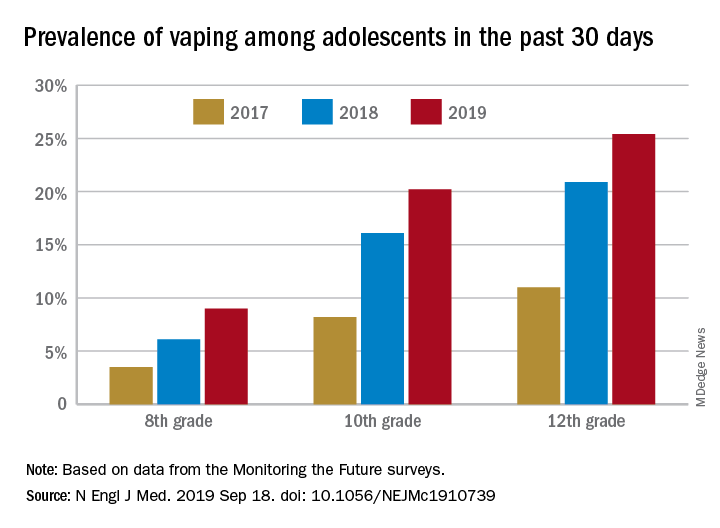
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
and in 2019 almost 12% of high school seniors reported that they were vaping every day, according to data from the Monitoring the Future surveys.
Daily use – defined as vaping on 20 or more of the previous 30 days – was reported by 6.9% of 10th-grade and 1.9% of 8th-grade respondents in the 2019 survey, which was the first time use in these age groups was assessed. “The substantial levels of daily vaping suggest the development of nicotine addiction,” Richard Miech, PhD, and associates said Sept. 18 in the New England Journal of Medicine.
From 2017 to 2019, e-cigarette use over the previous 30 days increased from 11.0% to 25.4% among 12th graders, from 8.2% to 20.2% in 10th graders, and from 3.5% to 9.0% of 8th graders, suggesting that “current efforts by the vaping industry, government agencies, and schools have thus far proved insufficient to stop the rapid spread of nicotine vaping among adolescents,” the investigators wrote.
By 2019, over 40% of 12th-grade students reported ever using e-cigarettes, along with more than 36% of 10th graders and almost 21% of 8th graders. Corresponding figures for past 12-month use were 35.1%, 31.1%, and 16.1%, they reported.
“New efforts are needed to protect youth from using nicotine during adolescence, when the developing brain is particularly susceptible to permanent changes from nicotine use and when almost all nicotine addiction is established,” the investigators wrote.
The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
SOURCE: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Adolescents who use e-cigarettes every day may be developing nicotine addiction.
Major finding: In 2019, almost 12% of high school seniors were vaping every day.
Study details: Monitoring the Future surveys nationally representative samples of 8th-, 10th-, and 12th-grade students each year.
Disclosures: The analysis was funded by a grant from the National Institute on Drug Abuse to Dr. Miech.
Source: Miech R et al. N Engl J Med. 2019 Sep 18. doi: 10.1056/NEJMc1910739.
Benefits of peanut desensitization may not last
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.

Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.
Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
based on data from a phase 2 randomized trial of individuals with confirmed peanut allergies.
Previous studies have shown that desensitization to peanuts can be successful, but sustained response to oral immunotherapy after treatment reduction or discontinuation has not been well studied, wrote R. Sharon Chinthrajah, MD, of Stanford (Calif.)University, and colleagues.
“We found that OIT with peanut was able to desensitise people with peanut allergy to 4,000 mg of peanut protein, but that discontinuation of peanut, or even a reduction to 300 mg daily, increased the likelihood of regaining clinical reactivity to peanut,” they wrote. “With peanut allergy therapies in varying stages of clinical development, and some nearing [Food and Drug Administration] approval, vital questions remain regarding the durability of treatment effects and the appropriate maintenance doses.”
In the Peanut Oral Immunotherapy Study: Safety Efficacy and Discovery (POISED), published in The Lancet, the researchers randomized 120 participants to three groups:
• 60 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by total discontinuation (peanut-0).
• 35 patients built up to a maintenance dose of 4,000 mg of peanut protein for 104 weeks followed by a 300-mg maintenance dose of peanut protein in the form of peanut flour (peanut-300).
• 25 patients to an oat flour placebo.
All participants were trained on how and when to use epinephrine autoinjector devices to treat allergic symptoms such as respiratory problems (cough, shortness of breath, or change in voice), widespread hives or erythema, repetitive vomiting, persistent abdominal pain, angioedema of the face, or feeling faint.
The primary outcome was passing a double-blind, placebo-controlled, food challenge (DBPCFC) to 4,000 mg of peanut protein, which was measured at baseline and at weeks 104, 117, 130, 143, and 156.
Overall, 35% of the peanut-0 group passed the challenge at 104 and 117 weeks, compared with 4% of the placebo group. At week 156 after discontinuing OIT, 13% of the peanut-0 group met the DBPCFC challenge, compared with 4% of the placebo group. However, 37% of participants randomized to a reduced peanut protein dose of 300 mg passed the challenge at 156 weeks, suggesting that more data are needed on optimal maintenance dosing strategies.
Baseline demographics were similar across all groups. The median age at study enrollment was 11 years and the median allergy duration was 9 years. The most common adverse events were mild gastrointestinal and respiratory problems. Adverse events decreased over time in all three groups.
“Higher levels of peanut-specific IgE to total IgE ratio, peanut sIgE, Ara h 1, Ara h 2, and Ara h 1 IgE to peanut-specific IgE ratio at baseline in participants were associated with increased frequencies of adverse events during active peanut OIT,” the researchers noted.
The study findings were limited by several factors including the ability of participants to tolerate 4,000 mg of peanut protein after achieving a maintenance dose but conducting serial testing only for those who passed the challenge. In addition, the results may be limited to peanut and not generalizable to other food allergies, the researchers said.
However, the results suggest that OIT remains a promising treatment for peanut allergies, and the association of biomarkers with clinical outcomes “might help the practitioner in identifying good candidates for OIT and those individuals who warrant increased vigilance against allergic reactions during OIT,” they said.
The National Institutes of Health supported the study. The researchers had no financial conflicts to disclose.
SOURCE: Chinthrajah RS et al. Lancet. 2019 Sep 12. doi: 10.1016/S0140-6736(19)31793-3.
FROM THE LANCET
Prolonged Antibiotic Treatment in Newborns May Promote Multidrug Resistance
Antibiotics given to preterm infants can set them up for health problems later in life; research has shown, including allergies, psoriasis, diabetes, and inflammatory bowel disease. Researchers who conducted a National Institutes of Health (NIH)-funded study have added to that body of knowledge with their finding that treating preterm infants with long-term antibiotics could have lasting effects by promoting multidrug-resistant gut bacteria.
They used high-speed DNA sequencing and advanced computational analysis to study stool samples from 32 infants born very preterm who received antibiotic treatment for 21 months in the hospital and after discharge, then compared those with results from 9 very preterm infants treated with antibiotics for > 1 week and 17 healthy term and late-term infants who had not received antibiotics.
The infants on long-term antibiotics had less diverse bacterial populations in their gut, and those bacteria contained more antibiotic-resistant genes.
Strikingly, the genomes of the high-antibiotic-use samples contained genes for resistance to antibiotics typically not given to newborns, such as ciprofloxacin and chloramphenicol. The researchers say this may mean that the genes originate in multidrug-resistant bacteria. Using a particular antibiotic may trigger resistance to other antibiotics even if they were not used.
“The collateral damage of early-life antibiotic treatment and hospitalization in preterm infants is long lasting,” the researchers say. They urge development of strategies to protect these highly vulnerable patients.
Antibiotics given to preterm infants can set them up for health problems later in life; research has shown, including allergies, psoriasis, diabetes, and inflammatory bowel disease. Researchers who conducted a National Institutes of Health (NIH)-funded study have added to that body of knowledge with their finding that treating preterm infants with long-term antibiotics could have lasting effects by promoting multidrug-resistant gut bacteria.
They used high-speed DNA sequencing and advanced computational analysis to study stool samples from 32 infants born very preterm who received antibiotic treatment for 21 months in the hospital and after discharge, then compared those with results from 9 very preterm infants treated with antibiotics for > 1 week and 17 healthy term and late-term infants who had not received antibiotics.
The infants on long-term antibiotics had less diverse bacterial populations in their gut, and those bacteria contained more antibiotic-resistant genes.
Strikingly, the genomes of the high-antibiotic-use samples contained genes for resistance to antibiotics typically not given to newborns, such as ciprofloxacin and chloramphenicol. The researchers say this may mean that the genes originate in multidrug-resistant bacteria. Using a particular antibiotic may trigger resistance to other antibiotics even if they were not used.
“The collateral damage of early-life antibiotic treatment and hospitalization in preterm infants is long lasting,” the researchers say. They urge development of strategies to protect these highly vulnerable patients.
Antibiotics given to preterm infants can set them up for health problems later in life; research has shown, including allergies, psoriasis, diabetes, and inflammatory bowel disease. Researchers who conducted a National Institutes of Health (NIH)-funded study have added to that body of knowledge with their finding that treating preterm infants with long-term antibiotics could have lasting effects by promoting multidrug-resistant gut bacteria.
They used high-speed DNA sequencing and advanced computational analysis to study stool samples from 32 infants born very preterm who received antibiotic treatment for 21 months in the hospital and after discharge, then compared those with results from 9 very preterm infants treated with antibiotics for > 1 week and 17 healthy term and late-term infants who had not received antibiotics.
The infants on long-term antibiotics had less diverse bacterial populations in their gut, and those bacteria contained more antibiotic-resistant genes.
Strikingly, the genomes of the high-antibiotic-use samples contained genes for resistance to antibiotics typically not given to newborns, such as ciprofloxacin and chloramphenicol. The researchers say this may mean that the genes originate in multidrug-resistant bacteria. Using a particular antibiotic may trigger resistance to other antibiotics even if they were not used.
“The collateral damage of early-life antibiotic treatment and hospitalization in preterm infants is long lasting,” the researchers say. They urge development of strategies to protect these highly vulnerable patients.
CDC activates Emergency Operations Center to investigate vaping-associated lung injury
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting FDAVapingSampleInquiries@fda.hhs.gov.
To communicate with CDC about this public health response, clinicians and health officials can contact LungDiseaseOutbreak@cdc.gov.
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting FDAVapingSampleInquiries@fda.hhs.gov.
To communicate with CDC about this public health response, clinicians and health officials can contact LungDiseaseOutbreak@cdc.gov.
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
![]()
This move allows the CDC “to provide increased operational support” to CDC staff to meet the evolving challenges of the outbreak of vaping-related injuries and deaths, says a statement from the CDC.
“CDC has made it a priority to find out what is causing this outbreak,” noted CDC Director Robert Redfield, MD, in the statement.
The agency “continues to work closely with the U.S. Food and Drug Administration to collect information about recent e-cigarette product use, or vaping, among patients and to test the substances or chemicals within e-cigarette products used by case patients,” according to the statement.
The CDC provided email addresses and site addresses for gathering information and communicating about e-cigarettes.
Information about the collection of e-cigarettes for possible testing by FDA can be obtained through contacting FDAVapingSampleInquiries@fda.hhs.gov.
To communicate with CDC about this public health response, clinicians and health officials can contact LungDiseaseOutbreak@cdc.gov.
More information on the current outbreak related to e-cigarettes is available at https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html.
General information on electronic cigarette products, can be found at www.cdc.gov/e-cigarettes.
Individuals concerned about health risks of vaping should consider refraining from e-cigarette use while the cases of lung injury are being investigated, the CDC said.
Pulegone levels in e-liquids, smokeless tobacco products exceed FDA limits
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
lfranki@mdedge.com
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
lfranki@mdedge.com
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
A group of mint- and menthol-flavored e-liquids and smokeless tobacco products contained significantly more pulegone – a known carcinogen that causes hepatic carcinomas, pulmonary metaplasia, and other neoplasms – than the Food and Drug Administration considers acceptable, according to new findings.
Pulegone, an oil extract from mint plants such as peppermint, spearmint, and pennyroyal, was banned as a food additive by the agency in 2018, and the tobacco industry has taken steps to minimize pulegone levels in cigarettes because of the toxicity concerns.
Studies from the Centers for Disease Control and Prevention, however, have indicated that mint- and menthol-flavored e-cigarette liquids and smokeless tobacco products marketed in the United States contain substantial amounts of the substance, Sairam V. Jabba, DVM, PhD, and Sven-Eric Jordt, PhD, said in a research letter published in JAMA Internal Medicine.
Dr. Jabba and Dr. Jordt, both with the department of anesthesiology at Duke University, Durham, N.C., calculated the margin of exposure in five e-liquids (V2 Menthol, V2 Peppermint, Premium Menthol, South Beach Smoke Menthol, and South Beach Smoke Peppermint) and one smokeless tobacco product (Skoal Xtra Mint snuff) by dividing the no–observed adverse event level (13.39 mg/kg of bodyweight per day) by the mean human exposure to e-liquids or smokeless tobacco. The FDA considers margin-of-exposure values of 10,000 or less to require mitigation strategies.
The six products included in the analysis had pulegone concentration levels ranging from 25.7 to 119.0 mcg/g (a menthol cigarette has a pulegone concentration of 0.037-0.290 mcg/g). Based on those levels, light daily use (5 mL e-liquid, 10 g smokeless tobacco, half a pack of cigarettes) exposed e-cigarette users to 44-198 times more pulegone, compared with menthol cigarettes, and exposed smokeless tobacco users to 168-1,319 times as much pulegone. The margin of exposure ranged from 1,298 to 6,012, all below 10,000 threshhold the FDA deems acceptable.
For heavy daily use (20 mL e-liquid, 30 g smokeless tobacco, two packs of cigarettes), e-cigarette users were exposed to 282-1,608 times more pulegone, compared with menthol cigarettes; smokeless tobacco users were exposed to 126-990 times more pulegone. The margin of exposure ranged from 325 to 1,503.
The study findings “appear to establish health risks associated with pulegone intake and concerns that the FDA should address before suggesting mint- and menthol-flavored e-cigarettes and smokeless tobacco products as alternatives for people who use combustible tobacco products,” Dr. Jabba and Dr. Jordt concluded.
The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
lfranki@mdedge.com
SOURCE: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Mint- and menthol-flavored e-liquids and smokeless tobacco contain levels of pulegone that are much higher than those deemed acceptable by the Food and Drug Administration.
Major finding:
Study details: An assessment of pulegone in five e-liquids and one brand of smokeless tobacco.
Disclosures: The study was funded by a grant from the National Institute of Environmental Health Sciences. Dr. Jordt reported receiving grants from the NIEHS and the National Institute on Drug Abuse, personal fees from Hydra Biosciences and Sanofi, and nonfinancial support from GlaxoSmithKline. Dr. Jabba reported no disclosures.
Source: Jabba SV, Jordt S-E. JAMA Intern Med. 2019 Sep 16. doi: 10.1001/jamainternmed.2019.3649.
His Heart’s Awkward Timing
ANSWER
The correct answer is atrial flutter with 2:1 atrioventricular (AV) conduction. The QRS complexes are narrow and regular, indicating the rhythm originates within the atria or AV node, with conduction down the normal His-Purkinje system, and not from the ventricles.
The regular rate of the P waves and QRS complexes rules out atrial fibrillation with a rapid ventricular response. If you look carefully, you’ll see a P wave immediately before each QRS complex, and you’ll also see a P wave at the onset of the T wave (best seen in leads II, V3, and V6) resulting in what looks like a notched T wave. If you measure the duration of the P at the onset of the T wave to the P wave prior to the QRS complex, you’ll see the intervals are regular and march through the QRS complexes.
With 2 P waves for every QRS complex, the atria are contracting at 310 beats/min (193 ms), a rate consistent with atrial flutter in a 2:1 conduction pattern, compared to the ventricular rate of 155 beats/min (387 ms).
ANSWER
The correct answer is atrial flutter with 2:1 atrioventricular (AV) conduction. The QRS complexes are narrow and regular, indicating the rhythm originates within the atria or AV node, with conduction down the normal His-Purkinje system, and not from the ventricles.
The regular rate of the P waves and QRS complexes rules out atrial fibrillation with a rapid ventricular response. If you look carefully, you’ll see a P wave immediately before each QRS complex, and you’ll also see a P wave at the onset of the T wave (best seen in leads II, V3, and V6) resulting in what looks like a notched T wave. If you measure the duration of the P at the onset of the T wave to the P wave prior to the QRS complex, you’ll see the intervals are regular and march through the QRS complexes.
With 2 P waves for every QRS complex, the atria are contracting at 310 beats/min (193 ms), a rate consistent with atrial flutter in a 2:1 conduction pattern, compared to the ventricular rate of 155 beats/min (387 ms).
ANSWER
The correct answer is atrial flutter with 2:1 atrioventricular (AV) conduction. The QRS complexes are narrow and regular, indicating the rhythm originates within the atria or AV node, with conduction down the normal His-Purkinje system, and not from the ventricles.
The regular rate of the P waves and QRS complexes rules out atrial fibrillation with a rapid ventricular response. If you look carefully, you’ll see a P wave immediately before each QRS complex, and you’ll also see a P wave at the onset of the T wave (best seen in leads II, V3, and V6) resulting in what looks like a notched T wave. If you measure the duration of the P at the onset of the T wave to the P wave prior to the QRS complex, you’ll see the intervals are regular and march through the QRS complexes.
With 2 P waves for every QRS complex, the atria are contracting at 310 beats/min (193 ms), a rate consistent with atrial flutter in a 2:1 conduction pattern, compared to the ventricular rate of 155 beats/min (387 ms).

Approximately 5
Feeling fine, he went about his normal workday—but in the afternoon, while sitting at his desk, his rapid heart rate returned. He called over a coworker, who observed that he was “pale” and “sweaty.” His pulse was 130 beats/min. After “a few minutes,” the rapid heart rate spontaneously terminated, and he decided to take off the rest of the day.
This morning, he again awoke with a rapid heart rate and lightheadedness—but he also felt like the room was spinning. At that point, he called 911. By the time the paramedics arrived, his rapid heart rate had spontaneously terminated. Understandably concerned, however, he requested transport to your facility.
The patient says he is in normal health, with no prior cardiac history. He denies any chest pain, dyspnea, shortness of breath, nausea, vomiting, syncope, or near-syncope associated with his recent episodes.
Medical history is remarkable for hypertension, hyperlipidemia, and type 2 diabetes. He has had no surgical procedures. His medications include aspirin, lisinopril, and lovastatin; he says he takes his medications as prescribed and there have been no recent changes to the drugs or dosages. He has no known drug allergies.
Family history includes myocardial infarction in both parents; they are alive and well. The patient’s younger brother has Wolff-Parkinson-White syndrome and underwent an ablation at age 24.
The patient is a practicing attorney for a local firm. He is married with 2 children. He has no history of alcohol, tobacco, or illicit drug use.
Review of systems is positive for a 10-lb weight gain over the past 6 months and new-onset nocturia.
During the physical exam, the patient informs you that his heart is racing again. The exam is suspended, and a 12-lead ECG is quickly performed. It shows a ventricular rate of 155 beats/min; no measurable PR interval; QRS duration, 78 ms; QT/QTc interval, 272/437 ms; P axis, unmeasurable; R axis, 34°; and T axis, –50°.
The physical exam is completed after the tachycardia spontaneously terminates. The patient’s blood pressure is 148/88 mm Hg; pulse, 94 beats/min and regular; respiratory rate, 18 breaths/min-1; and temperature, 97.9°F. He appears frightened but otherwise healthy. Pertinent findings of the physical exam include a normal fundoscopic examination with sharp disc margins, clear breath sounds bilaterally, normal heart sounds with no murmur or rub, a soft abdomen with no palpable masses, strong and equal pulses bilaterally in both upper and lower extremities, and a normal neurologic exam with no cognitive deficits.
Now that the physical exam is complete, what is your interpretation of this ECG?

FDA issues warning for CDK 4/6 inhibitors
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.

“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.
“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
The Food and Drug Administration is warning that the entire class of the cyclin-dependent kinase 4/6 (CDK 4/6) inhibitors used to treat advanced breast cancer may cause rare but severe inflammation of the lungs.
“We reviewed CDK 4/6 inhibitors cases from completed and ongoing clinical trials undertaken by manufacturers and their postmarket safety databases that described specific types of inflammation of the lungs, called interstitial lung disease and pneumonitis. Across the entire drug class, there were reports of serious cases, including fatalities,” the FDA said in a press statement.
The overall benefit of CDK 4/6 inhibitors, however, is still greater than the risks when used as prescribed, the agency said.
CDK 4/6 inhibitors are used in combination with hormone therapies to treat adults with hormone receptor–positive, human epidermal growth factor 2–negative advanced or metastatic breast cancer that has spread to other parts of the body. The FDA approved the CDK 4/6 inhibitors palbociclib (Ibrance) in 2015 and ribociclib (Kisqali) and abemaciclib (Verzenio) in 2017, based on improvements in progression-free survival.
Health care professionals should monitor patients regularly for pulmonary symptoms indicative of interstitial lung disease and/or pneumonitis. Signs and symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams in patients in whom infectious, neoplastic, and other causes have been excluded. Interrupt CDK 4/6 inhibitor treatment in patients who have new or worsening respiratory symptoms, and permanently discontinue treatment in patients with severe interstitial lung disease and/or pneumonitis, the FDA said.
FDA approves mepolizumab for severe eosinophilic asthma in younger kids
according to a release from GlaxoSmithKline, which developed the drug. This is the first targeted biologic approved for this condition in this age group.

The approval is supported by both an open-label study in children aged 6-11 years and evidence from other trials conducted in adults and adolescents. The 52-week, long-term study in these younger patients investigated pharmacokinetics, pharmacodynamics, and safety, the last of which was shown to be similar to that seen in older patients.
Hypersensitivity reactions, such as anaphylaxis, rash, and bronchospasm, have been associated with mepolizumab. It should not be used to treat acute bronchospasm or status asthmaticus, nor should systemic or inhaled corticosteroids be stopped abruptly after initiating mepolizumab treatment. Common adverse events include headache, injection-site reactions, back pain, and fatigue. Injection site reactions (such as pain, erythema, and itching) occurred in 8% of mepolizumab patients treated with 100 mg of the drug versus 3% of placebo patients.
The monoclonal antibody targeting interleukin-5 was first approved for severe eosinophilic asthma in 2015 for ages 12 years and older and in ages 6 years and older in the European Union in August 2018. It inhibits IL-5 from binding to eosinophils, which reduces the presence of eosinophils in blood without completely eliminating them.
according to a release from GlaxoSmithKline, which developed the drug. This is the first targeted biologic approved for this condition in this age group.
The approval is supported by both an open-label study in children aged 6-11 years and evidence from other trials conducted in adults and adolescents. The 52-week, long-term study in these younger patients investigated pharmacokinetics, pharmacodynamics, and safety, the last of which was shown to be similar to that seen in older patients.
Hypersensitivity reactions, such as anaphylaxis, rash, and bronchospasm, have been associated with mepolizumab. It should not be used to treat acute bronchospasm or status asthmaticus, nor should systemic or inhaled corticosteroids be stopped abruptly after initiating mepolizumab treatment. Common adverse events include headache, injection-site reactions, back pain, and fatigue. Injection site reactions (such as pain, erythema, and itching) occurred in 8% of mepolizumab patients treated with 100 mg of the drug versus 3% of placebo patients.
The monoclonal antibody targeting interleukin-5 was first approved for severe eosinophilic asthma in 2015 for ages 12 years and older and in ages 6 years and older in the European Union in August 2018. It inhibits IL-5 from binding to eosinophils, which reduces the presence of eosinophils in blood without completely eliminating them.
according to a release from GlaxoSmithKline, which developed the drug. This is the first targeted biologic approved for this condition in this age group.
The approval is supported by both an open-label study in children aged 6-11 years and evidence from other trials conducted in adults and adolescents. The 52-week, long-term study in these younger patients investigated pharmacokinetics, pharmacodynamics, and safety, the last of which was shown to be similar to that seen in older patients.
Hypersensitivity reactions, such as anaphylaxis, rash, and bronchospasm, have been associated with mepolizumab. It should not be used to treat acute bronchospasm or status asthmaticus, nor should systemic or inhaled corticosteroids be stopped abruptly after initiating mepolizumab treatment. Common adverse events include headache, injection-site reactions, back pain, and fatigue. Injection site reactions (such as pain, erythema, and itching) occurred in 8% of mepolizumab patients treated with 100 mg of the drug versus 3% of placebo patients.
The monoclonal antibody targeting interleukin-5 was first approved for severe eosinophilic asthma in 2015 for ages 12 years and older and in ages 6 years and older in the European Union in August 2018. It inhibits IL-5 from binding to eosinophils, which reduces the presence of eosinophils in blood without completely eliminating them.