User login
Right Paraduodenal Hernia
Paraduodenal hernia, also called mesocolic hernia, is a type of internal hernia that is thought to be caused by a congenital defect involving abnormal retroperitoneal fixation of the mesentery due to abnormal rotation of the midgut.1 Internal hernias account for only 1% of all hernias, and paraduodenal hernias make up 50% of those.2
Paraduodenal hernias can be classified as left or right with left being far more common than right, 75% and 25%, respectively.2 Due to the fixation abnormalities in the midgut, fossae are formed that help to classify left vs right paraduodenal hernias. Herniation through Landzert fossae results in a left paraduodenal hernia with the primary constituents of the hernia sac being the inferior mesenteric artery and vein.1 This result is due to an in utero defect of the small intestine herniated between the inferior mesenteric vein and posterior parietal attachments of the descending mesocolon to the retroperitoneal.3
In a right paraduodenal hernia, herniation occurs through Waldeyer fossae with the main contents of the hernia sac being the iliocolic, right colic, and middle colic vessels within the anterior wall and the superior mesenteric artery along the medial border of the hernia.1 Since there is a failure of rotation around the superior mesenteric artery, the majority of the small intestine remains to the right of the superior mesenteric artery, resulting in the small intestine being trapped between the posteriolateral peritoneum.3 Regardless of the type of paraduodenal hernia, patients usually will present with symptoms of small bowel obstruction. In these types of hernias, a computed tomography (CT) scan with IV contrast may suggest evidence of obstruction between the duodenum and jejunum, but this may be unclear. Although rare, clinical suspicion of paraduodenal hernia is necessary to prevent ensuing complications and mortality.
Case Presentation
A 43-year-old man presented to the emergency department with symptoms that included nausea, vomiting, intermittent epigastric abdominal pain, and obstipation, which were suggestive of a small bowel obstruction. The patient reported similar intermittent episodes over the past 10 years that had resolved without surgery. The patient had no history of abdominal surgeries. A nasogastric tube was inserted and immediately drew out a significant amount of bilious contents. A CT scan indicated an obstruction at the proximal jejunum with suspicion of an internal hernia.
The patient underwent exploratory laparotomy soon after, which confirmed a right paraduodenal hernia (Figure). The surgery began laproscopically by retracting the omentum and transverse colon cranially to expose the ligament of Treitz. The hernia defect was identified on the mesentery where the proximal jejunum twisted on itself in a loop. The hernia was untwisted, and adhesions were removed. The posterior attachment of the hernia sac was freed with harmonic cautery and blunt dissection along with its attachment to the ligament of Treitz. In the process of freeing the herniation, a 1-cm enterotomy ensued, which did not contain succus or spillage of luminal contents at that time. Due to difficulties in visualizing the remainder of the small bowel, the procedure was converted to a laparotomy. This allowed complete freeing of the twisted loop of bowel.
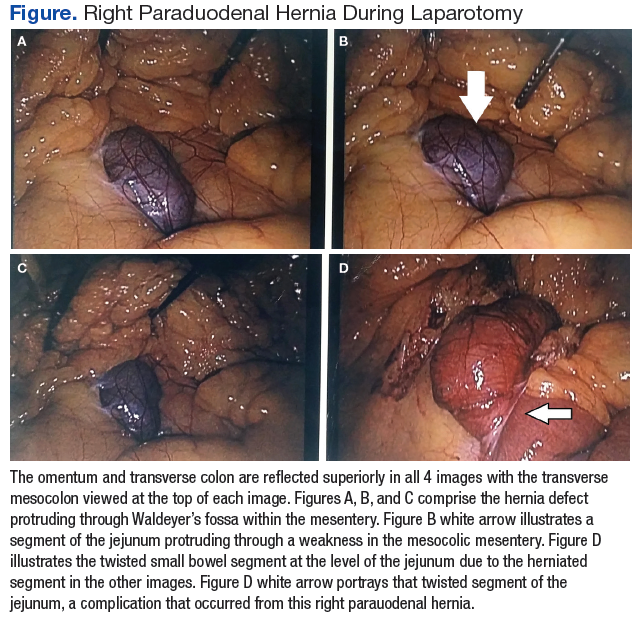
Afterward, there was succus and bile draining from the enterotomy, so it was closed transversely in 2 layers, making sure there was a lumen between the layers. The first and second parts of the duodenum were examined followed by palpitation of the duodenal sweep. The remainder of the small bowel was visualized to the cecum, and the retroperitoneal space was dissected out of the hernia sac space. The abdomen was irrigated, and the omentum was draped back over the intestines. The fascia was closed followed by skin reapproximation with staples. The patient experienced an uneventful recovery and was discharged on day 6 with resolution of his symptoms.
Discussion
Paraduodenal hernias are a type of internal hernia and a rare cause of intestinal obstruction accounting for about 0.5% of all hernias. Right paraduodenal hernias are far less common than left paraduodenal hernias and occur due to a defect in the jejunum mesentery called Waldeyer fossae.4 This is located at the third part of the duodenum and behind the superior mesenteric artery.4 Symptoms of paraduodenal hernias are nonspecific and may include nausea, vomiting, and intermittent cramping. Symptoms of obstruction can be intermittent due to the small bowel herniating through the fossae and then retracting.1 Computed tomography has good specificity and aides in the diagnosis of an internal hernia, but physicians must have a high index of suspicion as well.5
Definitive diagnosis and treatment of paraduodenal hernias involves laparoscopy or exploratory laparotomy to visualize the internal hernia and its surrounding sac.4,5 All hernias should be repaired to prevent strangulation of the bowel, but internal hernias are even more important to fix because these hernias may not present until there is severe injury to the bowel.5 On identification of the paraduodenal hernia, it is important to release the bowel from the hernia sac, free up adhesions, and place small bowel segments back into the correct anatomical position.4,5
In the event of bowel injury, resection with reanastomosis is indicated. Careful dissection is important to prevent injury to the superior mesenteric artery, which supplies most of the small bowel and ascending colon.4,5 Injury to the superior mesenteric artery could lead to ischemia and gangrenous bowel.2 Immediate detection and early surgery intervention of these congenital hernias can prevent such complications.2 The literature includes reports of paraduodenal hernias with complications of gangrenous bowel that required small bowel resection.2 These complications further emphasize the need to proceed immediately with surgery if a paraduodenal hernia is suspected.
Conclusion
This rare cause of bowel obstruction was documented in order to emphasize the importance of having a high clinical suspicion for a paraduodenal hernia. This particular patient with no history of abdominal surgeries had previously dealt with bowel obstruction and would likely have this complication again without surgical intervention. Patients with paraduodenal hernias also are at risk for bowel ischemia, other high-risk complications, and even death.5 Although a CT scan provided information about an approximate location of the obstruction, laparoscopy confirmed the diagnosis. Going into the operation with paraduodenal hernia in the differential allowed the surgeon to be prepared for the appropriate anatomy involved with this procedure to minimize damage to important structures, such as the superior mesenteric artery and its branches.
1. Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Saunders; 2012.
2. Fukada T, Mukai H, Shimamura F, Furukawa T, Miyazaki M. A causal relationship between right paraduodenal hernia and superior mesenteric artery syndrome: a case report. J Med Case Rep. 2010;4:159.
3. Skandalakis JE. Peritoneum, omenta, and internal hernias. In: Skandalakis JE, Colborn GL, eds. Skandalakis Surgical Anatomy: The Embryologic and Anatomic Basis of Modern Surgery. 1st ed. Athens, Greece: Paschalidis Medical Publications; 2004:chap 10.
4. Papaziogas B, Souparis A, Makris J, Alexandrakis A, Papaziogas T. Surgical images: soft tissue. Right paraduodenal hernia. Can J Surg. 2004;47(3):195-196.
5. Manfredelli S, Andrea Z, Stefano P, et al. Rare small bowel obstruction: right paraduodenal hernia. Case report. Int J Surg Case Rep. 2013;4(4):412-415.
Paraduodenal hernia, also called mesocolic hernia, is a type of internal hernia that is thought to be caused by a congenital defect involving abnormal retroperitoneal fixation of the mesentery due to abnormal rotation of the midgut.1 Internal hernias account for only 1% of all hernias, and paraduodenal hernias make up 50% of those.2
Paraduodenal hernias can be classified as left or right with left being far more common than right, 75% and 25%, respectively.2 Due to the fixation abnormalities in the midgut, fossae are formed that help to classify left vs right paraduodenal hernias. Herniation through Landzert fossae results in a left paraduodenal hernia with the primary constituents of the hernia sac being the inferior mesenteric artery and vein.1 This result is due to an in utero defect of the small intestine herniated between the inferior mesenteric vein and posterior parietal attachments of the descending mesocolon to the retroperitoneal.3
In a right paraduodenal hernia, herniation occurs through Waldeyer fossae with the main contents of the hernia sac being the iliocolic, right colic, and middle colic vessels within the anterior wall and the superior mesenteric artery along the medial border of the hernia.1 Since there is a failure of rotation around the superior mesenteric artery, the majority of the small intestine remains to the right of the superior mesenteric artery, resulting in the small intestine being trapped between the posteriolateral peritoneum.3 Regardless of the type of paraduodenal hernia, patients usually will present with symptoms of small bowel obstruction. In these types of hernias, a computed tomography (CT) scan with IV contrast may suggest evidence of obstruction between the duodenum and jejunum, but this may be unclear. Although rare, clinical suspicion of paraduodenal hernia is necessary to prevent ensuing complications and mortality.
Case Presentation
A 43-year-old man presented to the emergency department with symptoms that included nausea, vomiting, intermittent epigastric abdominal pain, and obstipation, which were suggestive of a small bowel obstruction. The patient reported similar intermittent episodes over the past 10 years that had resolved without surgery. The patient had no history of abdominal surgeries. A nasogastric tube was inserted and immediately drew out a significant amount of bilious contents. A CT scan indicated an obstruction at the proximal jejunum with suspicion of an internal hernia.
The patient underwent exploratory laparotomy soon after, which confirmed a right paraduodenal hernia (Figure). The surgery began laproscopically by retracting the omentum and transverse colon cranially to expose the ligament of Treitz. The hernia defect was identified on the mesentery where the proximal jejunum twisted on itself in a loop. The hernia was untwisted, and adhesions were removed. The posterior attachment of the hernia sac was freed with harmonic cautery and blunt dissection along with its attachment to the ligament of Treitz. In the process of freeing the herniation, a 1-cm enterotomy ensued, which did not contain succus or spillage of luminal contents at that time. Due to difficulties in visualizing the remainder of the small bowel, the procedure was converted to a laparotomy. This allowed complete freeing of the twisted loop of bowel.
Afterward, there was succus and bile draining from the enterotomy, so it was closed transversely in 2 layers, making sure there was a lumen between the layers. The first and second parts of the duodenum were examined followed by palpitation of the duodenal sweep. The remainder of the small bowel was visualized to the cecum, and the retroperitoneal space was dissected out of the hernia sac space. The abdomen was irrigated, and the omentum was draped back over the intestines. The fascia was closed followed by skin reapproximation with staples. The patient experienced an uneventful recovery and was discharged on day 6 with resolution of his symptoms.
Discussion
Paraduodenal hernias are a type of internal hernia and a rare cause of intestinal obstruction accounting for about 0.5% of all hernias. Right paraduodenal hernias are far less common than left paraduodenal hernias and occur due to a defect in the jejunum mesentery called Waldeyer fossae.4 This is located at the third part of the duodenum and behind the superior mesenteric artery.4 Symptoms of paraduodenal hernias are nonspecific and may include nausea, vomiting, and intermittent cramping. Symptoms of obstruction can be intermittent due to the small bowel herniating through the fossae and then retracting.1 Computed tomography has good specificity and aides in the diagnosis of an internal hernia, but physicians must have a high index of suspicion as well.5
Definitive diagnosis and treatment of paraduodenal hernias involves laparoscopy or exploratory laparotomy to visualize the internal hernia and its surrounding sac.4,5 All hernias should be repaired to prevent strangulation of the bowel, but internal hernias are even more important to fix because these hernias may not present until there is severe injury to the bowel.5 On identification of the paraduodenal hernia, it is important to release the bowel from the hernia sac, free up adhesions, and place small bowel segments back into the correct anatomical position.4,5
In the event of bowel injury, resection with reanastomosis is indicated. Careful dissection is important to prevent injury to the superior mesenteric artery, which supplies most of the small bowel and ascending colon.4,5 Injury to the superior mesenteric artery could lead to ischemia and gangrenous bowel.2 Immediate detection and early surgery intervention of these congenital hernias can prevent such complications.2 The literature includes reports of paraduodenal hernias with complications of gangrenous bowel that required small bowel resection.2 These complications further emphasize the need to proceed immediately with surgery if a paraduodenal hernia is suspected.
Conclusion
This rare cause of bowel obstruction was documented in order to emphasize the importance of having a high clinical suspicion for a paraduodenal hernia. This particular patient with no history of abdominal surgeries had previously dealt with bowel obstruction and would likely have this complication again without surgical intervention. Patients with paraduodenal hernias also are at risk for bowel ischemia, other high-risk complications, and even death.5 Although a CT scan provided information about an approximate location of the obstruction, laparoscopy confirmed the diagnosis. Going into the operation with paraduodenal hernia in the differential allowed the surgeon to be prepared for the appropriate anatomy involved with this procedure to minimize damage to important structures, such as the superior mesenteric artery and its branches.
Paraduodenal hernia, also called mesocolic hernia, is a type of internal hernia that is thought to be caused by a congenital defect involving abnormal retroperitoneal fixation of the mesentery due to abnormal rotation of the midgut.1 Internal hernias account for only 1% of all hernias, and paraduodenal hernias make up 50% of those.2
Paraduodenal hernias can be classified as left or right with left being far more common than right, 75% and 25%, respectively.2 Due to the fixation abnormalities in the midgut, fossae are formed that help to classify left vs right paraduodenal hernias. Herniation through Landzert fossae results in a left paraduodenal hernia with the primary constituents of the hernia sac being the inferior mesenteric artery and vein.1 This result is due to an in utero defect of the small intestine herniated between the inferior mesenteric vein and posterior parietal attachments of the descending mesocolon to the retroperitoneal.3
In a right paraduodenal hernia, herniation occurs through Waldeyer fossae with the main contents of the hernia sac being the iliocolic, right colic, and middle colic vessels within the anterior wall and the superior mesenteric artery along the medial border of the hernia.1 Since there is a failure of rotation around the superior mesenteric artery, the majority of the small intestine remains to the right of the superior mesenteric artery, resulting in the small intestine being trapped between the posteriolateral peritoneum.3 Regardless of the type of paraduodenal hernia, patients usually will present with symptoms of small bowel obstruction. In these types of hernias, a computed tomography (CT) scan with IV contrast may suggest evidence of obstruction between the duodenum and jejunum, but this may be unclear. Although rare, clinical suspicion of paraduodenal hernia is necessary to prevent ensuing complications and mortality.
Case Presentation
A 43-year-old man presented to the emergency department with symptoms that included nausea, vomiting, intermittent epigastric abdominal pain, and obstipation, which were suggestive of a small bowel obstruction. The patient reported similar intermittent episodes over the past 10 years that had resolved without surgery. The patient had no history of abdominal surgeries. A nasogastric tube was inserted and immediately drew out a significant amount of bilious contents. A CT scan indicated an obstruction at the proximal jejunum with suspicion of an internal hernia.
The patient underwent exploratory laparotomy soon after, which confirmed a right paraduodenal hernia (Figure). The surgery began laproscopically by retracting the omentum and transverse colon cranially to expose the ligament of Treitz. The hernia defect was identified on the mesentery where the proximal jejunum twisted on itself in a loop. The hernia was untwisted, and adhesions were removed. The posterior attachment of the hernia sac was freed with harmonic cautery and blunt dissection along with its attachment to the ligament of Treitz. In the process of freeing the herniation, a 1-cm enterotomy ensued, which did not contain succus or spillage of luminal contents at that time. Due to difficulties in visualizing the remainder of the small bowel, the procedure was converted to a laparotomy. This allowed complete freeing of the twisted loop of bowel.
Afterward, there was succus and bile draining from the enterotomy, so it was closed transversely in 2 layers, making sure there was a lumen between the layers. The first and second parts of the duodenum were examined followed by palpitation of the duodenal sweep. The remainder of the small bowel was visualized to the cecum, and the retroperitoneal space was dissected out of the hernia sac space. The abdomen was irrigated, and the omentum was draped back over the intestines. The fascia was closed followed by skin reapproximation with staples. The patient experienced an uneventful recovery and was discharged on day 6 with resolution of his symptoms.
Discussion
Paraduodenal hernias are a type of internal hernia and a rare cause of intestinal obstruction accounting for about 0.5% of all hernias. Right paraduodenal hernias are far less common than left paraduodenal hernias and occur due to a defect in the jejunum mesentery called Waldeyer fossae.4 This is located at the third part of the duodenum and behind the superior mesenteric artery.4 Symptoms of paraduodenal hernias are nonspecific and may include nausea, vomiting, and intermittent cramping. Symptoms of obstruction can be intermittent due to the small bowel herniating through the fossae and then retracting.1 Computed tomography has good specificity and aides in the diagnosis of an internal hernia, but physicians must have a high index of suspicion as well.5
Definitive diagnosis and treatment of paraduodenal hernias involves laparoscopy or exploratory laparotomy to visualize the internal hernia and its surrounding sac.4,5 All hernias should be repaired to prevent strangulation of the bowel, but internal hernias are even more important to fix because these hernias may not present until there is severe injury to the bowel.5 On identification of the paraduodenal hernia, it is important to release the bowel from the hernia sac, free up adhesions, and place small bowel segments back into the correct anatomical position.4,5
In the event of bowel injury, resection with reanastomosis is indicated. Careful dissection is important to prevent injury to the superior mesenteric artery, which supplies most of the small bowel and ascending colon.4,5 Injury to the superior mesenteric artery could lead to ischemia and gangrenous bowel.2 Immediate detection and early surgery intervention of these congenital hernias can prevent such complications.2 The literature includes reports of paraduodenal hernias with complications of gangrenous bowel that required small bowel resection.2 These complications further emphasize the need to proceed immediately with surgery if a paraduodenal hernia is suspected.
Conclusion
This rare cause of bowel obstruction was documented in order to emphasize the importance of having a high clinical suspicion for a paraduodenal hernia. This particular patient with no history of abdominal surgeries had previously dealt with bowel obstruction and would likely have this complication again without surgical intervention. Patients with paraduodenal hernias also are at risk for bowel ischemia, other high-risk complications, and even death.5 Although a CT scan provided information about an approximate location of the obstruction, laparoscopy confirmed the diagnosis. Going into the operation with paraduodenal hernia in the differential allowed the surgeon to be prepared for the appropriate anatomy involved with this procedure to minimize damage to important structures, such as the superior mesenteric artery and its branches.
1. Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Saunders; 2012.
2. Fukada T, Mukai H, Shimamura F, Furukawa T, Miyazaki M. A causal relationship between right paraduodenal hernia and superior mesenteric artery syndrome: a case report. J Med Case Rep. 2010;4:159.
3. Skandalakis JE. Peritoneum, omenta, and internal hernias. In: Skandalakis JE, Colborn GL, eds. Skandalakis Surgical Anatomy: The Embryologic and Anatomic Basis of Modern Surgery. 1st ed. Athens, Greece: Paschalidis Medical Publications; 2004:chap 10.
4. Papaziogas B, Souparis A, Makris J, Alexandrakis A, Papaziogas T. Surgical images: soft tissue. Right paraduodenal hernia. Can J Surg. 2004;47(3):195-196.
5. Manfredelli S, Andrea Z, Stefano P, et al. Rare small bowel obstruction: right paraduodenal hernia. Case report. Int J Surg Case Rep. 2013;4(4):412-415.
1. Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Saunders; 2012.
2. Fukada T, Mukai H, Shimamura F, Furukawa T, Miyazaki M. A causal relationship between right paraduodenal hernia and superior mesenteric artery syndrome: a case report. J Med Case Rep. 2010;4:159.
3. Skandalakis JE. Peritoneum, omenta, and internal hernias. In: Skandalakis JE, Colborn GL, eds. Skandalakis Surgical Anatomy: The Embryologic and Anatomic Basis of Modern Surgery. 1st ed. Athens, Greece: Paschalidis Medical Publications; 2004:chap 10.
4. Papaziogas B, Souparis A, Makris J, Alexandrakis A, Papaziogas T. Surgical images: soft tissue. Right paraduodenal hernia. Can J Surg. 2004;47(3):195-196.
5. Manfredelli S, Andrea Z, Stefano P, et al. Rare small bowel obstruction: right paraduodenal hernia. Case report. Int J Surg Case Rep. 2013;4(4):412-415.
Multiple Primary Atypical Vascular Lesions Occurring in the Same Breast
Atypical vascular lesions (AVLs) of the breast are rare cutaneous vascular proliferations that present as erythematous, violaceous, or flesh-colored papules, patches, or plaques in women who have undergone radiation treatment for breast carcinoma.1,2 These lesions most commonly develop in the irradiated area within 3 to 6 years following radiation treatment.3
Various terms have been used to describe AVLs in the literature, including atypical hemangiomas, benign lymphangiomatous papules, benign lymphangioendotheliomas, lymphangioma circumscriptum, and acquired progressive lymphangiomas, suggesting benign behavior.4-10 However, their identity as benign lesions has been a source of controversy, with some investigators proposing that AVLs may be a precursor lesion to postirradiation angiosarcoma.2 Research has addressed if there are markers that can predict AVL types that are more likely to develop into angiosarcomas.1 Although most clinicians treat AVLs with complete excision, there currently are no specific guidelines to direct this practice.
We report the case of a patient with a history of 1 AVL that was excised who developed 3 additional AVLs in the same breast over the course of 15 months.
Case Report
A 55-year-old woman with a history of obesity, hypertension, and infiltrating ductal carcinoma in situ of the right breast (grade 2, estrogen receptor and progesterone receptor positive) underwent a right breast lumpectomy and sentinel lymph node dissection. Three months later, she underwent re-excision for positive margins and started adjuvant hormonal therapy with tamoxifen. One month later, she began external beam radiation therapy and received a total dose of 6040 cGy over the course of 9 weeks (34 total treatments).
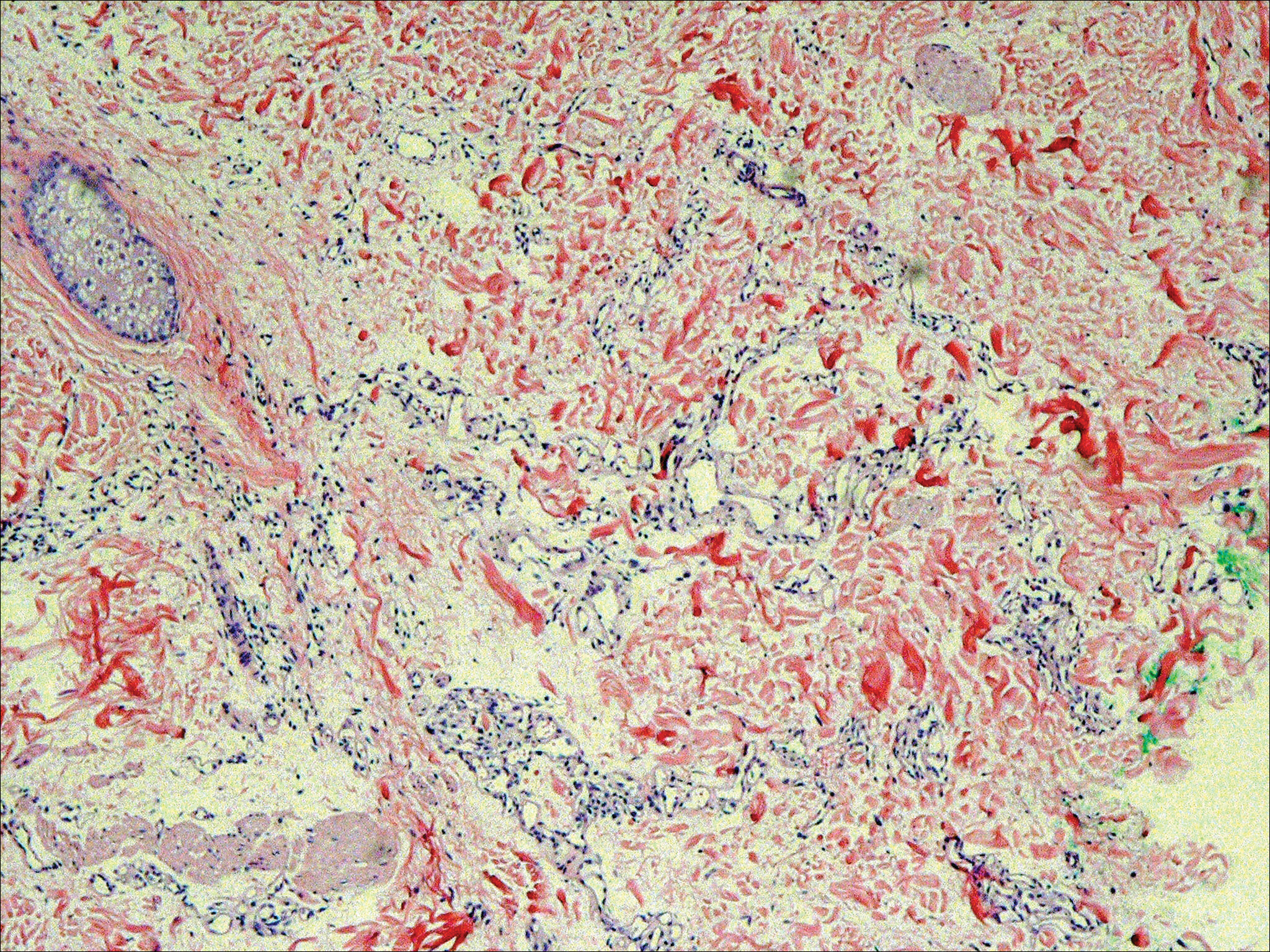
The patient presented to an outside dermatology clinic 2 years after completing external beam radiation therapy for evaluation of a new pink nodule on the right mid breast. The nodule was biopsied and discovered to be an AVL. Pathology showed an anastomosing proliferation of thin-walled vascular channels mainly located in the superficial dermis with notable endothelial nuclear atypia and hyperchromasia. There were several tiny foci with the beginnings of multilayering with prominent endothelial atypia (Figure 1). She underwent complete excision for this AVL with negative margins.
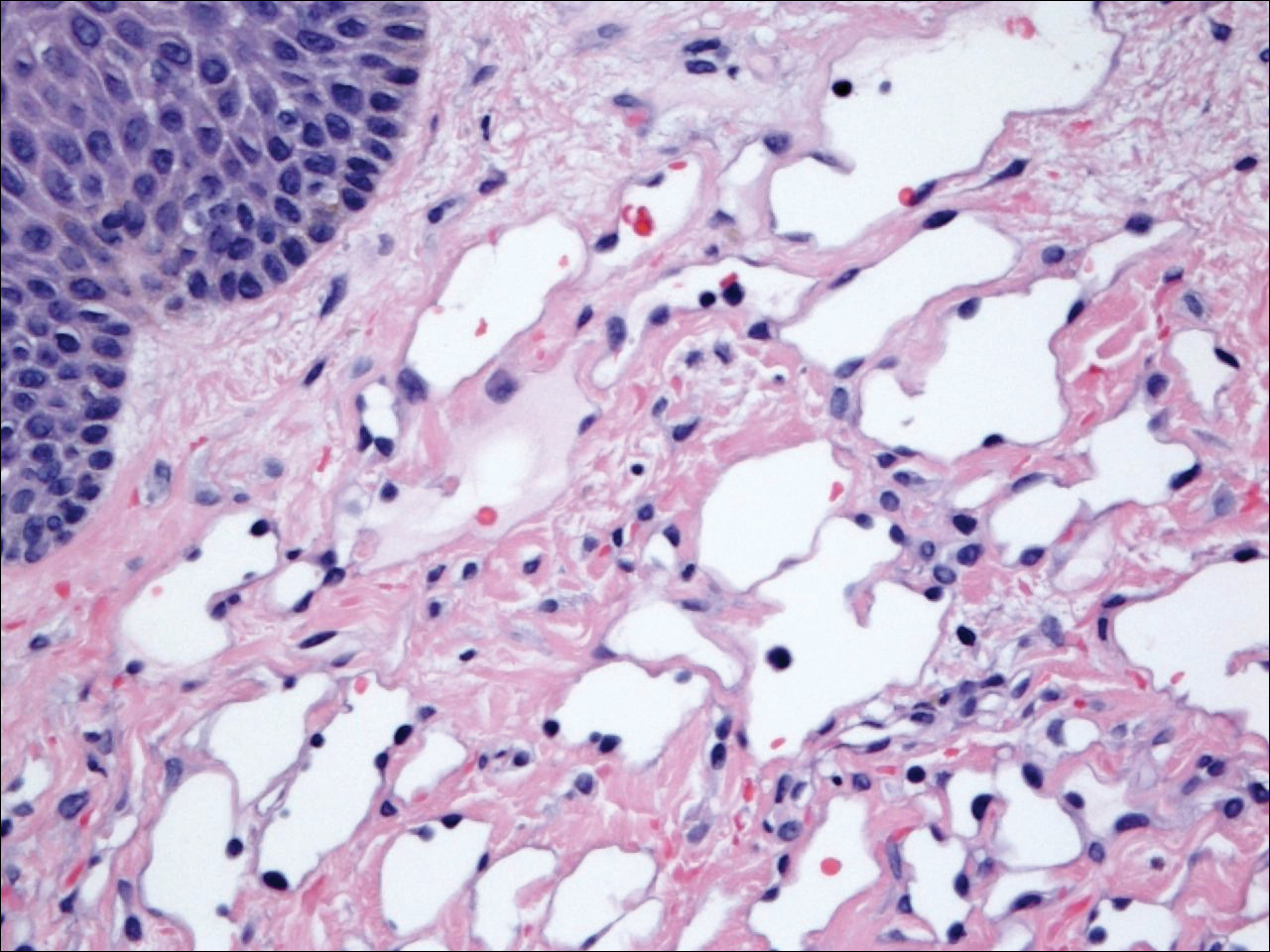
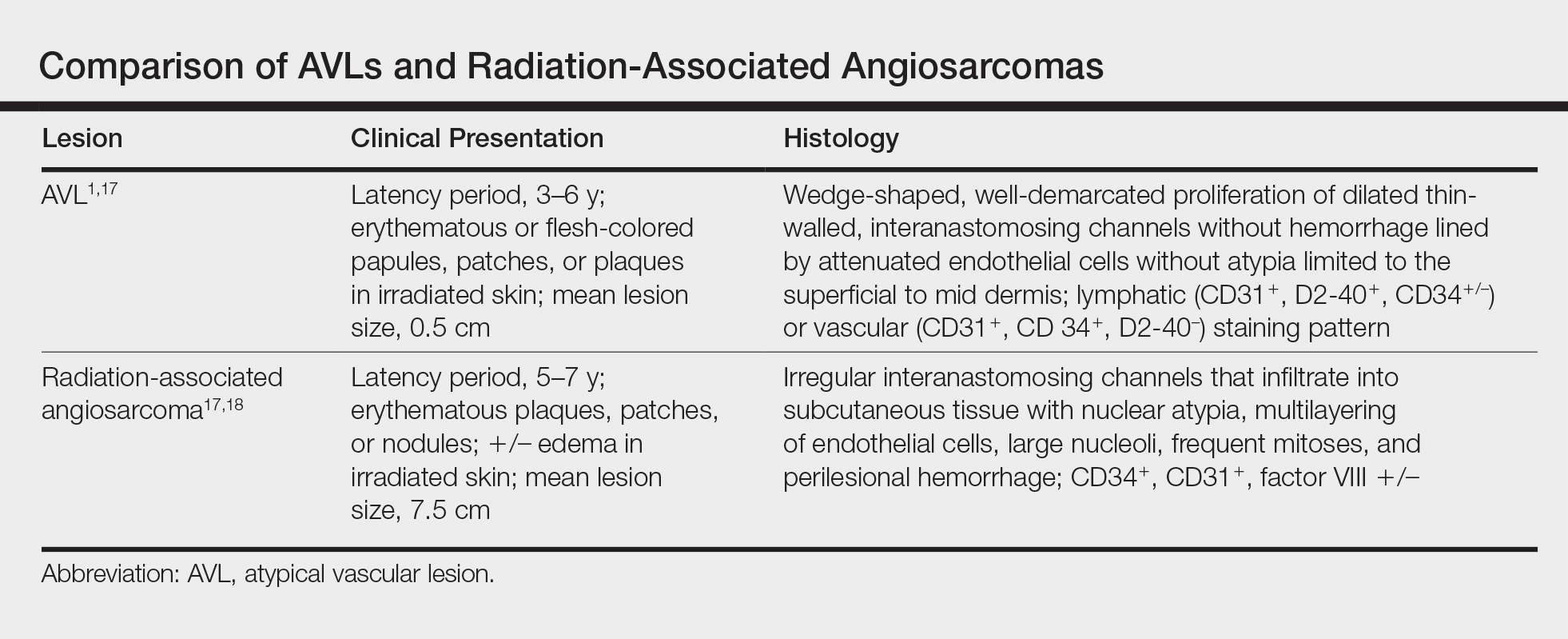
Six months after the initial AVL diagnosis, she presented to our dermatology clinic with another asymptomatic red bump on the right breast. On physical examination, a 4-mm firm, erythematous, well-circumscribed papule was noted on the medial aspect of the right breast along with a similar-appearing 4-mm papule on the right lateral aspect of the right breast (Figure 2). The patient was unsure of the duration of the second lesion but felt that it had been present at least as long as the other lesion. Both lesions clinically resembled typical capillary hemangiomas. A 6-mm punch biopsy of the right medial breast was performed and revealed enlarged vessels and capillaries in the upper dermis lined by endothelial cells with focal prominent nuclei without necrosis, overt atypia, mitosis, or tufting (Figure 3). Immunostaining was positive for CD34, factor VIII antigen, podoplanin (D2-40), and CD31, and negative for cytokeratin 7 and pankeratin. This staining was compatible with a lymphatic-type AVL.1 A diagnosis of AVL was made and complete excision with clear margins was performed. At the time of this excision, a biopsy of the right lateral breast was performed revealing thin-walled, dilated vascular channels in the superficial dermis with architecturally atypical angulated outlines, mild endothelial nuclear atypia, and hyperchromasia without endothelial multilayering. Clear margins were noted on the biopsy, but the patient subsequently declined re-excision of this third AVL.
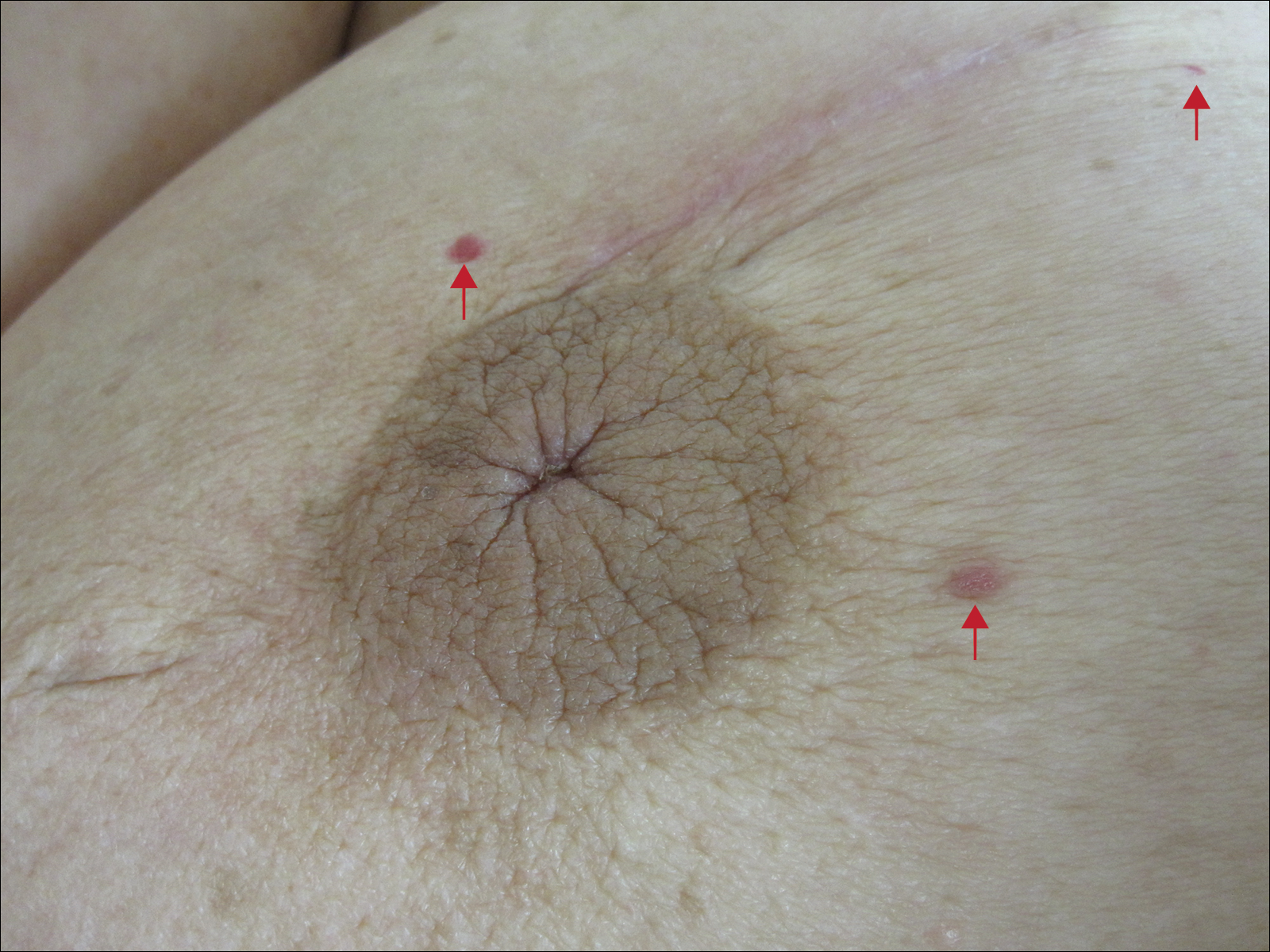
During a subsequent follow-up visit 9 months later, the patient was noted to have a 2-mm red, vascular-appearing papule on the right upper medial breast (Figure 2). A 6-mm biopsy was performed and revealed thin-walled vascular channels in the superficial dermis with endothelial nuclear atypia consistent with an AVL.
Comment
Fineberg and Rosen8 were the first to describe AVLs in their 1994 study of 4 women with cutaneous vascular proliferations that developed after radiation and chemotherapy for breast cancer. They concluded that these AVLs were benign lesions distinct from angiosarcomas.8 However, further research has challenged the benign nature of AVLs. In 2005, Brenn and Fletcher2 studied 42 women diagnosed with either angiosarcoma or atypical radiation-associated cutaneous vascular lesions. They suggested that AVLs resided on the same spectrum as angiosarcomas and that AVLs may be precursor lesions to angiosarcomas.2 Furthermore, Hildebrandt et al11 in 2001 and Di Tommaso and Fabbri12 in 2003 published case reports of individual patients who developed an angiosarcoma from a preexisting AVL.
The controversy continued when Patton et al1 published a study in 2008 in which 32 cases of AVLs were reviewed. In this study, 2 histologic types of AVLs were described: vascular type and lymphatic type. Vascular-type AVLs are characterized by irregularly dispersed, pericyte-invested, capillary-sized vessels within the papillary or reticular dermis that often are associated with extravasated erythrocytes or hemosiderin. On the other hand, lymphatic-type AVLs display thin-walled, variably anastomosing, lymphatic vessels lined by attenuated or slightly protuberant endothelial cells. These subtypes have been suggested based on the antigens known to be present in certain tissues, specifically vascular and lymphatic tissue. Despite these seemingly distinct histologies, 6 lesions classified as vascular type displayed some histologic overlap with the lymphatic-type AVLs. The authors concluded that the vascular type showed greater potential to develop into an angiosarcoma based on the degree of endothelial atypia.1
In 2011, Santi et al13 found that both AVLs and angiosarcomas share inactivation mutations in the tumor suppressor gene TP53, providing further evidence to suggest that AVLs may be precursors to angiosarcomas.
Although the malignant potential of AVLs remains questionable, research has shown that they do have a propensity to recur.3 In 2007, Gengler et al3 determined that 20% of patients with AVLs experienced recurrence after a biopsy or excision with varying margins; however, the group stated that these new vascular lesions might not be recurrences but rather entirely new lesions in the same irradiated field (field-effect phenomenon). Several other studies demonstrated that more than 30% of patients with 1 AVL developed more lesions within the same irradiated area.3,14-16 Despite the high rate of recurrence documented in the literature, only 5 of more than 100 diagnosed AVLs have progressed to angiosarcoma.1,3
Many differences can be noted when comparing the histology of AVLs versus angiosarcomas, though some are subtle (Table). Angiosarcomas display poorly circumscribed vascular infiltration into the subcutaneous tissue, multilayering of endothelial cells, prominent nucleoli, hemorrhage, mitoses, and notable aytpia. Atypical vascular lesions lack these features and tend to be wedge shaped and display chronic inflammation.8,15,17-19 Atypical vascular lesions show superficial localized growth without destruction of adjacent adnexa, display dilated vascular spaces, and exhibit large endothelial cells.5,6,8,14,15,19,20 However, there is overlap between AVLs and angiosarcomas that can make diagnosis difficult.2,14,16,17,19 Areas within or just outside of an angiosarcoma, especially in well-differentiated angiosarcomas, can appear histologically identical to AVLs, and multiple biopsies may be required for diagnosis.17,19,21
Conclusion
More research is needed in the arenas of classification, diagnosis, treatment, and follow-up recommendations for AVLs. In particular, more specific histologic markers may be needed to identify those AVLs that may progress to angiosarcomas. Although most AVLs are treated with excision, a consensus needs to be reached on adequate surgical margins. Lastly, due to the tendency of AVLs to recur coupled with their unknown malignant potential, recommendations are needed for consistent follow-up examinations.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A, et al. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process; a study from the French sarcoma group. Cancer. 2007;109:1584-1598.
- Hoda SA, Cranor ML, Rosen PP. Hemangiomas of the breast with atypical histological features: further analysis of histological subtypes confirming their benign character. Am J Surg Pathol. 1992;16:553-560.
- Wagamon K, Ranchoff RE, Rosenberg AS, et al. Benign lymphangiomatous papules of the skin. J Am Acad Dermatol. 2005;52:912-913.
- Diaz-Cascajo C, Borghi S, Weyers W, et al. Benign lymphangiomatous papules of the skin following radiotherapy: a report of five new cases and review of the literature. Histopathology. 1999;35:319-327.
- Martín-González T, Sanz-Trelles A, Del Boz J, et al. Benign lymphangiomatous papules and plaques after radiotherapy [in Spanish]. Actas Dermosifiliogr. 2008;99:84-86.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Guillou L, Fletcher CD. Benign lymphangioendothelioma (acquired progressive lymphangioma): a lesion not to be confused with well-differentiated angiosarcoma and patch stage Kaposi’s sarcoma: clinicopathologic analysis of a series. Am J Surg Pathol. 2000;24:1047-1057.
- Rosso R, Gianelli U, Carnevali L. Acquired progressive lymphangioma of the skin following radiotherapy for breast carcinoma. J Cutan Pathol. 1995;22:164-167.
- Hildebrandt G, Mittag M, Gutz U, et al. Cutaneous breast angiosarcoma after conservative treatment of breast cancer. Eur J Dermatol. 2001;11:580-583.
- Di Tommaso L, Fabbri A. Cutaneous angiosarcoma arising after radiotherapy treatment of a breast carcinoma: description of a case and review of the literature [in Italian]. Pathologica. 2003;95:196-202.
- Santi R, Cetica V, Franchi A, et al. Tumour suppressor gene TP53 mutations in atypical vascular lesions of breast skin following radiotherapy. Histopathology. 2011;58:455-466.
- Requena L, Kutzner H, Mentzel T, et al. Benign vascular proliferations in irradiated skin. Am J Surg Pathol. 2002;26:328-337.
- Brodie C, Provenzano E. Vascular proliferations of the breast. Histopathology. 2008;52:30-44.
- Brenn T, Fletcher CD. Postradiation vascular proliferations: an increasing problem. Histopathology. 2006;48:106-114.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Kardum-Skelin I, Jelić-Puskarić B, Pazur M, et al. A case report of breast angiosarcoma. Coll Antropol. 2010;34:645-648.
- Mattoch IW, Robbins JB, Kempson RL, et al. Post-radiotherapy vascular proliferations in mammary skin: a clinicopathologic study of 11 cases. J Am Acad Dermatol. 2007;57:126-133.
- Bodet D, Rodríguez-Cano L, Bartralot R, et al. Benign lymphangiomatous papules of the skin associated with ovarian fibroma. J Am Acad Dermatol. 2007;56(2 suppl):S41-S44.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
Atypical vascular lesions (AVLs) of the breast are rare cutaneous vascular proliferations that present as erythematous, violaceous, or flesh-colored papules, patches, or plaques in women who have undergone radiation treatment for breast carcinoma.1,2 These lesions most commonly develop in the irradiated area within 3 to 6 years following radiation treatment.3
Various terms have been used to describe AVLs in the literature, including atypical hemangiomas, benign lymphangiomatous papules, benign lymphangioendotheliomas, lymphangioma circumscriptum, and acquired progressive lymphangiomas, suggesting benign behavior.4-10 However, their identity as benign lesions has been a source of controversy, with some investigators proposing that AVLs may be a precursor lesion to postirradiation angiosarcoma.2 Research has addressed if there are markers that can predict AVL types that are more likely to develop into angiosarcomas.1 Although most clinicians treat AVLs with complete excision, there currently are no specific guidelines to direct this practice.
We report the case of a patient with a history of 1 AVL that was excised who developed 3 additional AVLs in the same breast over the course of 15 months.
Case Report
A 55-year-old woman with a history of obesity, hypertension, and infiltrating ductal carcinoma in situ of the right breast (grade 2, estrogen receptor and progesterone receptor positive) underwent a right breast lumpectomy and sentinel lymph node dissection. Three months later, she underwent re-excision for positive margins and started adjuvant hormonal therapy with tamoxifen. One month later, she began external beam radiation therapy and received a total dose of 6040 cGy over the course of 9 weeks (34 total treatments).
The patient presented to an outside dermatology clinic 2 years after completing external beam radiation therapy for evaluation of a new pink nodule on the right mid breast. The nodule was biopsied and discovered to be an AVL. Pathology showed an anastomosing proliferation of thin-walled vascular channels mainly located in the superficial dermis with notable endothelial nuclear atypia and hyperchromasia. There were several tiny foci with the beginnings of multilayering with prominent endothelial atypia (Figure 1). She underwent complete excision for this AVL with negative margins.
Six months after the initial AVL diagnosis, she presented to our dermatology clinic with another asymptomatic red bump on the right breast. On physical examination, a 4-mm firm, erythematous, well-circumscribed papule was noted on the medial aspect of the right breast along with a similar-appearing 4-mm papule on the right lateral aspect of the right breast (Figure 2). The patient was unsure of the duration of the second lesion but felt that it had been present at least as long as the other lesion. Both lesions clinically resembled typical capillary hemangiomas. A 6-mm punch biopsy of the right medial breast was performed and revealed enlarged vessels and capillaries in the upper dermis lined by endothelial cells with focal prominent nuclei without necrosis, overt atypia, mitosis, or tufting (Figure 3). Immunostaining was positive for CD34, factor VIII antigen, podoplanin (D2-40), and CD31, and negative for cytokeratin 7 and pankeratin. This staining was compatible with a lymphatic-type AVL.1 A diagnosis of AVL was made and complete excision with clear margins was performed. At the time of this excision, a biopsy of the right lateral breast was performed revealing thin-walled, dilated vascular channels in the superficial dermis with architecturally atypical angulated outlines, mild endothelial nuclear atypia, and hyperchromasia without endothelial multilayering. Clear margins were noted on the biopsy, but the patient subsequently declined re-excision of this third AVL.
During a subsequent follow-up visit 9 months later, the patient was noted to have a 2-mm red, vascular-appearing papule on the right upper medial breast (Figure 2). A 6-mm biopsy was performed and revealed thin-walled vascular channels in the superficial dermis with endothelial nuclear atypia consistent with an AVL.
Comment
Fineberg and Rosen8 were the first to describe AVLs in their 1994 study of 4 women with cutaneous vascular proliferations that developed after radiation and chemotherapy for breast cancer. They concluded that these AVLs were benign lesions distinct from angiosarcomas.8 However, further research has challenged the benign nature of AVLs. In 2005, Brenn and Fletcher2 studied 42 women diagnosed with either angiosarcoma or atypical radiation-associated cutaneous vascular lesions. They suggested that AVLs resided on the same spectrum as angiosarcomas and that AVLs may be precursor lesions to angiosarcomas.2 Furthermore, Hildebrandt et al11 in 2001 and Di Tommaso and Fabbri12 in 2003 published case reports of individual patients who developed an angiosarcoma from a preexisting AVL.
The controversy continued when Patton et al1 published a study in 2008 in which 32 cases of AVLs were reviewed. In this study, 2 histologic types of AVLs were described: vascular type and lymphatic type. Vascular-type AVLs are characterized by irregularly dispersed, pericyte-invested, capillary-sized vessels within the papillary or reticular dermis that often are associated with extravasated erythrocytes or hemosiderin. On the other hand, lymphatic-type AVLs display thin-walled, variably anastomosing, lymphatic vessels lined by attenuated or slightly protuberant endothelial cells. These subtypes have been suggested based on the antigens known to be present in certain tissues, specifically vascular and lymphatic tissue. Despite these seemingly distinct histologies, 6 lesions classified as vascular type displayed some histologic overlap with the lymphatic-type AVLs. The authors concluded that the vascular type showed greater potential to develop into an angiosarcoma based on the degree of endothelial atypia.1
In 2011, Santi et al13 found that both AVLs and angiosarcomas share inactivation mutations in the tumor suppressor gene TP53, providing further evidence to suggest that AVLs may be precursors to angiosarcomas.
Although the malignant potential of AVLs remains questionable, research has shown that they do have a propensity to recur.3 In 2007, Gengler et al3 determined that 20% of patients with AVLs experienced recurrence after a biopsy or excision with varying margins; however, the group stated that these new vascular lesions might not be recurrences but rather entirely new lesions in the same irradiated field (field-effect phenomenon). Several other studies demonstrated that more than 30% of patients with 1 AVL developed more lesions within the same irradiated area.3,14-16 Despite the high rate of recurrence documented in the literature, only 5 of more than 100 diagnosed AVLs have progressed to angiosarcoma.1,3
Many differences can be noted when comparing the histology of AVLs versus angiosarcomas, though some are subtle (Table). Angiosarcomas display poorly circumscribed vascular infiltration into the subcutaneous tissue, multilayering of endothelial cells, prominent nucleoli, hemorrhage, mitoses, and notable aytpia. Atypical vascular lesions lack these features and tend to be wedge shaped and display chronic inflammation.8,15,17-19 Atypical vascular lesions show superficial localized growth without destruction of adjacent adnexa, display dilated vascular spaces, and exhibit large endothelial cells.5,6,8,14,15,19,20 However, there is overlap between AVLs and angiosarcomas that can make diagnosis difficult.2,14,16,17,19 Areas within or just outside of an angiosarcoma, especially in well-differentiated angiosarcomas, can appear histologically identical to AVLs, and multiple biopsies may be required for diagnosis.17,19,21
Conclusion
More research is needed in the arenas of classification, diagnosis, treatment, and follow-up recommendations for AVLs. In particular, more specific histologic markers may be needed to identify those AVLs that may progress to angiosarcomas. Although most AVLs are treated with excision, a consensus needs to be reached on adequate surgical margins. Lastly, due to the tendency of AVLs to recur coupled with their unknown malignant potential, recommendations are needed for consistent follow-up examinations.
Atypical vascular lesions (AVLs) of the breast are rare cutaneous vascular proliferations that present as erythematous, violaceous, or flesh-colored papules, patches, or plaques in women who have undergone radiation treatment for breast carcinoma.1,2 These lesions most commonly develop in the irradiated area within 3 to 6 years following radiation treatment.3
Various terms have been used to describe AVLs in the literature, including atypical hemangiomas, benign lymphangiomatous papules, benign lymphangioendotheliomas, lymphangioma circumscriptum, and acquired progressive lymphangiomas, suggesting benign behavior.4-10 However, their identity as benign lesions has been a source of controversy, with some investigators proposing that AVLs may be a precursor lesion to postirradiation angiosarcoma.2 Research has addressed if there are markers that can predict AVL types that are more likely to develop into angiosarcomas.1 Although most clinicians treat AVLs with complete excision, there currently are no specific guidelines to direct this practice.
We report the case of a patient with a history of 1 AVL that was excised who developed 3 additional AVLs in the same breast over the course of 15 months.
Case Report
A 55-year-old woman with a history of obesity, hypertension, and infiltrating ductal carcinoma in situ of the right breast (grade 2, estrogen receptor and progesterone receptor positive) underwent a right breast lumpectomy and sentinel lymph node dissection. Three months later, she underwent re-excision for positive margins and started adjuvant hormonal therapy with tamoxifen. One month later, she began external beam radiation therapy and received a total dose of 6040 cGy over the course of 9 weeks (34 total treatments).
The patient presented to an outside dermatology clinic 2 years after completing external beam radiation therapy for evaluation of a new pink nodule on the right mid breast. The nodule was biopsied and discovered to be an AVL. Pathology showed an anastomosing proliferation of thin-walled vascular channels mainly located in the superficial dermis with notable endothelial nuclear atypia and hyperchromasia. There were several tiny foci with the beginnings of multilayering with prominent endothelial atypia (Figure 1). She underwent complete excision for this AVL with negative margins.
Six months after the initial AVL diagnosis, she presented to our dermatology clinic with another asymptomatic red bump on the right breast. On physical examination, a 4-mm firm, erythematous, well-circumscribed papule was noted on the medial aspect of the right breast along with a similar-appearing 4-mm papule on the right lateral aspect of the right breast (Figure 2). The patient was unsure of the duration of the second lesion but felt that it had been present at least as long as the other lesion. Both lesions clinically resembled typical capillary hemangiomas. A 6-mm punch biopsy of the right medial breast was performed and revealed enlarged vessels and capillaries in the upper dermis lined by endothelial cells with focal prominent nuclei without necrosis, overt atypia, mitosis, or tufting (Figure 3). Immunostaining was positive for CD34, factor VIII antigen, podoplanin (D2-40), and CD31, and negative for cytokeratin 7 and pankeratin. This staining was compatible with a lymphatic-type AVL.1 A diagnosis of AVL was made and complete excision with clear margins was performed. At the time of this excision, a biopsy of the right lateral breast was performed revealing thin-walled, dilated vascular channels in the superficial dermis with architecturally atypical angulated outlines, mild endothelial nuclear atypia, and hyperchromasia without endothelial multilayering. Clear margins were noted on the biopsy, but the patient subsequently declined re-excision of this third AVL.
During a subsequent follow-up visit 9 months later, the patient was noted to have a 2-mm red, vascular-appearing papule on the right upper medial breast (Figure 2). A 6-mm biopsy was performed and revealed thin-walled vascular channels in the superficial dermis with endothelial nuclear atypia consistent with an AVL.
Comment
Fineberg and Rosen8 were the first to describe AVLs in their 1994 study of 4 women with cutaneous vascular proliferations that developed after radiation and chemotherapy for breast cancer. They concluded that these AVLs were benign lesions distinct from angiosarcomas.8 However, further research has challenged the benign nature of AVLs. In 2005, Brenn and Fletcher2 studied 42 women diagnosed with either angiosarcoma or atypical radiation-associated cutaneous vascular lesions. They suggested that AVLs resided on the same spectrum as angiosarcomas and that AVLs may be precursor lesions to angiosarcomas.2 Furthermore, Hildebrandt et al11 in 2001 and Di Tommaso and Fabbri12 in 2003 published case reports of individual patients who developed an angiosarcoma from a preexisting AVL.
The controversy continued when Patton et al1 published a study in 2008 in which 32 cases of AVLs were reviewed. In this study, 2 histologic types of AVLs were described: vascular type and lymphatic type. Vascular-type AVLs are characterized by irregularly dispersed, pericyte-invested, capillary-sized vessels within the papillary or reticular dermis that often are associated with extravasated erythrocytes or hemosiderin. On the other hand, lymphatic-type AVLs display thin-walled, variably anastomosing, lymphatic vessels lined by attenuated or slightly protuberant endothelial cells. These subtypes have been suggested based on the antigens known to be present in certain tissues, specifically vascular and lymphatic tissue. Despite these seemingly distinct histologies, 6 lesions classified as vascular type displayed some histologic overlap with the lymphatic-type AVLs. The authors concluded that the vascular type showed greater potential to develop into an angiosarcoma based on the degree of endothelial atypia.1
In 2011, Santi et al13 found that both AVLs and angiosarcomas share inactivation mutations in the tumor suppressor gene TP53, providing further evidence to suggest that AVLs may be precursors to angiosarcomas.
Although the malignant potential of AVLs remains questionable, research has shown that they do have a propensity to recur.3 In 2007, Gengler et al3 determined that 20% of patients with AVLs experienced recurrence after a biopsy or excision with varying margins; however, the group stated that these new vascular lesions might not be recurrences but rather entirely new lesions in the same irradiated field (field-effect phenomenon). Several other studies demonstrated that more than 30% of patients with 1 AVL developed more lesions within the same irradiated area.3,14-16 Despite the high rate of recurrence documented in the literature, only 5 of more than 100 diagnosed AVLs have progressed to angiosarcoma.1,3
Many differences can be noted when comparing the histology of AVLs versus angiosarcomas, though some are subtle (Table). Angiosarcomas display poorly circumscribed vascular infiltration into the subcutaneous tissue, multilayering of endothelial cells, prominent nucleoli, hemorrhage, mitoses, and notable aytpia. Atypical vascular lesions lack these features and tend to be wedge shaped and display chronic inflammation.8,15,17-19 Atypical vascular lesions show superficial localized growth without destruction of adjacent adnexa, display dilated vascular spaces, and exhibit large endothelial cells.5,6,8,14,15,19,20 However, there is overlap between AVLs and angiosarcomas that can make diagnosis difficult.2,14,16,17,19 Areas within or just outside of an angiosarcoma, especially in well-differentiated angiosarcomas, can appear histologically identical to AVLs, and multiple biopsies may be required for diagnosis.17,19,21
Conclusion
More research is needed in the arenas of classification, diagnosis, treatment, and follow-up recommendations for AVLs. In particular, more specific histologic markers may be needed to identify those AVLs that may progress to angiosarcomas. Although most AVLs are treated with excision, a consensus needs to be reached on adequate surgical margins. Lastly, due to the tendency of AVLs to recur coupled with their unknown malignant potential, recommendations are needed for consistent follow-up examinations.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A, et al. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process; a study from the French sarcoma group. Cancer. 2007;109:1584-1598.
- Hoda SA, Cranor ML, Rosen PP. Hemangiomas of the breast with atypical histological features: further analysis of histological subtypes confirming their benign character. Am J Surg Pathol. 1992;16:553-560.
- Wagamon K, Ranchoff RE, Rosenberg AS, et al. Benign lymphangiomatous papules of the skin. J Am Acad Dermatol. 2005;52:912-913.
- Diaz-Cascajo C, Borghi S, Weyers W, et al. Benign lymphangiomatous papules of the skin following radiotherapy: a report of five new cases and review of the literature. Histopathology. 1999;35:319-327.
- Martín-González T, Sanz-Trelles A, Del Boz J, et al. Benign lymphangiomatous papules and plaques after radiotherapy [in Spanish]. Actas Dermosifiliogr. 2008;99:84-86.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Guillou L, Fletcher CD. Benign lymphangioendothelioma (acquired progressive lymphangioma): a lesion not to be confused with well-differentiated angiosarcoma and patch stage Kaposi’s sarcoma: clinicopathologic analysis of a series. Am J Surg Pathol. 2000;24:1047-1057.
- Rosso R, Gianelli U, Carnevali L. Acquired progressive lymphangioma of the skin following radiotherapy for breast carcinoma. J Cutan Pathol. 1995;22:164-167.
- Hildebrandt G, Mittag M, Gutz U, et al. Cutaneous breast angiosarcoma after conservative treatment of breast cancer. Eur J Dermatol. 2001;11:580-583.
- Di Tommaso L, Fabbri A. Cutaneous angiosarcoma arising after radiotherapy treatment of a breast carcinoma: description of a case and review of the literature [in Italian]. Pathologica. 2003;95:196-202.
- Santi R, Cetica V, Franchi A, et al. Tumour suppressor gene TP53 mutations in atypical vascular lesions of breast skin following radiotherapy. Histopathology. 2011;58:455-466.
- Requena L, Kutzner H, Mentzel T, et al. Benign vascular proliferations in irradiated skin. Am J Surg Pathol. 2002;26:328-337.
- Brodie C, Provenzano E. Vascular proliferations of the breast. Histopathology. 2008;52:30-44.
- Brenn T, Fletcher CD. Postradiation vascular proliferations: an increasing problem. Histopathology. 2006;48:106-114.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Kardum-Skelin I, Jelić-Puskarić B, Pazur M, et al. A case report of breast angiosarcoma. Coll Antropol. 2010;34:645-648.
- Mattoch IW, Robbins JB, Kempson RL, et al. Post-radiotherapy vascular proliferations in mammary skin: a clinicopathologic study of 11 cases. J Am Acad Dermatol. 2007;57:126-133.
- Bodet D, Rodríguez-Cano L, Bartralot R, et al. Benign lymphangiomatous papules of the skin associated with ovarian fibroma. J Am Acad Dermatol. 2007;56(2 suppl):S41-S44.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A, et al. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process; a study from the French sarcoma group. Cancer. 2007;109:1584-1598.
- Hoda SA, Cranor ML, Rosen PP. Hemangiomas of the breast with atypical histological features: further analysis of histological subtypes confirming their benign character. Am J Surg Pathol. 1992;16:553-560.
- Wagamon K, Ranchoff RE, Rosenberg AS, et al. Benign lymphangiomatous papules of the skin. J Am Acad Dermatol. 2005;52:912-913.
- Diaz-Cascajo C, Borghi S, Weyers W, et al. Benign lymphangiomatous papules of the skin following radiotherapy: a report of five new cases and review of the literature. Histopathology. 1999;35:319-327.
- Martín-González T, Sanz-Trelles A, Del Boz J, et al. Benign lymphangiomatous papules and plaques after radiotherapy [in Spanish]. Actas Dermosifiliogr. 2008;99:84-86.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Guillou L, Fletcher CD. Benign lymphangioendothelioma (acquired progressive lymphangioma): a lesion not to be confused with well-differentiated angiosarcoma and patch stage Kaposi’s sarcoma: clinicopathologic analysis of a series. Am J Surg Pathol. 2000;24:1047-1057.
- Rosso R, Gianelli U, Carnevali L. Acquired progressive lymphangioma of the skin following radiotherapy for breast carcinoma. J Cutan Pathol. 1995;22:164-167.
- Hildebrandt G, Mittag M, Gutz U, et al. Cutaneous breast angiosarcoma after conservative treatment of breast cancer. Eur J Dermatol. 2001;11:580-583.
- Di Tommaso L, Fabbri A. Cutaneous angiosarcoma arising after radiotherapy treatment of a breast carcinoma: description of a case and review of the literature [in Italian]. Pathologica. 2003;95:196-202.
- Santi R, Cetica V, Franchi A, et al. Tumour suppressor gene TP53 mutations in atypical vascular lesions of breast skin following radiotherapy. Histopathology. 2011;58:455-466.
- Requena L, Kutzner H, Mentzel T, et al. Benign vascular proliferations in irradiated skin. Am J Surg Pathol. 2002;26:328-337.
- Brodie C, Provenzano E. Vascular proliferations of the breast. Histopathology. 2008;52:30-44.
- Brenn T, Fletcher CD. Postradiation vascular proliferations: an increasing problem. Histopathology. 2006;48:106-114.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Kardum-Skelin I, Jelić-Puskarić B, Pazur M, et al. A case report of breast angiosarcoma. Coll Antropol. 2010;34:645-648.
- Mattoch IW, Robbins JB, Kempson RL, et al. Post-radiotherapy vascular proliferations in mammary skin: a clinicopathologic study of 11 cases. J Am Acad Dermatol. 2007;57:126-133.
- Bodet D, Rodríguez-Cano L, Bartralot R, et al. Benign lymphangiomatous papules of the skin associated with ovarian fibroma. J Am Acad Dermatol. 2007;56(2 suppl):S41-S44.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
Practice Points
- Atypical vascular lesions (AVLs) of the breast can appear an average of 5 years following radiation therapy.
- Although the malignant potential of AVLs remains debatable, excision generally is recommended, as lesions tend to recur.
Approach to the Multitrauma Patient With Sternoclavicular Joint Dislocation
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.

Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.
Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.
Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.

Emergency Imaging: Severe Left Testicular Swelling
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.

Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.

“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.
Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.
“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.
Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.
“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
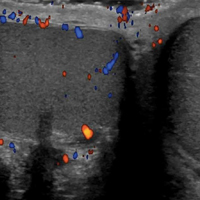
Parkinsonism and Vitamin C Deficiency
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9

Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
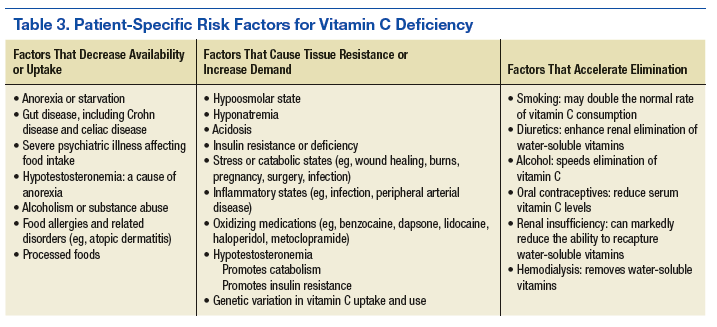
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Hyaluronic Acid Gel Filler for Nipple Enhancement Following Breast Reconstruction
The most frequently used surgical techniques in nipple-areola complex (NAC) reconstruction involve the use of local tissue flaps and yield the fewest complications, though these techniques can be associated with up to a 75% loss in nipple projection over time.1 In a best-case scenario for both the surgeon and the patient, the NAC is preserved during mastectomy; however, even when the tissues are spared, an eventual loss of nipple projection is expected due to atrophy and contraction of the healing skin.2 Loss of nipple projection is the most common attribute that patients dislike regarding their NAC reconstruction results.Additional efforts made to restore the natural look and feel of the NAC provides undeniable benefit to the patient in the form of improved body image and psychosocial well-being.3
Augmentation with a grafted material can include cartilage or fat (autologous grafts), calcium hydroxylapatite or polymethyl methacrylate (PMMA)(alloplastic grafts), and acellular dermal matrix or biologic collagen (allografts). Among these options, successive treatment with autologous fat has been shown to provide satisfactory projections over time with minimal complications.4 However, an additional consideration associated with graft augmentation is the need for an additional surgical site (autologous grafts) or the possibility that graft material may not be compatible with subsequent breast examination techniques. For example, calcium hydroxylapatite is a radiopaque material that may interfere with the interpretation of radiography and mammography.5
The use of injectable hyaluronic acid (HA) dermal fillers to enhance nipple projection represents a noninvasive procedure with immediate and adjustable results. A variety of dermal fillers that do not interfere with subsequent breast imaging needs have already been successfully used for nipple reconstruction including HA 60% plus acrylic hydrogel 40%, PMMA microspheres in a bovine collagen 3.5% gel, and poly-L-lactic acid.5-7
The results achieved with HA 60% plus acrylic hydrogel 40% were as much as a 2.5-mm mean increase in nipple projection after 12 months for 70 nipples reconstructed using a small wedge from the labia minora.5 In these treatments, an initial injection of 0.1 to 0.3 mL of filler into each nipple along with a 0.2-mL injection at the base of each nipple was made. Further optional treatments at 2 and 4 months after the initial injection were made using up to 0.3 mL additional volume depending on filler reabsorption.5 Results achieved with PMMA microspheres in a bovine collagen 3.5% gel included a 1.6-mm mean increase in nipple projection at 9 months versus baseline for 33 nipples in 23 patients, which involved up to 2 injections at baseline and again at 3 months.6 Treatment with poly-L-lactic acid provided a 2.3-mm mean increase in nipple projection for 12 patients after 1 year of treatment, which involved 0.5-mL injections every 4 weeks over a series of 4 treatments.7
This report describes the technique and cosmetic outcome using an injectable HA gel to postoperatively restore the 3-dimensional contour of the nipple following surgical breast reconstruction. This chemically cross-linked, stabilized HA gel suspended in phosphate-buffered saline at a pH of 7 and a concentration of 20 mg/mL with lidocaine 0.3% is indicated for mid to deep dermal implantation for the correction of moderate to severe facial wrinkles and folds, such as the nasolabial folds.8
Case Report
A 49-year-old woman with a history of breast cancer with a focal, high-grade ductal carcinoma in situ underwent a complete bilateral mastectomy. The sentinel lymph nodes were negative at the time of mastectomy. One year later, the patient elected to have breast and nipple-areola (flap) reconstruction. Following the reconstructive surgery, her nipples had become visibly atrophic and flat, and she was interested in cosmetic enhancement.
After informed consent had been obtained from the patient, a baseline measurement of each nipple was made while the patient was standing. Each nipple was then injected with up to 0.1 to 0.2 mL of HA gel filler using a 30-gauge needle inserted 2-mm deep (bilaterally) into each nipple. The patient tolerated the procedure well with no pain, bleeding, or bruising. Although HA gel filler contains lidocaine 0.3% and tricaine can further be used to ensure patient comfort, the nipple reconstruction surgery left the patient with little sensation in the treatment area. Rubbing alcohol was used to prepare the skin prior to the procedure, and fractionated coconut oil spray with a nonadherent dressing was used postprocedure.
Following the injection, an immediate increase of 1.6 and 1.5 mm in nipple projection in the right and left breasts, respectively, was achieved with HA gel. The nipple projection of the right breast was 1.7 mm before injection (Figure, A) and 3.3 mm immediately postinjection (Figure, C). The nipple projection of the left breast was 1.8 mm before injection (Figure, B) and 3.3 mm immediately postinjection (Figure, D).

Comment
With a single treatment consisting of 0.2 mL or less of filler volume, the HA gel used in this procedure provided an immediate mean increase in nipple projection of 1.5 mm. Although our assessment involved a single patient evaluated at baseline and immediately post-injection of HA filler only, it is reasonable to assume that subsequent reinjections would provide results comparable to other fillers. Although other fillers that are semipermanent (acrylic hydrogel) and nonbiodegradable (PMMA) make them more durable, these properties also make the augmentation less reversible in the case of overfilling. As with all dermal fillers, rare side effects associated with injection of HA gel filler could potentially include injection-site inflammation, extrusion of filler at the needle insertion site, minimal pain or discomfort during or after injections, bruising, swelling, or delayed-type hypersensitivity reaction. Ideally, HA gel is a soft transparent filler that is reversible with hyaluronidase, an advantage not shared by other filler materials.9
Conclusion
Nipple augmentation with HA gel is a simple noninvasive
- Sisti A, Grimaldi L, Tassinari J, et al. Nipple-areola complex reconstruction techniques: a literature review. Eur J Surg Oncol. 2016;42:441-465.
- Murthy V, Chamberlain RS. Defining a place for nipple sparing mastectomy in modern breast care: an evidence based review. Breast J. 2013;19:571-581.
- Jabor MA, Shayani P, Collins DR Jr, et al. Nipple-areola reconstruction: satisfaction and clinical determinants. Plast Reconstr Surg. 2002;110:457-463.
- Kaoutzanis C, Xin M, Ballard TN, et al. Autologous fat grafting after breast reconstruction in postmastectomy patients: complications, biopsy rates, and locoregional cancer recurrence rates. Ann Plast Surg. 2016;76:270-275.
- Panettiere P, Marchetti L, Accorsi D. Filler injection enhances the projection of the reconstructed nipple: an original easy technique. Aesthet Plast Surg. 2005;29:287-294.
- McCarthy CM, Van Laeken N, Lennox P, et al. The efficacy of Artecoll injections for the augmentation of nipple projection in breast reconstruction. Eplasty. 2010;10:E7.
- Dessy LA, Troccola A, Ranno RL, et al. The use of Poly-lactic acid to improve projection of reconstructed nipple. Breast. 2011;20:220-224.
- Restylane L [package insert]. Fort Worth, TX: Galderma Laboratories, LP; 2016.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosmet Investig Dermatol. 2013;6:295-316.
The most frequently used surgical techniques in nipple-areola complex (NAC) reconstruction involve the use of local tissue flaps and yield the fewest complications, though these techniques can be associated with up to a 75% loss in nipple projection over time.1 In a best-case scenario for both the surgeon and the patient, the NAC is preserved during mastectomy; however, even when the tissues are spared, an eventual loss of nipple projection is expected due to atrophy and contraction of the healing skin.2 Loss of nipple projection is the most common attribute that patients dislike regarding their NAC reconstruction results.Additional efforts made to restore the natural look and feel of the NAC provides undeniable benefit to the patient in the form of improved body image and psychosocial well-being.3
Augmentation with a grafted material can include cartilage or fat (autologous grafts), calcium hydroxylapatite or polymethyl methacrylate (PMMA)(alloplastic grafts), and acellular dermal matrix or biologic collagen (allografts). Among these options, successive treatment with autologous fat has been shown to provide satisfactory projections over time with minimal complications.4 However, an additional consideration associated with graft augmentation is the need for an additional surgical site (autologous grafts) or the possibility that graft material may not be compatible with subsequent breast examination techniques. For example, calcium hydroxylapatite is a radiopaque material that may interfere with the interpretation of radiography and mammography.5
The use of injectable hyaluronic acid (HA) dermal fillers to enhance nipple projection represents a noninvasive procedure with immediate and adjustable results. A variety of dermal fillers that do not interfere with subsequent breast imaging needs have already been successfully used for nipple reconstruction including HA 60% plus acrylic hydrogel 40%, PMMA microspheres in a bovine collagen 3.5% gel, and poly-L-lactic acid.5-7
The results achieved with HA 60% plus acrylic hydrogel 40% were as much as a 2.5-mm mean increase in nipple projection after 12 months for 70 nipples reconstructed using a small wedge from the labia minora.5 In these treatments, an initial injection of 0.1 to 0.3 mL of filler into each nipple along with a 0.2-mL injection at the base of each nipple was made. Further optional treatments at 2 and 4 months after the initial injection were made using up to 0.3 mL additional volume depending on filler reabsorption.5 Results achieved with PMMA microspheres in a bovine collagen 3.5% gel included a 1.6-mm mean increase in nipple projection at 9 months versus baseline for 33 nipples in 23 patients, which involved up to 2 injections at baseline and again at 3 months.6 Treatment with poly-L-lactic acid provided a 2.3-mm mean increase in nipple projection for 12 patients after 1 year of treatment, which involved 0.5-mL injections every 4 weeks over a series of 4 treatments.7
This report describes the technique and cosmetic outcome using an injectable HA gel to postoperatively restore the 3-dimensional contour of the nipple following surgical breast reconstruction. This chemically cross-linked, stabilized HA gel suspended in phosphate-buffered saline at a pH of 7 and a concentration of 20 mg/mL with lidocaine 0.3% is indicated for mid to deep dermal implantation for the correction of moderate to severe facial wrinkles and folds, such as the nasolabial folds.8
Case Report
A 49-year-old woman with a history of breast cancer with a focal, high-grade ductal carcinoma in situ underwent a complete bilateral mastectomy. The sentinel lymph nodes were negative at the time of mastectomy. One year later, the patient elected to have breast and nipple-areola (flap) reconstruction. Following the reconstructive surgery, her nipples had become visibly atrophic and flat, and she was interested in cosmetic enhancement.
After informed consent had been obtained from the patient, a baseline measurement of each nipple was made while the patient was standing. Each nipple was then injected with up to 0.1 to 0.2 mL of HA gel filler using a 30-gauge needle inserted 2-mm deep (bilaterally) into each nipple. The patient tolerated the procedure well with no pain, bleeding, or bruising. Although HA gel filler contains lidocaine 0.3% and tricaine can further be used to ensure patient comfort, the nipple reconstruction surgery left the patient with little sensation in the treatment area. Rubbing alcohol was used to prepare the skin prior to the procedure, and fractionated coconut oil spray with a nonadherent dressing was used postprocedure.
Following the injection, an immediate increase of 1.6 and 1.5 mm in nipple projection in the right and left breasts, respectively, was achieved with HA gel. The nipple projection of the right breast was 1.7 mm before injection (Figure, A) and 3.3 mm immediately postinjection (Figure, C). The nipple projection of the left breast was 1.8 mm before injection (Figure, B) and 3.3 mm immediately postinjection (Figure, D).
Comment
With a single treatment consisting of 0.2 mL or less of filler volume, the HA gel used in this procedure provided an immediate mean increase in nipple projection of 1.5 mm. Although our assessment involved a single patient evaluated at baseline and immediately post-injection of HA filler only, it is reasonable to assume that subsequent reinjections would provide results comparable to other fillers. Although other fillers that are semipermanent (acrylic hydrogel) and nonbiodegradable (PMMA) make them more durable, these properties also make the augmentation less reversible in the case of overfilling. As with all dermal fillers, rare side effects associated with injection of HA gel filler could potentially include injection-site inflammation, extrusion of filler at the needle insertion site, minimal pain or discomfort during or after injections, bruising, swelling, or delayed-type hypersensitivity reaction. Ideally, HA gel is a soft transparent filler that is reversible with hyaluronidase, an advantage not shared by other filler materials.9
Conclusion
Nipple augmentation with HA gel is a simple noninvasive
The most frequently used surgical techniques in nipple-areola complex (NAC) reconstruction involve the use of local tissue flaps and yield the fewest complications, though these techniques can be associated with up to a 75% loss in nipple projection over time.1 In a best-case scenario for both the surgeon and the patient, the NAC is preserved during mastectomy; however, even when the tissues are spared, an eventual loss of nipple projection is expected due to atrophy and contraction of the healing skin.2 Loss of nipple projection is the most common attribute that patients dislike regarding their NAC reconstruction results.Additional efforts made to restore the natural look and feel of the NAC provides undeniable benefit to the patient in the form of improved body image and psychosocial well-being.3
Augmentation with a grafted material can include cartilage or fat (autologous grafts), calcium hydroxylapatite or polymethyl methacrylate (PMMA)(alloplastic grafts), and acellular dermal matrix or biologic collagen (allografts). Among these options, successive treatment with autologous fat has been shown to provide satisfactory projections over time with minimal complications.4 However, an additional consideration associated with graft augmentation is the need for an additional surgical site (autologous grafts) or the possibility that graft material may not be compatible with subsequent breast examination techniques. For example, calcium hydroxylapatite is a radiopaque material that may interfere with the interpretation of radiography and mammography.5
The use of injectable hyaluronic acid (HA) dermal fillers to enhance nipple projection represents a noninvasive procedure with immediate and adjustable results. A variety of dermal fillers that do not interfere with subsequent breast imaging needs have already been successfully used for nipple reconstruction including HA 60% plus acrylic hydrogel 40%, PMMA microspheres in a bovine collagen 3.5% gel, and poly-L-lactic acid.5-7
The results achieved with HA 60% plus acrylic hydrogel 40% were as much as a 2.5-mm mean increase in nipple projection after 12 months for 70 nipples reconstructed using a small wedge from the labia minora.5 In these treatments, an initial injection of 0.1 to 0.3 mL of filler into each nipple along with a 0.2-mL injection at the base of each nipple was made. Further optional treatments at 2 and 4 months after the initial injection were made using up to 0.3 mL additional volume depending on filler reabsorption.5 Results achieved with PMMA microspheres in a bovine collagen 3.5% gel included a 1.6-mm mean increase in nipple projection at 9 months versus baseline for 33 nipples in 23 patients, which involved up to 2 injections at baseline and again at 3 months.6 Treatment with poly-L-lactic acid provided a 2.3-mm mean increase in nipple projection for 12 patients after 1 year of treatment, which involved 0.5-mL injections every 4 weeks over a series of 4 treatments.7
This report describes the technique and cosmetic outcome using an injectable HA gel to postoperatively restore the 3-dimensional contour of the nipple following surgical breast reconstruction. This chemically cross-linked, stabilized HA gel suspended in phosphate-buffered saline at a pH of 7 and a concentration of 20 mg/mL with lidocaine 0.3% is indicated for mid to deep dermal implantation for the correction of moderate to severe facial wrinkles and folds, such as the nasolabial folds.8
Case Report
A 49-year-old woman with a history of breast cancer with a focal, high-grade ductal carcinoma in situ underwent a complete bilateral mastectomy. The sentinel lymph nodes were negative at the time of mastectomy. One year later, the patient elected to have breast and nipple-areola (flap) reconstruction. Following the reconstructive surgery, her nipples had become visibly atrophic and flat, and she was interested in cosmetic enhancement.
After informed consent had been obtained from the patient, a baseline measurement of each nipple was made while the patient was standing. Each nipple was then injected with up to 0.1 to 0.2 mL of HA gel filler using a 30-gauge needle inserted 2-mm deep (bilaterally) into each nipple. The patient tolerated the procedure well with no pain, bleeding, or bruising. Although HA gel filler contains lidocaine 0.3% and tricaine can further be used to ensure patient comfort, the nipple reconstruction surgery left the patient with little sensation in the treatment area. Rubbing alcohol was used to prepare the skin prior to the procedure, and fractionated coconut oil spray with a nonadherent dressing was used postprocedure.
Following the injection, an immediate increase of 1.6 and 1.5 mm in nipple projection in the right and left breasts, respectively, was achieved with HA gel. The nipple projection of the right breast was 1.7 mm before injection (Figure, A) and 3.3 mm immediately postinjection (Figure, C). The nipple projection of the left breast was 1.8 mm before injection (Figure, B) and 3.3 mm immediately postinjection (Figure, D).
Comment
With a single treatment consisting of 0.2 mL or less of filler volume, the HA gel used in this procedure provided an immediate mean increase in nipple projection of 1.5 mm. Although our assessment involved a single patient evaluated at baseline and immediately post-injection of HA filler only, it is reasonable to assume that subsequent reinjections would provide results comparable to other fillers. Although other fillers that are semipermanent (acrylic hydrogel) and nonbiodegradable (PMMA) make them more durable, these properties also make the augmentation less reversible in the case of overfilling. As with all dermal fillers, rare side effects associated with injection of HA gel filler could potentially include injection-site inflammation, extrusion of filler at the needle insertion site, minimal pain or discomfort during or after injections, bruising, swelling, or delayed-type hypersensitivity reaction. Ideally, HA gel is a soft transparent filler that is reversible with hyaluronidase, an advantage not shared by other filler materials.9
Conclusion
Nipple augmentation with HA gel is a simple noninvasive
- Sisti A, Grimaldi L, Tassinari J, et al. Nipple-areola complex reconstruction techniques: a literature review. Eur J Surg Oncol. 2016;42:441-465.
- Murthy V, Chamberlain RS. Defining a place for nipple sparing mastectomy in modern breast care: an evidence based review. Breast J. 2013;19:571-581.
- Jabor MA, Shayani P, Collins DR Jr, et al. Nipple-areola reconstruction: satisfaction and clinical determinants. Plast Reconstr Surg. 2002;110:457-463.
- Kaoutzanis C, Xin M, Ballard TN, et al. Autologous fat grafting after breast reconstruction in postmastectomy patients: complications, biopsy rates, and locoregional cancer recurrence rates. Ann Plast Surg. 2016;76:270-275.
- Panettiere P, Marchetti L, Accorsi D. Filler injection enhances the projection of the reconstructed nipple: an original easy technique. Aesthet Plast Surg. 2005;29:287-294.
- McCarthy CM, Van Laeken N, Lennox P, et al. The efficacy of Artecoll injections for the augmentation of nipple projection in breast reconstruction. Eplasty. 2010;10:E7.
- Dessy LA, Troccola A, Ranno RL, et al. The use of Poly-lactic acid to improve projection of reconstructed nipple. Breast. 2011;20:220-224.
- Restylane L [package insert]. Fort Worth, TX: Galderma Laboratories, LP; 2016.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosmet Investig Dermatol. 2013;6:295-316.
- Sisti A, Grimaldi L, Tassinari J, et al. Nipple-areola complex reconstruction techniques: a literature review. Eur J Surg Oncol. 2016;42:441-465.
- Murthy V, Chamberlain RS. Defining a place for nipple sparing mastectomy in modern breast care: an evidence based review. Breast J. 2013;19:571-581.
- Jabor MA, Shayani P, Collins DR Jr, et al. Nipple-areola reconstruction: satisfaction and clinical determinants. Plast Reconstr Surg. 2002;110:457-463.
- Kaoutzanis C, Xin M, Ballard TN, et al. Autologous fat grafting after breast reconstruction in postmastectomy patients: complications, biopsy rates, and locoregional cancer recurrence rates. Ann Plast Surg. 2016;76:270-275.
- Panettiere P, Marchetti L, Accorsi D. Filler injection enhances the projection of the reconstructed nipple: an original easy technique. Aesthet Plast Surg. 2005;29:287-294.
- McCarthy CM, Van Laeken N, Lennox P, et al. The efficacy of Artecoll injections for the augmentation of nipple projection in breast reconstruction. Eplasty. 2010;10:E7.
- Dessy LA, Troccola A, Ranno RL, et al. The use of Poly-lactic acid to improve projection of reconstructed nipple. Breast. 2011;20:220-224.
- Restylane L [package insert]. Fort Worth, TX: Galderma Laboratories, LP; 2016.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosmet Investig Dermatol. 2013;6:295-316.
Practice Points
- The use of injectable hyaluronic acid (HA) gel to restore 3-dimensional contour of the nipple following nipple-areola complex (NAC) reconstruction is a noninvasive procedure that contributes to patient well-being.
- The use of HA gel for NAC augmentation can be performed in an office setting and may eliminate the need for secondary reconstructive surgeries.
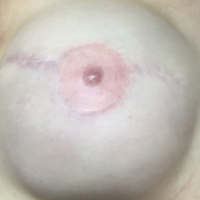
Aggressive Merkel Cell Carcinoma in a Liver Transplant Recipient
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
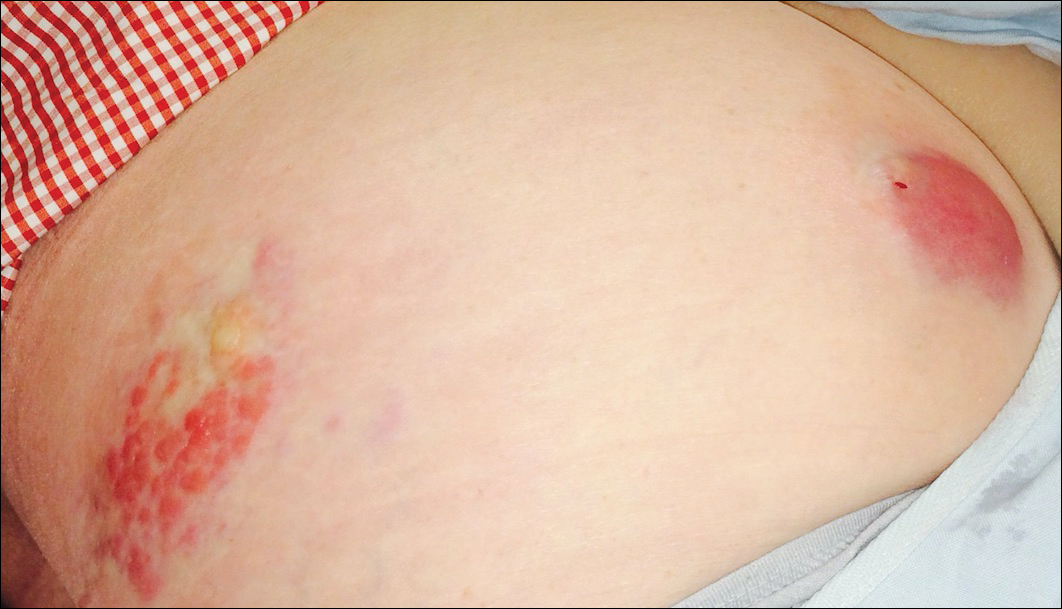
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
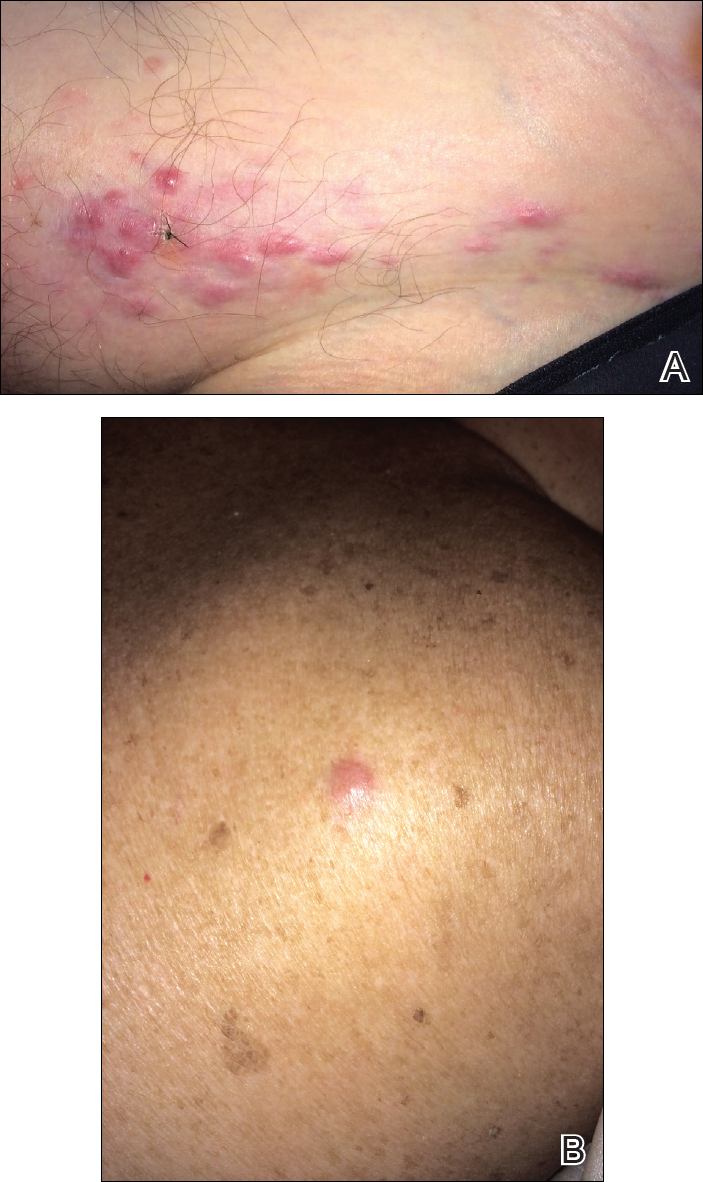
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
Practice Points
- Organ transplant recipients are at an increased risk for Merkel cell carcinoma (MCC).
- Early recognition and diagnosis of MCC is important to improve morbidity and mortality.
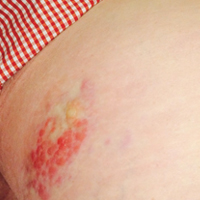
Annular Atrophic Lichen Planus Responds to Hydroxychloroquine and Acitretin
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.


The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).

The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
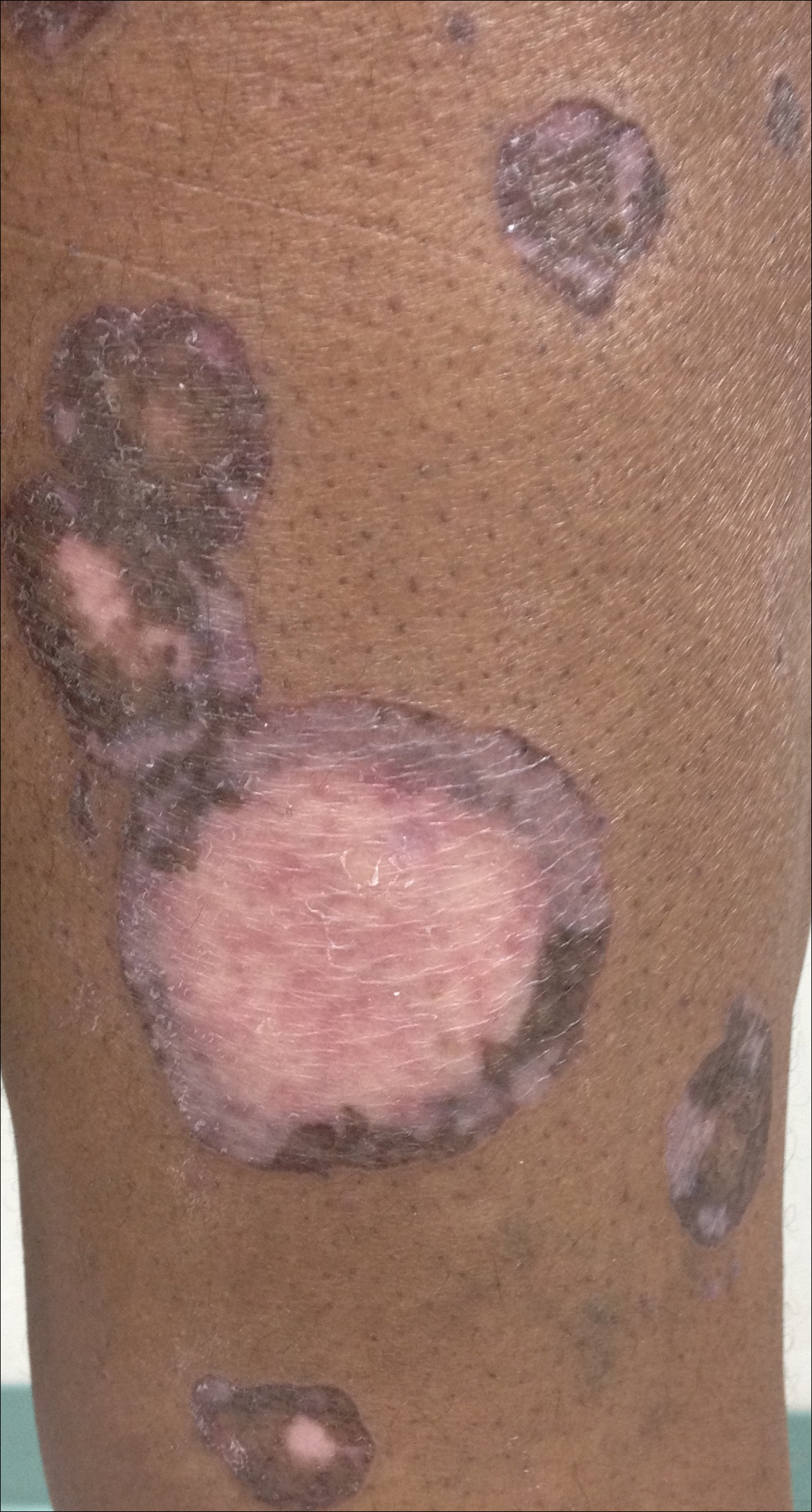
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
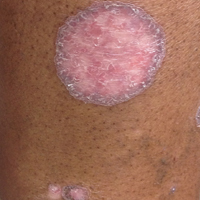
Asymptomatic Cutaneous Polyarteritis Nodosa: Treatment Options and Therapeutic Guidelines
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
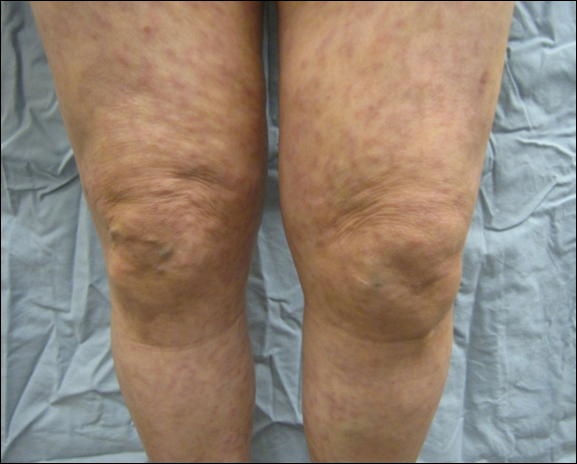
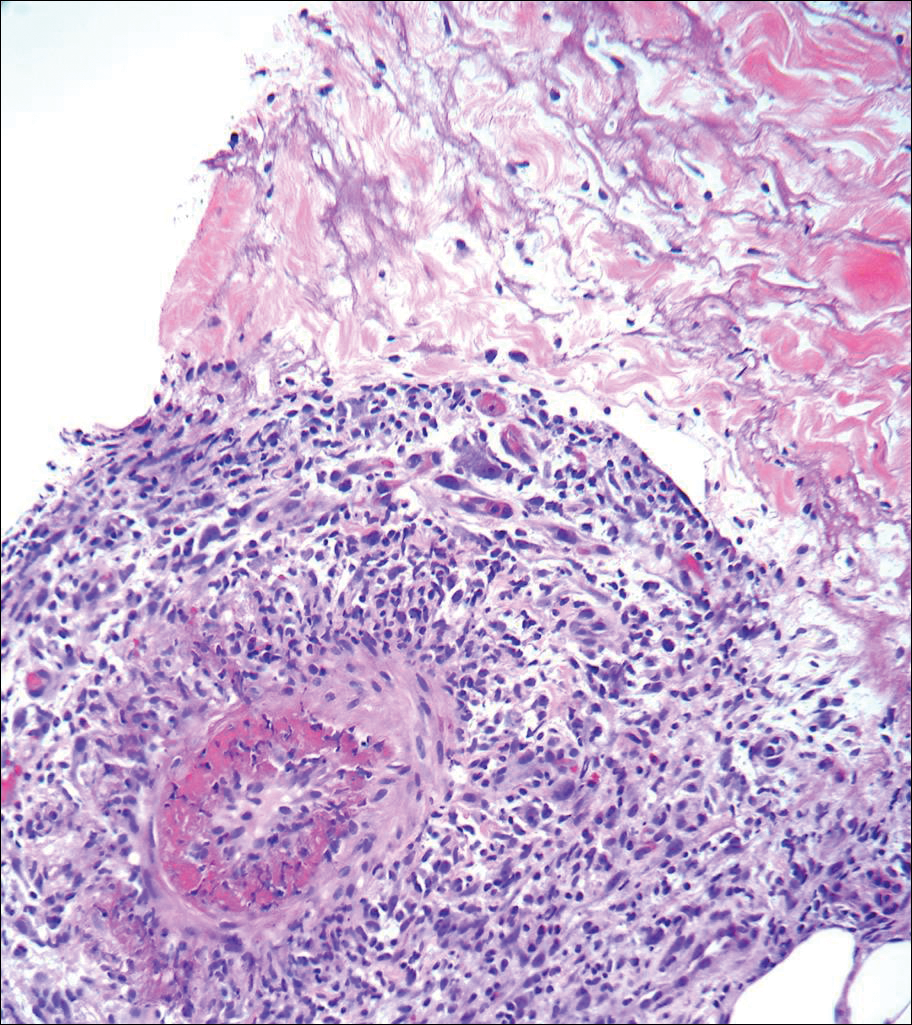
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.

First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
Practice Points
- Cutaneous polyarteritis nodosa should be in the differential of new-onset livedo reticularis.
- Workup with biopsy and specific blood work is important.
- Treatment options at this time are limited.
Low-grade fever, erythematous rash in pregnant woman • Dx?
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.
