User login
MDedge latest news is breaking news from medical conferences, journals, guidelines, the FDA and CDC.
Skin Stress Biomarker May Predict Nerve Damage in Early T2D
TOPLINE:
Increased cutaneous carbonyl stress is linked to slower nerve conduction in patients with metabolically well-controlled, recent-onset type 2 diabetes (T2D) and can predict the development of neuropathic deficits over 5 years.
METHODOLOGY:
- Accumulation of advanced glycation end products (AGEs), which results from endogenous carbonyl stress, may be a potential target for preventing and treating the diabetic sensorimotor polyneuropathy (DSPN) that is a common complication of T2D.
- Researchers investigated novel cutaneous biomarkers for the development and progression of DSPN in 160 individuals with recent-onset T2D (diagnosed within 12 months or less) and 144 individuals with normal glucose tolerance, all recruited consecutively from the German Diabetes Study baseline cohort.
- Peripheral nerve function was assessed through nerve conduction studies, quantitative sensory testing, and clinical neuropathy scores.
- Skin biopsies were used to analyze intraepidermal nerve fiber density, endothelial integrity, cutaneous oxidative stress markers, and cutaneous carbonyl stress markers, including AGE autofluorescence and argpyrimidine area.
- Skin autofluorescence was measured noninvasively using an AGE reader device.
- A subgroup of 80 patients with T2D were reassessed after 5 years to evaluate the progression of neurophysiological deficits.
TAKEAWAY:
- Patients with recent-onset T2D had greater AGE autofluorescence and argpyrimidine area (P ≤ .05 for both) and lower nerve fiber density (P ≤ .05) than individuals with normal glucose tolerance.
- In patients with T2D, AGE autofluorescence was inversely associated with nerve conduction (P = .0002, P = .002, and P = .001 for peroneal motor, median motor, and sural sensory nerve conduction velocity, respectively) and positively associated with AGE reader measurements (P < .05); no such associations were observed in those with normal glucose tolerance.
- In the prospective T2D cohort, associations were noted between cutaneous markers for AGEs and endothelial cells at baseline and changes in nerve function indices over a 5-year period.
IN PRACTICE:
“Prospective analyses revealed some predictive value of cutaneous AGEs and lower endothelial integrity for declining nerve function, supporting the role of carbonyl stress in the development and progression of DSPN, representing a potential therapeutic target,” the authors wrote.
SOURCE:
The study was led by Gidon J. Bönhof, Department of Endocrinology and Diabetology, Medical Faculty, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany. It was published online in Diabetes Care.
LIMITATIONS:
The observational design of the study limited the ability to draw causal conclusions. The groups were not matched for age or body mass index. Various mechanisms related to DSPN were analyzed; however, specific pathways of AGEs were not studied in detail. The relatively low number of individuals with clinically manifested DSPN limited the exploration of different stages of the condition.
DISCLOSURES:
The study was supported by a German Center for Diabetes Research grant. The German Diabetes Study was supported by the German Diabetes Center funded by the German Federal Ministry of Health (Berlin), the Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia (Düsseldorf, Germany), and grants from the German Federal Ministry of Education and Research to the German Center for Diabetes Research e.V. No relevant conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Increased cutaneous carbonyl stress is linked to slower nerve conduction in patients with metabolically well-controlled, recent-onset type 2 diabetes (T2D) and can predict the development of neuropathic deficits over 5 years.
METHODOLOGY:
- Accumulation of advanced glycation end products (AGEs), which results from endogenous carbonyl stress, may be a potential target for preventing and treating the diabetic sensorimotor polyneuropathy (DSPN) that is a common complication of T2D.
- Researchers investigated novel cutaneous biomarkers for the development and progression of DSPN in 160 individuals with recent-onset T2D (diagnosed within 12 months or less) and 144 individuals with normal glucose tolerance, all recruited consecutively from the German Diabetes Study baseline cohort.
- Peripheral nerve function was assessed through nerve conduction studies, quantitative sensory testing, and clinical neuropathy scores.
- Skin biopsies were used to analyze intraepidermal nerve fiber density, endothelial integrity, cutaneous oxidative stress markers, and cutaneous carbonyl stress markers, including AGE autofluorescence and argpyrimidine area.
- Skin autofluorescence was measured noninvasively using an AGE reader device.
- A subgroup of 80 patients with T2D were reassessed after 5 years to evaluate the progression of neurophysiological deficits.
TAKEAWAY:
- Patients with recent-onset T2D had greater AGE autofluorescence and argpyrimidine area (P ≤ .05 for both) and lower nerve fiber density (P ≤ .05) than individuals with normal glucose tolerance.
- In patients with T2D, AGE autofluorescence was inversely associated with nerve conduction (P = .0002, P = .002, and P = .001 for peroneal motor, median motor, and sural sensory nerve conduction velocity, respectively) and positively associated with AGE reader measurements (P < .05); no such associations were observed in those with normal glucose tolerance.
- In the prospective T2D cohort, associations were noted between cutaneous markers for AGEs and endothelial cells at baseline and changes in nerve function indices over a 5-year period.
IN PRACTICE:
“Prospective analyses revealed some predictive value of cutaneous AGEs and lower endothelial integrity for declining nerve function, supporting the role of carbonyl stress in the development and progression of DSPN, representing a potential therapeutic target,” the authors wrote.
SOURCE:
The study was led by Gidon J. Bönhof, Department of Endocrinology and Diabetology, Medical Faculty, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany. It was published online in Diabetes Care.
LIMITATIONS:
The observational design of the study limited the ability to draw causal conclusions. The groups were not matched for age or body mass index. Various mechanisms related to DSPN were analyzed; however, specific pathways of AGEs were not studied in detail. The relatively low number of individuals with clinically manifested DSPN limited the exploration of different stages of the condition.
DISCLOSURES:
The study was supported by a German Center for Diabetes Research grant. The German Diabetes Study was supported by the German Diabetes Center funded by the German Federal Ministry of Health (Berlin), the Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia (Düsseldorf, Germany), and grants from the German Federal Ministry of Education and Research to the German Center for Diabetes Research e.V. No relevant conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Increased cutaneous carbonyl stress is linked to slower nerve conduction in patients with metabolically well-controlled, recent-onset type 2 diabetes (T2D) and can predict the development of neuropathic deficits over 5 years.
METHODOLOGY:
- Accumulation of advanced glycation end products (AGEs), which results from endogenous carbonyl stress, may be a potential target for preventing and treating the diabetic sensorimotor polyneuropathy (DSPN) that is a common complication of T2D.
- Researchers investigated novel cutaneous biomarkers for the development and progression of DSPN in 160 individuals with recent-onset T2D (diagnosed within 12 months or less) and 144 individuals with normal glucose tolerance, all recruited consecutively from the German Diabetes Study baseline cohort.
- Peripheral nerve function was assessed through nerve conduction studies, quantitative sensory testing, and clinical neuropathy scores.
- Skin biopsies were used to analyze intraepidermal nerve fiber density, endothelial integrity, cutaneous oxidative stress markers, and cutaneous carbonyl stress markers, including AGE autofluorescence and argpyrimidine area.
- Skin autofluorescence was measured noninvasively using an AGE reader device.
- A subgroup of 80 patients with T2D were reassessed after 5 years to evaluate the progression of neurophysiological deficits.
TAKEAWAY:
- Patients with recent-onset T2D had greater AGE autofluorescence and argpyrimidine area (P ≤ .05 for both) and lower nerve fiber density (P ≤ .05) than individuals with normal glucose tolerance.
- In patients with T2D, AGE autofluorescence was inversely associated with nerve conduction (P = .0002, P = .002, and P = .001 for peroneal motor, median motor, and sural sensory nerve conduction velocity, respectively) and positively associated with AGE reader measurements (P < .05); no such associations were observed in those with normal glucose tolerance.
- In the prospective T2D cohort, associations were noted between cutaneous markers for AGEs and endothelial cells at baseline and changes in nerve function indices over a 5-year period.
IN PRACTICE:
“Prospective analyses revealed some predictive value of cutaneous AGEs and lower endothelial integrity for declining nerve function, supporting the role of carbonyl stress in the development and progression of DSPN, representing a potential therapeutic target,” the authors wrote.
SOURCE:
The study was led by Gidon J. Bönhof, Department of Endocrinology and Diabetology, Medical Faculty, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany. It was published online in Diabetes Care.
LIMITATIONS:
The observational design of the study limited the ability to draw causal conclusions. The groups were not matched for age or body mass index. Various mechanisms related to DSPN were analyzed; however, specific pathways of AGEs were not studied in detail. The relatively low number of individuals with clinically manifested DSPN limited the exploration of different stages of the condition.
DISCLOSURES:
The study was supported by a German Center for Diabetes Research grant. The German Diabetes Study was supported by the German Diabetes Center funded by the German Federal Ministry of Health (Berlin), the Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia (Düsseldorf, Germany), and grants from the German Federal Ministry of Education and Research to the German Center for Diabetes Research e.V. No relevant conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
Untreated Infertility Linked to Higher Risk for Systemic Autoimmune Rheumatic Disease After Childbirth
TOPLINE:
The association persists even after accounting for adverse pregnancy outcomes. Women who have experienced infertility without fertility treatment show a 25% higher risk for systemic autoimmune rheumatic disease (SARD) up to 9 years after delivery, compared with those without infertility.
METHODOLOGY:
- Population-based cohort study analyzed 568,053 singleton births among 465,078 women aged 18-50 years without pre-existing SARD in Ontario, Canada, from 2012 to 2021.
- Participants were categorized into four groups: No infertility with unassisted conception (88.0%), infertility without fertility treatment (9.2%), infertility with noninvasive fertility treatment (1.4%), and infertility with invasive fertility treatment (1.4%).
- Researchers used marginal structural Cox proportional hazards models to generate hazard ratios and 95% CIs, adjusting for sociodemographic characteristics, comorbidities, smoking, and adverse pregnancy outcomes.
- Analysis included a median follow-up duration of 6.5 years (interquartile range: 4-9 years) from delivery date until SARD diagnosis, death, loss of health insurance, or study end.
TAKEAWAY:
- The incidence rate of SARD was 12.5 per 10,000 person-years in women with untreated infertility, compared with 9.3 per 10,000 person-years in women without infertility.
- Women with untreated infertility showed an elevated risk for SARD (controlled direct effect hazard ratio [HR], 1.25; 95% CI, 1.12-1.40) even after accounting for adverse pregnancy outcomes.
- Neither noninvasive fertility treatment (total effect HR, 1.06; 95% CI, 0.79-1.42) nor invasive fertility treatment (total effect HR, 0.97; 95% CI, 0.69-1.36) were associated with increased SARD risk.
- The association between untreated infertility and SARD persisted in analyses restricted to women aged < 38 years and in those without endometriosis or other autoimmune diseases.
IN PRACTICE:
“Future research efforts should seek to corroborate this association by infertility cause, with a focus on possible mechanisms related to ovulatory, ovarian, and sexual dysfunction. Greater health provider awareness of SARD symptoms and related gynecological issues that may present in women with infertility could facilitate earlier detection and treatment of SARD during the reproductive years,” wrote the authors of the study.
SOURCE:
The study was led by Natalie V. Scime of the Department of Health and Society, University of Toronto Scarborough in Ontario, Canada. It was published online in Human Reproduction.
LIMITATIONS:
Exposure and outcome misclassification was possible due to the use of published algorithms in health administrative data with unknown or imperfect sensitivity and specificity. The researchers noted that individual-level social and lifestyle factors and underlying causes of infertility were not available, and thus, were not included in the analysis.
DISCLOSURES:
This research received funding through a Banting Postdoctoral Fellowship to Scime and Canada Research Chair to Hilary K. Brown (2019-00158), with support from ICES, funded by an annual grant from the Ontario Ministry of Health and the Ministry of Long-Term Care. One coauthor disclosed consulting for Celltrion, Werfen, Organon, MitogenDx, AstraZeneca, Mallinckrodt Canada, and GlaxoSmithKline. All other authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
The association persists even after accounting for adverse pregnancy outcomes. Women who have experienced infertility without fertility treatment show a 25% higher risk for systemic autoimmune rheumatic disease (SARD) up to 9 years after delivery, compared with those without infertility.
METHODOLOGY:
- Population-based cohort study analyzed 568,053 singleton births among 465,078 women aged 18-50 years without pre-existing SARD in Ontario, Canada, from 2012 to 2021.
- Participants were categorized into four groups: No infertility with unassisted conception (88.0%), infertility without fertility treatment (9.2%), infertility with noninvasive fertility treatment (1.4%), and infertility with invasive fertility treatment (1.4%).
- Researchers used marginal structural Cox proportional hazards models to generate hazard ratios and 95% CIs, adjusting for sociodemographic characteristics, comorbidities, smoking, and adverse pregnancy outcomes.
- Analysis included a median follow-up duration of 6.5 years (interquartile range: 4-9 years) from delivery date until SARD diagnosis, death, loss of health insurance, or study end.
TAKEAWAY:
- The incidence rate of SARD was 12.5 per 10,000 person-years in women with untreated infertility, compared with 9.3 per 10,000 person-years in women without infertility.
- Women with untreated infertility showed an elevated risk for SARD (controlled direct effect hazard ratio [HR], 1.25; 95% CI, 1.12-1.40) even after accounting for adverse pregnancy outcomes.
- Neither noninvasive fertility treatment (total effect HR, 1.06; 95% CI, 0.79-1.42) nor invasive fertility treatment (total effect HR, 0.97; 95% CI, 0.69-1.36) were associated with increased SARD risk.
- The association between untreated infertility and SARD persisted in analyses restricted to women aged < 38 years and in those without endometriosis or other autoimmune diseases.
IN PRACTICE:
“Future research efforts should seek to corroborate this association by infertility cause, with a focus on possible mechanisms related to ovulatory, ovarian, and sexual dysfunction. Greater health provider awareness of SARD symptoms and related gynecological issues that may present in women with infertility could facilitate earlier detection and treatment of SARD during the reproductive years,” wrote the authors of the study.
SOURCE:
The study was led by Natalie V. Scime of the Department of Health and Society, University of Toronto Scarborough in Ontario, Canada. It was published online in Human Reproduction.
LIMITATIONS:
Exposure and outcome misclassification was possible due to the use of published algorithms in health administrative data with unknown or imperfect sensitivity and specificity. The researchers noted that individual-level social and lifestyle factors and underlying causes of infertility were not available, and thus, were not included in the analysis.
DISCLOSURES:
This research received funding through a Banting Postdoctoral Fellowship to Scime and Canada Research Chair to Hilary K. Brown (2019-00158), with support from ICES, funded by an annual grant from the Ontario Ministry of Health and the Ministry of Long-Term Care. One coauthor disclosed consulting for Celltrion, Werfen, Organon, MitogenDx, AstraZeneca, Mallinckrodt Canada, and GlaxoSmithKline. All other authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
The association persists even after accounting for adverse pregnancy outcomes. Women who have experienced infertility without fertility treatment show a 25% higher risk for systemic autoimmune rheumatic disease (SARD) up to 9 years after delivery, compared with those without infertility.
METHODOLOGY:
- Population-based cohort study analyzed 568,053 singleton births among 465,078 women aged 18-50 years without pre-existing SARD in Ontario, Canada, from 2012 to 2021.
- Participants were categorized into four groups: No infertility with unassisted conception (88.0%), infertility without fertility treatment (9.2%), infertility with noninvasive fertility treatment (1.4%), and infertility with invasive fertility treatment (1.4%).
- Researchers used marginal structural Cox proportional hazards models to generate hazard ratios and 95% CIs, adjusting for sociodemographic characteristics, comorbidities, smoking, and adverse pregnancy outcomes.
- Analysis included a median follow-up duration of 6.5 years (interquartile range: 4-9 years) from delivery date until SARD diagnosis, death, loss of health insurance, or study end.
TAKEAWAY:
- The incidence rate of SARD was 12.5 per 10,000 person-years in women with untreated infertility, compared with 9.3 per 10,000 person-years in women without infertility.
- Women with untreated infertility showed an elevated risk for SARD (controlled direct effect hazard ratio [HR], 1.25; 95% CI, 1.12-1.40) even after accounting for adverse pregnancy outcomes.
- Neither noninvasive fertility treatment (total effect HR, 1.06; 95% CI, 0.79-1.42) nor invasive fertility treatment (total effect HR, 0.97; 95% CI, 0.69-1.36) were associated with increased SARD risk.
- The association between untreated infertility and SARD persisted in analyses restricted to women aged < 38 years and in those without endometriosis or other autoimmune diseases.
IN PRACTICE:
“Future research efforts should seek to corroborate this association by infertility cause, with a focus on possible mechanisms related to ovulatory, ovarian, and sexual dysfunction. Greater health provider awareness of SARD symptoms and related gynecological issues that may present in women with infertility could facilitate earlier detection and treatment of SARD during the reproductive years,” wrote the authors of the study.
SOURCE:
The study was led by Natalie V. Scime of the Department of Health and Society, University of Toronto Scarborough in Ontario, Canada. It was published online in Human Reproduction.
LIMITATIONS:
Exposure and outcome misclassification was possible due to the use of published algorithms in health administrative data with unknown or imperfect sensitivity and specificity. The researchers noted that individual-level social and lifestyle factors and underlying causes of infertility were not available, and thus, were not included in the analysis.
DISCLOSURES:
This research received funding through a Banting Postdoctoral Fellowship to Scime and Canada Research Chair to Hilary K. Brown (2019-00158), with support from ICES, funded by an annual grant from the Ontario Ministry of Health and the Ministry of Long-Term Care. One coauthor disclosed consulting for Celltrion, Werfen, Organon, MitogenDx, AstraZeneca, Mallinckrodt Canada, and GlaxoSmithKline. All other authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
When Is the Best Time to Deliver for Pregnant Patients With Chronic Hypertension?
TOPLINE:
Among pregnant patients with chronic hypertension, delivery at 39 weeks of gestation provides an optimal balance between stillbirth risk and neonatal outcomes. Analysis of 227,977 term singleton deliveries shows consistent findings across different patient subgroups.
METHODOLOGY:
- A population-based retrospective cohort study analyzed 227,977 nonanomalous singleton term births in the United States from 2014 to 2018 among patients with chronic hypertension.
- Researchers excluded pregnancies with superimposed preeclampsia, eclampsia, pregestational diabetes, and deliveries occurring before 37 weeks or at 43 or more weeks of gestation.
- Analysis compared rates of stillbirth, infant death within 1 year of life, and neonatal morbidity at each week of term pregnancy.
- Neonatal morbidity was defined as a composite of neonatal intensive care unit admission, ventilation for 6 hours or longer, a low 5-minute Apgar score (≤ 3), and seizures.
TAKEAWAY:
- The rate of stillbirth per 10,000 ongoing pregnancies increased with gestational age and was lowest at 38 weeks (6.5; 95% CI, 5.4-7.7).
- Rates of infant death and neonatal morbidity were lowest at 40 weeks (18.0/10,000 live births; 95% CI, 13.7-23.6) and 39 weeks (637/10,000 live births; 95% CI, 619-654), respectively.
- At 39 weeks of gestation, the risk for delivery was lower (651/10,000; 95% CI, 633-670) than the composite risk for expectant management (750/10,000; 95% CI, 720-781).
- According to the authors, findings were consistent for non-Hispanic Black patients and pregnancies complicated by fetal growth restriction.
IN PRACTICE:
“To prevent one case of stillbirth, infant death, or neonatal morbidity, an estimated 101 patients with chronic hypertension would need to deliver at 39 weeks of gestation as opposed to 40 weeks. Given the approximately 45,000 patients with chronic hypertension who deliver at term each year in the United States, a policy of delivery at 39 weeks of gestation theoretically would prevent 450 adverse perinatal events per year,” wrote the authors of the study.
SOURCE:
The study was led by Ira Hamilton, James Liu, Labeena Wajahat, and Robert Rossi, University of Cincinnati College of Medicine in Cincinnati. It was published online in O&G Open.
LIMITATIONS:
According to the authors, the study could not stratify chronic hypertension based on medication use, number of medications, or degree of control. The researchers note that exact timing of delivery in weeks and days was not reported, limiting precise understanding of optimal delivery timing. Additionally, the study could not examine rates of neonatal morbidity and mortality in patients who developed superimposed preeclampsia during expectant management.
DISCLOSURES:
The authors did not report any potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Among pregnant patients with chronic hypertension, delivery at 39 weeks of gestation provides an optimal balance between stillbirth risk and neonatal outcomes. Analysis of 227,977 term singleton deliveries shows consistent findings across different patient subgroups.
METHODOLOGY:
- A population-based retrospective cohort study analyzed 227,977 nonanomalous singleton term births in the United States from 2014 to 2018 among patients with chronic hypertension.
- Researchers excluded pregnancies with superimposed preeclampsia, eclampsia, pregestational diabetes, and deliveries occurring before 37 weeks or at 43 or more weeks of gestation.
- Analysis compared rates of stillbirth, infant death within 1 year of life, and neonatal morbidity at each week of term pregnancy.
- Neonatal morbidity was defined as a composite of neonatal intensive care unit admission, ventilation for 6 hours or longer, a low 5-minute Apgar score (≤ 3), and seizures.
TAKEAWAY:
- The rate of stillbirth per 10,000 ongoing pregnancies increased with gestational age and was lowest at 38 weeks (6.5; 95% CI, 5.4-7.7).
- Rates of infant death and neonatal morbidity were lowest at 40 weeks (18.0/10,000 live births; 95% CI, 13.7-23.6) and 39 weeks (637/10,000 live births; 95% CI, 619-654), respectively.
- At 39 weeks of gestation, the risk for delivery was lower (651/10,000; 95% CI, 633-670) than the composite risk for expectant management (750/10,000; 95% CI, 720-781).
- According to the authors, findings were consistent for non-Hispanic Black patients and pregnancies complicated by fetal growth restriction.
IN PRACTICE:
“To prevent one case of stillbirth, infant death, or neonatal morbidity, an estimated 101 patients with chronic hypertension would need to deliver at 39 weeks of gestation as opposed to 40 weeks. Given the approximately 45,000 patients with chronic hypertension who deliver at term each year in the United States, a policy of delivery at 39 weeks of gestation theoretically would prevent 450 adverse perinatal events per year,” wrote the authors of the study.
SOURCE:
The study was led by Ira Hamilton, James Liu, Labeena Wajahat, and Robert Rossi, University of Cincinnati College of Medicine in Cincinnati. It was published online in O&G Open.
LIMITATIONS:
According to the authors, the study could not stratify chronic hypertension based on medication use, number of medications, or degree of control. The researchers note that exact timing of delivery in weeks and days was not reported, limiting precise understanding of optimal delivery timing. Additionally, the study could not examine rates of neonatal morbidity and mortality in patients who developed superimposed preeclampsia during expectant management.
DISCLOSURES:
The authors did not report any potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Among pregnant patients with chronic hypertension, delivery at 39 weeks of gestation provides an optimal balance between stillbirth risk and neonatal outcomes. Analysis of 227,977 term singleton deliveries shows consistent findings across different patient subgroups.
METHODOLOGY:
- A population-based retrospective cohort study analyzed 227,977 nonanomalous singleton term births in the United States from 2014 to 2018 among patients with chronic hypertension.
- Researchers excluded pregnancies with superimposed preeclampsia, eclampsia, pregestational diabetes, and deliveries occurring before 37 weeks or at 43 or more weeks of gestation.
- Analysis compared rates of stillbirth, infant death within 1 year of life, and neonatal morbidity at each week of term pregnancy.
- Neonatal morbidity was defined as a composite of neonatal intensive care unit admission, ventilation for 6 hours or longer, a low 5-minute Apgar score (≤ 3), and seizures.
TAKEAWAY:
- The rate of stillbirth per 10,000 ongoing pregnancies increased with gestational age and was lowest at 38 weeks (6.5; 95% CI, 5.4-7.7).
- Rates of infant death and neonatal morbidity were lowest at 40 weeks (18.0/10,000 live births; 95% CI, 13.7-23.6) and 39 weeks (637/10,000 live births; 95% CI, 619-654), respectively.
- At 39 weeks of gestation, the risk for delivery was lower (651/10,000; 95% CI, 633-670) than the composite risk for expectant management (750/10,000; 95% CI, 720-781).
- According to the authors, findings were consistent for non-Hispanic Black patients and pregnancies complicated by fetal growth restriction.
IN PRACTICE:
“To prevent one case of stillbirth, infant death, or neonatal morbidity, an estimated 101 patients with chronic hypertension would need to deliver at 39 weeks of gestation as opposed to 40 weeks. Given the approximately 45,000 patients with chronic hypertension who deliver at term each year in the United States, a policy of delivery at 39 weeks of gestation theoretically would prevent 450 adverse perinatal events per year,” wrote the authors of the study.
SOURCE:
The study was led by Ira Hamilton, James Liu, Labeena Wajahat, and Robert Rossi, University of Cincinnati College of Medicine in Cincinnati. It was published online in O&G Open.
LIMITATIONS:
According to the authors, the study could not stratify chronic hypertension based on medication use, number of medications, or degree of control. The researchers note that exact timing of delivery in weeks and days was not reported, limiting precise understanding of optimal delivery timing. Additionally, the study could not examine rates of neonatal morbidity and mortality in patients who developed superimposed preeclampsia during expectant management.
DISCLOSURES:
The authors did not report any potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Noise and Artificial Light
If you’ve ever taken a red-eye flight you have probably received a little packet of items the airline hopes will make your night flight more comfortable. If you had shelled out for “extra leg room” or “more comfort” seating, your little kit may have included some one-size-never-fits-all socks, a toothbrush large enough to brush one tooth at a time, and a miniature tube of toothpaste the GEICO gecko would laugh at. I have no personal knowledge what the folks in first class are getting, but I suspect it comes in a calf skin Gucci pouch. But, regardless of where you are sitting, at a minimum your night comfort kit will come with an eye mask and ear plugs. Unfortunately, these freebies are wasted on me because I already use a sleep mask every night and simply turn off my hearing aids to mute the noise. But I appreciate their effort.
Light and sound are well-known sleep disruptors. Temperature gets less attention, but is nonetheless a potent contributor to a poor night’s sleep in my experience. Just by chance while I was recovering from my most recent jet lag, I encountered two papers from investigators who were curious about the association between healthy sleep and ambient light and noise.

The first paper looked at the relationship between artificial light at night (ALAN) and the incidence of insomnia. Looking at more than 300 Chinese cities, the investigators measured ALAN using satellite images and correlated the data with insomnia-related posts on social media. The researchers found when ALAN increased insomnia, related posts also increased. Not surprisingly, this relationship was greater in less populated cities during extreme temperatures and when air quality was poor.
The second paper came from University of Texas at Houston. Using Fitbit data from more than 3000 adolescents, the researchers looked for correlations between blood pressure, sleep health, and “median nighttime anthropogenic noise levels by ZIP code.” Turns out the Federal Highway Administration has a readily available map of these noise levels.
What the investigators found was that adequate sleep significantly reduces the risk of hypertension in adolescents. Not an unexpected finding to an ex-pediatrician like myself who is obsessed with the importance of sleep deprivation. However, the investigators and I were surprised that they had found no association between neighborhood noise alone or in combination with sleep health. I still suspect there is an association lurking there in the weeds of their data, but obviously it is not robust enough to float to the surface. It may be that in an acute situation noise can contribute to hypertension, but over time individuals adjust to the new sound level and their blood pressure settles down. Sleep is such a critical factor that it is not something our cardiovascular system can adapt to so easily. For various reasons most of us may already be functioning at the margins of sleep deprivation.
How then do we respond to observations by these two research teams? Do we take an approach similar to that the airlines have taken and prescribe, hand out, or sell ear plugs and sleep masks to every patient, or at least those with hypertension? This is what we could call the put-the-onus-on-the-patient approach, which seems to be the default when we lack the political will to take a bolder step.
The other path we could call the socio-environmental approach. The airlines have made a passing attempt at this by turning the cabin lights down on red-eye flights. I recently wrote about the “exposome,” which some investigators define as the total non-genetic exposures an individual endures during a lifetime and which in many situations has a negative effect on the individual’s health. These two papers clearly demonstrate that noise and nighttime artificial light are potent features of an uncountable number of individuals’ exposomes.
Unfortunately, it is going to require something far beyond these two relatively obscure studies to move the needle in the direction of a healthier population. It’s is not a stretch to put obesity and the attention deficit phenomenon under this same umbrella where our society needs to look at itself for the answers.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
If you’ve ever taken a red-eye flight you have probably received a little packet of items the airline hopes will make your night flight more comfortable. If you had shelled out for “extra leg room” or “more comfort” seating, your little kit may have included some one-size-never-fits-all socks, a toothbrush large enough to brush one tooth at a time, and a miniature tube of toothpaste the GEICO gecko would laugh at. I have no personal knowledge what the folks in first class are getting, but I suspect it comes in a calf skin Gucci pouch. But, regardless of where you are sitting, at a minimum your night comfort kit will come with an eye mask and ear plugs. Unfortunately, these freebies are wasted on me because I already use a sleep mask every night and simply turn off my hearing aids to mute the noise. But I appreciate their effort.
Light and sound are well-known sleep disruptors. Temperature gets less attention, but is nonetheless a potent contributor to a poor night’s sleep in my experience. Just by chance while I was recovering from my most recent jet lag, I encountered two papers from investigators who were curious about the association between healthy sleep and ambient light and noise.
The first paper looked at the relationship between artificial light at night (ALAN) and the incidence of insomnia. Looking at more than 300 Chinese cities, the investigators measured ALAN using satellite images and correlated the data with insomnia-related posts on social media. The researchers found when ALAN increased insomnia, related posts also increased. Not surprisingly, this relationship was greater in less populated cities during extreme temperatures and when air quality was poor.
The second paper came from University of Texas at Houston. Using Fitbit data from more than 3000 adolescents, the researchers looked for correlations between blood pressure, sleep health, and “median nighttime anthropogenic noise levels by ZIP code.” Turns out the Federal Highway Administration has a readily available map of these noise levels.
What the investigators found was that adequate sleep significantly reduces the risk of hypertension in adolescents. Not an unexpected finding to an ex-pediatrician like myself who is obsessed with the importance of sleep deprivation. However, the investigators and I were surprised that they had found no association between neighborhood noise alone or in combination with sleep health. I still suspect there is an association lurking there in the weeds of their data, but obviously it is not robust enough to float to the surface. It may be that in an acute situation noise can contribute to hypertension, but over time individuals adjust to the new sound level and their blood pressure settles down. Sleep is such a critical factor that it is not something our cardiovascular system can adapt to so easily. For various reasons most of us may already be functioning at the margins of sleep deprivation.
How then do we respond to observations by these two research teams? Do we take an approach similar to that the airlines have taken and prescribe, hand out, or sell ear plugs and sleep masks to every patient, or at least those with hypertension? This is what we could call the put-the-onus-on-the-patient approach, which seems to be the default when we lack the political will to take a bolder step.
The other path we could call the socio-environmental approach. The airlines have made a passing attempt at this by turning the cabin lights down on red-eye flights. I recently wrote about the “exposome,” which some investigators define as the total non-genetic exposures an individual endures during a lifetime and which in many situations has a negative effect on the individual’s health. These two papers clearly demonstrate that noise and nighttime artificial light are potent features of an uncountable number of individuals’ exposomes.
Unfortunately, it is going to require something far beyond these two relatively obscure studies to move the needle in the direction of a healthier population. It’s is not a stretch to put obesity and the attention deficit phenomenon under this same umbrella where our society needs to look at itself for the answers.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
If you’ve ever taken a red-eye flight you have probably received a little packet of items the airline hopes will make your night flight more comfortable. If you had shelled out for “extra leg room” or “more comfort” seating, your little kit may have included some one-size-never-fits-all socks, a toothbrush large enough to brush one tooth at a time, and a miniature tube of toothpaste the GEICO gecko would laugh at. I have no personal knowledge what the folks in first class are getting, but I suspect it comes in a calf skin Gucci pouch. But, regardless of where you are sitting, at a minimum your night comfort kit will come with an eye mask and ear plugs. Unfortunately, these freebies are wasted on me because I already use a sleep mask every night and simply turn off my hearing aids to mute the noise. But I appreciate their effort.
Light and sound are well-known sleep disruptors. Temperature gets less attention, but is nonetheless a potent contributor to a poor night’s sleep in my experience. Just by chance while I was recovering from my most recent jet lag, I encountered two papers from investigators who were curious about the association between healthy sleep and ambient light and noise.
The first paper looked at the relationship between artificial light at night (ALAN) and the incidence of insomnia. Looking at more than 300 Chinese cities, the investigators measured ALAN using satellite images and correlated the data with insomnia-related posts on social media. The researchers found when ALAN increased insomnia, related posts also increased. Not surprisingly, this relationship was greater in less populated cities during extreme temperatures and when air quality was poor.
The second paper came from University of Texas at Houston. Using Fitbit data from more than 3000 adolescents, the researchers looked for correlations between blood pressure, sleep health, and “median nighttime anthropogenic noise levels by ZIP code.” Turns out the Federal Highway Administration has a readily available map of these noise levels.
What the investigators found was that adequate sleep significantly reduces the risk of hypertension in adolescents. Not an unexpected finding to an ex-pediatrician like myself who is obsessed with the importance of sleep deprivation. However, the investigators and I were surprised that they had found no association between neighborhood noise alone or in combination with sleep health. I still suspect there is an association lurking there in the weeds of their data, but obviously it is not robust enough to float to the surface. It may be that in an acute situation noise can contribute to hypertension, but over time individuals adjust to the new sound level and their blood pressure settles down. Sleep is such a critical factor that it is not something our cardiovascular system can adapt to so easily. For various reasons most of us may already be functioning at the margins of sleep deprivation.
How then do we respond to observations by these two research teams? Do we take an approach similar to that the airlines have taken and prescribe, hand out, or sell ear plugs and sleep masks to every patient, or at least those with hypertension? This is what we could call the put-the-onus-on-the-patient approach, which seems to be the default when we lack the political will to take a bolder step.
The other path we could call the socio-environmental approach. The airlines have made a passing attempt at this by turning the cabin lights down on red-eye flights. I recently wrote about the “exposome,” which some investigators define as the total non-genetic exposures an individual endures during a lifetime and which in many situations has a negative effect on the individual’s health. These two papers clearly demonstrate that noise and nighttime artificial light are potent features of an uncountable number of individuals’ exposomes.
Unfortunately, it is going to require something far beyond these two relatively obscure studies to move the needle in the direction of a healthier population. It’s is not a stretch to put obesity and the attention deficit phenomenon under this same umbrella where our society needs to look at itself for the answers.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
The Cause of All That Stress: Tonsillectomy?
This transcript has been edited for clarity.
You know those times in your life when you’re just feeling ... stressed? You’re on the edge; you have no chill; everything just sort of gets to you. If you can step away from the anxiety for a moment, you might ask yourself where it’s all coming from. Is it really the stuff in your inbox at work or is it money issues at home? Is it something with your relationship, or maybe it’s your sleep quality or your diet? One thing you probably won’t blame for those acute stress reactions is the tonsillectomy you had as a kid. But according to new research, maybe you should.
Tonsillectomy and adenoidectomy are among the most common surgical procedures young people in the United States undergo, with about 300,000 cases a year, according to recent numbers. That’s down a bit from numbers a decade or so ago, but suffice it to say, a good chunk of the population is walking around right now without their tonsils.
The data supporting tonsillectomy have never been great. The two big indications for the surgery are recurrent sore throat — data show that tonsillectomy reduces this by about 0.7 sore throats per year— and obstructive sleep apnea (OSA). The data for improvement of OSA are a bit better than the data for sore throats.
Also, tonsillectomy is a relatively quick, relatively well-reimbursed surgery with indications that are — let’s be honest — somewhat subjective, and so variation is high. One study found that in a single Vermont town, nearly 60% of the population had had their tonsils removed by the time they turned 18. A few towns over, the rate was 20%.
A few factors have led to the decline of tonsillectomy in recent years. Reimbursement rates have gone down a bit. Additionally, better data collection and statistical analysis have shown that the benefits of the procedure are relatively modest.
And then there is a body of medical literature that at first struck me as surprising and almost bizarre: data linking tonsillectomy to subsequent physical and psychiatric disorders.
I teach a course on interpretation of the medical literature, and one of the first things I teach my students is to check their gut when they see the conclusion of a study.
Basically, even before you read the data, have a sense in your own mind if the hypothesis seems reasonable. If a paper is going to conclude that smoking leads to increased risk for bone cancer, I’d say that seems like a reasonable thing to study. If a paper purports to show a link between eating poultry and bone cancer, I’m going to be reading it with quite a bit more skepticism.
The technical term for that process is assessing “biologic plausibility.” If we’re talking tonsils, we have to ask ourselves: Is it plausible that removing someone’s tonsils when they are young should lead to major problems in the future?
At first blush, it didn’t seem very plausible to me.
But the truth is, there are quite a few studies out there demonstrating links like this: links between tonsillectomy and irritable bowel syndrome; links between tonsillectomy and cancer; links between tonsillectomy and depression.
And this week, appearing in JAMA Network Open, is a study linking tonsillectomy with stress disorders.
Researchers leveraged Sweden’s health database, which contains longitudinal data on basically every person who has lived in Sweden since 1981. This database let them know who had a tonsillectomy or adenoidectomy, and when, and what happened to them later in life.
I think the best way to present these data is to show you what they found, and then challenge that finding, and then show you what they did in anticipation of the challenges we would have to their findings. It’s a pretty thorough study.
So, topline results here. The researchers first identified 83,957 individuals who had their tonsils removed. They matched each of them with 10 controls who did not have their tonsils removed but were the same age and sex.
Over around 30 years of follow-up, those people who had their tonsils removed were 43% more likely to develop a stress-related disorder. Among the specific disorders, the risk for PTSD was substantially higher: 55% higher in the tonsillectomy group.
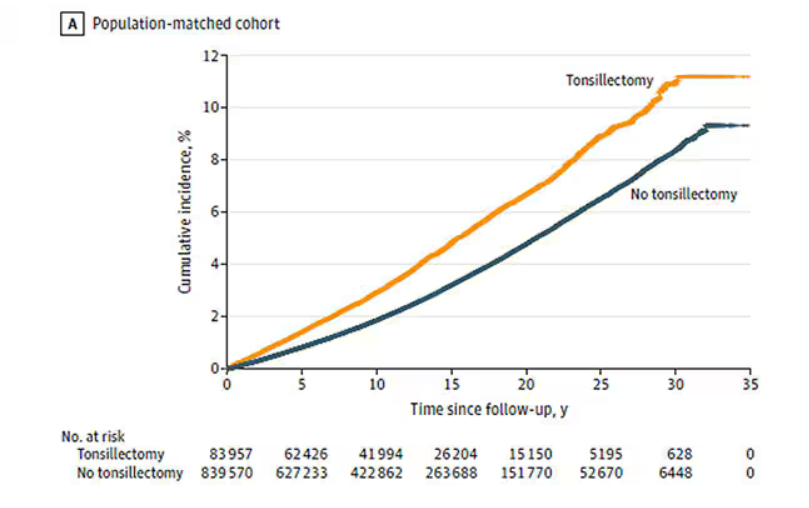
That’s pretty surprising, but I bet you already want to push back against this. Sure, the control group was the same age and sex, but other factors might be different between the two groups. You’d be right to think so. People who got their tonsils out were more likely to have parents with a history of stress-related disorders and who had lower educational attainment. But the primary results were adjusted for those factors.
There’s more to a family than parental educational attainment, of course. To account for household factors that might be harder to measure, the researchers created a second control group, this one comprising the siblings of people who had their tonsils removed but who hadn’t themselves had their tonsils removed.
The relationship between tonsillectomy and stress disorders in this population was not quite as robust but still present: a 34% increase in any stress disorder and a 41% increase in the risk for PTSD.
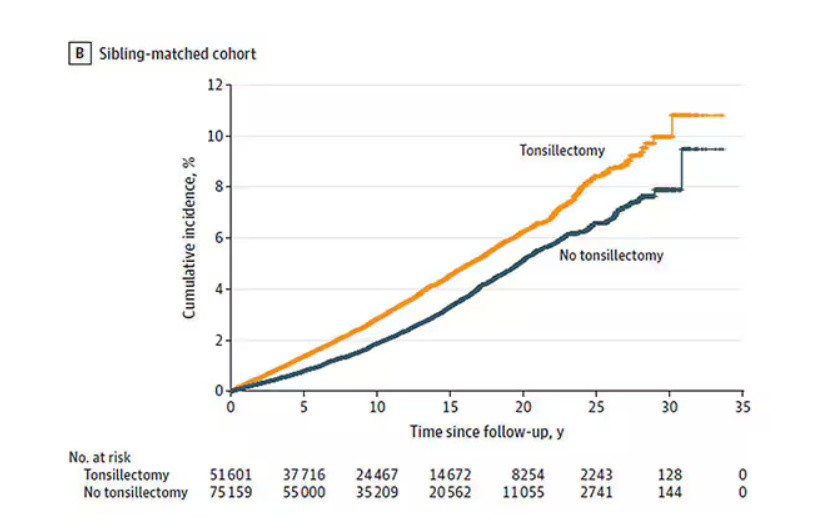
Maybe kids who get their tonsils out are just followed more closely thereafter, so doctors might notice a stress disorder and document it in the medical record; whereas with other kids it might go unnoticed. This is known as ascertainment bias. The researchers addressed this in a sensitivity analysis where they excluded new diagnoses of stress disorders that occurred in the first 3 years after tonsillectomy. The results were largely unchanged.
So how do we explain these data? We observe a correlation between tonsillectomy in youth and stress disorders in later life. But correlation is not causation. One possibility, perhaps even the most likely possibility, is that tonsillectomy is a marker of some other problem. Maybe these kids are more prone to infections and are therefore more likely to need their tonsils removed. Then, after a lifetime of more infections than average, their stress responses are higher. Or maybe kids with a higher BMI are more likely to have their tonsils removed due to sleep apnea concerns, and it’s that elevated BMI that leads to higher stress in later life.
Or maybe this is causal. Maybe there actually is biological plausibility here. The authors suggest that removal of tonsils might lead to broader changes in the immune system; after all, tonsillar tissue is on the front line of our defense against pathogens that might enter our bodies through our mouths or noses. Immunologic changes lead to greater inflammation over time, and there is decent evidence to link chronic inflammation to a variety of physical and psychological disorders.
In support of this, the authors show that the kids with tonsillectomy were more likely to be hospitalized for an infectious disease in the future as well, in magnitudes similar to the increased risk for stress. But they don’t actually show that the relationship between tonsillectomy and stress is mediated by that increased risk for infectious disease.
In the end, I find these data really intriguing. Before I dug into the literature, it seemed highly unlikely that removal of these small lumps of tissue would have much of an effect on anything. Now I’m not so sure. A few things can be removed from the human body without any consequences, but it can be hard to know exactly what those consequences are.
That said, given the rather marginal benefits of tonsillectomy and the growing number of studies expanding on the risks, I expect that we’ll see the rates of the surgery decline even further in the future.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Connecticut. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
You know those times in your life when you’re just feeling ... stressed? You’re on the edge; you have no chill; everything just sort of gets to you. If you can step away from the anxiety for a moment, you might ask yourself where it’s all coming from. Is it really the stuff in your inbox at work or is it money issues at home? Is it something with your relationship, or maybe it’s your sleep quality or your diet? One thing you probably won’t blame for those acute stress reactions is the tonsillectomy you had as a kid. But according to new research, maybe you should.
Tonsillectomy and adenoidectomy are among the most common surgical procedures young people in the United States undergo, with about 300,000 cases a year, according to recent numbers. That’s down a bit from numbers a decade or so ago, but suffice it to say, a good chunk of the population is walking around right now without their tonsils.
The data supporting tonsillectomy have never been great. The two big indications for the surgery are recurrent sore throat — data show that tonsillectomy reduces this by about 0.7 sore throats per year— and obstructive sleep apnea (OSA). The data for improvement of OSA are a bit better than the data for sore throats.
Also, tonsillectomy is a relatively quick, relatively well-reimbursed surgery with indications that are — let’s be honest — somewhat subjective, and so variation is high. One study found that in a single Vermont town, nearly 60% of the population had had their tonsils removed by the time they turned 18. A few towns over, the rate was 20%.
A few factors have led to the decline of tonsillectomy in recent years. Reimbursement rates have gone down a bit. Additionally, better data collection and statistical analysis have shown that the benefits of the procedure are relatively modest.
And then there is a body of medical literature that at first struck me as surprising and almost bizarre: data linking tonsillectomy to subsequent physical and psychiatric disorders.
I teach a course on interpretation of the medical literature, and one of the first things I teach my students is to check their gut when they see the conclusion of a study.
Basically, even before you read the data, have a sense in your own mind if the hypothesis seems reasonable. If a paper is going to conclude that smoking leads to increased risk for bone cancer, I’d say that seems like a reasonable thing to study. If a paper purports to show a link between eating poultry and bone cancer, I’m going to be reading it with quite a bit more skepticism.
The technical term for that process is assessing “biologic plausibility.” If we’re talking tonsils, we have to ask ourselves: Is it plausible that removing someone’s tonsils when they are young should lead to major problems in the future?
At first blush, it didn’t seem very plausible to me.
But the truth is, there are quite a few studies out there demonstrating links like this: links between tonsillectomy and irritable bowel syndrome; links between tonsillectomy and cancer; links between tonsillectomy and depression.
And this week, appearing in JAMA Network Open, is a study linking tonsillectomy with stress disorders.
Researchers leveraged Sweden’s health database, which contains longitudinal data on basically every person who has lived in Sweden since 1981. This database let them know who had a tonsillectomy or adenoidectomy, and when, and what happened to them later in life.
I think the best way to present these data is to show you what they found, and then challenge that finding, and then show you what they did in anticipation of the challenges we would have to their findings. It’s a pretty thorough study.
So, topline results here. The researchers first identified 83,957 individuals who had their tonsils removed. They matched each of them with 10 controls who did not have their tonsils removed but were the same age and sex.
Over around 30 years of follow-up, those people who had their tonsils removed were 43% more likely to develop a stress-related disorder. Among the specific disorders, the risk for PTSD was substantially higher: 55% higher in the tonsillectomy group.
That’s pretty surprising, but I bet you already want to push back against this. Sure, the control group was the same age and sex, but other factors might be different between the two groups. You’d be right to think so. People who got their tonsils out were more likely to have parents with a history of stress-related disorders and who had lower educational attainment. But the primary results were adjusted for those factors.
There’s more to a family than parental educational attainment, of course. To account for household factors that might be harder to measure, the researchers created a second control group, this one comprising the siblings of people who had their tonsils removed but who hadn’t themselves had their tonsils removed.
The relationship between tonsillectomy and stress disorders in this population was not quite as robust but still present: a 34% increase in any stress disorder and a 41% increase in the risk for PTSD.
Maybe kids who get their tonsils out are just followed more closely thereafter, so doctors might notice a stress disorder and document it in the medical record; whereas with other kids it might go unnoticed. This is known as ascertainment bias. The researchers addressed this in a sensitivity analysis where they excluded new diagnoses of stress disorders that occurred in the first 3 years after tonsillectomy. The results were largely unchanged.
So how do we explain these data? We observe a correlation between tonsillectomy in youth and stress disorders in later life. But correlation is not causation. One possibility, perhaps even the most likely possibility, is that tonsillectomy is a marker of some other problem. Maybe these kids are more prone to infections and are therefore more likely to need their tonsils removed. Then, after a lifetime of more infections than average, their stress responses are higher. Or maybe kids with a higher BMI are more likely to have their tonsils removed due to sleep apnea concerns, and it’s that elevated BMI that leads to higher stress in later life.
Or maybe this is causal. Maybe there actually is biological plausibility here. The authors suggest that removal of tonsils might lead to broader changes in the immune system; after all, tonsillar tissue is on the front line of our defense against pathogens that might enter our bodies through our mouths or noses. Immunologic changes lead to greater inflammation over time, and there is decent evidence to link chronic inflammation to a variety of physical and psychological disorders.
In support of this, the authors show that the kids with tonsillectomy were more likely to be hospitalized for an infectious disease in the future as well, in magnitudes similar to the increased risk for stress. But they don’t actually show that the relationship between tonsillectomy and stress is mediated by that increased risk for infectious disease.
In the end, I find these data really intriguing. Before I dug into the literature, it seemed highly unlikely that removal of these small lumps of tissue would have much of an effect on anything. Now I’m not so sure. A few things can be removed from the human body without any consequences, but it can be hard to know exactly what those consequences are.
That said, given the rather marginal benefits of tonsillectomy and the growing number of studies expanding on the risks, I expect that we’ll see the rates of the surgery decline even further in the future.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Connecticut. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
You know those times in your life when you’re just feeling ... stressed? You’re on the edge; you have no chill; everything just sort of gets to you. If you can step away from the anxiety for a moment, you might ask yourself where it’s all coming from. Is it really the stuff in your inbox at work or is it money issues at home? Is it something with your relationship, or maybe it’s your sleep quality or your diet? One thing you probably won’t blame for those acute stress reactions is the tonsillectomy you had as a kid. But according to new research, maybe you should.
Tonsillectomy and adenoidectomy are among the most common surgical procedures young people in the United States undergo, with about 300,000 cases a year, according to recent numbers. That’s down a bit from numbers a decade or so ago, but suffice it to say, a good chunk of the population is walking around right now without their tonsils.
The data supporting tonsillectomy have never been great. The two big indications for the surgery are recurrent sore throat — data show that tonsillectomy reduces this by about 0.7 sore throats per year— and obstructive sleep apnea (OSA). The data for improvement of OSA are a bit better than the data for sore throats.
Also, tonsillectomy is a relatively quick, relatively well-reimbursed surgery with indications that are — let’s be honest — somewhat subjective, and so variation is high. One study found that in a single Vermont town, nearly 60% of the population had had their tonsils removed by the time they turned 18. A few towns over, the rate was 20%.
A few factors have led to the decline of tonsillectomy in recent years. Reimbursement rates have gone down a bit. Additionally, better data collection and statistical analysis have shown that the benefits of the procedure are relatively modest.
And then there is a body of medical literature that at first struck me as surprising and almost bizarre: data linking tonsillectomy to subsequent physical and psychiatric disorders.
I teach a course on interpretation of the medical literature, and one of the first things I teach my students is to check their gut when they see the conclusion of a study.
Basically, even before you read the data, have a sense in your own mind if the hypothesis seems reasonable. If a paper is going to conclude that smoking leads to increased risk for bone cancer, I’d say that seems like a reasonable thing to study. If a paper purports to show a link between eating poultry and bone cancer, I’m going to be reading it with quite a bit more skepticism.
The technical term for that process is assessing “biologic plausibility.” If we’re talking tonsils, we have to ask ourselves: Is it plausible that removing someone’s tonsils when they are young should lead to major problems in the future?
At first blush, it didn’t seem very plausible to me.
But the truth is, there are quite a few studies out there demonstrating links like this: links between tonsillectomy and irritable bowel syndrome; links between tonsillectomy and cancer; links between tonsillectomy and depression.
And this week, appearing in JAMA Network Open, is a study linking tonsillectomy with stress disorders.
Researchers leveraged Sweden’s health database, which contains longitudinal data on basically every person who has lived in Sweden since 1981. This database let them know who had a tonsillectomy or adenoidectomy, and when, and what happened to them later in life.
I think the best way to present these data is to show you what they found, and then challenge that finding, and then show you what they did in anticipation of the challenges we would have to their findings. It’s a pretty thorough study.
So, topline results here. The researchers first identified 83,957 individuals who had their tonsils removed. They matched each of them with 10 controls who did not have their tonsils removed but were the same age and sex.
Over around 30 years of follow-up, those people who had their tonsils removed were 43% more likely to develop a stress-related disorder. Among the specific disorders, the risk for PTSD was substantially higher: 55% higher in the tonsillectomy group.
That’s pretty surprising, but I bet you already want to push back against this. Sure, the control group was the same age and sex, but other factors might be different between the two groups. You’d be right to think so. People who got their tonsils out were more likely to have parents with a history of stress-related disorders and who had lower educational attainment. But the primary results were adjusted for those factors.
There’s more to a family than parental educational attainment, of course. To account for household factors that might be harder to measure, the researchers created a second control group, this one comprising the siblings of people who had their tonsils removed but who hadn’t themselves had their tonsils removed.
The relationship between tonsillectomy and stress disorders in this population was not quite as robust but still present: a 34% increase in any stress disorder and a 41% increase in the risk for PTSD.
Maybe kids who get their tonsils out are just followed more closely thereafter, so doctors might notice a stress disorder and document it in the medical record; whereas with other kids it might go unnoticed. This is known as ascertainment bias. The researchers addressed this in a sensitivity analysis where they excluded new diagnoses of stress disorders that occurred in the first 3 years after tonsillectomy. The results were largely unchanged.
So how do we explain these data? We observe a correlation between tonsillectomy in youth and stress disorders in later life. But correlation is not causation. One possibility, perhaps even the most likely possibility, is that tonsillectomy is a marker of some other problem. Maybe these kids are more prone to infections and are therefore more likely to need their tonsils removed. Then, after a lifetime of more infections than average, their stress responses are higher. Or maybe kids with a higher BMI are more likely to have their tonsils removed due to sleep apnea concerns, and it’s that elevated BMI that leads to higher stress in later life.
Or maybe this is causal. Maybe there actually is biological plausibility here. The authors suggest that removal of tonsils might lead to broader changes in the immune system; after all, tonsillar tissue is on the front line of our defense against pathogens that might enter our bodies through our mouths or noses. Immunologic changes lead to greater inflammation over time, and there is decent evidence to link chronic inflammation to a variety of physical and psychological disorders.
In support of this, the authors show that the kids with tonsillectomy were more likely to be hospitalized for an infectious disease in the future as well, in magnitudes similar to the increased risk for stress. But they don’t actually show that the relationship between tonsillectomy and stress is mediated by that increased risk for infectious disease.
In the end, I find these data really intriguing. Before I dug into the literature, it seemed highly unlikely that removal of these small lumps of tissue would have much of an effect on anything. Now I’m not so sure. A few things can be removed from the human body without any consequences, but it can be hard to know exactly what those consequences are.
That said, given the rather marginal benefits of tonsillectomy and the growing number of studies expanding on the risks, I expect that we’ll see the rates of the surgery decline even further in the future.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Connecticut. He reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Is Vitamin E Beneficial for Bone Health?
Vitamin E may be best known for boosting skin and eye health as well as immune function. In recent years, researchers have explored the potential benefits of vitamin E on bone loss, especially in women with menopause-related osteoporosis. While data are beginning to roll in from these studies, evidence supporting a positive impact of vitamin E on osteoporosis and hip fracture risk in perimenopausal women remains elusive.
For osteoporosis, the rationale for using vitamin E is based on its antioxidant activity, which can scavenge potentially damaging free radicals. Researchers have asked whether vitamin E can help maintain the integrity of bone matrix and stimulate bone formation while minimizing bone resorption, particularly in trabecular (spongy) bone, the bone compartment preferentially affected in perimenopausal bone loss.
Vitamin E mostly consists of two isomers: alpha-tocopherol and gamma-tocopherol. Alpha-tocopherol has higher antioxidant activity and is found in nuts, seeds, vegetable oils, green leafy vegetables, fortified cereals, and vitamin E supplements. Gamma-tocopherol is known for its superior anti-inflammatory properties and accounts for about 70% of the total vitamin E intake in a typical American diet, largely sourced from soybean and other vegetable oils.
Benefits and Risks in Bone Loss Studies
Perimenopausal bone loss is caused, to a great extent, by the decrease in sex hormones. Studies of vitamin E in ovariectomized rats have yielded mixed results. This animal model lacks sex hormones and has similar bone changes to those of postmenopausal women. Some animal studies have suggested a positive effect of vitamin E on bone while others have reported no effect.
Studies in humans also have produced conflicting reports of positive, neutral, and negative associations of vitamin E with bone health. For example, the Women’s Health Initiative examined the relationship between vitamin and mineral antioxidants and bone health in postmenopausal women and found no significant association between antioxidants and bone mineral density.
Another study examining data from children and adolescents enrolled in the National Health and Nutrition Examination Survey (NHANES) database found an inverse association between alpha-tocopherol and lumbar spine bone density, suggesting a deleterious effect on bone. Inverse associations also have been reported in certain studies of postmenopausal women.
High doses of alpha-tocopherol have been linked to a risk for impaired bone health through a variety of mechanisms, such as interference with vitamin K metabolism; competitive binding for alpha-tocopherol transfer protein, inhibiting the entry of beneficial vitamin E isomers, including gamma-tocopherol; and pro-oxidant effects that harm bone. Thus, postmenopausal women taking vitamin E supplements primarily as high doses of alpha-tocopherol might be hindering their bone health.
Data for gamma-tocopherol are more promising. Some studies hypothesize that gamma-tocopherol might uncouple bone turnover, leading to increased bone formation without affecting bone resorption. Further, a randomized controlled study of mixed tocopherols (rather than alpha-tocopherol) vs placebo reported a protective effect of this preparation on bone outcomes by suppressing bone resorption. This raises the importance of considering the specific forms of vitamin E when evaluating its role in bone health.
Limitations of Current Studies
Researchers acknowledge several limitations in studies to date. For example, there are very few randomized controlled trials assessing the impact of vitamin E on bone health. Most studies are cross-sectional or observational, even when longitudinal. Cross-sectional and observational designs prevent us from establishing a causal relationship between vitamin E and bone endpoints.
Such designs also run the risk of additional confounders that may affect associations between vitamin E and bone, or the lack thereof. These could include both known and unknown confounders. Of note, gamma-tocopherol intake data were not available for certain NHANES studies.
Further, people often consume multiple nutrients and supplements, complicating the identification of specific nutrient-disease associations. Most human studies estimate tocopherol intake by dietary questionnaires or measure serum tocopherol levels, which reflect short-term dietary intake, while bone mineral density is probably influenced by long-term dietary patterns.
Too Soon to Prescribe Vitamin E for Bone Health
Some nutrition experts advocate for vitamin E supplements containing mixed tocopherols, specifically suggesting a ratio of 50-100 IU of gamma-tocopherol per 400 IU of D-alpha-tocopherol. Additional research is essential to confirm and further clarify the role of gamma-tocopherol in bone formation and resorption. In fact, it is also important to explore the influence of other compounds in the vitamin E family on skeletal health.
Until more data are available, we would recommend following the Institute of Medicine’s guidelines for the recommended daily allowance (RDA) of vitamin E. This is age dependent, ranging from 4 to 11 mg/d between the ages of 0 and 13 years, and 15 mg/d thereafter.
Overall, evidence of vitamin E’s impact on osteoporosis and hip fracture risk in perimenopausal women remains inconclusive. Although some observational and interventional studies suggest potential benefits, more interventional studies, particularly randomized controlled trials, are necessary to explore the risks and benefits of vitamin E supplementation and serum vitamin E levels on bone density and fracture risk more thoroughly.
Dr. Pani, Assistant Professor, Department of Internal Medicine, UVA School of Medicine; Medical Director, Department of General Medicine, Same Day Care Clinic, both in Charlottesville, has disclosed no relevant financial relationships. Dr. Misra, Professor, Chair, Physician-in-Chief, Department of Pediatrics, University of Virginia, and UVA Health Children’s, Charlottesville, has disclosed being a key opinion leader for Lumos Pharma.
A version of this article appeared on Medscape.com.
Vitamin E may be best known for boosting skin and eye health as well as immune function. In recent years, researchers have explored the potential benefits of vitamin E on bone loss, especially in women with menopause-related osteoporosis. While data are beginning to roll in from these studies, evidence supporting a positive impact of vitamin E on osteoporosis and hip fracture risk in perimenopausal women remains elusive.
For osteoporosis, the rationale for using vitamin E is based on its antioxidant activity, which can scavenge potentially damaging free radicals. Researchers have asked whether vitamin E can help maintain the integrity of bone matrix and stimulate bone formation while minimizing bone resorption, particularly in trabecular (spongy) bone, the bone compartment preferentially affected in perimenopausal bone loss.
Vitamin E mostly consists of two isomers: alpha-tocopherol and gamma-tocopherol. Alpha-tocopherol has higher antioxidant activity and is found in nuts, seeds, vegetable oils, green leafy vegetables, fortified cereals, and vitamin E supplements. Gamma-tocopherol is known for its superior anti-inflammatory properties and accounts for about 70% of the total vitamin E intake in a typical American diet, largely sourced from soybean and other vegetable oils.
Benefits and Risks in Bone Loss Studies
Perimenopausal bone loss is caused, to a great extent, by the decrease in sex hormones. Studies of vitamin E in ovariectomized rats have yielded mixed results. This animal model lacks sex hormones and has similar bone changes to those of postmenopausal women. Some animal studies have suggested a positive effect of vitamin E on bone while others have reported no effect.
Studies in humans also have produced conflicting reports of positive, neutral, and negative associations of vitamin E with bone health. For example, the Women’s Health Initiative examined the relationship between vitamin and mineral antioxidants and bone health in postmenopausal women and found no significant association between antioxidants and bone mineral density.
Another study examining data from children and adolescents enrolled in the National Health and Nutrition Examination Survey (NHANES) database found an inverse association between alpha-tocopherol and lumbar spine bone density, suggesting a deleterious effect on bone. Inverse associations also have been reported in certain studies of postmenopausal women.
High doses of alpha-tocopherol have been linked to a risk for impaired bone health through a variety of mechanisms, such as interference with vitamin K metabolism; competitive binding for alpha-tocopherol transfer protein, inhibiting the entry of beneficial vitamin E isomers, including gamma-tocopherol; and pro-oxidant effects that harm bone. Thus, postmenopausal women taking vitamin E supplements primarily as high doses of alpha-tocopherol might be hindering their bone health.
Data for gamma-tocopherol are more promising. Some studies hypothesize that gamma-tocopherol might uncouple bone turnover, leading to increased bone formation without affecting bone resorption. Further, a randomized controlled study of mixed tocopherols (rather than alpha-tocopherol) vs placebo reported a protective effect of this preparation on bone outcomes by suppressing bone resorption. This raises the importance of considering the specific forms of vitamin E when evaluating its role in bone health.
Limitations of Current Studies
Researchers acknowledge several limitations in studies to date. For example, there are very few randomized controlled trials assessing the impact of vitamin E on bone health. Most studies are cross-sectional or observational, even when longitudinal. Cross-sectional and observational designs prevent us from establishing a causal relationship between vitamin E and bone endpoints.
Such designs also run the risk of additional confounders that may affect associations between vitamin E and bone, or the lack thereof. These could include both known and unknown confounders. Of note, gamma-tocopherol intake data were not available for certain NHANES studies.
Further, people often consume multiple nutrients and supplements, complicating the identification of specific nutrient-disease associations. Most human studies estimate tocopherol intake by dietary questionnaires or measure serum tocopherol levels, which reflect short-term dietary intake, while bone mineral density is probably influenced by long-term dietary patterns.
Too Soon to Prescribe Vitamin E for Bone Health
Some nutrition experts advocate for vitamin E supplements containing mixed tocopherols, specifically suggesting a ratio of 50-100 IU of gamma-tocopherol per 400 IU of D-alpha-tocopherol. Additional research is essential to confirm and further clarify the role of gamma-tocopherol in bone formation and resorption. In fact, it is also important to explore the influence of other compounds in the vitamin E family on skeletal health.
Until more data are available, we would recommend following the Institute of Medicine’s guidelines for the recommended daily allowance (RDA) of vitamin E. This is age dependent, ranging from 4 to 11 mg/d between the ages of 0 and 13 years, and 15 mg/d thereafter.
Overall, evidence of vitamin E’s impact on osteoporosis and hip fracture risk in perimenopausal women remains inconclusive. Although some observational and interventional studies suggest potential benefits, more interventional studies, particularly randomized controlled trials, are necessary to explore the risks and benefits of vitamin E supplementation and serum vitamin E levels on bone density and fracture risk more thoroughly.
Dr. Pani, Assistant Professor, Department of Internal Medicine, UVA School of Medicine; Medical Director, Department of General Medicine, Same Day Care Clinic, both in Charlottesville, has disclosed no relevant financial relationships. Dr. Misra, Professor, Chair, Physician-in-Chief, Department of Pediatrics, University of Virginia, and UVA Health Children’s, Charlottesville, has disclosed being a key opinion leader for Lumos Pharma.
A version of this article appeared on Medscape.com.
Vitamin E may be best known for boosting skin and eye health as well as immune function. In recent years, researchers have explored the potential benefits of vitamin E on bone loss, especially in women with menopause-related osteoporosis. While data are beginning to roll in from these studies, evidence supporting a positive impact of vitamin E on osteoporosis and hip fracture risk in perimenopausal women remains elusive.
For osteoporosis, the rationale for using vitamin E is based on its antioxidant activity, which can scavenge potentially damaging free radicals. Researchers have asked whether vitamin E can help maintain the integrity of bone matrix and stimulate bone formation while minimizing bone resorption, particularly in trabecular (spongy) bone, the bone compartment preferentially affected in perimenopausal bone loss.
Vitamin E mostly consists of two isomers: alpha-tocopherol and gamma-tocopherol. Alpha-tocopherol has higher antioxidant activity and is found in nuts, seeds, vegetable oils, green leafy vegetables, fortified cereals, and vitamin E supplements. Gamma-tocopherol is known for its superior anti-inflammatory properties and accounts for about 70% of the total vitamin E intake in a typical American diet, largely sourced from soybean and other vegetable oils.
Benefits and Risks in Bone Loss Studies
Perimenopausal bone loss is caused, to a great extent, by the decrease in sex hormones. Studies of vitamin E in ovariectomized rats have yielded mixed results. This animal model lacks sex hormones and has similar bone changes to those of postmenopausal women. Some animal studies have suggested a positive effect of vitamin E on bone while others have reported no effect.
Studies in humans also have produced conflicting reports of positive, neutral, and negative associations of vitamin E with bone health. For example, the Women’s Health Initiative examined the relationship between vitamin and mineral antioxidants and bone health in postmenopausal women and found no significant association between antioxidants and bone mineral density.
Another study examining data from children and adolescents enrolled in the National Health and Nutrition Examination Survey (NHANES) database found an inverse association between alpha-tocopherol and lumbar spine bone density, suggesting a deleterious effect on bone. Inverse associations also have been reported in certain studies of postmenopausal women.
High doses of alpha-tocopherol have been linked to a risk for impaired bone health through a variety of mechanisms, such as interference with vitamin K metabolism; competitive binding for alpha-tocopherol transfer protein, inhibiting the entry of beneficial vitamin E isomers, including gamma-tocopherol; and pro-oxidant effects that harm bone. Thus, postmenopausal women taking vitamin E supplements primarily as high doses of alpha-tocopherol might be hindering their bone health.
Data for gamma-tocopherol are more promising. Some studies hypothesize that gamma-tocopherol might uncouple bone turnover, leading to increased bone formation without affecting bone resorption. Further, a randomized controlled study of mixed tocopherols (rather than alpha-tocopherol) vs placebo reported a protective effect of this preparation on bone outcomes by suppressing bone resorption. This raises the importance of considering the specific forms of vitamin E when evaluating its role in bone health.
Limitations of Current Studies
Researchers acknowledge several limitations in studies to date. For example, there are very few randomized controlled trials assessing the impact of vitamin E on bone health. Most studies are cross-sectional or observational, even when longitudinal. Cross-sectional and observational designs prevent us from establishing a causal relationship between vitamin E and bone endpoints.
Such designs also run the risk of additional confounders that may affect associations between vitamin E and bone, or the lack thereof. These could include both known and unknown confounders. Of note, gamma-tocopherol intake data were not available for certain NHANES studies.
Further, people often consume multiple nutrients and supplements, complicating the identification of specific nutrient-disease associations. Most human studies estimate tocopherol intake by dietary questionnaires or measure serum tocopherol levels, which reflect short-term dietary intake, while bone mineral density is probably influenced by long-term dietary patterns.
Too Soon to Prescribe Vitamin E for Bone Health
Some nutrition experts advocate for vitamin E supplements containing mixed tocopherols, specifically suggesting a ratio of 50-100 IU of gamma-tocopherol per 400 IU of D-alpha-tocopherol. Additional research is essential to confirm and further clarify the role of gamma-tocopherol in bone formation and resorption. In fact, it is also important to explore the influence of other compounds in the vitamin E family on skeletal health.
Until more data are available, we would recommend following the Institute of Medicine’s guidelines for the recommended daily allowance (RDA) of vitamin E. This is age dependent, ranging from 4 to 11 mg/d between the ages of 0 and 13 years, and 15 mg/d thereafter.
Overall, evidence of vitamin E’s impact on osteoporosis and hip fracture risk in perimenopausal women remains inconclusive. Although some observational and interventional studies suggest potential benefits, more interventional studies, particularly randomized controlled trials, are necessary to explore the risks and benefits of vitamin E supplementation and serum vitamin E levels on bone density and fracture risk more thoroughly.
Dr. Pani, Assistant Professor, Department of Internal Medicine, UVA School of Medicine; Medical Director, Department of General Medicine, Same Day Care Clinic, both in Charlottesville, has disclosed no relevant financial relationships. Dr. Misra, Professor, Chair, Physician-in-Chief, Department of Pediatrics, University of Virginia, and UVA Health Children’s, Charlottesville, has disclosed being a key opinion leader for Lumos Pharma.
A version of this article appeared on Medscape.com.
Acne Outcome Measures: Do they Incorporate LGBTQ+ Inclusive Language?
TOPLINE:
with heteronormative terms used in three of six measures addressing intimate relationships.
METHODOLOGY:
- Researchers conducted an inductive thematic analysis of 22 PROMs for acne, identified through a PubMed search.
- LGBTQ+-inclusive language was defined per the National Institutes of Health style guide.
- The analysis included 16 PROMs: Nine were acne-specific with 56 relevant items, 4 were dermatology-specific with 28 items, and 4 were health-related with 43 items.
TAKEAWAY:
- LGBTQ+-noninclusive language was identified in four of nine acne-specific PROMs — the Acne Disability Index (ADI), Acne Quality of Life Scale (AQOL), Acne-Quality of Life (Acne-QoL), and Cardiff Acne Disability Index (CADI) — but not in health-related or dermatology-specific PROMs.
- Among PROMs addressing intimate relationships, three of six acne-specific measures (CADI, ADI, and Acne-QoL) used heteronormative language, while three acne-specific PROMs, three dermatology-specific PROMs, and one health-related PROM used nonheteronormative terminology (such as “partner”).
- All PROMs contained items with nongendered pronouns (such as “I” or “you” instead of “he” or “she”). However, the AQOL included gendered language (“brothers” and “sisters,” rather than “siblings”).
- Two acne-specific PROMs demonstrated partial LGBTQ+ inclusivity, incorporating some but not all LGBTQ+ identities.
IN PRACTICE:
“Using LGBTQ+-inclusive language may promote the acquisition of accurate and relevant data for patient care and clinical trials and even enhance patient-clinician relationships,” the authors of the study wrote. “While demographics such as sex, age, race, and ethnicity are commonly considered during patient-reported outcome development and validation,” wrote the authors of an accompanying editorial, the study highlights that “sexual orientation and gender identity should also be considered to ensure these measures have similar performance across diverse populations.”
SOURCE:
The study was led by Twan Sia, BA, Department of Dermatology, Stanford University School of Medicine in California. The authors of the editorial were John S. Barbieri, MD, MBA, Department of Dermatology, Brigham and Women’s Hospital, Boston, Massachusetts, and Mya L. Roberson, MSPH, PhD, University of North Carolina at Chapel Hill.
LIMITATIONS:
The study was limited to the analysis of only English-language PROMs.
DISCLOSURES:
Two study authors disclosed receiving grants or personal fees from various sources, including pharmaceutical companies outside the submitted work. Barbieri disclosed receiving consulting fees from Dexcel Pharma and Honeydew Care; Roberson disclosed receiving consulting fees from the National Committee for Quality Assurance.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
with heteronormative terms used in three of six measures addressing intimate relationships.
METHODOLOGY:
- Researchers conducted an inductive thematic analysis of 22 PROMs for acne, identified through a PubMed search.
- LGBTQ+-inclusive language was defined per the National Institutes of Health style guide.
- The analysis included 16 PROMs: Nine were acne-specific with 56 relevant items, 4 were dermatology-specific with 28 items, and 4 were health-related with 43 items.
TAKEAWAY:
- LGBTQ+-noninclusive language was identified in four of nine acne-specific PROMs — the Acne Disability Index (ADI), Acne Quality of Life Scale (AQOL), Acne-Quality of Life (Acne-QoL), and Cardiff Acne Disability Index (CADI) — but not in health-related or dermatology-specific PROMs.
- Among PROMs addressing intimate relationships, three of six acne-specific measures (CADI, ADI, and Acne-QoL) used heteronormative language, while three acne-specific PROMs, three dermatology-specific PROMs, and one health-related PROM used nonheteronormative terminology (such as “partner”).
- All PROMs contained items with nongendered pronouns (such as “I” or “you” instead of “he” or “she”). However, the AQOL included gendered language (“brothers” and “sisters,” rather than “siblings”).
- Two acne-specific PROMs demonstrated partial LGBTQ+ inclusivity, incorporating some but not all LGBTQ+ identities.
IN PRACTICE:
“Using LGBTQ+-inclusive language may promote the acquisition of accurate and relevant data for patient care and clinical trials and even enhance patient-clinician relationships,” the authors of the study wrote. “While demographics such as sex, age, race, and ethnicity are commonly considered during patient-reported outcome development and validation,” wrote the authors of an accompanying editorial, the study highlights that “sexual orientation and gender identity should also be considered to ensure these measures have similar performance across diverse populations.”
SOURCE:
The study was led by Twan Sia, BA, Department of Dermatology, Stanford University School of Medicine in California. The authors of the editorial were John S. Barbieri, MD, MBA, Department of Dermatology, Brigham and Women’s Hospital, Boston, Massachusetts, and Mya L. Roberson, MSPH, PhD, University of North Carolina at Chapel Hill.
LIMITATIONS:
The study was limited to the analysis of only English-language PROMs.
DISCLOSURES:
Two study authors disclosed receiving grants or personal fees from various sources, including pharmaceutical companies outside the submitted work. Barbieri disclosed receiving consulting fees from Dexcel Pharma and Honeydew Care; Roberson disclosed receiving consulting fees from the National Committee for Quality Assurance.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
with heteronormative terms used in three of six measures addressing intimate relationships.
METHODOLOGY:
- Researchers conducted an inductive thematic analysis of 22 PROMs for acne, identified through a PubMed search.
- LGBTQ+-inclusive language was defined per the National Institutes of Health style guide.
- The analysis included 16 PROMs: Nine were acne-specific with 56 relevant items, 4 were dermatology-specific with 28 items, and 4 were health-related with 43 items.
TAKEAWAY:
- LGBTQ+-noninclusive language was identified in four of nine acne-specific PROMs — the Acne Disability Index (ADI), Acne Quality of Life Scale (AQOL), Acne-Quality of Life (Acne-QoL), and Cardiff Acne Disability Index (CADI) — but not in health-related or dermatology-specific PROMs.
- Among PROMs addressing intimate relationships, three of six acne-specific measures (CADI, ADI, and Acne-QoL) used heteronormative language, while three acne-specific PROMs, three dermatology-specific PROMs, and one health-related PROM used nonheteronormative terminology (such as “partner”).
- All PROMs contained items with nongendered pronouns (such as “I” or “you” instead of “he” or “she”). However, the AQOL included gendered language (“brothers” and “sisters,” rather than “siblings”).
- Two acne-specific PROMs demonstrated partial LGBTQ+ inclusivity, incorporating some but not all LGBTQ+ identities.
IN PRACTICE:
“Using LGBTQ+-inclusive language may promote the acquisition of accurate and relevant data for patient care and clinical trials and even enhance patient-clinician relationships,” the authors of the study wrote. “While demographics such as sex, age, race, and ethnicity are commonly considered during patient-reported outcome development and validation,” wrote the authors of an accompanying editorial, the study highlights that “sexual orientation and gender identity should also be considered to ensure these measures have similar performance across diverse populations.”
SOURCE:
The study was led by Twan Sia, BA, Department of Dermatology, Stanford University School of Medicine in California. The authors of the editorial were John S. Barbieri, MD, MBA, Department of Dermatology, Brigham and Women’s Hospital, Boston, Massachusetts, and Mya L. Roberson, MSPH, PhD, University of North Carolina at Chapel Hill.
LIMITATIONS:
The study was limited to the analysis of only English-language PROMs.
DISCLOSURES:
Two study authors disclosed receiving grants or personal fees from various sources, including pharmaceutical companies outside the submitted work. Barbieri disclosed receiving consulting fees from Dexcel Pharma and Honeydew Care; Roberson disclosed receiving consulting fees from the National Committee for Quality Assurance.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Cutaneous Lupus Associated with Greater Risk for Atherosclerotic Cardiovascular Disease
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Rilzabrutinib Shines in Phase 3 Trial of Tough-to-Treat Immune Thrombocytopenia
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FROM ASH 2024
More Biologics May Be Breaking Through for COPD
New biologic drugs for chronic obstructive pulmonary disease (COPD) are finally here, said Stephen Rennard, MD, in a presentation in a session on new drugs at the 2024 GOLD International COPD Conference.
The therapeutic goals of biologics remain the same as with other treatments for COPD, namely restoration of normal inflammatory response and alteration of disease progression, as well as restoration of lost structure and function and improvement of systemic effects, Rennard said in his presentation. Most studies of new and up-and-coming drugs have improvement in acute exacerbation of COPD as the primary outcome.
The Biology Behind the Biologics
T2 inflammation is “an inflammatory cascade led by IL [interleukin]-4, IL-13, and IL-5,” Mona Bafadhel, MD, chair of Respiratory Medicine at King’s College London in England, said in her presentation during the session.
Bafadhel, who served as one of the investigators on the BOREAS and NOTUS studies, explained some of the science behind the development of the new biologics.
Eosinophils are powerful regulators of immune response and inflammation by stimulating T-cell production and affecting other immune cell types, she noted.
In the context of COPD and drug development, high blood eosinophil counts have been associated with increased COPD-related exacerbations, Bafadhel said. She cited data from a Dutch study of more than 7000 patients with COPD (with and without clinical diagnoses), in which absolute eosinophil counts ≥ 3.3% were associated with increased risk for severe exacerbations of 32% and 84% across all patients with COPD and clinical COPD, respectively.
Understanding the mechanisms of the eosinophil in COPD is important for research and development, Bafadhel said. Along with standardizing measurement of T2 inflammatory markers (IL-4, IL-13, and IL-5), more research is needed to fully understand the role of eosinophils in immunoregulation and repair.
Fitting the Biologic to the Patient
Several recent studies of up-and-coming biologics have focused on subsets of COPD patients, said Dave Singh, MD, professor of clinical pharmacology and respiratory medicine at The University of Manchester in England, in his presentation at the meeting. In September 2024, the Food and Drug Administration approved dupilumab as the first biologic treatment for patients with uncontrolled COPD and type 2 inflammation on the basis of eosinophil counts. Singh cited data from the BOREAS and NOTUS studies in which dupilumab significantly reduced exacerbations and improved lung function in these patients, compared with a placebo.
Mepolizumab, a biologic approved for asthma, is not currently approved for COPD, but data from a 2017 study showed a trend toward reduced exacerbations, compared with placebo, in a subset of patients with high blood eosinophil counts, Singh said.
In addition, a recent unpublished phase 3 study (MATINEE) showed a reduction in the annualized rate of exacerbations, compared with placebo, on the basis of up to 2 years’ follow-up.
Singh also highlighted data from a phase 2a study of astegolimab, a biologic drug that focuses on the IL-33 receptor, in which COPD exacerbation rates were not significantly different between treatment and placebo groups. However, astegolimab has shown safety and efficacy in adults with severe asthma and is under development in phase 3 trials for COPD.
Tezepelumab, which was approved by the FDA in 2021 as an add-on therapy for severe asthma in patients aged 12 years or older, is also in development as a therapy for COPD exacerbations, Singh said.
In a study presented at the 2024 American Thoracic Society annual meeting, Singh and colleagues found that tezepelumab at a subcutaneous dose of 420 mg every 4 weeks reduced the annualized rate of moderate or severe COPD exacerbations compared with placebo based on data from approximately 300 patients, although the difference was not statistically significant.
Itepekimab, another biologic, showed promise in a phase 2a genetic association study involving current and former smokers with moderate to severe COPD, Singh said.
In that study, published in 2022 in The Lancet Respiratory Medicine, itepekimab failed to meet the primary endpoint in the overall study population of reduced annualized rate of moderate to severe exacerbations; however, a subgroup analysis of former smokers showed a significant (42%) reduction in exacerbations, Singh said in his presentation. Two phase 3 clinical studies (AERIFY-1/2) are ongoing to confirm the safety and efficacy of itepekimab in former smokers with COPD.
Takeaways and Next Steps
“These therapies provide the first new classes of medications approved for COPD in nearly 20 years,” said David M. Mannino, MD, of the University of Kentucky, Lexington, in an interview. “Dupilumab will be available to a subset of patients who are poorly controlled and have evidence of high eosinophils in their blood and is only used once every 2 weeks,” added Mannino, who has served as a consultant to companies developing COPD drugs.
Both dupilumab and ensifentrine, a phosphodiesterase (PDE) 3 and PDE4 inhibitor also recently approved for maintenance treatment of COPD, have been shown in clinical trials to reduce exacerbations and improve symptoms, said Mannino. Both offer additional options for patients who continue to have symptoms and exacerbations in spite of their current therapy.
Some barriers to the use of biologics in practice include the high cost. “Access and overcoming insurance-related issues such as preauthorization and high copays will be a challenge,” he said. Also, because dupilumab is an injectable drug, some patient training will be required.
Newer biologic therapies in development are also injectables, but some studies are examining longer time intervals as long as every 6 months, which could be a major advancement for some patients. The newer therapies in development are similar to dupilumab in that they will be injected therapies. Some in development are looking at longer time intervals as long as every 6 months, which may be a major advancement for some patients. “All of these therapies, however, are currently targeting more advanced or serious disease,” he said.
Looking ahead, more therapies are needed for the treatment of early COPD, as well as therapies that can be administered to a large number of patients at a reasonable cost, Mannino added.
Rennard disclosed serving as a consultant for Verona Pharma, Sanofi, Beyond Air, RS BioTherapeutics, RespirAI, and Roche, as well as speaker fees from Sanofi and temporary ownership interest while employed by AstraZeneca. Rennard is also the founder of Great Plains Biometrix. Bafadhel disclosed funding from the National Institute for Health Research (NIHR), grants from Asthma + Lung UK, Horizon Europe, NIHR, and AstraZeneca to her institution, and honoraria from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, and Pfizer. Singh disclosed relationships including speaking sponsorships, honoraria, and advisory board memberships for Adovate, Aerogen, Almirall, Apogee, Arrowhead, AstraZeneca, Bial, Boehringer Ingelheim, Chiesi, Cipla, Connect Biopharm, Covis, CSL Behring, DevPro Biopharm, Elpen, Empirico, EpiEndo, Genentech, Generate Biomedicines, GlaxoSmithKline, Glenmark, Kamada, Kinaset Therapeutics, Kymera, Menarini, MicroA, OM Pharma, Orion, Pieris Pharmaceuticals, Pulmatrix, Revolo, Roivant Sciences, Sanofi, Synairgen, Tetherex, Teva, Theravance Biopharma, Upstream, and Verona Pharma. Mannino disclosed serving as a consultant to multiple companies currently developing COPD therapies (AstraZeneca, GlaxoSmithKline, Roche, Regeneron, Sanofi, Genentech, Amgen, and Chiesi).
A version of this article appeared on Medscape.com.
New biologic drugs for chronic obstructive pulmonary disease (COPD) are finally here, said Stephen Rennard, MD, in a presentation in a session on new drugs at the 2024 GOLD International COPD Conference.
The therapeutic goals of biologics remain the same as with other treatments for COPD, namely restoration of normal inflammatory response and alteration of disease progression, as well as restoration of lost structure and function and improvement of systemic effects, Rennard said in his presentation. Most studies of new and up-and-coming drugs have improvement in acute exacerbation of COPD as the primary outcome.
The Biology Behind the Biologics
T2 inflammation is “an inflammatory cascade led by IL [interleukin]-4, IL-13, and IL-5,” Mona Bafadhel, MD, chair of Respiratory Medicine at King’s College London in England, said in her presentation during the session.
Bafadhel, who served as one of the investigators on the BOREAS and NOTUS studies, explained some of the science behind the development of the new biologics.
Eosinophils are powerful regulators of immune response and inflammation by stimulating T-cell production and affecting other immune cell types, she noted.
In the context of COPD and drug development, high blood eosinophil counts have been associated with increased COPD-related exacerbations, Bafadhel said. She cited data from a Dutch study of more than 7000 patients with COPD (with and without clinical diagnoses), in which absolute eosinophil counts ≥ 3.3% were associated with increased risk for severe exacerbations of 32% and 84% across all patients with COPD and clinical COPD, respectively.
Understanding the mechanisms of the eosinophil in COPD is important for research and development, Bafadhel said. Along with standardizing measurement of T2 inflammatory markers (IL-4, IL-13, and IL-5), more research is needed to fully understand the role of eosinophils in immunoregulation and repair.
Fitting the Biologic to the Patient
Several recent studies of up-and-coming biologics have focused on subsets of COPD patients, said Dave Singh, MD, professor of clinical pharmacology and respiratory medicine at The University of Manchester in England, in his presentation at the meeting. In September 2024, the Food and Drug Administration approved dupilumab as the first biologic treatment for patients with uncontrolled COPD and type 2 inflammation on the basis of eosinophil counts. Singh cited data from the BOREAS and NOTUS studies in which dupilumab significantly reduced exacerbations and improved lung function in these patients, compared with a placebo.
Mepolizumab, a biologic approved for asthma, is not currently approved for COPD, but data from a 2017 study showed a trend toward reduced exacerbations, compared with placebo, in a subset of patients with high blood eosinophil counts, Singh said.
In addition, a recent unpublished phase 3 study (MATINEE) showed a reduction in the annualized rate of exacerbations, compared with placebo, on the basis of up to 2 years’ follow-up.
Singh also highlighted data from a phase 2a study of astegolimab, a biologic drug that focuses on the IL-33 receptor, in which COPD exacerbation rates were not significantly different between treatment and placebo groups. However, astegolimab has shown safety and efficacy in adults with severe asthma and is under development in phase 3 trials for COPD.
Tezepelumab, which was approved by the FDA in 2021 as an add-on therapy for severe asthma in patients aged 12 years or older, is also in development as a therapy for COPD exacerbations, Singh said.
In a study presented at the 2024 American Thoracic Society annual meeting, Singh and colleagues found that tezepelumab at a subcutaneous dose of 420 mg every 4 weeks reduced the annualized rate of moderate or severe COPD exacerbations compared with placebo based on data from approximately 300 patients, although the difference was not statistically significant.
Itepekimab, another biologic, showed promise in a phase 2a genetic association study involving current and former smokers with moderate to severe COPD, Singh said.
In that study, published in 2022 in The Lancet Respiratory Medicine, itepekimab failed to meet the primary endpoint in the overall study population of reduced annualized rate of moderate to severe exacerbations; however, a subgroup analysis of former smokers showed a significant (42%) reduction in exacerbations, Singh said in his presentation. Two phase 3 clinical studies (AERIFY-1/2) are ongoing to confirm the safety and efficacy of itepekimab in former smokers with COPD.
Takeaways and Next Steps
“These therapies provide the first new classes of medications approved for COPD in nearly 20 years,” said David M. Mannino, MD, of the University of Kentucky, Lexington, in an interview. “Dupilumab will be available to a subset of patients who are poorly controlled and have evidence of high eosinophils in their blood and is only used once every 2 weeks,” added Mannino, who has served as a consultant to companies developing COPD drugs.
Both dupilumab and ensifentrine, a phosphodiesterase (PDE) 3 and PDE4 inhibitor also recently approved for maintenance treatment of COPD, have been shown in clinical trials to reduce exacerbations and improve symptoms, said Mannino. Both offer additional options for patients who continue to have symptoms and exacerbations in spite of their current therapy.
Some barriers to the use of biologics in practice include the high cost. “Access and overcoming insurance-related issues such as preauthorization and high copays will be a challenge,” he said. Also, because dupilumab is an injectable drug, some patient training will be required.
Newer biologic therapies in development are also injectables, but some studies are examining longer time intervals as long as every 6 months, which could be a major advancement for some patients. The newer therapies in development are similar to dupilumab in that they will be injected therapies. Some in development are looking at longer time intervals as long as every 6 months, which may be a major advancement for some patients. “All of these therapies, however, are currently targeting more advanced or serious disease,” he said.
Looking ahead, more therapies are needed for the treatment of early COPD, as well as therapies that can be administered to a large number of patients at a reasonable cost, Mannino added.
Rennard disclosed serving as a consultant for Verona Pharma, Sanofi, Beyond Air, RS BioTherapeutics, RespirAI, and Roche, as well as speaker fees from Sanofi and temporary ownership interest while employed by AstraZeneca. Rennard is also the founder of Great Plains Biometrix. Bafadhel disclosed funding from the National Institute for Health Research (NIHR), grants from Asthma + Lung UK, Horizon Europe, NIHR, and AstraZeneca to her institution, and honoraria from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, and Pfizer. Singh disclosed relationships including speaking sponsorships, honoraria, and advisory board memberships for Adovate, Aerogen, Almirall, Apogee, Arrowhead, AstraZeneca, Bial, Boehringer Ingelheim, Chiesi, Cipla, Connect Biopharm, Covis, CSL Behring, DevPro Biopharm, Elpen, Empirico, EpiEndo, Genentech, Generate Biomedicines, GlaxoSmithKline, Glenmark, Kamada, Kinaset Therapeutics, Kymera, Menarini, MicroA, OM Pharma, Orion, Pieris Pharmaceuticals, Pulmatrix, Revolo, Roivant Sciences, Sanofi, Synairgen, Tetherex, Teva, Theravance Biopharma, Upstream, and Verona Pharma. Mannino disclosed serving as a consultant to multiple companies currently developing COPD therapies (AstraZeneca, GlaxoSmithKline, Roche, Regeneron, Sanofi, Genentech, Amgen, and Chiesi).
A version of this article appeared on Medscape.com.
New biologic drugs for chronic obstructive pulmonary disease (COPD) are finally here, said Stephen Rennard, MD, in a presentation in a session on new drugs at the 2024 GOLD International COPD Conference.
The therapeutic goals of biologics remain the same as with other treatments for COPD, namely restoration of normal inflammatory response and alteration of disease progression, as well as restoration of lost structure and function and improvement of systemic effects, Rennard said in his presentation. Most studies of new and up-and-coming drugs have improvement in acute exacerbation of COPD as the primary outcome.
The Biology Behind the Biologics
T2 inflammation is “an inflammatory cascade led by IL [interleukin]-4, IL-13, and IL-5,” Mona Bafadhel, MD, chair of Respiratory Medicine at King’s College London in England, said in her presentation during the session.
Bafadhel, who served as one of the investigators on the BOREAS and NOTUS studies, explained some of the science behind the development of the new biologics.
Eosinophils are powerful regulators of immune response and inflammation by stimulating T-cell production and affecting other immune cell types, she noted.
In the context of COPD and drug development, high blood eosinophil counts have been associated with increased COPD-related exacerbations, Bafadhel said. She cited data from a Dutch study of more than 7000 patients with COPD (with and without clinical diagnoses), in which absolute eosinophil counts ≥ 3.3% were associated with increased risk for severe exacerbations of 32% and 84% across all patients with COPD and clinical COPD, respectively.
Understanding the mechanisms of the eosinophil in COPD is important for research and development, Bafadhel said. Along with standardizing measurement of T2 inflammatory markers (IL-4, IL-13, and IL-5), more research is needed to fully understand the role of eosinophils in immunoregulation and repair.
Fitting the Biologic to the Patient
Several recent studies of up-and-coming biologics have focused on subsets of COPD patients, said Dave Singh, MD, professor of clinical pharmacology and respiratory medicine at The University of Manchester in England, in his presentation at the meeting. In September 2024, the Food and Drug Administration approved dupilumab as the first biologic treatment for patients with uncontrolled COPD and type 2 inflammation on the basis of eosinophil counts. Singh cited data from the BOREAS and NOTUS studies in which dupilumab significantly reduced exacerbations and improved lung function in these patients, compared with a placebo.
Mepolizumab, a biologic approved for asthma, is not currently approved for COPD, but data from a 2017 study showed a trend toward reduced exacerbations, compared with placebo, in a subset of patients with high blood eosinophil counts, Singh said.
In addition, a recent unpublished phase 3 study (MATINEE) showed a reduction in the annualized rate of exacerbations, compared with placebo, on the basis of up to 2 years’ follow-up.
Singh also highlighted data from a phase 2a study of astegolimab, a biologic drug that focuses on the IL-33 receptor, in which COPD exacerbation rates were not significantly different between treatment and placebo groups. However, astegolimab has shown safety and efficacy in adults with severe asthma and is under development in phase 3 trials for COPD.
Tezepelumab, which was approved by the FDA in 2021 as an add-on therapy for severe asthma in patients aged 12 years or older, is also in development as a therapy for COPD exacerbations, Singh said.
In a study presented at the 2024 American Thoracic Society annual meeting, Singh and colleagues found that tezepelumab at a subcutaneous dose of 420 mg every 4 weeks reduced the annualized rate of moderate or severe COPD exacerbations compared with placebo based on data from approximately 300 patients, although the difference was not statistically significant.
Itepekimab, another biologic, showed promise in a phase 2a genetic association study involving current and former smokers with moderate to severe COPD, Singh said.
In that study, published in 2022 in The Lancet Respiratory Medicine, itepekimab failed to meet the primary endpoint in the overall study population of reduced annualized rate of moderate to severe exacerbations; however, a subgroup analysis of former smokers showed a significant (42%) reduction in exacerbations, Singh said in his presentation. Two phase 3 clinical studies (AERIFY-1/2) are ongoing to confirm the safety and efficacy of itepekimab in former smokers with COPD.
Takeaways and Next Steps
“These therapies provide the first new classes of medications approved for COPD in nearly 20 years,” said David M. Mannino, MD, of the University of Kentucky, Lexington, in an interview. “Dupilumab will be available to a subset of patients who are poorly controlled and have evidence of high eosinophils in their blood and is only used once every 2 weeks,” added Mannino, who has served as a consultant to companies developing COPD drugs.
Both dupilumab and ensifentrine, a phosphodiesterase (PDE) 3 and PDE4 inhibitor also recently approved for maintenance treatment of COPD, have been shown in clinical trials to reduce exacerbations and improve symptoms, said Mannino. Both offer additional options for patients who continue to have symptoms and exacerbations in spite of their current therapy.
Some barriers to the use of biologics in practice include the high cost. “Access and overcoming insurance-related issues such as preauthorization and high copays will be a challenge,” he said. Also, because dupilumab is an injectable drug, some patient training will be required.
Newer biologic therapies in development are also injectables, but some studies are examining longer time intervals as long as every 6 months, which could be a major advancement for some patients. The newer therapies in development are similar to dupilumab in that they will be injected therapies. Some in development are looking at longer time intervals as long as every 6 months, which may be a major advancement for some patients. “All of these therapies, however, are currently targeting more advanced or serious disease,” he said.
Looking ahead, more therapies are needed for the treatment of early COPD, as well as therapies that can be administered to a large number of patients at a reasonable cost, Mannino added.
Rennard disclosed serving as a consultant for Verona Pharma, Sanofi, Beyond Air, RS BioTherapeutics, RespirAI, and Roche, as well as speaker fees from Sanofi and temporary ownership interest while employed by AstraZeneca. Rennard is also the founder of Great Plains Biometrix. Bafadhel disclosed funding from the National Institute for Health Research (NIHR), grants from Asthma + Lung UK, Horizon Europe, NIHR, and AstraZeneca to her institution, and honoraria from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, and Pfizer. Singh disclosed relationships including speaking sponsorships, honoraria, and advisory board memberships for Adovate, Aerogen, Almirall, Apogee, Arrowhead, AstraZeneca, Bial, Boehringer Ingelheim, Chiesi, Cipla, Connect Biopharm, Covis, CSL Behring, DevPro Biopharm, Elpen, Empirico, EpiEndo, Genentech, Generate Biomedicines, GlaxoSmithKline, Glenmark, Kamada, Kinaset Therapeutics, Kymera, Menarini, MicroA, OM Pharma, Orion, Pieris Pharmaceuticals, Pulmatrix, Revolo, Roivant Sciences, Sanofi, Synairgen, Tetherex, Teva, Theravance Biopharma, Upstream, and Verona Pharma. Mannino disclosed serving as a consultant to multiple companies currently developing COPD therapies (AstraZeneca, GlaxoSmithKline, Roche, Regeneron, Sanofi, Genentech, Amgen, and Chiesi).
A version of this article appeared on Medscape.com.