User login
IPH4102 on fast track for Sézary syndrome
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
FDA approves ibrutinib plus obinutuzumab for CLL/SLL
The Food and Drug Administration has approved ibrutinib (Imbruvica) for use in combination with obinutuzumab to treat adults with previously untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL).
This is the tenth FDA approval for ibrutinib, a Bruton tyrosine kinase inhibitor jointly developed and commercialized by Pharmacyclics, an AbbVie company, and Janssen Biotech.
The approval is supported by the phase 3 iLLUMINATE trial (NCT02264574).
Results from this study were recently presented at the annual meeting of the American Society of Hematology (Blood. 2018;132:691) and published in the Lancet Oncology (2019 Jan;20[1]:43-56).
The iLLUMINATE trial enrolled newly diagnosed CLL patients who were randomized to receive ibrutinib plus obinutuzumab (n = 113) or chlorambucil plus obinutuzumab (n = 116).
The median follow-up was 31.3 months. The overall response rate was 88% in the ibrutinib arm and 73% in the chlorambucil arm. The complete response rate, including complete response with incomplete marrow recovery, was 19% and 8%, respectively.
The median progression-free survival was not reached in the ibrutinib arm and was 19.0 months in the chlorambucil arm (hazard ratio, 0.23; 95% confidence interval, 0.15-0.37; P less than .0001). The estimated 30-month progression-free survival was 79% and 31%, respectively.
The most common grade 3/4 adverse events in both arms were neutropenia (36% in the ibrutinib arm and 46% in the chlorambucil arm) and thrombocytopenia (19% and 10%, respectively).
There were 10 deaths caused by adverse events in the ibrutinib arm and 3 in the chlorambucil arm. One death was considered possibly related to ibrutinib (sudden death), and another was considered possibly related to chlorambucil (neuroendocrine carcinoma of the skin).
The Food and Drug Administration has approved ibrutinib (Imbruvica) for use in combination with obinutuzumab to treat adults with previously untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL).
This is the tenth FDA approval for ibrutinib, a Bruton tyrosine kinase inhibitor jointly developed and commercialized by Pharmacyclics, an AbbVie company, and Janssen Biotech.
The approval is supported by the phase 3 iLLUMINATE trial (NCT02264574).
Results from this study were recently presented at the annual meeting of the American Society of Hematology (Blood. 2018;132:691) and published in the Lancet Oncology (2019 Jan;20[1]:43-56).
The iLLUMINATE trial enrolled newly diagnosed CLL patients who were randomized to receive ibrutinib plus obinutuzumab (n = 113) or chlorambucil plus obinutuzumab (n = 116).
The median follow-up was 31.3 months. The overall response rate was 88% in the ibrutinib arm and 73% in the chlorambucil arm. The complete response rate, including complete response with incomplete marrow recovery, was 19% and 8%, respectively.
The median progression-free survival was not reached in the ibrutinib arm and was 19.0 months in the chlorambucil arm (hazard ratio, 0.23; 95% confidence interval, 0.15-0.37; P less than .0001). The estimated 30-month progression-free survival was 79% and 31%, respectively.
The most common grade 3/4 adverse events in both arms were neutropenia (36% in the ibrutinib arm and 46% in the chlorambucil arm) and thrombocytopenia (19% and 10%, respectively).
There were 10 deaths caused by adverse events in the ibrutinib arm and 3 in the chlorambucil arm. One death was considered possibly related to ibrutinib (sudden death), and another was considered possibly related to chlorambucil (neuroendocrine carcinoma of the skin).
The Food and Drug Administration has approved ibrutinib (Imbruvica) for use in combination with obinutuzumab to treat adults with previously untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL).
This is the tenth FDA approval for ibrutinib, a Bruton tyrosine kinase inhibitor jointly developed and commercialized by Pharmacyclics, an AbbVie company, and Janssen Biotech.
The approval is supported by the phase 3 iLLUMINATE trial (NCT02264574).
Results from this study were recently presented at the annual meeting of the American Society of Hematology (Blood. 2018;132:691) and published in the Lancet Oncology (2019 Jan;20[1]:43-56).
The iLLUMINATE trial enrolled newly diagnosed CLL patients who were randomized to receive ibrutinib plus obinutuzumab (n = 113) or chlorambucil plus obinutuzumab (n = 116).
The median follow-up was 31.3 months. The overall response rate was 88% in the ibrutinib arm and 73% in the chlorambucil arm. The complete response rate, including complete response with incomplete marrow recovery, was 19% and 8%, respectively.
The median progression-free survival was not reached in the ibrutinib arm and was 19.0 months in the chlorambucil arm (hazard ratio, 0.23; 95% confidence interval, 0.15-0.37; P less than .0001). The estimated 30-month progression-free survival was 79% and 31%, respectively.
The most common grade 3/4 adverse events in both arms were neutropenia (36% in the ibrutinib arm and 46% in the chlorambucil arm) and thrombocytopenia (19% and 10%, respectively).
There were 10 deaths caused by adverse events in the ibrutinib arm and 3 in the chlorambucil arm. One death was considered possibly related to ibrutinib (sudden death), and another was considered possibly related to chlorambucil (neuroendocrine carcinoma of the skin).
Flu activity increases after 2 weeks of declines
according to the Centers for Disease Control and Prevention.
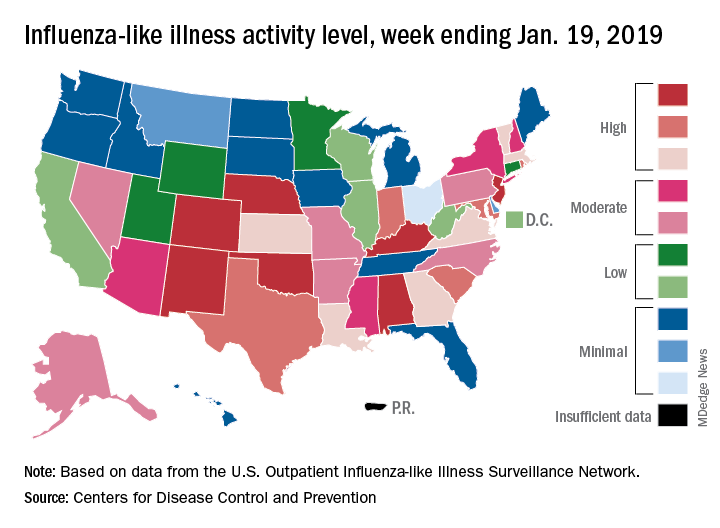
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
FDA: Nitrosamine-contaminated ARBs marketed for 4 years
Although first detected in summer 2018, angiotensin II receptor blockers (ARBs) contaminated with nitrosamines have been on the market in the United States for 4 years, according to the Food and Drug Administration.
“FDA scientists estimate that, if 8,000 people took the highest daily valsartan dose (320 mg) that contained NDMA [N-Nitrosodimethylamine] for 4 years (the time we think the affected products had been on the U.S. market), there may be one additional case of cancer beyond the average cancer rate among those 8,000 Americans,” the agency said in a Jan. 25 update from Commissioner Scott Gottlieb, MD, and Janet Woodcock, MD, director of the Center for Drug Evaluation and Research.
“The vast majority of patients exposed to NDMA through ARBs received much smaller amounts of the impurity than this worst-case scenario. Since not all ARBs are affected, it’s very likely that a patient taking an ARB for 4 years would not have always received one of the affected products. We’re still seeking to similarly quantify the risk from NDEA [N-Nitrosodiethylamine] and plan to communicate our findings as soon as possible,” they said.
“While the total exposure to these impurities for most patients was small, we are deeply concerned that patients were exposed to this impurity in the first place and that the presence of nitrosamines went undetected for a period of time,” Dr. Gottlieb and Dr. Woodcock said in the statement. Through ongoing and “exhaustive” efforts, they pledged the agency will resolve the problem and ensure it never happens again.
Meanwhile, the ongoing recalls have led to a shortage of valsartan, and other ARBs may soon follow suit. The agency hoped the one case of cancer per 8,000 patients analysis would help providers “balance the risk of patients ingesting low levels of the impurities ... for a short period of time” during shortages until a suitable replacement or alternative is found.
The problem surfaced last year when FDA was alerted to nitrosamine contamination in valsartan manufactured in China and marketed in the U.S. by generic pharmaceutical companies. Contamination has since been detected in generic irbesartan and losartan.
The impurities are generated “when specific chemicals and reaction conditions are present in the manufacturing process ... and may also result from the reuse of materials, such as solvents,” something “neither regulators nor industry fully understood” before. The agency has developed and shared new tests to detect nitrosamines. “Manufacturers using processes at risk for these impurities are expected to test for them to ensure that active ingredients and finished products are free of detectable levels,” it said.
Meanwhile, the recalls keep coming, the latest for losartan and hydrochlorothiazide combination tablets from Torrent Pharmaceuticals. The recalls are listed on FDA’s website.
Although first detected in summer 2018, angiotensin II receptor blockers (ARBs) contaminated with nitrosamines have been on the market in the United States for 4 years, according to the Food and Drug Administration.
“FDA scientists estimate that, if 8,000 people took the highest daily valsartan dose (320 mg) that contained NDMA [N-Nitrosodimethylamine] for 4 years (the time we think the affected products had been on the U.S. market), there may be one additional case of cancer beyond the average cancer rate among those 8,000 Americans,” the agency said in a Jan. 25 update from Commissioner Scott Gottlieb, MD, and Janet Woodcock, MD, director of the Center for Drug Evaluation and Research.
“The vast majority of patients exposed to NDMA through ARBs received much smaller amounts of the impurity than this worst-case scenario. Since not all ARBs are affected, it’s very likely that a patient taking an ARB for 4 years would not have always received one of the affected products. We’re still seeking to similarly quantify the risk from NDEA [N-Nitrosodiethylamine] and plan to communicate our findings as soon as possible,” they said.
“While the total exposure to these impurities for most patients was small, we are deeply concerned that patients were exposed to this impurity in the first place and that the presence of nitrosamines went undetected for a period of time,” Dr. Gottlieb and Dr. Woodcock said in the statement. Through ongoing and “exhaustive” efforts, they pledged the agency will resolve the problem and ensure it never happens again.
Meanwhile, the ongoing recalls have led to a shortage of valsartan, and other ARBs may soon follow suit. The agency hoped the one case of cancer per 8,000 patients analysis would help providers “balance the risk of patients ingesting low levels of the impurities ... for a short period of time” during shortages until a suitable replacement or alternative is found.
The problem surfaced last year when FDA was alerted to nitrosamine contamination in valsartan manufactured in China and marketed in the U.S. by generic pharmaceutical companies. Contamination has since been detected in generic irbesartan and losartan.
The impurities are generated “when specific chemicals and reaction conditions are present in the manufacturing process ... and may also result from the reuse of materials, such as solvents,” something “neither regulators nor industry fully understood” before. The agency has developed and shared new tests to detect nitrosamines. “Manufacturers using processes at risk for these impurities are expected to test for them to ensure that active ingredients and finished products are free of detectable levels,” it said.
Meanwhile, the recalls keep coming, the latest for losartan and hydrochlorothiazide combination tablets from Torrent Pharmaceuticals. The recalls are listed on FDA’s website.
Although first detected in summer 2018, angiotensin II receptor blockers (ARBs) contaminated with nitrosamines have been on the market in the United States for 4 years, according to the Food and Drug Administration.
“FDA scientists estimate that, if 8,000 people took the highest daily valsartan dose (320 mg) that contained NDMA [N-Nitrosodimethylamine] for 4 years (the time we think the affected products had been on the U.S. market), there may be one additional case of cancer beyond the average cancer rate among those 8,000 Americans,” the agency said in a Jan. 25 update from Commissioner Scott Gottlieb, MD, and Janet Woodcock, MD, director of the Center for Drug Evaluation and Research.
“The vast majority of patients exposed to NDMA through ARBs received much smaller amounts of the impurity than this worst-case scenario. Since not all ARBs are affected, it’s very likely that a patient taking an ARB for 4 years would not have always received one of the affected products. We’re still seeking to similarly quantify the risk from NDEA [N-Nitrosodiethylamine] and plan to communicate our findings as soon as possible,” they said.
“While the total exposure to these impurities for most patients was small, we are deeply concerned that patients were exposed to this impurity in the first place and that the presence of nitrosamines went undetected for a period of time,” Dr. Gottlieb and Dr. Woodcock said in the statement. Through ongoing and “exhaustive” efforts, they pledged the agency will resolve the problem and ensure it never happens again.
Meanwhile, the ongoing recalls have led to a shortage of valsartan, and other ARBs may soon follow suit. The agency hoped the one case of cancer per 8,000 patients analysis would help providers “balance the risk of patients ingesting low levels of the impurities ... for a short period of time” during shortages until a suitable replacement or alternative is found.
The problem surfaced last year when FDA was alerted to nitrosamine contamination in valsartan manufactured in China and marketed in the U.S. by generic pharmaceutical companies. Contamination has since been detected in generic irbesartan and losartan.
The impurities are generated “when specific chemicals and reaction conditions are present in the manufacturing process ... and may also result from the reuse of materials, such as solvents,” something “neither regulators nor industry fully understood” before. The agency has developed and shared new tests to detect nitrosamines. “Manufacturers using processes at risk for these impurities are expected to test for them to ensure that active ingredients and finished products are free of detectable levels,” it said.
Meanwhile, the recalls keep coming, the latest for losartan and hydrochlorothiazide combination tablets from Torrent Pharmaceuticals. The recalls are listed on FDA’s website.
SGLT inhibitor still possible for T1DM, despite FDA committee vote
on recommendation for approval from the Food and Drug Administration’s Endocrinologic and Metabolic Drugs Advisory Committee.

In the company’s three trials, involving about 3,000 insulin-dependent adults treated for up to a year, the drug lowered hemoglobin A1c a respectable 0.5% without increasing hypoglycemia risk; reduced glucose variability; and increased time in range, with some modest benefits in both weight loss and lower blood pressure. There was no sign of the increased amputation risk that has bedeviled the sodium-glucose cotransporter 2 (SGLT2) inhibitor canagliflozin (Invokana), already on the market for type 2 diabetes mellitus.
The fly in the ointment was diabetic ketoacidosis (DKA); the drug increased the risk eightfold versus placebo, and, although there were no DKA deaths and over 60% of patients resumed sotagliflozin after recovering, the cases were serious and sometimes occurred in patients with glucose levels as low as 150 mg/dL. Younger people and women seemed to be at higher risk, according to the data.
DKA risk was 4 cases per 100 patients/year, a 4% risk, and that was in the ideal setting of a trial, not everyday practice. The annual background risk of DKA is 1% or less in type 1 diabetes mellitus (T1DM).
“It’s got to be safer than this,” said committee chair Peter Wilson, MD, professor of cardiology and public health at Emory University, Atlanta.
Dr. Wilson voted to recommend approval but with the major caveat that Sanofi have a strong risk mitigation program in place, perhaps based on ketone monitoring to catch emerging DKA before people end up in the ED. That was a universal request among others who voted for recommendation; among those who voted against, the concern in large part was that, even with such a program, the risk of DKA was still too high.
“If they had already developed a mitigation program that had been piloted, and they showed us some data, there would have been more enthusiasm, but we didn’t have that,” he said in an interview after the hearing.
Sanofi did suggest possible risk mitigation strategies during the meeting. In a statement afterwards, spokesman Nicolas Kressmann said, “While we acknowledge the increase in incidence of DKA observed with the addition of sotagliflozin to insulin, we believe that the risks may be mitigated and managed with proper patient selection and education regarding appropriate ketone monitoring. We will continue to work with the FDA to ensure the agency has the data it needs to evaluate the safety and efficacy of sotagliflozin when used as an oral treatment together with insulin by adults with T1DM. We are confident in the data of our T1DM clinical program.”
Meanwhile, the company’s development for T2DMs is ongoing, with results from a number of trials expected later in 2019. Sotagliflozin would join canagliflozin and two other SGLT2 inhibitors already on the market for T2DM, none of which have been approved for T1DM disease. The approved drugs work by increasing renal glucose excretion.

A significant proportion of DKA cases in sotagliflozin’s T1DM trials were preceded by infections and other well-known triggers, “but there were a proportion of patients where they couldn’t identify the cause; it just kind of came out of the blue. Something about the medication lowers the threshold,” said panelist and endocrinologist Cecilia Low Wang, MD, director of the glucose management team at the University of Colorado Anschutz Medical Campus, Aurora, who voted against recommending approval.
“There’s definitely an increased risk” with other SGLT2 inhibitors, as well, when used off label for T1DM. “No one really knows why,” she said.
Dr. Wilson was also concerned that insulin wasn’t more tightly titrated in the placebo groups, which might have led to the 0.5% improvement seen with sotagliflozin, but “they wanted to have trials that were likely to be beneficial, so it’s reasonable to do what they did,” he said.
Overall, “we don’t really have many options for type 1, and many of us were sympathetic to the idea of increasing options.” In T1DM, “you can lose your concentration” on insulin dosing for a couple hours, “and the next thing you know you are going too high or too low and going off the road. These pills help smooth out your ups and downs. I would like to think [sotagliflozin] might be approved for a restricted group, for which we’ve really sorted out the ketone data,” he said.
Dr. Wilson and Dr. Low Wang did not have any disclosures.
on recommendation for approval from the Food and Drug Administration’s Endocrinologic and Metabolic Drugs Advisory Committee.
In the company’s three trials, involving about 3,000 insulin-dependent adults treated for up to a year, the drug lowered hemoglobin A1c a respectable 0.5% without increasing hypoglycemia risk; reduced glucose variability; and increased time in range, with some modest benefits in both weight loss and lower blood pressure. There was no sign of the increased amputation risk that has bedeviled the sodium-glucose cotransporter 2 (SGLT2) inhibitor canagliflozin (Invokana), already on the market for type 2 diabetes mellitus.
The fly in the ointment was diabetic ketoacidosis (DKA); the drug increased the risk eightfold versus placebo, and, although there were no DKA deaths and over 60% of patients resumed sotagliflozin after recovering, the cases were serious and sometimes occurred in patients with glucose levels as low as 150 mg/dL. Younger people and women seemed to be at higher risk, according to the data.
DKA risk was 4 cases per 100 patients/year, a 4% risk, and that was in the ideal setting of a trial, not everyday practice. The annual background risk of DKA is 1% or less in type 1 diabetes mellitus (T1DM).
“It’s got to be safer than this,” said committee chair Peter Wilson, MD, professor of cardiology and public health at Emory University, Atlanta.
Dr. Wilson voted to recommend approval but with the major caveat that Sanofi have a strong risk mitigation program in place, perhaps based on ketone monitoring to catch emerging DKA before people end up in the ED. That was a universal request among others who voted for recommendation; among those who voted against, the concern in large part was that, even with such a program, the risk of DKA was still too high.
“If they had already developed a mitigation program that had been piloted, and they showed us some data, there would have been more enthusiasm, but we didn’t have that,” he said in an interview after the hearing.
Sanofi did suggest possible risk mitigation strategies during the meeting. In a statement afterwards, spokesman Nicolas Kressmann said, “While we acknowledge the increase in incidence of DKA observed with the addition of sotagliflozin to insulin, we believe that the risks may be mitigated and managed with proper patient selection and education regarding appropriate ketone monitoring. We will continue to work with the FDA to ensure the agency has the data it needs to evaluate the safety and efficacy of sotagliflozin when used as an oral treatment together with insulin by adults with T1DM. We are confident in the data of our T1DM clinical program.”
Meanwhile, the company’s development for T2DMs is ongoing, with results from a number of trials expected later in 2019. Sotagliflozin would join canagliflozin and two other SGLT2 inhibitors already on the market for T2DM, none of which have been approved for T1DM disease. The approved drugs work by increasing renal glucose excretion.
A significant proportion of DKA cases in sotagliflozin’s T1DM trials were preceded by infections and other well-known triggers, “but there were a proportion of patients where they couldn’t identify the cause; it just kind of came out of the blue. Something about the medication lowers the threshold,” said panelist and endocrinologist Cecilia Low Wang, MD, director of the glucose management team at the University of Colorado Anschutz Medical Campus, Aurora, who voted against recommending approval.
“There’s definitely an increased risk” with other SGLT2 inhibitors, as well, when used off label for T1DM. “No one really knows why,” she said.
Dr. Wilson was also concerned that insulin wasn’t more tightly titrated in the placebo groups, which might have led to the 0.5% improvement seen with sotagliflozin, but “they wanted to have trials that were likely to be beneficial, so it’s reasonable to do what they did,” he said.
Overall, “we don’t really have many options for type 1, and many of us were sympathetic to the idea of increasing options.” In T1DM, “you can lose your concentration” on insulin dosing for a couple hours, “and the next thing you know you are going too high or too low and going off the road. These pills help smooth out your ups and downs. I would like to think [sotagliflozin] might be approved for a restricted group, for which we’ve really sorted out the ketone data,” he said.
Dr. Wilson and Dr. Low Wang did not have any disclosures.
on recommendation for approval from the Food and Drug Administration’s Endocrinologic and Metabolic Drugs Advisory Committee.
In the company’s three trials, involving about 3,000 insulin-dependent adults treated for up to a year, the drug lowered hemoglobin A1c a respectable 0.5% without increasing hypoglycemia risk; reduced glucose variability; and increased time in range, with some modest benefits in both weight loss and lower blood pressure. There was no sign of the increased amputation risk that has bedeviled the sodium-glucose cotransporter 2 (SGLT2) inhibitor canagliflozin (Invokana), already on the market for type 2 diabetes mellitus.
The fly in the ointment was diabetic ketoacidosis (DKA); the drug increased the risk eightfold versus placebo, and, although there were no DKA deaths and over 60% of patients resumed sotagliflozin after recovering, the cases were serious and sometimes occurred in patients with glucose levels as low as 150 mg/dL. Younger people and women seemed to be at higher risk, according to the data.
DKA risk was 4 cases per 100 patients/year, a 4% risk, and that was in the ideal setting of a trial, not everyday practice. The annual background risk of DKA is 1% or less in type 1 diabetes mellitus (T1DM).
“It’s got to be safer than this,” said committee chair Peter Wilson, MD, professor of cardiology and public health at Emory University, Atlanta.
Dr. Wilson voted to recommend approval but with the major caveat that Sanofi have a strong risk mitigation program in place, perhaps based on ketone monitoring to catch emerging DKA before people end up in the ED. That was a universal request among others who voted for recommendation; among those who voted against, the concern in large part was that, even with such a program, the risk of DKA was still too high.
“If they had already developed a mitigation program that had been piloted, and they showed us some data, there would have been more enthusiasm, but we didn’t have that,” he said in an interview after the hearing.
Sanofi did suggest possible risk mitigation strategies during the meeting. In a statement afterwards, spokesman Nicolas Kressmann said, “While we acknowledge the increase in incidence of DKA observed with the addition of sotagliflozin to insulin, we believe that the risks may be mitigated and managed with proper patient selection and education regarding appropriate ketone monitoring. We will continue to work with the FDA to ensure the agency has the data it needs to evaluate the safety and efficacy of sotagliflozin when used as an oral treatment together with insulin by adults with T1DM. We are confident in the data of our T1DM clinical program.”
Meanwhile, the company’s development for T2DMs is ongoing, with results from a number of trials expected later in 2019. Sotagliflozin would join canagliflozin and two other SGLT2 inhibitors already on the market for T2DM, none of which have been approved for T1DM disease. The approved drugs work by increasing renal glucose excretion.
A significant proportion of DKA cases in sotagliflozin’s T1DM trials were preceded by infections and other well-known triggers, “but there were a proportion of patients where they couldn’t identify the cause; it just kind of came out of the blue. Something about the medication lowers the threshold,” said panelist and endocrinologist Cecilia Low Wang, MD, director of the glucose management team at the University of Colorado Anschutz Medical Campus, Aurora, who voted against recommending approval.
“There’s definitely an increased risk” with other SGLT2 inhibitors, as well, when used off label for T1DM. “No one really knows why,” she said.
Dr. Wilson was also concerned that insulin wasn’t more tightly titrated in the placebo groups, which might have led to the 0.5% improvement seen with sotagliflozin, but “they wanted to have trials that were likely to be beneficial, so it’s reasonable to do what they did,” he said.
Overall, “we don’t really have many options for type 1, and many of us were sympathetic to the idea of increasing options.” In T1DM, “you can lose your concentration” on insulin dosing for a couple hours, “and the next thing you know you are going too high or too low and going off the road. These pills help smooth out your ups and downs. I would like to think [sotagliflozin] might be approved for a restricted group, for which we’ve really sorted out the ketone data,” he said.
Dr. Wilson and Dr. Low Wang did not have any disclosures.
EC approves blinatumomab for MRD-positive BCP-ALL
The European Commission (EC) has expanded the approved indication for blinatumomab (Blincyto).
The drug is now approved in Europe to treat adults with Philadelphia chromosome–negative (Ph–), CD19-positive B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with minimal residual disease (MRD) of at least 0.1%.
Blinatumomab is already approved in Europe to treat adults with Ph–, CD19-positive relapsed/refractory BCP-ALL and children aged 1 year or older who have relapsed/refractory Ph–, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
The drug was approved in the United States in March 2018 for the treatment of adults and children with BCP-ALL in first or second complete remission with MRD of at least 0.1%.
The EC’s decision to approve blinatumomab in MRD-positive patients was supported by the phase 2 BLAST trial (Blood. 2018;131[14]:1522-31).
The EC’s approval is also based on a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP).
That opinion, issued in November 2018, was a reversal of the opinion the committee issued in July 2018. At that time, the CHMP said the available data did not support approval for blinatumomab to treat MRD-positive BCP-ALL.
The CHMP acknowledged that blinatumomab produced MRD negativity in many patients in the BLAST trial but said there was no strong evidence that this led to improved survival. As a result, the CHMP said the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen requested a reexamination of the CHMP’s opinion. During the reexamination, the CHMP reviewed all the data and consulted a group of experts.
The experts echoed the CHMP’s prior sentiment that there was no strong evidence of improved survival in MRD-positive patients treated with blinatumomab. However, they also said the data indicate a good response to blinatumomab, with around 78% of patients becoming negative for MRD after treatment.
Noting that MRD-positive patients have a high risk of relapse and few treatment options, the CHMP concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended expanding the approved indication for blinatumomab but also requested that Amgen provide data from ongoing studies of the drug in MRD-positive patients.
The European Commission (EC) has expanded the approved indication for blinatumomab (Blincyto).
The drug is now approved in Europe to treat adults with Philadelphia chromosome–negative (Ph–), CD19-positive B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with minimal residual disease (MRD) of at least 0.1%.
Blinatumomab is already approved in Europe to treat adults with Ph–, CD19-positive relapsed/refractory BCP-ALL and children aged 1 year or older who have relapsed/refractory Ph–, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
The drug was approved in the United States in March 2018 for the treatment of adults and children with BCP-ALL in first or second complete remission with MRD of at least 0.1%.
The EC’s decision to approve blinatumomab in MRD-positive patients was supported by the phase 2 BLAST trial (Blood. 2018;131[14]:1522-31).
The EC’s approval is also based on a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP).
That opinion, issued in November 2018, was a reversal of the opinion the committee issued in July 2018. At that time, the CHMP said the available data did not support approval for blinatumomab to treat MRD-positive BCP-ALL.
The CHMP acknowledged that blinatumomab produced MRD negativity in many patients in the BLAST trial but said there was no strong evidence that this led to improved survival. As a result, the CHMP said the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen requested a reexamination of the CHMP’s opinion. During the reexamination, the CHMP reviewed all the data and consulted a group of experts.
The experts echoed the CHMP’s prior sentiment that there was no strong evidence of improved survival in MRD-positive patients treated with blinatumomab. However, they also said the data indicate a good response to blinatumomab, with around 78% of patients becoming negative for MRD after treatment.
Noting that MRD-positive patients have a high risk of relapse and few treatment options, the CHMP concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended expanding the approved indication for blinatumomab but also requested that Amgen provide data from ongoing studies of the drug in MRD-positive patients.
The European Commission (EC) has expanded the approved indication for blinatumomab (Blincyto).
The drug is now approved in Europe to treat adults with Philadelphia chromosome–negative (Ph–), CD19-positive B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in first or second complete remission with minimal residual disease (MRD) of at least 0.1%.
Blinatumomab is already approved in Europe to treat adults with Ph–, CD19-positive relapsed/refractory BCP-ALL and children aged 1 year or older who have relapsed/refractory Ph–, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
The drug was approved in the United States in March 2018 for the treatment of adults and children with BCP-ALL in first or second complete remission with MRD of at least 0.1%.
The EC’s decision to approve blinatumomab in MRD-positive patients was supported by the phase 2 BLAST trial (Blood. 2018;131[14]:1522-31).
The EC’s approval is also based on a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP).
That opinion, issued in November 2018, was a reversal of the opinion the committee issued in July 2018. At that time, the CHMP said the available data did not support approval for blinatumomab to treat MRD-positive BCP-ALL.
The CHMP acknowledged that blinatumomab produced MRD negativity in many patients in the BLAST trial but said there was no strong evidence that this led to improved survival. As a result, the CHMP said the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen requested a reexamination of the CHMP’s opinion. During the reexamination, the CHMP reviewed all the data and consulted a group of experts.
The experts echoed the CHMP’s prior sentiment that there was no strong evidence of improved survival in MRD-positive patients treated with blinatumomab. However, they also said the data indicate a good response to blinatumomab, with around 78% of patients becoming negative for MRD after treatment.
Noting that MRD-positive patients have a high risk of relapse and few treatment options, the CHMP concluded that the benefits of blinatumomab outweigh its risks in this patient population.
The CHMP recommended expanding the approved indication for blinatumomab but also requested that Amgen provide data from ongoing studies of the drug in MRD-positive patients.
Umbralisib gains FDA breakthrough designation for MZL
The who have received at least one prior anti-CD20 regimen.
Umbralisib (formerly TGR-1202) is a PI3K-delta inhibitor being developed by TG Therapeutics.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance and other actions that may expedite FDA review. For a treatment to earn breakthrough designation, early clinical results must show that it provides improvement over available therapies or fulfills an unmet need.
The breakthrough designation for umbralisib was based on interim data from the MZL cohort in the ongoing, phase 2b UNITY-NHL trial (NCT02793583).
In this trial, researchers are testing umbralisib alone or in combination with ublituximab, with or without bendamustine, in patients with previously treated non-Hodgkin lymphoma.
“The MZL single-agent umbralisib cohort of the UNITY-NHL study is fully enrolled, and we look forward to reporting topline results from this cohort by mid-year and presenting the data at a major medical meeting in 2019,” Michael S. Weiss, chief executive officer of TG Therapeutics, said in a statement.
Umbralisib monotherapy was previously evaluated in a phase 1 trial (NCT01767766) of patients with relapsed or refractory B-cell malignancies (Lancet Oncol. 2018 Apr;19[4]:486-96).
The trial enrolled 90 patients, and five of them had MZL. A total of 33 patients achieved a response to umbralisib. This includes one MZL patient who achieved a partial response.
The most common treatment-emergent adverse events (AEs) in this trial were diarrhea, nausea, and fatigue. The most common grade 3/4 AEs were neutropenia, anemia, and thrombocytopenia.
Serious AEs considered at least possibly related to umbralisib were pneumonia, lung infection, febrile neutropenia, and colitis. There were no treatment-related deaths.
The who have received at least one prior anti-CD20 regimen.
Umbralisib (formerly TGR-1202) is a PI3K-delta inhibitor being developed by TG Therapeutics.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance and other actions that may expedite FDA review. For a treatment to earn breakthrough designation, early clinical results must show that it provides improvement over available therapies or fulfills an unmet need.
The breakthrough designation for umbralisib was based on interim data from the MZL cohort in the ongoing, phase 2b UNITY-NHL trial (NCT02793583).
In this trial, researchers are testing umbralisib alone or in combination with ublituximab, with or without bendamustine, in patients with previously treated non-Hodgkin lymphoma.
“The MZL single-agent umbralisib cohort of the UNITY-NHL study is fully enrolled, and we look forward to reporting topline results from this cohort by mid-year and presenting the data at a major medical meeting in 2019,” Michael S. Weiss, chief executive officer of TG Therapeutics, said in a statement.
Umbralisib monotherapy was previously evaluated in a phase 1 trial (NCT01767766) of patients with relapsed or refractory B-cell malignancies (Lancet Oncol. 2018 Apr;19[4]:486-96).
The trial enrolled 90 patients, and five of them had MZL. A total of 33 patients achieved a response to umbralisib. This includes one MZL patient who achieved a partial response.
The most common treatment-emergent adverse events (AEs) in this trial were diarrhea, nausea, and fatigue. The most common grade 3/4 AEs were neutropenia, anemia, and thrombocytopenia.
Serious AEs considered at least possibly related to umbralisib were pneumonia, lung infection, febrile neutropenia, and colitis. There were no treatment-related deaths.
The who have received at least one prior anti-CD20 regimen.
Umbralisib (formerly TGR-1202) is a PI3K-delta inhibitor being developed by TG Therapeutics.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance and other actions that may expedite FDA review. For a treatment to earn breakthrough designation, early clinical results must show that it provides improvement over available therapies or fulfills an unmet need.
The breakthrough designation for umbralisib was based on interim data from the MZL cohort in the ongoing, phase 2b UNITY-NHL trial (NCT02793583).
In this trial, researchers are testing umbralisib alone or in combination with ublituximab, with or without bendamustine, in patients with previously treated non-Hodgkin lymphoma.
“The MZL single-agent umbralisib cohort of the UNITY-NHL study is fully enrolled, and we look forward to reporting topline results from this cohort by mid-year and presenting the data at a major medical meeting in 2019,” Michael S. Weiss, chief executive officer of TG Therapeutics, said in a statement.
Umbralisib monotherapy was previously evaluated in a phase 1 trial (NCT01767766) of patients with relapsed or refractory B-cell malignancies (Lancet Oncol. 2018 Apr;19[4]:486-96).
The trial enrolled 90 patients, and five of them had MZL. A total of 33 patients achieved a response to umbralisib. This includes one MZL patient who achieved a partial response.
The most common treatment-emergent adverse events (AEs) in this trial were diarrhea, nausea, and fatigue. The most common grade 3/4 AEs were neutropenia, anemia, and thrombocytopenia.
Serious AEs considered at least possibly related to umbralisib were pneumonia, lung infection, febrile neutropenia, and colitis. There were no treatment-related deaths.
FDA: Benefits still outweigh risks from paclitaxel-coated devices for PAD
The Food and Drug Administration has issued a letter alerting health care providers that it is aware of and examining recent data on an increase in long-term mortality rates for patients receiving paclitaxel-coated balloons and paclitaxel-eluting stents for treatment of peripheral artery disease.
“Currently, the FDA believes that the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use,” William Maisel, MD, MPH, chief medical officer of the Center for Devices and Radiological Health at the FDA, wrote in a letter to health care providers.
The FDA letter was in response to a recent systematic review of paclitaxel-coated balloons and stents recently published in the Journal of the American Heart Association. Konstantinos Katsanos, MD, PhD, from Patras University Hospital in Rion, Greece, and colleagues performed the systematic review and meta-analysis of 28 randomized controlled trials with 4,663 patients who received paclitaxel-coated devices in the femoral and/or popliteal arteries and found similar 1-year risk of all-cause patient mortality (2.3%; risk ratio, 1.08; 95% confidence interval, 0.72-1.61). However, there was an increased risk of all-cause mortality for patients with paclitaxel-coated devices at 2 years (7.2% vs. 3.8%; RR, 1.68; 95% CI, 1.15-2.47) and at 5 years (14.7% vs. 8.1%; RR, 1.93; 95% CI, 1.27-2.93), compared with control groups. The number needed to harm at 2 years was 29 patients (95% CI, 19-59) and 14 patients (95% CI, 9-32) at 5 years. Their meta regression analysis found a significant link between paclitaxel exposure and absolute risk of death.
“Actual causes for this serious late side effect remain unknown, and further investigations with longer-term follow-up are urgently warranted,” Dr. Katsanos and colleagues wrote in their review.
The FDA told health care providers of patients with paclitaxel-coated balloons and paclitaxel-eluting stents to continue surveillance of these patients per standard of care, to discuss the risks and benefits of PAD treatment options with patients, and to report any adverse or suspected adverse events to MedWatch.
The FDA said they are currently evaluating long-term data on paclitaxel-coated products to determine whether the devices carry an increased risk of death or other long-term risks, and noted there were several paclitaxel-coated balloons or paclitaxel-eluting stents that have either been approved or are under study in the United States.
SOURCE: Katsanos K et al. J Am Heart Assoc. 2018. doi: 10.1161/JAHA.118.011245.
The Food and Drug Administration has issued a letter alerting health care providers that it is aware of and examining recent data on an increase in long-term mortality rates for patients receiving paclitaxel-coated balloons and paclitaxel-eluting stents for treatment of peripheral artery disease.
“Currently, the FDA believes that the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use,” William Maisel, MD, MPH, chief medical officer of the Center for Devices and Radiological Health at the FDA, wrote in a letter to health care providers.
The FDA letter was in response to a recent systematic review of paclitaxel-coated balloons and stents recently published in the Journal of the American Heart Association. Konstantinos Katsanos, MD, PhD, from Patras University Hospital in Rion, Greece, and colleagues performed the systematic review and meta-analysis of 28 randomized controlled trials with 4,663 patients who received paclitaxel-coated devices in the femoral and/or popliteal arteries and found similar 1-year risk of all-cause patient mortality (2.3%; risk ratio, 1.08; 95% confidence interval, 0.72-1.61). However, there was an increased risk of all-cause mortality for patients with paclitaxel-coated devices at 2 years (7.2% vs. 3.8%; RR, 1.68; 95% CI, 1.15-2.47) and at 5 years (14.7% vs. 8.1%; RR, 1.93; 95% CI, 1.27-2.93), compared with control groups. The number needed to harm at 2 years was 29 patients (95% CI, 19-59) and 14 patients (95% CI, 9-32) at 5 years. Their meta regression analysis found a significant link between paclitaxel exposure and absolute risk of death.
“Actual causes for this serious late side effect remain unknown, and further investigations with longer-term follow-up are urgently warranted,” Dr. Katsanos and colleagues wrote in their review.
The FDA told health care providers of patients with paclitaxel-coated balloons and paclitaxel-eluting stents to continue surveillance of these patients per standard of care, to discuss the risks and benefits of PAD treatment options with patients, and to report any adverse or suspected adverse events to MedWatch.
The FDA said they are currently evaluating long-term data on paclitaxel-coated products to determine whether the devices carry an increased risk of death or other long-term risks, and noted there were several paclitaxel-coated balloons or paclitaxel-eluting stents that have either been approved or are under study in the United States.
SOURCE: Katsanos K et al. J Am Heart Assoc. 2018. doi: 10.1161/JAHA.118.011245.
The Food and Drug Administration has issued a letter alerting health care providers that it is aware of and examining recent data on an increase in long-term mortality rates for patients receiving paclitaxel-coated balloons and paclitaxel-eluting stents for treatment of peripheral artery disease.
“Currently, the FDA believes that the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use,” William Maisel, MD, MPH, chief medical officer of the Center for Devices and Radiological Health at the FDA, wrote in a letter to health care providers.
The FDA letter was in response to a recent systematic review of paclitaxel-coated balloons and stents recently published in the Journal of the American Heart Association. Konstantinos Katsanos, MD, PhD, from Patras University Hospital in Rion, Greece, and colleagues performed the systematic review and meta-analysis of 28 randomized controlled trials with 4,663 patients who received paclitaxel-coated devices in the femoral and/or popliteal arteries and found similar 1-year risk of all-cause patient mortality (2.3%; risk ratio, 1.08; 95% confidence interval, 0.72-1.61). However, there was an increased risk of all-cause mortality for patients with paclitaxel-coated devices at 2 years (7.2% vs. 3.8%; RR, 1.68; 95% CI, 1.15-2.47) and at 5 years (14.7% vs. 8.1%; RR, 1.93; 95% CI, 1.27-2.93), compared with control groups. The number needed to harm at 2 years was 29 patients (95% CI, 19-59) and 14 patients (95% CI, 9-32) at 5 years. Their meta regression analysis found a significant link between paclitaxel exposure and absolute risk of death.
“Actual causes for this serious late side effect remain unknown, and further investigations with longer-term follow-up are urgently warranted,” Dr. Katsanos and colleagues wrote in their review.
The FDA told health care providers of patients with paclitaxel-coated balloons and paclitaxel-eluting stents to continue surveillance of these patients per standard of care, to discuss the risks and benefits of PAD treatment options with patients, and to report any adverse or suspected adverse events to MedWatch.
The FDA said they are currently evaluating long-term data on paclitaxel-coated products to determine whether the devices carry an increased risk of death or other long-term risks, and noted there were several paclitaxel-coated balloons or paclitaxel-eluting stents that have either been approved or are under study in the United States.
SOURCE: Katsanos K et al. J Am Heart Assoc. 2018. doi: 10.1161/JAHA.118.011245.
Key clinical point: In a letter to health care providers, FDA said it was investigating data from a recent meta-analysis of increased long-term mortality risk from paclitaxel-coated balloons and paclitaxel-eluting stents for treatment of peripheral artery disease.
Major finding: All-cause mortality increased significantly after 2 years (7.2% vs. 3.8%) and 5 years (14.7% vs. 8.1%) compared with control groups.
Study details: A systematic review and meta-analysis of 28 randomized controlled trials with 4,663 patients.
Source: Katsanos K et al. J Am Heart Assoc. 2018. doi: 10.1161/JAHA.118.011245.
FDA approves third trastuzumab biosimilar
The Food and Drug Administration .
Ontruzant (trastuzumab-dttb), marketed by Samsung Bioepis, is the third approved biosimilar to Genentech’s Herceptin in the United States.
Patients should be selected for treatment with Ontruzant using an FDA-approved companion diagnostic for a trastuzumab product.
The prescribing information for the newly approved biosimilar includes a boxed warning highlighting the risk of cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity associated with the drug.
Ontruzant was shown to be equivalent to Herceptin in a phase 3 study (Eur J Cancer. 2018 Apr;93:19-27).
The trial included 875 patients with HER2-positive early breast cancer. Patients were randomized to receive Ontruzant or Herceptin for eight cycles concurrently with chemotherapy. The chemotherapy consisted of four cycles of docetaxel followed by four cycles of 5-fluorouracil/epirubicin/cyclophosphamide.
The patients then underwent surgery, which was followed by 10 cycles of Ontruzant (n=380) or Herceptin (n=384).
The median follow-up was 437 days in the Ontruzant arm and 438 days in the Herceptin arm. Safety and efficacy results were similar between the treatment arms.
Treatment-emergent adverse events occurred in 97.5% of patients in the Ontruzant arm and 96.1% of those in the Herceptin arm.
The 12-month event-free survival rate was 93.7% in the Ontruzant arm and 93.4% in the Herceptin arm.
The Food and Drug Administration .
Ontruzant (trastuzumab-dttb), marketed by Samsung Bioepis, is the third approved biosimilar to Genentech’s Herceptin in the United States.
Patients should be selected for treatment with Ontruzant using an FDA-approved companion diagnostic for a trastuzumab product.
The prescribing information for the newly approved biosimilar includes a boxed warning highlighting the risk of cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity associated with the drug.
Ontruzant was shown to be equivalent to Herceptin in a phase 3 study (Eur J Cancer. 2018 Apr;93:19-27).
The trial included 875 patients with HER2-positive early breast cancer. Patients were randomized to receive Ontruzant or Herceptin for eight cycles concurrently with chemotherapy. The chemotherapy consisted of four cycles of docetaxel followed by four cycles of 5-fluorouracil/epirubicin/cyclophosphamide.
The patients then underwent surgery, which was followed by 10 cycles of Ontruzant (n=380) or Herceptin (n=384).
The median follow-up was 437 days in the Ontruzant arm and 438 days in the Herceptin arm. Safety and efficacy results were similar between the treatment arms.
Treatment-emergent adverse events occurred in 97.5% of patients in the Ontruzant arm and 96.1% of those in the Herceptin arm.
The 12-month event-free survival rate was 93.7% in the Ontruzant arm and 93.4% in the Herceptin arm.
The Food and Drug Administration .
Ontruzant (trastuzumab-dttb), marketed by Samsung Bioepis, is the third approved biosimilar to Genentech’s Herceptin in the United States.
Patients should be selected for treatment with Ontruzant using an FDA-approved companion diagnostic for a trastuzumab product.
The prescribing information for the newly approved biosimilar includes a boxed warning highlighting the risk of cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity associated with the drug.
Ontruzant was shown to be equivalent to Herceptin in a phase 3 study (Eur J Cancer. 2018 Apr;93:19-27).
The trial included 875 patients with HER2-positive early breast cancer. Patients were randomized to receive Ontruzant or Herceptin for eight cycles concurrently with chemotherapy. The chemotherapy consisted of four cycles of docetaxel followed by four cycles of 5-fluorouracil/epirubicin/cyclophosphamide.
The patients then underwent surgery, which was followed by 10 cycles of Ontruzant (n=380) or Herceptin (n=384).
The median follow-up was 437 days in the Ontruzant arm and 438 days in the Herceptin arm. Safety and efficacy results were similar between the treatment arms.
Treatment-emergent adverse events occurred in 97.5% of patients in the Ontruzant arm and 96.1% of those in the Herceptin arm.
The 12-month event-free survival rate was 93.7% in the Ontruzant arm and 93.4% in the Herceptin arm.
Flu activity down for second consecutive week
The second week of the new year brought a second straight week of declining activity for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
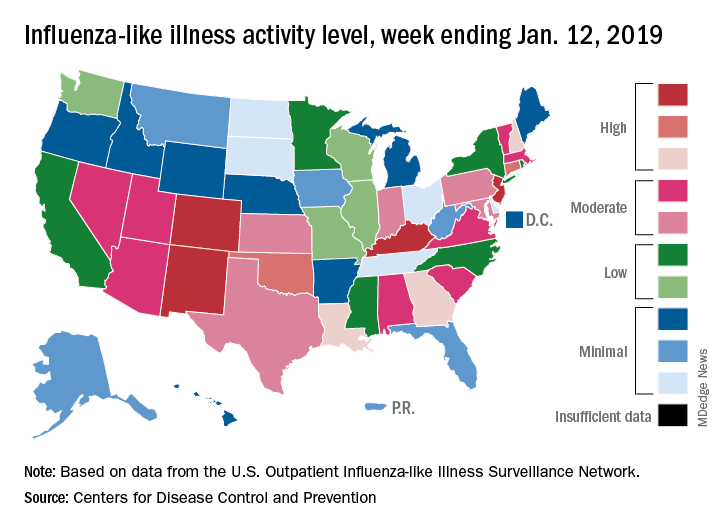
The proportion of outpatient visits for influenza-like illness (ILI) was 3.1% for the week ending Jan. 12, 2019, down from 3.5% the previous week but still above the national baseline level of 2.2%, the CDC’s influenza division reported Jan. 18.
Activity was also down at the state level. There were 4 states – Colorado, Kentucky, New Jersey, and New Mexico – at level 10 on the CDC’s 1-10 scale for ILI activity, compared with 10 the week before, and a total of 9 were in the high range from 8 to 10, compared with 15 the previous week, data from the influenza division show.
Reports of total influenza deaths, which lag a week behind other measures, continue to rise: 111 for the week ending Jan. 5, although reporting is only 72% complete. There were 89 deaths during the previous week, with reporting 82% complete so far. Total flu-related deaths among children are up to 19 for the 2018-2019 season after three more were reported during the week ending Jan. 12, the CDC said. Influenza deaths from the comparable weeks of the much more severe 2017-2018 season were 1,163 for all ages and 10 for children.
The second week of the new year brought a second straight week of declining activity for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.1% for the week ending Jan. 12, 2019, down from 3.5% the previous week but still above the national baseline level of 2.2%, the CDC’s influenza division reported Jan. 18.
Activity was also down at the state level. There were 4 states – Colorado, Kentucky, New Jersey, and New Mexico – at level 10 on the CDC’s 1-10 scale for ILI activity, compared with 10 the week before, and a total of 9 were in the high range from 8 to 10, compared with 15 the previous week, data from the influenza division show.
Reports of total influenza deaths, which lag a week behind other measures, continue to rise: 111 for the week ending Jan. 5, although reporting is only 72% complete. There were 89 deaths during the previous week, with reporting 82% complete so far. Total flu-related deaths among children are up to 19 for the 2018-2019 season after three more were reported during the week ending Jan. 12, the CDC said. Influenza deaths from the comparable weeks of the much more severe 2017-2018 season were 1,163 for all ages and 10 for children.
The second week of the new year brought a second straight week of declining activity for the 2018-2019 flu season, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.1% for the week ending Jan. 12, 2019, down from 3.5% the previous week but still above the national baseline level of 2.2%, the CDC’s influenza division reported Jan. 18.
Activity was also down at the state level. There were 4 states – Colorado, Kentucky, New Jersey, and New Mexico – at level 10 on the CDC’s 1-10 scale for ILI activity, compared with 10 the week before, and a total of 9 were in the high range from 8 to 10, compared with 15 the previous week, data from the influenza division show.
Reports of total influenza deaths, which lag a week behind other measures, continue to rise: 111 for the week ending Jan. 5, although reporting is only 72% complete. There were 89 deaths during the previous week, with reporting 82% complete so far. Total flu-related deaths among children are up to 19 for the 2018-2019 season after three more were reported during the week ending Jan. 12, the CDC said. Influenza deaths from the comparable weeks of the much more severe 2017-2018 season were 1,163 for all ages and 10 for children.