User login
Priority review granted to lenalidomide for FL, MZL
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).

Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).
Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).
Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
FDA approves subcutaneous trastuzumab for HER2-positive breast cancers
The Food and Drug Administration has approved a subcutaneous formulation of trastuzumab – trastuzumab and hyaluronidase-oysk (Herceptin Hylecta) – for the treatment of patients with HER2-overexpressing breast cancer.
Approval of the subcutaneous formulation is based on results from two clinical studies in HER2-positive early breast cancer – HannaH and SafeHER, the FDA said in a press release.
In the phase 3 HannaH study (NCT00950300), researchers compared neoadjuvant and adjuvant trastuzumab and hyaluronidase-oysk with intravenous trastuzumab, both in combination with chemotherapy. Trastuzumab and hyaluronidase-oysk proved noninferior to intravenous trastuzumab in this trial (Lancet Oncol. 2012 Sep;13[9]:869-78).
In the phase 3 SafeHER study (NCT01566721), the safety profile of trastuzumab and hyaluronidase-oysk was deemed consistent with the known safety profiles of intravenous trastuzumab and trastuzumab and hyaluronidase-oysk (Eur J Cancer. 2017 Sep;82:237-246).
The most common adverse reactions observed in patients receiving the subcutaneous formulation were fatigue, arthralgia, diarrhea, injection-site reaction, upper respiratory tract infection, rash, myalgia, nausea, headache, edema, flushing, pyrexia, cough, and pain in extremity, the FDA said.
Trastuzumab plus hyaluronidase-oysk was approved with a black box warning detailing the risk of cardiomyopathy, pulmonary toxicity, and embryo-fetal toxicity associated with the product.
The recommended dose is 600 mg/10,000 units (600 mg trastuzumab and 10,000 units hyaluronidase) administered subcutaneously over approximately 2-5 minutes once every 3 weeks.
Additional details can be found in the prescribing information.
The Food and Drug Administration has approved a subcutaneous formulation of trastuzumab – trastuzumab and hyaluronidase-oysk (Herceptin Hylecta) – for the treatment of patients with HER2-overexpressing breast cancer.
Approval of the subcutaneous formulation is based on results from two clinical studies in HER2-positive early breast cancer – HannaH and SafeHER, the FDA said in a press release.
In the phase 3 HannaH study (NCT00950300), researchers compared neoadjuvant and adjuvant trastuzumab and hyaluronidase-oysk with intravenous trastuzumab, both in combination with chemotherapy. Trastuzumab and hyaluronidase-oysk proved noninferior to intravenous trastuzumab in this trial (Lancet Oncol. 2012 Sep;13[9]:869-78).
In the phase 3 SafeHER study (NCT01566721), the safety profile of trastuzumab and hyaluronidase-oysk was deemed consistent with the known safety profiles of intravenous trastuzumab and trastuzumab and hyaluronidase-oysk (Eur J Cancer. 2017 Sep;82:237-246).
The most common adverse reactions observed in patients receiving the subcutaneous formulation were fatigue, arthralgia, diarrhea, injection-site reaction, upper respiratory tract infection, rash, myalgia, nausea, headache, edema, flushing, pyrexia, cough, and pain in extremity, the FDA said.
Trastuzumab plus hyaluronidase-oysk was approved with a black box warning detailing the risk of cardiomyopathy, pulmonary toxicity, and embryo-fetal toxicity associated with the product.
The recommended dose is 600 mg/10,000 units (600 mg trastuzumab and 10,000 units hyaluronidase) administered subcutaneously over approximately 2-5 minutes once every 3 weeks.
Additional details can be found in the prescribing information.
The Food and Drug Administration has approved a subcutaneous formulation of trastuzumab – trastuzumab and hyaluronidase-oysk (Herceptin Hylecta) – for the treatment of patients with HER2-overexpressing breast cancer.
Approval of the subcutaneous formulation is based on results from two clinical studies in HER2-positive early breast cancer – HannaH and SafeHER, the FDA said in a press release.
In the phase 3 HannaH study (NCT00950300), researchers compared neoadjuvant and adjuvant trastuzumab and hyaluronidase-oysk with intravenous trastuzumab, both in combination with chemotherapy. Trastuzumab and hyaluronidase-oysk proved noninferior to intravenous trastuzumab in this trial (Lancet Oncol. 2012 Sep;13[9]:869-78).
In the phase 3 SafeHER study (NCT01566721), the safety profile of trastuzumab and hyaluronidase-oysk was deemed consistent with the known safety profiles of intravenous trastuzumab and trastuzumab and hyaluronidase-oysk (Eur J Cancer. 2017 Sep;82:237-246).
The most common adverse reactions observed in patients receiving the subcutaneous formulation were fatigue, arthralgia, diarrhea, injection-site reaction, upper respiratory tract infection, rash, myalgia, nausea, headache, edema, flushing, pyrexia, cough, and pain in extremity, the FDA said.
Trastuzumab plus hyaluronidase-oysk was approved with a black box warning detailing the risk of cardiomyopathy, pulmonary toxicity, and embryo-fetal toxicity associated with the product.
The recommended dose is 600 mg/10,000 units (600 mg trastuzumab and 10,000 units hyaluronidase) administered subcutaneously over approximately 2-5 minutes once every 3 weeks.
Additional details can be found in the prescribing information.
Anthrax booster expanded to 3 years for moderate-risk groups
A booster dose for pre-exposure prophylaxis with an anthrax vaccine may be given at 3 years after an initial series for individuals not currently at risk who wish to maintain protection, according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
In a unanimous 15-0 vote at the February meeting, ACIP committee members agreed on the recommendation after adjusting the wording to reflect a permissive, rather than mandated, guidance.
William Bower, MD, of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), presented data on Anthrax Vaccine Adsorbed (AVA) to support its protective effects over a longer booster dose interval.
The recommendations apply to persons aged 18 years or older who are not currently at high risk of exposure to Bacillus anthracis, but who might need to deploy to a high-risk area quickly, such as military personnel, Dr. Bower said.
In addition, data suggest that adults who have started, but not completed the pre-exposure priming series, can transition to the postexposure schedule prior to entering a high-risk area, he noted.
The previous pre-exposure anthrax vaccination schedule was a three-dose priming series at 0, 1, and 3 months, followed by a booster at 12 months and 18 months, then annually.
with “sustained immunological memory to at least month 42,” and suggested that even longer intervals between boosters may be possible, Dr. Bower said.
A dosing schedule of intramuscular injections at 0 and at 1 month and 6 months, with a booster at 42 months yielded survival estimates of approximately 84%-93%.
Dr. Bower noted that a new vaccine, AV7909, has demonstrated safety and effectiveness similar to AVA and could be used for pre-exposure prophylaxis if AVA is not available. AVA remains the preferred option, but ultimately will be replaced by AV7909, when the current AVA stockpile is exhausted.
Additional safety data on AV7909 will be reviewed by ACIP as they become available, and future guidance from the CDC will include statements on dosing for special populations including pregnant and breastfeeding women, said Dr. Bower.
“We anticipate that this [anthrax vaccine] work group will reconvene in 2021 to review data from pending studies” of AV7909, he said.
The ACIP members had no financial conflicts to disclose.
A booster dose for pre-exposure prophylaxis with an anthrax vaccine may be given at 3 years after an initial series for individuals not currently at risk who wish to maintain protection, according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
In a unanimous 15-0 vote at the February meeting, ACIP committee members agreed on the recommendation after adjusting the wording to reflect a permissive, rather than mandated, guidance.
William Bower, MD, of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), presented data on Anthrax Vaccine Adsorbed (AVA) to support its protective effects over a longer booster dose interval.
The recommendations apply to persons aged 18 years or older who are not currently at high risk of exposure to Bacillus anthracis, but who might need to deploy to a high-risk area quickly, such as military personnel, Dr. Bower said.
In addition, data suggest that adults who have started, but not completed the pre-exposure priming series, can transition to the postexposure schedule prior to entering a high-risk area, he noted.
The previous pre-exposure anthrax vaccination schedule was a three-dose priming series at 0, 1, and 3 months, followed by a booster at 12 months and 18 months, then annually.
with “sustained immunological memory to at least month 42,” and suggested that even longer intervals between boosters may be possible, Dr. Bower said.
A dosing schedule of intramuscular injections at 0 and at 1 month and 6 months, with a booster at 42 months yielded survival estimates of approximately 84%-93%.
Dr. Bower noted that a new vaccine, AV7909, has demonstrated safety and effectiveness similar to AVA and could be used for pre-exposure prophylaxis if AVA is not available. AVA remains the preferred option, but ultimately will be replaced by AV7909, when the current AVA stockpile is exhausted.
Additional safety data on AV7909 will be reviewed by ACIP as they become available, and future guidance from the CDC will include statements on dosing for special populations including pregnant and breastfeeding women, said Dr. Bower.
“We anticipate that this [anthrax vaccine] work group will reconvene in 2021 to review data from pending studies” of AV7909, he said.
The ACIP members had no financial conflicts to disclose.
A booster dose for pre-exposure prophylaxis with an anthrax vaccine may be given at 3 years after an initial series for individuals not currently at risk who wish to maintain protection, according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
In a unanimous 15-0 vote at the February meeting, ACIP committee members agreed on the recommendation after adjusting the wording to reflect a permissive, rather than mandated, guidance.
William Bower, MD, of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), presented data on Anthrax Vaccine Adsorbed (AVA) to support its protective effects over a longer booster dose interval.
The recommendations apply to persons aged 18 years or older who are not currently at high risk of exposure to Bacillus anthracis, but who might need to deploy to a high-risk area quickly, such as military personnel, Dr. Bower said.
In addition, data suggest that adults who have started, but not completed the pre-exposure priming series, can transition to the postexposure schedule prior to entering a high-risk area, he noted.
The previous pre-exposure anthrax vaccination schedule was a three-dose priming series at 0, 1, and 3 months, followed by a booster at 12 months and 18 months, then annually.
with “sustained immunological memory to at least month 42,” and suggested that even longer intervals between boosters may be possible, Dr. Bower said.
A dosing schedule of intramuscular injections at 0 and at 1 month and 6 months, with a booster at 42 months yielded survival estimates of approximately 84%-93%.
Dr. Bower noted that a new vaccine, AV7909, has demonstrated safety and effectiveness similar to AVA and could be used for pre-exposure prophylaxis if AVA is not available. AVA remains the preferred option, but ultimately will be replaced by AV7909, when the current AVA stockpile is exhausted.
Additional safety data on AV7909 will be reviewed by ACIP as they become available, and future guidance from the CDC will include statements on dosing for special populations including pregnant and breastfeeding women, said Dr. Bower.
“We anticipate that this [anthrax vaccine] work group will reconvene in 2021 to review data from pending studies” of AV7909, he said.
The ACIP members had no financial conflicts to disclose.
FROM AN ACIP MEETING
ACIP unanimously supports updates to Japanese encephalitis vaccination
according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.![]()
Japanese encephalitis (JE) virus is a mosquito-borne flavivirus and those at risk for infection include travelers to countries where JE is endemic, as well as laboratory personnel who work with the virus.
The committee voted unanimously 15-0 in favor of the recommendations, which also advised vaccination for those whose travels in endemic areas are uncertain, but not for travelers with low-risk itineraries “such as shorter term travel limited to urban areas or travel that occurs outside of a well-defined JE virus transmission season.”
Susan Hills, MD, of the of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases, presented data in support of the recommendations.
A second unanimous vote confirmed recommendations for a primary series schedule for JE vaccination for adults aged 18-65 years as “two doses of vaccine administered on days 0 and 7-28.”
The third vote, also a unanimous 15-0, updated recommendations for a JE booster dose. The new recommendation is that adults and children receive a booster dose (a third dose) at least a year after completion of the primary JE vaccine series “if ongoing exposure or re-exposure to JE virus is expected.”
The currently available Japanese encephalitis vaccine in the United States is an inactivated Vero cell culture-derived vaccine marketed as IXIARO that was approved in March 2009 for individuals aged 17 years and older and approved in May 2013 for children aged 2 months through 16 years.
The ACIP members had no financial conflicts to disclose.
according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.![]()
Japanese encephalitis (JE) virus is a mosquito-borne flavivirus and those at risk for infection include travelers to countries where JE is endemic, as well as laboratory personnel who work with the virus.
The committee voted unanimously 15-0 in favor of the recommendations, which also advised vaccination for those whose travels in endemic areas are uncertain, but not for travelers with low-risk itineraries “such as shorter term travel limited to urban areas or travel that occurs outside of a well-defined JE virus transmission season.”
Susan Hills, MD, of the of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases, presented data in support of the recommendations.
A second unanimous vote confirmed recommendations for a primary series schedule for JE vaccination for adults aged 18-65 years as “two doses of vaccine administered on days 0 and 7-28.”
The third vote, also a unanimous 15-0, updated recommendations for a JE booster dose. The new recommendation is that adults and children receive a booster dose (a third dose) at least a year after completion of the primary JE vaccine series “if ongoing exposure or re-exposure to JE virus is expected.”
The currently available Japanese encephalitis vaccine in the United States is an inactivated Vero cell culture-derived vaccine marketed as IXIARO that was approved in March 2009 for individuals aged 17 years and older and approved in May 2013 for children aged 2 months through 16 years.
The ACIP members had no financial conflicts to disclose.
according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.![]()
Japanese encephalitis (JE) virus is a mosquito-borne flavivirus and those at risk for infection include travelers to countries where JE is endemic, as well as laboratory personnel who work with the virus.
The committee voted unanimously 15-0 in favor of the recommendations, which also advised vaccination for those whose travels in endemic areas are uncertain, but not for travelers with low-risk itineraries “such as shorter term travel limited to urban areas or travel that occurs outside of a well-defined JE virus transmission season.”
Susan Hills, MD, of the of the CDC’s National Center for Emerging and Zoonotic Infectious Diseases, presented data in support of the recommendations.
A second unanimous vote confirmed recommendations for a primary series schedule for JE vaccination for adults aged 18-65 years as “two doses of vaccine administered on days 0 and 7-28.”
The third vote, also a unanimous 15-0, updated recommendations for a JE booster dose. The new recommendation is that adults and children receive a booster dose (a third dose) at least a year after completion of the primary JE vaccine series “if ongoing exposure or re-exposure to JE virus is expected.”
The currently available Japanese encephalitis vaccine in the United States is an inactivated Vero cell culture-derived vaccine marketed as IXIARO that was approved in March 2009 for individuals aged 17 years and older and approved in May 2013 for children aged 2 months through 16 years.
The ACIP members had no financial conflicts to disclose.
FROM AN ACIP MEETING
New BinaxNOW influenza test gets CLIA waiver
The Food and Drug Administration has granted the reformulated BinaxNOW Influenza A & B Card 2 waived status under the Clinical Laboratory Improvements Amendments for use with Abbott’s DIGIVAL reader, which means the system is relatively simple and has a low risk of erroneous results.
According to Abbott’s press release, the DIGIVAL reader (formerly known as the Alere Reader) automatically interprets this rapid influenza diagnostic test’s results in seconds and is intended to remove subjectivity from the diagnostic process. The BinaxNOW Influenza Card A & B Card 2 is a Class II assay and complies with the FDA’s new reclassification requirements for rapid influenza diagnostic tests.
The Food and Drug Administration has granted the reformulated BinaxNOW Influenza A & B Card 2 waived status under the Clinical Laboratory Improvements Amendments for use with Abbott’s DIGIVAL reader, which means the system is relatively simple and has a low risk of erroneous results.
According to Abbott’s press release, the DIGIVAL reader (formerly known as the Alere Reader) automatically interprets this rapid influenza diagnostic test’s results in seconds and is intended to remove subjectivity from the diagnostic process. The BinaxNOW Influenza Card A & B Card 2 is a Class II assay and complies with the FDA’s new reclassification requirements for rapid influenza diagnostic tests.
The Food and Drug Administration has granted the reformulated BinaxNOW Influenza A & B Card 2 waived status under the Clinical Laboratory Improvements Amendments for use with Abbott’s DIGIVAL reader, which means the system is relatively simple and has a low risk of erroneous results.
According to Abbott’s press release, the DIGIVAL reader (formerly known as the Alere Reader) automatically interprets this rapid influenza diagnostic test’s results in seconds and is intended to remove subjectivity from the diagnostic process. The BinaxNOW Influenza Card A & B Card 2 is a Class II assay and complies with the FDA’s new reclassification requirements for rapid influenza diagnostic tests.
CDC: United States has hit a plateau with HIV
The annual number of new HIV infections has remained stable in recent years, and the Centers for Disease Control and Prevention says that’s not good – but solutions are at hand.
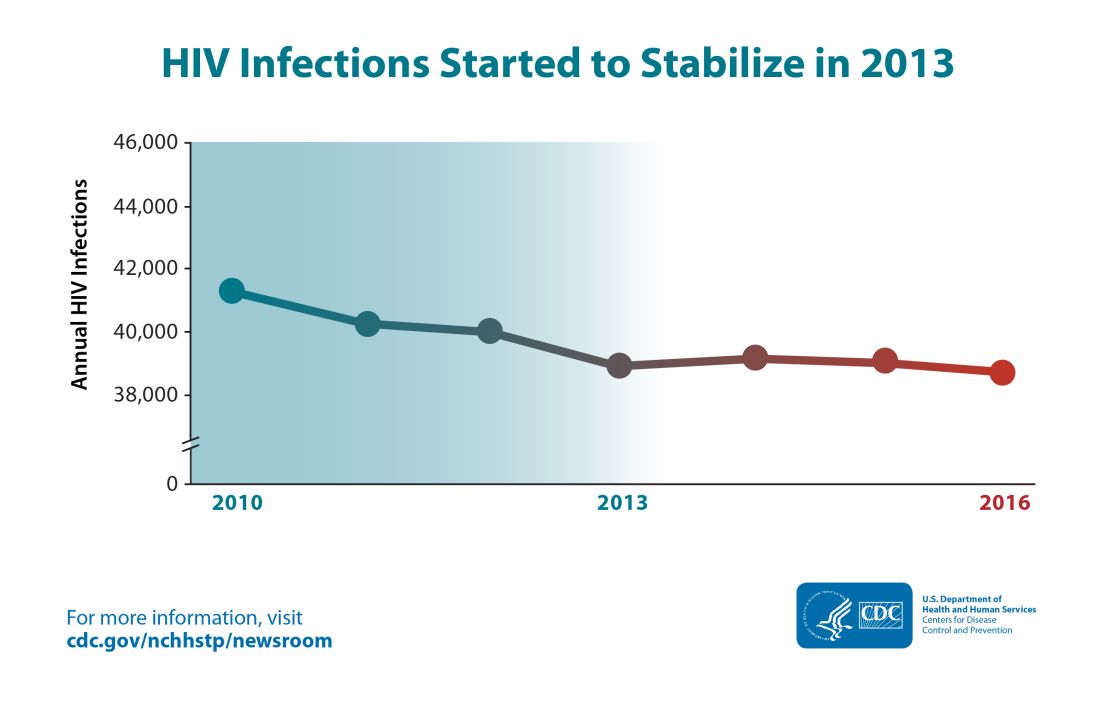
Though the estimated number of new HIV infections declined from just under 42,000 per year in 2010 to about 39,000 annually in 2013, that figure was essentially unchanged by 2016, with 38,700 new HIV infections seen that year.
“CDC estimates that the decline in HIV infections has plateaued because effective HIV prevention and treatment are not adequately reaching those who could most benefit from them. These gaps remain particularly troublesome in rural areas and in the South and among disproportionately affected populations like African Americans and Latinos,” said the CDC in a press release accompanying the report.
The report comes soon after President Trump’s State of the Union address, which announced a new multiagency initiative to eliminate the HIV epidemic in the United States, with the goal of reducing new HIV infections by 90% over the next 10 years. The multipronged initiative will implement geographically targeted HIV elimination teams in areas with high HIV prevalence, pulling together federal agencies, local and state governments, and community-level resources.
The initiative, called “Ending the Epidemic: A Plan for America” will combine an intensified approach to early diagnosis and treatment with efforts to boost uptake of pre-exposure prophylaxis for individuals at high risk for HIV infection.
The new CDC report used CD4 counts reported to the National HIV Surveillance System at the time of diagnosis to identify new (incident) cases and to track prevalence. Much of the report is devoted to finely detailed reporting of HIV incidence across sex, age, race/ethnicity, and transmission mode.
Though some groups, such as people who inject drugs, have seen a decrease of about 30% in the annual rate of new HIV cases, new cases have jumped for other groups. In particular, Latino gay and bisexual men saw new cases climb from 6,400 per year in 2010 to 8,300 in 2016. The incidence rate has stayed high and stable among African American gay and bisexual men, with 9,800 new cases reported in 2010; the same number was seen in 2016.
Among gay and bisexual men overall, the rate has also stayed stable, with about 26,000 new HIV infections reported at the beginning and end of the studied period. White heterosexual women saw about 1,000 new cases per year in 2010 and in 2016.
Some groups saw declines in new cases: African American and Latina heterosexual women each saw a falling incidence of new HIV cases. For the former group, new cases fell from 4,700 to 4,000, while the latter group of women saw new cases drop from 1,200 to 980 per year from 2010 to 2016.
Within these broad groups, HIV incidence also rose among some age groups and fell among others. Decreases were seen for younger African American gay and bisexual men (those aged 13-24 years), but rates increased by about two-thirds for men in this group aged 25-34 years. A similar increase was seen for Latino men in the 25-34 years age group, a change which drove the overall 30% increase in new infections for Latino gay and bisexual men.
White gay and bisexual men saw across-the-board decreases in new infections, though the overall decrease was less than 20%.
For heterosexual individuals as a group, new infections dropped by about 17%, from 10,900 to 9,100 annually. This change was driven mostly by decreases in women identifying as heterosexual.
“After a decades-long struggle, the path to eliminate America’s HIV epidemic is clear,” said Eugene McCray, MD, director of CDC’s Division of HIV/AIDS Prevention, in the press release. “Expanding efforts across the country will close gaps, overcome threats, and turn around troublesome trends.”
The press release cited local work in Washington and New York as evidence that targeted resources can make a difference in reducing new HIV cases. In these two areas, new infections dropped by 23% and 40% respectively from 2010 to 2016.
SOURCE: Centers for Disease Control. CDC Report: www.cdc.gov/hiv/library/reports/hiv-surveillance.html.
The annual number of new HIV infections has remained stable in recent years, and the Centers for Disease Control and Prevention says that’s not good – but solutions are at hand.
Though the estimated number of new HIV infections declined from just under 42,000 per year in 2010 to about 39,000 annually in 2013, that figure was essentially unchanged by 2016, with 38,700 new HIV infections seen that year.
“CDC estimates that the decline in HIV infections has plateaued because effective HIV prevention and treatment are not adequately reaching those who could most benefit from them. These gaps remain particularly troublesome in rural areas and in the South and among disproportionately affected populations like African Americans and Latinos,” said the CDC in a press release accompanying the report.
The report comes soon after President Trump’s State of the Union address, which announced a new multiagency initiative to eliminate the HIV epidemic in the United States, with the goal of reducing new HIV infections by 90% over the next 10 years. The multipronged initiative will implement geographically targeted HIV elimination teams in areas with high HIV prevalence, pulling together federal agencies, local and state governments, and community-level resources.
The initiative, called “Ending the Epidemic: A Plan for America” will combine an intensified approach to early diagnosis and treatment with efforts to boost uptake of pre-exposure prophylaxis for individuals at high risk for HIV infection.
The new CDC report used CD4 counts reported to the National HIV Surveillance System at the time of diagnosis to identify new (incident) cases and to track prevalence. Much of the report is devoted to finely detailed reporting of HIV incidence across sex, age, race/ethnicity, and transmission mode.
Though some groups, such as people who inject drugs, have seen a decrease of about 30% in the annual rate of new HIV cases, new cases have jumped for other groups. In particular, Latino gay and bisexual men saw new cases climb from 6,400 per year in 2010 to 8,300 in 2016. The incidence rate has stayed high and stable among African American gay and bisexual men, with 9,800 new cases reported in 2010; the same number was seen in 2016.
Among gay and bisexual men overall, the rate has also stayed stable, with about 26,000 new HIV infections reported at the beginning and end of the studied period. White heterosexual women saw about 1,000 new cases per year in 2010 and in 2016.
Some groups saw declines in new cases: African American and Latina heterosexual women each saw a falling incidence of new HIV cases. For the former group, new cases fell from 4,700 to 4,000, while the latter group of women saw new cases drop from 1,200 to 980 per year from 2010 to 2016.
Within these broad groups, HIV incidence also rose among some age groups and fell among others. Decreases were seen for younger African American gay and bisexual men (those aged 13-24 years), but rates increased by about two-thirds for men in this group aged 25-34 years. A similar increase was seen for Latino men in the 25-34 years age group, a change which drove the overall 30% increase in new infections for Latino gay and bisexual men.
White gay and bisexual men saw across-the-board decreases in new infections, though the overall decrease was less than 20%.
For heterosexual individuals as a group, new infections dropped by about 17%, from 10,900 to 9,100 annually. This change was driven mostly by decreases in women identifying as heterosexual.
“After a decades-long struggle, the path to eliminate America’s HIV epidemic is clear,” said Eugene McCray, MD, director of CDC’s Division of HIV/AIDS Prevention, in the press release. “Expanding efforts across the country will close gaps, overcome threats, and turn around troublesome trends.”
The press release cited local work in Washington and New York as evidence that targeted resources can make a difference in reducing new HIV cases. In these two areas, new infections dropped by 23% and 40% respectively from 2010 to 2016.
SOURCE: Centers for Disease Control. CDC Report: www.cdc.gov/hiv/library/reports/hiv-surveillance.html.
The annual number of new HIV infections has remained stable in recent years, and the Centers for Disease Control and Prevention says that’s not good – but solutions are at hand.
Though the estimated number of new HIV infections declined from just under 42,000 per year in 2010 to about 39,000 annually in 2013, that figure was essentially unchanged by 2016, with 38,700 new HIV infections seen that year.
“CDC estimates that the decline in HIV infections has plateaued because effective HIV prevention and treatment are not adequately reaching those who could most benefit from them. These gaps remain particularly troublesome in rural areas and in the South and among disproportionately affected populations like African Americans and Latinos,” said the CDC in a press release accompanying the report.
The report comes soon after President Trump’s State of the Union address, which announced a new multiagency initiative to eliminate the HIV epidemic in the United States, with the goal of reducing new HIV infections by 90% over the next 10 years. The multipronged initiative will implement geographically targeted HIV elimination teams in areas with high HIV prevalence, pulling together federal agencies, local and state governments, and community-level resources.
The initiative, called “Ending the Epidemic: A Plan for America” will combine an intensified approach to early diagnosis and treatment with efforts to boost uptake of pre-exposure prophylaxis for individuals at high risk for HIV infection.
The new CDC report used CD4 counts reported to the National HIV Surveillance System at the time of diagnosis to identify new (incident) cases and to track prevalence. Much of the report is devoted to finely detailed reporting of HIV incidence across sex, age, race/ethnicity, and transmission mode.
Though some groups, such as people who inject drugs, have seen a decrease of about 30% in the annual rate of new HIV cases, new cases have jumped for other groups. In particular, Latino gay and bisexual men saw new cases climb from 6,400 per year in 2010 to 8,300 in 2016. The incidence rate has stayed high and stable among African American gay and bisexual men, with 9,800 new cases reported in 2010; the same number was seen in 2016.
Among gay and bisexual men overall, the rate has also stayed stable, with about 26,000 new HIV infections reported at the beginning and end of the studied period. White heterosexual women saw about 1,000 new cases per year in 2010 and in 2016.
Some groups saw declines in new cases: African American and Latina heterosexual women each saw a falling incidence of new HIV cases. For the former group, new cases fell from 4,700 to 4,000, while the latter group of women saw new cases drop from 1,200 to 980 per year from 2010 to 2016.
Within these broad groups, HIV incidence also rose among some age groups and fell among others. Decreases were seen for younger African American gay and bisexual men (those aged 13-24 years), but rates increased by about two-thirds for men in this group aged 25-34 years. A similar increase was seen for Latino men in the 25-34 years age group, a change which drove the overall 30% increase in new infections for Latino gay and bisexual men.
White gay and bisexual men saw across-the-board decreases in new infections, though the overall decrease was less than 20%.
For heterosexual individuals as a group, new infections dropped by about 17%, from 10,900 to 9,100 annually. This change was driven mostly by decreases in women identifying as heterosexual.
“After a decades-long struggle, the path to eliminate America’s HIV epidemic is clear,” said Eugene McCray, MD, director of CDC’s Division of HIV/AIDS Prevention, in the press release. “Expanding efforts across the country will close gaps, overcome threats, and turn around troublesome trends.”
The press release cited local work in Washington and New York as evidence that targeted resources can make a difference in reducing new HIV cases. In these two areas, new infections dropped by 23% and 40% respectively from 2010 to 2016.
SOURCE: Centers for Disease Control. CDC Report: www.cdc.gov/hiv/library/reports/hiv-surveillance.html.
FDA approves label extension for dapagliflozin
The Food and Drug Administration has approved a label extension for Farxiga (dapagliflozin) and Xigduo XR (extended-release dapagliflozin and metformin HCl) for use in patients with type 2 diabetes and moderate renal impairment, lowering the estimated glomerular filtration rate (eGFR) threshold to 45 mL/min per 1.73 m2 from the current60 mL/min per 1.73 m2.
The update is based on results from DERIVE, a phase 3 study in patients with inadequately controlled diabetes and an eGFR of 45-59 mL/min per 1.73 m2 who received either dapagliflozin 10 mg or placebo during a 24-week period. After that time, patients who received dapagliflozin had significant reductions in glycosylated hemoglobin, compared with placebo. The safety profile was similar to that in other studies with dapagliflozin.
The most common adverse events associated with Farxiga are female genital mycotic infections, nasopharyngitis, and urinary tract infections. For Xigduo XR, the most common adverse events are female genital mycotic infection, nasopharyngitis, urinary tract infection, diarrhea, and headache.
“The DERIVE study, which further confirmed the well-established efficacy and safety profile for Farxiga and Xigduo XR, has resulted in important label changes for patients with type 2 diabetes that enable a broader population with impaired renal function to potentially benefit from these important treatment options,” Jim McDermott, PhD, vice president, U.S. medical affairs, diabetes, at AstraZeneca, said in the press release.
Find the full press release on the AstraZeneca website.
The Food and Drug Administration has approved a label extension for Farxiga (dapagliflozin) and Xigduo XR (extended-release dapagliflozin and metformin HCl) for use in patients with type 2 diabetes and moderate renal impairment, lowering the estimated glomerular filtration rate (eGFR) threshold to 45 mL/min per 1.73 m2 from the current60 mL/min per 1.73 m2.
The update is based on results from DERIVE, a phase 3 study in patients with inadequately controlled diabetes and an eGFR of 45-59 mL/min per 1.73 m2 who received either dapagliflozin 10 mg or placebo during a 24-week period. After that time, patients who received dapagliflozin had significant reductions in glycosylated hemoglobin, compared with placebo. The safety profile was similar to that in other studies with dapagliflozin.
The most common adverse events associated with Farxiga are female genital mycotic infections, nasopharyngitis, and urinary tract infections. For Xigduo XR, the most common adverse events are female genital mycotic infection, nasopharyngitis, urinary tract infection, diarrhea, and headache.
“The DERIVE study, which further confirmed the well-established efficacy and safety profile for Farxiga and Xigduo XR, has resulted in important label changes for patients with type 2 diabetes that enable a broader population with impaired renal function to potentially benefit from these important treatment options,” Jim McDermott, PhD, vice president, U.S. medical affairs, diabetes, at AstraZeneca, said in the press release.
Find the full press release on the AstraZeneca website.
The Food and Drug Administration has approved a label extension for Farxiga (dapagliflozin) and Xigduo XR (extended-release dapagliflozin and metformin HCl) for use in patients with type 2 diabetes and moderate renal impairment, lowering the estimated glomerular filtration rate (eGFR) threshold to 45 mL/min per 1.73 m2 from the current60 mL/min per 1.73 m2.
The update is based on results from DERIVE, a phase 3 study in patients with inadequately controlled diabetes and an eGFR of 45-59 mL/min per 1.73 m2 who received either dapagliflozin 10 mg or placebo during a 24-week period. After that time, patients who received dapagliflozin had significant reductions in glycosylated hemoglobin, compared with placebo. The safety profile was similar to that in other studies with dapagliflozin.
The most common adverse events associated with Farxiga are female genital mycotic infections, nasopharyngitis, and urinary tract infections. For Xigduo XR, the most common adverse events are female genital mycotic infection, nasopharyngitis, urinary tract infection, diarrhea, and headache.
“The DERIVE study, which further confirmed the well-established efficacy and safety profile for Farxiga and Xigduo XR, has resulted in important label changes for patients with type 2 diabetes that enable a broader population with impaired renal function to potentially benefit from these important treatment options,” Jim McDermott, PhD, vice president, U.S. medical affairs, diabetes, at AstraZeneca, said in the press release.
Find the full press release on the AstraZeneca website.
FDA accepts supplemental New Drug Application to expand lorcaserin label
The Food and Drug Administration has accepted a supplemental New Drug Application from Eisai to update the label for the weight-loss drug lorcaserin (Belviq) with long-term efficacy and safety data from the CAMELLIA-TIMI 61 trial.
In CAMELLIA-TIMI 61, a double-blind, placebo-controlled, parallel-group, phase 3b/4 clinical trial, about 12,000 people who were overweight or obese and had cardiovascular disease or CVD risk factors, such as type 2 diabetes, received lorcaserin or placebo. In a study published in the New England Journal of Medicine last fall, lorcaserin, a serotonin 2C receptor agonist, met the primary safety endpoint of noninferiority to placebo in the rate of major adverse cardiovascular events (hazard ratio, 0.99; 95% confidence interval, 0.85-1.14; P less than .001).
After meeting the primary endpoint, the trial investigators continued the study, assessing for a broader composite endpoint consisting of cardiovascular death, nonfatal MI, nonfatal stroke, hospitalization caused by unstable angina, heart failure, or coronary revascularization. Although lorcaserin was not superior to placebo in this expanded study, it remained noninferior.
Lorcaserin is contraindicated in women who are pregnant and in patients hypersensitive to lorcaserin. The most common adverse events in patients without diabetes were headache, dizziness, fatigue, nausea, dry mouth, and constipation; in patients with diabetes, the most common adverse events were hypoglycemia, headache, back pain, cough, and fatigue.
“This sNDA file acceptance brings us one step closer to potentially incorporating data into our label based on the first completed large-scale cardiovascular outcomes trial for a weight loss agent,” Lynn Kramer, MD, chief clinical officer and chief medical officer of the Neurology Business Group at Eisai, said in the press release.
Find the full press release on the Eisai website.
The Food and Drug Administration has accepted a supplemental New Drug Application from Eisai to update the label for the weight-loss drug lorcaserin (Belviq) with long-term efficacy and safety data from the CAMELLIA-TIMI 61 trial.
In CAMELLIA-TIMI 61, a double-blind, placebo-controlled, parallel-group, phase 3b/4 clinical trial, about 12,000 people who were overweight or obese and had cardiovascular disease or CVD risk factors, such as type 2 diabetes, received lorcaserin or placebo. In a study published in the New England Journal of Medicine last fall, lorcaserin, a serotonin 2C receptor agonist, met the primary safety endpoint of noninferiority to placebo in the rate of major adverse cardiovascular events (hazard ratio, 0.99; 95% confidence interval, 0.85-1.14; P less than .001).
After meeting the primary endpoint, the trial investigators continued the study, assessing for a broader composite endpoint consisting of cardiovascular death, nonfatal MI, nonfatal stroke, hospitalization caused by unstable angina, heart failure, or coronary revascularization. Although lorcaserin was not superior to placebo in this expanded study, it remained noninferior.
Lorcaserin is contraindicated in women who are pregnant and in patients hypersensitive to lorcaserin. The most common adverse events in patients without diabetes were headache, dizziness, fatigue, nausea, dry mouth, and constipation; in patients with diabetes, the most common adverse events were hypoglycemia, headache, back pain, cough, and fatigue.
“This sNDA file acceptance brings us one step closer to potentially incorporating data into our label based on the first completed large-scale cardiovascular outcomes trial for a weight loss agent,” Lynn Kramer, MD, chief clinical officer and chief medical officer of the Neurology Business Group at Eisai, said in the press release.
Find the full press release on the Eisai website.
The Food and Drug Administration has accepted a supplemental New Drug Application from Eisai to update the label for the weight-loss drug lorcaserin (Belviq) with long-term efficacy and safety data from the CAMELLIA-TIMI 61 trial.
In CAMELLIA-TIMI 61, a double-blind, placebo-controlled, parallel-group, phase 3b/4 clinical trial, about 12,000 people who were overweight or obese and had cardiovascular disease or CVD risk factors, such as type 2 diabetes, received lorcaserin or placebo. In a study published in the New England Journal of Medicine last fall, lorcaserin, a serotonin 2C receptor agonist, met the primary safety endpoint of noninferiority to placebo in the rate of major adverse cardiovascular events (hazard ratio, 0.99; 95% confidence interval, 0.85-1.14; P less than .001).
After meeting the primary endpoint, the trial investigators continued the study, assessing for a broader composite endpoint consisting of cardiovascular death, nonfatal MI, nonfatal stroke, hospitalization caused by unstable angina, heart failure, or coronary revascularization. Although lorcaserin was not superior to placebo in this expanded study, it remained noninferior.
Lorcaserin is contraindicated in women who are pregnant and in patients hypersensitive to lorcaserin. The most common adverse events in patients without diabetes were headache, dizziness, fatigue, nausea, dry mouth, and constipation; in patients with diabetes, the most common adverse events were hypoglycemia, headache, back pain, cough, and fatigue.
“This sNDA file acceptance brings us one step closer to potentially incorporating data into our label based on the first completed large-scale cardiovascular outcomes trial for a weight loss agent,” Lynn Kramer, MD, chief clinical officer and chief medical officer of the Neurology Business Group at Eisai, said in the press release.
Find the full press release on the Eisai website.
Selinexor hits FDA stumbling block
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”
FDA: Safety signal emerged with higher dose of tofacitinib in RA study
the Food and Drug Administration reported.
The trial’s Data Safety and Monitoring Board identified the signal in patients taking a 10-mg dose of tofacitinib twice daily, the FDA said in a safety announcement.
Pfizer, the trial’s sponsor, took “immediate action” to transition patients in the ongoing trial from the 10-mg, twice-daily dose to 5 mg twice daily, which is the approved dose for adult patients with moderate to severe rheumatoid arthritis, the agency said. The 10-mg, twice-daily dose is approved only in the dosing regimen for patients with ulcerative colitis. Xeljanz is also approved to treat psoriatic arthritis. The 11-mg, once-daily dose of Xeljanz XR that is approved to treat rheumatoid arthritis and psoriatic arthritis was not tested in the trial.
The ongoing study was designed to assess risks of cardiovascular events, cancer, and opportunistic infections with tofacitinib 10 mg twice daily or 5 mg twice daily versus the risks in a control group treated with a tumor necrosis factor (TNF) inhibitor, according to the statement.
Patients had to be 50 years of age or older and have at least one cardiovascular risk factor to be eligible for the study, which was required by the agency in 2012 when it approved tofacitinib, the statement says.
The FDA is reviewing trial data and working with Pfizer to better understand the safety signal, its effect on patients, and how tofacitinib should be used, Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a news release. The trial will continue and is expected to be completed by the end of 2019.
“The agency will take appropriate action, as warranted, to ensure patients enrolled in this and other trials are protected and that health care professionals and clinical trial researchers understand the risks associated with this use,” she added.
Health care professionals should follow tofacitinib prescribing information, monitor patients for the signs and symptoms of pulmonary embolism, and advise patients to seek medical attention immediately if they experience those signs and symptoms, according to the statement.
“We are communicating now, given the serious nature of the safety issue, to ensure that patients taking tofacitinib are aware that the FDA still believes the benefits of taking tofacitinib for its approved uses continue to outweigh the risks,” Dr. Woodcock said in the release.
While not approved in rheumatoid arthritis, the 10-mg, twice-daily dose of tofacitinib is approved in the dosing regimen for patients with ulcerative colitis, the release says.
the Food and Drug Administration reported.
The trial’s Data Safety and Monitoring Board identified the signal in patients taking a 10-mg dose of tofacitinib twice daily, the FDA said in a safety announcement.
Pfizer, the trial’s sponsor, took “immediate action” to transition patients in the ongoing trial from the 10-mg, twice-daily dose to 5 mg twice daily, which is the approved dose for adult patients with moderate to severe rheumatoid arthritis, the agency said. The 10-mg, twice-daily dose is approved only in the dosing regimen for patients with ulcerative colitis. Xeljanz is also approved to treat psoriatic arthritis. The 11-mg, once-daily dose of Xeljanz XR that is approved to treat rheumatoid arthritis and psoriatic arthritis was not tested in the trial.
The ongoing study was designed to assess risks of cardiovascular events, cancer, and opportunistic infections with tofacitinib 10 mg twice daily or 5 mg twice daily versus the risks in a control group treated with a tumor necrosis factor (TNF) inhibitor, according to the statement.
Patients had to be 50 years of age or older and have at least one cardiovascular risk factor to be eligible for the study, which was required by the agency in 2012 when it approved tofacitinib, the statement says.
The FDA is reviewing trial data and working with Pfizer to better understand the safety signal, its effect on patients, and how tofacitinib should be used, Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a news release. The trial will continue and is expected to be completed by the end of 2019.
“The agency will take appropriate action, as warranted, to ensure patients enrolled in this and other trials are protected and that health care professionals and clinical trial researchers understand the risks associated with this use,” she added.
Health care professionals should follow tofacitinib prescribing information, monitor patients for the signs and symptoms of pulmonary embolism, and advise patients to seek medical attention immediately if they experience those signs and symptoms, according to the statement.
“We are communicating now, given the serious nature of the safety issue, to ensure that patients taking tofacitinib are aware that the FDA still believes the benefits of taking tofacitinib for its approved uses continue to outweigh the risks,” Dr. Woodcock said in the release.
While not approved in rheumatoid arthritis, the 10-mg, twice-daily dose of tofacitinib is approved in the dosing regimen for patients with ulcerative colitis, the release says.
the Food and Drug Administration reported.
The trial’s Data Safety and Monitoring Board identified the signal in patients taking a 10-mg dose of tofacitinib twice daily, the FDA said in a safety announcement.
Pfizer, the trial’s sponsor, took “immediate action” to transition patients in the ongoing trial from the 10-mg, twice-daily dose to 5 mg twice daily, which is the approved dose for adult patients with moderate to severe rheumatoid arthritis, the agency said. The 10-mg, twice-daily dose is approved only in the dosing regimen for patients with ulcerative colitis. Xeljanz is also approved to treat psoriatic arthritis. The 11-mg, once-daily dose of Xeljanz XR that is approved to treat rheumatoid arthritis and psoriatic arthritis was not tested in the trial.
The ongoing study was designed to assess risks of cardiovascular events, cancer, and opportunistic infections with tofacitinib 10 mg twice daily or 5 mg twice daily versus the risks in a control group treated with a tumor necrosis factor (TNF) inhibitor, according to the statement.
Patients had to be 50 years of age or older and have at least one cardiovascular risk factor to be eligible for the study, which was required by the agency in 2012 when it approved tofacitinib, the statement says.
The FDA is reviewing trial data and working with Pfizer to better understand the safety signal, its effect on patients, and how tofacitinib should be used, Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a news release. The trial will continue and is expected to be completed by the end of 2019.
“The agency will take appropriate action, as warranted, to ensure patients enrolled in this and other trials are protected and that health care professionals and clinical trial researchers understand the risks associated with this use,” she added.
Health care professionals should follow tofacitinib prescribing information, monitor patients for the signs and symptoms of pulmonary embolism, and advise patients to seek medical attention immediately if they experience those signs and symptoms, according to the statement.
“We are communicating now, given the serious nature of the safety issue, to ensure that patients taking tofacitinib are aware that the FDA still believes the benefits of taking tofacitinib for its approved uses continue to outweigh the risks,” Dr. Woodcock said in the release.
While not approved in rheumatoid arthritis, the 10-mg, twice-daily dose of tofacitinib is approved in the dosing regimen for patients with ulcerative colitis, the release says.