User login
Uninjured athlete with edematous arm • Dx?
THE CASE
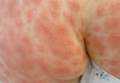
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
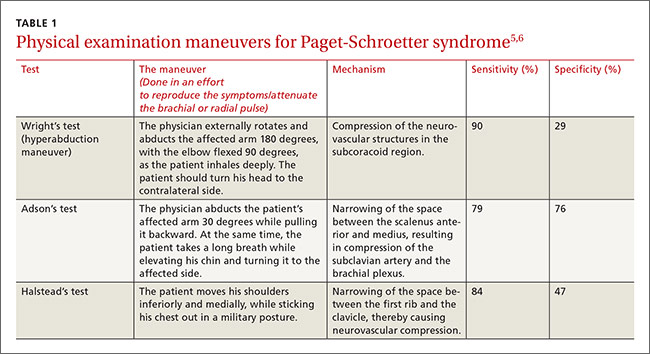
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
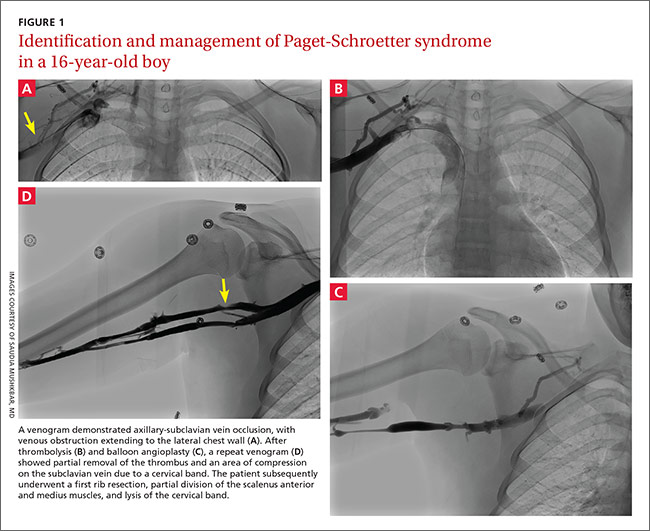
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
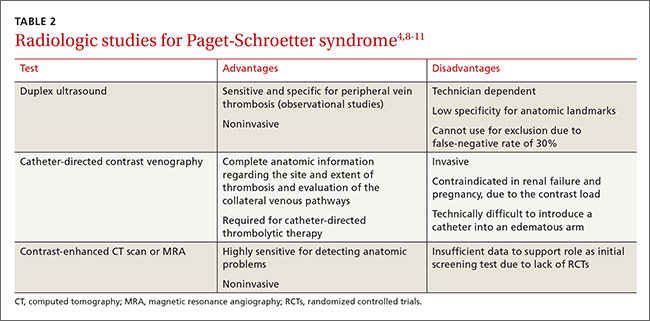
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.
THE CASE
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
THE CASE
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.

Acute promyelocytic leukemia presenting as a paraspinal mass
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
An uncommon presentation of non-small-cell lung cancer with acrometastases to the great toe and index finger
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.
Night of the Living Thrips: An Unusual Outbreak of Thysanoptera Dermatitis
Case Reports
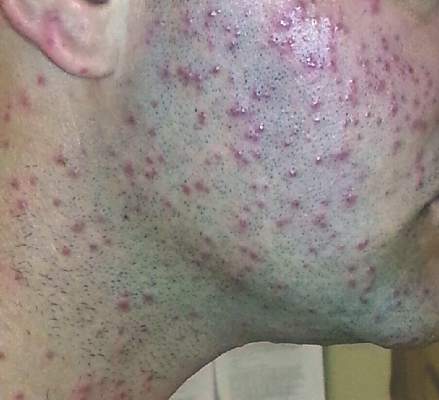
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |

|
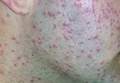
| Figure 2. Papules with classic anemic halos. |
Referral
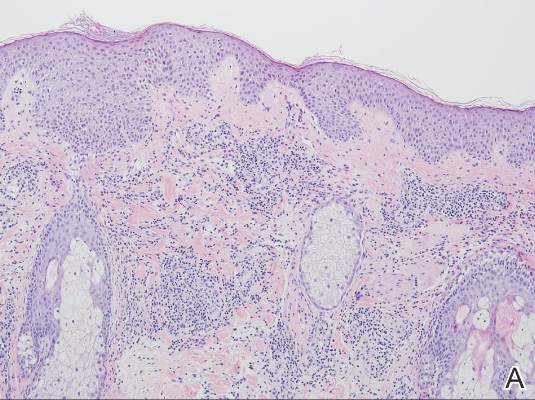
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
| 
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
Case Reports
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
|
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |
|
|
| Figure 2. Papules with classic anemic halos. |
Referral
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
|
|
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
Case Reports
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
|
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |
|
|
| Figure 2. Papules with classic anemic halos. |
Referral
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
|
|
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
Practice Points
- Thysanoptera dermatitis presents as a diffuse cutaneous eruption consisting of scattered pruritic papules to exposed skin surfaces.
- The importance of considering the environmental component of a cutaneous eruption via a thorough understanding of local flora and fauna cannot be underestimated.
- The role of a dermatologist in the rapid identification of a cutaneous eruption in the setting of an acute cluster outbreak is of utmost importance to assist with eliminating infectious and environmental public health threats from the differential diagnosis.
Complete Atrioventricular Nodal Block Due to Malignancy-Related Hypercalcemia
Complete atrioventricular (AV) block can occur due to structural or functional causes. Common structural etiologies include sclerodegenerative disease of the conduction system, ischemic heart disease in the acute or chronic setting, infiltrative myocardial disease, congenital heart disease, and cardiac surgery. Reversible etiologies of complete AV block include drug overdose and electrolyte abnormalities. In the following case study, the authors present a rare case of complete AV block caused by severe hypercalcemia related to malignancy that completely normalized after treatment of the hypercalcemia.
Case Report
A 63-year-old African-American man with metastatic carcinoma of the lungs (Figure 1) with unknown primary cancer was found to have a serum calcium level of 17.5 mg/dL (reference range:8.4-10.2 mg/dL) on routine preoperative laboratory testing prior to placement of a surgical port for chemotherapy. The patient also was noted to have a slow heart rate, and his electrocardiogram revealed a third-degree AV block with an escape rhythm at 29 bpm with a prolonged corrected QT (QTc) of 556 ms (Figure 2).
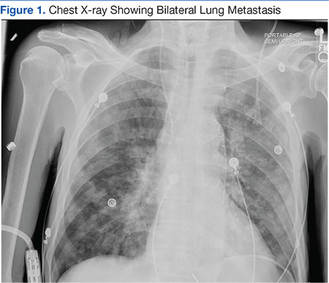
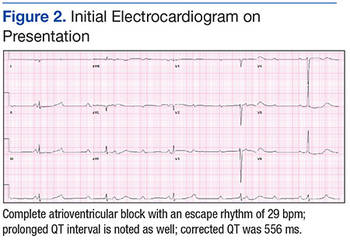
Although the patient reported nonspecific symptoms of fatigue, anorexia, dysphagia, and weight loss for 3 months, there were no new symptoms of dizziness, chest discomfort, or syncope. His past medical history included hypertension, hyperlipidemia, chronic kidney disease, obstructive sleep apnea, and the recently discovered bilateral lung metastasis. The patient reported no prior history of cardiac arrhythmias, coronary artery disease, or structural heart defects. His outpatient medications included aspirin, amlodipine, bupropion, hydralazine, and simvastatin.
At the physical examination the patient was cachectic but in no apparent distress. His heart rate escape rhythm was 29 bpm, with no murmurs and mildly reduced breath sounds. The patient’s blood pressure was 110/70. After correction for albumin, the serum calcium level was 17.8 mg/dL; ionized calcium level was 8.6 mg/dL; parathyroid hormone was 7.6 pg/mL (normal range, 12-88 pg/mL); parathyroid hormone-related protein was 6.4 pmol/L (normal range, < 2.0 pmol/L); potassium was 3.4 mmol/L (normal range, 3.5 – 5.1 mmol/L); and magnesium was 2.01 mg/dL. The patient’s thyroid stimulating hormone level was normal, and serial cardiac enzymes stayed within the reference range.
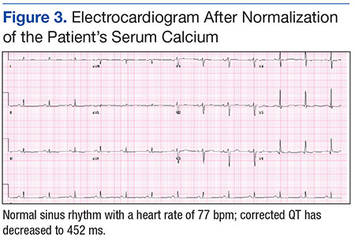
The patient was admitted to a cardiac care unit. A temporary transvenous pacemaker was placed, and the hypercalcemia was treated with aggressive fluid hydration, calcitonin, and zoledronic acid. Serum calcium gradually decreased to 14.6 mg/dL the following day and 9.6 mg/dL the subsequent day. The normalization of calcium resulted in resolution of complete heart block (Figure 3). The patient did not experience recurrence of AV nodal dysfunction and eventually died 3 months later due to his advanced metastatic disease.
Discussion
The reported cardiovascular effects of hypercalcemia include hypertension, arrhythmias, increased myocardial contractility at serum calcium level below 15 mg/dL, and myocardial depression above that level. Electrocardiographic manifestations of hypercalcemia include a shortened ST segment leading to a short corrected QT interval (QTc), slight increase in T wave duration, and rarely, Osborn waves or J waves.1-3 However, its influence on the AV node is less clear.
One small study assessed the prevalence of cardiac arrhythmias and conduction disturbances in 20 patients with hyperparathyroidism and moderate hypercalcemia and found no increase in the frequency of arrhythmias or high grade AV block.4
There are reports of conduction abnormality secondary to experimentally induced hypercalcemia in the literature. Hoff and colleagues described findings of AV block generated by the injection of IV calcium in dogs.5 In 2 human subjects, sinus bradycardia was precipitated after they received IV infusion of calcium gluconate.6 Shah and colleagues described 2 patients with sinus node dysfunction attributed to hypercalcemia secondary to hyperparathyroidism.7
Case reports of AV nodal dysfunction provoked by hypercalcemia have primarily occurred in the setting of primary hyperparathyroidism.8,9 Milk-alkali syndrome and vitamin D related hypercalcemia also have been reported to cause complete heart block.10,11 Reports of malignancy-related hypercalcemia causing conduction abnormalities are rare. The authors also found one case report of marked sinus bradycardia due to hypercalcemia related to breast cancer.12The case study presented in this report is rare because the patient developed complete AV block due to malignancy-related hypercalcemia that resolved completely with resolution of hypercalcemia. The prolongation of the QTc interval was another unique electrocardiographic change observed in this case. Calcium levels are inversely proportional to the QTc interval, and hypercalcemia is typically associated with a shortened QTc interval. However, this patient had a prolonged QTc without any other clear-cut cause. His hypokalemia was of a mild degree and not severe enough to produce such a long QTc interval. A possible explanation of QTc prolongation may be an increase in the T wave width associated with a serum calcium level above 16 mg/dL.
The pathophysiology of hypercalcemia-induced AV nodal conduction system disease is unknown. Calcium deposition in AV nodes of elderly patients has been associated with paroxysmal 2:1 AV block.8 It could be postulated that elevated serum calcium levels predispose to calcium deposition in cardiac conduction tissue, leading to progressive dysfunction. Although this theory may be applicable in a chronic setting, the mechanism in an acute setting likely relates to elevated serum levels of calcium that causes an alteration in electrochemical gradients. These elevated serum levels also increase intracellular calcium. This rise may result in increased calmodulin activation on the intracellular portion of the myocyte cell membrane and consequent enhanced sodium channel activation, which may then inhibit AV nodal conduction.13
Conclusion
Physicians should be aware that severe hypercalcemia can cause significant conduction system alterations, including complete AV block. A short QTc interval is typical, but a prolonged QTc interval also may be seen. While temporary support with a transvenous pacemaker may be needed, the conduction system abnormality is expected to resolve by treatment of the underlying hypercalcemia.
1. Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol. 1979;44(2):243-248.
2. Bronsky D, Dubin A, Waldstein SS, Kushner DS. Calcium and the electrocardiogram II. The electrocardiographic manifestations of hyperparathyroidism and of marked hypercalcemia from various other etiologies. Am J Cardiol. 1961;7(6):833-839.
3. Otero J, Lenihan DJ. The "normothermic" Osborn wave induced by severe hypercalcemia. Tex Heart Inst J. 2000;27(3):316-317.
4. Rosenqvist M, Nordenström J, Andersson M, Edhag OK. Cardiac conduction inpatients with hypercalcaemia due to primary hyperparathyroidism. Clin Endocrinol (Oxf). 1992;37(1):29-33.
5. Hoff H, Smith P, Winkler A. Electrocardiographic changes and concentration of calcium in serum following injection of calcium chloride. Am J Physiol. 1939;125:162-171.
6. Howard JE, Hopkins TR, Connor TB. The use of intravenous calcium as a measure of activity of the parathyroid glands. Trans Assoc Am Physicians. 1952;65:351-358.
7. Shah AP, Lopez A, Wachsner RY, Meymandi SK, El-Bialy AK, Ichiuji AM. Sinus node dysfunction secondary to hyperparathyroidism. J Cardiovasc Pharmacol Ther. 2004;9(2):145-147.
8. Vosnakidis A, Polymeropoulos K, Zaragoulidis P, Zarifis I. Atrioventricular nodal dysfunction secondary to hyperparathyroidism. J Thoracic Dis. 2013;5(3):E90-E92.
9. Crum WB, Till HJ. Hyperparathyroidism with Wenckebach's phenomenon. Am J Cardiol. 1960;6:838-840.
10. Ginsberg H, Schwarz KV. Letter: hypercalcemia and complete heart block. Ann Intern Med. 1973;79(6):903.
11. Garg G, Khadgwat R, Khandelwal D, Gupta N. Vitamin D toxicity presenting as hypercalcemia and complete heart block: an interesting case report. Indian J Endocrinol Metab. 2012;16 (suppl 2):S423-S425.
12. Badertscher E, Warnica JW, Ernst DS. Acute hypercalcemia and severe bradycardia in a patient with breast cancer. CMAJ. 1993;148(9):1506-1508.
13. Potet F, Chagot B, Anghelescu M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009;284(13):8846-8854.
Complete atrioventricular (AV) block can occur due to structural or functional causes. Common structural etiologies include sclerodegenerative disease of the conduction system, ischemic heart disease in the acute or chronic setting, infiltrative myocardial disease, congenital heart disease, and cardiac surgery. Reversible etiologies of complete AV block include drug overdose and electrolyte abnormalities. In the following case study, the authors present a rare case of complete AV block caused by severe hypercalcemia related to malignancy that completely normalized after treatment of the hypercalcemia.
Case Report
A 63-year-old African-American man with metastatic carcinoma of the lungs (Figure 1) with unknown primary cancer was found to have a serum calcium level of 17.5 mg/dL (reference range:8.4-10.2 mg/dL) on routine preoperative laboratory testing prior to placement of a surgical port for chemotherapy. The patient also was noted to have a slow heart rate, and his electrocardiogram revealed a third-degree AV block with an escape rhythm at 29 bpm with a prolonged corrected QT (QTc) of 556 ms (Figure 2).
Although the patient reported nonspecific symptoms of fatigue, anorexia, dysphagia, and weight loss for 3 months, there were no new symptoms of dizziness, chest discomfort, or syncope. His past medical history included hypertension, hyperlipidemia, chronic kidney disease, obstructive sleep apnea, and the recently discovered bilateral lung metastasis. The patient reported no prior history of cardiac arrhythmias, coronary artery disease, or structural heart defects. His outpatient medications included aspirin, amlodipine, bupropion, hydralazine, and simvastatin.
At the physical examination the patient was cachectic but in no apparent distress. His heart rate escape rhythm was 29 bpm, with no murmurs and mildly reduced breath sounds. The patient’s blood pressure was 110/70. After correction for albumin, the serum calcium level was 17.8 mg/dL; ionized calcium level was 8.6 mg/dL; parathyroid hormone was 7.6 pg/mL (normal range, 12-88 pg/mL); parathyroid hormone-related protein was 6.4 pmol/L (normal range, < 2.0 pmol/L); potassium was 3.4 mmol/L (normal range, 3.5 – 5.1 mmol/L); and magnesium was 2.01 mg/dL. The patient’s thyroid stimulating hormone level was normal, and serial cardiac enzymes stayed within the reference range.
The patient was admitted to a cardiac care unit. A temporary transvenous pacemaker was placed, and the hypercalcemia was treated with aggressive fluid hydration, calcitonin, and zoledronic acid. Serum calcium gradually decreased to 14.6 mg/dL the following day and 9.6 mg/dL the subsequent day. The normalization of calcium resulted in resolution of complete heart block (Figure 3). The patient did not experience recurrence of AV nodal dysfunction and eventually died 3 months later due to his advanced metastatic disease.
Discussion
The reported cardiovascular effects of hypercalcemia include hypertension, arrhythmias, increased myocardial contractility at serum calcium level below 15 mg/dL, and myocardial depression above that level. Electrocardiographic manifestations of hypercalcemia include a shortened ST segment leading to a short corrected QT interval (QTc), slight increase in T wave duration, and rarely, Osborn waves or J waves.1-3 However, its influence on the AV node is less clear.
One small study assessed the prevalence of cardiac arrhythmias and conduction disturbances in 20 patients with hyperparathyroidism and moderate hypercalcemia and found no increase in the frequency of arrhythmias or high grade AV block.4
There are reports of conduction abnormality secondary to experimentally induced hypercalcemia in the literature. Hoff and colleagues described findings of AV block generated by the injection of IV calcium in dogs.5 In 2 human subjects, sinus bradycardia was precipitated after they received IV infusion of calcium gluconate.6 Shah and colleagues described 2 patients with sinus node dysfunction attributed to hypercalcemia secondary to hyperparathyroidism.7
Case reports of AV nodal dysfunction provoked by hypercalcemia have primarily occurred in the setting of primary hyperparathyroidism.8,9 Milk-alkali syndrome and vitamin D related hypercalcemia also have been reported to cause complete heart block.10,11 Reports of malignancy-related hypercalcemia causing conduction abnormalities are rare. The authors also found one case report of marked sinus bradycardia due to hypercalcemia related to breast cancer.12The case study presented in this report is rare because the patient developed complete AV block due to malignancy-related hypercalcemia that resolved completely with resolution of hypercalcemia. The prolongation of the QTc interval was another unique electrocardiographic change observed in this case. Calcium levels are inversely proportional to the QTc interval, and hypercalcemia is typically associated with a shortened QTc interval. However, this patient had a prolonged QTc without any other clear-cut cause. His hypokalemia was of a mild degree and not severe enough to produce such a long QTc interval. A possible explanation of QTc prolongation may be an increase in the T wave width associated with a serum calcium level above 16 mg/dL.
The pathophysiology of hypercalcemia-induced AV nodal conduction system disease is unknown. Calcium deposition in AV nodes of elderly patients has been associated with paroxysmal 2:1 AV block.8 It could be postulated that elevated serum calcium levels predispose to calcium deposition in cardiac conduction tissue, leading to progressive dysfunction. Although this theory may be applicable in a chronic setting, the mechanism in an acute setting likely relates to elevated serum levels of calcium that causes an alteration in electrochemical gradients. These elevated serum levels also increase intracellular calcium. This rise may result in increased calmodulin activation on the intracellular portion of the myocyte cell membrane and consequent enhanced sodium channel activation, which may then inhibit AV nodal conduction.13
Conclusion
Physicians should be aware that severe hypercalcemia can cause significant conduction system alterations, including complete AV block. A short QTc interval is typical, but a prolonged QTc interval also may be seen. While temporary support with a transvenous pacemaker may be needed, the conduction system abnormality is expected to resolve by treatment of the underlying hypercalcemia.
Complete atrioventricular (AV) block can occur due to structural or functional causes. Common structural etiologies include sclerodegenerative disease of the conduction system, ischemic heart disease in the acute or chronic setting, infiltrative myocardial disease, congenital heart disease, and cardiac surgery. Reversible etiologies of complete AV block include drug overdose and electrolyte abnormalities. In the following case study, the authors present a rare case of complete AV block caused by severe hypercalcemia related to malignancy that completely normalized after treatment of the hypercalcemia.
Case Report
A 63-year-old African-American man with metastatic carcinoma of the lungs (Figure 1) with unknown primary cancer was found to have a serum calcium level of 17.5 mg/dL (reference range:8.4-10.2 mg/dL) on routine preoperative laboratory testing prior to placement of a surgical port for chemotherapy. The patient also was noted to have a slow heart rate, and his electrocardiogram revealed a third-degree AV block with an escape rhythm at 29 bpm with a prolonged corrected QT (QTc) of 556 ms (Figure 2).
Although the patient reported nonspecific symptoms of fatigue, anorexia, dysphagia, and weight loss for 3 months, there were no new symptoms of dizziness, chest discomfort, or syncope. His past medical history included hypertension, hyperlipidemia, chronic kidney disease, obstructive sleep apnea, and the recently discovered bilateral lung metastasis. The patient reported no prior history of cardiac arrhythmias, coronary artery disease, or structural heart defects. His outpatient medications included aspirin, amlodipine, bupropion, hydralazine, and simvastatin.
At the physical examination the patient was cachectic but in no apparent distress. His heart rate escape rhythm was 29 bpm, with no murmurs and mildly reduced breath sounds. The patient’s blood pressure was 110/70. After correction for albumin, the serum calcium level was 17.8 mg/dL; ionized calcium level was 8.6 mg/dL; parathyroid hormone was 7.6 pg/mL (normal range, 12-88 pg/mL); parathyroid hormone-related protein was 6.4 pmol/L (normal range, < 2.0 pmol/L); potassium was 3.4 mmol/L (normal range, 3.5 – 5.1 mmol/L); and magnesium was 2.01 mg/dL. The patient’s thyroid stimulating hormone level was normal, and serial cardiac enzymes stayed within the reference range.
The patient was admitted to a cardiac care unit. A temporary transvenous pacemaker was placed, and the hypercalcemia was treated with aggressive fluid hydration, calcitonin, and zoledronic acid. Serum calcium gradually decreased to 14.6 mg/dL the following day and 9.6 mg/dL the subsequent day. The normalization of calcium resulted in resolution of complete heart block (Figure 3). The patient did not experience recurrence of AV nodal dysfunction and eventually died 3 months later due to his advanced metastatic disease.
Discussion
The reported cardiovascular effects of hypercalcemia include hypertension, arrhythmias, increased myocardial contractility at serum calcium level below 15 mg/dL, and myocardial depression above that level. Electrocardiographic manifestations of hypercalcemia include a shortened ST segment leading to a short corrected QT interval (QTc), slight increase in T wave duration, and rarely, Osborn waves or J waves.1-3 However, its influence on the AV node is less clear.
One small study assessed the prevalence of cardiac arrhythmias and conduction disturbances in 20 patients with hyperparathyroidism and moderate hypercalcemia and found no increase in the frequency of arrhythmias or high grade AV block.4
There are reports of conduction abnormality secondary to experimentally induced hypercalcemia in the literature. Hoff and colleagues described findings of AV block generated by the injection of IV calcium in dogs.5 In 2 human subjects, sinus bradycardia was precipitated after they received IV infusion of calcium gluconate.6 Shah and colleagues described 2 patients with sinus node dysfunction attributed to hypercalcemia secondary to hyperparathyroidism.7
Case reports of AV nodal dysfunction provoked by hypercalcemia have primarily occurred in the setting of primary hyperparathyroidism.8,9 Milk-alkali syndrome and vitamin D related hypercalcemia also have been reported to cause complete heart block.10,11 Reports of malignancy-related hypercalcemia causing conduction abnormalities are rare. The authors also found one case report of marked sinus bradycardia due to hypercalcemia related to breast cancer.12The case study presented in this report is rare because the patient developed complete AV block due to malignancy-related hypercalcemia that resolved completely with resolution of hypercalcemia. The prolongation of the QTc interval was another unique electrocardiographic change observed in this case. Calcium levels are inversely proportional to the QTc interval, and hypercalcemia is typically associated with a shortened QTc interval. However, this patient had a prolonged QTc without any other clear-cut cause. His hypokalemia was of a mild degree and not severe enough to produce such a long QTc interval. A possible explanation of QTc prolongation may be an increase in the T wave width associated with a serum calcium level above 16 mg/dL.
The pathophysiology of hypercalcemia-induced AV nodal conduction system disease is unknown. Calcium deposition in AV nodes of elderly patients has been associated with paroxysmal 2:1 AV block.8 It could be postulated that elevated serum calcium levels predispose to calcium deposition in cardiac conduction tissue, leading to progressive dysfunction. Although this theory may be applicable in a chronic setting, the mechanism in an acute setting likely relates to elevated serum levels of calcium that causes an alteration in electrochemical gradients. These elevated serum levels also increase intracellular calcium. This rise may result in increased calmodulin activation on the intracellular portion of the myocyte cell membrane and consequent enhanced sodium channel activation, which may then inhibit AV nodal conduction.13
Conclusion
Physicians should be aware that severe hypercalcemia can cause significant conduction system alterations, including complete AV block. A short QTc interval is typical, but a prolonged QTc interval also may be seen. While temporary support with a transvenous pacemaker may be needed, the conduction system abnormality is expected to resolve by treatment of the underlying hypercalcemia.
1. Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol. 1979;44(2):243-248.
2. Bronsky D, Dubin A, Waldstein SS, Kushner DS. Calcium and the electrocardiogram II. The electrocardiographic manifestations of hyperparathyroidism and of marked hypercalcemia from various other etiologies. Am J Cardiol. 1961;7(6):833-839.
3. Otero J, Lenihan DJ. The "normothermic" Osborn wave induced by severe hypercalcemia. Tex Heart Inst J. 2000;27(3):316-317.
4. Rosenqvist M, Nordenström J, Andersson M, Edhag OK. Cardiac conduction inpatients with hypercalcaemia due to primary hyperparathyroidism. Clin Endocrinol (Oxf). 1992;37(1):29-33.
5. Hoff H, Smith P, Winkler A. Electrocardiographic changes and concentration of calcium in serum following injection of calcium chloride. Am J Physiol. 1939;125:162-171.
6. Howard JE, Hopkins TR, Connor TB. The use of intravenous calcium as a measure of activity of the parathyroid glands. Trans Assoc Am Physicians. 1952;65:351-358.
7. Shah AP, Lopez A, Wachsner RY, Meymandi SK, El-Bialy AK, Ichiuji AM. Sinus node dysfunction secondary to hyperparathyroidism. J Cardiovasc Pharmacol Ther. 2004;9(2):145-147.
8. Vosnakidis A, Polymeropoulos K, Zaragoulidis P, Zarifis I. Atrioventricular nodal dysfunction secondary to hyperparathyroidism. J Thoracic Dis. 2013;5(3):E90-E92.
9. Crum WB, Till HJ. Hyperparathyroidism with Wenckebach's phenomenon. Am J Cardiol. 1960;6:838-840.
10. Ginsberg H, Schwarz KV. Letter: hypercalcemia and complete heart block. Ann Intern Med. 1973;79(6):903.
11. Garg G, Khadgwat R, Khandelwal D, Gupta N. Vitamin D toxicity presenting as hypercalcemia and complete heart block: an interesting case report. Indian J Endocrinol Metab. 2012;16 (suppl 2):S423-S425.
12. Badertscher E, Warnica JW, Ernst DS. Acute hypercalcemia and severe bradycardia in a patient with breast cancer. CMAJ. 1993;148(9):1506-1508.
13. Potet F, Chagot B, Anghelescu M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009;284(13):8846-8854.
1. Nierenberg DW, Ransil BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol. 1979;44(2):243-248.
2. Bronsky D, Dubin A, Waldstein SS, Kushner DS. Calcium and the electrocardiogram II. The electrocardiographic manifestations of hyperparathyroidism and of marked hypercalcemia from various other etiologies. Am J Cardiol. 1961;7(6):833-839.
3. Otero J, Lenihan DJ. The "normothermic" Osborn wave induced by severe hypercalcemia. Tex Heart Inst J. 2000;27(3):316-317.
4. Rosenqvist M, Nordenström J, Andersson M, Edhag OK. Cardiac conduction inpatients with hypercalcaemia due to primary hyperparathyroidism. Clin Endocrinol (Oxf). 1992;37(1):29-33.
5. Hoff H, Smith P, Winkler A. Electrocardiographic changes and concentration of calcium in serum following injection of calcium chloride. Am J Physiol. 1939;125:162-171.
6. Howard JE, Hopkins TR, Connor TB. The use of intravenous calcium as a measure of activity of the parathyroid glands. Trans Assoc Am Physicians. 1952;65:351-358.
7. Shah AP, Lopez A, Wachsner RY, Meymandi SK, El-Bialy AK, Ichiuji AM. Sinus node dysfunction secondary to hyperparathyroidism. J Cardiovasc Pharmacol Ther. 2004;9(2):145-147.
8. Vosnakidis A, Polymeropoulos K, Zaragoulidis P, Zarifis I. Atrioventricular nodal dysfunction secondary to hyperparathyroidism. J Thoracic Dis. 2013;5(3):E90-E92.
9. Crum WB, Till HJ. Hyperparathyroidism with Wenckebach's phenomenon. Am J Cardiol. 1960;6:838-840.
10. Ginsberg H, Schwarz KV. Letter: hypercalcemia and complete heart block. Ann Intern Med. 1973;79(6):903.
11. Garg G, Khadgwat R, Khandelwal D, Gupta N. Vitamin D toxicity presenting as hypercalcemia and complete heart block: an interesting case report. Indian J Endocrinol Metab. 2012;16 (suppl 2):S423-S425.
12. Badertscher E, Warnica JW, Ernst DS. Acute hypercalcemia and severe bradycardia in a patient with breast cancer. CMAJ. 1993;148(9):1506-1508.
13. Potet F, Chagot B, Anghelescu M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009;284(13):8846-8854.
Progressive Cardiomyopathy in a Patient With Elevated Cobalt Ion Levels and Bilateral Metal-on-Metal Hip Arthroplasties
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
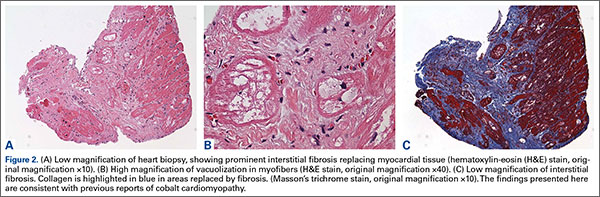
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
Systemic cobalt toxicity has been reported in the literature after hip arthroplasty revisions for failed ceramic components secondary to third-body abrasive wear of cobalt-chrome (CoCr) components, as well as with metal-on-metal (MOM) hip arthroplasty designs. There have been several cases of systemic cobalt toxicity after revision for fractured ceramic components.1,2 Of these 7 reported cases, all patients had neurologic complaints and 4 patients developed cardiomyopathy secondary to toxic cobalt levels, with 1 case being fatal.1 MOM hip prostheses have also been associated with local and systemic problems secondary to metal debris. Adverse local tissue reactions have been reported to occur in up to 59% of patients, and, in some registries, the failure rate of MOM arthroplasty caused by these soft-tissue reactions is 2 to 3 times that of conventional metal-on-polyethylene design failures.3,4 The occurrence of systemic complications from MOM total hip arthroplasty (THA) wear debris is much less common. There have been 6 cases of systemic cobalt toxicity reported in the literature resulting from MOM total hip prosthesis design.1,2
We present a case of biopsy-confirmed cardiomyopathy secondary to cobalt toxicity from a MOM THA design with subsequent requirement for left ventricular assist device (LVAD) implantation despite prosthesis removal. To our knowledge, this is the first report in the literature of this specific implant design causing systemic cobalt toxicity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a healthy nondiabetic man age 54 years who presented to our clinic 6 years after undergoing left THA and 5 years after undergoing right THA with the Biomet M2a-Magnum MOM prosthesis at an outside facility. The left-side components placed at the index procedure were a size 50 cup, 44 magnum head, 10 Taperloc stem (Biomet), and +9 neck. The right-side components were a size 52 cup, 46 magnum head, 10 Taperloc stem, and +3 neck. The patient emphasized that he was very happy with his hip prostheses and denied groin or thigh pain. His medical history was significant for exogenous obesity, and he denied any history of alcohol, tobacco, steroid, or recreational drug use.
The patient’s review of systems suggested that, approximately 11 months prior to presentation at our facility, he began having difficulty with his activities of daily living secondary to chest pressure with exertion, fatigue, and associated diaphoresis. He complained of decreased sensation in his feet bilaterally but denied any hearing loss, tinnitus, or vision changes. He underwent evaluation of the new-onset chest discomfort with a cardiac stress test that suggested no active cardiac ischemia. An echocardiogram revealed mitral regurgitation, stage II diastolic dysfunction with a left ventricular ejection fraction of 55%. Additionally, during this time period, the patient was being followed by his local orthopedic surgeon for an elevated cobalt level of 120 ppb and a chromium level of 109 ppb. The patient was referred to our clinic for recommendations regarding the elevated metal-ion levels. Upon initial evaluation, the patient denied any hip or groin pain. His physical examination revealed a nonantalgic gait with full range of motion and no signs of instability, tenderness, or masses. The patient was also noted to have no vibratory sensation in his feet bilaterally. The plain radiographs indicated bilateral MOM THA with acetabular inclination levels of 55º on the right and left sides. No cystic changes or other worrisome signs that would suggest implant loosening or failure were present (Figure 1). The serum metal levels were repeated and showed a cobalt level of 189 ppb and a chromium level of 71 ppb. Whole venous blood samples were drawn at our request using trace element tubes and were sent to Medtox Laboratories Inc. for analysis. Other pertinent laboratory values, including hematocrit and thyroid levels, were within normal limits. Because of concerns of systemic toxicity from significantly elevated cobalt and chromium levels, the patient elected to proceed with revision of the MOM components.
During the preoperative medical evaluation, the patient’s cardiac status was a concern, and the etiology of the cardiac dysfunction was unclear. Cardiac magnetic resonance imaging (MRI), which was performed to evaluate the extent and etiology of cardiac dysfunction, showed biventricular dysfunction. To evaluate the underlying myocardial tissue characteristics, delayed contrast imaging was performed and showed diffuse myocardial hyperenhancement of the anterior, lateral, and apical walls, with sparing of the base and midseptum. This type of extensive hyperenhancement is commonly seen with cardiac amyloidosis; however, the blood-pool kinetics during contrast administration is unusual for amyloidosis, as well as the diffuse edema noted on T2-weighted MRI. Importantly, cardiac MRI is very specific in excluding alternative diagnoses, such as postinfarct, infiltrative, acquired, viral, or alcoholic/drugs of abuse etiologies. In the absence of amyloidosis, the only other pattern that would be consistent with symptoms was diffuse, fulminant myocarditis of toxic origin lacking clinical evidence for an infectious origin. The patient’s prior exposure to cobalt was noted. Thus, the hyperenhancement and edema could be strong supportive evidence of cobalt infiltration, despite no reported cases in the literature of cobalt cardiomyopathy found on cardiac MRI.
Additional workup was initiated, and cardiac catheterization showed that the patient continued to decompensate, with worsening global left ventricular dysfunction with an ejection fraction of 30% without evidence of coronary artery disease. Also, he was noted to have mild renal impairment with a blood urea nitrogen level of 31 mg/dL and a creatinine level of 1.7 mg/dL. The etiology of the renal impairment was unknown and had not been established, according to the patient and his wife. The renal impairment was not thought to be caused by the elevated metal ions levels but likely resulted from prerenal azotemia secondary to decreased cardiac output. During catheterization, an endomyocardial biopsy was performed and the tissue sent to the Mayo Clinic pathology department for analysis. The sample showed myocyte hypertrophy and interstitial fibrosis with scattered myofibers containing large cytoplasmic vacuoles. Also present was karyomegaly consistent with myocyte hypertrophy (Figures 2A, 2B). Trichrome stain confirmed replacement of myofibers by collagen (Figure 2C). Electron microscopy performed on a paraffin block showed reduced contractile elements, vacuolar spaces, and increased lipofuscin. The findings were very consistent with, but not specific for, cardiomyopathy from cobalt toxicity. No evidence of an inflammatory infiltrate was identified. The diagnosis was cobalt cardiomyopathy based on biopsy, presentation, cobalt levels, and intraoperative findings.
The patient was admitted to the cardiac intensive care unit preoperatively and optimized with inotropic agents. A multidisciplinary consultation with the cardiology and anesthesia departments was obtained. Both recommended cardiac anesthesia with intraoperative Swan-Ganz catheter and transesophageal echo monitoring. Assuming that the patient remained hemodynamically stable with limited blood loss and the first hip was timely performed, the cardiology department recommended a single surgery, because fewer risks and complications could be expected than from a staged procedure. Subsequently, surgery was performed on the left hip via a conservative anterior approach on the fracture table. The patient remained stable with limited blood loss. During the same operating room time, revision of the right hip was performed using an anterior approach. The intraoperative findings showed evidence of pseudotumors in the adjacent soft tissues and abundant brown, creamy fluid upon entering the joint capsule, consistent with a metallic appearance. Both hips showed similar prosthetic findings. There was no significant visible wear of the large diameter metal heads or gross abnormality of the acetabular components. The trunnion area on both femoral implants was abnormal, revealing a black coating suggestive of marked corrosion. The components were all well fixed, without visible damage, and, because of his fragile cardiac status, the patient’s acetabular components were not revised. The trunnions were cleaned and the femoral heads were revised to active articulation dual-mobility metal-on-polyethylene constructs using 28-mm Biolox Option ceramic (CeramTec). The tissue specimens from the operation showed chronic inflammation with areas of fibroconnective tissue and bland fibrinoid necrosis with extensive brown pigment-laden macrophage reaction. The intraoperative cultures were negative.
The patient tolerated the surgery without complication, and his postoperative period was without incident. Nine months after surgery, the patient’s cobalt and chromium levels had declined to 16 ppb and 32 ppb, respectively (normal, <1 ppb). However, his cardiac status continued to worsen with significant shortness of breath and bilateral lower extremity edema despite diuresis. Follow-up cardiac MRI indicated progressive left and right dysfunction with ejection fractions of 23% and 25%, respectively. After progressive heart-failure symptoms, the patient was admitted to the hospital for severe congestive heart failure and underwent implantation of a HeartWare LVAD with tricuspid valve repair using an Edwards annuloplasty ring. He has since had a cardiac transplant and is doing well.
Discussion
To our knowledge, this is the first reported case of cardiomyopathy in a patient with elevated cobalt ion levels and a Biomet M2a-Magnum hip prosthesis. This is also the first reported case of cardiac MRI–defined cobalt cardiomyopathy. The cobalt levels seen in this patient were similar to those of other cases with systemic cobalt toxicity from a MOM hip construct. Mao and colleagues5 reported 2 cases of systemic cobalt toxicity in 2 patients with articular surface replacement hip prostheses.One patient presented with mild groin pain, neurologic symptoms, and a cobalt level of 410 ppb 5 years after her index procedure. The other patient presented with cardiac and neurologic symptoms but no hip complaints. The patient’s cobalt levels ranged from 185 ppb to 210 ppb. Both patients improved after their revision surgery, and their cobalt levels decreased. The 2 patients in Tower’s report6 were 49-year-old men who had articular surface replacement implants (DePuy). One patient who presented with progressive hip pain 11 months postoperatively developed neurologic symptoms and cardiomyopathy, with cobalt levels of 83 ppb before revision surgery 43 months after his index procedure. The other patient presented with hip pain and vertigo, headaches, fatigue, and dyspnea. He underwent hip revision 40 months postoperatively and required closed reduction under sedation for dislocation. Finally, and most recently, Allen and colleagues2 reported a 59-year-old woman with a cobalt level of 287 ppb whose symptoms did not resolve after implantation of an LVAD or cardiac transplantation but only after removal of her bilateral hip prosthesis. Our case is most similar to this report but significantly adds to the literature in 2 distinct manners: (1) Biomet M2a-Magnum has not been implicated in cobalt toxicity; and (2) this is the first reported use of dedicated cardiac MRI to noninvasively define underlying cardiac pathology.
The cardiac manifestations secondary to systemic cobalt toxicity in this patient represent a frightening consequence of MOM prosthetic wear. The effects of cobalt toxicity on cardiac tissues were first described in a series of alcoholic patients from Manchester in 1900;7 however, it was not until 1967, in a series of patients in Quebec, that cobalt was found to be the inciting factor. In the modern era, hip arthroplasty techniques resulting in excessive cobalt and chromium wear have demonstrated the same findings of myocyte hypertrophy, interstitial fibrosis, and scattered myofibers containing large cytoplasmic inclusions.8,9 The patient presented here has pathologic findings consistent with previous cases of cobalt cardiomyopathy; however, in the other cases of cardiomyopathy due to MOM total hip components, the patients’ cardiac conditions improved after the prostheses were revised and the cobalt levels began to diminish.5,6In our case, the patient has sustained permanent damage to his myocardium and a progressive decline in his cardiac status, which is a deviation from reported cases as of 2014.
While there is no guideline to unequivocally diagnose cobalt cardiomyopathy, the constellation of findings, including pathologic, biologic, blood levels, imaging, and surgical, all uniformly indicate a unifying diagnosis. The lack of improvement after prosthetic device removal supports a diagnosis of permanent myocardial damage, which is consistent with cardiomyopathy of advanced toxic etiology.
Conclusion
This case presents a patient with bilateral MOM THAs, acetabular cup inclinations of greater than 55º, renal impairment, and cobalt levels greater than 60 ppb, with occult cardiac failure leading to LVAD implantation as a prelude to cardiac transplantation in order to avoid certain death. These factors have been shown, in prior case reports, to be associated with cardiac damage that may be reversible.6 However; it is important for orthopedic surgeons to recognize that certain hip prostheses can be associated or lead to irreversible cardiac damage.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
1. Zywiel MG, Brandt JM, Overgaard CB, Cheung AC, Turgeon TR, Syed KA. Fatal cardiomyopathy after revision total hip replacement for fracture of a ceramic liner. Bone Joint J. 2013;95(1):31-37.
2. Allen LA, Ambardekar AV, Devaraj KM, Maleszewski JJ, Wolfel EE. Clinical problem-solving. Missing elements of the history. N Engl J Med. 2014;370(6):559-566.
3. Hart AJ, Satchihananda K, Liddle AD, et al. Pseudotumors in association with well-functioning metal-on-metal hip prostheses: a case-control study using three-dimensional tomography and magnetic resonance imaging. J Bone Joint Surg Am. 2012;94(4);317-325.
4. Kwon MK, Jacobs JJ, MacDonald SJ, Potter HG, Fehring TK, Lombardi AV. Evidence-based understanding of management perils for metal-on-metal hip arthroplasty patients. J Arthroplasty. 2012;27(8 suppl):20-25.
5. Mao X, Wong AA, Crawford RW. Cobalt toxicity- -an emerging clinical problem in patients with metal-on-metal hip prostheses? Med J Aust. 2011;194(12):649-651.
6. Tower SS. Arthroprosthetic cobaltism: neurological and cardiac manifestations in two patients with metal-on-metal arthroplasty: a case report. J Bone Joint Surg Am. 2010;92(17):2847-2851.
7. Morin Y, Daniel P. Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Can Med Assoc J. 1967;97(15):926-928.
8. Gilbert C, Cheung A, Butany J, et al. Hip pain and heart failure: the missing link. Can J Cardiol. 2013;29(5):639.e1-e2.
9. Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G. Cobalt myocardiopathy. A critical review of literature. Sci Total Environ. 1994;150(1-3):105-109.
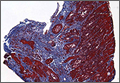
14-Year-Old Boy With Mild Antecedent Neck Pain in Setting of Acute Trauma: A Rare Case of Benign Fibrous Histiocytoma of the Spine
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
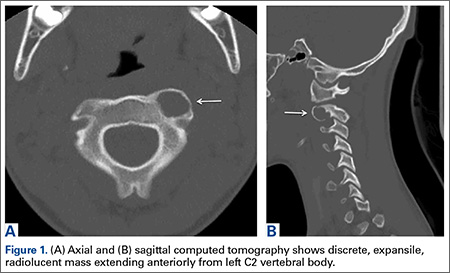
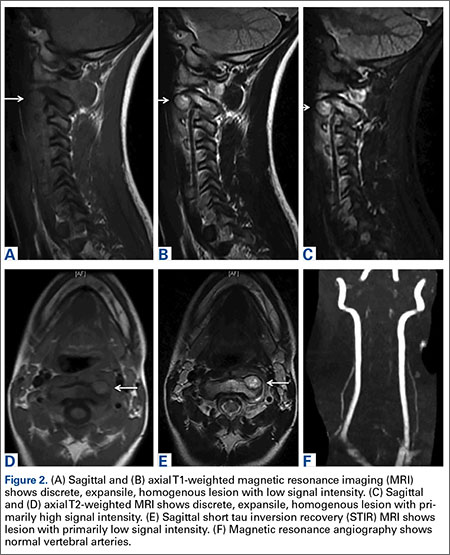
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
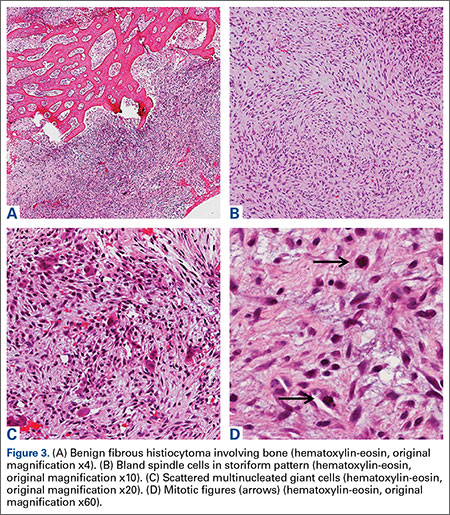
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
Necrotizing Cellulitis With Multiple Abscesses on the Leg Caused by Serratia marcescens
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
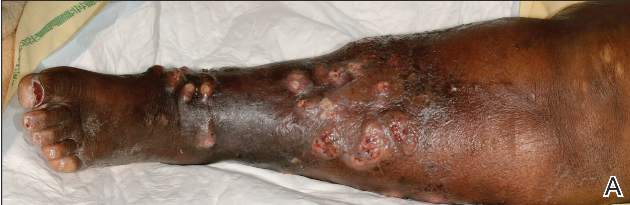
|
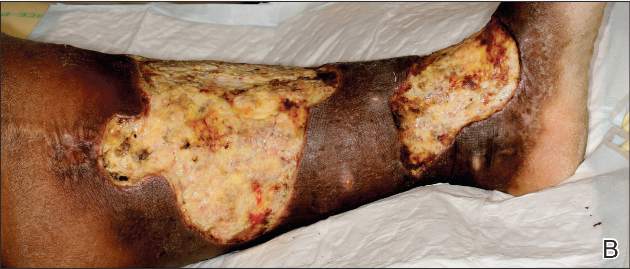
|
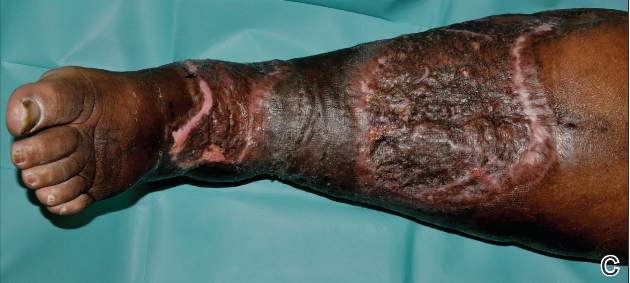
|
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |

Comment
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
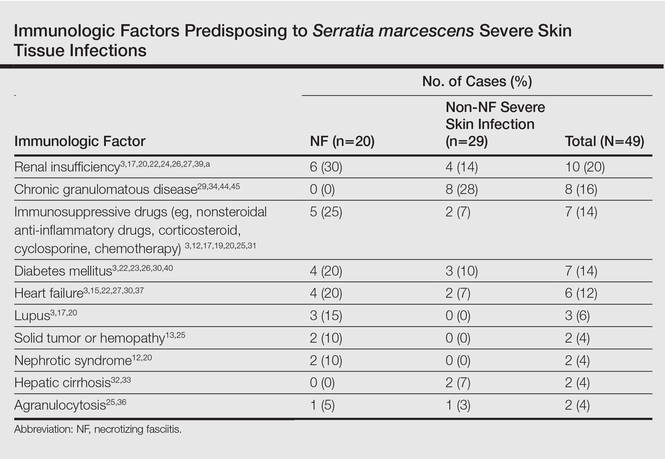
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
|
|
|
|
|
|
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |
Comment
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
|
|
|
|
|
|
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |
Comment
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
Practice Points
- Serratia marcescens skin infection should be considered in cases of cellulitis in immunocompromised patients when conventional antibiotics are not effective.
- Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered.
Radiation-Induced Pemphigus or Pemphigoid Disease in 3 Patients With Distinct Underlying Malignancies
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
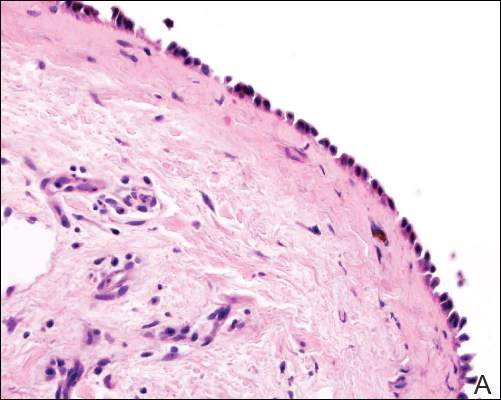
| 
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
|
|
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
|
|
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
Practice Points
- The use of radiation therapy is increasing because of its therapeutic benefit, especially in advanced-stage cancer patients.
- Although there is a wide range of adverse effects associated with radiation therapy, pemphigus or pemphigoid disease is rare and needs to be distinguished from other skin diseases or even recurrent underlying cancer.
- The precise mechanism of radiation-induced pemphigus or pemphigoid disease is unknown, but clinicians should be alert to this potentially serious complication, and all cutaneous eruptions developing during and after radiation therapy should be evaluated with routine histologic examination in conjunction with direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay.
Prolonged Pustular Eruption From Hydroxychloroquine: An Unusual Case of Acute Generalized Exanthematous Pustulosis
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
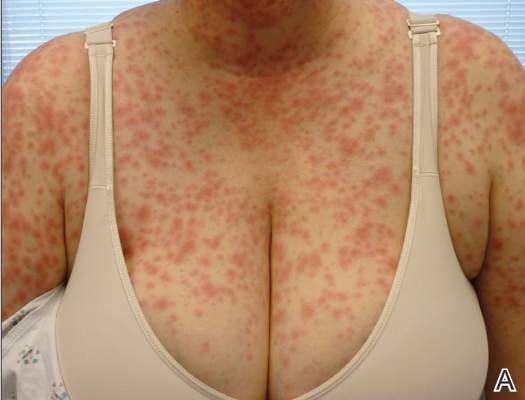

Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
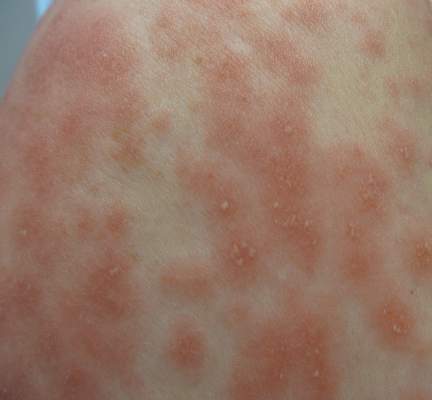
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Practice Points
- Acute generalized exanthematous pustulosis (AGEP) is most commonly caused by antibiotics (eg, aminopenicillins, macrolides, cephalosporins) followed by calcium channel blockers.
- The main treatment of AGEP is discontinuation of the culprit medication, which typically results in resolution within 2 weeks. Treatment also can symptomatically include topical or systemic corticosteroids and antipyretics.
- Hydroxychloroquine (HCQ) can be a culprit of AGEP with a prolonged recovery course. It is important to inform patients with HCQ-associated AGEP that the clearance of their lesions may take longer than the typical 2 weeks.