User login
In rheumatology, biosimilars are flatlining. Why?
Although biosimilar versions of tumor necrosis factor inhibitors (TNFis) have been available to U.S. rheumatologists and their patients for over 3 years, uptake has thus far been slow.
In an analysis of data from a large commercial payer, the two available biosimilars for infliximab (Remicade) accounted for less than 1% of TNFi prescribing since the first biosimilar to infliximab was approved in 2016.
The study, published in Arthritis & Rheumatology, involved a total of 1.1 million TNFi prescriptions or infusions received by 95,906 patients from 2016 to 2019. Investigators found that uptake of biosimilar infliximab was essentially flat, standing at 0.1% of prescribing in the second quarter of 2017, and topping out at 0.9% in the first quarter of 2019. For branded infliximab, prescribing was also stable, but accounted for about 20% of overall biologic dispensing in each quarter of the period studied.
There are currently two biosimilar medications to the originator infliximab, which is one of five originator biologics available to treat rheumatic diseases in the United States: infliximab-dyyb (Inflectra) and infliximab-abda (Renflexis). The former was approved in 2016 and the latter in 2017, said study author Seoyoung C. Kim, MD, ScD, of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital, Boston, and her coauthors.

“Our paper reports a disappointingly low uptake of biosimilar infliximab since the first quarter of 2017 using claims data from a large private health plan. The main and maybe the only reason to consider using a biosimilar is cost saving,” said Dr. Kim in an interview. “Our results suggest that current modest cost savings from infliximab biosimilars in the U.S. are not sufficient to promote their widespread use.”
In the payer database study conducted by Dr. Kim and colleagues, the insurer paid similar mean amounts per patient per quarter for originator and biosimilar infliximab in mid-2017 ($8,322 versus $8,656). By the end of 2018, a gap appeared, with the insurer paying a mean quarterly per-patient sum of $8,111 for biosimilar infliximab compared with $9,535 for the branded biologic.
“The lack of market penetration and very modest price reductions for biosimilars have left policymakers, payers, physicians, and the public frustrated, particularly because sales in Europe continue to rapidly expand and robust cost-savings have materialized,” wrote Jinoos Yazdany, MD, MPH, in an editorial accompanying the study.

Dr. Yazdany, professor and chief of the division of rheumatology at the University of California, San Francisco, noted that increased spending on biologics in the United States – which increased by 50% from 2014 to 2018 – has been driven by rising prices as well as increased uptake of biologic therapies.
At least in part, Europe has been able to reap cost savings where the United States hasn’t because fundamental differences in health care reimbursement can ease sweeping biosimilar adoption, Dr. Yazdany noted. “Countries like Denmark and Sweden, using the negotiating and purchasing power of their single-payer systems have instituted a winner-takes-all bidding system,” with Denmark seeing cost savings of up to two-thirds when bidding was combined with mandatory switching, she said.
The continued market dominance of originator infliximab means that savings from biosimilars have thus far amounted to about $91 million, far short of the $1 billion that the Congressional Budget Office had projected for this date, Dr. Yazdany said.
One problem in the adoption of biosimilars by U.S. rheumatologists may have been uneven marketing and pricing across different types of practice, Colin C. Edgerton, MD, a rheumatologist at Low Country Rheumatology in South Carolina and chair of the American College of Rheumatology’s Committee on Rheumatologic Care, said in an interview.

“Rheumatologists have generally developed comfort with biosimilars, although this is not universal. The core message, that all biologics vary and that this is OK, is getting out. In general, rheumatologists also understand the problem with high drug prices and the threat to patient access,” Dr. Edgerton said. But “the early marketing and pricing focus for biosimilars seemed to be on hospitals and facilities, and this did not work effectively for community rheumatologists, where the majority of care is delivered. We have been pleased to see a manufacturer pivot toward community rheumatology where additional efforts need to be made to bend the curve on biosimilar adoption. It is critical for practices with experience using biosimilars to educate peers, and this is where networks of practicing rheumatologists are important.”
In Dr. Yazdany’s editorial, she cited four structural factors impeding biosimilar uptake and downstream savings.
First, she cites ongoing actions by pharmaceutical companies, which create a “patent thicket” that has the effect of fencing off originator biologics from biosimilars long beyond the original 12-year exclusivity period. Supporting the notion that “patent thickets” are a common strategy, Dr. Yazdany noted that almost half of the patent applications that AbbVie has filed for adalimumab (Humira) have come in after the original exclusivity period expired in 2014. Humira’s price has risen 18% yearly during this period.
The complicated role played by pharmacy benefit managers (PBMs) is another factor in slow adoption, said Dr. Yazdany: When manufacturers offer rebates to PBMs, the price of the originator biologic may be less than its biosimilar. Further, manufacturers may sign multiyear rebate agreements just before a biosimilar launch; PBMs are also sometimes threatened with the withdrawal of rebates if they offer biosimilars, she noted.
Third, prescriber inertia may also be at play, Dr. Yazdany noted, not least because patients often see little difference in out-of-pocket costs when they make the switch to a biosimilar – PBM rebates are not necessarily passed on to patients. Payers may not reimburse a biosimilar, or formularies can be built without them, influencing prescribing, and there’s usually no reimbursement incentive for biosimilar prescribing in the nonpublic sector, she said. To the contrary, infusing a drug with a higher price often means higher reimbursement for the administering clinician, since commercial insurance reimbursement is often calculated as a percent of the charge for the drug.
Further contributing to inertia is the extra time required for patient education and writing a new set of orders – all work that can’t be captured for extra reimbursement. Dr. Edgerton said that rheumatologists can talk with patients about the “nocebo effect” relating to biosimilars. “This is a phenomenon in which patients are thought to experience worsening symptoms associated with negative beliefs about biosimilars. There has been a study in Arthritis Care & Research addressing this concern. The authors found that positive framing of biosimilars led to more participants being willing to switch than negative framing. This suggests that clinicians have an important role in informing patients about biosimilars, and addressing hesitancy.”
Finally, Dr. Yazdany pointed out that for a pharmaceutical company pursuing biosimilar approval, the regulatory pathway itself can provide its own set of complications and confusion. Biosimilars are not exact molecular replicas of the originator biologic, and these differences can change efficacy and immunogenicity, and also affect stability. Hence, a company wishing to market a biosimilar has to show the Food and Drug Administration that safety and efficacy aren’t affected by a switch to biosimilar from an originator biologic. Extrapolation from one indication to another can be made – with scientific justification.
Rheumatologists are mindful of the potential differences between biosimilars and the originator biologic, as evinced in a recent position statement from the American College of Rheumatology. The position statement advises that “extrapolation should be pursued with caution,” and asks for clear labeling when biosimilars have been designated “interchangeable” with their biosimilar. Interchangeability can clear the way for pharmacy substitution of a prescribed biologic, though Dr. Yazdany noted that 40 states have passed legislation requiring prescriber notification.
The FDA is currently using postmarketing pharmacovigilance to monitor biosimilar performance in the real world, and a recent systematic review “should provide some reassurance,” wrote Dr. Yazdany, citing the study, which looked at 14,000 patients who had a total of 14 disease indications for biosimilar use. The 90-article review largely found no differences in safety, efficacy, or immunogenicity between originators and their biosimilars. Dr. Yazdany recommended greater openness to incorporating the European experience in the FDA’s ongoing reassessment.
A further way forward can come through tackling the patent thicket with the proposed bipartisan Biologic Patent Transparency Act, which would require publication of biologic patents in a one-stop publicly searchable database. Going further with legislation to address anticompetitive activity by pharmaceutical companies could shorten the runway to biosimilar launching considerably, she noted.
The complicated landscape of PBMs and rebates affects many sectors of health care, and new policy efforts are needed here as well, she said. Reimbursement strategies – and much-needed continuing medical education – can both ease prescriber unfamiliarity with biosimilars and provide incentives for their use, she concluded.
Dr. Kim concurred that change is needed before the United States is likely to reap significant economic benefit from biosimilars. “The uptake of biosimilars and their impact on overall health care cost needs to be reevaluated when we have more biosimilars available in the next 3-4 years. However, for now, it appears that substantial savings achieved in some European countries – for example, Denmark – may not be possible without systemic reform of the U.S. pharmaceutical market,” she said.
Dr. Yazdany is supported by the Alice Betts Endowed Chair in Arthritis Research, the Russel/Engleman Research Center at the University of California, San Francisco, and the National Institutes of Health. She has received independent research grants from Pfizer and Genentech and research consulting fees from Eli Lilly and AstraZeneca.
Dr. Kim’s study was supported by the division of pharmacoepidemiology and pharmacoeconomics, department of medicine, Brigham and Women’s Hospital, and Arnold Ventures. Dr. Kim has received research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Bristol-Myers Squibb, and Roche.
SOURCES: Kim SC et al. Arthritis Rheumatol. 2020 Jan 13. doi: 10.1002/art.41201; Yazdany J. Arthritis Rheumatol. 2020 Jan 10. doi: 10.1002/art.41203.
Although biosimilar versions of tumor necrosis factor inhibitors (TNFis) have been available to U.S. rheumatologists and their patients for over 3 years, uptake has thus far been slow.
In an analysis of data from a large commercial payer, the two available biosimilars for infliximab (Remicade) accounted for less than 1% of TNFi prescribing since the first biosimilar to infliximab was approved in 2016.
The study, published in Arthritis & Rheumatology, involved a total of 1.1 million TNFi prescriptions or infusions received by 95,906 patients from 2016 to 2019. Investigators found that uptake of biosimilar infliximab was essentially flat, standing at 0.1% of prescribing in the second quarter of 2017, and topping out at 0.9% in the first quarter of 2019. For branded infliximab, prescribing was also stable, but accounted for about 20% of overall biologic dispensing in each quarter of the period studied.
There are currently two biosimilar medications to the originator infliximab, which is one of five originator biologics available to treat rheumatic diseases in the United States: infliximab-dyyb (Inflectra) and infliximab-abda (Renflexis). The former was approved in 2016 and the latter in 2017, said study author Seoyoung C. Kim, MD, ScD, of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital, Boston, and her coauthors.
“Our paper reports a disappointingly low uptake of biosimilar infliximab since the first quarter of 2017 using claims data from a large private health plan. The main and maybe the only reason to consider using a biosimilar is cost saving,” said Dr. Kim in an interview. “Our results suggest that current modest cost savings from infliximab biosimilars in the U.S. are not sufficient to promote their widespread use.”
In the payer database study conducted by Dr. Kim and colleagues, the insurer paid similar mean amounts per patient per quarter for originator and biosimilar infliximab in mid-2017 ($8,322 versus $8,656). By the end of 2018, a gap appeared, with the insurer paying a mean quarterly per-patient sum of $8,111 for biosimilar infliximab compared with $9,535 for the branded biologic.
“The lack of market penetration and very modest price reductions for biosimilars have left policymakers, payers, physicians, and the public frustrated, particularly because sales in Europe continue to rapidly expand and robust cost-savings have materialized,” wrote Jinoos Yazdany, MD, MPH, in an editorial accompanying the study.
Dr. Yazdany, professor and chief of the division of rheumatology at the University of California, San Francisco, noted that increased spending on biologics in the United States – which increased by 50% from 2014 to 2018 – has been driven by rising prices as well as increased uptake of biologic therapies.
At least in part, Europe has been able to reap cost savings where the United States hasn’t because fundamental differences in health care reimbursement can ease sweeping biosimilar adoption, Dr. Yazdany noted. “Countries like Denmark and Sweden, using the negotiating and purchasing power of their single-payer systems have instituted a winner-takes-all bidding system,” with Denmark seeing cost savings of up to two-thirds when bidding was combined with mandatory switching, she said.
The continued market dominance of originator infliximab means that savings from biosimilars have thus far amounted to about $91 million, far short of the $1 billion that the Congressional Budget Office had projected for this date, Dr. Yazdany said.
One problem in the adoption of biosimilars by U.S. rheumatologists may have been uneven marketing and pricing across different types of practice, Colin C. Edgerton, MD, a rheumatologist at Low Country Rheumatology in South Carolina and chair of the American College of Rheumatology’s Committee on Rheumatologic Care, said in an interview.
“Rheumatologists have generally developed comfort with biosimilars, although this is not universal. The core message, that all biologics vary and that this is OK, is getting out. In general, rheumatologists also understand the problem with high drug prices and the threat to patient access,” Dr. Edgerton said. But “the early marketing and pricing focus for biosimilars seemed to be on hospitals and facilities, and this did not work effectively for community rheumatologists, where the majority of care is delivered. We have been pleased to see a manufacturer pivot toward community rheumatology where additional efforts need to be made to bend the curve on biosimilar adoption. It is critical for practices with experience using biosimilars to educate peers, and this is where networks of practicing rheumatologists are important.”
In Dr. Yazdany’s editorial, she cited four structural factors impeding biosimilar uptake and downstream savings.
First, she cites ongoing actions by pharmaceutical companies, which create a “patent thicket” that has the effect of fencing off originator biologics from biosimilars long beyond the original 12-year exclusivity period. Supporting the notion that “patent thickets” are a common strategy, Dr. Yazdany noted that almost half of the patent applications that AbbVie has filed for adalimumab (Humira) have come in after the original exclusivity period expired in 2014. Humira’s price has risen 18% yearly during this period.
The complicated role played by pharmacy benefit managers (PBMs) is another factor in slow adoption, said Dr. Yazdany: When manufacturers offer rebates to PBMs, the price of the originator biologic may be less than its biosimilar. Further, manufacturers may sign multiyear rebate agreements just before a biosimilar launch; PBMs are also sometimes threatened with the withdrawal of rebates if they offer biosimilars, she noted.
Third, prescriber inertia may also be at play, Dr. Yazdany noted, not least because patients often see little difference in out-of-pocket costs when they make the switch to a biosimilar – PBM rebates are not necessarily passed on to patients. Payers may not reimburse a biosimilar, or formularies can be built without them, influencing prescribing, and there’s usually no reimbursement incentive for biosimilar prescribing in the nonpublic sector, she said. To the contrary, infusing a drug with a higher price often means higher reimbursement for the administering clinician, since commercial insurance reimbursement is often calculated as a percent of the charge for the drug.
Further contributing to inertia is the extra time required for patient education and writing a new set of orders – all work that can’t be captured for extra reimbursement. Dr. Edgerton said that rheumatologists can talk with patients about the “nocebo effect” relating to biosimilars. “This is a phenomenon in which patients are thought to experience worsening symptoms associated with negative beliefs about biosimilars. There has been a study in Arthritis Care & Research addressing this concern. The authors found that positive framing of biosimilars led to more participants being willing to switch than negative framing. This suggests that clinicians have an important role in informing patients about biosimilars, and addressing hesitancy.”
Finally, Dr. Yazdany pointed out that for a pharmaceutical company pursuing biosimilar approval, the regulatory pathway itself can provide its own set of complications and confusion. Biosimilars are not exact molecular replicas of the originator biologic, and these differences can change efficacy and immunogenicity, and also affect stability. Hence, a company wishing to market a biosimilar has to show the Food and Drug Administration that safety and efficacy aren’t affected by a switch to biosimilar from an originator biologic. Extrapolation from one indication to another can be made – with scientific justification.
Rheumatologists are mindful of the potential differences between biosimilars and the originator biologic, as evinced in a recent position statement from the American College of Rheumatology. The position statement advises that “extrapolation should be pursued with caution,” and asks for clear labeling when biosimilars have been designated “interchangeable” with their biosimilar. Interchangeability can clear the way for pharmacy substitution of a prescribed biologic, though Dr. Yazdany noted that 40 states have passed legislation requiring prescriber notification.
The FDA is currently using postmarketing pharmacovigilance to monitor biosimilar performance in the real world, and a recent systematic review “should provide some reassurance,” wrote Dr. Yazdany, citing the study, which looked at 14,000 patients who had a total of 14 disease indications for biosimilar use. The 90-article review largely found no differences in safety, efficacy, or immunogenicity between originators and their biosimilars. Dr. Yazdany recommended greater openness to incorporating the European experience in the FDA’s ongoing reassessment.
A further way forward can come through tackling the patent thicket with the proposed bipartisan Biologic Patent Transparency Act, which would require publication of biologic patents in a one-stop publicly searchable database. Going further with legislation to address anticompetitive activity by pharmaceutical companies could shorten the runway to biosimilar launching considerably, she noted.
The complicated landscape of PBMs and rebates affects many sectors of health care, and new policy efforts are needed here as well, she said. Reimbursement strategies – and much-needed continuing medical education – can both ease prescriber unfamiliarity with biosimilars and provide incentives for their use, she concluded.
Dr. Kim concurred that change is needed before the United States is likely to reap significant economic benefit from biosimilars. “The uptake of biosimilars and their impact on overall health care cost needs to be reevaluated when we have more biosimilars available in the next 3-4 years. However, for now, it appears that substantial savings achieved in some European countries – for example, Denmark – may not be possible without systemic reform of the U.S. pharmaceutical market,” she said.
Dr. Yazdany is supported by the Alice Betts Endowed Chair in Arthritis Research, the Russel/Engleman Research Center at the University of California, San Francisco, and the National Institutes of Health. She has received independent research grants from Pfizer and Genentech and research consulting fees from Eli Lilly and AstraZeneca.
Dr. Kim’s study was supported by the division of pharmacoepidemiology and pharmacoeconomics, department of medicine, Brigham and Women’s Hospital, and Arnold Ventures. Dr. Kim has received research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Bristol-Myers Squibb, and Roche.
SOURCES: Kim SC et al. Arthritis Rheumatol. 2020 Jan 13. doi: 10.1002/art.41201; Yazdany J. Arthritis Rheumatol. 2020 Jan 10. doi: 10.1002/art.41203.
Although biosimilar versions of tumor necrosis factor inhibitors (TNFis) have been available to U.S. rheumatologists and their patients for over 3 years, uptake has thus far been slow.
In an analysis of data from a large commercial payer, the two available biosimilars for infliximab (Remicade) accounted for less than 1% of TNFi prescribing since the first biosimilar to infliximab was approved in 2016.
The study, published in Arthritis & Rheumatology, involved a total of 1.1 million TNFi prescriptions or infusions received by 95,906 patients from 2016 to 2019. Investigators found that uptake of biosimilar infliximab was essentially flat, standing at 0.1% of prescribing in the second quarter of 2017, and topping out at 0.9% in the first quarter of 2019. For branded infliximab, prescribing was also stable, but accounted for about 20% of overall biologic dispensing in each quarter of the period studied.
There are currently two biosimilar medications to the originator infliximab, which is one of five originator biologics available to treat rheumatic diseases in the United States: infliximab-dyyb (Inflectra) and infliximab-abda (Renflexis). The former was approved in 2016 and the latter in 2017, said study author Seoyoung C. Kim, MD, ScD, of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital, Boston, and her coauthors.
“Our paper reports a disappointingly low uptake of biosimilar infliximab since the first quarter of 2017 using claims data from a large private health plan. The main and maybe the only reason to consider using a biosimilar is cost saving,” said Dr. Kim in an interview. “Our results suggest that current modest cost savings from infliximab biosimilars in the U.S. are not sufficient to promote their widespread use.”
In the payer database study conducted by Dr. Kim and colleagues, the insurer paid similar mean amounts per patient per quarter for originator and biosimilar infliximab in mid-2017 ($8,322 versus $8,656). By the end of 2018, a gap appeared, with the insurer paying a mean quarterly per-patient sum of $8,111 for biosimilar infliximab compared with $9,535 for the branded biologic.
“The lack of market penetration and very modest price reductions for biosimilars have left policymakers, payers, physicians, and the public frustrated, particularly because sales in Europe continue to rapidly expand and robust cost-savings have materialized,” wrote Jinoos Yazdany, MD, MPH, in an editorial accompanying the study.
Dr. Yazdany, professor and chief of the division of rheumatology at the University of California, San Francisco, noted that increased spending on biologics in the United States – which increased by 50% from 2014 to 2018 – has been driven by rising prices as well as increased uptake of biologic therapies.
At least in part, Europe has been able to reap cost savings where the United States hasn’t because fundamental differences in health care reimbursement can ease sweeping biosimilar adoption, Dr. Yazdany noted. “Countries like Denmark and Sweden, using the negotiating and purchasing power of their single-payer systems have instituted a winner-takes-all bidding system,” with Denmark seeing cost savings of up to two-thirds when bidding was combined with mandatory switching, she said.
The continued market dominance of originator infliximab means that savings from biosimilars have thus far amounted to about $91 million, far short of the $1 billion that the Congressional Budget Office had projected for this date, Dr. Yazdany said.
One problem in the adoption of biosimilars by U.S. rheumatologists may have been uneven marketing and pricing across different types of practice, Colin C. Edgerton, MD, a rheumatologist at Low Country Rheumatology in South Carolina and chair of the American College of Rheumatology’s Committee on Rheumatologic Care, said in an interview.
“Rheumatologists have generally developed comfort with biosimilars, although this is not universal. The core message, that all biologics vary and that this is OK, is getting out. In general, rheumatologists also understand the problem with high drug prices and the threat to patient access,” Dr. Edgerton said. But “the early marketing and pricing focus for biosimilars seemed to be on hospitals and facilities, and this did not work effectively for community rheumatologists, where the majority of care is delivered. We have been pleased to see a manufacturer pivot toward community rheumatology where additional efforts need to be made to bend the curve on biosimilar adoption. It is critical for practices with experience using biosimilars to educate peers, and this is where networks of practicing rheumatologists are important.”
In Dr. Yazdany’s editorial, she cited four structural factors impeding biosimilar uptake and downstream savings.
First, she cites ongoing actions by pharmaceutical companies, which create a “patent thicket” that has the effect of fencing off originator biologics from biosimilars long beyond the original 12-year exclusivity period. Supporting the notion that “patent thickets” are a common strategy, Dr. Yazdany noted that almost half of the patent applications that AbbVie has filed for adalimumab (Humira) have come in after the original exclusivity period expired in 2014. Humira’s price has risen 18% yearly during this period.
The complicated role played by pharmacy benefit managers (PBMs) is another factor in slow adoption, said Dr. Yazdany: When manufacturers offer rebates to PBMs, the price of the originator biologic may be less than its biosimilar. Further, manufacturers may sign multiyear rebate agreements just before a biosimilar launch; PBMs are also sometimes threatened with the withdrawal of rebates if they offer biosimilars, she noted.
Third, prescriber inertia may also be at play, Dr. Yazdany noted, not least because patients often see little difference in out-of-pocket costs when they make the switch to a biosimilar – PBM rebates are not necessarily passed on to patients. Payers may not reimburse a biosimilar, or formularies can be built without them, influencing prescribing, and there’s usually no reimbursement incentive for biosimilar prescribing in the nonpublic sector, she said. To the contrary, infusing a drug with a higher price often means higher reimbursement for the administering clinician, since commercial insurance reimbursement is often calculated as a percent of the charge for the drug.
Further contributing to inertia is the extra time required for patient education and writing a new set of orders – all work that can’t be captured for extra reimbursement. Dr. Edgerton said that rheumatologists can talk with patients about the “nocebo effect” relating to biosimilars. “This is a phenomenon in which patients are thought to experience worsening symptoms associated with negative beliefs about biosimilars. There has been a study in Arthritis Care & Research addressing this concern. The authors found that positive framing of biosimilars led to more participants being willing to switch than negative framing. This suggests that clinicians have an important role in informing patients about biosimilars, and addressing hesitancy.”
Finally, Dr. Yazdany pointed out that for a pharmaceutical company pursuing biosimilar approval, the regulatory pathway itself can provide its own set of complications and confusion. Biosimilars are not exact molecular replicas of the originator biologic, and these differences can change efficacy and immunogenicity, and also affect stability. Hence, a company wishing to market a biosimilar has to show the Food and Drug Administration that safety and efficacy aren’t affected by a switch to biosimilar from an originator biologic. Extrapolation from one indication to another can be made – with scientific justification.
Rheumatologists are mindful of the potential differences between biosimilars and the originator biologic, as evinced in a recent position statement from the American College of Rheumatology. The position statement advises that “extrapolation should be pursued with caution,” and asks for clear labeling when biosimilars have been designated “interchangeable” with their biosimilar. Interchangeability can clear the way for pharmacy substitution of a prescribed biologic, though Dr. Yazdany noted that 40 states have passed legislation requiring prescriber notification.
The FDA is currently using postmarketing pharmacovigilance to monitor biosimilar performance in the real world, and a recent systematic review “should provide some reassurance,” wrote Dr. Yazdany, citing the study, which looked at 14,000 patients who had a total of 14 disease indications for biosimilar use. The 90-article review largely found no differences in safety, efficacy, or immunogenicity between originators and their biosimilars. Dr. Yazdany recommended greater openness to incorporating the European experience in the FDA’s ongoing reassessment.
A further way forward can come through tackling the patent thicket with the proposed bipartisan Biologic Patent Transparency Act, which would require publication of biologic patents in a one-stop publicly searchable database. Going further with legislation to address anticompetitive activity by pharmaceutical companies could shorten the runway to biosimilar launching considerably, she noted.
The complicated landscape of PBMs and rebates affects many sectors of health care, and new policy efforts are needed here as well, she said. Reimbursement strategies – and much-needed continuing medical education – can both ease prescriber unfamiliarity with biosimilars and provide incentives for their use, she concluded.
Dr. Kim concurred that change is needed before the United States is likely to reap significant economic benefit from biosimilars. “The uptake of biosimilars and their impact on overall health care cost needs to be reevaluated when we have more biosimilars available in the next 3-4 years. However, for now, it appears that substantial savings achieved in some European countries – for example, Denmark – may not be possible without systemic reform of the U.S. pharmaceutical market,” she said.
Dr. Yazdany is supported by the Alice Betts Endowed Chair in Arthritis Research, the Russel/Engleman Research Center at the University of California, San Francisco, and the National Institutes of Health. She has received independent research grants from Pfizer and Genentech and research consulting fees from Eli Lilly and AstraZeneca.
Dr. Kim’s study was supported by the division of pharmacoepidemiology and pharmacoeconomics, department of medicine, Brigham and Women’s Hospital, and Arnold Ventures. Dr. Kim has received research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Bristol-Myers Squibb, and Roche.
SOURCES: Kim SC et al. Arthritis Rheumatol. 2020 Jan 13. doi: 10.1002/art.41201; Yazdany J. Arthritis Rheumatol. 2020 Jan 10. doi: 10.1002/art.41203.
FROM ARTHRITIS & RHEUMATOLOGY
Doctor wins $4.75 million award in defamation suit against hospital
Jurors awarded Carmel, Ind.–based ob.gyn. Rebecca Denman, MD, $4.75 million in damages against St. Vincent Carmel Hospital and St. Vincent Carmel Medical Group on Jan. 16, 2020, after a 4-day trial in Indiana Commercial Court. Dr. Denman sued after the hospital and medical group took a series of actions in response to a nurse practitioner’s claim that Dr. Denman smelled of alcohol while on duty. The doctor’s lawsuit alleged the NP’s claim was unproven; that administrators failed to conduct a proper peer-review investigation; and that repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.

Indianapolis attorney Kathleen DeLaney, who represented Dr. Denman in the case, said that her client was pleased with the verdict.
“Dr. Denman feels vindicated that a group of jurors spent 4 days listening to all the evidence and gave her a resounding victory,” she said in an interview.
Dr. Denman declined to comment for this story through her attorney.
In a statement, a spokesman for Ascension, the hospital’s parent company, said the hospital was disappointed by the verdict and that it was “exploring all options available to us, including appeal.” The spokesman declined to answer further questions about the case or its peer-review process.
The case stems from an NP’s claim that Dr. Denman’s breath smelled of alcohol during an evening shift on Dec. 11, 2017. Dr. Denman was not informed of the allegation on Dec. 11 and was not tested for alcohol at the time, according to Dr. Denman’s lawsuit. Under hospital policy, if a physician is suspected of being under the influence of alcohol at work, the employer must immediately assess the doctor, relieve the doctor of duty, and request the physician submit to immediate blood testing at an external facility.
The NP reported the allegation to her supervisor through an email on Dec. 12, 2017. The supervisor relayed the information to the hospital’s chief medical officer who met with other administrators and physicians to discuss the claim. During the discussions, a previous concern about Dr. Denman’s drinking was raised, according to deposition information included in court documents. In 2015, two physicians had suggested Dr. Denman consider an assistance program after expressing concerns that she was arriving late to work and missing partner meetings. At the time, Dr. Denman did not enter an assistance program, but she changed her drinking habits, began seeing a therapist, and started arriving on-time to work and to partner meetings, according to court documents. No other criticism or complaints regarding her drinking or workplace behavior had been reported since, according to court documents.
When confronted with the NP’s claim on Dec. 13, 2017, Dr. Denman denied consuming alcohol on Dec. 11, 2017, and questioned why the hospital’s substance abuse protocol was not followed.
St. Vincent Carmel Hospital conducted a preliminary review of the allegation through its peer-review process and turned the matter over to St. Vincent Medical Group for further review, according to court documents. St. Vincent Medical Group later informed Dr. Denman they had reviewed the allegation through its peer-review process and that she was suspended with partial pay until she underwent an evaluation for alcohol abuse through the Indiana State Medical Association, according to the lawsuit.
“They falsely misrepresented to her that peer review had been done,” Ms. DeLaney said in an interview. “In spite of that statement, they never offered her a hearing before a peer-review committee, they never shared with her the substance of any evidence they had against her, they never gave her an opportunity to respond to the allegations. In fact, she wasn’t interviewed at all until the deposition in the lawsuit.”
According to the Indiana Peer Review law, a health care provider under investigation is permitted to see any records accumulated by a peer-review committee pertaining to the provider’s personal practice, and the provider shall be offered the opportunity to appear before the peer-review committee with adequate representation to hear all charges and findings concerning the provider and to offer rebuttal information. The rebuttal shall be part of the record before any disclosure of the charges and before any findings can be made, according to the statute.
Dr. Denman was referred by the medical association to an addiction treatment center that evaluated Dr. Denman and diagnosed her with alcohol use disorder, according to the lawsuit. As a result of the report and as a condition of retaining her medical license, the medical association and St. Vincent Medical Group required Dr. Denman to enter a treatment program at the same addiction treatment center. Dr. Denman was also required to sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit.
“The actions had life-changing consequences,” Ms. DeLaney said. “As a result, she was required to sign a contract that mandates she do a breathalyzer test four times a day for the first year and then three times a day for 4 more years. She has to go for random drug screenings. For the first year, she had to go to four [Alcoholics Anonymous] meetings a week. Now that number has been reduced, but she’s on a 5-year monitoring contract because of all of this.”
Dr. Denman sued the hospital, the medical group, and the NP in July 2018 alleging fraud, defamation, tortuous interference with an employment relationship, and negligent misrepresentation. The NP was dismissed from the case shortly before trial.
In its response to the lawsuit, attorneys for St. Vincent wrote that Dr. Denman’s action was frivolous, vexatious, and executed in bad faith. The defendants requested that a judge dismiss the lawsuit, noting that they were entitled to immunity pursuant to Indiana state and federal laws, including protection by Indiana’s Peer Review statute. In October 2019, a judge denied the hospital’s request to dismiss the lawsuit and allowed the case to proceed.
In their verdict, jurors awarded Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
Dr. Denman remains employed by the medical group and must continue the conditions of her 5-year monitoring contract, Ms. DeLaney said. She hopes Dr. Denman’s case raises awareness about physicians’ due process rights.
“We hope that Dr. Denman’s case emboldens physicians to stand up for themselves in the face of false accusations and rushes to judgment,” she said. “We hope the verdict leads to fair, prompt, and unbiased investigations by hospital and medical practice administrators, which include due process for accused physicians.”
Jurors awarded Carmel, Ind.–based ob.gyn. Rebecca Denman, MD, $4.75 million in damages against St. Vincent Carmel Hospital and St. Vincent Carmel Medical Group on Jan. 16, 2020, after a 4-day trial in Indiana Commercial Court. Dr. Denman sued after the hospital and medical group took a series of actions in response to a nurse practitioner’s claim that Dr. Denman smelled of alcohol while on duty. The doctor’s lawsuit alleged the NP’s claim was unproven; that administrators failed to conduct a proper peer-review investigation; and that repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.
Indianapolis attorney Kathleen DeLaney, who represented Dr. Denman in the case, said that her client was pleased with the verdict.
“Dr. Denman feels vindicated that a group of jurors spent 4 days listening to all the evidence and gave her a resounding victory,” she said in an interview.
Dr. Denman declined to comment for this story through her attorney.
In a statement, a spokesman for Ascension, the hospital’s parent company, said the hospital was disappointed by the verdict and that it was “exploring all options available to us, including appeal.” The spokesman declined to answer further questions about the case or its peer-review process.
The case stems from an NP’s claim that Dr. Denman’s breath smelled of alcohol during an evening shift on Dec. 11, 2017. Dr. Denman was not informed of the allegation on Dec. 11 and was not tested for alcohol at the time, according to Dr. Denman’s lawsuit. Under hospital policy, if a physician is suspected of being under the influence of alcohol at work, the employer must immediately assess the doctor, relieve the doctor of duty, and request the physician submit to immediate blood testing at an external facility.
The NP reported the allegation to her supervisor through an email on Dec. 12, 2017. The supervisor relayed the information to the hospital’s chief medical officer who met with other administrators and physicians to discuss the claim. During the discussions, a previous concern about Dr. Denman’s drinking was raised, according to deposition information included in court documents. In 2015, two physicians had suggested Dr. Denman consider an assistance program after expressing concerns that she was arriving late to work and missing partner meetings. At the time, Dr. Denman did not enter an assistance program, but she changed her drinking habits, began seeing a therapist, and started arriving on-time to work and to partner meetings, according to court documents. No other criticism or complaints regarding her drinking or workplace behavior had been reported since, according to court documents.
When confronted with the NP’s claim on Dec. 13, 2017, Dr. Denman denied consuming alcohol on Dec. 11, 2017, and questioned why the hospital’s substance abuse protocol was not followed.
St. Vincent Carmel Hospital conducted a preliminary review of the allegation through its peer-review process and turned the matter over to St. Vincent Medical Group for further review, according to court documents. St. Vincent Medical Group later informed Dr. Denman they had reviewed the allegation through its peer-review process and that she was suspended with partial pay until she underwent an evaluation for alcohol abuse through the Indiana State Medical Association, according to the lawsuit.
“They falsely misrepresented to her that peer review had been done,” Ms. DeLaney said in an interview. “In spite of that statement, they never offered her a hearing before a peer-review committee, they never shared with her the substance of any evidence they had against her, they never gave her an opportunity to respond to the allegations. In fact, she wasn’t interviewed at all until the deposition in the lawsuit.”
According to the Indiana Peer Review law, a health care provider under investigation is permitted to see any records accumulated by a peer-review committee pertaining to the provider’s personal practice, and the provider shall be offered the opportunity to appear before the peer-review committee with adequate representation to hear all charges and findings concerning the provider and to offer rebuttal information. The rebuttal shall be part of the record before any disclosure of the charges and before any findings can be made, according to the statute.
Dr. Denman was referred by the medical association to an addiction treatment center that evaluated Dr. Denman and diagnosed her with alcohol use disorder, according to the lawsuit. As a result of the report and as a condition of retaining her medical license, the medical association and St. Vincent Medical Group required Dr. Denman to enter a treatment program at the same addiction treatment center. Dr. Denman was also required to sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit.
“The actions had life-changing consequences,” Ms. DeLaney said. “As a result, she was required to sign a contract that mandates she do a breathalyzer test four times a day for the first year and then three times a day for 4 more years. She has to go for random drug screenings. For the first year, she had to go to four [Alcoholics Anonymous] meetings a week. Now that number has been reduced, but she’s on a 5-year monitoring contract because of all of this.”
Dr. Denman sued the hospital, the medical group, and the NP in July 2018 alleging fraud, defamation, tortuous interference with an employment relationship, and negligent misrepresentation. The NP was dismissed from the case shortly before trial.
In its response to the lawsuit, attorneys for St. Vincent wrote that Dr. Denman’s action was frivolous, vexatious, and executed in bad faith. The defendants requested that a judge dismiss the lawsuit, noting that they were entitled to immunity pursuant to Indiana state and federal laws, including protection by Indiana’s Peer Review statute. In October 2019, a judge denied the hospital’s request to dismiss the lawsuit and allowed the case to proceed.
In their verdict, jurors awarded Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
Dr. Denman remains employed by the medical group and must continue the conditions of her 5-year monitoring contract, Ms. DeLaney said. She hopes Dr. Denman’s case raises awareness about physicians’ due process rights.
“We hope that Dr. Denman’s case emboldens physicians to stand up for themselves in the face of false accusations and rushes to judgment,” she said. “We hope the verdict leads to fair, prompt, and unbiased investigations by hospital and medical practice administrators, which include due process for accused physicians.”
Jurors awarded Carmel, Ind.–based ob.gyn. Rebecca Denman, MD, $4.75 million in damages against St. Vincent Carmel Hospital and St. Vincent Carmel Medical Group on Jan. 16, 2020, after a 4-day trial in Indiana Commercial Court. Dr. Denman sued after the hospital and medical group took a series of actions in response to a nurse practitioner’s claim that Dr. Denman smelled of alcohol while on duty. The doctor’s lawsuit alleged the NP’s claim was unproven; that administrators failed to conduct a proper peer-review investigation; and that repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.
Indianapolis attorney Kathleen DeLaney, who represented Dr. Denman in the case, said that her client was pleased with the verdict.
“Dr. Denman feels vindicated that a group of jurors spent 4 days listening to all the evidence and gave her a resounding victory,” she said in an interview.
Dr. Denman declined to comment for this story through her attorney.
In a statement, a spokesman for Ascension, the hospital’s parent company, said the hospital was disappointed by the verdict and that it was “exploring all options available to us, including appeal.” The spokesman declined to answer further questions about the case or its peer-review process.
The case stems from an NP’s claim that Dr. Denman’s breath smelled of alcohol during an evening shift on Dec. 11, 2017. Dr. Denman was not informed of the allegation on Dec. 11 and was not tested for alcohol at the time, according to Dr. Denman’s lawsuit. Under hospital policy, if a physician is suspected of being under the influence of alcohol at work, the employer must immediately assess the doctor, relieve the doctor of duty, and request the physician submit to immediate blood testing at an external facility.
The NP reported the allegation to her supervisor through an email on Dec. 12, 2017. The supervisor relayed the information to the hospital’s chief medical officer who met with other administrators and physicians to discuss the claim. During the discussions, a previous concern about Dr. Denman’s drinking was raised, according to deposition information included in court documents. In 2015, two physicians had suggested Dr. Denman consider an assistance program after expressing concerns that she was arriving late to work and missing partner meetings. At the time, Dr. Denman did not enter an assistance program, but she changed her drinking habits, began seeing a therapist, and started arriving on-time to work and to partner meetings, according to court documents. No other criticism or complaints regarding her drinking or workplace behavior had been reported since, according to court documents.
When confronted with the NP’s claim on Dec. 13, 2017, Dr. Denman denied consuming alcohol on Dec. 11, 2017, and questioned why the hospital’s substance abuse protocol was not followed.
St. Vincent Carmel Hospital conducted a preliminary review of the allegation through its peer-review process and turned the matter over to St. Vincent Medical Group for further review, according to court documents. St. Vincent Medical Group later informed Dr. Denman they had reviewed the allegation through its peer-review process and that she was suspended with partial pay until she underwent an evaluation for alcohol abuse through the Indiana State Medical Association, according to the lawsuit.
“They falsely misrepresented to her that peer review had been done,” Ms. DeLaney said in an interview. “In spite of that statement, they never offered her a hearing before a peer-review committee, they never shared with her the substance of any evidence they had against her, they never gave her an opportunity to respond to the allegations. In fact, she wasn’t interviewed at all until the deposition in the lawsuit.”
According to the Indiana Peer Review law, a health care provider under investigation is permitted to see any records accumulated by a peer-review committee pertaining to the provider’s personal practice, and the provider shall be offered the opportunity to appear before the peer-review committee with adequate representation to hear all charges and findings concerning the provider and to offer rebuttal information. The rebuttal shall be part of the record before any disclosure of the charges and before any findings can be made, according to the statute.
Dr. Denman was referred by the medical association to an addiction treatment center that evaluated Dr. Denman and diagnosed her with alcohol use disorder, according to the lawsuit. As a result of the report and as a condition of retaining her medical license, the medical association and St. Vincent Medical Group required Dr. Denman to enter a treatment program at the same addiction treatment center. Dr. Denman was also required to sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit.
“The actions had life-changing consequences,” Ms. DeLaney said. “As a result, she was required to sign a contract that mandates she do a breathalyzer test four times a day for the first year and then three times a day for 4 more years. She has to go for random drug screenings. For the first year, she had to go to four [Alcoholics Anonymous] meetings a week. Now that number has been reduced, but she’s on a 5-year monitoring contract because of all of this.”
Dr. Denman sued the hospital, the medical group, and the NP in July 2018 alleging fraud, defamation, tortuous interference with an employment relationship, and negligent misrepresentation. The NP was dismissed from the case shortly before trial.
In its response to the lawsuit, attorneys for St. Vincent wrote that Dr. Denman’s action was frivolous, vexatious, and executed in bad faith. The defendants requested that a judge dismiss the lawsuit, noting that they were entitled to immunity pursuant to Indiana state and federal laws, including protection by Indiana’s Peer Review statute. In October 2019, a judge denied the hospital’s request to dismiss the lawsuit and allowed the case to proceed.
In their verdict, jurors awarded Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
Dr. Denman remains employed by the medical group and must continue the conditions of her 5-year monitoring contract, Ms. DeLaney said. She hopes Dr. Denman’s case raises awareness about physicians’ due process rights.
“We hope that Dr. Denman’s case emboldens physicians to stand up for themselves in the face of false accusations and rushes to judgment,” she said. “We hope the verdict leads to fair, prompt, and unbiased investigations by hospital and medical practice administrators, which include due process for accused physicians.”
Wuhan virus: What clinicians need to know
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.

“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.

The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.

“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.

Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.

WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.
“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.
The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.
“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.
Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.
WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
As the Wuhan coronavirus story unfolds, , according to infectious disease experts.
“We are asking that of everyone with fever and respiratory symptoms who comes to our clinics, hospital, or emergency room. It’s a powerful screening tool,” said William Schaffner, MD, professor of preventive medicine and infectious diseases at Vanderbilt University Medical Center, Nashville, Tenn.
In addition to fever, common signs of infection include cough, shortness of breath, and breathing difficulties. Some patients have had diarrhea, vomiting, and other gastrointestinal symptoms. In more severe cases, infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and death. The incubation period appears to be up to 2 weeks, according to the World Health Organization (WHO).
If patients exhibit symptoms and either they or a close contact has returned from China recently, take standard airborne precautions and send specimens – a serum sample, oral and nasal pharyngeal swabs, and lower respiratory tract specimens if available – to the local health department, which will forward them to the Centers for Disease Control and Prevention (CDC) for testing. Turnaround time is 24-48 hours.
The 2019 Novel Coronavirus (2019-nCoV), identified as the cause of an outbreak of respiratory illness first detected in December in association with a live animal market in Wuhan, China, has been implicated in almost 2,000 cases and 56 deaths in that country. Cases have been reported in 13 countries besides China. Five cases of 2019-nCoV infection have been confirmed in the United States, all in people recently returned from Wuhan. As the virus spreads in China, however, it’s almost certain more cases will show up in the United States. Travel history is key, Dr. Schaffner and others said.
Plan and rehearse
The first step to prepare is to use the CDC’s Interim Guidance for Healthcare Professionals to make a written plan specific to your practice to respond to a potential case. The plan must include notifying the local health department, the CDC liaison for testing, and tracking down patient contacts.
“It’s not good enough to just download CDC’s guidance; use it to make your own local plan and know what to do 24/7,” said Daniel Lucey, MD, an infectious disease expert at Georgetown University Medical Center, Washington, D.C.
“Know who is on call at the health department on weekends and nights,” he said. Know where the patient is going to be isolated; figure out what to do if there’s more than one, and tests come back positive. Have masks on hand, and rehearse the response. “Make a coronavirus team, and absolutely have the nurses involved,” as well as other providers who may come into contact with a case, he added.
“You want to be able to do as well as your counterparts in Washington state and Chicago,” where the first two U.S. cases emerged. “They were prepared. They knew what to do,” Dr. Lucey said.
Those first two U.S. patients – a man in Everett, Wash., and a Chicago woman – developed symptoms after returning from Wuhan, a city of 11 million just over 400 miles inland from the port city of Shanghai. On Jan. 26 three more cases were confirmed by the CDC, two in California and one in Arizona, and each had recently traveled to Wuhan. All five patients remain hospitalized, and there’s no evidence they spread the infection further. There is also no evidence of human-to-human transmission of other cases exported from China to any other countries, according to the WHO.
WHO declined to declare a global health emergency – a Public Health Emergency of International Concern, in its parlance – on Jan. 23. The step would have triggered travel and trade restrictions in member states, including the United States. For now, at least, the group said it wasn’t warranted at this point.
Fatality rates
The focus right now is China. The outbreak has spread beyond Wuhan to other parts of the country, and there’s evidence of fourth-generation spread.
Transportation into and out of Wuhan and other cities has been curtailed, Lunar New Year festivals have been canceled, and the Shanghai Disneyland has been closed, among other measures taken by Chinese officials.
The government could be taking drastic measures in part to prevent the public criticism it took in the early 2000’s for the delayed response and lack of transparency during the global outbreak of another wildlife market coronavirus epidemic, severe acute respiratory syndrome (SARS). In a press conference Jan. 22, WHO officials commended the government’s containment efforts but did not say they recommended them.
According to WHO, serious cases in China have mostly been in people over 40 years old with significant comorbidities and have skewed towards men. Spread seems to be limited to family members, health care providers, and other close contacts, probably by respiratory droplets. If that pattern holds, WHO officials said, the outbreak is containable.
The fatality rate appears to be around 3%, a good deal lower than the 10% reported for SARS and much lower than the nearly 40% reported for Middle East respiratory syndrome (MERS), another recent coronavirus mutation from the animal trade.
The Wuhan virus fatality rate might drop as milder cases are detected and added to the denominator. “It definitely appears to be less severe than SARS and MERS,” said Amesh Adalja, MD, an infectious disease physician in Pittsburgh and emerging infectious disease researcher at Johns Hopkins University, Baltimore.
SARS: Lessons learned
In general, the world is much better equipped for coronavirus outbreaks than when SARS, in particular, emerged in 2003.
WHO officials in their press conference lauded China for it openness with the current outbreak, and for isolating and sequencing the virus immediately, which gave the world a diagnostic test in the first days of the outbreak, something that wasn’t available for SARS. China and other countries also are cooperating and working closely to contain the Wuhan virus.
“What we know today might change tomorrow, so we have to keep tuned in to new information, but we learned a lot from SARS,” Dr. Shaffner said. Overall, it’s likely “the impact on the United States of this new coronavirus is going to be trivial,” he predicted.
Dr. Lucey, however, recalled that the SARS outbreak in Toronto in 2003 started with one missed case. A woman returned asymptomatic from Hong Kong and spread the infection to her family members before she died. Her cause of death wasn’t immediately recognized, nor was the reason her family members were sick, since they hadn’t been to Hong Kong recently.
The infection ultimately spread to more than 200 people, about half of them health care workers. A few people died.
If a virus is sufficiently contagious, “it just takes one. You don’t want to be the one who misses that first patient,” Dr. Lucey said.
Currently, there are no antivirals or vaccines for coronaviruses; researchers are working on both, but for now, care is supportive.
This article was updated with new case numbers on 1/26/20.
What will it take to lower the cost of insulin in the United States?
Mayo Clinic hematologist S. Vincent Rajkumar, MD, argues in a new commentary.
The High Cost of Insulin in the United States: An Urgent Call to Action was published in the January 2020 issue of the Mayo Clinic Proceedings by Dr. Rajkumar, professor of medicine at the Mayo Clinic, Rochester, Minn., who specializes in treating myeloma and, more recently, has become an expert in drug pricing.
He also presented the information in a YouTube video.
As has been widely reported and examined by Congress in the past few years, the cost of insulin in the United States has risen at a far higher rate than inflation. For example, the list price of a single vial of Humalog jumped from $21 in 1999 to $275 in 2019, a far higher price than anywhere else in the world.
Stories of one in four patients having to ration their insulin use because of cost, and of some dying, have fueled protests, leading to legislative efforts and to a few initiatives by some of the manufacturers to address the cost problem.
Collective advocacy
Much of the blame has been placed on the manufacturers for charging such high prices and on the pharmacy benefit managers (PBMs) – also known as “middlemen” – for incentivizing higher-priced products on formularies through rebates. Those are major factors, Dr. Rajkumar argues, but they are not the only ones.
“There is no one reason why this is happening, and no one solution. It’s very complicated. It’s multiple factors all playing together. The only way to tackle it is to really understand it 360,” he said in an interview.
This is true of drug prices overall in the United States, but insulin is a special case. The current analog formulations have not changed in more than 20 years, yet only in the past 5 years have a handful of biosimilar and generic versions started to appear from the same manufacturers as the branded products.
“Insulin is a window into what’s wrong with the pharma industry. ... It’s the best example of how the system is broken,” Dr. Rajkumar stressed.
Physicians can help ease the problem, he said, by becoming educated about drug prices, taking cost into account when prescribing, and routinely discussing prescription drug affordability with patients.
Resources such as www.goodrx.com and www.blinkhealth.com provide information about drug prices and pharmacies that offer drugs at the lowest prices.
“Doctors need to not be suspicious of biosimilars and generics,” he emphasized.
Physicians can also advocate for policies that will lower insulin prices, and their institutions can establish preferences for lower-cost biosimilars in formularies.
“Our individual and collective advocacy gives voice to the needs of our patients,” Dr. Rajkumar emphasized.
‘Everyone in the supply chain benefits’
In his commentary, Dr. Rajkumar lists six major reasons for the high cost of insulin:
1. People with type 1 diabetes are a “vulnerable population” who will die without insulin and are therefore willing to pay a high price to stay alive.
2. Just three manufacturers – Eli Lilly, Novo Nordisk, and Sanofi-Aventis – control nearly the entire insulin market in the United States, with no regulations to cap or control the prices they can charge.
3. The manufacturers continually file new patents for existing insulin products – 70 in the case of Lantus, for example – that provide additional years of monopoly protection from competition.
4. Although the Food and Drug Administration has been receptive to approving insulin biosimilars, it still requires manufacturers to go through a long and cumbersome process to obtain licensure, sometimes taking as long as 10 years.
5.PBMs, paid by insurance companies to negotiate prices with retail pharmacies and pharmaceutical companies (through rebates), stand to benefit from higher, not lower, list prices.
6. Pharmaceutical companies have vast lobbying power.
In regard to the fifth point, about PBMs, Dr. Rajkumar said, “It’s not just the PBMs – it’s the whole supply chain. It’s hard to put a finger on the source of the problem. There’s no transparency in any of the arrangements for you to know why only certain drugs are on a given formulary of an insurance company or PBM. But we do know that, in general, the whole supply chain benefits from the higher list price. Everyone.”
‘Authorized generic’ insulins
That is why Dr. Rajkumar is not convinced that Eli Lilly’s recent launch of half-priced “authorized generic” insulins – first the Lispro injection in March 2019, and then two combination pen products in January 2020 – or Novo Nordisk’s My$99Insulin program and “follow-on” authorized generic versions of Novolog and NovoLog Mix, launched Jan. 2, 2020, will have a huge impact.
“It’s common sense. If Apple made the same iPhone for two different prices, who would pay the full price? It gives you a window into asking what is wrong with the system that allows that? To pay for the higher-priced product, somebody is being paid,” he said.
Indeed, in December 2019, the offices of Sen. Elizabeth Warren (D-MA) and Sen. Richard Blumenthal (D-CT) issued a report from a survey of 400 pharmacies nationwide that found that 83% of the less expensive authorized generic Insulin Lispro was not in stock.
More than two-thirds of pharmacies reported they could not order the product.
“Eli Lilly has failed to take consequential steps – such as simply lowering the list price of Humalog, as it has in foreign markets – to provide lower-cost access to this important diabetes drug,” according to the senators’ report, which concludes by urging Eli Lilly to lower the list price of its insulin and calling for Congress to take steps to enact systemic change to reduce drug prices nationwide.
Asked for comment, Dani Barnhizer, Eli Lilly’s manager of global diabetes communications, said, “Like Senators Warren and Blumenthal, Lilly would like to see even broader use of Insulin Lispro injection because it’s a real solution that can lower copays for people living with diabetes.”
“But the Senators’ paper failed to identify the system challenges that have inhibited access to this lower list price product,” Ms. Barnhizer said. “Payers determine an individual’s premiums and copays, which are subsidized by rebates that pharmaceutical companies pay. And many payers prioritize these rebates to lower premiums instead of offering low copays for chronic medications such as insulin.
“It’s why only one in four people using Medicare Part D, and one in five with commercial plans, have coverage for Insulin Lispro injection. That will not change until payers prioritize providing consistent, affordable insulin copays,” she added.
However, Ms. Barnhizer also noted, “It is not unusual for pharmacies to not stock a medicine. Any pharmacy can place an order for Insulin Lispro injection, with delivery in 1-2 days. All major U.S. wholesalers are now distributing Insulin Lispro injection.”
She also said that people who earn 400% or less of the federal poverty level may be eligible for free insulin, and that Lilly can provide free insulin to anyone in emergency situations. (Novo Nordisk offers that as well.)
Asked why Lilly does not simply lower the price of all their insulins, Ms. Barnhizer responded: “Cutting the list prices would significantly disrupt access to branded insulins, which thousands of insured patients depend on. Launching lower-priced insulin options is a less disruptive approach to help reduce the amount people pay at the pharmacy for people who need the help.”
Government role needed
In addition to the recommendations for physicians and their institutions, Dr. Rajkumar listed several potential policy-level solutions:
1. At the state and federal level, regulations should protect against excessive drug launch prices through the same type of value-based pricing approaches used in other developed countries. “Capping the maximum price increases to the rate of inflation is needed and can happen only through state and/or federal legislation,” he wrote.
Although some may see this as antithetical to “free market” economics, Dr. Rajkumar points out that, in the case of many prescription drugs, including insulin, “we not only have an unregulated monopoly, but it’s a prolonged unregulated monopoly for a lifesaving product, not a luxury item.”
“When you grant monopoly protection and put a barrier on competition, that’s not a free market. People have always had regulations on monopolies. Unregulated monopoly is a recipe for high prices. You have to have some regulation that protects citizens from exploitation.”
And here, he believes, the United States could adopt price negotiations as practiced in Europe and other parts of the world, but which are currently forbidden in the United States.
“Other countries negotiate the price in exchange for monopoly protection. You don’t have to reinvent the wheel,” he said.
2. Reform of the regulatory and legal processes to ease the path for approval of generics and biosimilars to enter the market. This could include reciprocal approval so that biosimilars approved in Canada or the European Union could automatically be granted FDA approval in the United States.
3. Reform of the patent system to prevent overpatenting and patent abuse, by capping patent life to 7-10 years and forbidding use of additional patents as a way of prolonging market exclusivity.
To this point, Barnhizer pointed out that “none of the Lilly insulins are patent protected. Our most commonly used insulin, Humalog U-100, lost patent protection in 2014.”
4. A nongovernmental agency should oversee pricing and make recommendations to Medicare and insurers on the maximum price of new and existing drugs, including insulin. One body poised to do that work is the Institute for Clinical and Economic Review, which receives 77% of its funding from nonprofit foundations. “I have worked with them and they are the best there is,” Dr. Rajkumar said.
5. Any rebates paid by manufacturers to PBMs should be transparent to all stakeholders, including patients.
6. Nonprofit generic manufacturing should be established. The Mayo Clinic has recently partnered with Intermountain Healthcare and several other organizations in creating Civica Rx, a nonprofit generic company.
7. Measures and laws that provide access to insulin in emergency situations are needed, particularly for people with type 1 diabetes.
Recent proposed and enacted legislation has been aimed at some of these goals.
The Insulin Price Reduction Act, introduced in October 2019 and just endorsed by the American Diabetes Association, would reduce insulin costs by providing incentives for manufacturers to revert to the 2006 list price of all insulins.
The Affordable Insulin Approvals Now Act, introduced in July 2019, aims to speed up approvals of generic and biosimilar insulins.
And in May 2019, the state of Colorado passed a bill to cap patient copays for insulin at $100 a month, and similar legislation has been introduced in several other states.
Dr. Rajkumar says that an overall fix will require changes at the federal level.
But, he said, they do not need to happen all at once.
“There are a number of changes that need to happen, but we can do one legislation at a time. ... I’m optimistic that something will happen. This is a national conversation. Both sides of the aisle want to do something about it. People are indeed feeling the pinch, so maybe something will get done,” he said.
Dr. Rajkumar has reported no relevant financial relationships.
This article first appeared on Medscape.com.
Mayo Clinic hematologist S. Vincent Rajkumar, MD, argues in a new commentary.
The High Cost of Insulin in the United States: An Urgent Call to Action was published in the January 2020 issue of the Mayo Clinic Proceedings by Dr. Rajkumar, professor of medicine at the Mayo Clinic, Rochester, Minn., who specializes in treating myeloma and, more recently, has become an expert in drug pricing.
He also presented the information in a YouTube video.
As has been widely reported and examined by Congress in the past few years, the cost of insulin in the United States has risen at a far higher rate than inflation. For example, the list price of a single vial of Humalog jumped from $21 in 1999 to $275 in 2019, a far higher price than anywhere else in the world.
Stories of one in four patients having to ration their insulin use because of cost, and of some dying, have fueled protests, leading to legislative efforts and to a few initiatives by some of the manufacturers to address the cost problem.
Collective advocacy
Much of the blame has been placed on the manufacturers for charging such high prices and on the pharmacy benefit managers (PBMs) – also known as “middlemen” – for incentivizing higher-priced products on formularies through rebates. Those are major factors, Dr. Rajkumar argues, but they are not the only ones.
“There is no one reason why this is happening, and no one solution. It’s very complicated. It’s multiple factors all playing together. The only way to tackle it is to really understand it 360,” he said in an interview.
This is true of drug prices overall in the United States, but insulin is a special case. The current analog formulations have not changed in more than 20 years, yet only in the past 5 years have a handful of biosimilar and generic versions started to appear from the same manufacturers as the branded products.
“Insulin is a window into what’s wrong with the pharma industry. ... It’s the best example of how the system is broken,” Dr. Rajkumar stressed.
Physicians can help ease the problem, he said, by becoming educated about drug prices, taking cost into account when prescribing, and routinely discussing prescription drug affordability with patients.
Resources such as www.goodrx.com and www.blinkhealth.com provide information about drug prices and pharmacies that offer drugs at the lowest prices.
“Doctors need to not be suspicious of biosimilars and generics,” he emphasized.
Physicians can also advocate for policies that will lower insulin prices, and their institutions can establish preferences for lower-cost biosimilars in formularies.
“Our individual and collective advocacy gives voice to the needs of our patients,” Dr. Rajkumar emphasized.
‘Everyone in the supply chain benefits’
In his commentary, Dr. Rajkumar lists six major reasons for the high cost of insulin:
1. People with type 1 diabetes are a “vulnerable population” who will die without insulin and are therefore willing to pay a high price to stay alive.
2. Just three manufacturers – Eli Lilly, Novo Nordisk, and Sanofi-Aventis – control nearly the entire insulin market in the United States, with no regulations to cap or control the prices they can charge.
3. The manufacturers continually file new patents for existing insulin products – 70 in the case of Lantus, for example – that provide additional years of monopoly protection from competition.
4. Although the Food and Drug Administration has been receptive to approving insulin biosimilars, it still requires manufacturers to go through a long and cumbersome process to obtain licensure, sometimes taking as long as 10 years.
5.PBMs, paid by insurance companies to negotiate prices with retail pharmacies and pharmaceutical companies (through rebates), stand to benefit from higher, not lower, list prices.
6. Pharmaceutical companies have vast lobbying power.
In regard to the fifth point, about PBMs, Dr. Rajkumar said, “It’s not just the PBMs – it’s the whole supply chain. It’s hard to put a finger on the source of the problem. There’s no transparency in any of the arrangements for you to know why only certain drugs are on a given formulary of an insurance company or PBM. But we do know that, in general, the whole supply chain benefits from the higher list price. Everyone.”
‘Authorized generic’ insulins
That is why Dr. Rajkumar is not convinced that Eli Lilly’s recent launch of half-priced “authorized generic” insulins – first the Lispro injection in March 2019, and then two combination pen products in January 2020 – or Novo Nordisk’s My$99Insulin program and “follow-on” authorized generic versions of Novolog and NovoLog Mix, launched Jan. 2, 2020, will have a huge impact.
“It’s common sense. If Apple made the same iPhone for two different prices, who would pay the full price? It gives you a window into asking what is wrong with the system that allows that? To pay for the higher-priced product, somebody is being paid,” he said.
Indeed, in December 2019, the offices of Sen. Elizabeth Warren (D-MA) and Sen. Richard Blumenthal (D-CT) issued a report from a survey of 400 pharmacies nationwide that found that 83% of the less expensive authorized generic Insulin Lispro was not in stock.
More than two-thirds of pharmacies reported they could not order the product.
“Eli Lilly has failed to take consequential steps – such as simply lowering the list price of Humalog, as it has in foreign markets – to provide lower-cost access to this important diabetes drug,” according to the senators’ report, which concludes by urging Eli Lilly to lower the list price of its insulin and calling for Congress to take steps to enact systemic change to reduce drug prices nationwide.
Asked for comment, Dani Barnhizer, Eli Lilly’s manager of global diabetes communications, said, “Like Senators Warren and Blumenthal, Lilly would like to see even broader use of Insulin Lispro injection because it’s a real solution that can lower copays for people living with diabetes.”
“But the Senators’ paper failed to identify the system challenges that have inhibited access to this lower list price product,” Ms. Barnhizer said. “Payers determine an individual’s premiums and copays, which are subsidized by rebates that pharmaceutical companies pay. And many payers prioritize these rebates to lower premiums instead of offering low copays for chronic medications such as insulin.
“It’s why only one in four people using Medicare Part D, and one in five with commercial plans, have coverage for Insulin Lispro injection. That will not change until payers prioritize providing consistent, affordable insulin copays,” she added.
However, Ms. Barnhizer also noted, “It is not unusual for pharmacies to not stock a medicine. Any pharmacy can place an order for Insulin Lispro injection, with delivery in 1-2 days. All major U.S. wholesalers are now distributing Insulin Lispro injection.”
She also said that people who earn 400% or less of the federal poverty level may be eligible for free insulin, and that Lilly can provide free insulin to anyone in emergency situations. (Novo Nordisk offers that as well.)
Asked why Lilly does not simply lower the price of all their insulins, Ms. Barnhizer responded: “Cutting the list prices would significantly disrupt access to branded insulins, which thousands of insured patients depend on. Launching lower-priced insulin options is a less disruptive approach to help reduce the amount people pay at the pharmacy for people who need the help.”
Government role needed
In addition to the recommendations for physicians and their institutions, Dr. Rajkumar listed several potential policy-level solutions:
1. At the state and federal level, regulations should protect against excessive drug launch prices through the same type of value-based pricing approaches used in other developed countries. “Capping the maximum price increases to the rate of inflation is needed and can happen only through state and/or federal legislation,” he wrote.
Although some may see this as antithetical to “free market” economics, Dr. Rajkumar points out that, in the case of many prescription drugs, including insulin, “we not only have an unregulated monopoly, but it’s a prolonged unregulated monopoly for a lifesaving product, not a luxury item.”
“When you grant monopoly protection and put a barrier on competition, that’s not a free market. People have always had regulations on monopolies. Unregulated monopoly is a recipe for high prices. You have to have some regulation that protects citizens from exploitation.”
And here, he believes, the United States could adopt price negotiations as practiced in Europe and other parts of the world, but which are currently forbidden in the United States.
“Other countries negotiate the price in exchange for monopoly protection. You don’t have to reinvent the wheel,” he said.
2. Reform of the regulatory and legal processes to ease the path for approval of generics and biosimilars to enter the market. This could include reciprocal approval so that biosimilars approved in Canada or the European Union could automatically be granted FDA approval in the United States.
3. Reform of the patent system to prevent overpatenting and patent abuse, by capping patent life to 7-10 years and forbidding use of additional patents as a way of prolonging market exclusivity.
To this point, Barnhizer pointed out that “none of the Lilly insulins are patent protected. Our most commonly used insulin, Humalog U-100, lost patent protection in 2014.”
4. A nongovernmental agency should oversee pricing and make recommendations to Medicare and insurers on the maximum price of new and existing drugs, including insulin. One body poised to do that work is the Institute for Clinical and Economic Review, which receives 77% of its funding from nonprofit foundations. “I have worked with them and they are the best there is,” Dr. Rajkumar said.
5. Any rebates paid by manufacturers to PBMs should be transparent to all stakeholders, including patients.
6. Nonprofit generic manufacturing should be established. The Mayo Clinic has recently partnered with Intermountain Healthcare and several other organizations in creating Civica Rx, a nonprofit generic company.
7. Measures and laws that provide access to insulin in emergency situations are needed, particularly for people with type 1 diabetes.
Recent proposed and enacted legislation has been aimed at some of these goals.
The Insulin Price Reduction Act, introduced in October 2019 and just endorsed by the American Diabetes Association, would reduce insulin costs by providing incentives for manufacturers to revert to the 2006 list price of all insulins.
The Affordable Insulin Approvals Now Act, introduced in July 2019, aims to speed up approvals of generic and biosimilar insulins.
And in May 2019, the state of Colorado passed a bill to cap patient copays for insulin at $100 a month, and similar legislation has been introduced in several other states.
Dr. Rajkumar says that an overall fix will require changes at the federal level.
But, he said, they do not need to happen all at once.
“There are a number of changes that need to happen, but we can do one legislation at a time. ... I’m optimistic that something will happen. This is a national conversation. Both sides of the aisle want to do something about it. People are indeed feeling the pinch, so maybe something will get done,” he said.
Dr. Rajkumar has reported no relevant financial relationships.
This article first appeared on Medscape.com.
Mayo Clinic hematologist S. Vincent Rajkumar, MD, argues in a new commentary.
The High Cost of Insulin in the United States: An Urgent Call to Action was published in the January 2020 issue of the Mayo Clinic Proceedings by Dr. Rajkumar, professor of medicine at the Mayo Clinic, Rochester, Minn., who specializes in treating myeloma and, more recently, has become an expert in drug pricing.
He also presented the information in a YouTube video.
As has been widely reported and examined by Congress in the past few years, the cost of insulin in the United States has risen at a far higher rate than inflation. For example, the list price of a single vial of Humalog jumped from $21 in 1999 to $275 in 2019, a far higher price than anywhere else in the world.
Stories of one in four patients having to ration their insulin use because of cost, and of some dying, have fueled protests, leading to legislative efforts and to a few initiatives by some of the manufacturers to address the cost problem.
Collective advocacy
Much of the blame has been placed on the manufacturers for charging such high prices and on the pharmacy benefit managers (PBMs) – also known as “middlemen” – for incentivizing higher-priced products on formularies through rebates. Those are major factors, Dr. Rajkumar argues, but they are not the only ones.
“There is no one reason why this is happening, and no one solution. It’s very complicated. It’s multiple factors all playing together. The only way to tackle it is to really understand it 360,” he said in an interview.
This is true of drug prices overall in the United States, but insulin is a special case. The current analog formulations have not changed in more than 20 years, yet only in the past 5 years have a handful of biosimilar and generic versions started to appear from the same manufacturers as the branded products.
“Insulin is a window into what’s wrong with the pharma industry. ... It’s the best example of how the system is broken,” Dr. Rajkumar stressed.
Physicians can help ease the problem, he said, by becoming educated about drug prices, taking cost into account when prescribing, and routinely discussing prescription drug affordability with patients.
Resources such as www.goodrx.com and www.blinkhealth.com provide information about drug prices and pharmacies that offer drugs at the lowest prices.
“Doctors need to not be suspicious of biosimilars and generics,” he emphasized.
Physicians can also advocate for policies that will lower insulin prices, and their institutions can establish preferences for lower-cost biosimilars in formularies.
“Our individual and collective advocacy gives voice to the needs of our patients,” Dr. Rajkumar emphasized.
‘Everyone in the supply chain benefits’
In his commentary, Dr. Rajkumar lists six major reasons for the high cost of insulin:
1. People with type 1 diabetes are a “vulnerable population” who will die without insulin and are therefore willing to pay a high price to stay alive.
2. Just three manufacturers – Eli Lilly, Novo Nordisk, and Sanofi-Aventis – control nearly the entire insulin market in the United States, with no regulations to cap or control the prices they can charge.
3. The manufacturers continually file new patents for existing insulin products – 70 in the case of Lantus, for example – that provide additional years of monopoly protection from competition.
4. Although the Food and Drug Administration has been receptive to approving insulin biosimilars, it still requires manufacturers to go through a long and cumbersome process to obtain licensure, sometimes taking as long as 10 years.
5.PBMs, paid by insurance companies to negotiate prices with retail pharmacies and pharmaceutical companies (through rebates), stand to benefit from higher, not lower, list prices.
6. Pharmaceutical companies have vast lobbying power.
In regard to the fifth point, about PBMs, Dr. Rajkumar said, “It’s not just the PBMs – it’s the whole supply chain. It’s hard to put a finger on the source of the problem. There’s no transparency in any of the arrangements for you to know why only certain drugs are on a given formulary of an insurance company or PBM. But we do know that, in general, the whole supply chain benefits from the higher list price. Everyone.”
‘Authorized generic’ insulins
That is why Dr. Rajkumar is not convinced that Eli Lilly’s recent launch of half-priced “authorized generic” insulins – first the Lispro injection in March 2019, and then two combination pen products in January 2020 – or Novo Nordisk’s My$99Insulin program and “follow-on” authorized generic versions of Novolog and NovoLog Mix, launched Jan. 2, 2020, will have a huge impact.
“It’s common sense. If Apple made the same iPhone for two different prices, who would pay the full price? It gives you a window into asking what is wrong with the system that allows that? To pay for the higher-priced product, somebody is being paid,” he said.
Indeed, in December 2019, the offices of Sen. Elizabeth Warren (D-MA) and Sen. Richard Blumenthal (D-CT) issued a report from a survey of 400 pharmacies nationwide that found that 83% of the less expensive authorized generic Insulin Lispro was not in stock.
More than two-thirds of pharmacies reported they could not order the product.
“Eli Lilly has failed to take consequential steps – such as simply lowering the list price of Humalog, as it has in foreign markets – to provide lower-cost access to this important diabetes drug,” according to the senators’ report, which concludes by urging Eli Lilly to lower the list price of its insulin and calling for Congress to take steps to enact systemic change to reduce drug prices nationwide.
Asked for comment, Dani Barnhizer, Eli Lilly’s manager of global diabetes communications, said, “Like Senators Warren and Blumenthal, Lilly would like to see even broader use of Insulin Lispro injection because it’s a real solution that can lower copays for people living with diabetes.”
“But the Senators’ paper failed to identify the system challenges that have inhibited access to this lower list price product,” Ms. Barnhizer said. “Payers determine an individual’s premiums and copays, which are subsidized by rebates that pharmaceutical companies pay. And many payers prioritize these rebates to lower premiums instead of offering low copays for chronic medications such as insulin.
“It’s why only one in four people using Medicare Part D, and one in five with commercial plans, have coverage for Insulin Lispro injection. That will not change until payers prioritize providing consistent, affordable insulin copays,” she added.
However, Ms. Barnhizer also noted, “It is not unusual for pharmacies to not stock a medicine. Any pharmacy can place an order for Insulin Lispro injection, with delivery in 1-2 days. All major U.S. wholesalers are now distributing Insulin Lispro injection.”
She also said that people who earn 400% or less of the federal poverty level may be eligible for free insulin, and that Lilly can provide free insulin to anyone in emergency situations. (Novo Nordisk offers that as well.)
Asked why Lilly does not simply lower the price of all their insulins, Ms. Barnhizer responded: “Cutting the list prices would significantly disrupt access to branded insulins, which thousands of insured patients depend on. Launching lower-priced insulin options is a less disruptive approach to help reduce the amount people pay at the pharmacy for people who need the help.”
Government role needed
In addition to the recommendations for physicians and their institutions, Dr. Rajkumar listed several potential policy-level solutions:
1. At the state and federal level, regulations should protect against excessive drug launch prices through the same type of value-based pricing approaches used in other developed countries. “Capping the maximum price increases to the rate of inflation is needed and can happen only through state and/or federal legislation,” he wrote.
Although some may see this as antithetical to “free market” economics, Dr. Rajkumar points out that, in the case of many prescription drugs, including insulin, “we not only have an unregulated monopoly, but it’s a prolonged unregulated monopoly for a lifesaving product, not a luxury item.”
“When you grant monopoly protection and put a barrier on competition, that’s not a free market. People have always had regulations on monopolies. Unregulated monopoly is a recipe for high prices. You have to have some regulation that protects citizens from exploitation.”
And here, he believes, the United States could adopt price negotiations as practiced in Europe and other parts of the world, but which are currently forbidden in the United States.
“Other countries negotiate the price in exchange for monopoly protection. You don’t have to reinvent the wheel,” he said.
2. Reform of the regulatory and legal processes to ease the path for approval of generics and biosimilars to enter the market. This could include reciprocal approval so that biosimilars approved in Canada or the European Union could automatically be granted FDA approval in the United States.
3. Reform of the patent system to prevent overpatenting and patent abuse, by capping patent life to 7-10 years and forbidding use of additional patents as a way of prolonging market exclusivity.
To this point, Barnhizer pointed out that “none of the Lilly insulins are patent protected. Our most commonly used insulin, Humalog U-100, lost patent protection in 2014.”
4. A nongovernmental agency should oversee pricing and make recommendations to Medicare and insurers on the maximum price of new and existing drugs, including insulin. One body poised to do that work is the Institute for Clinical and Economic Review, which receives 77% of its funding from nonprofit foundations. “I have worked with them and they are the best there is,” Dr. Rajkumar said.
5. Any rebates paid by manufacturers to PBMs should be transparent to all stakeholders, including patients.
6. Nonprofit generic manufacturing should be established. The Mayo Clinic has recently partnered with Intermountain Healthcare and several other organizations in creating Civica Rx, a nonprofit generic company.
7. Measures and laws that provide access to insulin in emergency situations are needed, particularly for people with type 1 diabetes.
Recent proposed and enacted legislation has been aimed at some of these goals.
The Insulin Price Reduction Act, introduced in October 2019 and just endorsed by the American Diabetes Association, would reduce insulin costs by providing incentives for manufacturers to revert to the 2006 list price of all insulins.
The Affordable Insulin Approvals Now Act, introduced in July 2019, aims to speed up approvals of generic and biosimilar insulins.
And in May 2019, the state of Colorado passed a bill to cap patient copays for insulin at $100 a month, and similar legislation has been introduced in several other states.
Dr. Rajkumar says that an overall fix will require changes at the federal level.
But, he said, they do not need to happen all at once.
“There are a number of changes that need to happen, but we can do one legislation at a time. ... I’m optimistic that something will happen. This is a national conversation. Both sides of the aisle want to do something about it. People are indeed feeling the pinch, so maybe something will get done,” he said.
Dr. Rajkumar has reported no relevant financial relationships.
This article first appeared on Medscape.com.
FDA advisers set high bar for new opioids
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?

On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?
On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?
On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
The rise of U.S. dermatology: A brief history from the 1800s to 1970
As Dermatology News (formerly Skin and Allergy News) reaches its 50th-year milestone, a reflection on the history of the discipline, especially in the United States, up to the time of the launch of this publication is in order. Such an overview must, of course, be cursory in this context. Yet, for those who want to learn more, a large body of historical references and research has been created to fill in the gaps, as modern dermatology has always been cognizant of the importance of its history, with many individuals and groups drawn to the subject.

Two excellent sources for the history of the field can be found in work by William Allen Pusey, MD (1865-1940), and Herbert Rattner, MD (1900-1962), “The History of Dermatology” published in 1933 and research by members of the History of Dermatology Society, founded in 1973 in New York.
Modern dermatology
The development of the field of modern dermatology can be traced back to the early to mid-19th century. During the first half of the 19th century, England and France dominated the study of dermatology, but by the middle of the century, the German revolution in microparasitology shifted that focus “with remarkable German discoveries,” according to Bernard S. Potter, MD, in his review of bibliographic landmarks of the history of dermatology (J Am Acad Dermatol 2003;48:919-32). For example, Johann Lucas Schoenlein (1793-1864) in 1839 discovered the fungal origin of favus, and in 1841 Jacob Henle (1809-1885) discovered Demodex folliculorum. Karl Ferdinand Eichstedt (1816-1892) in 1846 followed with the discovery of the causative agent of pityriasis versicolor, and Friedrich Wilhelm Felix von Barensprung (1822-1864) in 1862 coined the term erythrasma and named the organism responsible for this condition Microsporum minutissimum.
Dr. Potter described how American dermatology originated in New York City in 1836 when Henry Daggett Bulkley, MD, (1803-1872) opened the first dispensary for skin diseases, the Broome Street Infirmary for Diseases of the Skin, thus creating the first institution in the United States for the treatment of cutaneous disease. As the first American dermatologist, he was also the first in the United States to lecture on and to exclusively practice dermatology.
The rise of interest in the importance of dermatology led to the organization of the early American Dermatological Association in 1886.
However, the state of dermatology as a science in the 19th century was not always looked upon favorably, even by its practitioners, especially in the United States. In 1871, in a “Review on Modern Dermatology,” given as a series of lectures on skin disease at Harvard University, James C. White, MD (1833-1916) of Massachusetts General Hospital, stated that: “Were the literature of skin diseases previous to that of the last half-century absolutely annihilated, and with it, the influence it has exercised upon that of the present day, it would be an immense gain to dermatology, although much of real value would perish.” He lamented that America had contributed little so far to the study of dermatology, and that the discipline was only taught in some of its largest schools, and he urged that this be changed. He also lamented that The American Journal of Syphilography and Dermatology, established the year before, had so far proved itself heavy on syphilis, but light on dermatology, a situation he also hoped would change dramatically.
By the late-19th century, the conviction that diseases of the skin needed to be connected to the overall metabolism and physiology of the patient as a whole was becoming more mainstream.
“It has been, and still is, too much the custom to study diseases of the skin in the light of pathological pictures, to name the local manifestation and to so label it as disease. It is much easier to give the disease name and to label it than it is to comprehend the process at work. The former is comparatively unimportant for the patient, the latter point upon which recovery may depend. The nature and meaning of the process in connection with the cutaneous symptoms has not received enough attention, and I believe this to be one reason why the treatment of many of these diseases in the past has been so notoriously unsatisfactory,” Louis A. Duhring, MD (1845-1913) chided his colleagues in the Section of Dermatology and Syphilography, at the Forty-fourth Annual Meeting of the American Medical Association in 1894. (collections.nlm.nih.gov/ext/dw/101489447/PDF/101489447.pdf)
In the early-20th century, German dermatology influenced American dermatology more than any other, according to Karl Holubar, MD, of the Institute for the History of Medicine, University of Vienna, in his lecture on the history of European dermatopathology.
He stated that, with regard to dermatopathology, it was Oscar Gans, MD (1888-1983) who brought the latest knowledge into the United States by delivering a series of lectures at Mayo Clinic in the late 1920s upon the invitation of Paul A. O’Leary, MD, (1891-1955) who then headed the Mayo section of dermatology.
By the 1930s, a flurry of organizational activity overtook American dermatology. In 1932, the American Board of Dermatology was established, with its first exams given in 1933 (20 students passed, 7 failed). The Society for Investigative Dermatology was founded in 1937, and the American Academy of Dermatology and Syphilology (now the American Academy of Dermatology), founded in 1938.
The 1930s also saw a major influx of German and other European Jews fleeing Nazi oppression who would forever change the face of American dermatology. “Between 1933 and 1938, a series of repressive measures eliminated them from the practice of medicine in Germany and other countries. Although some died in concentration camps and others committed suicide, many were able to emigrate from Europe. Dermatology in the United States particularly benefited from the influx of several stellar Jewish dermatologists who were major contributors to the subsequent flowering of academic dermatology in the United States” (JAMA Derm. 2013;149[9]:1090-4).
“The overtures of the holocaust and the rising power of Hitler in Europe finally brought over to the United States the flower of dermatologists and investigators of the German School, e.g., Alexander and Walter Lever, Felix and Hermann Pinkus, the Epsteins, Erich Auerbach, Stephen Rothman, to name just a few. With this exodus and transfer of brain power, Europe lost its leading role to never again regain it,” according to Dr. Holubar. Walter F. Lever, MD (1909-1992) was especially well-known for his landmark textbook on dermatology, “Histopathology of the Skin,” published in the United States in 1949.
The therapeutic era
Throughout the 19th century, a variety of soaps and patent medicines were touted as cure-alls for a host of skin diseases. Other than their benefits to surface cleanliness and their antiseptic properties, however, they were of little effect.
It wasn’t until the 20th century that truly effective therapeutics entered the dermatologic pharmacopoeia. In their 1989 review, Diane Quintal, MD, and Robert Jackson, MD, discussed the origins of the most important of these drugs and pointed out that, “Until this century, the essence of dermatology resided in the realm of morphology. Early contributors largely confined their activities to the classification of skin diseases and to the elaboration of clinical dermatologic entities based on morphologic features. ... but “in the last 50 years, there have been significant scientific discoveries in the field of therapeutics that have revolutionized the practice of dermatology.“ (Clin Dermatol. 1989;7[3]38-47).
These key drugs comprised:
- Quinacrine was introduced in 1932 by Walter Kikuth, MD, as an antimalarial drug. But it was not until 1940, that A.J. Prokoptchouksi, MD, reported on its effectiveness 35 patients with lupus erythematosus.
- Para-aminobenzoic acid (PABA) came into prominence in 1942, when Stephen Rothman, MD, and Jack Rubin, MD, at the University of Chicago, published the results of their experiment, showing that when PABA was incorporated in an ointment base and applied to the skin, it could protect against sunburn.
- Dapsone. The effectiveness of sulfapyridine was demonstrated in 1947 by M.J. Costello, MD, who reported its usefulness in a patient with dermatitis herpetiformis, which he believed to be caused by a bacterial allergy. Sulfapyridine controlled the disease, but gastrointestinal intolerance and sulfonamide sensitivity were side effects. Ultimately, in 1951, Theodore Cornbleet, MD, introduced the use of sulfoxones in his article entitled “Sulfoxone (diasones) sodium for dermatitis herpetiformis,” considered more effective than sulfapyridine. Dapsone is the active principal ingredient.
- Hydrocortisone. In August 1952, Marion Sulzberger, MD, and Victor H. Witten, MD (both members of the first Skin & Allergy News editorial advisory board), described use of Compound F (17-hydroxycorticosterone-21-acetate, hydrocortisone) in seven cases of atopic dermatitis and one case of discoid or subacute lupus erythematosus, reporting improvement in all of these cases.
- Benzoyl peroxide. Canadian dermatologist William E. Pace, MD, reported on the beneficial effects of benzoyl peroxide on acne in 1953. The product had originally been used for chronic Staphylococcus aureus folliculitis of the beard.
- Griseofulvin, a metabolic byproduct of a number of species of Penicillium, was first isolated in 1939. But in 1958, Harvey Blank, MD, at the University of Miami (also on the first Skin & Allergy News editorial advisory board), and Stanley I. Cullen, MD, administered the drug to a patient with Trichophyton rubrum granuloma, in the first human trial. In 1959, they reported the drug’s benefits on 31 patients with various fungal infections.
- Methotrexate. In 1951, R. Gubner, MD, and colleagues noticed the rapid clearing of skin lesions in a patient with rheumatoid arthritis who had been treated with the folic acid antagonist, aminopterin. And in 1958, W.F. Edmundson, MD, and W.B. Guy, MD, reported on the oral use of the folic acid antagonist, methotrexate. This was followed by multiple reports on the successful use of methotrexate in psoriasis.
- 5-fluorouracil (5-FU). In 1957, 5-FU, an antimetabolite of uracil, was first synthesized. In 1962, G. Falkson, MD, and E.J. Schulz, MD, reported on skin changes observed in 85 patients being treated with systemic 5-FU for advanced carcinomatosis. They found that 31 of the 85 patients developed sensitivity to sunlight and subsequent disappearance of actinic keratoses in these same patients.

Technology in skin care also was developing in the era just before the launch of Skin & Allergy News. For example, Leon Goldman, MD, then chairman of the department of dermatology at the University of Cincinnati, was the first physician to use a laser for tattoo removal. His publication in 1965 helped to solidify its use, leading him to be “regarded by many in the dermatologic community as the ‘godfather of lasers in medicine and surgery’ ” (Clin Dermatol. 2007;25:434-42).
So, by 1970, dermatology as a field had established itself fully with a strong societal infrastructure, a vibrant base of journals and books, and an evolving set of scientific and technical tools. The launch of Skin & Allergy News (now Dermatology News) that year would chronicle dermatology’s commitment to the development of new therapeutics and technologies in service of patient needs – the stories of which would grace the newspaper’s pages for 5 decades and counting.
As Dermatology News (formerly Skin and Allergy News) reaches its 50th-year milestone, a reflection on the history of the discipline, especially in the United States, up to the time of the launch of this publication is in order. Such an overview must, of course, be cursory in this context. Yet, for those who want to learn more, a large body of historical references and research has been created to fill in the gaps, as modern dermatology has always been cognizant of the importance of its history, with many individuals and groups drawn to the subject.
Two excellent sources for the history of the field can be found in work by William Allen Pusey, MD (1865-1940), and Herbert Rattner, MD (1900-1962), “The History of Dermatology” published in 1933 and research by members of the History of Dermatology Society, founded in 1973 in New York.
Modern dermatology
The development of the field of modern dermatology can be traced back to the early to mid-19th century. During the first half of the 19th century, England and France dominated the study of dermatology, but by the middle of the century, the German revolution in microparasitology shifted that focus “with remarkable German discoveries,” according to Bernard S. Potter, MD, in his review of bibliographic landmarks of the history of dermatology (J Am Acad Dermatol 2003;48:919-32). For example, Johann Lucas Schoenlein (1793-1864) in 1839 discovered the fungal origin of favus, and in 1841 Jacob Henle (1809-1885) discovered Demodex folliculorum. Karl Ferdinand Eichstedt (1816-1892) in 1846 followed with the discovery of the causative agent of pityriasis versicolor, and Friedrich Wilhelm Felix von Barensprung (1822-1864) in 1862 coined the term erythrasma and named the organism responsible for this condition Microsporum minutissimum.
Dr. Potter described how American dermatology originated in New York City in 1836 when Henry Daggett Bulkley, MD, (1803-1872) opened the first dispensary for skin diseases, the Broome Street Infirmary for Diseases of the Skin, thus creating the first institution in the United States for the treatment of cutaneous disease. As the first American dermatologist, he was also the first in the United States to lecture on and to exclusively practice dermatology.
The rise of interest in the importance of dermatology led to the organization of the early American Dermatological Association in 1886.
However, the state of dermatology as a science in the 19th century was not always looked upon favorably, even by its practitioners, especially in the United States. In 1871, in a “Review on Modern Dermatology,” given as a series of lectures on skin disease at Harvard University, James C. White, MD (1833-1916) of Massachusetts General Hospital, stated that: “Were the literature of skin diseases previous to that of the last half-century absolutely annihilated, and with it, the influence it has exercised upon that of the present day, it would be an immense gain to dermatology, although much of real value would perish.” He lamented that America had contributed little so far to the study of dermatology, and that the discipline was only taught in some of its largest schools, and he urged that this be changed. He also lamented that The American Journal of Syphilography and Dermatology, established the year before, had so far proved itself heavy on syphilis, but light on dermatology, a situation he also hoped would change dramatically.
By the late-19th century, the conviction that diseases of the skin needed to be connected to the overall metabolism and physiology of the patient as a whole was becoming more mainstream.
“It has been, and still is, too much the custom to study diseases of the skin in the light of pathological pictures, to name the local manifestation and to so label it as disease. It is much easier to give the disease name and to label it than it is to comprehend the process at work. The former is comparatively unimportant for the patient, the latter point upon which recovery may depend. The nature and meaning of the process in connection with the cutaneous symptoms has not received enough attention, and I believe this to be one reason why the treatment of many of these diseases in the past has been so notoriously unsatisfactory,” Louis A. Duhring, MD (1845-1913) chided his colleagues in the Section of Dermatology and Syphilography, at the Forty-fourth Annual Meeting of the American Medical Association in 1894. (collections.nlm.nih.gov/ext/dw/101489447/PDF/101489447.pdf)
In the early-20th century, German dermatology influenced American dermatology more than any other, according to Karl Holubar, MD, of the Institute for the History of Medicine, University of Vienna, in his lecture on the history of European dermatopathology.
He stated that, with regard to dermatopathology, it was Oscar Gans, MD (1888-1983) who brought the latest knowledge into the United States by delivering a series of lectures at Mayo Clinic in the late 1920s upon the invitation of Paul A. O’Leary, MD, (1891-1955) who then headed the Mayo section of dermatology.
By the 1930s, a flurry of organizational activity overtook American dermatology. In 1932, the American Board of Dermatology was established, with its first exams given in 1933 (20 students passed, 7 failed). The Society for Investigative Dermatology was founded in 1937, and the American Academy of Dermatology and Syphilology (now the American Academy of Dermatology), founded in 1938.
The 1930s also saw a major influx of German and other European Jews fleeing Nazi oppression who would forever change the face of American dermatology. “Between 1933 and 1938, a series of repressive measures eliminated them from the practice of medicine in Germany and other countries. Although some died in concentration camps and others committed suicide, many were able to emigrate from Europe. Dermatology in the United States particularly benefited from the influx of several stellar Jewish dermatologists who were major contributors to the subsequent flowering of academic dermatology in the United States” (JAMA Derm. 2013;149[9]:1090-4).
“The overtures of the holocaust and the rising power of Hitler in Europe finally brought over to the United States the flower of dermatologists and investigators of the German School, e.g., Alexander and Walter Lever, Felix and Hermann Pinkus, the Epsteins, Erich Auerbach, Stephen Rothman, to name just a few. With this exodus and transfer of brain power, Europe lost its leading role to never again regain it,” according to Dr. Holubar. Walter F. Lever, MD (1909-1992) was especially well-known for his landmark textbook on dermatology, “Histopathology of the Skin,” published in the United States in 1949.
The therapeutic era
Throughout the 19th century, a variety of soaps and patent medicines were touted as cure-alls for a host of skin diseases. Other than their benefits to surface cleanliness and their antiseptic properties, however, they were of little effect.
It wasn’t until the 20th century that truly effective therapeutics entered the dermatologic pharmacopoeia. In their 1989 review, Diane Quintal, MD, and Robert Jackson, MD, discussed the origins of the most important of these drugs and pointed out that, “Until this century, the essence of dermatology resided in the realm of morphology. Early contributors largely confined their activities to the classification of skin diseases and to the elaboration of clinical dermatologic entities based on morphologic features. ... but “in the last 50 years, there have been significant scientific discoveries in the field of therapeutics that have revolutionized the practice of dermatology.“ (Clin Dermatol. 1989;7[3]38-47).
These key drugs comprised:
- Quinacrine was introduced in 1932 by Walter Kikuth, MD, as an antimalarial drug. But it was not until 1940, that A.J. Prokoptchouksi, MD, reported on its effectiveness 35 patients with lupus erythematosus.
- Para-aminobenzoic acid (PABA) came into prominence in 1942, when Stephen Rothman, MD, and Jack Rubin, MD, at the University of Chicago, published the results of their experiment, showing that when PABA was incorporated in an ointment base and applied to the skin, it could protect against sunburn.
- Dapsone. The effectiveness of sulfapyridine was demonstrated in 1947 by M.J. Costello, MD, who reported its usefulness in a patient with dermatitis herpetiformis, which he believed to be caused by a bacterial allergy. Sulfapyridine controlled the disease, but gastrointestinal intolerance and sulfonamide sensitivity were side effects. Ultimately, in 1951, Theodore Cornbleet, MD, introduced the use of sulfoxones in his article entitled “Sulfoxone (diasones) sodium for dermatitis herpetiformis,” considered more effective than sulfapyridine. Dapsone is the active principal ingredient.
- Hydrocortisone. In August 1952, Marion Sulzberger, MD, and Victor H. Witten, MD (both members of the first Skin & Allergy News editorial advisory board), described use of Compound F (17-hydroxycorticosterone-21-acetate, hydrocortisone) in seven cases of atopic dermatitis and one case of discoid or subacute lupus erythematosus, reporting improvement in all of these cases.
- Benzoyl peroxide. Canadian dermatologist William E. Pace, MD, reported on the beneficial effects of benzoyl peroxide on acne in 1953. The product had originally been used for chronic Staphylococcus aureus folliculitis of the beard.
- Griseofulvin, a metabolic byproduct of a number of species of Penicillium, was first isolated in 1939. But in 1958, Harvey Blank, MD, at the University of Miami (also on the first Skin & Allergy News editorial advisory board), and Stanley I. Cullen, MD, administered the drug to a patient with Trichophyton rubrum granuloma, in the first human trial. In 1959, they reported the drug’s benefits on 31 patients with various fungal infections.
- Methotrexate. In 1951, R. Gubner, MD, and colleagues noticed the rapid clearing of skin lesions in a patient with rheumatoid arthritis who had been treated with the folic acid antagonist, aminopterin. And in 1958, W.F. Edmundson, MD, and W.B. Guy, MD, reported on the oral use of the folic acid antagonist, methotrexate. This was followed by multiple reports on the successful use of methotrexate in psoriasis.
- 5-fluorouracil (5-FU). In 1957, 5-FU, an antimetabolite of uracil, was first synthesized. In 1962, G. Falkson, MD, and E.J. Schulz, MD, reported on skin changes observed in 85 patients being treated with systemic 5-FU for advanced carcinomatosis. They found that 31 of the 85 patients developed sensitivity to sunlight and subsequent disappearance of actinic keratoses in these same patients.
Technology in skin care also was developing in the era just before the launch of Skin & Allergy News. For example, Leon Goldman, MD, then chairman of the department of dermatology at the University of Cincinnati, was the first physician to use a laser for tattoo removal. His publication in 1965 helped to solidify its use, leading him to be “regarded by many in the dermatologic community as the ‘godfather of lasers in medicine and surgery’ ” (Clin Dermatol. 2007;25:434-42).
So, by 1970, dermatology as a field had established itself fully with a strong societal infrastructure, a vibrant base of journals and books, and an evolving set of scientific and technical tools. The launch of Skin & Allergy News (now Dermatology News) that year would chronicle dermatology’s commitment to the development of new therapeutics and technologies in service of patient needs – the stories of which would grace the newspaper’s pages for 5 decades and counting.
As Dermatology News (formerly Skin and Allergy News) reaches its 50th-year milestone, a reflection on the history of the discipline, especially in the United States, up to the time of the launch of this publication is in order. Such an overview must, of course, be cursory in this context. Yet, for those who want to learn more, a large body of historical references and research has been created to fill in the gaps, as modern dermatology has always been cognizant of the importance of its history, with many individuals and groups drawn to the subject.
Two excellent sources for the history of the field can be found in work by William Allen Pusey, MD (1865-1940), and Herbert Rattner, MD (1900-1962), “The History of Dermatology” published in 1933 and research by members of the History of Dermatology Society, founded in 1973 in New York.
Modern dermatology
The development of the field of modern dermatology can be traced back to the early to mid-19th century. During the first half of the 19th century, England and France dominated the study of dermatology, but by the middle of the century, the German revolution in microparasitology shifted that focus “with remarkable German discoveries,” according to Bernard S. Potter, MD, in his review of bibliographic landmarks of the history of dermatology (J Am Acad Dermatol 2003;48:919-32). For example, Johann Lucas Schoenlein (1793-1864) in 1839 discovered the fungal origin of favus, and in 1841 Jacob Henle (1809-1885) discovered Demodex folliculorum. Karl Ferdinand Eichstedt (1816-1892) in 1846 followed with the discovery of the causative agent of pityriasis versicolor, and Friedrich Wilhelm Felix von Barensprung (1822-1864) in 1862 coined the term erythrasma and named the organism responsible for this condition Microsporum minutissimum.
Dr. Potter described how American dermatology originated in New York City in 1836 when Henry Daggett Bulkley, MD, (1803-1872) opened the first dispensary for skin diseases, the Broome Street Infirmary for Diseases of the Skin, thus creating the first institution in the United States for the treatment of cutaneous disease. As the first American dermatologist, he was also the first in the United States to lecture on and to exclusively practice dermatology.
The rise of interest in the importance of dermatology led to the organization of the early American Dermatological Association in 1886.
However, the state of dermatology as a science in the 19th century was not always looked upon favorably, even by its practitioners, especially in the United States. In 1871, in a “Review on Modern Dermatology,” given as a series of lectures on skin disease at Harvard University, James C. White, MD (1833-1916) of Massachusetts General Hospital, stated that: “Were the literature of skin diseases previous to that of the last half-century absolutely annihilated, and with it, the influence it has exercised upon that of the present day, it would be an immense gain to dermatology, although much of real value would perish.” He lamented that America had contributed little so far to the study of dermatology, and that the discipline was only taught in some of its largest schools, and he urged that this be changed. He also lamented that The American Journal of Syphilography and Dermatology, established the year before, had so far proved itself heavy on syphilis, but light on dermatology, a situation he also hoped would change dramatically.
By the late-19th century, the conviction that diseases of the skin needed to be connected to the overall metabolism and physiology of the patient as a whole was becoming more mainstream.
“It has been, and still is, too much the custom to study diseases of the skin in the light of pathological pictures, to name the local manifestation and to so label it as disease. It is much easier to give the disease name and to label it than it is to comprehend the process at work. The former is comparatively unimportant for the patient, the latter point upon which recovery may depend. The nature and meaning of the process in connection with the cutaneous symptoms has not received enough attention, and I believe this to be one reason why the treatment of many of these diseases in the past has been so notoriously unsatisfactory,” Louis A. Duhring, MD (1845-1913) chided his colleagues in the Section of Dermatology and Syphilography, at the Forty-fourth Annual Meeting of the American Medical Association in 1894. (collections.nlm.nih.gov/ext/dw/101489447/PDF/101489447.pdf)
In the early-20th century, German dermatology influenced American dermatology more than any other, according to Karl Holubar, MD, of the Institute for the History of Medicine, University of Vienna, in his lecture on the history of European dermatopathology.
He stated that, with regard to dermatopathology, it was Oscar Gans, MD (1888-1983) who brought the latest knowledge into the United States by delivering a series of lectures at Mayo Clinic in the late 1920s upon the invitation of Paul A. O’Leary, MD, (1891-1955) who then headed the Mayo section of dermatology.
By the 1930s, a flurry of organizational activity overtook American dermatology. In 1932, the American Board of Dermatology was established, with its first exams given in 1933 (20 students passed, 7 failed). The Society for Investigative Dermatology was founded in 1937, and the American Academy of Dermatology and Syphilology (now the American Academy of Dermatology), founded in 1938.
The 1930s also saw a major influx of German and other European Jews fleeing Nazi oppression who would forever change the face of American dermatology. “Between 1933 and 1938, a series of repressive measures eliminated them from the practice of medicine in Germany and other countries. Although some died in concentration camps and others committed suicide, many were able to emigrate from Europe. Dermatology in the United States particularly benefited from the influx of several stellar Jewish dermatologists who were major contributors to the subsequent flowering of academic dermatology in the United States” (JAMA Derm. 2013;149[9]:1090-4).
“The overtures of the holocaust and the rising power of Hitler in Europe finally brought over to the United States the flower of dermatologists and investigators of the German School, e.g., Alexander and Walter Lever, Felix and Hermann Pinkus, the Epsteins, Erich Auerbach, Stephen Rothman, to name just a few. With this exodus and transfer of brain power, Europe lost its leading role to never again regain it,” according to Dr. Holubar. Walter F. Lever, MD (1909-1992) was especially well-known for his landmark textbook on dermatology, “Histopathology of the Skin,” published in the United States in 1949.
The therapeutic era
Throughout the 19th century, a variety of soaps and patent medicines were touted as cure-alls for a host of skin diseases. Other than their benefits to surface cleanliness and their antiseptic properties, however, they were of little effect.
It wasn’t until the 20th century that truly effective therapeutics entered the dermatologic pharmacopoeia. In their 1989 review, Diane Quintal, MD, and Robert Jackson, MD, discussed the origins of the most important of these drugs and pointed out that, “Until this century, the essence of dermatology resided in the realm of morphology. Early contributors largely confined their activities to the classification of skin diseases and to the elaboration of clinical dermatologic entities based on morphologic features. ... but “in the last 50 years, there have been significant scientific discoveries in the field of therapeutics that have revolutionized the practice of dermatology.“ (Clin Dermatol. 1989;7[3]38-47).
These key drugs comprised:
- Quinacrine was introduced in 1932 by Walter Kikuth, MD, as an antimalarial drug. But it was not until 1940, that A.J. Prokoptchouksi, MD, reported on its effectiveness 35 patients with lupus erythematosus.
- Para-aminobenzoic acid (PABA) came into prominence in 1942, when Stephen Rothman, MD, and Jack Rubin, MD, at the University of Chicago, published the results of their experiment, showing that when PABA was incorporated in an ointment base and applied to the skin, it could protect against sunburn.
- Dapsone. The effectiveness of sulfapyridine was demonstrated in 1947 by M.J. Costello, MD, who reported its usefulness in a patient with dermatitis herpetiformis, which he believed to be caused by a bacterial allergy. Sulfapyridine controlled the disease, but gastrointestinal intolerance and sulfonamide sensitivity were side effects. Ultimately, in 1951, Theodore Cornbleet, MD, introduced the use of sulfoxones in his article entitled “Sulfoxone (diasones) sodium for dermatitis herpetiformis,” considered more effective than sulfapyridine. Dapsone is the active principal ingredient.
- Hydrocortisone. In August 1952, Marion Sulzberger, MD, and Victor H. Witten, MD (both members of the first Skin & Allergy News editorial advisory board), described use of Compound F (17-hydroxycorticosterone-21-acetate, hydrocortisone) in seven cases of atopic dermatitis and one case of discoid or subacute lupus erythematosus, reporting improvement in all of these cases.
- Benzoyl peroxide. Canadian dermatologist William E. Pace, MD, reported on the beneficial effects of benzoyl peroxide on acne in 1953. The product had originally been used for chronic Staphylococcus aureus folliculitis of the beard.
- Griseofulvin, a metabolic byproduct of a number of species of Penicillium, was first isolated in 1939. But in 1958, Harvey Blank, MD, at the University of Miami (also on the first Skin & Allergy News editorial advisory board), and Stanley I. Cullen, MD, administered the drug to a patient with Trichophyton rubrum granuloma, in the first human trial. In 1959, they reported the drug’s benefits on 31 patients with various fungal infections.
- Methotrexate. In 1951, R. Gubner, MD, and colleagues noticed the rapid clearing of skin lesions in a patient with rheumatoid arthritis who had been treated with the folic acid antagonist, aminopterin. And in 1958, W.F. Edmundson, MD, and W.B. Guy, MD, reported on the oral use of the folic acid antagonist, methotrexate. This was followed by multiple reports on the successful use of methotrexate in psoriasis.
- 5-fluorouracil (5-FU). In 1957, 5-FU, an antimetabolite of uracil, was first synthesized. In 1962, G. Falkson, MD, and E.J. Schulz, MD, reported on skin changes observed in 85 patients being treated with systemic 5-FU for advanced carcinomatosis. They found that 31 of the 85 patients developed sensitivity to sunlight and subsequent disappearance of actinic keratoses in these same patients.
Technology in skin care also was developing in the era just before the launch of Skin & Allergy News. For example, Leon Goldman, MD, then chairman of the department of dermatology at the University of Cincinnati, was the first physician to use a laser for tattoo removal. His publication in 1965 helped to solidify its use, leading him to be “regarded by many in the dermatologic community as the ‘godfather of lasers in medicine and surgery’ ” (Clin Dermatol. 2007;25:434-42).
So, by 1970, dermatology as a field had established itself fully with a strong societal infrastructure, a vibrant base of journals and books, and an evolving set of scientific and technical tools. The launch of Skin & Allergy News (now Dermatology News) that year would chronicle dermatology’s commitment to the development of new therapeutics and technologies in service of patient needs – the stories of which would grace the newspaper’s pages for 5 decades and counting.
ACP maps two potential paths to universal health care
The American College of Physicians is recommending either a single-payer system or a public option within a regulated private insurance system to help deliver universal and affordable access to health care for all Americans.
“We came to the conclusion that two directions or approaches could get us to where we need to be,” ACP President Robert McLean, MD, said in an interview. “We need ... a system that provides universal, affordable access to care.”
After examining the evidence, ACP discarded one option: a direct market-based approach.
“Direct market-based approaches won’t work,” Dr. McLean explained. “If you look at where direct marketplace approaches ... have been implemented, they just will not get you to a place where you are going to get universal coverage, portability, essential benefits, and preexisting condition protection and administrative simplification.”
Dr. McLean highlighted two paths that could achieve universal coverage and better access to health care: a single-payer–financed system, or a publicly financed coverage option within a system of regulated private insurance.
It’s the first time ACP has endorsed a single-payer approach. The college supported the public option that wasn’t included as part of the Affordable Care Act. But ACP’s latest publicly financed proposal offers a deeper level of detail on how to make that option work in the context of a private insurance system.
While the health reform conversation may be a political, ACP doesn’t want to make it a partisan one. ACP’s policy recommendations represent a carefully researched series of ideas backed by evidence-based research, Dr. McLean said.
“There is a lot of nuance behind” the two recommendations, he noted, and those nuances are explored in a series of articles and editorials published Jan. 21 in Annals of Internal Medicine.
Sizing up single payer
The ACP acknowledges that for its single-payer system, the transition could be “politically difficult and strain the federal budget,” according to Ryan A. Crowley, senior analyst at ACP, and colleagues in an article outlining the organization’s vision. “Taxes would probably replace premiums, and private insurance would have a reduced role or be eliminated altogether.”
However, the authors note that a single-payer system could be designed to address concerns from a generally skeptical public, such as providing bulk funding or setting minimum standards to guide state operations. It also could include private insurance to provide supplemental coverage.
Even so, “adopting a single-payer system would be highly disruptive and could lead to price controls that would perpetuate flaws in the current Medicare payment system, including the undervaluation of primary care,” Mr. Crowley and colleagues wrote. “If prices are set too low, it could lead to shortages and longer wait times for services. Without sufficient cost controls, however, the cost of a single-payer system could be too high to be feasible.”
Pondering the public option
Given a single-payer plan’s potential challenges, ACP also is endorsing a public option model, which provides the choice of a government-sponsored health insurance plan to compete with existing private insurance options.
“Depending on its structure and implementation, a public choice (or public option) model available to all could help to achieve universal coverage, better access, and improved outcomes without the disruption of a single-payer approach,” the ACP authors noted.
The public option has its own drawbacks, they acknowledge. Those include an inability to achieve better savings on prescription drugs, compared with a single-payer system. The public option approach also doesn’t do away with the current administrative burden, and access issues related to narrow provider networks would persist.
Dr. McLean noted that a more highly regulated insurance market would be needed to help make the public option model work.
“Insurance companies don’t have regulation in a lot of things that they do,” Dr. McLean said. “We see that as quite problematic. They are kind of running amok at this point.”
Expanding the role of primary care
In either reform scenario, primary care would play a much greater role.
“We need to promote primary care,” Dr. McLean said. That includes better incentives to draw physicians to it. “We have to pay them enough,” he added.
The health care models will need to move away from higher pay to specialties for high-cost, high-volume procedural reimbursement. And they’ll need to recognize the need for placing a higher value on the cognitive services provided at the primary care level.
Also in need of change: physicians’ administrative burdens. Reforms need to address the burden created by value-based care and the poor application and misapplication of quality measures.
Migration to a single-payer environment could would make reducing the administrative burden a lot easier, Dr. McLean said. But it also could be done with a public option approach.
That’s where regulators can play a big role in working with insurers to help address administrative burden – streamlining prior authorization of procedures, the types of forms used, and other policies, Dr. McLean explained.
“The number of insurers and their ability to have their own rules and regulations [make it] incredibly complex for patients as well as physicians trying to figure out how to deliver the care that they need,” he noted.
Dr. McLean hopes that the ACP’s papers will spark conversation, particularly among legislators and regulators.
“The bottom line is we cannot afford to not do something bold,” he cautioned. “It is just not working. Our patients deserve better, and we can do better.”
The American College of Physicians is recommending either a single-payer system or a public option within a regulated private insurance system to help deliver universal and affordable access to health care for all Americans.
“We came to the conclusion that two directions or approaches could get us to where we need to be,” ACP President Robert McLean, MD, said in an interview. “We need ... a system that provides universal, affordable access to care.”
After examining the evidence, ACP discarded one option: a direct market-based approach.
“Direct market-based approaches won’t work,” Dr. McLean explained. “If you look at where direct marketplace approaches ... have been implemented, they just will not get you to a place where you are going to get universal coverage, portability, essential benefits, and preexisting condition protection and administrative simplification.”
Dr. McLean highlighted two paths that could achieve universal coverage and better access to health care: a single-payer–financed system, or a publicly financed coverage option within a system of regulated private insurance.
It’s the first time ACP has endorsed a single-payer approach. The college supported the public option that wasn’t included as part of the Affordable Care Act. But ACP’s latest publicly financed proposal offers a deeper level of detail on how to make that option work in the context of a private insurance system.
While the health reform conversation may be a political, ACP doesn’t want to make it a partisan one. ACP’s policy recommendations represent a carefully researched series of ideas backed by evidence-based research, Dr. McLean said.
“There is a lot of nuance behind” the two recommendations, he noted, and those nuances are explored in a series of articles and editorials published Jan. 21 in Annals of Internal Medicine.
Sizing up single payer
The ACP acknowledges that for its single-payer system, the transition could be “politically difficult and strain the federal budget,” according to Ryan A. Crowley, senior analyst at ACP, and colleagues in an article outlining the organization’s vision. “Taxes would probably replace premiums, and private insurance would have a reduced role or be eliminated altogether.”
However, the authors note that a single-payer system could be designed to address concerns from a generally skeptical public, such as providing bulk funding or setting minimum standards to guide state operations. It also could include private insurance to provide supplemental coverage.
Even so, “adopting a single-payer system would be highly disruptive and could lead to price controls that would perpetuate flaws in the current Medicare payment system, including the undervaluation of primary care,” Mr. Crowley and colleagues wrote. “If prices are set too low, it could lead to shortages and longer wait times for services. Without sufficient cost controls, however, the cost of a single-payer system could be too high to be feasible.”
Pondering the public option
Given a single-payer plan’s potential challenges, ACP also is endorsing a public option model, which provides the choice of a government-sponsored health insurance plan to compete with existing private insurance options.
“Depending on its structure and implementation, a public choice (or public option) model available to all could help to achieve universal coverage, better access, and improved outcomes without the disruption of a single-payer approach,” the ACP authors noted.
The public option has its own drawbacks, they acknowledge. Those include an inability to achieve better savings on prescription drugs, compared with a single-payer system. The public option approach also doesn’t do away with the current administrative burden, and access issues related to narrow provider networks would persist.
Dr. McLean noted that a more highly regulated insurance market would be needed to help make the public option model work.
“Insurance companies don’t have regulation in a lot of things that they do,” Dr. McLean said. “We see that as quite problematic. They are kind of running amok at this point.”
Expanding the role of primary care
In either reform scenario, primary care would play a much greater role.
“We need to promote primary care,” Dr. McLean said. That includes better incentives to draw physicians to it. “We have to pay them enough,” he added.
The health care models will need to move away from higher pay to specialties for high-cost, high-volume procedural reimbursement. And they’ll need to recognize the need for placing a higher value on the cognitive services provided at the primary care level.
Also in need of change: physicians’ administrative burdens. Reforms need to address the burden created by value-based care and the poor application and misapplication of quality measures.
Migration to a single-payer environment could would make reducing the administrative burden a lot easier, Dr. McLean said. But it also could be done with a public option approach.
That’s where regulators can play a big role in working with insurers to help address administrative burden – streamlining prior authorization of procedures, the types of forms used, and other policies, Dr. McLean explained.
“The number of insurers and their ability to have their own rules and regulations [make it] incredibly complex for patients as well as physicians trying to figure out how to deliver the care that they need,” he noted.
Dr. McLean hopes that the ACP’s papers will spark conversation, particularly among legislators and regulators.
“The bottom line is we cannot afford to not do something bold,” he cautioned. “It is just not working. Our patients deserve better, and we can do better.”
The American College of Physicians is recommending either a single-payer system or a public option within a regulated private insurance system to help deliver universal and affordable access to health care for all Americans.
“We came to the conclusion that two directions or approaches could get us to where we need to be,” ACP President Robert McLean, MD, said in an interview. “We need ... a system that provides universal, affordable access to care.”
After examining the evidence, ACP discarded one option: a direct market-based approach.
“Direct market-based approaches won’t work,” Dr. McLean explained. “If you look at where direct marketplace approaches ... have been implemented, they just will not get you to a place where you are going to get universal coverage, portability, essential benefits, and preexisting condition protection and administrative simplification.”
Dr. McLean highlighted two paths that could achieve universal coverage and better access to health care: a single-payer–financed system, or a publicly financed coverage option within a system of regulated private insurance.
It’s the first time ACP has endorsed a single-payer approach. The college supported the public option that wasn’t included as part of the Affordable Care Act. But ACP’s latest publicly financed proposal offers a deeper level of detail on how to make that option work in the context of a private insurance system.
While the health reform conversation may be a political, ACP doesn’t want to make it a partisan one. ACP’s policy recommendations represent a carefully researched series of ideas backed by evidence-based research, Dr. McLean said.
“There is a lot of nuance behind” the two recommendations, he noted, and those nuances are explored in a series of articles and editorials published Jan. 21 in Annals of Internal Medicine.
Sizing up single payer
The ACP acknowledges that for its single-payer system, the transition could be “politically difficult and strain the federal budget,” according to Ryan A. Crowley, senior analyst at ACP, and colleagues in an article outlining the organization’s vision. “Taxes would probably replace premiums, and private insurance would have a reduced role or be eliminated altogether.”
However, the authors note that a single-payer system could be designed to address concerns from a generally skeptical public, such as providing bulk funding or setting minimum standards to guide state operations. It also could include private insurance to provide supplemental coverage.
Even so, “adopting a single-payer system would be highly disruptive and could lead to price controls that would perpetuate flaws in the current Medicare payment system, including the undervaluation of primary care,” Mr. Crowley and colleagues wrote. “If prices are set too low, it could lead to shortages and longer wait times for services. Without sufficient cost controls, however, the cost of a single-payer system could be too high to be feasible.”
Pondering the public option
Given a single-payer plan’s potential challenges, ACP also is endorsing a public option model, which provides the choice of a government-sponsored health insurance plan to compete with existing private insurance options.
“Depending on its structure and implementation, a public choice (or public option) model available to all could help to achieve universal coverage, better access, and improved outcomes without the disruption of a single-payer approach,” the ACP authors noted.
The public option has its own drawbacks, they acknowledge. Those include an inability to achieve better savings on prescription drugs, compared with a single-payer system. The public option approach also doesn’t do away with the current administrative burden, and access issues related to narrow provider networks would persist.
Dr. McLean noted that a more highly regulated insurance market would be needed to help make the public option model work.
“Insurance companies don’t have regulation in a lot of things that they do,” Dr. McLean said. “We see that as quite problematic. They are kind of running amok at this point.”
Expanding the role of primary care
In either reform scenario, primary care would play a much greater role.
“We need to promote primary care,” Dr. McLean said. That includes better incentives to draw physicians to it. “We have to pay them enough,” he added.
The health care models will need to move away from higher pay to specialties for high-cost, high-volume procedural reimbursement. And they’ll need to recognize the need for placing a higher value on the cognitive services provided at the primary care level.
Also in need of change: physicians’ administrative burdens. Reforms need to address the burden created by value-based care and the poor application and misapplication of quality measures.
Migration to a single-payer environment could would make reducing the administrative burden a lot easier, Dr. McLean said. But it also could be done with a public option approach.
That’s where regulators can play a big role in working with insurers to help address administrative burden – streamlining prior authorization of procedures, the types of forms used, and other policies, Dr. McLean explained.
“The number of insurers and their ability to have their own rules and regulations [make it] incredibly complex for patients as well as physicians trying to figure out how to deliver the care that they need,” he noted.
Dr. McLean hopes that the ACP’s papers will spark conversation, particularly among legislators and regulators.
“The bottom line is we cannot afford to not do something bold,” he cautioned. “It is just not working. Our patients deserve better, and we can do better.”
FROM ANNALS OF INTERNAL MEDICINE
Medscape survey points to generational differences in physician burnout
Burnout among physicians appears to have decreased slightly in the past few years, but remains a significant problem for the medical profession, according to the Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide.

A survey of more than 15,000 physicians revealed that 42% reported being burned out, down from 46% who responded to the survey 5 years ago. However, there are variations in the rates based on certain demographic factors such as specialty, age, and gender.
Urology sits at the top of the list as the specialty that is experiencing the highest rate of burnout, with 54% of urologists responding to the survey reporting burnout. Neurology and nephrology followed with rates of burnout at 50% and 49%, respectively. The next five specialties on the list all reported burnout rates of 46%: diabetes and endocrinology, family medicine, radiology, ob.gyn., and rheumatology. Pulmonology specialists reported a burnout rate of 41%. Gastroenterologists reported burnout rates of 37%.
The survey divided participants into three age categories – Millennial (ages 25-39 years), Generation X (ages 40-54 years), and Baby Boomer (ages 55-73 years). Both Millennials and Baby Boomers reported similar rates of burnout (38% and 39%, respectively) and those in Generation X reported a higher rate of burnout (48%).
This higher rate is not unexpected. The survey results cite Carol Bernstein, MD, of the Albert Einstein College of Medicine, New York, as noting that midcareer “is typically the time of highest burnout, which is where Gen Xers are in their career trajectory, suggesting a number of factors outside of work such as caring for children and elderly parents, planning for retirement, can play a role in contributing to burnout.”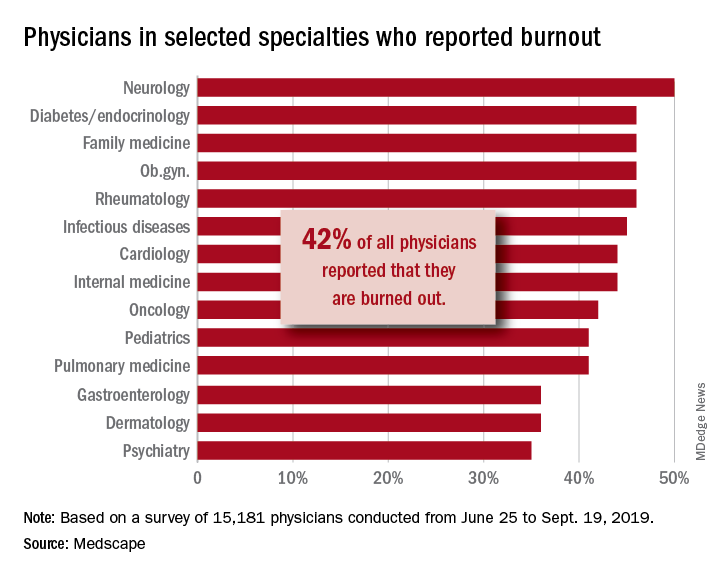
Women also reported a higher rate of burnout, although the rate has dropped from the survey conducted 5 years ago. The rate of burnout among women reported for the 2020 survey was 48%, down from 51% reported 5 years ago. By comparison, the rate of burnout for men was 37% in 2020, down from 43% in 2015.
In terms of what is causing burnout, the biggest contributor is the bureaucratic tasks (charting and paperwork, for example) that physicians must complete, which 55% of respondents to the survey said was the leading cause of burnout. Next was spending too many hours at work (33%); lack of respect from administrators, employers, colleagues, and staff (32%); and the increased computerization of the practice, including the use of electronic health records (30%).
When broken down by age category, the bureaucratic tasks was tops in all three groups (57% for Millennials, 56% for Generation X, and 54% for Baby Boomers), but what ranks next differs slightly by age group. For Millennials, the next two factors were too many hours at work (38%) and lack of respect (35%). Generation X respondents cited the same two factors, both at 33%. Baby Boomers cited computerization as their second-highest factor (41%) and spending too many hours at work as the third-highest factor (31%).
The generations had different approaches to coping with burnout. Millennials (56%) reported sleep as their top-ranked coping strategy, while Gen Xers and Baby Boomers ranked exercise and personal isolation as their top choice. For these two older groups, sleep was ranked last, after other activities such as talking with family and friends.
The survey also asked about depression, and respondents reported a similar rate across all age groups (15%, 18%, and 16%, respectively). Among those who said they were depressed, the three age groups had similar rates of suicidal thoughts (21%, 24%, and 22%).
Perhaps the most striking finding of the survey is the number of physicians who would take a pay cut to achieve a better work-life balance. Among Millennials, 52% would accept a pay cut, compared with 48% of Generation X and 49% of Baby Boomers. A surprising number (36%, 34%, and 31%, respectively, reported that they would accept a $10,000-$20,000 pay cut to have a 20% reduction in work hours. gtwachtman@mdedge.com
*This story was updated on 1/22/2020.
SOURCE: Kane L et al. Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide. Medscape. 2020 Jan 15.
Burnout among physicians appears to have decreased slightly in the past few years, but remains a significant problem for the medical profession, according to the Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide.
A survey of more than 15,000 physicians revealed that 42% reported being burned out, down from 46% who responded to the survey 5 years ago. However, there are variations in the rates based on certain demographic factors such as specialty, age, and gender.
Urology sits at the top of the list as the specialty that is experiencing the highest rate of burnout, with 54% of urologists responding to the survey reporting burnout. Neurology and nephrology followed with rates of burnout at 50% and 49%, respectively. The next five specialties on the list all reported burnout rates of 46%: diabetes and endocrinology, family medicine, radiology, ob.gyn., and rheumatology. Pulmonology specialists reported a burnout rate of 41%. Gastroenterologists reported burnout rates of 37%.
The survey divided participants into three age categories – Millennial (ages 25-39 years), Generation X (ages 40-54 years), and Baby Boomer (ages 55-73 years). Both Millennials and Baby Boomers reported similar rates of burnout (38% and 39%, respectively) and those in Generation X reported a higher rate of burnout (48%).
This higher rate is not unexpected. The survey results cite Carol Bernstein, MD, of the Albert Einstein College of Medicine, New York, as noting that midcareer “is typically the time of highest burnout, which is where Gen Xers are in their career trajectory, suggesting a number of factors outside of work such as caring for children and elderly parents, planning for retirement, can play a role in contributing to burnout.”
Women also reported a higher rate of burnout, although the rate has dropped from the survey conducted 5 years ago. The rate of burnout among women reported for the 2020 survey was 48%, down from 51% reported 5 years ago. By comparison, the rate of burnout for men was 37% in 2020, down from 43% in 2015.
In terms of what is causing burnout, the biggest contributor is the bureaucratic tasks (charting and paperwork, for example) that physicians must complete, which 55% of respondents to the survey said was the leading cause of burnout. Next was spending too many hours at work (33%); lack of respect from administrators, employers, colleagues, and staff (32%); and the increased computerization of the practice, including the use of electronic health records (30%).
When broken down by age category, the bureaucratic tasks was tops in all three groups (57% for Millennials, 56% for Generation X, and 54% for Baby Boomers), but what ranks next differs slightly by age group. For Millennials, the next two factors were too many hours at work (38%) and lack of respect (35%). Generation X respondents cited the same two factors, both at 33%. Baby Boomers cited computerization as their second-highest factor (41%) and spending too many hours at work as the third-highest factor (31%).
The generations had different approaches to coping with burnout. Millennials (56%) reported sleep as their top-ranked coping strategy, while Gen Xers and Baby Boomers ranked exercise and personal isolation as their top choice. For these two older groups, sleep was ranked last, after other activities such as talking with family and friends.
The survey also asked about depression, and respondents reported a similar rate across all age groups (15%, 18%, and 16%, respectively). Among those who said they were depressed, the three age groups had similar rates of suicidal thoughts (21%, 24%, and 22%).
Perhaps the most striking finding of the survey is the number of physicians who would take a pay cut to achieve a better work-life balance. Among Millennials, 52% would accept a pay cut, compared with 48% of Generation X and 49% of Baby Boomers. A surprising number (36%, 34%, and 31%, respectively, reported that they would accept a $10,000-$20,000 pay cut to have a 20% reduction in work hours. gtwachtman@mdedge.com
*This story was updated on 1/22/2020.
SOURCE: Kane L et al. Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide. Medscape. 2020 Jan 15.
Burnout among physicians appears to have decreased slightly in the past few years, but remains a significant problem for the medical profession, according to the Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide.
A survey of more than 15,000 physicians revealed that 42% reported being burned out, down from 46% who responded to the survey 5 years ago. However, there are variations in the rates based on certain demographic factors such as specialty, age, and gender.
Urology sits at the top of the list as the specialty that is experiencing the highest rate of burnout, with 54% of urologists responding to the survey reporting burnout. Neurology and nephrology followed with rates of burnout at 50% and 49%, respectively. The next five specialties on the list all reported burnout rates of 46%: diabetes and endocrinology, family medicine, radiology, ob.gyn., and rheumatology. Pulmonology specialists reported a burnout rate of 41%. Gastroenterologists reported burnout rates of 37%.
The survey divided participants into three age categories – Millennial (ages 25-39 years), Generation X (ages 40-54 years), and Baby Boomer (ages 55-73 years). Both Millennials and Baby Boomers reported similar rates of burnout (38% and 39%, respectively) and those in Generation X reported a higher rate of burnout (48%).
This higher rate is not unexpected. The survey results cite Carol Bernstein, MD, of the Albert Einstein College of Medicine, New York, as noting that midcareer “is typically the time of highest burnout, which is where Gen Xers are in their career trajectory, suggesting a number of factors outside of work such as caring for children and elderly parents, planning for retirement, can play a role in contributing to burnout.”
Women also reported a higher rate of burnout, although the rate has dropped from the survey conducted 5 years ago. The rate of burnout among women reported for the 2020 survey was 48%, down from 51% reported 5 years ago. By comparison, the rate of burnout for men was 37% in 2020, down from 43% in 2015.
In terms of what is causing burnout, the biggest contributor is the bureaucratic tasks (charting and paperwork, for example) that physicians must complete, which 55% of respondents to the survey said was the leading cause of burnout. Next was spending too many hours at work (33%); lack of respect from administrators, employers, colleagues, and staff (32%); and the increased computerization of the practice, including the use of electronic health records (30%).
When broken down by age category, the bureaucratic tasks was tops in all three groups (57% for Millennials, 56% for Generation X, and 54% for Baby Boomers), but what ranks next differs slightly by age group. For Millennials, the next two factors were too many hours at work (38%) and lack of respect (35%). Generation X respondents cited the same two factors, both at 33%. Baby Boomers cited computerization as their second-highest factor (41%) and spending too many hours at work as the third-highest factor (31%).
The generations had different approaches to coping with burnout. Millennials (56%) reported sleep as their top-ranked coping strategy, while Gen Xers and Baby Boomers ranked exercise and personal isolation as their top choice. For these two older groups, sleep was ranked last, after other activities such as talking with family and friends.
The survey also asked about depression, and respondents reported a similar rate across all age groups (15%, 18%, and 16%, respectively). Among those who said they were depressed, the three age groups had similar rates of suicidal thoughts (21%, 24%, and 22%).
Perhaps the most striking finding of the survey is the number of physicians who would take a pay cut to achieve a better work-life balance. Among Millennials, 52% would accept a pay cut, compared with 48% of Generation X and 49% of Baby Boomers. A surprising number (36%, 34%, and 31%, respectively, reported that they would accept a $10,000-$20,000 pay cut to have a 20% reduction in work hours. gtwachtman@mdedge.com
*This story was updated on 1/22/2020.
SOURCE: Kane L et al. Medscape National Physician Burnout & Suicide Report 2020: The Generational Divide. Medscape. 2020 Jan 15.
Alan Alda, Scripps Research join forces to improve science communication
LA JOLLA, CALIF. – The first time that legendary actor Alan Alda conducted an interview for “Scientific American Frontiers” on PBS, an award-winning series that ran for more than a decade, he remembers learning a lesson in humility.

“I wasn’t as smart as I thought I was,” he told a crowd of largely scientists and medical professionals who gathered in a small auditorium on the campus of Scripps Research on Jan. 16, 2020. “I didn’t realize the value of ignorance. I have a natural supply of it. I began to use it and say [to interviewees]: ‘I don’t understand what that means.’ Sometimes it would be basic physics and they’d look at me like I was a school child. I am a very curious person. What I discovered was, I was bringing out their humanity by my own curiosity, by the way I related to them, which I developed through studying improvisation as an actor, and relating as an actor to other actors.”
Mr. Alda, 83, appeared on the research campus to announce that Scripps Research is the new West Coast home of Alda Communication Training, which will work in tandem with the Alan Alda Center for Communicating Science at the State University of New York at Stony Brook, a nonprofit organization that Mr. Alda helped found in 2009.
Immersive training experience
“This will be a center where people can come to get training in effective communication,” said Mr. Alda, who is the winner of six Emmy Awards and six Golden Globe awards. “It’s an experiential kind of training. We don’t give tips. We don’t give lectures. We put you through exercises that are fun and actually make you laugh, but turn you into a better communicator, so you’re better able to connect to the people you’re talking to.”
To date, the Alan Alda Center for Communicating Science has trained more than 15,000 scientific leaders in the United States and other countries. The location at Scripps Research makes it more convenient for West Coast–based researchers and industry leaders to participate. “One of the things we wished, for years, we had was a place where we could train scientists and researchers and medical professionals all up and down the West Coast,” he said.
Recently, more than 30 of Scripps Research scientists participated in Mr. Alda’s training program, an immersive and engaging experience that helps participants learn to empathize with an audience and present their work in a way that connects with different stakeholders. The skills and strategies can help participants relate to prospective investors and philanthropists, government officials, members of the media, peers across scientific disciplines, and the general public.
Earlier in the day that he spoke on the Scripps campus, Mr. Alda encountered some of the Scripps researchers who had participated in that training. “One group of scientists came in and we shook hands,” he said. “They introduced themselves and said: ‘We’re working on infectious diseases.’ I said: ‘Oh my God; I just shook hands with you!’ No matter what I asked them, they had a clear way to express what they did. Then I realized they had studied with Alda Communications.”
Why communication matters
During the early stages of forming what became the Alan Alda Center for Communicating Science, one Nobel Prize winner at a major university dismissed the importance of improving the communication skills of young scientists. “He said to me: ‘We don’t have time for that; we have too much science to teach,’ ” said Mr. Alda, who played Army surgeon “Hawkeye” Pierce on the TV series “M*A*S*H”. “But communication is the essence of science. How can you do science unless you communicate with other scientists? There’s a stereotype that scientists are not as good at communicating as other people are. It’s true that they often speak a language that a lot of us don’t understand, but we all speak a language that is hard for other people to understand if we know something in great depth. We want to tell all the details; we want to speak in our special language because it makes us feel good.”
He underscored the importance of scientists being able to effectively communicate with the general public, “because the public needs to understand how important science is to their lives. It matters because at a place like [Scripps Research], understanding how nature works is put to work to keep our health secure.” Members of the public, he continued, “are busy living their lives; they’re busy working and bringing up their children. They haven’t spent 20, 30, 40 years devoted to a single aspect of nature the way scientists have. We can’t expect them to know as much as professional scientists, so we have to help them understand it. I hope we find ways to increase curiosity. I don’t know how to do that. I wish somebody would do a study on it, how you can take someone with a modicum of curiosity and help them enlarge it so it gives them the pleasure of discovering things about nature or understanding things about nature that other people don’t discover. Curiosity is the key to staying alive. That would bring us to a point of more people understanding science.”
Cultivating a sense of responsibility is another key to effective communication. “It’s the job of the person leading the discussion to make clear to the person listening,” Mr. Alda said. “You get the impression that ‘this person is my responsibility. I have to take care of them, so they understand what’s going on.’ ”
Parkinson’s disease diagnosis
During a question-and-answer session, Mr. Alda opened up about his Parkinson’s disease, which he said was diagnosed about 5 years ago. In 2018, he decided to speak publicly about his diagnosis for the first time.
“The reason was that I wanted to communicate to people who had recently been diagnosed not to believe or give into the stereotype that when you get a diagnosis, your life is over,” said Mr. Alda, who received the Public Welfare Medal from the National Academy of Sciences in 2016. “Under the burden of that belief, some people won’t tell their family or workplace colleagues. There are exercises you can do and medications you can take to prolong the time it takes before Parkinson’s gets much more serious. It’s not to diminish the fact that it can get really bad; but to think that your life is over as soon as you get a diagnosis is wrong.”
He added: “I’ve gone 5 years and I’m almost busier than I’ve ever been. I’m getting a lot accomplished and I look forward to I don’t know how many years. As long as I have them, I’m going to be grateful. It’s amazing how great it feels not to keep the diagnosis a secret.”
The first 2-day training session at Scripps Research will be held in June 2020. Additional sessions are scheduled to take place in October and December. Registration is available at aldacommunicationtraining.com/workshops.
LA JOLLA, CALIF. – The first time that legendary actor Alan Alda conducted an interview for “Scientific American Frontiers” on PBS, an award-winning series that ran for more than a decade, he remembers learning a lesson in humility.
“I wasn’t as smart as I thought I was,” he told a crowd of largely scientists and medical professionals who gathered in a small auditorium on the campus of Scripps Research on Jan. 16, 2020. “I didn’t realize the value of ignorance. I have a natural supply of it. I began to use it and say [to interviewees]: ‘I don’t understand what that means.’ Sometimes it would be basic physics and they’d look at me like I was a school child. I am a very curious person. What I discovered was, I was bringing out their humanity by my own curiosity, by the way I related to them, which I developed through studying improvisation as an actor, and relating as an actor to other actors.”
Mr. Alda, 83, appeared on the research campus to announce that Scripps Research is the new West Coast home of Alda Communication Training, which will work in tandem with the Alan Alda Center for Communicating Science at the State University of New York at Stony Brook, a nonprofit organization that Mr. Alda helped found in 2009.
Immersive training experience
“This will be a center where people can come to get training in effective communication,” said Mr. Alda, who is the winner of six Emmy Awards and six Golden Globe awards. “It’s an experiential kind of training. We don’t give tips. We don’t give lectures. We put you through exercises that are fun and actually make you laugh, but turn you into a better communicator, so you’re better able to connect to the people you’re talking to.”
To date, the Alan Alda Center for Communicating Science has trained more than 15,000 scientific leaders in the United States and other countries. The location at Scripps Research makes it more convenient for West Coast–based researchers and industry leaders to participate. “One of the things we wished, for years, we had was a place where we could train scientists and researchers and medical professionals all up and down the West Coast,” he said.
Recently, more than 30 of Scripps Research scientists participated in Mr. Alda’s training program, an immersive and engaging experience that helps participants learn to empathize with an audience and present their work in a way that connects with different stakeholders. The skills and strategies can help participants relate to prospective investors and philanthropists, government officials, members of the media, peers across scientific disciplines, and the general public.
Earlier in the day that he spoke on the Scripps campus, Mr. Alda encountered some of the Scripps researchers who had participated in that training. “One group of scientists came in and we shook hands,” he said. “They introduced themselves and said: ‘We’re working on infectious diseases.’ I said: ‘Oh my God; I just shook hands with you!’ No matter what I asked them, they had a clear way to express what they did. Then I realized they had studied with Alda Communications.”
Why communication matters
During the early stages of forming what became the Alan Alda Center for Communicating Science, one Nobel Prize winner at a major university dismissed the importance of improving the communication skills of young scientists. “He said to me: ‘We don’t have time for that; we have too much science to teach,’ ” said Mr. Alda, who played Army surgeon “Hawkeye” Pierce on the TV series “M*A*S*H”. “But communication is the essence of science. How can you do science unless you communicate with other scientists? There’s a stereotype that scientists are not as good at communicating as other people are. It’s true that they often speak a language that a lot of us don’t understand, but we all speak a language that is hard for other people to understand if we know something in great depth. We want to tell all the details; we want to speak in our special language because it makes us feel good.”
He underscored the importance of scientists being able to effectively communicate with the general public, “because the public needs to understand how important science is to their lives. It matters because at a place like [Scripps Research], understanding how nature works is put to work to keep our health secure.” Members of the public, he continued, “are busy living their lives; they’re busy working and bringing up their children. They haven’t spent 20, 30, 40 years devoted to a single aspect of nature the way scientists have. We can’t expect them to know as much as professional scientists, so we have to help them understand it. I hope we find ways to increase curiosity. I don’t know how to do that. I wish somebody would do a study on it, how you can take someone with a modicum of curiosity and help them enlarge it so it gives them the pleasure of discovering things about nature or understanding things about nature that other people don’t discover. Curiosity is the key to staying alive. That would bring us to a point of more people understanding science.”
Cultivating a sense of responsibility is another key to effective communication. “It’s the job of the person leading the discussion to make clear to the person listening,” Mr. Alda said. “You get the impression that ‘this person is my responsibility. I have to take care of them, so they understand what’s going on.’ ”
Parkinson’s disease diagnosis
During a question-and-answer session, Mr. Alda opened up about his Parkinson’s disease, which he said was diagnosed about 5 years ago. In 2018, he decided to speak publicly about his diagnosis for the first time.
“The reason was that I wanted to communicate to people who had recently been diagnosed not to believe or give into the stereotype that when you get a diagnosis, your life is over,” said Mr. Alda, who received the Public Welfare Medal from the National Academy of Sciences in 2016. “Under the burden of that belief, some people won’t tell their family or workplace colleagues. There are exercises you can do and medications you can take to prolong the time it takes before Parkinson’s gets much more serious. It’s not to diminish the fact that it can get really bad; but to think that your life is over as soon as you get a diagnosis is wrong.”
He added: “I’ve gone 5 years and I’m almost busier than I’ve ever been. I’m getting a lot accomplished and I look forward to I don’t know how many years. As long as I have them, I’m going to be grateful. It’s amazing how great it feels not to keep the diagnosis a secret.”
The first 2-day training session at Scripps Research will be held in June 2020. Additional sessions are scheduled to take place in October and December. Registration is available at aldacommunicationtraining.com/workshops.
LA JOLLA, CALIF. – The first time that legendary actor Alan Alda conducted an interview for “Scientific American Frontiers” on PBS, an award-winning series that ran for more than a decade, he remembers learning a lesson in humility.
“I wasn’t as smart as I thought I was,” he told a crowd of largely scientists and medical professionals who gathered in a small auditorium on the campus of Scripps Research on Jan. 16, 2020. “I didn’t realize the value of ignorance. I have a natural supply of it. I began to use it and say [to interviewees]: ‘I don’t understand what that means.’ Sometimes it would be basic physics and they’d look at me like I was a school child. I am a very curious person. What I discovered was, I was bringing out their humanity by my own curiosity, by the way I related to them, which I developed through studying improvisation as an actor, and relating as an actor to other actors.”
Mr. Alda, 83, appeared on the research campus to announce that Scripps Research is the new West Coast home of Alda Communication Training, which will work in tandem with the Alan Alda Center for Communicating Science at the State University of New York at Stony Brook, a nonprofit organization that Mr. Alda helped found in 2009.
Immersive training experience
“This will be a center where people can come to get training in effective communication,” said Mr. Alda, who is the winner of six Emmy Awards and six Golden Globe awards. “It’s an experiential kind of training. We don’t give tips. We don’t give lectures. We put you through exercises that are fun and actually make you laugh, but turn you into a better communicator, so you’re better able to connect to the people you’re talking to.”
To date, the Alan Alda Center for Communicating Science has trained more than 15,000 scientific leaders in the United States and other countries. The location at Scripps Research makes it more convenient for West Coast–based researchers and industry leaders to participate. “One of the things we wished, for years, we had was a place where we could train scientists and researchers and medical professionals all up and down the West Coast,” he said.
Recently, more than 30 of Scripps Research scientists participated in Mr. Alda’s training program, an immersive and engaging experience that helps participants learn to empathize with an audience and present their work in a way that connects with different stakeholders. The skills and strategies can help participants relate to prospective investors and philanthropists, government officials, members of the media, peers across scientific disciplines, and the general public.
Earlier in the day that he spoke on the Scripps campus, Mr. Alda encountered some of the Scripps researchers who had participated in that training. “One group of scientists came in and we shook hands,” he said. “They introduced themselves and said: ‘We’re working on infectious diseases.’ I said: ‘Oh my God; I just shook hands with you!’ No matter what I asked them, they had a clear way to express what they did. Then I realized they had studied with Alda Communications.”
Why communication matters
During the early stages of forming what became the Alan Alda Center for Communicating Science, one Nobel Prize winner at a major university dismissed the importance of improving the communication skills of young scientists. “He said to me: ‘We don’t have time for that; we have too much science to teach,’ ” said Mr. Alda, who played Army surgeon “Hawkeye” Pierce on the TV series “M*A*S*H”. “But communication is the essence of science. How can you do science unless you communicate with other scientists? There’s a stereotype that scientists are not as good at communicating as other people are. It’s true that they often speak a language that a lot of us don’t understand, but we all speak a language that is hard for other people to understand if we know something in great depth. We want to tell all the details; we want to speak in our special language because it makes us feel good.”
He underscored the importance of scientists being able to effectively communicate with the general public, “because the public needs to understand how important science is to their lives. It matters because at a place like [Scripps Research], understanding how nature works is put to work to keep our health secure.” Members of the public, he continued, “are busy living their lives; they’re busy working and bringing up their children. They haven’t spent 20, 30, 40 years devoted to a single aspect of nature the way scientists have. We can’t expect them to know as much as professional scientists, so we have to help them understand it. I hope we find ways to increase curiosity. I don’t know how to do that. I wish somebody would do a study on it, how you can take someone with a modicum of curiosity and help them enlarge it so it gives them the pleasure of discovering things about nature or understanding things about nature that other people don’t discover. Curiosity is the key to staying alive. That would bring us to a point of more people understanding science.”
Cultivating a sense of responsibility is another key to effective communication. “It’s the job of the person leading the discussion to make clear to the person listening,” Mr. Alda said. “You get the impression that ‘this person is my responsibility. I have to take care of them, so they understand what’s going on.’ ”
Parkinson’s disease diagnosis
During a question-and-answer session, Mr. Alda opened up about his Parkinson’s disease, which he said was diagnosed about 5 years ago. In 2018, he decided to speak publicly about his diagnosis for the first time.
“The reason was that I wanted to communicate to people who had recently been diagnosed not to believe or give into the stereotype that when you get a diagnosis, your life is over,” said Mr. Alda, who received the Public Welfare Medal from the National Academy of Sciences in 2016. “Under the burden of that belief, some people won’t tell their family or workplace colleagues. There are exercises you can do and medications you can take to prolong the time it takes before Parkinson’s gets much more serious. It’s not to diminish the fact that it can get really bad; but to think that your life is over as soon as you get a diagnosis is wrong.”
He added: “I’ve gone 5 years and I’m almost busier than I’ve ever been. I’m getting a lot accomplished and I look forward to I don’t know how many years. As long as I have them, I’m going to be grateful. It’s amazing how great it feels not to keep the diagnosis a secret.”
The first 2-day training session at Scripps Research will be held in June 2020. Additional sessions are scheduled to take place in October and December. Registration is available at aldacommunicationtraining.com/workshops.
Value analysis of JAK inhibitors for RA hampered by limited data
Adequate evidence shows that adding a Janus kinase (JAK) inhibitor to conventional disease-modifying antirheumatic drug therapy provides a net health benefit for patients with rheumatoid arthritis, compared with conventional drugs alone, according to a report by an independent research institute. But the long-term economic value of JAK inhibitors for rheumatoid arthritis is less clear, the report by the Institute for Clinical and Economic Review (ICER) indicates.
ICER on Jan. 9 released a finalized report and policy recommendations on JAK inhibitors and biosimilars for rheumatoid arthritis. The report reviews current evidence for JAK inhibitors for adults with moderately active to severely active rheumatoid arthritis.
Since the nonprofit’s 2017 review of targeted immune modulators for rheumatoid arthritis, two JAK inhibitors, baricitinib (Olumiant) and upadacitinib (Rinvoq), were approved by the Food and Drug Administration. At a December 2019 public meeting of the California Technology Assessment Forum (CTAF), one of ICER’s independent evidence appraisal committees, panelists reviewed recent evidence.
A pricey comparator
In ICER’s analysis, the JAK inhibitor upadacitinib reached common thresholds for cost-effectiveness when compared with adalimumab (Humira). Nevertheless, the 14 members of the independent evidence appraisal committee voted that upadacitinib’s long-term economic value was “low” (8 votes) or “intermediate” (6 votes). Concerns about the generalizability of phase 3 clinical trial data to patients in the real world were among the reservations noted by panelists. Furthermore, “legitimate questions remain about whether or not adalimumab, launched 17 years ago, is fairly priced to begin with,” Pamela Bradt, MD, MPH, ICER’s chief scientific officer, said in a news release.
The panel did not vote on the economic value of tofacitinib (Xeljanz) or baricitinib, the two other JAK inhibitors that are approved for rheumatoid arthritis, because head-to-head evidence against adalimumab was insufficient, ICER said.
“Rheumatoid arthritis is a progressively disabling condition, and patients are fortunate to have multiple therapy options – including biosimilars – that effectively slow disease progression,” Dr. Bradt said. “Many economists might expect medicines to become more affordable in an increasingly crowded therapeutic class; however, because the current rebate structure has erected barriers between patients and several emerging RA therapies, traditional market dynamics have been unable to drive down prices.”
Weighing efficacy and cost
Panelists found that the net health benefit provided by upadacitinib is superior to that provided by adalimumab. At the same time, they said that there is insufficient head-to-head evidence to distinguish between the net health benefit of upadacitinib and tofacitinib or to demonstrate that tofacitinib is superior to adalimumab. Evidence comparing baricitinib to adalimumab does not exist.
CTAF members unanimously agreed that adequate evidence demonstrates that the biosimilar infliximab-dyyb (Inflectra) is clinically equivalent to its reference biologic, infliximab (Remicade).
Economic modeling demonstrated that upadacitinib plus a conventional drug achieves marginally higher quality of life than adalimumab plus a conventional drug does, at similar costs. “Based on this comparison with adalimumab, ICER’s value-based price benchmark range for upadacitinib is between $44,000 and $45,000,” according to the ICER news release. “This benchmark represents a 25% discount off of upadacitinib’s annual list price of $59,860, a suggested discount that is consistent with the rebates we assume the manufacturer is currently offering.”
After the voting session, various experts, including clinicians, patient advocates, and representatives from manufacturers and insurance companies, made the following policy recommendations:
- Regulatory intervention may be needed to ensure that drug prices do not continue to increase further from reasonable alignment with added benefits for patients.
- Insurers, pharmacy benefit managers, and employers should increase transparency around the role of discounts and rebates in formulary design.
- Policymakers should aim to create a system that rewards lower-priced biosimilar treatment options.
The findings of the clinical review by the Institute for Clinical and Economic Review (ICER) are generally in line with our clinical perceptions. We have an increasing number of treatment options for our RA patients, and the results of this review support the efficacy of tofacitinib and upadacitinib, compared with currently available biologic treatments. While ICER’s voting panel did find the data supported the superiority of upadacitinib over adalimumab, the cost analysis notes a WAC (wholesale acquisition cost) for upadacitinib of $59,860. While at expected discounted rates it is felt to be cost effective when compared with adalimumab, it is difficult to know what this means since ICER found adalimumab itself not to be cost effective, compared with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), in its 2017 review.

ICER’s focus is drug pricing and cost effectiveness, so obviously our biologic drugs are in the institute’s crosshairs. This review provided context for a policy roundtable discussion that included patient, payer, and manufacturer input, as well as American College of Rheumatology (ACR) input. We are thankful that ACR had a seat at the table, and thankful ICER is attempting to bring light to the important issues and barriers that perpetuate high drug prices in our marketplace. The discussion was wide ranging but focused on step-edit policies, the role of pharmacy benefit managers (PBMs) in perpetuating high drug prices and the relatively slow uptake of biosimilars in our marketplace.
These issues are critical to every practicing rheumatologist because we each deal daily with the hassles of step-edit/fail-first policies, which hijack our otherwise thoughtful and evidence-based decision making regarding the best treatments for our patients. We know how much (unreimbursed) time it takes our staff to sort through these step edits and prior authorizations, and we have seen recent data regarding how these policies delay care and harm patients. We were thankful to see ICER validate these concerns and note that their suggested guidelines for rational step therapy somewhat mirror those in the Safe Step Act, which ACR supports on a federal legislative level. ACR continues to vigorously support the grandfathering of any patient on an effective treatment, regardless of changes in insurance or formulary; this was an issue of robust debate at their meeting, and this patient-centric position is not uniformly held among policymakers, unfortunately.
ACR agrees with ICER’s conclusion that transparency in the PBM system regarding rebates should be promoted and that opaque rebate negotiations between PBMs and manufacturers both incentivize higher prices and block access to the marketplace for cheaper biosimilar options.
Additionally, ICER and ACR agree about the critical role that biosimilar uptake will play in controlling drug costs. While we do not yet have any biosimilars that have been deemed interchangeable by the Food and Drug Administration, we agree with ICER that data regarding comparable efficacy and safety of biosimilars to their originator products is very reassuring. While the decision to switch to a biosimilar should be an individual decision between a provider and patient, and while we recognize with frustration that many FDA-approved biosimilars are not commercially available because of patent law, it is clear that the current costs of our biologic drugs are not sustainable and the uptake of biosimilars will be critical if we hope our health care economy can continue to support coverage of these life-changing drugs in years to come. We agree with ICER that it is incumbent upon prescribers to reassure our patients regarding the safety and efficacy of these drugs.
Christopher Phillips, MD , is a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the ACR, under the guidance of the Committee on Rheumatologic Care. He attended the initial ICER rheumatoid arthritis review meeting in 2017 on behalf of ACR. In 2019, Dr. Phillips served as a reviewer and clinical expert to the ICER panel and participated in the policy roundtable discussion.
The findings of the clinical review by the Institute for Clinical and Economic Review (ICER) are generally in line with our clinical perceptions. We have an increasing number of treatment options for our RA patients, and the results of this review support the efficacy of tofacitinib and upadacitinib, compared with currently available biologic treatments. While ICER’s voting panel did find the data supported the superiority of upadacitinib over adalimumab, the cost analysis notes a WAC (wholesale acquisition cost) for upadacitinib of $59,860. While at expected discounted rates it is felt to be cost effective when compared with adalimumab, it is difficult to know what this means since ICER found adalimumab itself not to be cost effective, compared with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), in its 2017 review.
ICER’s focus is drug pricing and cost effectiveness, so obviously our biologic drugs are in the institute’s crosshairs. This review provided context for a policy roundtable discussion that included patient, payer, and manufacturer input, as well as American College of Rheumatology (ACR) input. We are thankful that ACR had a seat at the table, and thankful ICER is attempting to bring light to the important issues and barriers that perpetuate high drug prices in our marketplace. The discussion was wide ranging but focused on step-edit policies, the role of pharmacy benefit managers (PBMs) in perpetuating high drug prices and the relatively slow uptake of biosimilars in our marketplace.
These issues are critical to every practicing rheumatologist because we each deal daily with the hassles of step-edit/fail-first policies, which hijack our otherwise thoughtful and evidence-based decision making regarding the best treatments for our patients. We know how much (unreimbursed) time it takes our staff to sort through these step edits and prior authorizations, and we have seen recent data regarding how these policies delay care and harm patients. We were thankful to see ICER validate these concerns and note that their suggested guidelines for rational step therapy somewhat mirror those in the Safe Step Act, which ACR supports on a federal legislative level. ACR continues to vigorously support the grandfathering of any patient on an effective treatment, regardless of changes in insurance or formulary; this was an issue of robust debate at their meeting, and this patient-centric position is not uniformly held among policymakers, unfortunately.
ACR agrees with ICER’s conclusion that transparency in the PBM system regarding rebates should be promoted and that opaque rebate negotiations between PBMs and manufacturers both incentivize higher prices and block access to the marketplace for cheaper biosimilar options.
Additionally, ICER and ACR agree about the critical role that biosimilar uptake will play in controlling drug costs. While we do not yet have any biosimilars that have been deemed interchangeable by the Food and Drug Administration, we agree with ICER that data regarding comparable efficacy and safety of biosimilars to their originator products is very reassuring. While the decision to switch to a biosimilar should be an individual decision between a provider and patient, and while we recognize with frustration that many FDA-approved biosimilars are not commercially available because of patent law, it is clear that the current costs of our biologic drugs are not sustainable and the uptake of biosimilars will be critical if we hope our health care economy can continue to support coverage of these life-changing drugs in years to come. We agree with ICER that it is incumbent upon prescribers to reassure our patients regarding the safety and efficacy of these drugs.
Christopher Phillips, MD , is a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the ACR, under the guidance of the Committee on Rheumatologic Care. He attended the initial ICER rheumatoid arthritis review meeting in 2017 on behalf of ACR. In 2019, Dr. Phillips served as a reviewer and clinical expert to the ICER panel and participated in the policy roundtable discussion.
The findings of the clinical review by the Institute for Clinical and Economic Review (ICER) are generally in line with our clinical perceptions. We have an increasing number of treatment options for our RA patients, and the results of this review support the efficacy of tofacitinib and upadacitinib, compared with currently available biologic treatments. While ICER’s voting panel did find the data supported the superiority of upadacitinib over adalimumab, the cost analysis notes a WAC (wholesale acquisition cost) for upadacitinib of $59,860. While at expected discounted rates it is felt to be cost effective when compared with adalimumab, it is difficult to know what this means since ICER found adalimumab itself not to be cost effective, compared with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), in its 2017 review.
ICER’s focus is drug pricing and cost effectiveness, so obviously our biologic drugs are in the institute’s crosshairs. This review provided context for a policy roundtable discussion that included patient, payer, and manufacturer input, as well as American College of Rheumatology (ACR) input. We are thankful that ACR had a seat at the table, and thankful ICER is attempting to bring light to the important issues and barriers that perpetuate high drug prices in our marketplace. The discussion was wide ranging but focused on step-edit policies, the role of pharmacy benefit managers (PBMs) in perpetuating high drug prices and the relatively slow uptake of biosimilars in our marketplace.
These issues are critical to every practicing rheumatologist because we each deal daily with the hassles of step-edit/fail-first policies, which hijack our otherwise thoughtful and evidence-based decision making regarding the best treatments for our patients. We know how much (unreimbursed) time it takes our staff to sort through these step edits and prior authorizations, and we have seen recent data regarding how these policies delay care and harm patients. We were thankful to see ICER validate these concerns and note that their suggested guidelines for rational step therapy somewhat mirror those in the Safe Step Act, which ACR supports on a federal legislative level. ACR continues to vigorously support the grandfathering of any patient on an effective treatment, regardless of changes in insurance or formulary; this was an issue of robust debate at their meeting, and this patient-centric position is not uniformly held among policymakers, unfortunately.
ACR agrees with ICER’s conclusion that transparency in the PBM system regarding rebates should be promoted and that opaque rebate negotiations between PBMs and manufacturers both incentivize higher prices and block access to the marketplace for cheaper biosimilar options.
Additionally, ICER and ACR agree about the critical role that biosimilar uptake will play in controlling drug costs. While we do not yet have any biosimilars that have been deemed interchangeable by the Food and Drug Administration, we agree with ICER that data regarding comparable efficacy and safety of biosimilars to their originator products is very reassuring. While the decision to switch to a biosimilar should be an individual decision between a provider and patient, and while we recognize with frustration that many FDA-approved biosimilars are not commercially available because of patent law, it is clear that the current costs of our biologic drugs are not sustainable and the uptake of biosimilars will be critical if we hope our health care economy can continue to support coverage of these life-changing drugs in years to come. We agree with ICER that it is incumbent upon prescribers to reassure our patients regarding the safety and efficacy of these drugs.
Christopher Phillips, MD , is a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the ACR, under the guidance of the Committee on Rheumatologic Care. He attended the initial ICER rheumatoid arthritis review meeting in 2017 on behalf of ACR. In 2019, Dr. Phillips served as a reviewer and clinical expert to the ICER panel and participated in the policy roundtable discussion.
Adequate evidence shows that adding a Janus kinase (JAK) inhibitor to conventional disease-modifying antirheumatic drug therapy provides a net health benefit for patients with rheumatoid arthritis, compared with conventional drugs alone, according to a report by an independent research institute. But the long-term economic value of JAK inhibitors for rheumatoid arthritis is less clear, the report by the Institute for Clinical and Economic Review (ICER) indicates.
ICER on Jan. 9 released a finalized report and policy recommendations on JAK inhibitors and biosimilars for rheumatoid arthritis. The report reviews current evidence for JAK inhibitors for adults with moderately active to severely active rheumatoid arthritis.
Since the nonprofit’s 2017 review of targeted immune modulators for rheumatoid arthritis, two JAK inhibitors, baricitinib (Olumiant) and upadacitinib (Rinvoq), were approved by the Food and Drug Administration. At a December 2019 public meeting of the California Technology Assessment Forum (CTAF), one of ICER’s independent evidence appraisal committees, panelists reviewed recent evidence.
A pricey comparator
In ICER’s analysis, the JAK inhibitor upadacitinib reached common thresholds for cost-effectiveness when compared with adalimumab (Humira). Nevertheless, the 14 members of the independent evidence appraisal committee voted that upadacitinib’s long-term economic value was “low” (8 votes) or “intermediate” (6 votes). Concerns about the generalizability of phase 3 clinical trial data to patients in the real world were among the reservations noted by panelists. Furthermore, “legitimate questions remain about whether or not adalimumab, launched 17 years ago, is fairly priced to begin with,” Pamela Bradt, MD, MPH, ICER’s chief scientific officer, said in a news release.
The panel did not vote on the economic value of tofacitinib (Xeljanz) or baricitinib, the two other JAK inhibitors that are approved for rheumatoid arthritis, because head-to-head evidence against adalimumab was insufficient, ICER said.
“Rheumatoid arthritis is a progressively disabling condition, and patients are fortunate to have multiple therapy options – including biosimilars – that effectively slow disease progression,” Dr. Bradt said. “Many economists might expect medicines to become more affordable in an increasingly crowded therapeutic class; however, because the current rebate structure has erected barriers between patients and several emerging RA therapies, traditional market dynamics have been unable to drive down prices.”
Weighing efficacy and cost
Panelists found that the net health benefit provided by upadacitinib is superior to that provided by adalimumab. At the same time, they said that there is insufficient head-to-head evidence to distinguish between the net health benefit of upadacitinib and tofacitinib or to demonstrate that tofacitinib is superior to adalimumab. Evidence comparing baricitinib to adalimumab does not exist.
CTAF members unanimously agreed that adequate evidence demonstrates that the biosimilar infliximab-dyyb (Inflectra) is clinically equivalent to its reference biologic, infliximab (Remicade).
Economic modeling demonstrated that upadacitinib plus a conventional drug achieves marginally higher quality of life than adalimumab plus a conventional drug does, at similar costs. “Based on this comparison with adalimumab, ICER’s value-based price benchmark range for upadacitinib is between $44,000 and $45,000,” according to the ICER news release. “This benchmark represents a 25% discount off of upadacitinib’s annual list price of $59,860, a suggested discount that is consistent with the rebates we assume the manufacturer is currently offering.”
After the voting session, various experts, including clinicians, patient advocates, and representatives from manufacturers and insurance companies, made the following policy recommendations:
- Regulatory intervention may be needed to ensure that drug prices do not continue to increase further from reasonable alignment with added benefits for patients.
- Insurers, pharmacy benefit managers, and employers should increase transparency around the role of discounts and rebates in formulary design.
- Policymakers should aim to create a system that rewards lower-priced biosimilar treatment options.
Adequate evidence shows that adding a Janus kinase (JAK) inhibitor to conventional disease-modifying antirheumatic drug therapy provides a net health benefit for patients with rheumatoid arthritis, compared with conventional drugs alone, according to a report by an independent research institute. But the long-term economic value of JAK inhibitors for rheumatoid arthritis is less clear, the report by the Institute for Clinical and Economic Review (ICER) indicates.
ICER on Jan. 9 released a finalized report and policy recommendations on JAK inhibitors and biosimilars for rheumatoid arthritis. The report reviews current evidence for JAK inhibitors for adults with moderately active to severely active rheumatoid arthritis.
Since the nonprofit’s 2017 review of targeted immune modulators for rheumatoid arthritis, two JAK inhibitors, baricitinib (Olumiant) and upadacitinib (Rinvoq), were approved by the Food and Drug Administration. At a December 2019 public meeting of the California Technology Assessment Forum (CTAF), one of ICER’s independent evidence appraisal committees, panelists reviewed recent evidence.
A pricey comparator
In ICER’s analysis, the JAK inhibitor upadacitinib reached common thresholds for cost-effectiveness when compared with adalimumab (Humira). Nevertheless, the 14 members of the independent evidence appraisal committee voted that upadacitinib’s long-term economic value was “low” (8 votes) or “intermediate” (6 votes). Concerns about the generalizability of phase 3 clinical trial data to patients in the real world were among the reservations noted by panelists. Furthermore, “legitimate questions remain about whether or not adalimumab, launched 17 years ago, is fairly priced to begin with,” Pamela Bradt, MD, MPH, ICER’s chief scientific officer, said in a news release.
The panel did not vote on the economic value of tofacitinib (Xeljanz) or baricitinib, the two other JAK inhibitors that are approved for rheumatoid arthritis, because head-to-head evidence against adalimumab was insufficient, ICER said.
“Rheumatoid arthritis is a progressively disabling condition, and patients are fortunate to have multiple therapy options – including biosimilars – that effectively slow disease progression,” Dr. Bradt said. “Many economists might expect medicines to become more affordable in an increasingly crowded therapeutic class; however, because the current rebate structure has erected barriers between patients and several emerging RA therapies, traditional market dynamics have been unable to drive down prices.”
Weighing efficacy and cost
Panelists found that the net health benefit provided by upadacitinib is superior to that provided by adalimumab. At the same time, they said that there is insufficient head-to-head evidence to distinguish between the net health benefit of upadacitinib and tofacitinib or to demonstrate that tofacitinib is superior to adalimumab. Evidence comparing baricitinib to adalimumab does not exist.
CTAF members unanimously agreed that adequate evidence demonstrates that the biosimilar infliximab-dyyb (Inflectra) is clinically equivalent to its reference biologic, infliximab (Remicade).
Economic modeling demonstrated that upadacitinib plus a conventional drug achieves marginally higher quality of life than adalimumab plus a conventional drug does, at similar costs. “Based on this comparison with adalimumab, ICER’s value-based price benchmark range for upadacitinib is between $44,000 and $45,000,” according to the ICER news release. “This benchmark represents a 25% discount off of upadacitinib’s annual list price of $59,860, a suggested discount that is consistent with the rebates we assume the manufacturer is currently offering.”
After the voting session, various experts, including clinicians, patient advocates, and representatives from manufacturers and insurance companies, made the following policy recommendations:
- Regulatory intervention may be needed to ensure that drug prices do not continue to increase further from reasonable alignment with added benefits for patients.
- Insurers, pharmacy benefit managers, and employers should increase transparency around the role of discounts and rebates in formulary design.
- Policymakers should aim to create a system that rewards lower-priced biosimilar treatment options.