User login
Eosinophilic Pustular Folliculitis With Underlying Mantle Cell Lymphoma
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
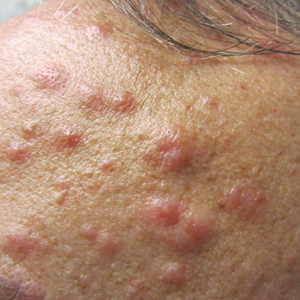
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
, neck, and chest (B), as well as edematous pink papules and plaques on the forehead (C and D).")
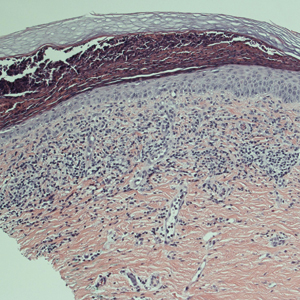
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
(H&E, original magnifications ×10 and ×20).")
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Practice Points
- Recalcitrant folliculocentric papules and pustules involving the head, trunk, arms, and legs should raise suspicion of possible eosinophilic pustular folliculitis (EPF).
- Underlying hematopoietic malignancy may be associated with cases of EPF.
Acrodermatitis Enteropathica From Zinc-Deficient Total Parenteral Nutrition
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).

 and dorsum of the right foot (B).")
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).

After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).

Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7 Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7 Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7 Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Practice Points
- Acrodermatitis enteropathica (AE) may be acquired or due to a rare autosomal-recessive disorder of zinc absorption.
- Hereditary AE typically becomes symptomatic during infancy, while acquired AE may develop during hypozincemia in patients of any age.
- Both acquired and hereditary AE improve with zinc supplementation.
Uncommon Presentation of Chromoblastomycosis
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.

Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
 in the dermis (H&E, original magnification ×100).")
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
 grew olive green, rugose, velvety, leathery colonies within 48 hours.")
.")
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.

Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.

Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Practice Points
- Chromoblastomycosis is limited to skin and subcutaneous tissue, most commonly of the lower limb, especially the foot; it does not directly invade muscle or bone. Secondary bacterial infection is common.
- Chromoblastomycosis is a therapeutic challenge due to its recalcitrant nature. Itraconazole and terbinafine are considered drugs of first choice, but consensus and evidence are lacking on a standard of care.
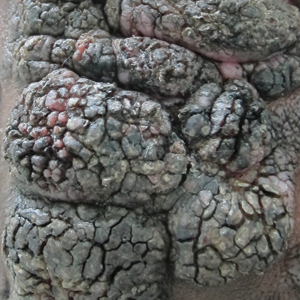
Delayed Cutaneous Reactions to Iodinated Contrast
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
 and left arm (B).")
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
Practice Points
- Delayed cutaneous reactions to iodinated contrast (IC) are common, but patients frequently are misdiagnosed and inadvertently readministered the offending agent.
- The most common IC-induced delayed reactions are self-limited exanthematous eruptions that develop within 1 week of exposure.
- Risk factors for delayed reactions to IC include atopy, contrast exposure during high pollen season, use of the agent iodixanol, a history of other cutaneous drug eruptions, elevated serum creatinine levels, and treatment with recombinant interleukin 2.
- Dermatologists can help prevent recurrent reactions in patients who require repeated exposure to IC by recommending gadolinium-based contrast agents and/or premedication.
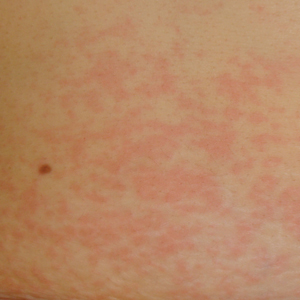
Unusual Presentation of Ectopic Extramammary Paget Disease
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
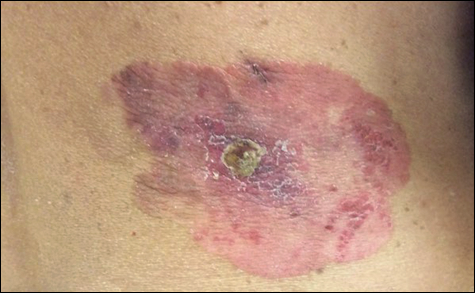
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
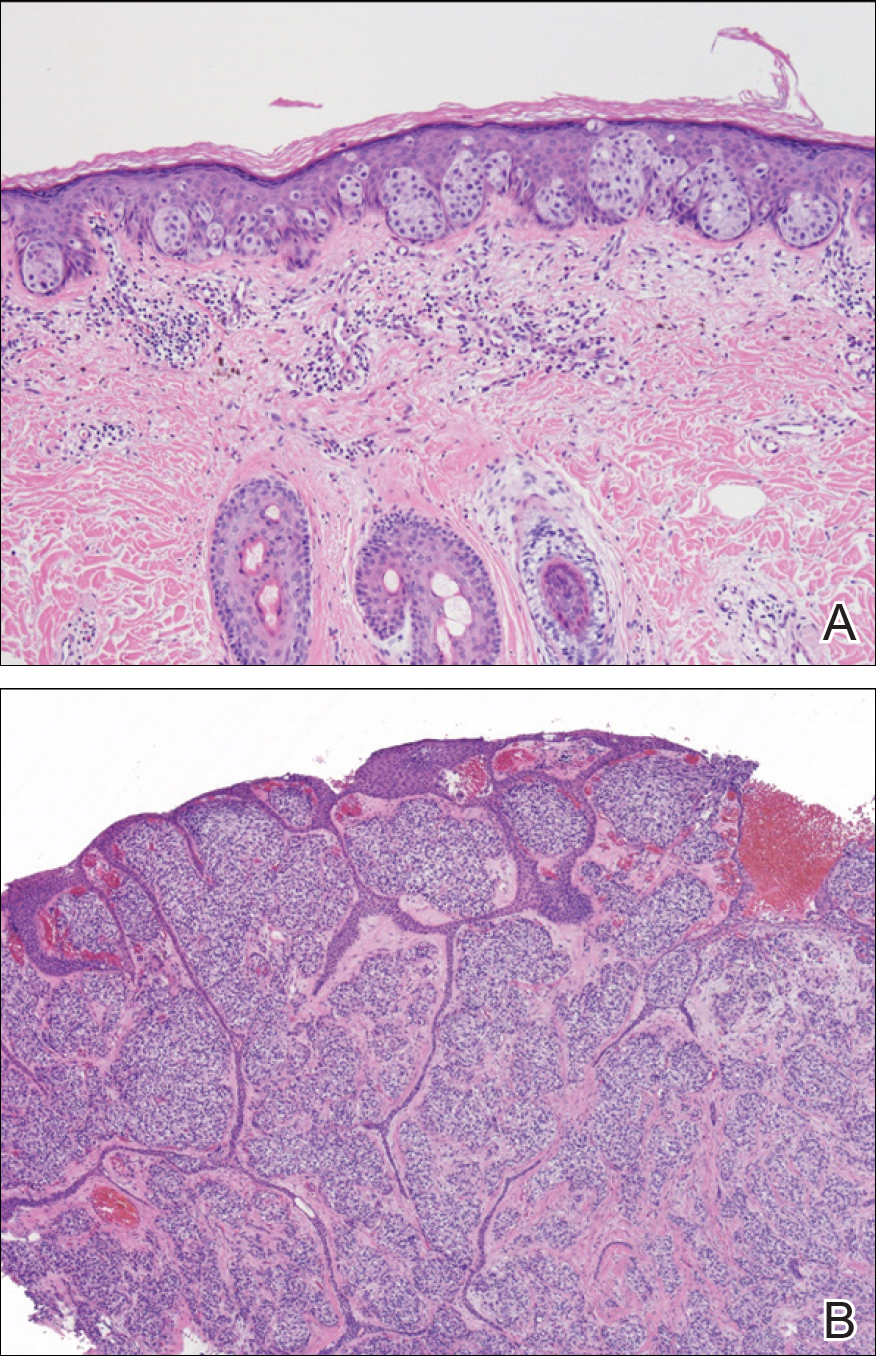
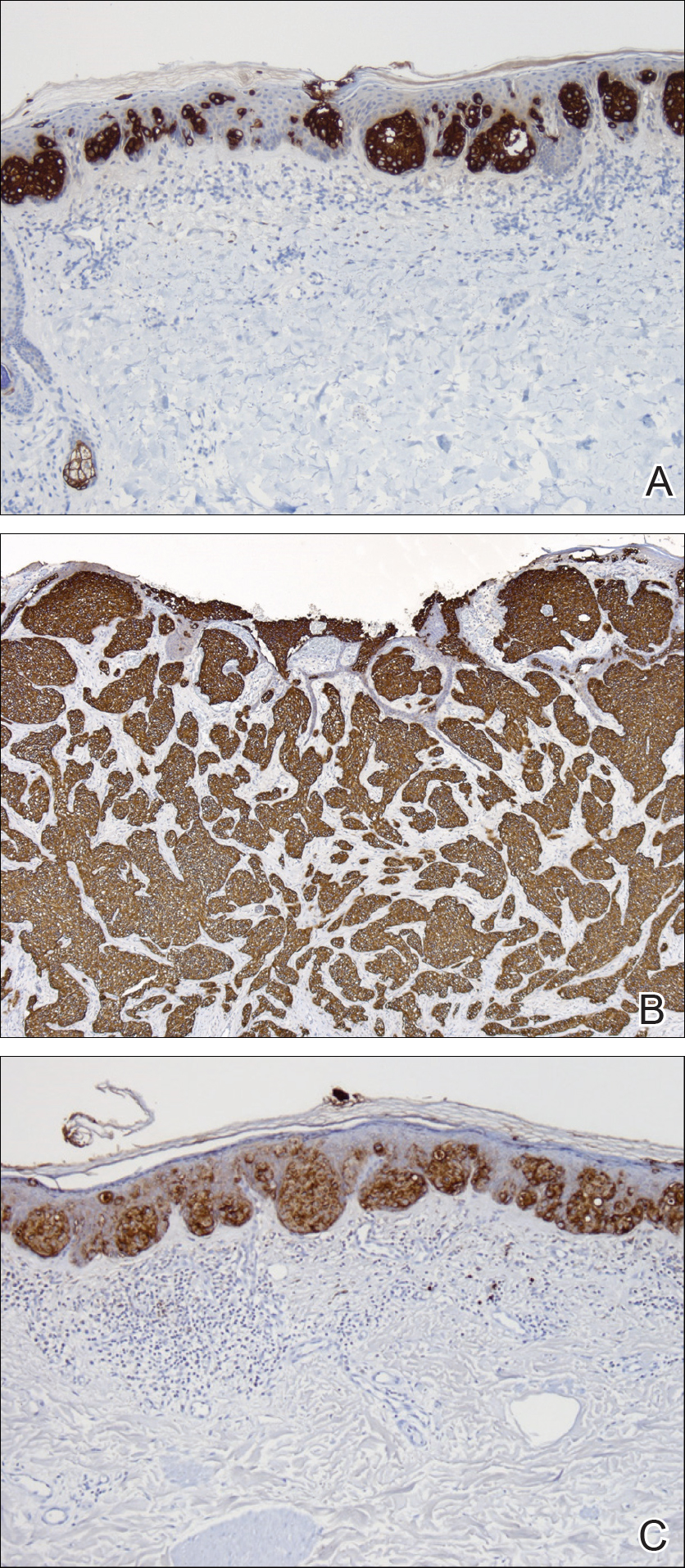
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
Practice Points
- Ectopic extramammary Paget disease (EMPD) is a rare presentation of EMPD that is histologically identical to EMPD.
- Ectopic EMPD can be associated with underlying malignancy and therefore warrants a thorough workup.
Electrocardiography: Flecainide Toxicity
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
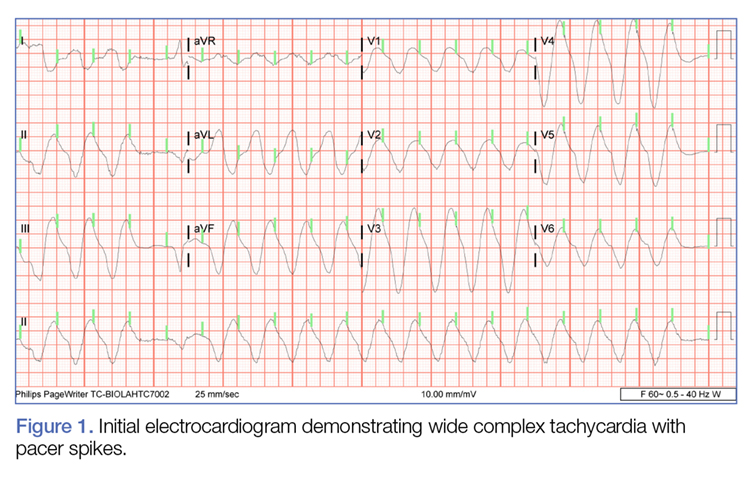
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
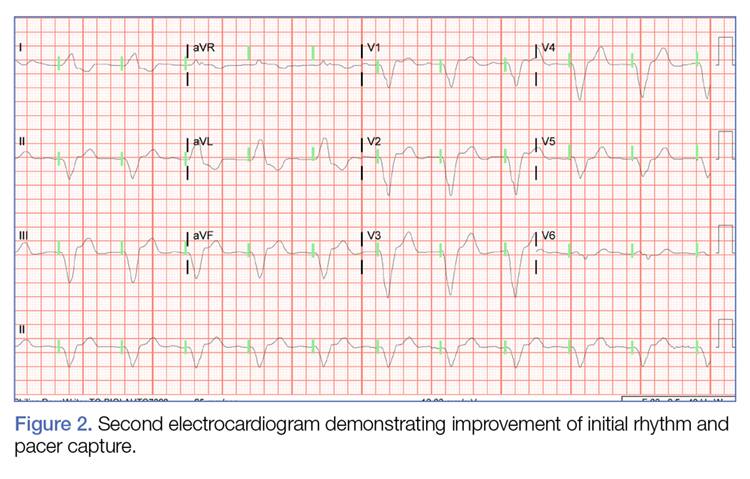
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
Sarcoidosis Resulting in Exsanguinating Esophageal Variceal Hemorrhage
Sarcoidosis is a systemic disorder of unknown etiology and is characterized by the formation of granulomas throughout various organs in the body. The most common form is pulmonary sarcoidosis, which affects 90% of patients; the second most common form is oculocutaneous sarcoidosis;1 and the third most common form is hepatic sarcoidosis, which affects 63% to 90% of patients.2 Although the liver is frequently involved in all forms of sarcoidosis, only a fraction of patients present with clinically evident liver disease.1 Approximately 20% to 30% of patients have abnormalities on liver function tests, whereas only about 1% of patients show evidence of portal hypertension and cirrhosis.3 In fact, in the English literature, there were 35 reported cases of portal hypertension due to sarcoidosis between 1949 to 2001, of which 16 of the patients had no evidence of cirrhosis.4
The diagnosis of sarcoidosis is usually made by a compilation of clinical signs and symptoms, imaging studies, and biopsies demonstrating noncaseating granulomas. This case report describes a patient who presented with portal hypertension and esophageal variceal bleeding secondary to sarcoidosis of the liver without cirrhotic changes.
Case
A 47-year-old woman presented to the ED via emergency medical services with a 1-hour history of hematemesis and melena. The patient stated that she felt fatigued, nauseated, and light-headed, but had no pain or focal weakness. Her medical history was significant for pulmonary and renal sarcoidosis. She underwent a liver biopsy 1 week prior to presentation, with a 6-day hospitalization period, due to new ascites found on examination.
The patient’s vital signs at presentation were: blood pressure (BP), 72/56 mm Hg; heart rate (HR), 133 beats/min, respiratory rate, 24 breaths/min; and temperature, 97.0oF. Oxygen saturation was 99% on room air. Physical examination revealed an alert and oriented middle-aged woman in extremis who was vomiting dark-colored blood. The cardiac and pulmonary examination revealed no extraneous sounds; the abdominal examination showed ascites with a liver edge palpable 4 cm beneath the right costal margin. The patient had no scleral icterus, palmar erythema, spider angiomata, fetor hepaticus, caput medusa, cutaneous ecchymoses, or any other stigmata of cirrhosis.
Two large-bore peripheral intravenous (IV) catheters were placed and a massive blood transfusion protocol was initiated. Packed red blood cells (PRBCs) from the resuscitation-area refrigerator were infused immediately via a pressurized fluid warmer.
After consultation with gastroenterology and general surgery services, the patient was given 1 g ceftriaxone IV, 1 g tranexamic acid IV, 20 mcg desmopressin IV, 50 mcg octreotide IV, 40 mg pantoprazole IV, 8 mg ondansetron IV, 4 g calcium gluconate IV, and 100 mg hydrocortisone IV.
Throughout the patient’s first 10 minutes in the ED, she remained persistently hypotensive and continued to vomit. Since the patient’s sensorium was intact, the team quickly discussed goals of care with her. The patient’s wishes were for maximal life-sustaining therapy, including endotracheal intubation and chest compressions, if necessary.
After this discussion, the patient was given IV etomidate and rocuronium and was intubated using video-assisted laryngoscopy. Following intubation, she was sedated with an infusion of fentanyl and underwent orogastric tube placement to aspirate stomach contents. A total of 2.5 L of frank blood were drained from the patient’s stomach.
A size 9 French single lumen left-femoral central venous catheter also was placed, through which additional blood products were infused. The patient received a total of 28 U PRBCs, fresh frozen plasma, and platelets over a 3-hour period. During transfusion, the patient’s vital signs improved to a systolic BP ranging between 110 to 120 mm Hg and an HR ranging between 90 to 110 beats/min; she did not experience any further hypotensive episodes throughout her stay in the ED.
Laboratory studies were significant for metabolic acidosis, hyperkalemia, acute on chronic anemia, leukocytosis, and acute on chronic renal failure. Synthetic function of the liver and transaminases appeared normal (Table).
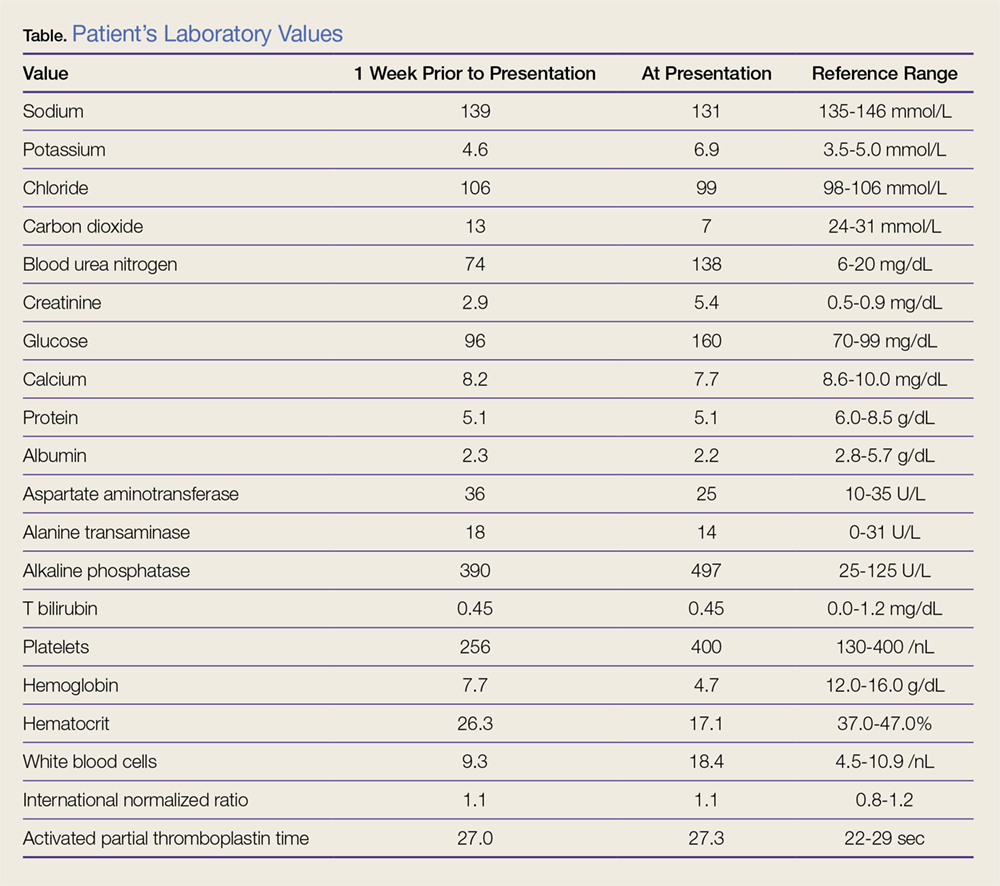
The patient’s hyperkalemia was treated with 1 g calcium chloride IV, 50 g dextrose IV, and 10 U regular insulin IV. A portable chest radiograph showed an appropriately positioned endotracheal tube, and an electrocardiogram revealed sinus tachycardia without signs of hyperkalemia. A computed tomography (CT) scan of the abdomen and pelvis from the patient’s recent hospitalization, 1 week prior to presentation, showed hepatomegaly, liver granulomas, ascites, and periportal lymphadenopathy (Figure 1).
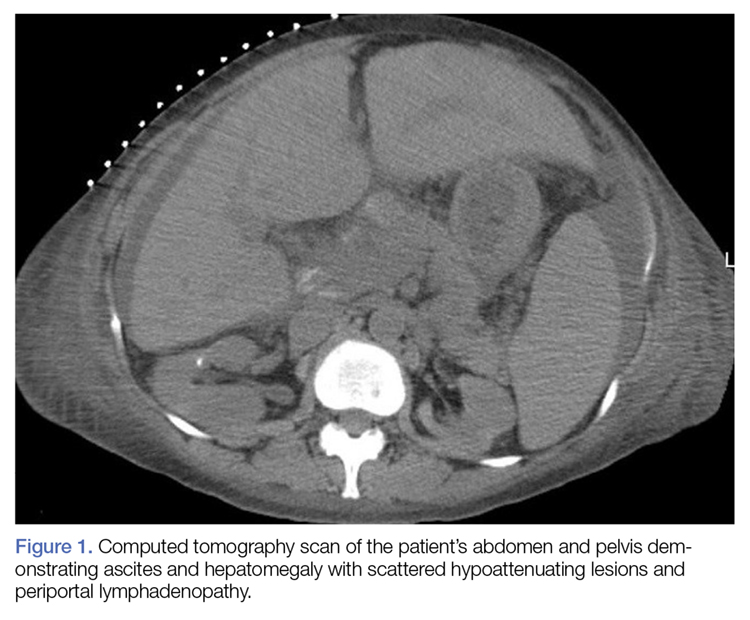
A review of the patient’s recent liver biopsy and ascitic fluid analysis revealed noncaseating granulomas compressing the hepatic sinusoids, and a serum ascites albumin gradient greater than 1.1 g/dL, implying portal hypertension without cirrhosis. The surgical team attempted to place a Sengstaken-Blakemore tube, but the device could not be positioned properly due to the patient’s narrowed esophagus.
The ED nurses cleaned the patient, preserving her dignity; thereafter the patient’s adult children visited with her briefly before she was taken for an upper endoscopy, which was performed in the ED. The endoscopy revealed actively hemorrhaging esophageal varices at the gastroesophageal junction (Figure 2). The varices were treated with endoscopic ligation; the gastroenterologist placed a total of 11 bands, resulting in cessation of bleeding.

After the endoscopy, the patient was admitted to the medical intensive care unit (ICU). Approximately 1.5 hours after arriving at the ICU, she developed renewed hematemesis. Despite efforts to control bleeding and provide hemodynamic support, the patient died 1 hour later.
Discussion
Etiology
Esophageal variceal hemorrhage is caused by pressure elevation in the portal venous system, leading to engorged esophageal veins that can bleed spontaneously. Approximately 90% of portal hypertension is due to liver cirrhosis.5 The remaining 10% of cases are primarily vascular in etiology, with endothelial dysfunction and thrombosis leading to increased portal resistance. Noncirrhotic causes of portal hypertension include malignancy, congenital diseases, viral hepatitides, vascular thromboses or fistulae, constrictive pericarditis, fatty liver of pregnancy, drugs, radiation injury, and infiltrative diseases.5
Sarcoidosis may cause noncaseating granulomas to form in the liver, leading to portal hypertension and fatal exsanguination from esophageal variceal hemorrhage. Although the lesions of sarcoidosis classically form in the lungs, any organ system may be affected.6,7 Frank cirrhosis of the liver occurs in only 1% of sarcoidosis patients; however, radiographic involvement of the liver is seen in 5% to 15% of patients.8
There are several mechanisms which may be responsible for portal hypertension in patients with sarcoidosis, including granulomas causing mass effect on the hepatic sinusoids; arteriovenous shunts within the granuloma; granulomatous phlebitis within the sinusoids; or compressive periportal lymphadenopathy.9 Regardless of the mechanism, a review of the literature demonstrates an association between sarcoidosis and symptomatic portal hypertension.2,4,10,11Although our patient ultimately died, early initiation of massive blood transfusion protocol, airway protection, attention to electrolytes, and endoscopic control of the hemorrhage source provided the best chance for survival.
Medical Therapy
The first priority in managing and treating esophageal varices is to secure the patient’s airways to prevent aspiration. Two large bore IV lines should be placed to permit rapid infusion of crystalloid fluids or blood products. Initiating antibiotics, specifically IV ceftriaxone, to patients with variceal bleeding is a class I recommendation, as this is the only intervention shown to increase patient survival.12 Although proton pump inhibitors (PPI) and somatostatin analogues (typically octreotide) are frequently given, they are both class II recommendations because there is limited evidence supporting the benefit of their use.12 However, current guidelines recommend treating patients for variceal bleeding with an initial bolus of a PPI, followed by a continuous infusion of PPI for 72 hours. As previously noted, multiple studies, have failed to show any decrease in mortality associated with this treatment.12
Other agents that are used to treat variceal bleeding include octreotide and vasopressin. Octreotide, a somatostatin analog, is generally given as an initial IV bolus followed by continuous infusion, and has been shown to decrease transfusion requirements without mortality benefit.12 Vasopressin is generally given to critically ill patients, and is considered a third-line treatment for variceal bleeding.
Since our patient had a history of chronic kidney disease, desmopressin was empirically administered in the event platelet dysfunction was a contributing factor to bleeding.13 The absence of cirrhosis was significant because our patient was unlikely to have a bleeding diathesis caused by coagulation factor deficiency. Therefore, the goal transfusion ratio of blood products should be balanced, similar to that in traumatic exsanguination, rather than favoring an increased ratio of plasma to other blood products. Similarly, tranexamic acid was administered because insufficient tamponade rather than coagulopathy was the presumed cause of sustained hemorrhage.
An additional complicating factor in our patient’s care was the potential effect of the massive transfusion on electrolytes. Packed RBCs have a pH of approximately 6.8 and may carry up to 25 mmol/L of potassium, which may have exacerbated our patient’s underlying hyperkalemia.14 Rapid blood transfusion also places patients at risk for acute hypocalcemia secondary to citrate toxicity; this did not occur in our patient in part because the metabolic function of her liver was preserved and citrate could be broken down in the hepatocyte Krebs cycle.15 Calcium therapy doubled as treatment for the hyperkalemia and as prophylaxis against further hypocalcemia. No dysrhythmias were observed.
Surgical Intervention
Emergency physicians should consult with gastroenterology services so that an endoscopy can be performed as soon as possible to evaluate for and control bleeding. When an endoscopy cannot be performed rapidly, there are multiple balloon tamponade devices available that can be used to temporize the bleeding, such as the Sengstaken-Blakemore tube.12
Although balloon tamponade devices are typically reserved for the last line of therapy, endoscopy rather than transjugular intrahepatic portosystemic shunt (TIPS) was the preferred method of hemorrhage source control in our patient for several reasons. First, although the working diagnosis of varices was based on the patient’s history, we wanted to evaluate for other causes of upper gastrointestinal bleeding since our patient had no history of endoscopy. Therefore, endoscopy had both a therapeutic and diagnostic value. Secondly, though TIPS may decrease pressure within the bleeding varix, only endoscopy permits direct hemostasis. Also, endoscopy also was preferred over TIPS because our patient was too unstable to move to the interventional radiology suite.16
Conclusion
Although life-threatening esophageal variceal hemorrhage is a rare manifestation of an uncommon disease, it should be considered in the differential diagnosis of a patient who has sarcoidosis and presents with gastrointestinal bleeding. Additionally, when caring for a patient with massive hematemesis without evidence of liver cirrhosis, other etiologies of portal hypertension and esophageal varices, such as sarcoidosis, should be considered.
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39(2):277-297. doi:10.1016/j.rdc.2013.02.007.
2. Mistilis SP, Green JR, Schiff L. Hepatic sarcoidosis with portal hypertension. Am J Med. 1964;36(3):470-475. doi:10.1016/0002-9343(64)90175-5.
3. Tekeste H, Latour F, Levitt RE. Portal hypertension complicating sarcoid liver disease: case report and review of the literature. Am J Gastroenterol. 1984;79(5):389-396.
4. Ivonye C, Elhammali B, Henriques-Forsythe M, Bennett-Gittens R, Oderinde A. Disseminated sarcoidosis resulting in portal hypertension and gastrointestinal bleeding: a rare presentation. Can J Gastroenterol. 2012;26(8):508-509. http://www.ncbi.nlm.nih.gov/pubmed/22891173. Accessed May 16, 2018.
5. Tetangco EP, Silva RG, Lerma EV. Portal hypertension: etiology, evaluation, and management. Dis Mon. 2016;62(12):411-426. doi:10.1016/j.disamonth.2016.08.001.
6. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155-1167. doi:10.1016/S0140-6736(13)60680-7.
7. Al-Kofahi K, Korsten P, Ascoli C, et al. Management of extrapulmonary sarcoidosis: challenges and solutions. Ther Clin Risk Manag. 2016;12:1623-1634. doi:10.2147/TCRM.S74476.
8. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153-2165. doi:10.1056/NEJMra071714.
9. Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol. 2008;103(12):3184-3192. doi:10.1111/j.1572-0241.2008.02202.x.
10. Fraimow W, Myerson RM. Portal hypertension and bleeding esophageal varices secondary to sarcoidosis of the liver. Am J Med. 1957;23(6):995-998.
11. Saito H, Ohmori M, Iwamuro M, et al. Hepatic and gastric involvement in a case of systemic sarcoidosis presenting with rupture of esophageal varices. Intern Med. 2018;56(19):2583-2588. doi:10.2169/internalmedicine.8768-16.
12. DeLaney M, Greene CJ. Emergency Department evaluation and management of patients with upper gastrointestinal bleeding. Emerg Med Pract. 2015;17(4):1-18; quiz 19.
13. Ozgönenel B, Rajpurkar M, Lusher JM. How do you treat bleeding disorders with desmopressin? Postgrad Med J. 2007;83(977):159-163. doi:10.1136/pgmj.2006.052118.
14. Sümpelmann R, Schürholz T, Thorns E, Hausdörfer J. Acid-base, electrolyte and metabolite concentrations in packed red blood cells for major transfusion in infants. Paediatr Anaesth. 2001;11(2):169-173. doi:10.1046/j.1460-9592.2001.00637.x.
15. Monchi M. Citrate pathophysiology and metabolism. Transfus Apher Sci. 2018;56(1):28-30. doi:10.1016/j.transci.2016.12.013.
16. Shah RP, Sze DY. Complications during transjugular intrahepatic portosystemic shunt creation. Tech Vasc Interv Radiol. 2016;19(1):61-73. doi:10.1053/j.tvir.2016.01.007.
Sarcoidosis is a systemic disorder of unknown etiology and is characterized by the formation of granulomas throughout various organs in the body. The most common form is pulmonary sarcoidosis, which affects 90% of patients; the second most common form is oculocutaneous sarcoidosis;1 and the third most common form is hepatic sarcoidosis, which affects 63% to 90% of patients.2 Although the liver is frequently involved in all forms of sarcoidosis, only a fraction of patients present with clinically evident liver disease.1 Approximately 20% to 30% of patients have abnormalities on liver function tests, whereas only about 1% of patients show evidence of portal hypertension and cirrhosis.3 In fact, in the English literature, there were 35 reported cases of portal hypertension due to sarcoidosis between 1949 to 2001, of which 16 of the patients had no evidence of cirrhosis.4
The diagnosis of sarcoidosis is usually made by a compilation of clinical signs and symptoms, imaging studies, and biopsies demonstrating noncaseating granulomas. This case report describes a patient who presented with portal hypertension and esophageal variceal bleeding secondary to sarcoidosis of the liver without cirrhotic changes.
Case
A 47-year-old woman presented to the ED via emergency medical services with a 1-hour history of hematemesis and melena. The patient stated that she felt fatigued, nauseated, and light-headed, but had no pain or focal weakness. Her medical history was significant for pulmonary and renal sarcoidosis. She underwent a liver biopsy 1 week prior to presentation, with a 6-day hospitalization period, due to new ascites found on examination.
The patient’s vital signs at presentation were: blood pressure (BP), 72/56 mm Hg; heart rate (HR), 133 beats/min, respiratory rate, 24 breaths/min; and temperature, 97.0oF. Oxygen saturation was 99% on room air. Physical examination revealed an alert and oriented middle-aged woman in extremis who was vomiting dark-colored blood. The cardiac and pulmonary examination revealed no extraneous sounds; the abdominal examination showed ascites with a liver edge palpable 4 cm beneath the right costal margin. The patient had no scleral icterus, palmar erythema, spider angiomata, fetor hepaticus, caput medusa, cutaneous ecchymoses, or any other stigmata of cirrhosis.
Two large-bore peripheral intravenous (IV) catheters were placed and a massive blood transfusion protocol was initiated. Packed red blood cells (PRBCs) from the resuscitation-area refrigerator were infused immediately via a pressurized fluid warmer.
After consultation with gastroenterology and general surgery services, the patient was given 1 g ceftriaxone IV, 1 g tranexamic acid IV, 20 mcg desmopressin IV, 50 mcg octreotide IV, 40 mg pantoprazole IV, 8 mg ondansetron IV, 4 g calcium gluconate IV, and 100 mg hydrocortisone IV.
Throughout the patient’s first 10 minutes in the ED, she remained persistently hypotensive and continued to vomit. Since the patient’s sensorium was intact, the team quickly discussed goals of care with her. The patient’s wishes were for maximal life-sustaining therapy, including endotracheal intubation and chest compressions, if necessary.
After this discussion, the patient was given IV etomidate and rocuronium and was intubated using video-assisted laryngoscopy. Following intubation, she was sedated with an infusion of fentanyl and underwent orogastric tube placement to aspirate stomach contents. A total of 2.5 L of frank blood were drained from the patient’s stomach.
A size 9 French single lumen left-femoral central venous catheter also was placed, through which additional blood products were infused. The patient received a total of 28 U PRBCs, fresh frozen plasma, and platelets over a 3-hour period. During transfusion, the patient’s vital signs improved to a systolic BP ranging between 110 to 120 mm Hg and an HR ranging between 90 to 110 beats/min; she did not experience any further hypotensive episodes throughout her stay in the ED.
Laboratory studies were significant for metabolic acidosis, hyperkalemia, acute on chronic anemia, leukocytosis, and acute on chronic renal failure. Synthetic function of the liver and transaminases appeared normal (Table).
The patient’s hyperkalemia was treated with 1 g calcium chloride IV, 50 g dextrose IV, and 10 U regular insulin IV. A portable chest radiograph showed an appropriately positioned endotracheal tube, and an electrocardiogram revealed sinus tachycardia without signs of hyperkalemia. A computed tomography (CT) scan of the abdomen and pelvis from the patient’s recent hospitalization, 1 week prior to presentation, showed hepatomegaly, liver granulomas, ascites, and periportal lymphadenopathy (Figure 1).
A review of the patient’s recent liver biopsy and ascitic fluid analysis revealed noncaseating granulomas compressing the hepatic sinusoids, and a serum ascites albumin gradient greater than 1.1 g/dL, implying portal hypertension without cirrhosis. The surgical team attempted to place a Sengstaken-Blakemore tube, but the device could not be positioned properly due to the patient’s narrowed esophagus.
The ED nurses cleaned the patient, preserving her dignity; thereafter the patient’s adult children visited with her briefly before she was taken for an upper endoscopy, which was performed in the ED. The endoscopy revealed actively hemorrhaging esophageal varices at the gastroesophageal junction (Figure 2). The varices were treated with endoscopic ligation; the gastroenterologist placed a total of 11 bands, resulting in cessation of bleeding.
After the endoscopy, the patient was admitted to the medical intensive care unit (ICU). Approximately 1.5 hours after arriving at the ICU, she developed renewed hematemesis. Despite efforts to control bleeding and provide hemodynamic support, the patient died 1 hour later.
Discussion
Etiology
Esophageal variceal hemorrhage is caused by pressure elevation in the portal venous system, leading to engorged esophageal veins that can bleed spontaneously. Approximately 90% of portal hypertension is due to liver cirrhosis.5 The remaining 10% of cases are primarily vascular in etiology, with endothelial dysfunction and thrombosis leading to increased portal resistance. Noncirrhotic causes of portal hypertension include malignancy, congenital diseases, viral hepatitides, vascular thromboses or fistulae, constrictive pericarditis, fatty liver of pregnancy, drugs, radiation injury, and infiltrative diseases.5
Sarcoidosis may cause noncaseating granulomas to form in the liver, leading to portal hypertension and fatal exsanguination from esophageal variceal hemorrhage. Although the lesions of sarcoidosis classically form in the lungs, any organ system may be affected.6,7 Frank cirrhosis of the liver occurs in only 1% of sarcoidosis patients; however, radiographic involvement of the liver is seen in 5% to 15% of patients.8
There are several mechanisms which may be responsible for portal hypertension in patients with sarcoidosis, including granulomas causing mass effect on the hepatic sinusoids; arteriovenous shunts within the granuloma; granulomatous phlebitis within the sinusoids; or compressive periportal lymphadenopathy.9 Regardless of the mechanism, a review of the literature demonstrates an association between sarcoidosis and symptomatic portal hypertension.2,4,10,11Although our patient ultimately died, early initiation of massive blood transfusion protocol, airway protection, attention to electrolytes, and endoscopic control of the hemorrhage source provided the best chance for survival.
Medical Therapy
The first priority in managing and treating esophageal varices is to secure the patient’s airways to prevent aspiration. Two large bore IV lines should be placed to permit rapid infusion of crystalloid fluids or blood products. Initiating antibiotics, specifically IV ceftriaxone, to patients with variceal bleeding is a class I recommendation, as this is the only intervention shown to increase patient survival.12 Although proton pump inhibitors (PPI) and somatostatin analogues (typically octreotide) are frequently given, they are both class II recommendations because there is limited evidence supporting the benefit of their use.12 However, current guidelines recommend treating patients for variceal bleeding with an initial bolus of a PPI, followed by a continuous infusion of PPI for 72 hours. As previously noted, multiple studies, have failed to show any decrease in mortality associated with this treatment.12
Other agents that are used to treat variceal bleeding include octreotide and vasopressin. Octreotide, a somatostatin analog, is generally given as an initial IV bolus followed by continuous infusion, and has been shown to decrease transfusion requirements without mortality benefit.12 Vasopressin is generally given to critically ill patients, and is considered a third-line treatment for variceal bleeding.
Since our patient had a history of chronic kidney disease, desmopressin was empirically administered in the event platelet dysfunction was a contributing factor to bleeding.13 The absence of cirrhosis was significant because our patient was unlikely to have a bleeding diathesis caused by coagulation factor deficiency. Therefore, the goal transfusion ratio of blood products should be balanced, similar to that in traumatic exsanguination, rather than favoring an increased ratio of plasma to other blood products. Similarly, tranexamic acid was administered because insufficient tamponade rather than coagulopathy was the presumed cause of sustained hemorrhage.
An additional complicating factor in our patient’s care was the potential effect of the massive transfusion on electrolytes. Packed RBCs have a pH of approximately 6.8 and may carry up to 25 mmol/L of potassium, which may have exacerbated our patient’s underlying hyperkalemia.14 Rapid blood transfusion also places patients at risk for acute hypocalcemia secondary to citrate toxicity; this did not occur in our patient in part because the metabolic function of her liver was preserved and citrate could be broken down in the hepatocyte Krebs cycle.15 Calcium therapy doubled as treatment for the hyperkalemia and as prophylaxis against further hypocalcemia. No dysrhythmias were observed.
Surgical Intervention
Emergency physicians should consult with gastroenterology services so that an endoscopy can be performed as soon as possible to evaluate for and control bleeding. When an endoscopy cannot be performed rapidly, there are multiple balloon tamponade devices available that can be used to temporize the bleeding, such as the Sengstaken-Blakemore tube.12
Although balloon tamponade devices are typically reserved for the last line of therapy, endoscopy rather than transjugular intrahepatic portosystemic shunt (TIPS) was the preferred method of hemorrhage source control in our patient for several reasons. First, although the working diagnosis of varices was based on the patient’s history, we wanted to evaluate for other causes of upper gastrointestinal bleeding since our patient had no history of endoscopy. Therefore, endoscopy had both a therapeutic and diagnostic value. Secondly, though TIPS may decrease pressure within the bleeding varix, only endoscopy permits direct hemostasis. Also, endoscopy also was preferred over TIPS because our patient was too unstable to move to the interventional radiology suite.16
Conclusion
Although life-threatening esophageal variceal hemorrhage is a rare manifestation of an uncommon disease, it should be considered in the differential diagnosis of a patient who has sarcoidosis and presents with gastrointestinal bleeding. Additionally, when caring for a patient with massive hematemesis without evidence of liver cirrhosis, other etiologies of portal hypertension and esophageal varices, such as sarcoidosis, should be considered.
Sarcoidosis is a systemic disorder of unknown etiology and is characterized by the formation of granulomas throughout various organs in the body. The most common form is pulmonary sarcoidosis, which affects 90% of patients; the second most common form is oculocutaneous sarcoidosis;1 and the third most common form is hepatic sarcoidosis, which affects 63% to 90% of patients.2 Although the liver is frequently involved in all forms of sarcoidosis, only a fraction of patients present with clinically evident liver disease.1 Approximately 20% to 30% of patients have abnormalities on liver function tests, whereas only about 1% of patients show evidence of portal hypertension and cirrhosis.3 In fact, in the English literature, there were 35 reported cases of portal hypertension due to sarcoidosis between 1949 to 2001, of which 16 of the patients had no evidence of cirrhosis.4
The diagnosis of sarcoidosis is usually made by a compilation of clinical signs and symptoms, imaging studies, and biopsies demonstrating noncaseating granulomas. This case report describes a patient who presented with portal hypertension and esophageal variceal bleeding secondary to sarcoidosis of the liver without cirrhotic changes.
Case
A 47-year-old woman presented to the ED via emergency medical services with a 1-hour history of hematemesis and melena. The patient stated that she felt fatigued, nauseated, and light-headed, but had no pain or focal weakness. Her medical history was significant for pulmonary and renal sarcoidosis. She underwent a liver biopsy 1 week prior to presentation, with a 6-day hospitalization period, due to new ascites found on examination.
The patient’s vital signs at presentation were: blood pressure (BP), 72/56 mm Hg; heart rate (HR), 133 beats/min, respiratory rate, 24 breaths/min; and temperature, 97.0oF. Oxygen saturation was 99% on room air. Physical examination revealed an alert and oriented middle-aged woman in extremis who was vomiting dark-colored blood. The cardiac and pulmonary examination revealed no extraneous sounds; the abdominal examination showed ascites with a liver edge palpable 4 cm beneath the right costal margin. The patient had no scleral icterus, palmar erythema, spider angiomata, fetor hepaticus, caput medusa, cutaneous ecchymoses, or any other stigmata of cirrhosis.
Two large-bore peripheral intravenous (IV) catheters were placed and a massive blood transfusion protocol was initiated. Packed red blood cells (PRBCs) from the resuscitation-area refrigerator were infused immediately via a pressurized fluid warmer.
After consultation with gastroenterology and general surgery services, the patient was given 1 g ceftriaxone IV, 1 g tranexamic acid IV, 20 mcg desmopressin IV, 50 mcg octreotide IV, 40 mg pantoprazole IV, 8 mg ondansetron IV, 4 g calcium gluconate IV, and 100 mg hydrocortisone IV.
Throughout the patient’s first 10 minutes in the ED, she remained persistently hypotensive and continued to vomit. Since the patient’s sensorium was intact, the team quickly discussed goals of care with her. The patient’s wishes were for maximal life-sustaining therapy, including endotracheal intubation and chest compressions, if necessary.
After this discussion, the patient was given IV etomidate and rocuronium and was intubated using video-assisted laryngoscopy. Following intubation, she was sedated with an infusion of fentanyl and underwent orogastric tube placement to aspirate stomach contents. A total of 2.5 L of frank blood were drained from the patient’s stomach.
A size 9 French single lumen left-femoral central venous catheter also was placed, through which additional blood products were infused. The patient received a total of 28 U PRBCs, fresh frozen plasma, and platelets over a 3-hour period. During transfusion, the patient’s vital signs improved to a systolic BP ranging between 110 to 120 mm Hg and an HR ranging between 90 to 110 beats/min; she did not experience any further hypotensive episodes throughout her stay in the ED.
Laboratory studies were significant for metabolic acidosis, hyperkalemia, acute on chronic anemia, leukocytosis, and acute on chronic renal failure. Synthetic function of the liver and transaminases appeared normal (Table).
The patient’s hyperkalemia was treated with 1 g calcium chloride IV, 50 g dextrose IV, and 10 U regular insulin IV. A portable chest radiograph showed an appropriately positioned endotracheal tube, and an electrocardiogram revealed sinus tachycardia without signs of hyperkalemia. A computed tomography (CT) scan of the abdomen and pelvis from the patient’s recent hospitalization, 1 week prior to presentation, showed hepatomegaly, liver granulomas, ascites, and periportal lymphadenopathy (Figure 1).
A review of the patient’s recent liver biopsy and ascitic fluid analysis revealed noncaseating granulomas compressing the hepatic sinusoids, and a serum ascites albumin gradient greater than 1.1 g/dL, implying portal hypertension without cirrhosis. The surgical team attempted to place a Sengstaken-Blakemore tube, but the device could not be positioned properly due to the patient’s narrowed esophagus.
The ED nurses cleaned the patient, preserving her dignity; thereafter the patient’s adult children visited with her briefly before she was taken for an upper endoscopy, which was performed in the ED. The endoscopy revealed actively hemorrhaging esophageal varices at the gastroesophageal junction (Figure 2). The varices were treated with endoscopic ligation; the gastroenterologist placed a total of 11 bands, resulting in cessation of bleeding.
After the endoscopy, the patient was admitted to the medical intensive care unit (ICU). Approximately 1.5 hours after arriving at the ICU, she developed renewed hematemesis. Despite efforts to control bleeding and provide hemodynamic support, the patient died 1 hour later.
Discussion
Etiology
Esophageal variceal hemorrhage is caused by pressure elevation in the portal venous system, leading to engorged esophageal veins that can bleed spontaneously. Approximately 90% of portal hypertension is due to liver cirrhosis.5 The remaining 10% of cases are primarily vascular in etiology, with endothelial dysfunction and thrombosis leading to increased portal resistance. Noncirrhotic causes of portal hypertension include malignancy, congenital diseases, viral hepatitides, vascular thromboses or fistulae, constrictive pericarditis, fatty liver of pregnancy, drugs, radiation injury, and infiltrative diseases.5
Sarcoidosis may cause noncaseating granulomas to form in the liver, leading to portal hypertension and fatal exsanguination from esophageal variceal hemorrhage. Although the lesions of sarcoidosis classically form in the lungs, any organ system may be affected.6,7 Frank cirrhosis of the liver occurs in only 1% of sarcoidosis patients; however, radiographic involvement of the liver is seen in 5% to 15% of patients.8
There are several mechanisms which may be responsible for portal hypertension in patients with sarcoidosis, including granulomas causing mass effect on the hepatic sinusoids; arteriovenous shunts within the granuloma; granulomatous phlebitis within the sinusoids; or compressive periportal lymphadenopathy.9 Regardless of the mechanism, a review of the literature demonstrates an association between sarcoidosis and symptomatic portal hypertension.2,4,10,11Although our patient ultimately died, early initiation of massive blood transfusion protocol, airway protection, attention to electrolytes, and endoscopic control of the hemorrhage source provided the best chance for survival.
Medical Therapy
The first priority in managing and treating esophageal varices is to secure the patient’s airways to prevent aspiration. Two large bore IV lines should be placed to permit rapid infusion of crystalloid fluids or blood products. Initiating antibiotics, specifically IV ceftriaxone, to patients with variceal bleeding is a class I recommendation, as this is the only intervention shown to increase patient survival.12 Although proton pump inhibitors (PPI) and somatostatin analogues (typically octreotide) are frequently given, they are both class II recommendations because there is limited evidence supporting the benefit of their use.12 However, current guidelines recommend treating patients for variceal bleeding with an initial bolus of a PPI, followed by a continuous infusion of PPI for 72 hours. As previously noted, multiple studies, have failed to show any decrease in mortality associated with this treatment.12
Other agents that are used to treat variceal bleeding include octreotide and vasopressin. Octreotide, a somatostatin analog, is generally given as an initial IV bolus followed by continuous infusion, and has been shown to decrease transfusion requirements without mortality benefit.12 Vasopressin is generally given to critically ill patients, and is considered a third-line treatment for variceal bleeding.
Since our patient had a history of chronic kidney disease, desmopressin was empirically administered in the event platelet dysfunction was a contributing factor to bleeding.13 The absence of cirrhosis was significant because our patient was unlikely to have a bleeding diathesis caused by coagulation factor deficiency. Therefore, the goal transfusion ratio of blood products should be balanced, similar to that in traumatic exsanguination, rather than favoring an increased ratio of plasma to other blood products. Similarly, tranexamic acid was administered because insufficient tamponade rather than coagulopathy was the presumed cause of sustained hemorrhage.
An additional complicating factor in our patient’s care was the potential effect of the massive transfusion on electrolytes. Packed RBCs have a pH of approximately 6.8 and may carry up to 25 mmol/L of potassium, which may have exacerbated our patient’s underlying hyperkalemia.14 Rapid blood transfusion also places patients at risk for acute hypocalcemia secondary to citrate toxicity; this did not occur in our patient in part because the metabolic function of her liver was preserved and citrate could be broken down in the hepatocyte Krebs cycle.15 Calcium therapy doubled as treatment for the hyperkalemia and as prophylaxis against further hypocalcemia. No dysrhythmias were observed.
Surgical Intervention
Emergency physicians should consult with gastroenterology services so that an endoscopy can be performed as soon as possible to evaluate for and control bleeding. When an endoscopy cannot be performed rapidly, there are multiple balloon tamponade devices available that can be used to temporize the bleeding, such as the Sengstaken-Blakemore tube.12
Although balloon tamponade devices are typically reserved for the last line of therapy, endoscopy rather than transjugular intrahepatic portosystemic shunt (TIPS) was the preferred method of hemorrhage source control in our patient for several reasons. First, although the working diagnosis of varices was based on the patient’s history, we wanted to evaluate for other causes of upper gastrointestinal bleeding since our patient had no history of endoscopy. Therefore, endoscopy had both a therapeutic and diagnostic value. Secondly, though TIPS may decrease pressure within the bleeding varix, only endoscopy permits direct hemostasis. Also, endoscopy also was preferred over TIPS because our patient was too unstable to move to the interventional radiology suite.16
Conclusion
Although life-threatening esophageal variceal hemorrhage is a rare manifestation of an uncommon disease, it should be considered in the differential diagnosis of a patient who has sarcoidosis and presents with gastrointestinal bleeding. Additionally, when caring for a patient with massive hematemesis without evidence of liver cirrhosis, other etiologies of portal hypertension and esophageal varices, such as sarcoidosis, should be considered.
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39(2):277-297. doi:10.1016/j.rdc.2013.02.007.
2. Mistilis SP, Green JR, Schiff L. Hepatic sarcoidosis with portal hypertension. Am J Med. 1964;36(3):470-475. doi:10.1016/0002-9343(64)90175-5.
3. Tekeste H, Latour F, Levitt RE. Portal hypertension complicating sarcoid liver disease: case report and review of the literature. Am J Gastroenterol. 1984;79(5):389-396.
4. Ivonye C, Elhammali B, Henriques-Forsythe M, Bennett-Gittens R, Oderinde A. Disseminated sarcoidosis resulting in portal hypertension and gastrointestinal bleeding: a rare presentation. Can J Gastroenterol. 2012;26(8):508-509. http://www.ncbi.nlm.nih.gov/pubmed/22891173. Accessed May 16, 2018.
5. Tetangco EP, Silva RG, Lerma EV. Portal hypertension: etiology, evaluation, and management. Dis Mon. 2016;62(12):411-426. doi:10.1016/j.disamonth.2016.08.001.
6. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155-1167. doi:10.1016/S0140-6736(13)60680-7.
7. Al-Kofahi K, Korsten P, Ascoli C, et al. Management of extrapulmonary sarcoidosis: challenges and solutions. Ther Clin Risk Manag. 2016;12:1623-1634. doi:10.2147/TCRM.S74476.
8. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153-2165. doi:10.1056/NEJMra071714.
9. Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol. 2008;103(12):3184-3192. doi:10.1111/j.1572-0241.2008.02202.x.
10. Fraimow W, Myerson RM. Portal hypertension and bleeding esophageal varices secondary to sarcoidosis of the liver. Am J Med. 1957;23(6):995-998.
11. Saito H, Ohmori M, Iwamuro M, et al. Hepatic and gastric involvement in a case of systemic sarcoidosis presenting with rupture of esophageal varices. Intern Med. 2018;56(19):2583-2588. doi:10.2169/internalmedicine.8768-16.
12. DeLaney M, Greene CJ. Emergency Department evaluation and management of patients with upper gastrointestinal bleeding. Emerg Med Pract. 2015;17(4):1-18; quiz 19.
13. Ozgönenel B, Rajpurkar M, Lusher JM. How do you treat bleeding disorders with desmopressin? Postgrad Med J. 2007;83(977):159-163. doi:10.1136/pgmj.2006.052118.
14. Sümpelmann R, Schürholz T, Thorns E, Hausdörfer J. Acid-base, electrolyte and metabolite concentrations in packed red blood cells for major transfusion in infants. Paediatr Anaesth. 2001;11(2):169-173. doi:10.1046/j.1460-9592.2001.00637.x.
15. Monchi M. Citrate pathophysiology and metabolism. Transfus Apher Sci. 2018;56(1):28-30. doi:10.1016/j.transci.2016.12.013.
16. Shah RP, Sze DY. Complications during transjugular intrahepatic portosystemic shunt creation. Tech Vasc Interv Radiol. 2016;19(1):61-73. doi:10.1053/j.tvir.2016.01.007.
1. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013;39(2):277-297. doi:10.1016/j.rdc.2013.02.007.
2. Mistilis SP, Green JR, Schiff L. Hepatic sarcoidosis with portal hypertension. Am J Med. 1964;36(3):470-475. doi:10.1016/0002-9343(64)90175-5.
3. Tekeste H, Latour F, Levitt RE. Portal hypertension complicating sarcoid liver disease: case report and review of the literature. Am J Gastroenterol. 1984;79(5):389-396.
4. Ivonye C, Elhammali B, Henriques-Forsythe M, Bennett-Gittens R, Oderinde A. Disseminated sarcoidosis resulting in portal hypertension and gastrointestinal bleeding: a rare presentation. Can J Gastroenterol. 2012;26(8):508-509. http://www.ncbi.nlm.nih.gov/pubmed/22891173. Accessed May 16, 2018.
5. Tetangco EP, Silva RG, Lerma EV. Portal hypertension: etiology, evaluation, and management. Dis Mon. 2016;62(12):411-426. doi:10.1016/j.disamonth.2016.08.001.
6. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155-1167. doi:10.1016/S0140-6736(13)60680-7.
7. Al-Kofahi K, Korsten P, Ascoli C, et al. Management of extrapulmonary sarcoidosis: challenges and solutions. Ther Clin Risk Manag. 2016;12:1623-1634. doi:10.2147/TCRM.S74476.
8. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153-2165. doi:10.1056/NEJMra071714.
9. Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol. 2008;103(12):3184-3192. doi:10.1111/j.1572-0241.2008.02202.x.
10. Fraimow W, Myerson RM. Portal hypertension and bleeding esophageal varices secondary to sarcoidosis of the liver. Am J Med. 1957;23(6):995-998.
11. Saito H, Ohmori M, Iwamuro M, et al. Hepatic and gastric involvement in a case of systemic sarcoidosis presenting with rupture of esophageal varices. Intern Med. 2018;56(19):2583-2588. doi:10.2169/internalmedicine.8768-16.
12. DeLaney M, Greene CJ. Emergency Department evaluation and management of patients with upper gastrointestinal bleeding. Emerg Med Pract. 2015;17(4):1-18; quiz 19.
13. Ozgönenel B, Rajpurkar M, Lusher JM. How do you treat bleeding disorders with desmopressin? Postgrad Med J. 2007;83(977):159-163. doi:10.1136/pgmj.2006.052118.
14. Sümpelmann R, Schürholz T, Thorns E, Hausdörfer J. Acid-base, electrolyte and metabolite concentrations in packed red blood cells for major transfusion in infants. Paediatr Anaesth. 2001;11(2):169-173. doi:10.1046/j.1460-9592.2001.00637.x.
15. Monchi M. Citrate pathophysiology and metabolism. Transfus Apher Sci. 2018;56(1):28-30. doi:10.1016/j.transci.2016.12.013.
16. Shah RP, Sze DY. Complications during transjugular intrahepatic portosystemic shunt creation. Tech Vasc Interv Radiol. 2016;19(1):61-73. doi:10.1053/j.tvir.2016.01.007.

Enlarging nodule under the toenail • no history of trauma • unremarkable medical history • Dx?
THE CASE
A 28-year-old woman with an unremarkable medical history presented with an enlarging nodule that had been growing under her left great toenail for 6 months. The patient monitored the nodule, hoping that it would resolve on its own, but found that it steadily increased in size and began to displace the nail, causing pain. At the time of presentation, the nodule measured approximately 10 mm in diameter, and there was significant (~80°) superior displacement of the nail (FIGURE 1).

An initial radiograph identified a 5.5-mm bony density arising from the dorsal surface of the left first distal phalanx with no significant degenerative changes (FIGURE 2). A subsequent magnetic resonance image confirmed the bony excrescence and noted marrow continuity. A thin amount of T2 bright signal was also observed, suggesting either a cartilaginous cap or soft tissue edema secondary to pressure on the nail bed (FIGURE 3).

THE DIAGNOSIS
Histologic examination demonstrated a thin (3 mm) cartilaginous cap overlying an area of mature fibrocartilage with no definite periosteum. The osseous component appeared to mature from the cartilage, and the marrow was focally fatty and fibrosed (FIGURES 4A and 4B). Expert consultation with the Joint Pathology Center confirmed a benign osteochondromatous lesion.

The histologic differential diagnosis of this patient’s lesion included subungual exostosis and osteochondroma. Based on the patient’s age, location of the lesion, and histologic findings, the final diagnosis was subungual exostosis.

DISCUSSION
Subungual exostoses are benign osteocartilaginous tumors that most commonly affect children and young adults. They predominantly manifest on the dorsomedial aspect of the tip of the great toe (~80%), but can occur on other digits of the foot or hand.1 They are caused by a proliferation of fibrous tissue under the nail bed. The fibrocartilage cap then undergoes endochondral ossification to woven bone and lamellar bone trabeculae. As these lesions mature, they establish continuity with the underlying bone in the phalanx.2 Subungual exostoses were once thought to represent a proliferative response to trauma, but further research has identified a recurrent t(X;6) (q22;q13-14) translocation, suggesting a neoplastic origin.3
Osteochondromas are also common benign tumors formed by endochondral ossification, although secondary transformation into low-grade chondrosarcomas is well-documented.1 Osteochondromas commonly affect younger patients. They occur at epiphyseal areas of developing bone and have a hyaline matrix and chondrocyte pattern similar to that of a normal epiphyseal area, with confluence to the underlying trabecular and cortical bone. They are not caused by previous trauma and generally only become symptomatic after they have grown large enough to cause mechanical problems.1
Continue to: More diagnoses to consider
More diagnoses to consider
Other potential diagnoses for benign osteochondromatous lesions include bizarre parosteal osteochondromatous proliferations (BPOP) and digital mucous cysts.
BPOPs, also known as Nora’s lesions (crediting preliminary research performed by Nora and colleagues in 19834), are irregular formations of hypercellular cartilage, bone, and large chondrocytes. They predominantly occur in the small bones of the hands and feet, but may involve the skull and long bones.3 Unlike subungual exostoses and osteochondromas, BPOPs tend to occur in the third and fourth decades of life and generally do not alter, or have continuity with, the underlying bone.4
Histologically, BPOPs undergo irregular maturation, leaving a characteristic blue tint at the border of the newly formed trabecular bone. As with subungual exostoses, these lesions were traditionally believed to be reactive in nature. However, cytogenetic studies have identified variant translocations involving 1q32 (most commonly t[1;17] [q32;q21]) that are unique and common to these lesions.5
Digital mucous cysts are benign ganglion cysts that typically appear in the distal interphalangeal joints or at the proximal nail fold. They are believed to result from mucoid degeneration of connective tissue. Although generally associated with the hands, these cysts can also occur on the feet.6
Continue to: Our patient's outcome
Our patient’s outcome
After orthopedic consultation, the lesion and a 5 × 5-mm portion of the adherent germinal nail matrix were resected operatively through a medial excision. A small flap of the lateral nail matrix was rotated to cover the matrix defect, and the wound was closed. Postoperatively, the patient experienced slow wound healing (a total of 3 weeks), but there was no recurrence of the lesion at the 2-month follow-up.
THE TAKEAWAY
Osteocartilaginous tumors present as rapidly growing lesions on the distal tips of fingers and toes, but they may also occur on long bones and on the skull. Rarely malignant in nature, most of these lesions can be differentiated by location, histopathologic features, and patient age at onset. Consider surgical consultation and excision for relief of pain and/or cosmetic reasons. Recurrence is rare.
CORRESPONDENCE
Michael Barna, MD, Naval Hospital Camp Lejeune, Department of Family Medicine, 100 Brewster Blvd, Camp Lejeune, NC 28547; Michael.m.barna.mil@mail.mil.
1. Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
2. DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259.
3. Meneses MF, Unni KK, Swee RG. Bizarre parosteal osteochondromatous proliferation of bone (Nora’s lesion). Am J Surg Pathol. 1993;17:691-697.
4. Nora FE, Dahlin DC, Beabout JW. Bizarre parosteal osteochondromatous proliferations of the hand and feet. Am J Surg Pathol. 1983;7:245-250.
5. Zambrano E, Nosé V, Perez-Atayde AR, et al. Distinct chromosomal rearrangements in subungual (Dupuytren) exostosis and bizarre parosteal osteochondromatous proliferation (Nora lesion). Am J Surg Pathol. 2004;28:1033-1039.
6. Salerni G, Alonso C. Images in clinical medicine. Digital mucous cyst. N Engl J Med. 2012;366:1335.
THE CASE
A 28-year-old woman with an unremarkable medical history presented with an enlarging nodule that had been growing under her left great toenail for 6 months. The patient monitored the nodule, hoping that it would resolve on its own, but found that it steadily increased in size and began to displace the nail, causing pain. At the time of presentation, the nodule measured approximately 10 mm in diameter, and there was significant (~80°) superior displacement of the nail (FIGURE 1).
An initial radiograph identified a 5.5-mm bony density arising from the dorsal surface of the left first distal phalanx with no significant degenerative changes (FIGURE 2). A subsequent magnetic resonance image confirmed the bony excrescence and noted marrow continuity. A thin amount of T2 bright signal was also observed, suggesting either a cartilaginous cap or soft tissue edema secondary to pressure on the nail bed (FIGURE 3).
THE DIAGNOSIS
Histologic examination demonstrated a thin (3 mm) cartilaginous cap overlying an area of mature fibrocartilage with no definite periosteum. The osseous component appeared to mature from the cartilage, and the marrow was focally fatty and fibrosed (FIGURES 4A and 4B). Expert consultation with the Joint Pathology Center confirmed a benign osteochondromatous lesion.
The histologic differential diagnosis of this patient’s lesion included subungual exostosis and osteochondroma. Based on the patient’s age, location of the lesion, and histologic findings, the final diagnosis was subungual exostosis.
DISCUSSION
Subungual exostoses are benign osteocartilaginous tumors that most commonly affect children and young adults. They predominantly manifest on the dorsomedial aspect of the tip of the great toe (~80%), but can occur on other digits of the foot or hand.1 They are caused by a proliferation of fibrous tissue under the nail bed. The fibrocartilage cap then undergoes endochondral ossification to woven bone and lamellar bone trabeculae. As these lesions mature, they establish continuity with the underlying bone in the phalanx.2 Subungual exostoses were once thought to represent a proliferative response to trauma, but further research has identified a recurrent t(X;6) (q22;q13-14) translocation, suggesting a neoplastic origin.3
Osteochondromas are also common benign tumors formed by endochondral ossification, although secondary transformation into low-grade chondrosarcomas is well-documented.1 Osteochondromas commonly affect younger patients. They occur at epiphyseal areas of developing bone and have a hyaline matrix and chondrocyte pattern similar to that of a normal epiphyseal area, with confluence to the underlying trabecular and cortical bone. They are not caused by previous trauma and generally only become symptomatic after they have grown large enough to cause mechanical problems.1
Continue to: More diagnoses to consider
More diagnoses to consider
Other potential diagnoses for benign osteochondromatous lesions include bizarre parosteal osteochondromatous proliferations (BPOP) and digital mucous cysts.
BPOPs, also known as Nora’s lesions (crediting preliminary research performed by Nora and colleagues in 19834), are irregular formations of hypercellular cartilage, bone, and large chondrocytes. They predominantly occur in the small bones of the hands and feet, but may involve the skull and long bones.3 Unlike subungual exostoses and osteochondromas, BPOPs tend to occur in the third and fourth decades of life and generally do not alter, or have continuity with, the underlying bone.4
Histologically, BPOPs undergo irregular maturation, leaving a characteristic blue tint at the border of the newly formed trabecular bone. As with subungual exostoses, these lesions were traditionally believed to be reactive in nature. However, cytogenetic studies have identified variant translocations involving 1q32 (most commonly t[1;17] [q32;q21]) that are unique and common to these lesions.5
Digital mucous cysts are benign ganglion cysts that typically appear in the distal interphalangeal joints or at the proximal nail fold. They are believed to result from mucoid degeneration of connective tissue. Although generally associated with the hands, these cysts can also occur on the feet.6
Continue to: Our patient's outcome
Our patient’s outcome
After orthopedic consultation, the lesion and a 5 × 5-mm portion of the adherent germinal nail matrix were resected operatively through a medial excision. A small flap of the lateral nail matrix was rotated to cover the matrix defect, and the wound was closed. Postoperatively, the patient experienced slow wound healing (a total of 3 weeks), but there was no recurrence of the lesion at the 2-month follow-up.
THE TAKEAWAY
Osteocartilaginous tumors present as rapidly growing lesions on the distal tips of fingers and toes, but they may also occur on long bones and on the skull. Rarely malignant in nature, most of these lesions can be differentiated by location, histopathologic features, and patient age at onset. Consider surgical consultation and excision for relief of pain and/or cosmetic reasons. Recurrence is rare.
CORRESPONDENCE
Michael Barna, MD, Naval Hospital Camp Lejeune, Department of Family Medicine, 100 Brewster Blvd, Camp Lejeune, NC 28547; Michael.m.barna.mil@mail.mil.
THE CASE
A 28-year-old woman with an unremarkable medical history presented with an enlarging nodule that had been growing under her left great toenail for 6 months. The patient monitored the nodule, hoping that it would resolve on its own, but found that it steadily increased in size and began to displace the nail, causing pain. At the time of presentation, the nodule measured approximately 10 mm in diameter, and there was significant (~80°) superior displacement of the nail (FIGURE 1).
An initial radiograph identified a 5.5-mm bony density arising from the dorsal surface of the left first distal phalanx with no significant degenerative changes (FIGURE 2). A subsequent magnetic resonance image confirmed the bony excrescence and noted marrow continuity. A thin amount of T2 bright signal was also observed, suggesting either a cartilaginous cap or soft tissue edema secondary to pressure on the nail bed (FIGURE 3).
THE DIAGNOSIS
Histologic examination demonstrated a thin (3 mm) cartilaginous cap overlying an area of mature fibrocartilage with no definite periosteum. The osseous component appeared to mature from the cartilage, and the marrow was focally fatty and fibrosed (FIGURES 4A and 4B). Expert consultation with the Joint Pathology Center confirmed a benign osteochondromatous lesion.
The histologic differential diagnosis of this patient’s lesion included subungual exostosis and osteochondroma. Based on the patient’s age, location of the lesion, and histologic findings, the final diagnosis was subungual exostosis.
DISCUSSION
Subungual exostoses are benign osteocartilaginous tumors that most commonly affect children and young adults. They predominantly manifest on the dorsomedial aspect of the tip of the great toe (~80%), but can occur on other digits of the foot or hand.1 They are caused by a proliferation of fibrous tissue under the nail bed. The fibrocartilage cap then undergoes endochondral ossification to woven bone and lamellar bone trabeculae. As these lesions mature, they establish continuity with the underlying bone in the phalanx.2 Subungual exostoses were once thought to represent a proliferative response to trauma, but further research has identified a recurrent t(X;6) (q22;q13-14) translocation, suggesting a neoplastic origin.3
Osteochondromas are also common benign tumors formed by endochondral ossification, although secondary transformation into low-grade chondrosarcomas is well-documented.1 Osteochondromas commonly affect younger patients. They occur at epiphyseal areas of developing bone and have a hyaline matrix and chondrocyte pattern similar to that of a normal epiphyseal area, with confluence to the underlying trabecular and cortical bone. They are not caused by previous trauma and generally only become symptomatic after they have grown large enough to cause mechanical problems.1
Continue to: More diagnoses to consider
More diagnoses to consider
Other potential diagnoses for benign osteochondromatous lesions include bizarre parosteal osteochondromatous proliferations (BPOP) and digital mucous cysts.
BPOPs, also known as Nora’s lesions (crediting preliminary research performed by Nora and colleagues in 19834), are irregular formations of hypercellular cartilage, bone, and large chondrocytes. They predominantly occur in the small bones of the hands and feet, but may involve the skull and long bones.3 Unlike subungual exostoses and osteochondromas, BPOPs tend to occur in the third and fourth decades of life and generally do not alter, or have continuity with, the underlying bone.4
Histologically, BPOPs undergo irregular maturation, leaving a characteristic blue tint at the border of the newly formed trabecular bone. As with subungual exostoses, these lesions were traditionally believed to be reactive in nature. However, cytogenetic studies have identified variant translocations involving 1q32 (most commonly t[1;17] [q32;q21]) that are unique and common to these lesions.5
Digital mucous cysts are benign ganglion cysts that typically appear in the distal interphalangeal joints or at the proximal nail fold. They are believed to result from mucoid degeneration of connective tissue. Although generally associated with the hands, these cysts can also occur on the feet.6
Continue to: Our patient's outcome
Our patient’s outcome
After orthopedic consultation, the lesion and a 5 × 5-mm portion of the adherent germinal nail matrix were resected operatively through a medial excision. A small flap of the lateral nail matrix was rotated to cover the matrix defect, and the wound was closed. Postoperatively, the patient experienced slow wound healing (a total of 3 weeks), but there was no recurrence of the lesion at the 2-month follow-up.
THE TAKEAWAY
Osteocartilaginous tumors present as rapidly growing lesions on the distal tips of fingers and toes, but they may also occur on long bones and on the skull. Rarely malignant in nature, most of these lesions can be differentiated by location, histopathologic features, and patient age at onset. Consider surgical consultation and excision for relief of pain and/or cosmetic reasons. Recurrence is rare.
CORRESPONDENCE
Michael Barna, MD, Naval Hospital Camp Lejeune, Department of Family Medicine, 100 Brewster Blvd, Camp Lejeune, NC 28547; Michael.m.barna.mil@mail.mil.
1. Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
2. DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259.
3. Meneses MF, Unni KK, Swee RG. Bizarre parosteal osteochondromatous proliferation of bone (Nora’s lesion). Am J Surg Pathol. 1993;17:691-697.
4. Nora FE, Dahlin DC, Beabout JW. Bizarre parosteal osteochondromatous proliferations of the hand and feet. Am J Surg Pathol. 1983;7:245-250.
5. Zambrano E, Nosé V, Perez-Atayde AR, et al. Distinct chromosomal rearrangements in subungual (Dupuytren) exostosis and bizarre parosteal osteochondromatous proliferation (Nora lesion). Am J Surg Pathol. 2004;28:1033-1039.
6. Salerni G, Alonso C. Images in clinical medicine. Digital mucous cyst. N Engl J Med. 2012;366:1335.
1. Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
2. DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259.
3. Meneses MF, Unni KK, Swee RG. Bizarre parosteal osteochondromatous proliferation of bone (Nora’s lesion). Am J Surg Pathol. 1993;17:691-697.
4. Nora FE, Dahlin DC, Beabout JW. Bizarre parosteal osteochondromatous proliferations of the hand and feet. Am J Surg Pathol. 1983;7:245-250.
5. Zambrano E, Nosé V, Perez-Atayde AR, et al. Distinct chromosomal rearrangements in subungual (Dupuytren) exostosis and bizarre parosteal osteochondromatous proliferation (Nora lesion). Am J Surg Pathol. 2004;28:1033-1039.
6. Salerni G, Alonso C. Images in clinical medicine. Digital mucous cyst. N Engl J Med. 2012;366:1335.

History of posttraumatic stress disorder • priapism • Dx?
THE CASE
A 35-year-old African-American man, who was an active duty service member, presented to the Troop Medical Clinic with a 4-hour history of priapism. He had been taking sertraline 100 mg and prazosin 10 mg nightly for 4 months to treat his posttraumatic stress disorder (PTSD) with no reported adverse effects. These doses were titrated 2 months prior to presentation. The patient reported that he took his usual medication doses before bed and awoke at 3 am with a penile erection. At 7 am, he presented to the clinic because of pain from the continued erection.
THE DIAGNOSIS
A penile erection was present on physical exam. All medications were reviewed for adverse effects. A work-up for anemia, sickle cell disease, thalassemia, and platelet abnormalities was negative. A blood gas analysis performed on blood aspirated from the corpus cavernosum showed hypoxemia, hypercarbia, and acidosis, confirming a diagnosis of ischemic priapism.
DISCUSSION
Priapism is a prolonged erection of the penis that is usually not associated with sexual activity or stimulation. It is considered a urologic emergency and requires prompt treatment to prevent long-term complications, such as permanent erectile dysfunction.
Priapism is classified as one of 2 types: ischemic (“low flow”) or nonischemic (“high flow”).
Ischemic priapism is the most common type. It is caused by dysfunctional cavernosal smooth muscle, which creates a compartment-like syndrome in the cavernous tissue that leads to hypoxia and acidosis.1 Nonischemic priapism is often caused by a fistula between the cavernosal artery and corpus cavernosum and is common with traumatic injuries. Nonischemic priapism has a lower risk for long-term complications (due to the blood being well-oxygenated) and often resolves spontaneously without treatment.2,3
Certain medications can cause priapism
Our patient’s ischemic priapism was most likely caused by the combined antagonistic properties of prazosin and sertraline on alpha-1 adrenergic receptors.3,4 Adrenergic alpha-blockers block the sympathetic system, which can in turn inhibit penile detumescence and cause priapism.4
An increasingly common Tx combination. Selective serotonin reuptake inhibitors (SSRIs) such as sertraline are considered first-line treatment for the symptoms of PTSD, and prazosin has been found to be effective in the treatment of nightmares associated with PTSD. (Treatment of PTSD-related nightmares with prazosin is an off-label but frequent use of the medication.) This combination of medications is becoming increasingly common for the treatment of PTSD and its associated symptoms.5-7
Continue to: Cases to date provide interesting insight into this adverse effect
Cases to date provide interesting insight into this adverse effect
In our literature review, no documented cases of priapism were attributed to prazosin when it was used for the treatment of nightmares, but there are multiple case reports of priapism linked to the drug’s use for hypertension.
In the majority of these case reports, the dosage exceeded 10 mg/d and was much higher than the dosage typically used to treat nightmares.7 Many of the affected patients also had associated comorbidities such as diabetes or chronic kidney disease.4
Sertraline has been associated with priapism when used as monotherapy and in combination therapy with antipsychotics. All SSRIs have antagonistic properties to alpha-1 adrenergic receptors, but sertraline appears to have more than a 10-fold increase in affinity when compared to other SSRIs.3
Treatment: An injection and aspiration
Our patient was treated with phenylephrine injection and aspiration, which resolved the priapism. Prazosin was stopped, and the patient was weaned off of sertraline. He continued to follow up closely with Behavioral Health for further management of his PTSD and associated symptoms.
Continue to: THE TAKEAWAY
THE TAKEAWAY
PTSD is being diagnosed more frequently, especially in active duty soldiers, veterans, members of the National Guard, and reservists.8 Because nightmares are a common symptom of PTSD and SSRIs are first-line treatment for PTSD, the combination of prazosin and an SSRI for the treatment of PTSD is frequently encountered.5-7 Providers who prescribe and/or care for patients treated with these medications need to counsel patients on the risk of priapism and the risks associated with a delay in seeking medical care.
If a patient who is taking these medications presents with priapism, contact Urology immediately for acute management. Both medications must be stopped to prevent future episodes; prazosin can be stopped immediately, but patients must be weaned off of sertraline to avoid experiencing withdrawal symptoms. Patients will need to follow up with a behavioral health team for continued management of their PTSD symptoms.
CORRESPONDENCE
Caleb Dickison, DO, Fort Belvoir Community Hospital, 9300 Dewitt Loop, Fort Belvoir, VA 22060; caleb.g.dickison.mil@mail.mil.
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1:116-120.
2. Broderick GA, Gordon D, Hypolite J, et al. Anoxia and corporal smooth muscle dysfunction: a model for ischemic priapism. J Urol. 1994;151:259-262.
3. Choua, R, Lee HC, Castro J, et al. Priapism associated with multiple psychotropics: a case report and review of the literature. 2007. Available at: http://primarypsychiatry.com/priapism-associated-with-multiple-psychotropics-a-case-report-and-review-of-the-literature/. Accessed on May 7, 2018.
4. Spagnul SJ, Cabral PH, Verndl DO, et al. Adrenergic alpha-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23:95-98.
5. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2006;CD002795.
6. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma PTSD: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
8. Grieger TA, Cozza SJ, Ursano RJ, et al. Posttraumatic stress disorder and depression in battle-injured soldiers. Am J Psychiatry. 2006;163:1777-1783.
THE CASE
A 35-year-old African-American man, who was an active duty service member, presented to the Troop Medical Clinic with a 4-hour history of priapism. He had been taking sertraline 100 mg and prazosin 10 mg nightly for 4 months to treat his posttraumatic stress disorder (PTSD) with no reported adverse effects. These doses were titrated 2 months prior to presentation. The patient reported that he took his usual medication doses before bed and awoke at 3 am with a penile erection. At 7 am, he presented to the clinic because of pain from the continued erection.
THE DIAGNOSIS
A penile erection was present on physical exam. All medications were reviewed for adverse effects. A work-up for anemia, sickle cell disease, thalassemia, and platelet abnormalities was negative. A blood gas analysis performed on blood aspirated from the corpus cavernosum showed hypoxemia, hypercarbia, and acidosis, confirming a diagnosis of ischemic priapism.
DISCUSSION
Priapism is a prolonged erection of the penis that is usually not associated with sexual activity or stimulation. It is considered a urologic emergency and requires prompt treatment to prevent long-term complications, such as permanent erectile dysfunction.
Priapism is classified as one of 2 types: ischemic (“low flow”) or nonischemic (“high flow”).
Ischemic priapism is the most common type. It is caused by dysfunctional cavernosal smooth muscle, which creates a compartment-like syndrome in the cavernous tissue that leads to hypoxia and acidosis.1 Nonischemic priapism is often caused by a fistula between the cavernosal artery and corpus cavernosum and is common with traumatic injuries. Nonischemic priapism has a lower risk for long-term complications (due to the blood being well-oxygenated) and often resolves spontaneously without treatment.2,3
Certain medications can cause priapism
Our patient’s ischemic priapism was most likely caused by the combined antagonistic properties of prazosin and sertraline on alpha-1 adrenergic receptors.3,4 Adrenergic alpha-blockers block the sympathetic system, which can in turn inhibit penile detumescence and cause priapism.4
An increasingly common Tx combination. Selective serotonin reuptake inhibitors (SSRIs) such as sertraline are considered first-line treatment for the symptoms of PTSD, and prazosin has been found to be effective in the treatment of nightmares associated with PTSD. (Treatment of PTSD-related nightmares with prazosin is an off-label but frequent use of the medication.) This combination of medications is becoming increasingly common for the treatment of PTSD and its associated symptoms.5-7
Continue to: Cases to date provide interesting insight into this adverse effect
Cases to date provide interesting insight into this adverse effect
In our literature review, no documented cases of priapism were attributed to prazosin when it was used for the treatment of nightmares, but there are multiple case reports of priapism linked to the drug’s use for hypertension.
In the majority of these case reports, the dosage exceeded 10 mg/d and was much higher than the dosage typically used to treat nightmares.7 Many of the affected patients also had associated comorbidities such as diabetes or chronic kidney disease.4
Sertraline has been associated with priapism when used as monotherapy and in combination therapy with antipsychotics. All SSRIs have antagonistic properties to alpha-1 adrenergic receptors, but sertraline appears to have more than a 10-fold increase in affinity when compared to other SSRIs.3
Treatment: An injection and aspiration
Our patient was treated with phenylephrine injection and aspiration, which resolved the priapism. Prazosin was stopped, and the patient was weaned off of sertraline. He continued to follow up closely with Behavioral Health for further management of his PTSD and associated symptoms.
Continue to: THE TAKEAWAY
THE TAKEAWAY
PTSD is being diagnosed more frequently, especially in active duty soldiers, veterans, members of the National Guard, and reservists.8 Because nightmares are a common symptom of PTSD and SSRIs are first-line treatment for PTSD, the combination of prazosin and an SSRI for the treatment of PTSD is frequently encountered.5-7 Providers who prescribe and/or care for patients treated with these medications need to counsel patients on the risk of priapism and the risks associated with a delay in seeking medical care.
If a patient who is taking these medications presents with priapism, contact Urology immediately for acute management. Both medications must be stopped to prevent future episodes; prazosin can be stopped immediately, but patients must be weaned off of sertraline to avoid experiencing withdrawal symptoms. Patients will need to follow up with a behavioral health team for continued management of their PTSD symptoms.
CORRESPONDENCE
Caleb Dickison, DO, Fort Belvoir Community Hospital, 9300 Dewitt Loop, Fort Belvoir, VA 22060; caleb.g.dickison.mil@mail.mil.
THE CASE
A 35-year-old African-American man, who was an active duty service member, presented to the Troop Medical Clinic with a 4-hour history of priapism. He had been taking sertraline 100 mg and prazosin 10 mg nightly for 4 months to treat his posttraumatic stress disorder (PTSD) with no reported adverse effects. These doses were titrated 2 months prior to presentation. The patient reported that he took his usual medication doses before bed and awoke at 3 am with a penile erection. At 7 am, he presented to the clinic because of pain from the continued erection.
THE DIAGNOSIS
A penile erection was present on physical exam. All medications were reviewed for adverse effects. A work-up for anemia, sickle cell disease, thalassemia, and platelet abnormalities was negative. A blood gas analysis performed on blood aspirated from the corpus cavernosum showed hypoxemia, hypercarbia, and acidosis, confirming a diagnosis of ischemic priapism.
DISCUSSION
Priapism is a prolonged erection of the penis that is usually not associated with sexual activity or stimulation. It is considered a urologic emergency and requires prompt treatment to prevent long-term complications, such as permanent erectile dysfunction.
Priapism is classified as one of 2 types: ischemic (“low flow”) or nonischemic (“high flow”).
Ischemic priapism is the most common type. It is caused by dysfunctional cavernosal smooth muscle, which creates a compartment-like syndrome in the cavernous tissue that leads to hypoxia and acidosis.1 Nonischemic priapism is often caused by a fistula between the cavernosal artery and corpus cavernosum and is common with traumatic injuries. Nonischemic priapism has a lower risk for long-term complications (due to the blood being well-oxygenated) and often resolves spontaneously without treatment.2,3
Certain medications can cause priapism
Our patient’s ischemic priapism was most likely caused by the combined antagonistic properties of prazosin and sertraline on alpha-1 adrenergic receptors.3,4 Adrenergic alpha-blockers block the sympathetic system, which can in turn inhibit penile detumescence and cause priapism.4
An increasingly common Tx combination. Selective serotonin reuptake inhibitors (SSRIs) such as sertraline are considered first-line treatment for the symptoms of PTSD, and prazosin has been found to be effective in the treatment of nightmares associated with PTSD. (Treatment of PTSD-related nightmares with prazosin is an off-label but frequent use of the medication.) This combination of medications is becoming increasingly common for the treatment of PTSD and its associated symptoms.5-7
Continue to: Cases to date provide interesting insight into this adverse effect
Cases to date provide interesting insight into this adverse effect
In our literature review, no documented cases of priapism were attributed to prazosin when it was used for the treatment of nightmares, but there are multiple case reports of priapism linked to the drug’s use for hypertension.
In the majority of these case reports, the dosage exceeded 10 mg/d and was much higher than the dosage typically used to treat nightmares.7 Many of the affected patients also had associated comorbidities such as diabetes or chronic kidney disease.4
Sertraline has been associated with priapism when used as monotherapy and in combination therapy with antipsychotics. All SSRIs have antagonistic properties to alpha-1 adrenergic receptors, but sertraline appears to have more than a 10-fold increase in affinity when compared to other SSRIs.3
Treatment: An injection and aspiration
Our patient was treated with phenylephrine injection and aspiration, which resolved the priapism. Prazosin was stopped, and the patient was weaned off of sertraline. He continued to follow up closely with Behavioral Health for further management of his PTSD and associated symptoms.
Continue to: THE TAKEAWAY
THE TAKEAWAY
PTSD is being diagnosed more frequently, especially in active duty soldiers, veterans, members of the National Guard, and reservists.8 Because nightmares are a common symptom of PTSD and SSRIs are first-line treatment for PTSD, the combination of prazosin and an SSRI for the treatment of PTSD is frequently encountered.5-7 Providers who prescribe and/or care for patients treated with these medications need to counsel patients on the risk of priapism and the risks associated with a delay in seeking medical care.
If a patient who is taking these medications presents with priapism, contact Urology immediately for acute management. Both medications must be stopped to prevent future episodes; prazosin can be stopped immediately, but patients must be weaned off of sertraline to avoid experiencing withdrawal symptoms. Patients will need to follow up with a behavioral health team for continued management of their PTSD symptoms.
CORRESPONDENCE
Caleb Dickison, DO, Fort Belvoir Community Hospital, 9300 Dewitt Loop, Fort Belvoir, VA 22060; caleb.g.dickison.mil@mail.mil.
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1:116-120.
2. Broderick GA, Gordon D, Hypolite J, et al. Anoxia and corporal smooth muscle dysfunction: a model for ischemic priapism. J Urol. 1994;151:259-262.
3. Choua, R, Lee HC, Castro J, et al. Priapism associated with multiple psychotropics: a case report and review of the literature. 2007. Available at: http://primarypsychiatry.com/priapism-associated-with-multiple-psychotropics-a-case-report-and-review-of-the-literature/. Accessed on May 7, 2018.
4. Spagnul SJ, Cabral PH, Verndl DO, et al. Adrenergic alpha-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23:95-98.
5. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2006;CD002795.
6. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma PTSD: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
8. Grieger TA, Cozza SJ, Ursano RJ, et al. Posttraumatic stress disorder and depression in battle-injured soldiers. Am J Psychiatry. 2006;163:1777-1783.
1. Pryor J, Akkus E, Alter G, et al. Priapism. J Sex Med. 2004;1:116-120.
2. Broderick GA, Gordon D, Hypolite J, et al. Anoxia and corporal smooth muscle dysfunction: a model for ischemic priapism. J Urol. 1994;151:259-262.
3. Choua, R, Lee HC, Castro J, et al. Priapism associated with multiple psychotropics: a case report and review of the literature. 2007. Available at: http://primarypsychiatry.com/priapism-associated-with-multiple-psychotropics-a-case-report-and-review-of-the-literature/. Accessed on May 7, 2018.
4. Spagnul SJ, Cabral PH, Verndl DO, et al. Adrenergic alpha-blockers: an infrequent and overlooked cause of priapism. Int J Impot Res. 2011;23:95-98.
5. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2006;CD002795.
6. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma PTSD: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
8. Grieger TA, Cozza SJ, Ursano RJ, et al. Posttraumatic stress disorder and depression in battle-injured soldiers. Am J Psychiatry. 2006;163:1777-1783.
