User login
Community mourns nurse knocked over, killed in Times Square
in New York City. The Mass was promoted by the local consulate general of the Philippines.
Maria Ambrocio, 58, was visiting Times Square with a friend on October 1 when she was shoved to the ground by a man who reportedly snatched a cellphone and was running away. He later collided with a police officer before being arrested.
Ms. Ambrosio, of Bayonne, N.J., was taken to Bellevue Hospital in Manhattan with a traumatic brain injury. She was taken off life support a day later.
Jermaine Foster, 26, was charged with murder and robbery and is scheduled to appear in New York Criminal Court Thursday, according to court records.
Ms. Ambrocio, who had been a nurse for 25 years at Bayonne Medical Center in New Jersey, treated cancer patients, even during the height of the pandemic. The medical community at the hospital posted on social media, “Maria’s untimely death is a profound loss to us all, especially those whose lives she touched each day at Bayonne Medical Center.”
New York organizations expressed sympathy after the incident. Tom Harris, president of the Times Square Alliance, said in a statement shared with this news organization, “Our deepest condolences to the family of Maria Ambrocio. The killing of Maria Ambrocio near Times Square highlights one of our city’s greatest public safety challenges, the proliferation of people with untreated mental illness and drug addictions on our streets committing crimes without an effective strategy to address them. Our city needs to come together and solve these problems and those of us who work in these areas are willing and able to help. Let her death not be in vain.”
The New York Philippine consulate reported on its Facebook page that Ms. Ambrocio had just visited its office when the incident occurred. “We grieve with the rest of the Filipino Community over the death of our kababayan [countryman], Maria Ambrocio, a 58-year-old health frontliner from Bayonne, New Jersey....
“Maria’s passing was announced shortly after she was removed from life support a few hours ago. She had been on life support for the head trauma she sustained on Friday afternoon after she was knocked down by someone who was described as a mentally disturbed homeless man. Maria was walking with a kababayan near Times Square after visiting the Philippine consulate general when she was struck by the suspect, who was reportedly being chased after grabbing a mobile phone from someone.”
On the day she passed away, Bayonne Mayor Jimmy Davis shared an emotional message on Facebook: “I’m asking for all Bayonne people to say a prayer for Maria Ambrocio. Maria, an Oncology nurse at Bayonne Medical Center, was viciously attacked in an unprovoked assault by a deranged man in Times Square yesterday.
“Please keep Maria and her family in your thoughts through these difficult days.”
A version of this article first appeared on Medscape.com.
in New York City. The Mass was promoted by the local consulate general of the Philippines.
Maria Ambrocio, 58, was visiting Times Square with a friend on October 1 when she was shoved to the ground by a man who reportedly snatched a cellphone and was running away. He later collided with a police officer before being arrested.
Ms. Ambrosio, of Bayonne, N.J., was taken to Bellevue Hospital in Manhattan with a traumatic brain injury. She was taken off life support a day later.
Jermaine Foster, 26, was charged with murder and robbery and is scheduled to appear in New York Criminal Court Thursday, according to court records.
Ms. Ambrocio, who had been a nurse for 25 years at Bayonne Medical Center in New Jersey, treated cancer patients, even during the height of the pandemic. The medical community at the hospital posted on social media, “Maria’s untimely death is a profound loss to us all, especially those whose lives she touched each day at Bayonne Medical Center.”
New York organizations expressed sympathy after the incident. Tom Harris, president of the Times Square Alliance, said in a statement shared with this news organization, “Our deepest condolences to the family of Maria Ambrocio. The killing of Maria Ambrocio near Times Square highlights one of our city’s greatest public safety challenges, the proliferation of people with untreated mental illness and drug addictions on our streets committing crimes without an effective strategy to address them. Our city needs to come together and solve these problems and those of us who work in these areas are willing and able to help. Let her death not be in vain.”
The New York Philippine consulate reported on its Facebook page that Ms. Ambrocio had just visited its office when the incident occurred. “We grieve with the rest of the Filipino Community over the death of our kababayan [countryman], Maria Ambrocio, a 58-year-old health frontliner from Bayonne, New Jersey....
“Maria’s passing was announced shortly after she was removed from life support a few hours ago. She had been on life support for the head trauma she sustained on Friday afternoon after she was knocked down by someone who was described as a mentally disturbed homeless man. Maria was walking with a kababayan near Times Square after visiting the Philippine consulate general when she was struck by the suspect, who was reportedly being chased after grabbing a mobile phone from someone.”
On the day she passed away, Bayonne Mayor Jimmy Davis shared an emotional message on Facebook: “I’m asking for all Bayonne people to say a prayer for Maria Ambrocio. Maria, an Oncology nurse at Bayonne Medical Center, was viciously attacked in an unprovoked assault by a deranged man in Times Square yesterday.
“Please keep Maria and her family in your thoughts through these difficult days.”
A version of this article first appeared on Medscape.com.
in New York City. The Mass was promoted by the local consulate general of the Philippines.
Maria Ambrocio, 58, was visiting Times Square with a friend on October 1 when she was shoved to the ground by a man who reportedly snatched a cellphone and was running away. He later collided with a police officer before being arrested.
Ms. Ambrosio, of Bayonne, N.J., was taken to Bellevue Hospital in Manhattan with a traumatic brain injury. She was taken off life support a day later.
Jermaine Foster, 26, was charged with murder and robbery and is scheduled to appear in New York Criminal Court Thursday, according to court records.
Ms. Ambrocio, who had been a nurse for 25 years at Bayonne Medical Center in New Jersey, treated cancer patients, even during the height of the pandemic. The medical community at the hospital posted on social media, “Maria’s untimely death is a profound loss to us all, especially those whose lives she touched each day at Bayonne Medical Center.”
New York organizations expressed sympathy after the incident. Tom Harris, president of the Times Square Alliance, said in a statement shared with this news organization, “Our deepest condolences to the family of Maria Ambrocio. The killing of Maria Ambrocio near Times Square highlights one of our city’s greatest public safety challenges, the proliferation of people with untreated mental illness and drug addictions on our streets committing crimes without an effective strategy to address them. Our city needs to come together and solve these problems and those of us who work in these areas are willing and able to help. Let her death not be in vain.”
The New York Philippine consulate reported on its Facebook page that Ms. Ambrocio had just visited its office when the incident occurred. “We grieve with the rest of the Filipino Community over the death of our kababayan [countryman], Maria Ambrocio, a 58-year-old health frontliner from Bayonne, New Jersey....
“Maria’s passing was announced shortly after she was removed from life support a few hours ago. She had been on life support for the head trauma she sustained on Friday afternoon after she was knocked down by someone who was described as a mentally disturbed homeless man. Maria was walking with a kababayan near Times Square after visiting the Philippine consulate general when she was struck by the suspect, who was reportedly being chased after grabbing a mobile phone from someone.”
On the day she passed away, Bayonne Mayor Jimmy Davis shared an emotional message on Facebook: “I’m asking for all Bayonne people to say a prayer for Maria Ambrocio. Maria, an Oncology nurse at Bayonne Medical Center, was viciously attacked in an unprovoked assault by a deranged man in Times Square yesterday.
“Please keep Maria and her family in your thoughts through these difficult days.”
A version of this article first appeared on Medscape.com.
FDA issues warning about use of dermal fillers with needle-free devices
.

Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
.
Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
.
Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
Psychiatrists shift stance on gender dysphoria, recommend therapy
A new position statement from the Royal Australian and New Zealand College of Psychiatrists (RANZCP) stresses the importance of a mental health evaluation for people with gender dysphoria – in particular for children and adolescents – before any firm decisions are made on whether to prescribe hormonal treatments to transition, or perform surgeries, often referred to as “gender-affirming care.”
“There is a paucity of quality evidence on the outcomes of those presenting with gender dysphoria. In particular, there is a need for better evidence in relation to outcomes for children and young people,” the guidance states.
Because gender dysphoria “is associated with significant distress ... each case should be assessed by a mental health professional, which will frequently be a psychiatrist, with the person at the center of care. It is important the psychological state and context in which gender dysphoria has arisen is explored to assess the most appropriate treatment,” it adds.
The move by the psychiatry body represents a big shift in the landscape regarding recommendations for the treatment of gender dysphoria in Australia and New Zealand.
Asked to explain the new RANZCP position, Philip Morris, MBBS, FRANZCP, said: “The College acknowledged the complexity of the issues and the legitimacy of different approaches.”
Exploration of a patient’s reasons for identifying as transgender is essential, he said in an interview, especially when it comes to young people.
“There may be other reasons for doing it, and we need to look for those, identify them and treat them. This needs to be done before initiating hormones and changing the whole physical nature of the child,” he said.
“A cautious psychotherapy-first approach makes sense. If we can do that with adolescents, then we will take a big step in the right direction,” stressed Dr. Morris, who is president of the National Association of Practising Psychiatrists in Australia.
Keira Bell case and Scandinavian stance lead to more open discussion
The rapid rise in gender dysphoria among adolescents in the Western world, referred to as “rapid-onset” or “late-onset” gender dysphoria, has seen a huge increase in the number of natal girls presenting and created frenzied debate that has intensified worldwide in the last 12 months about how to best treat youth with gender dysphoria.
Concerns have arisen that some transgender identification is due to social contagion, and there is a growing number of “detransitioners” – people who identified as transgender, transitioned to the opposite gender, but then regretted their decision, changed their minds, and “detransitioned” back to their birth sex. If they have had hormone therapy, and in some cases surgery, they are left with irreversible changes to their bodies.
As a result, Scandinavian countries, most notably Finland, once eager advocates of the gender-affirmative approach, have pulled back and issued new treatment guidelines in 2020 stating that psychotherapy, rather than gender reassignment, should be the first line of treatment for gender-dysphoric youth.
This, along with a landmark High Court decision in the U.K. regarding the use of puberty-blocking drugs for children with gender dysphoria, brought by detransitioner Keira Bell, which was recently overturned by the Appeal Court, but which Ms. Bell now says she will take to the Supreme Court, has led to a considerable shift in the conversation around treating transgender adolescents with hormonal therapy, says Dr. Morris.
“This [has moved from] ... a topic that could previously not be talked about freely to one that we can discuss more openly now. This is a big improvement. Previously, everyone thought it was all settled, but it’s not, certainly not from a medical angle,” he states.
At odds with prior Australian recommendations
The RANZCP had previously endorsed the standard guidelines of the Royal Children’s Hospital (RCH) Melbourne, followed by most gender-identity services in Australia and similar guidance from New Zealand, which both recommend gender-affirming care.
“Increasing evidence demonstrates that with supportive, gender-affirming care during childhood and adolescence, harms can be ameliorated and mental health and well-being outcomes can be significantly improved,” state the RCH guidelines.
But in 2019, RANZCP removed its endorsement of the RCH guidelines and started a consultation, which resulted in the new position statement.
However, Ken Pang, MD, of the Murdoch Children’s Research Institute in Melbourne and an author of the RCH guidelines, says the key recommendations of the new RANZCP position statement are consistent with their own guidelines.
The former note “the need for a skilled mental health clinician in providing comprehensive exploration of a child or adolescent’s biopsychosocial context,” Dr. Pang says.
However, it’s difficult not to see the contrast in stance when the new RANZCP statement maintains: “Research on gender dysphoria is still emerging. There are polarized views and mixed evidence regarding treatment options for people presenting with gender identity concerns, especially children and young people.”
Dr. Pang says the RCH guidelines do, however, recognize the need for further research in the field.
“I look forward to being able to incorporate such research, including from our own Trans20 study, into future revisions of our guidelines,” he told this news organization.
Watch your backs with affirmative therapy: Will there be a compromise?
Dr. Morris says there will obviously be cases where “the child might transition with a medical intervention, but that wouldn’t be the first step.”
And yet, he adds, “There are those who push the pro-trans view that everyone should be allowed to transition, and the doctors are only technicians that provide hormones with no questions asked.”
But from a doctor’s perspective, clinicians will still be held responsible in medical and legal terms for the treatments given, he stressed.
“I don’t think they will ever not be accountable for that. They will always need to determine in their own mind whether their actions have positive value that outweigh any disadvantages,” Dr. Morris continues.
The RANZCP statement does, in fact, stress just this.
All health care professionals need to “be aware of ethical and medicolegal dilemmas” pertaining to affirmative therapy, it indicates. “Psychiatrists should practice within the relevant laws and accepted professional standards in relation to assessing capacity and obtaining consent...”
Dr. Morris hopes there will ultimately be many more checks and balances in place and that courts and clinicians will need to step back and not assume every child who seeks to transition is doing it as a result of pure gender dysphoria.
He predicts that things will end in a compromise.
“In my view, this compromise will treat children with respect and approach them like any other patient that presents with a condition that requires proper assessment and treatment.”
“In the end, some cases will be transitioned, but there will be fewer than [are] transitioned at the moment,” he predicts.
Dr. Morris has reported no relevant financial relationships. Dr. Pang is a member of the Australian Professional Association for Trans Health and its research committee.
A version of this article first appeared on Medscape.com.
A new position statement from the Royal Australian and New Zealand College of Psychiatrists (RANZCP) stresses the importance of a mental health evaluation for people with gender dysphoria – in particular for children and adolescents – before any firm decisions are made on whether to prescribe hormonal treatments to transition, or perform surgeries, often referred to as “gender-affirming care.”
“There is a paucity of quality evidence on the outcomes of those presenting with gender dysphoria. In particular, there is a need for better evidence in relation to outcomes for children and young people,” the guidance states.
Because gender dysphoria “is associated with significant distress ... each case should be assessed by a mental health professional, which will frequently be a psychiatrist, with the person at the center of care. It is important the psychological state and context in which gender dysphoria has arisen is explored to assess the most appropriate treatment,” it adds.
The move by the psychiatry body represents a big shift in the landscape regarding recommendations for the treatment of gender dysphoria in Australia and New Zealand.
Asked to explain the new RANZCP position, Philip Morris, MBBS, FRANZCP, said: “The College acknowledged the complexity of the issues and the legitimacy of different approaches.”
Exploration of a patient’s reasons for identifying as transgender is essential, he said in an interview, especially when it comes to young people.
“There may be other reasons for doing it, and we need to look for those, identify them and treat them. This needs to be done before initiating hormones and changing the whole physical nature of the child,” he said.
“A cautious psychotherapy-first approach makes sense. If we can do that with adolescents, then we will take a big step in the right direction,” stressed Dr. Morris, who is president of the National Association of Practising Psychiatrists in Australia.
Keira Bell case and Scandinavian stance lead to more open discussion
The rapid rise in gender dysphoria among adolescents in the Western world, referred to as “rapid-onset” or “late-onset” gender dysphoria, has seen a huge increase in the number of natal girls presenting and created frenzied debate that has intensified worldwide in the last 12 months about how to best treat youth with gender dysphoria.
Concerns have arisen that some transgender identification is due to social contagion, and there is a growing number of “detransitioners” – people who identified as transgender, transitioned to the opposite gender, but then regretted their decision, changed their minds, and “detransitioned” back to their birth sex. If they have had hormone therapy, and in some cases surgery, they are left with irreversible changes to their bodies.
As a result, Scandinavian countries, most notably Finland, once eager advocates of the gender-affirmative approach, have pulled back and issued new treatment guidelines in 2020 stating that psychotherapy, rather than gender reassignment, should be the first line of treatment for gender-dysphoric youth.
This, along with a landmark High Court decision in the U.K. regarding the use of puberty-blocking drugs for children with gender dysphoria, brought by detransitioner Keira Bell, which was recently overturned by the Appeal Court, but which Ms. Bell now says she will take to the Supreme Court, has led to a considerable shift in the conversation around treating transgender adolescents with hormonal therapy, says Dr. Morris.
“This [has moved from] ... a topic that could previously not be talked about freely to one that we can discuss more openly now. This is a big improvement. Previously, everyone thought it was all settled, but it’s not, certainly not from a medical angle,” he states.
At odds with prior Australian recommendations
The RANZCP had previously endorsed the standard guidelines of the Royal Children’s Hospital (RCH) Melbourne, followed by most gender-identity services in Australia and similar guidance from New Zealand, which both recommend gender-affirming care.
“Increasing evidence demonstrates that with supportive, gender-affirming care during childhood and adolescence, harms can be ameliorated and mental health and well-being outcomes can be significantly improved,” state the RCH guidelines.
But in 2019, RANZCP removed its endorsement of the RCH guidelines and started a consultation, which resulted in the new position statement.
However, Ken Pang, MD, of the Murdoch Children’s Research Institute in Melbourne and an author of the RCH guidelines, says the key recommendations of the new RANZCP position statement are consistent with their own guidelines.
The former note “the need for a skilled mental health clinician in providing comprehensive exploration of a child or adolescent’s biopsychosocial context,” Dr. Pang says.
However, it’s difficult not to see the contrast in stance when the new RANZCP statement maintains: “Research on gender dysphoria is still emerging. There are polarized views and mixed evidence regarding treatment options for people presenting with gender identity concerns, especially children and young people.”
Dr. Pang says the RCH guidelines do, however, recognize the need for further research in the field.
“I look forward to being able to incorporate such research, including from our own Trans20 study, into future revisions of our guidelines,” he told this news organization.
Watch your backs with affirmative therapy: Will there be a compromise?
Dr. Morris says there will obviously be cases where “the child might transition with a medical intervention, but that wouldn’t be the first step.”
And yet, he adds, “There are those who push the pro-trans view that everyone should be allowed to transition, and the doctors are only technicians that provide hormones with no questions asked.”
But from a doctor’s perspective, clinicians will still be held responsible in medical and legal terms for the treatments given, he stressed.
“I don’t think they will ever not be accountable for that. They will always need to determine in their own mind whether their actions have positive value that outweigh any disadvantages,” Dr. Morris continues.
The RANZCP statement does, in fact, stress just this.
All health care professionals need to “be aware of ethical and medicolegal dilemmas” pertaining to affirmative therapy, it indicates. “Psychiatrists should practice within the relevant laws and accepted professional standards in relation to assessing capacity and obtaining consent...”
Dr. Morris hopes there will ultimately be many more checks and balances in place and that courts and clinicians will need to step back and not assume every child who seeks to transition is doing it as a result of pure gender dysphoria.
He predicts that things will end in a compromise.
“In my view, this compromise will treat children with respect and approach them like any other patient that presents with a condition that requires proper assessment and treatment.”
“In the end, some cases will be transitioned, but there will be fewer than [are] transitioned at the moment,” he predicts.
Dr. Morris has reported no relevant financial relationships. Dr. Pang is a member of the Australian Professional Association for Trans Health and its research committee.
A version of this article first appeared on Medscape.com.
A new position statement from the Royal Australian and New Zealand College of Psychiatrists (RANZCP) stresses the importance of a mental health evaluation for people with gender dysphoria – in particular for children and adolescents – before any firm decisions are made on whether to prescribe hormonal treatments to transition, or perform surgeries, often referred to as “gender-affirming care.”
“There is a paucity of quality evidence on the outcomes of those presenting with gender dysphoria. In particular, there is a need for better evidence in relation to outcomes for children and young people,” the guidance states.
Because gender dysphoria “is associated with significant distress ... each case should be assessed by a mental health professional, which will frequently be a psychiatrist, with the person at the center of care. It is important the psychological state and context in which gender dysphoria has arisen is explored to assess the most appropriate treatment,” it adds.
The move by the psychiatry body represents a big shift in the landscape regarding recommendations for the treatment of gender dysphoria in Australia and New Zealand.
Asked to explain the new RANZCP position, Philip Morris, MBBS, FRANZCP, said: “The College acknowledged the complexity of the issues and the legitimacy of different approaches.”
Exploration of a patient’s reasons for identifying as transgender is essential, he said in an interview, especially when it comes to young people.
“There may be other reasons for doing it, and we need to look for those, identify them and treat them. This needs to be done before initiating hormones and changing the whole physical nature of the child,” he said.
“A cautious psychotherapy-first approach makes sense. If we can do that with adolescents, then we will take a big step in the right direction,” stressed Dr. Morris, who is president of the National Association of Practising Psychiatrists in Australia.
Keira Bell case and Scandinavian stance lead to more open discussion
The rapid rise in gender dysphoria among adolescents in the Western world, referred to as “rapid-onset” or “late-onset” gender dysphoria, has seen a huge increase in the number of natal girls presenting and created frenzied debate that has intensified worldwide in the last 12 months about how to best treat youth with gender dysphoria.
Concerns have arisen that some transgender identification is due to social contagion, and there is a growing number of “detransitioners” – people who identified as transgender, transitioned to the opposite gender, but then regretted their decision, changed their minds, and “detransitioned” back to their birth sex. If they have had hormone therapy, and in some cases surgery, they are left with irreversible changes to their bodies.
As a result, Scandinavian countries, most notably Finland, once eager advocates of the gender-affirmative approach, have pulled back and issued new treatment guidelines in 2020 stating that psychotherapy, rather than gender reassignment, should be the first line of treatment for gender-dysphoric youth.
This, along with a landmark High Court decision in the U.K. regarding the use of puberty-blocking drugs for children with gender dysphoria, brought by detransitioner Keira Bell, which was recently overturned by the Appeal Court, but which Ms. Bell now says she will take to the Supreme Court, has led to a considerable shift in the conversation around treating transgender adolescents with hormonal therapy, says Dr. Morris.
“This [has moved from] ... a topic that could previously not be talked about freely to one that we can discuss more openly now. This is a big improvement. Previously, everyone thought it was all settled, but it’s not, certainly not from a medical angle,” he states.
At odds with prior Australian recommendations
The RANZCP had previously endorsed the standard guidelines of the Royal Children’s Hospital (RCH) Melbourne, followed by most gender-identity services in Australia and similar guidance from New Zealand, which both recommend gender-affirming care.
“Increasing evidence demonstrates that with supportive, gender-affirming care during childhood and adolescence, harms can be ameliorated and mental health and well-being outcomes can be significantly improved,” state the RCH guidelines.
But in 2019, RANZCP removed its endorsement of the RCH guidelines and started a consultation, which resulted in the new position statement.
However, Ken Pang, MD, of the Murdoch Children’s Research Institute in Melbourne and an author of the RCH guidelines, says the key recommendations of the new RANZCP position statement are consistent with their own guidelines.
The former note “the need for a skilled mental health clinician in providing comprehensive exploration of a child or adolescent’s biopsychosocial context,” Dr. Pang says.
However, it’s difficult not to see the contrast in stance when the new RANZCP statement maintains: “Research on gender dysphoria is still emerging. There are polarized views and mixed evidence regarding treatment options for people presenting with gender identity concerns, especially children and young people.”
Dr. Pang says the RCH guidelines do, however, recognize the need for further research in the field.
“I look forward to being able to incorporate such research, including from our own Trans20 study, into future revisions of our guidelines,” he told this news organization.
Watch your backs with affirmative therapy: Will there be a compromise?
Dr. Morris says there will obviously be cases where “the child might transition with a medical intervention, but that wouldn’t be the first step.”
And yet, he adds, “There are those who push the pro-trans view that everyone should be allowed to transition, and the doctors are only technicians that provide hormones with no questions asked.”
But from a doctor’s perspective, clinicians will still be held responsible in medical and legal terms for the treatments given, he stressed.
“I don’t think they will ever not be accountable for that. They will always need to determine in their own mind whether their actions have positive value that outweigh any disadvantages,” Dr. Morris continues.
The RANZCP statement does, in fact, stress just this.
All health care professionals need to “be aware of ethical and medicolegal dilemmas” pertaining to affirmative therapy, it indicates. “Psychiatrists should practice within the relevant laws and accepted professional standards in relation to assessing capacity and obtaining consent...”
Dr. Morris hopes there will ultimately be many more checks and balances in place and that courts and clinicians will need to step back and not assume every child who seeks to transition is doing it as a result of pure gender dysphoria.
He predicts that things will end in a compromise.
“In my view, this compromise will treat children with respect and approach them like any other patient that presents with a condition that requires proper assessment and treatment.”
“In the end, some cases will be transitioned, but there will be fewer than [are] transitioned at the moment,” he predicts.
Dr. Morris has reported no relevant financial relationships. Dr. Pang is a member of the Australian Professional Association for Trans Health and its research committee.
A version of this article first appeared on Medscape.com.
Benzene prompts recalls of spray antifungals and sunscreens

Bayer has voluntarily recalled batches of its Lotrimin and Tinactin products because of benzene detected in some samples, according to an Oct. 1 company announcement, available on the Food and Drug Administration website. “It is important to note that Bayer’s decision to voluntarily recall these products is a precautionary measure and that the levels detected are not expected to cause adverse health consequences in consumers,” the announcement said.
Benzene is classified as a human carcinogen present in the environment from both natural sources and human activity, and it has been shown to cause cancer with long-term exposure.
The products included in the recall – all in aerosol spray cans – are unexpired Lotrimin and Tinactin sprays with lot numbers starting with TN, CV, or NAA that were distributed to consumer venues between September 2018 and September 2021. The over-the-counter products are Lotrimin Anti-Fungal Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal Jock Itch (AFJI) Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal (AF) Athlete’s Foot Deodorant Powder Spray, Lotrimin AF Athlete’s Foot Liquid Spray, Lotrimin AF Athlete’s Foot Daily Prevention Deodorant Powder Spray, Tinactin Jock Itch (JI) Powder Spray, Tinactin Athlete’s Foot Deodorant Powder Spray, Tinactin Athlete’s Foot Powder Spray, and Tinactin Athlete’s Foot Liquid Spray.
Bayer has received no reports of adverse events related to the recall. The company also reported no concerns with its antifungal creams or other products.
In addition, Coppertone has issued a voluntary recall of specific lots of five spray sunscreen products because of the presence of benzene, according to a Sept. 30th company announcement, also posted on the FDA website. The recall includes Pure&Simple spray for babies, children, and adults; Coppertone Sport Mineral Spray; and Travel-sized Coppertone Sport spray. The specific lots were manufactured between January and June 2021, and are listed on the company announcement.
“Daily exposure to benzene at the levels detected in these affected Coppertone aerosol sunscreen spray products would not be expected to cause adverse health consequences based on generally accepted exposure modeling by numerous regulatory agencies,” according to the announcement. Coppertone has received no reports of adverse events related to the recall.
In the announcement, Coppertone advised consumers to discontinue use of the impacted products, dispose of the aerosol cans properly, and contact their physician or health care provider if they experience any problems related to the sunscreen sprays.
In May 2021, online pharmacy Valisure, which routinely tests their medications, petitioned the FDA to recall specific sunscreens after detecting high benzene levels in several brands and batches of sunscreen products. The FDA evaluated the petition, but the agency itself did not issue any recalls of sunscreens.
Clinicians are advised to report any adverse events to the FDA’s MedWatch Adverse Event Reporting program either online or by regular mail or fax using this form.
Bayer has voluntarily recalled batches of its Lotrimin and Tinactin products because of benzene detected in some samples, according to an Oct. 1 company announcement, available on the Food and Drug Administration website. “It is important to note that Bayer’s decision to voluntarily recall these products is a precautionary measure and that the levels detected are not expected to cause adverse health consequences in consumers,” the announcement said.
Benzene is classified as a human carcinogen present in the environment from both natural sources and human activity, and it has been shown to cause cancer with long-term exposure.
The products included in the recall – all in aerosol spray cans – are unexpired Lotrimin and Tinactin sprays with lot numbers starting with TN, CV, or NAA that were distributed to consumer venues between September 2018 and September 2021. The over-the-counter products are Lotrimin Anti-Fungal Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal Jock Itch (AFJI) Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal (AF) Athlete’s Foot Deodorant Powder Spray, Lotrimin AF Athlete’s Foot Liquid Spray, Lotrimin AF Athlete’s Foot Daily Prevention Deodorant Powder Spray, Tinactin Jock Itch (JI) Powder Spray, Tinactin Athlete’s Foot Deodorant Powder Spray, Tinactin Athlete’s Foot Powder Spray, and Tinactin Athlete’s Foot Liquid Spray.
Bayer has received no reports of adverse events related to the recall. The company also reported no concerns with its antifungal creams or other products.
In addition, Coppertone has issued a voluntary recall of specific lots of five spray sunscreen products because of the presence of benzene, according to a Sept. 30th company announcement, also posted on the FDA website. The recall includes Pure&Simple spray for babies, children, and adults; Coppertone Sport Mineral Spray; and Travel-sized Coppertone Sport spray. The specific lots were manufactured between January and June 2021, and are listed on the company announcement.
“Daily exposure to benzene at the levels detected in these affected Coppertone aerosol sunscreen spray products would not be expected to cause adverse health consequences based on generally accepted exposure modeling by numerous regulatory agencies,” according to the announcement. Coppertone has received no reports of adverse events related to the recall.
In the announcement, Coppertone advised consumers to discontinue use of the impacted products, dispose of the aerosol cans properly, and contact their physician or health care provider if they experience any problems related to the sunscreen sprays.
In May 2021, online pharmacy Valisure, which routinely tests their medications, petitioned the FDA to recall specific sunscreens after detecting high benzene levels in several brands and batches of sunscreen products. The FDA evaluated the petition, but the agency itself did not issue any recalls of sunscreens.
Clinicians are advised to report any adverse events to the FDA’s MedWatch Adverse Event Reporting program either online or by regular mail or fax using this form.
Bayer has voluntarily recalled batches of its Lotrimin and Tinactin products because of benzene detected in some samples, according to an Oct. 1 company announcement, available on the Food and Drug Administration website. “It is important to note that Bayer’s decision to voluntarily recall these products is a precautionary measure and that the levels detected are not expected to cause adverse health consequences in consumers,” the announcement said.
Benzene is classified as a human carcinogen present in the environment from both natural sources and human activity, and it has been shown to cause cancer with long-term exposure.
The products included in the recall – all in aerosol spray cans – are unexpired Lotrimin and Tinactin sprays with lot numbers starting with TN, CV, or NAA that were distributed to consumer venues between September 2018 and September 2021. The over-the-counter products are Lotrimin Anti-Fungal Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal Jock Itch (AFJI) Athlete’s Foot Powder Spray, Lotrimin Anti-Fungal (AF) Athlete’s Foot Deodorant Powder Spray, Lotrimin AF Athlete’s Foot Liquid Spray, Lotrimin AF Athlete’s Foot Daily Prevention Deodorant Powder Spray, Tinactin Jock Itch (JI) Powder Spray, Tinactin Athlete’s Foot Deodorant Powder Spray, Tinactin Athlete’s Foot Powder Spray, and Tinactin Athlete’s Foot Liquid Spray.
Bayer has received no reports of adverse events related to the recall. The company also reported no concerns with its antifungal creams or other products.
In addition, Coppertone has issued a voluntary recall of specific lots of five spray sunscreen products because of the presence of benzene, according to a Sept. 30th company announcement, also posted on the FDA website. The recall includes Pure&Simple spray for babies, children, and adults; Coppertone Sport Mineral Spray; and Travel-sized Coppertone Sport spray. The specific lots were manufactured between January and June 2021, and are listed on the company announcement.
“Daily exposure to benzene at the levels detected in these affected Coppertone aerosol sunscreen spray products would not be expected to cause adverse health consequences based on generally accepted exposure modeling by numerous regulatory agencies,” according to the announcement. Coppertone has received no reports of adverse events related to the recall.
In the announcement, Coppertone advised consumers to discontinue use of the impacted products, dispose of the aerosol cans properly, and contact their physician or health care provider if they experience any problems related to the sunscreen sprays.
In May 2021, online pharmacy Valisure, which routinely tests their medications, petitioned the FDA to recall specific sunscreens after detecting high benzene levels in several brands and batches of sunscreen products. The FDA evaluated the petition, but the agency itself did not issue any recalls of sunscreens.
Clinicians are advised to report any adverse events to the FDA’s MedWatch Adverse Event Reporting program either online or by regular mail or fax using this form.
Pfizer asks FDA to authorize COVID vaccine for kids 5-11
The request comes after the drugmaker submitted clinical trial data to the FDA on Sept. 28. Pfizer said the study of 2,268 children showed the vaccine was safe and produced a robust immune response.
Participants in the studies received a lower dose of the vaccine, 10 micrograms. Their response 2 weeks after a second dose was reportedly equal to the immune protection in a control group of 16- to 25-year-olds who received the fully approved 30-microgram doses.
Currently, the Pfizer EUA applies to 12- to 15-year-olds and people eligible for a Pfizer booster shot. The drugmaker received full FDA approval for the vaccine for Americans 16 years and older in August.
The filing for authorization in 5- to 11-year-olds comes as overall cases of COVID-19 in the United States continue to decline. The decrease includes a drop in new cases in children for the fourth consecutive week, according to analysis of data from the American Academy of Pediatrics and the Children’s Hospital Association.
The next step is an FDA decision on whether to expand the current emergency use authorization (EUA) for teenagers to the younger age group.
Timing of any official word from the agency is unknown. But possibly in anticipation of today’s filing, the FDA already scheduled a meeting of its Vaccines and Related Biological Products Advisory Committee for Oct. 25.
A version of this article first appeared on WebMD.com.
The request comes after the drugmaker submitted clinical trial data to the FDA on Sept. 28. Pfizer said the study of 2,268 children showed the vaccine was safe and produced a robust immune response.
Participants in the studies received a lower dose of the vaccine, 10 micrograms. Their response 2 weeks after a second dose was reportedly equal to the immune protection in a control group of 16- to 25-year-olds who received the fully approved 30-microgram doses.
Currently, the Pfizer EUA applies to 12- to 15-year-olds and people eligible for a Pfizer booster shot. The drugmaker received full FDA approval for the vaccine for Americans 16 years and older in August.
The filing for authorization in 5- to 11-year-olds comes as overall cases of COVID-19 in the United States continue to decline. The decrease includes a drop in new cases in children for the fourth consecutive week, according to analysis of data from the American Academy of Pediatrics and the Children’s Hospital Association.
The next step is an FDA decision on whether to expand the current emergency use authorization (EUA) for teenagers to the younger age group.
Timing of any official word from the agency is unknown. But possibly in anticipation of today’s filing, the FDA already scheduled a meeting of its Vaccines and Related Biological Products Advisory Committee for Oct. 25.
A version of this article first appeared on WebMD.com.
The request comes after the drugmaker submitted clinical trial data to the FDA on Sept. 28. Pfizer said the study of 2,268 children showed the vaccine was safe and produced a robust immune response.
Participants in the studies received a lower dose of the vaccine, 10 micrograms. Their response 2 weeks after a second dose was reportedly equal to the immune protection in a control group of 16- to 25-year-olds who received the fully approved 30-microgram doses.
Currently, the Pfizer EUA applies to 12- to 15-year-olds and people eligible for a Pfizer booster shot. The drugmaker received full FDA approval for the vaccine for Americans 16 years and older in August.
The filing for authorization in 5- to 11-year-olds comes as overall cases of COVID-19 in the United States continue to decline. The decrease includes a drop in new cases in children for the fourth consecutive week, according to analysis of data from the American Academy of Pediatrics and the Children’s Hospital Association.
The next step is an FDA decision on whether to expand the current emergency use authorization (EUA) for teenagers to the younger age group.
Timing of any official word from the agency is unknown. But possibly in anticipation of today’s filing, the FDA already scheduled a meeting of its Vaccines and Related Biological Products Advisory Committee for Oct. 25.
A version of this article first appeared on WebMD.com.
Merck’s new COVID-19 pill: ‘Game changer’ or just one more tool?
Soon after Merck announced on Oct. 1 that it would ask federal regulators for emergency use authorization (EUA) for its auspicious new COVID-19 pill, the accolades began.
Former Food and Drug Administration chief Scott Gottlieb, MD, told CNBC the drug was “a profound game changer.” Top infectious disease expert Anthony S. Fauci, MD, called the early data “impressive.” The World Health Organization termed it “certainly good news,” while saying it awaits more data.
Merck, partnering with Ridgeback Biotherapeutics on the investigational oral antiviral medicine molnupiravir, plans to submit applications to regulatory agencies worldwide, hoping to deliver the first oral antiviral medication for COVID-19.
Interim clinical trial results show that the drug may slash the risk for hospitalization or death by 50% in those with mild to moderate COVID-19.
When the results were found to be so favorable, the study was halted at the recommendation of an independent data-monitoring committee and in consultation with the FDA.
“This anticipated drug has gotten a little more hype than it deserves,” said William Schaffner, MD, professor of preventive medicine and infectious disease specialist at Vanderbilt University Medical Center in Nashville, Tenn. He and others suggest a reality check.
“It’s not exactly a home run, like penicillin for strep throat,” agreed Carl Fichtenbaum, MD, professor of infectious diseases at the University of Cincinnati, who is investigating a similar pill for a rival company, Atea, partnering with Roche.
“But it is encouraging,” he said. “It will probably be an incremental improvement on what we have.” The fact that it can be taken at home is a plus: “Anything we can do to keep people from getting sicker is a good thing.”
“The data show in this higher risk group [those who were studied had at least one risk factor for severe COVID-19, such as age or a medical condition], it reduces the risk of advancing to severe disease by 50%,” Dr. Schaffner said. While that’s a clear benefit for half, it of course leaves the other half without benefit, he said.
Others critiqued the predicted cost of the drug. The U.S. government has already agreed to pay about $700 per patient, according to a new report from Harvard T. H. Chan School of Public Health, Boston, and King’s College Hospital, London. That analysis concluded that the actual cost of production for the 5-day course is $17.74.
“We fully expect that having an oral treatment that reduces the risk of hospitalizations will be significantly cost effective for society,” Melissa Moody, a Merck spokesperson, told this news organization. “We are optimistic that molnupiravir can become an important medicine as part of the global effort to fight the pandemic.”
Merck expects to produce 10 million courses of treatment by the end of the year, with additional doses expected to be produced in 2022, according to a company press release. Earlier in 2021, Merck finalized its agreement with the U.S. government to supply about 1.7 million courses of the drug at the $700 price, once an EUA or FDA approval is given.
Merck also has supply and purchase agreements with other governments worldwide, pending regulatory approval.
Study details
Details about the study findings came from a Merck press release. In the planned interim analysis, Merck and Ridgeback evaluated data from 775 patients initially enrolled in the phase 3 MOVe-OUT trial.
All adults had lab-confirmed mild to moderate COVID-19, and reported onset of symptoms within 5 days of being randomly assigned to the drug or placebo. All had at least one risk factor linked with poor disease outcome (such as older age or obesity).
The drug is a ribonucleoside and works by creating mutations in the virus’s genome, halting the ability of the virus to replicate.
Through day 29 of the study, the drug reduced the risk or hospitalization or death by about 50%. While 7.3% of those who received the drug either died or were hospitalized by day 29, 14.1% of those on placebo did, a statistically significant difference (P = .0012).
Side effects were similar in both groups, with 35% of the drug-treated and 40% of the placebo group reporting some side effect, Merck reported. Adverse drug-related events were 12% in the drug group and 11% in the placebo group. While 1.3% of the drug-treated group quit the study because of an adverse event, 3.4% of the placebo group quit.
Pros, cons, and unknowns
The ability to take the drug orally, and at home, is a definite plus, Dr. Schaffner said, compared with the monoclonal antibody treatment currently approved that must be given intravenously or subcutaneously and in certain locations.
More people could be reached and helped with the option of an at-home, oral medicine, he and others agreed.
The regimen for molnupiravir is four pills, two times daily, for 5 days, even if symptoms are mild. As with other prescription drugs, “there will always be folks who don’t comply completely” with the prescribed regimen, Dr. Schaffner said. With this pill, that might be especially true if the symptoms are very mild.
The 50% reduction is not as effective as the benefit often quoted for monoclonal antibody treatment. In clinical trials of Regeneron’s monoclonal antibody treatment, the regimen reduced COVID-19–related hospitalization or death in high-risk patients by 70%.
Even so, the new pill could change the pandemic’s course, others say. “I think molnupiravir has the potential to change how we take care of people who have COVID and risk factors for developing severe disease,” Rajesh Tim Gandhi, MD, an infectious disease physician at Massachusetts General Hospital and Harvard Medical School in Boston, told this news organization.
“What we’ll need to do, however, is make sure that people get tested quickly after they develop symptoms and, if they’re confirmed to have COVID, start on the pills within 5 days of developing symptoms,” he said, while warning that more data are needed about the drug and the trial results.
Another concern is that the promise of a pill will stall vaccination rates, with some people figuring why get vaccinated when they can obtain the pill if they do get sick.
Relying on treatment alone won’t work, Dr. Schaffner said. “Let’s [also] focus on prevention, which is the vaccine. We have to keep working both sides of the street.”
Dr. Gandhi added: “It’s important to remember that even though molnupiravir reduced the likelihood of hospitalization and death, a number of people who received the drug still got sick enough to end up in the hospital.”
Also unknown, he said, is how severe their disease was and whether they will develop long COVID.
The Merck study included only unvaccinated people. Might it work for those vaccinated people who get a breakthrough infection? “From a purely scientific perspective, there is no reason to believe molnupiravir would not work in people who are vaccinated, but the overall efficacy on top of the vaccine is likely dependent on how well they were able to mount a protective immune response to the vaccine,” Ms. Moody said. Still, Merck believes the pill could be of benefit for these infections too, she added.
As for the expected cost, Ms. Moody said that the company takes into account a number of factors in setting pricing, “but fundamentally we look at the impact of the disease, the benefits that the drug delivers to patients and to society, and at supporting ongoing drug development.”
On Merck’s heels: Pfizer, Roche, Atea
Pfizer is studying an antiviral pill, PF-07321332, a protease inhibitor that blocks the protease enzymes and halts replication of the virus.
In addition to studying the drug in infected patients at high risk of severe illness and in those at typical risk, Pfizer launched a phase 2-3 study in late September that will enroll people who live in the same household as a person with a confirmed, symptomatic COVID-19 infection to see if the drug can prevent disease in those who have been exposed.
Atea and Roche’s COVID pill, AT527, is in phase 3 trials as well. AT527 is an inhibitor of polymerase, an enzyme many viruses have, to stop replications. Atea is evaluating the drug to reduce disease “burden” and for both pre- and postexposure prevention.
Big picture: Role of COVID-19 pills
It may be necessary to target the coronavirus with more than one antiviral agent, said Dr. Fichtenbaum, a principal investigator for the AT527 trials.
“Sometimes viruses require two or three active agents to control their replication,” he said, citing information gleaned from other viral research, such as HIV. For control of HIV infection, a cocktail or combination of antivirals is often recommended.
That may well be the case for COVID-19, Dr. Fichtenbaum said. The goal would be to attack the virus at more than one pathway.
A version of this article first appeared on Medscape.com.
Soon after Merck announced on Oct. 1 that it would ask federal regulators for emergency use authorization (EUA) for its auspicious new COVID-19 pill, the accolades began.
Former Food and Drug Administration chief Scott Gottlieb, MD, told CNBC the drug was “a profound game changer.” Top infectious disease expert Anthony S. Fauci, MD, called the early data “impressive.” The World Health Organization termed it “certainly good news,” while saying it awaits more data.
Merck, partnering with Ridgeback Biotherapeutics on the investigational oral antiviral medicine molnupiravir, plans to submit applications to regulatory agencies worldwide, hoping to deliver the first oral antiviral medication for COVID-19.
Interim clinical trial results show that the drug may slash the risk for hospitalization or death by 50% in those with mild to moderate COVID-19.
When the results were found to be so favorable, the study was halted at the recommendation of an independent data-monitoring committee and in consultation with the FDA.
“This anticipated drug has gotten a little more hype than it deserves,” said William Schaffner, MD, professor of preventive medicine and infectious disease specialist at Vanderbilt University Medical Center in Nashville, Tenn. He and others suggest a reality check.
“It’s not exactly a home run, like penicillin for strep throat,” agreed Carl Fichtenbaum, MD, professor of infectious diseases at the University of Cincinnati, who is investigating a similar pill for a rival company, Atea, partnering with Roche.
“But it is encouraging,” he said. “It will probably be an incremental improvement on what we have.” The fact that it can be taken at home is a plus: “Anything we can do to keep people from getting sicker is a good thing.”
“The data show in this higher risk group [those who were studied had at least one risk factor for severe COVID-19, such as age or a medical condition], it reduces the risk of advancing to severe disease by 50%,” Dr. Schaffner said. While that’s a clear benefit for half, it of course leaves the other half without benefit, he said.
Others critiqued the predicted cost of the drug. The U.S. government has already agreed to pay about $700 per patient, according to a new report from Harvard T. H. Chan School of Public Health, Boston, and King’s College Hospital, London. That analysis concluded that the actual cost of production for the 5-day course is $17.74.
“We fully expect that having an oral treatment that reduces the risk of hospitalizations will be significantly cost effective for society,” Melissa Moody, a Merck spokesperson, told this news organization. “We are optimistic that molnupiravir can become an important medicine as part of the global effort to fight the pandemic.”
Merck expects to produce 10 million courses of treatment by the end of the year, with additional doses expected to be produced in 2022, according to a company press release. Earlier in 2021, Merck finalized its agreement with the U.S. government to supply about 1.7 million courses of the drug at the $700 price, once an EUA or FDA approval is given.
Merck also has supply and purchase agreements with other governments worldwide, pending regulatory approval.
Study details
Details about the study findings came from a Merck press release. In the planned interim analysis, Merck and Ridgeback evaluated data from 775 patients initially enrolled in the phase 3 MOVe-OUT trial.
All adults had lab-confirmed mild to moderate COVID-19, and reported onset of symptoms within 5 days of being randomly assigned to the drug or placebo. All had at least one risk factor linked with poor disease outcome (such as older age or obesity).
The drug is a ribonucleoside and works by creating mutations in the virus’s genome, halting the ability of the virus to replicate.
Through day 29 of the study, the drug reduced the risk or hospitalization or death by about 50%. While 7.3% of those who received the drug either died or were hospitalized by day 29, 14.1% of those on placebo did, a statistically significant difference (P = .0012).
Side effects were similar in both groups, with 35% of the drug-treated and 40% of the placebo group reporting some side effect, Merck reported. Adverse drug-related events were 12% in the drug group and 11% in the placebo group. While 1.3% of the drug-treated group quit the study because of an adverse event, 3.4% of the placebo group quit.
Pros, cons, and unknowns
The ability to take the drug orally, and at home, is a definite plus, Dr. Schaffner said, compared with the monoclonal antibody treatment currently approved that must be given intravenously or subcutaneously and in certain locations.
More people could be reached and helped with the option of an at-home, oral medicine, he and others agreed.
The regimen for molnupiravir is four pills, two times daily, for 5 days, even if symptoms are mild. As with other prescription drugs, “there will always be folks who don’t comply completely” with the prescribed regimen, Dr. Schaffner said. With this pill, that might be especially true if the symptoms are very mild.
The 50% reduction is not as effective as the benefit often quoted for monoclonal antibody treatment. In clinical trials of Regeneron’s monoclonal antibody treatment, the regimen reduced COVID-19–related hospitalization or death in high-risk patients by 70%.
Even so, the new pill could change the pandemic’s course, others say. “I think molnupiravir has the potential to change how we take care of people who have COVID and risk factors for developing severe disease,” Rajesh Tim Gandhi, MD, an infectious disease physician at Massachusetts General Hospital and Harvard Medical School in Boston, told this news organization.
“What we’ll need to do, however, is make sure that people get tested quickly after they develop symptoms and, if they’re confirmed to have COVID, start on the pills within 5 days of developing symptoms,” he said, while warning that more data are needed about the drug and the trial results.
Another concern is that the promise of a pill will stall vaccination rates, with some people figuring why get vaccinated when they can obtain the pill if they do get sick.
Relying on treatment alone won’t work, Dr. Schaffner said. “Let’s [also] focus on prevention, which is the vaccine. We have to keep working both sides of the street.”
Dr. Gandhi added: “It’s important to remember that even though molnupiravir reduced the likelihood of hospitalization and death, a number of people who received the drug still got sick enough to end up in the hospital.”
Also unknown, he said, is how severe their disease was and whether they will develop long COVID.
The Merck study included only unvaccinated people. Might it work for those vaccinated people who get a breakthrough infection? “From a purely scientific perspective, there is no reason to believe molnupiravir would not work in people who are vaccinated, but the overall efficacy on top of the vaccine is likely dependent on how well they were able to mount a protective immune response to the vaccine,” Ms. Moody said. Still, Merck believes the pill could be of benefit for these infections too, she added.
As for the expected cost, Ms. Moody said that the company takes into account a number of factors in setting pricing, “but fundamentally we look at the impact of the disease, the benefits that the drug delivers to patients and to society, and at supporting ongoing drug development.”
On Merck’s heels: Pfizer, Roche, Atea
Pfizer is studying an antiviral pill, PF-07321332, a protease inhibitor that blocks the protease enzymes and halts replication of the virus.
In addition to studying the drug in infected patients at high risk of severe illness and in those at typical risk, Pfizer launched a phase 2-3 study in late September that will enroll people who live in the same household as a person with a confirmed, symptomatic COVID-19 infection to see if the drug can prevent disease in those who have been exposed.
Atea and Roche’s COVID pill, AT527, is in phase 3 trials as well. AT527 is an inhibitor of polymerase, an enzyme many viruses have, to stop replications. Atea is evaluating the drug to reduce disease “burden” and for both pre- and postexposure prevention.
Big picture: Role of COVID-19 pills
It may be necessary to target the coronavirus with more than one antiviral agent, said Dr. Fichtenbaum, a principal investigator for the AT527 trials.
“Sometimes viruses require two or three active agents to control their replication,” he said, citing information gleaned from other viral research, such as HIV. For control of HIV infection, a cocktail or combination of antivirals is often recommended.
That may well be the case for COVID-19, Dr. Fichtenbaum said. The goal would be to attack the virus at more than one pathway.
A version of this article first appeared on Medscape.com.
Soon after Merck announced on Oct. 1 that it would ask federal regulators for emergency use authorization (EUA) for its auspicious new COVID-19 pill, the accolades began.
Former Food and Drug Administration chief Scott Gottlieb, MD, told CNBC the drug was “a profound game changer.” Top infectious disease expert Anthony S. Fauci, MD, called the early data “impressive.” The World Health Organization termed it “certainly good news,” while saying it awaits more data.
Merck, partnering with Ridgeback Biotherapeutics on the investigational oral antiviral medicine molnupiravir, plans to submit applications to regulatory agencies worldwide, hoping to deliver the first oral antiviral medication for COVID-19.
Interim clinical trial results show that the drug may slash the risk for hospitalization or death by 50% in those with mild to moderate COVID-19.
When the results were found to be so favorable, the study was halted at the recommendation of an independent data-monitoring committee and in consultation with the FDA.
“This anticipated drug has gotten a little more hype than it deserves,” said William Schaffner, MD, professor of preventive medicine and infectious disease specialist at Vanderbilt University Medical Center in Nashville, Tenn. He and others suggest a reality check.
“It’s not exactly a home run, like penicillin for strep throat,” agreed Carl Fichtenbaum, MD, professor of infectious diseases at the University of Cincinnati, who is investigating a similar pill for a rival company, Atea, partnering with Roche.
“But it is encouraging,” he said. “It will probably be an incremental improvement on what we have.” The fact that it can be taken at home is a plus: “Anything we can do to keep people from getting sicker is a good thing.”
“The data show in this higher risk group [those who were studied had at least one risk factor for severe COVID-19, such as age or a medical condition], it reduces the risk of advancing to severe disease by 50%,” Dr. Schaffner said. While that’s a clear benefit for half, it of course leaves the other half without benefit, he said.
Others critiqued the predicted cost of the drug. The U.S. government has already agreed to pay about $700 per patient, according to a new report from Harvard T. H. Chan School of Public Health, Boston, and King’s College Hospital, London. That analysis concluded that the actual cost of production for the 5-day course is $17.74.
“We fully expect that having an oral treatment that reduces the risk of hospitalizations will be significantly cost effective for society,” Melissa Moody, a Merck spokesperson, told this news organization. “We are optimistic that molnupiravir can become an important medicine as part of the global effort to fight the pandemic.”
Merck expects to produce 10 million courses of treatment by the end of the year, with additional doses expected to be produced in 2022, according to a company press release. Earlier in 2021, Merck finalized its agreement with the U.S. government to supply about 1.7 million courses of the drug at the $700 price, once an EUA or FDA approval is given.
Merck also has supply and purchase agreements with other governments worldwide, pending regulatory approval.
Study details
Details about the study findings came from a Merck press release. In the planned interim analysis, Merck and Ridgeback evaluated data from 775 patients initially enrolled in the phase 3 MOVe-OUT trial.
All adults had lab-confirmed mild to moderate COVID-19, and reported onset of symptoms within 5 days of being randomly assigned to the drug or placebo. All had at least one risk factor linked with poor disease outcome (such as older age or obesity).
The drug is a ribonucleoside and works by creating mutations in the virus’s genome, halting the ability of the virus to replicate.
Through day 29 of the study, the drug reduced the risk or hospitalization or death by about 50%. While 7.3% of those who received the drug either died or were hospitalized by day 29, 14.1% of those on placebo did, a statistically significant difference (P = .0012).
Side effects were similar in both groups, with 35% of the drug-treated and 40% of the placebo group reporting some side effect, Merck reported. Adverse drug-related events were 12% in the drug group and 11% in the placebo group. While 1.3% of the drug-treated group quit the study because of an adverse event, 3.4% of the placebo group quit.
Pros, cons, and unknowns
The ability to take the drug orally, and at home, is a definite plus, Dr. Schaffner said, compared with the monoclonal antibody treatment currently approved that must be given intravenously or subcutaneously and in certain locations.
More people could be reached and helped with the option of an at-home, oral medicine, he and others agreed.
The regimen for molnupiravir is four pills, two times daily, for 5 days, even if symptoms are mild. As with other prescription drugs, “there will always be folks who don’t comply completely” with the prescribed regimen, Dr. Schaffner said. With this pill, that might be especially true if the symptoms are very mild.
The 50% reduction is not as effective as the benefit often quoted for monoclonal antibody treatment. In clinical trials of Regeneron’s monoclonal antibody treatment, the regimen reduced COVID-19–related hospitalization or death in high-risk patients by 70%.
Even so, the new pill could change the pandemic’s course, others say. “I think molnupiravir has the potential to change how we take care of people who have COVID and risk factors for developing severe disease,” Rajesh Tim Gandhi, MD, an infectious disease physician at Massachusetts General Hospital and Harvard Medical School in Boston, told this news organization.
“What we’ll need to do, however, is make sure that people get tested quickly after they develop symptoms and, if they’re confirmed to have COVID, start on the pills within 5 days of developing symptoms,” he said, while warning that more data are needed about the drug and the trial results.
Another concern is that the promise of a pill will stall vaccination rates, with some people figuring why get vaccinated when they can obtain the pill if they do get sick.
Relying on treatment alone won’t work, Dr. Schaffner said. “Let’s [also] focus on prevention, which is the vaccine. We have to keep working both sides of the street.”
Dr. Gandhi added: “It’s important to remember that even though molnupiravir reduced the likelihood of hospitalization and death, a number of people who received the drug still got sick enough to end up in the hospital.”
Also unknown, he said, is how severe their disease was and whether they will develop long COVID.
The Merck study included only unvaccinated people. Might it work for those vaccinated people who get a breakthrough infection? “From a purely scientific perspective, there is no reason to believe molnupiravir would not work in people who are vaccinated, but the overall efficacy on top of the vaccine is likely dependent on how well they were able to mount a protective immune response to the vaccine,” Ms. Moody said. Still, Merck believes the pill could be of benefit for these infections too, she added.
As for the expected cost, Ms. Moody said that the company takes into account a number of factors in setting pricing, “but fundamentally we look at the impact of the disease, the benefits that the drug delivers to patients and to society, and at supporting ongoing drug development.”
On Merck’s heels: Pfizer, Roche, Atea
Pfizer is studying an antiviral pill, PF-07321332, a protease inhibitor that blocks the protease enzymes and halts replication of the virus.
In addition to studying the drug in infected patients at high risk of severe illness and in those at typical risk, Pfizer launched a phase 2-3 study in late September that will enroll people who live in the same household as a person with a confirmed, symptomatic COVID-19 infection to see if the drug can prevent disease in those who have been exposed.
Atea and Roche’s COVID pill, AT527, is in phase 3 trials as well. AT527 is an inhibitor of polymerase, an enzyme many viruses have, to stop replications. Atea is evaluating the drug to reduce disease “burden” and for both pre- and postexposure prevention.
Big picture: Role of COVID-19 pills
It may be necessary to target the coronavirus with more than one antiviral agent, said Dr. Fichtenbaum, a principal investigator for the AT527 trials.
“Sometimes viruses require two or three active agents to control their replication,” he said, citing information gleaned from other viral research, such as HIV. For control of HIV infection, a cocktail or combination of antivirals is often recommended.
That may well be the case for COVID-19, Dr. Fichtenbaum said. The goal would be to attack the virus at more than one pathway.
A version of this article first appeared on Medscape.com.
New York’s largest health care provider fires 1,400 unvaccinated employees
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
ADHD med may reduce apathy in Alzheimer’s disease
Methylphenidate is safe and effective for treating apathy in patients with Alzheimer’s disease (AD), new research suggests.

Results from a phase 3 randomized trial showed that, after 6 months of treatment, mean score on the Neuropsychiatric Inventory (NPI) apathy subscale decreased by 4.5 points for patients who received methylphenidate vs. a decrease of 3.1 points for those who received placebo.
In addition, the safety profile showed no significant between-group differences.
“Methylphenidate offers a treatment approach providing a modest but potentially clinically significant benefit for patients and caregivers,” said the investigators, led by Jacobo E. Mintzer, MD, MBA, professor of health studies at the Medical University of South Carolina in Charleston.
The findings were published online Sept. 27 in JAMA Neurology.
Common problem
Apathy, which is common among patients with AD, is associated with increased risk for mortality, financial burden, and caregiver burden. No treatment has proved effective for apathy in this population.
Two trials of methylphenidate, a catecholaminergic agent, have provided preliminary evidence of efficacy. Findings from the Apathy in Dementia Methylphenidate trial (ADMET) suggested the drug was associated with improved cognition and few adverse events. However, both trials had small patient populations and short durations.
The current investigators conducted ADMET 2, a 6-month, phase 3 trial, to investigate methylphenidate further. They recruited 200 patients (mean age, 76 years; 66% men; 90% White) at nine clinical centers that specialized in dementia care in the United States and one in Canada.
Eligible patients had a diagnosis of possible or probable AD and a Mini-Mental State Examination (MMSE) score between 10 and 28. They also had clinically significant apathy for at least 4 weeks and an available caregiver who spent more than 10 hours a week with the patient.
The researchers randomly assigned patients to receive methylphenidate (n = 99) or placebo (n = 101). For 3 days, participants in the active group received 10 mg/day of methylphenidate. After that point, they received 20 mg/day of methylphenidate for the rest of the study.
Patients in both treatment groups were given the same number of identical-appearing capsules each day.
In-person follow-up visits took place monthly for 6 months. Participants also were contacted by telephone at days 15, 45, and 75 after treatment assignment.
Participants underwent cognitive testing at baseline and at 2, 4, and 6 months. The battery of tests included the MMSE, Hopkins Verbal Learning Test, and Wechsler Adult Intelligence Scale – Revised Digit Span.
The trial’s two primary outcomes were mean change in NPI apathy score from baseline to 6 months and the odds of an improved rating on the Alzheimer’s Disease Cooperative Study Clinical Global Impression of Change (ADCS-CGIC) between baseline and 6 months.
Significant change on either outcome was to be considered a signal of effective treatment.
Treatment-specific benefit
Ten patients in the methylphenidate group and seven in the placebo group withdrew during the study.
Mean baseline score on the NPI apathy subscale was 8.0 vs. 7.6, respectively.
In an adjusted, longitudinal model, mean between-group difference in change in NPI apathy score at 6 months was –1.25 (P = .002). The mean NPI apathy score decreased by 4.5 points in the methylphenidate group vs. 3.1 points in the placebo group.
The largest change in apathy score occurred during the first 2 months of treatment. At 6 months, 27% of the methylphenidate group vs. 14% of the placebo group had an NPI apathy score of 0.
In addition, 43.8% of the methylphenidate group had improvement on the ADCS-CGIC compared with 35.2% of the placebo group. The odds ratio (OR) for improvement on ADCS-CGIC for methylphenidate vs. placebo was 1.90 (P = .07).
There was also a strong association between score improvement on the NPI apathy subscale and improvement on the ADCS-CGIC subscale (OR, 2.95; P = .002).
“It is important to note that there were no group differences in any of the cognitive measures, suggesting that the effect of the treatment is specific to the treatment of apathy and not a secondary effect of improvement in cognition,” the researchers wrote.
In all, 17 serious adverse events occurred in the methylphenidate group and 10 occurred in the placebo group. However, all events were found to be hospitalizations for events not related to treatment.
‘Enduring effect’
Commenting on the findings, Jeffrey L. Cummings, MD, ScD, professor of brain sciences at the University of Nevada, Las Vegas, noted that the reduction in NPI apathy subscale score of more than 50% was clinically meaningful.

A more robust outcome on the ADCS-CGIC would have been desirable, he added, although that instrument is not designed specifically for apathy.
Methylphenidate’s effect on apathy observed at 2 months and remaining stable throughout the study makes it appear to be “an enduring effect, and not something that the patient accommodates to,” said Dr. Cummings, who was not involved with the research. Such a change may manifest itself in a patient’s greater willingness to help voluntarily with housework or to suggest going for a walk, he noted.
“These are not dramatic changes in cognition, of course, but they are changes in initiative and that is very important,” Dr. Cummings said. Decreased apathy also may improve quality of life for the patient’s caregiver, he added.
Overall, the findings raise the question of whether the Food and Drug Administration should recognize apathy as an indication for which drugs can be approved, said Dr. Cummings.
“For me, that would be the next major step in this line of investigation,” he concluded.
The study was funded by the National Institute on Aging. Dr. Mintzer has served as an adviser to Praxis Bioresearch and Cerevel Therapeutics on matters unrelated to this study. Dr. Cummings is the author of the Neuropsychiatric Inventory but does not receive payments for it from academic trials such as ADMET 2.
A version of this article first appeared on Medscape.com.
Methylphenidate is safe and effective for treating apathy in patients with Alzheimer’s disease (AD), new research suggests.
Results from a phase 3 randomized trial showed that, after 6 months of treatment, mean score on the Neuropsychiatric Inventory (NPI) apathy subscale decreased by 4.5 points for patients who received methylphenidate vs. a decrease of 3.1 points for those who received placebo.
In addition, the safety profile showed no significant between-group differences.
“Methylphenidate offers a treatment approach providing a modest but potentially clinically significant benefit for patients and caregivers,” said the investigators, led by Jacobo E. Mintzer, MD, MBA, professor of health studies at the Medical University of South Carolina in Charleston.
The findings were published online Sept. 27 in JAMA Neurology.
Common problem
Apathy, which is common among patients with AD, is associated with increased risk for mortality, financial burden, and caregiver burden. No treatment has proved effective for apathy in this population.
Two trials of methylphenidate, a catecholaminergic agent, have provided preliminary evidence of efficacy. Findings from the Apathy in Dementia Methylphenidate trial (ADMET) suggested the drug was associated with improved cognition and few adverse events. However, both trials had small patient populations and short durations.
The current investigators conducted ADMET 2, a 6-month, phase 3 trial, to investigate methylphenidate further. They recruited 200 patients (mean age, 76 years; 66% men; 90% White) at nine clinical centers that specialized in dementia care in the United States and one in Canada.
Eligible patients had a diagnosis of possible or probable AD and a Mini-Mental State Examination (MMSE) score between 10 and 28. They also had clinically significant apathy for at least 4 weeks and an available caregiver who spent more than 10 hours a week with the patient.
The researchers randomly assigned patients to receive methylphenidate (n = 99) or placebo (n = 101). For 3 days, participants in the active group received 10 mg/day of methylphenidate. After that point, they received 20 mg/day of methylphenidate for the rest of the study.
Patients in both treatment groups were given the same number of identical-appearing capsules each day.
In-person follow-up visits took place monthly for 6 months. Participants also were contacted by telephone at days 15, 45, and 75 after treatment assignment.
Participants underwent cognitive testing at baseline and at 2, 4, and 6 months. The battery of tests included the MMSE, Hopkins Verbal Learning Test, and Wechsler Adult Intelligence Scale – Revised Digit Span.
The trial’s two primary outcomes were mean change in NPI apathy score from baseline to 6 months and the odds of an improved rating on the Alzheimer’s Disease Cooperative Study Clinical Global Impression of Change (ADCS-CGIC) between baseline and 6 months.
Significant change on either outcome was to be considered a signal of effective treatment.
Treatment-specific benefit
Ten patients in the methylphenidate group and seven in the placebo group withdrew during the study.
Mean baseline score on the NPI apathy subscale was 8.0 vs. 7.6, respectively.
In an adjusted, longitudinal model, mean between-group difference in change in NPI apathy score at 6 months was –1.25 (P = .002). The mean NPI apathy score decreased by 4.5 points in the methylphenidate group vs. 3.1 points in the placebo group.
The largest change in apathy score occurred during the first 2 months of treatment. At 6 months, 27% of the methylphenidate group vs. 14% of the placebo group had an NPI apathy score of 0.
In addition, 43.8% of the methylphenidate group had improvement on the ADCS-CGIC compared with 35.2% of the placebo group. The odds ratio (OR) for improvement on ADCS-CGIC for methylphenidate vs. placebo was 1.90 (P = .07).
There was also a strong association between score improvement on the NPI apathy subscale and improvement on the ADCS-CGIC subscale (OR, 2.95; P = .002).
“It is important to note that there were no group differences in any of the cognitive measures, suggesting that the effect of the treatment is specific to the treatment of apathy and not a secondary effect of improvement in cognition,” the researchers wrote.
In all, 17 serious adverse events occurred in the methylphenidate group and 10 occurred in the placebo group. However, all events were found to be hospitalizations for events not related to treatment.
‘Enduring effect’
Commenting on the findings, Jeffrey L. Cummings, MD, ScD, professor of brain sciences at the University of Nevada, Las Vegas, noted that the reduction in NPI apathy subscale score of more than 50% was clinically meaningful.
A more robust outcome on the ADCS-CGIC would have been desirable, he added, although that instrument is not designed specifically for apathy.
Methylphenidate’s effect on apathy observed at 2 months and remaining stable throughout the study makes it appear to be “an enduring effect, and not something that the patient accommodates to,” said Dr. Cummings, who was not involved with the research. Such a change may manifest itself in a patient’s greater willingness to help voluntarily with housework or to suggest going for a walk, he noted.
“These are not dramatic changes in cognition, of course, but they are changes in initiative and that is very important,” Dr. Cummings said. Decreased apathy also may improve quality of life for the patient’s caregiver, he added.
Overall, the findings raise the question of whether the Food and Drug Administration should recognize apathy as an indication for which drugs can be approved, said Dr. Cummings.
“For me, that would be the next major step in this line of investigation,” he concluded.
The study was funded by the National Institute on Aging. Dr. Mintzer has served as an adviser to Praxis Bioresearch and Cerevel Therapeutics on matters unrelated to this study. Dr. Cummings is the author of the Neuropsychiatric Inventory but does not receive payments for it from academic trials such as ADMET 2.
A version of this article first appeared on Medscape.com.
Methylphenidate is safe and effective for treating apathy in patients with Alzheimer’s disease (AD), new research suggests.
Results from a phase 3 randomized trial showed that, after 6 months of treatment, mean score on the Neuropsychiatric Inventory (NPI) apathy subscale decreased by 4.5 points for patients who received methylphenidate vs. a decrease of 3.1 points for those who received placebo.
In addition, the safety profile showed no significant between-group differences.
“Methylphenidate offers a treatment approach providing a modest but potentially clinically significant benefit for patients and caregivers,” said the investigators, led by Jacobo E. Mintzer, MD, MBA, professor of health studies at the Medical University of South Carolina in Charleston.
The findings were published online Sept. 27 in JAMA Neurology.
Common problem
Apathy, which is common among patients with AD, is associated with increased risk for mortality, financial burden, and caregiver burden. No treatment has proved effective for apathy in this population.
Two trials of methylphenidate, a catecholaminergic agent, have provided preliminary evidence of efficacy. Findings from the Apathy in Dementia Methylphenidate trial (ADMET) suggested the drug was associated with improved cognition and few adverse events. However, both trials had small patient populations and short durations.
The current investigators conducted ADMET 2, a 6-month, phase 3 trial, to investigate methylphenidate further. They recruited 200 patients (mean age, 76 years; 66% men; 90% White) at nine clinical centers that specialized in dementia care in the United States and one in Canada.
Eligible patients had a diagnosis of possible or probable AD and a Mini-Mental State Examination (MMSE) score between 10 and 28. They also had clinically significant apathy for at least 4 weeks and an available caregiver who spent more than 10 hours a week with the patient.
The researchers randomly assigned patients to receive methylphenidate (n = 99) or placebo (n = 101). For 3 days, participants in the active group received 10 mg/day of methylphenidate. After that point, they received 20 mg/day of methylphenidate for the rest of the study.
Patients in both treatment groups were given the same number of identical-appearing capsules each day.
In-person follow-up visits took place monthly for 6 months. Participants also were contacted by telephone at days 15, 45, and 75 after treatment assignment.
Participants underwent cognitive testing at baseline and at 2, 4, and 6 months. The battery of tests included the MMSE, Hopkins Verbal Learning Test, and Wechsler Adult Intelligence Scale – Revised Digit Span.
The trial’s two primary outcomes were mean change in NPI apathy score from baseline to 6 months and the odds of an improved rating on the Alzheimer’s Disease Cooperative Study Clinical Global Impression of Change (ADCS-CGIC) between baseline and 6 months.
Significant change on either outcome was to be considered a signal of effective treatment.
Treatment-specific benefit
Ten patients in the methylphenidate group and seven in the placebo group withdrew during the study.
Mean baseline score on the NPI apathy subscale was 8.0 vs. 7.6, respectively.
In an adjusted, longitudinal model, mean between-group difference in change in NPI apathy score at 6 months was –1.25 (P = .002). The mean NPI apathy score decreased by 4.5 points in the methylphenidate group vs. 3.1 points in the placebo group.
The largest change in apathy score occurred during the first 2 months of treatment. At 6 months, 27% of the methylphenidate group vs. 14% of the placebo group had an NPI apathy score of 0.
In addition, 43.8% of the methylphenidate group had improvement on the ADCS-CGIC compared with 35.2% of the placebo group. The odds ratio (OR) for improvement on ADCS-CGIC for methylphenidate vs. placebo was 1.90 (P = .07).
There was also a strong association between score improvement on the NPI apathy subscale and improvement on the ADCS-CGIC subscale (OR, 2.95; P = .002).
“It is important to note that there were no group differences in any of the cognitive measures, suggesting that the effect of the treatment is specific to the treatment of apathy and not a secondary effect of improvement in cognition,” the researchers wrote.
In all, 17 serious adverse events occurred in the methylphenidate group and 10 occurred in the placebo group. However, all events were found to be hospitalizations for events not related to treatment.
‘Enduring effect’
Commenting on the findings, Jeffrey L. Cummings, MD, ScD, professor of brain sciences at the University of Nevada, Las Vegas, noted that the reduction in NPI apathy subscale score of more than 50% was clinically meaningful.
A more robust outcome on the ADCS-CGIC would have been desirable, he added, although that instrument is not designed specifically for apathy.
Methylphenidate’s effect on apathy observed at 2 months and remaining stable throughout the study makes it appear to be “an enduring effect, and not something that the patient accommodates to,” said Dr. Cummings, who was not involved with the research. Such a change may manifest itself in a patient’s greater willingness to help voluntarily with housework or to suggest going for a walk, he noted.
“These are not dramatic changes in cognition, of course, but they are changes in initiative and that is very important,” Dr. Cummings said. Decreased apathy also may improve quality of life for the patient’s caregiver, he added.
Overall, the findings raise the question of whether the Food and Drug Administration should recognize apathy as an indication for which drugs can be approved, said Dr. Cummings.
“For me, that would be the next major step in this line of investigation,” he concluded.
The study was funded by the National Institute on Aging. Dr. Mintzer has served as an adviser to Praxis Bioresearch and Cerevel Therapeutics on matters unrelated to this study. Dr. Cummings is the author of the Neuropsychiatric Inventory but does not receive payments for it from academic trials such as ADMET 2.
A version of this article first appeared on Medscape.com.
Children and COVID: Decline of summer surge continues
The continuing decline in COVID-19 incidence suggests the latest surge has peaked as new cases in children dropped for the 4th consecutive week, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
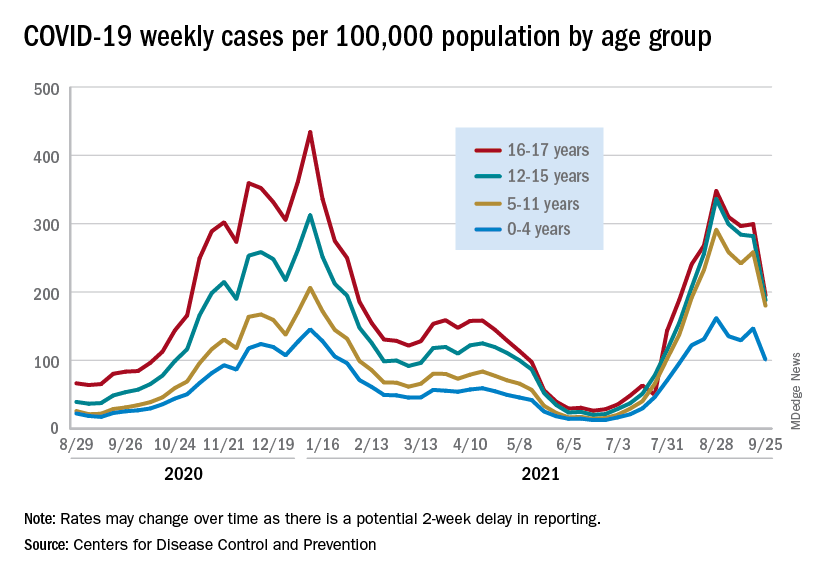
Preliminary data from the Centers for Disease Control and Prevention, however, show an uptick in new cases in late September, largely among younger children, that may indicate otherwise. Those data have a potential 2-week reporting delay, the CDC said on its COVID Data Tracker, so the most recent points on the graph (see above) could still go up.
. Those new cases made up almost 27% of all cases for the week, and the nearly 5.9 million child cases that have been reported since the start of the pandemic represent 16.2% of cases among Americans of all ages, the two groups said in their weekly COVID-19 report.
The CDC data on new cases by age group suggest that younger children have borne a heavier burden in the summer surge of COVID than they did last winter. The rate of new cases was not as high for 16- and 17-year-olds in the summer, but the other age groups all reached higher peaks than in the winter, including the 12- to 15-year-olds, who have been getting vaccinated since May, according to the COVID Data Tracker.
With vaccination approval getting closer for children under age 12 years, initiation in those already eligible continues to slide. Those aged 12-15 made up just 6.9% of new vaccinations during the 2 weeks from Sept. 21 to Oct. 4, and that figure has been dropping since July 13-26, when it was 14.1%. Vaccine initiation among 16- and 17-year-olds over that time has dropped by almost half, from 5.4% to 2.9%, the CDC data show.
All the vaccinations so far add up to this: Almost 55% of those aged 12-15 have gotten at least one dose of COVID vaccine, as have over 62% of those aged 16-17, and 52% of the older group is fully vaccinated, as is 44% of the younger group. Altogether, 10.8 million children were fully vaccinated as of Oct. 4, including those under 12 who may be participating in clinical trials or had a birth date entered incorrectly, the CDC said.
The continuing decline in COVID-19 incidence suggests the latest surge has peaked as new cases in children dropped for the 4th consecutive week, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
Preliminary data from the Centers for Disease Control and Prevention, however, show an uptick in new cases in late September, largely among younger children, that may indicate otherwise. Those data have a potential 2-week reporting delay, the CDC said on its COVID Data Tracker, so the most recent points on the graph (see above) could still go up.
. Those new cases made up almost 27% of all cases for the week, and the nearly 5.9 million child cases that have been reported since the start of the pandemic represent 16.2% of cases among Americans of all ages, the two groups said in their weekly COVID-19 report.
The CDC data on new cases by age group suggest that younger children have borne a heavier burden in the summer surge of COVID than they did last winter. The rate of new cases was not as high for 16- and 17-year-olds in the summer, but the other age groups all reached higher peaks than in the winter, including the 12- to 15-year-olds, who have been getting vaccinated since May, according to the COVID Data Tracker.
With vaccination approval getting closer for children under age 12 years, initiation in those already eligible continues to slide. Those aged 12-15 made up just 6.9% of new vaccinations during the 2 weeks from Sept. 21 to Oct. 4, and that figure has been dropping since July 13-26, when it was 14.1%. Vaccine initiation among 16- and 17-year-olds over that time has dropped by almost half, from 5.4% to 2.9%, the CDC data show.
All the vaccinations so far add up to this: Almost 55% of those aged 12-15 have gotten at least one dose of COVID vaccine, as have over 62% of those aged 16-17, and 52% of the older group is fully vaccinated, as is 44% of the younger group. Altogether, 10.8 million children were fully vaccinated as of Oct. 4, including those under 12 who may be participating in clinical trials or had a birth date entered incorrectly, the CDC said.
The continuing decline in COVID-19 incidence suggests the latest surge has peaked as new cases in children dropped for the 4th consecutive week, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
Preliminary data from the Centers for Disease Control and Prevention, however, show an uptick in new cases in late September, largely among younger children, that may indicate otherwise. Those data have a potential 2-week reporting delay, the CDC said on its COVID Data Tracker, so the most recent points on the graph (see above) could still go up.
. Those new cases made up almost 27% of all cases for the week, and the nearly 5.9 million child cases that have been reported since the start of the pandemic represent 16.2% of cases among Americans of all ages, the two groups said in their weekly COVID-19 report.
The CDC data on new cases by age group suggest that younger children have borne a heavier burden in the summer surge of COVID than they did last winter. The rate of new cases was not as high for 16- and 17-year-olds in the summer, but the other age groups all reached higher peaks than in the winter, including the 12- to 15-year-olds, who have been getting vaccinated since May, according to the COVID Data Tracker.
With vaccination approval getting closer for children under age 12 years, initiation in those already eligible continues to slide. Those aged 12-15 made up just 6.9% of new vaccinations during the 2 weeks from Sept. 21 to Oct. 4, and that figure has been dropping since July 13-26, when it was 14.1%. Vaccine initiation among 16- and 17-year-olds over that time has dropped by almost half, from 5.4% to 2.9%, the CDC data show.
All the vaccinations so far add up to this: Almost 55% of those aged 12-15 have gotten at least one dose of COVID vaccine, as have over 62% of those aged 16-17, and 52% of the older group is fully vaccinated, as is 44% of the younger group. Altogether, 10.8 million children were fully vaccinated as of Oct. 4, including those under 12 who may be participating in clinical trials or had a birth date entered incorrectly, the CDC said.
Johnson & Johnson requests FDA approval for vaccine booster doses
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.