User login
Palmoplantar Eruption in a Patient With Mercury Poisoning
Mercury poisoning affects multiple body systems, leading to variable clinical presentations. Mercury intoxication at low levels frequently presents with weakness, fatigue, weight loss, and abdominal pain. At higher levels of mercury intoxication, tremors and neurologic dysfunction are more prevalent.1 Dermatologic manifestations of mercury exposure vary and include pink disease (acrodynia), mercury exanthem, contact dermatitis, and cutaneous granulomas. Untreated mercury poisoning may result in severe complications, including renal tubular necrosis, pneumonitis, persistent neurologic dysfunction, and fatality in some cases.1,2
Pink disease is a rare disease that typically arises in infants and young children from chronic mercury exposure.3 We report a unique presentation of pink disease occurring in an 18-year-old woman following mercury exposure.
Case Report
An 18-year-old woman who was previously healthy presented to the hospital for evaluation of body aches and back pain. She reported a transient rash on the torso 2 weeks prior, but at the current presentation, only the distal upper and lower extremities were involved. A review of systems revealed myalgia, most severe in the lower back; muscle spasms; stiffness in the fingers; abdominal pain; constipation; paresthesia in the hands and feet; hyperhidrosis; and generalized weakness.
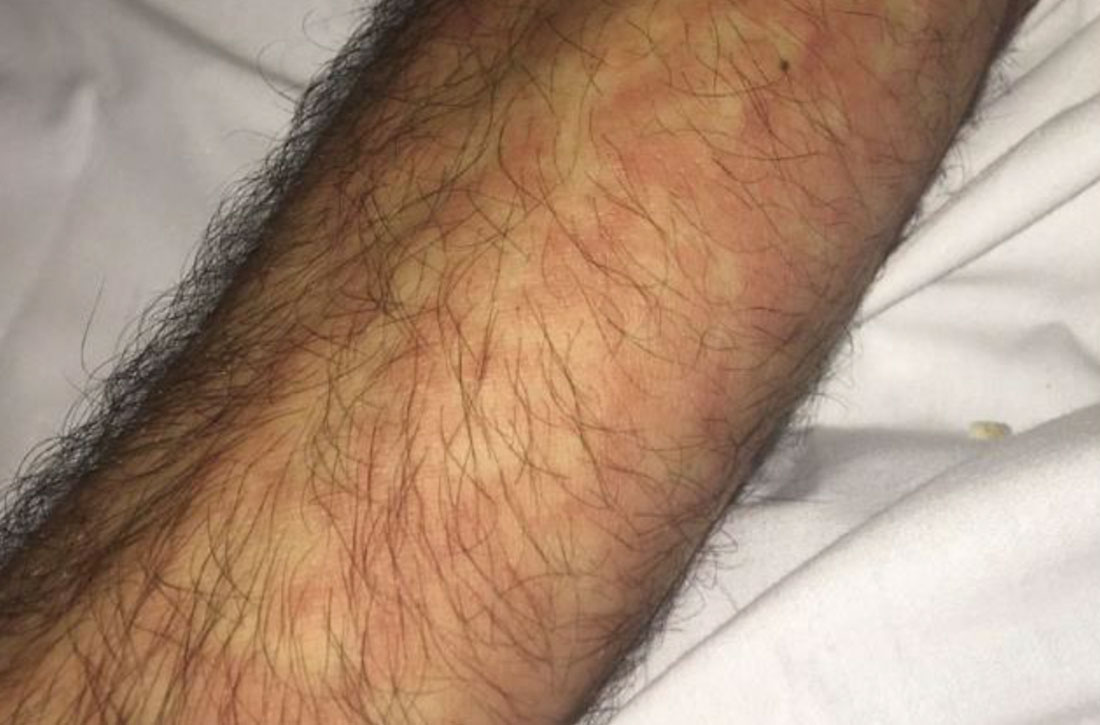
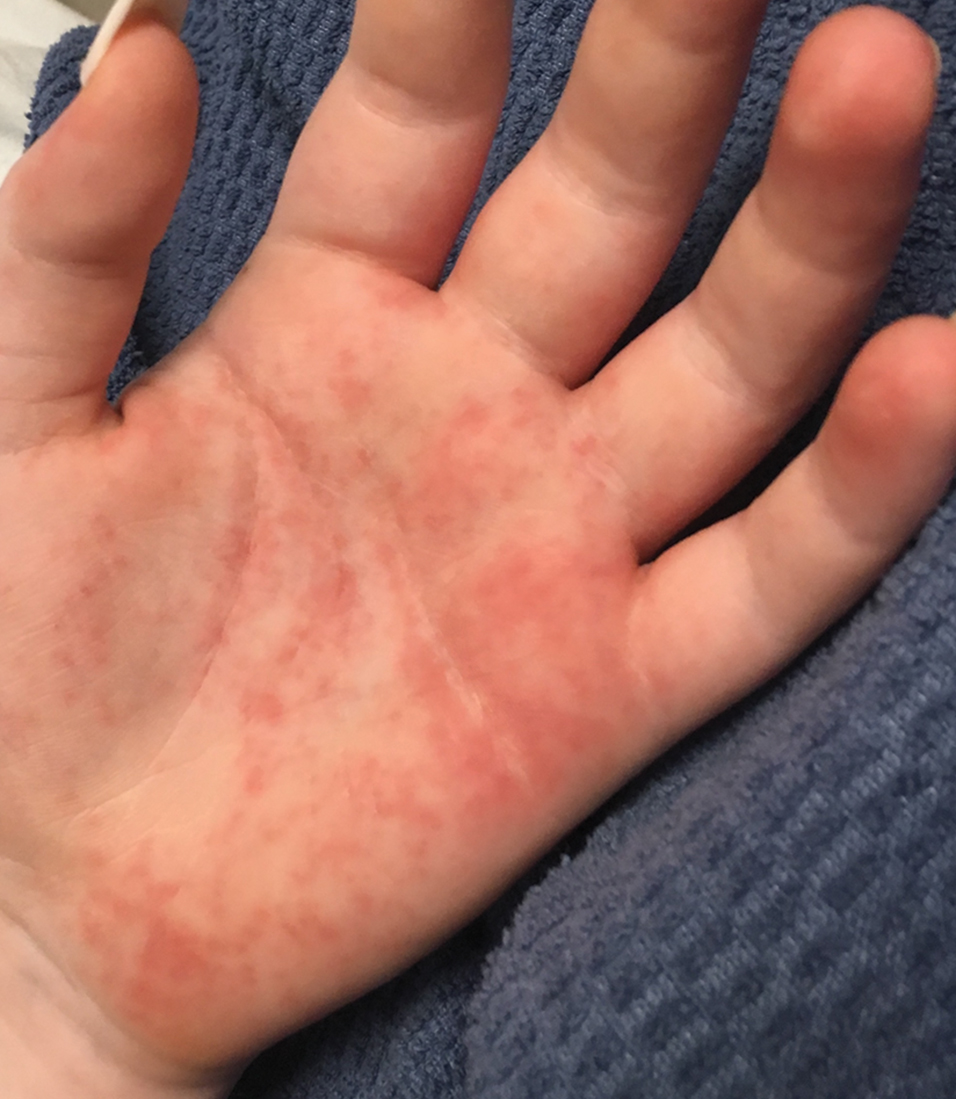
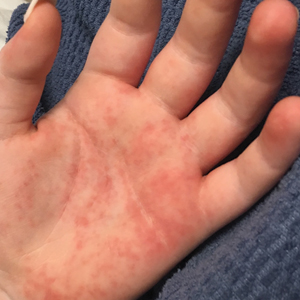
Vitals on admission revealed tachycardia (112 beats per minute). Physical examination revealed the patient was pale and fatigued; she appeared to be in pain, with observable facial grimacing and muscle spasms in the legs. She had poorly demarcated pink macules and papules scattered on the left palm (Figure 1), right forearm, right wrist, and dorsal aspects of the feet including the soles. A few pinpoint pustules were present on the left fifth digit.
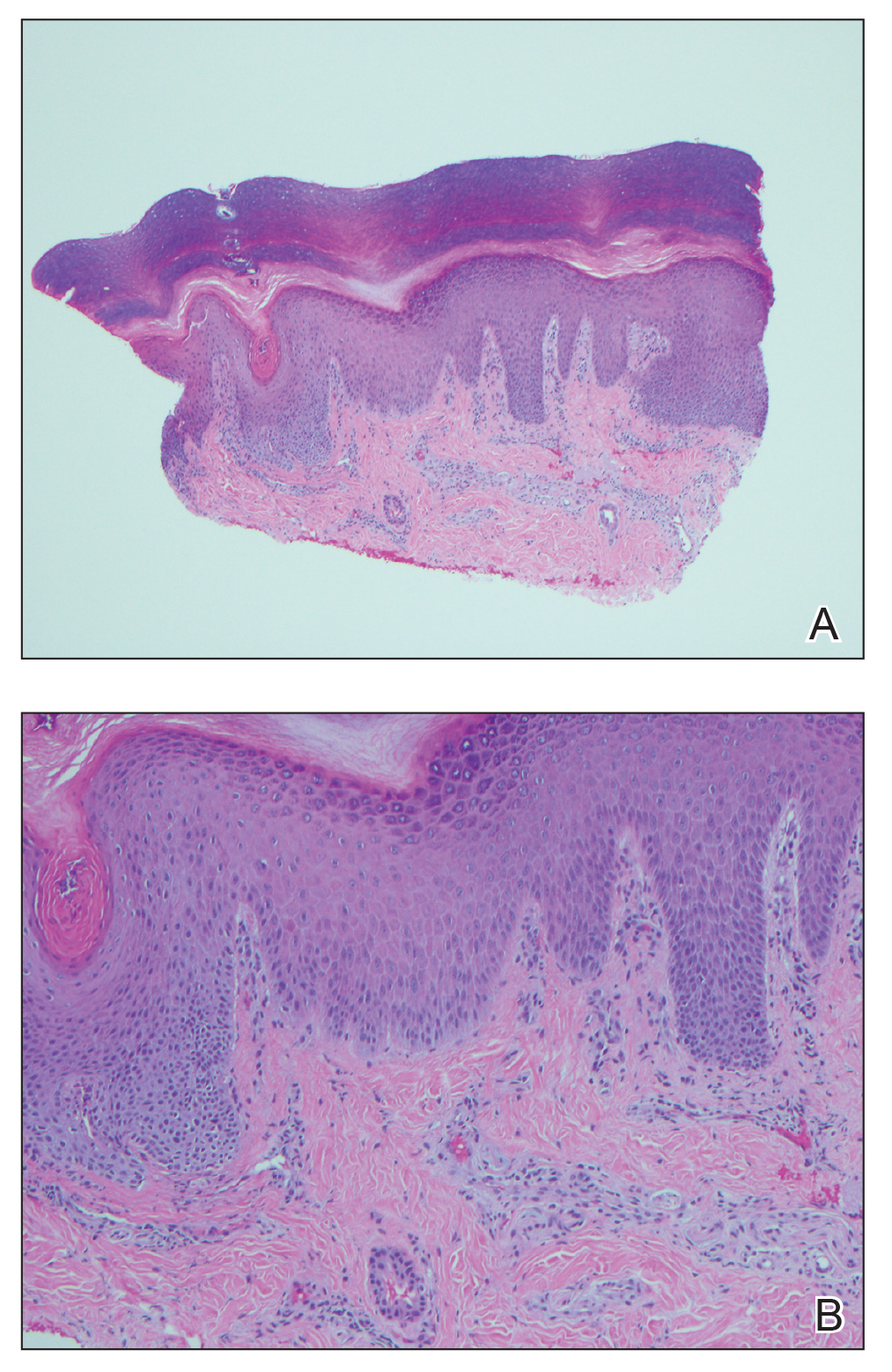
An extensive workup was initiated to rule out infectious, autoimmune, or toxic etiologies. Two 4-mm punch biopsies of the left palm were performed for hematoxylin and eosin staining and tissue culture. Findings on hematoxylin and eosin stain were nonspecific, showing acanthosis, orthokeratosis, and a mild interface and perivascular lymphocytic infiltrate (Figure 2); superficial bacterial colonization was present, but the tissue culture was negative.
Laboratory studies showed mild transaminitis, and stool was positive for Campylobacter antigen. Electromyography showed myokymia (fascicular muscle contractions). A heavy metal serum panel and urine screen were positive for elevated mercury levels, with a serum mercury level of 23 µg/L (reference range, 0.0–14.9 µg/L) and a urine mercury level of 76 µg/L (reference range, 0–19 µg/L).
Upon further questioning, it was discovered that the patient’s brother and neighbor found a glass bottle containing mercury in their house 10 days prior. They played with the mercury beads with their hands, throwing them around the room and spilling them around the house, which led to mercury exposure in multiple individuals, including our patient. Of note, her brother and neighbor also were hospitalized at the same time as our patient with similar symptoms.
A diagnosis of mercury poisoning was made along with a component of postinfectious reactive arthropathy due to Campylobacter. The myokymia and skin eruption were believed to be secondary to mercury poisoning. The patient was started on ciprofloxacin (750 mg twice daily), intravenous immunoglobulin for Campylobacter, a 2-week treatment regimen with the chelating agent succimer (500 mg twice daily) for mercury poisoning, and a 3-day regimen of pulse intravenous steroids (intravenous methylprednisolone 500 mg once daily) to reduce inflammation. Repeat mercury levels showed a downward trend, and the rash improved with time. All family members were advised to undergo testing for mercury exposure.
Comment
Manifestations of Mercury Poisoning
Dermatologic manifestations of mercury exposure are varied. The most common—allergic contact dermatitis—presents after repeat systemic or topical exposure.4 Mercury exanthem is an acute systemic contact dermatitis most commonly triggered by mercury vapor inhalation. It manifests as an erythematous maculopapular eruption predominantly involving the flexural areas and the anterior thighs in a V-shaped distribution.5 Purpura may be seen in severe cases. Cutaneous granulomas after direct injection of mercury also have been reported as well as cutaneous hyperpigmentation after chronic mercury absorption.6
Presentation of Pink Disease
Pink disease occurs in children after chronic mercury exposure. It was a common pediatric disorder in the 19th century due to the presence of mercury in certain anthelmintics and teething powders.7 However, prevalence drastically decreased after the removal of mercury from these products.3 Although pink disease classically was associated with mercury ingestion, cases also occurred secondary to external application of mercury.7 Additionally, in 1988 a case was reported in a 14-month-old girl after inhalation of mercury vapor from a spilled bottle of mercury.3
Pink disease begins with pink discoloration of the fingertips, nose, and toes, and later progresses to involvement of the hands and feet. Erythema, edema, and desquamation of the hands and feet are seen, along with irritability and autonomic dysfunction that manifests as profuse perspiration, tachycardia, and hypertension.3
Diagnosis of Pink Disease
The differential diagnosis of palmoplantar rash is broad and includes rickettsial disease; syphilis; scabies; toxic shock syndrome; infective endocarditis; meningococcal infection; hand-foot-and-mouth disease; dermatophytosis; and palmoplantar keratodermas. The involvement of the hands and feet in our patient, along with hyperhidrosis, tachycardia, and paresthesia, led us to believe that her condition was a variation of pink disease. The patient’s age at presentation (18 years) was unique, as it is atypical for pink disease. Although the polyarthropathy was attributed to Campylobacter, it is important to note that high levels of mercury exposure also have been associated with polyarthritis,8 polyneuropathy,4 and neuromuscular abnormalities on electromyography.4 Therefore, it is possible that the presence of these symptoms in our patient was either secondary to or compounded by mercury exposure.
Mercury Poisoning
Diagnosis of mercury poisoning can be made by assessing blood, urine, hair, or nail concentrations. However, as mercury deposits in multiple organs, individual concentrations do not correlate with total-body mercury levels.1 Currently, no universal diagnostic criteria for mercury toxicity exist, though a provocation test with the chelating agent 2,
Elemental mercury, as found in some thermometers, dental amalgams, and electrical appliances (eg, certain switches, fluorescent light bulbs), can be converted to inorganic mercury in the body.9 Elemental mercury is vaporized at room temperature; the predominant route of exposure is by subsequent inhalation and lung absorbtion.10 Cutaneous absorption of high concentrations of elementary mercury in either liquid or vapor form may occur, though the rate is slow and absorption is poor. In cases of accidental exposure, contaminated clothing should be removed and immediately decontaminated or disposed. Exposed skin should be washed with a mild soap and water and rinsed thoroughly.10
The treatment of inorganic mercury poisoning is accomplished with the chelating agents succimer, dimercaptopropanesulfonate, dimercaprol, or D-penicillamine.1 In symptomatic cases with high clinical suspicion, the first dose of chelation treatment should be initiated early without delay for laboratory confirmation, as treatment efficacy decreases with an increased interim between exposure and onset of chelation.11 Combination chelation therapy also may be used in treatment. Plasma exchange or hemodialysis are treatment options for extreme, life-threatening cases.1
Conclusion
Mercury exposure should be included in the differential diagnosis of patients presenting with a rash on the palms and soles, especially in young patients with systemic symptoms. A high level of suspicion and a thorough history can prevent a delay in treatment and an unnecessarily extensive and expensive workup. An emphasis on early diagnosis and treatment is important for optimal outcomes and can prevent the severe and potentially devastating consequences of mercury toxicity.
- Bernhoft RA. Mercury toxicity and treatment: a review of the literature. J Environ Public Health. 2012;2012:460508.
- Kamensky OL, Horton D, Kingsley DP, et al. A case of accidental mercury intoxication. J Emerg Med. 2019;56:275-278.
- Dinehart SM, Dillard R, Raimer SS, et al. Cutaneous manifestations of acrodynia (pink disease). Arch Dermatol. 1988;124:107-109.
- Malek A, Aouad K, El Khoury R, et al. Chronic mercury intoxication masquerading as systemic disease: a case report and review of the literature. Eur J Case Rep Intern Med. 2017;4:000632.
- Nakayama H, Niki F, Shono M, et al. Mercury exanthem. Contact Dermatitis. 1983;9:411-417.
- Boyd AS, Seger D, Vannucci S, et al. Mercury exposure and cutaneous disease. J Am Acad Dermatol. 2000;43:81-90.
- Warkany J. Acrodynia—postmortem of a disease. Am J Dis Child. 1966;112:147-156.
- Karatas¸ GK, Tosun AK, Karacehennem E, et al. Mercury poisoning: an unusual cause of polyarthritis. Clin Rheumatol. 2002;21:73-75.
- Mercury Factsheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/biomonitoring/Mercury_FactSheet.html. Reviewed April 7, 2017. Accessed October 21, 2020.
- Medical management guidelines for mercury. Agency for Toxic Substances & Disease Registry website. https://www.atsdr.cdc .gov/MMG/MMG.asp?id=106&tid=24. Update October 21, 2014. Accessed September 11, 2020.
- Kosnett MJ. The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol. 2013;9:347-354.
Mercury poisoning affects multiple body systems, leading to variable clinical presentations. Mercury intoxication at low levels frequently presents with weakness, fatigue, weight loss, and abdominal pain. At higher levels of mercury intoxication, tremors and neurologic dysfunction are more prevalent.1 Dermatologic manifestations of mercury exposure vary and include pink disease (acrodynia), mercury exanthem, contact dermatitis, and cutaneous granulomas. Untreated mercury poisoning may result in severe complications, including renal tubular necrosis, pneumonitis, persistent neurologic dysfunction, and fatality in some cases.1,2
Pink disease is a rare disease that typically arises in infants and young children from chronic mercury exposure.3 We report a unique presentation of pink disease occurring in an 18-year-old woman following mercury exposure.
Case Report
An 18-year-old woman who was previously healthy presented to the hospital for evaluation of body aches and back pain. She reported a transient rash on the torso 2 weeks prior, but at the current presentation, only the distal upper and lower extremities were involved. A review of systems revealed myalgia, most severe in the lower back; muscle spasms; stiffness in the fingers; abdominal pain; constipation; paresthesia in the hands and feet; hyperhidrosis; and generalized weakness.
Vitals on admission revealed tachycardia (112 beats per minute). Physical examination revealed the patient was pale and fatigued; she appeared to be in pain, with observable facial grimacing and muscle spasms in the legs. She had poorly demarcated pink macules and papules scattered on the left palm (Figure 1), right forearm, right wrist, and dorsal aspects of the feet including the soles. A few pinpoint pustules were present on the left fifth digit.
An extensive workup was initiated to rule out infectious, autoimmune, or toxic etiologies. Two 4-mm punch biopsies of the left palm were performed for hematoxylin and eosin staining and tissue culture. Findings on hematoxylin and eosin stain were nonspecific, showing acanthosis, orthokeratosis, and a mild interface and perivascular lymphocytic infiltrate (Figure 2); superficial bacterial colonization was present, but the tissue culture was negative.
Laboratory studies showed mild transaminitis, and stool was positive for Campylobacter antigen. Electromyography showed myokymia (fascicular muscle contractions). A heavy metal serum panel and urine screen were positive for elevated mercury levels, with a serum mercury level of 23 µg/L (reference range, 0.0–14.9 µg/L) and a urine mercury level of 76 µg/L (reference range, 0–19 µg/L).
Upon further questioning, it was discovered that the patient’s brother and neighbor found a glass bottle containing mercury in their house 10 days prior. They played with the mercury beads with their hands, throwing them around the room and spilling them around the house, which led to mercury exposure in multiple individuals, including our patient. Of note, her brother and neighbor also were hospitalized at the same time as our patient with similar symptoms.
A diagnosis of mercury poisoning was made along with a component of postinfectious reactive arthropathy due to Campylobacter. The myokymia and skin eruption were believed to be secondary to mercury poisoning. The patient was started on ciprofloxacin (750 mg twice daily), intravenous immunoglobulin for Campylobacter, a 2-week treatment regimen with the chelating agent succimer (500 mg twice daily) for mercury poisoning, and a 3-day regimen of pulse intravenous steroids (intravenous methylprednisolone 500 mg once daily) to reduce inflammation. Repeat mercury levels showed a downward trend, and the rash improved with time. All family members were advised to undergo testing for mercury exposure.
Comment
Manifestations of Mercury Poisoning
Dermatologic manifestations of mercury exposure are varied. The most common—allergic contact dermatitis—presents after repeat systemic or topical exposure.4 Mercury exanthem is an acute systemic contact dermatitis most commonly triggered by mercury vapor inhalation. It manifests as an erythematous maculopapular eruption predominantly involving the flexural areas and the anterior thighs in a V-shaped distribution.5 Purpura may be seen in severe cases. Cutaneous granulomas after direct injection of mercury also have been reported as well as cutaneous hyperpigmentation after chronic mercury absorption.6
Presentation of Pink Disease
Pink disease occurs in children after chronic mercury exposure. It was a common pediatric disorder in the 19th century due to the presence of mercury in certain anthelmintics and teething powders.7 However, prevalence drastically decreased after the removal of mercury from these products.3 Although pink disease classically was associated with mercury ingestion, cases also occurred secondary to external application of mercury.7 Additionally, in 1988 a case was reported in a 14-month-old girl after inhalation of mercury vapor from a spilled bottle of mercury.3
Pink disease begins with pink discoloration of the fingertips, nose, and toes, and later progresses to involvement of the hands and feet. Erythema, edema, and desquamation of the hands and feet are seen, along with irritability and autonomic dysfunction that manifests as profuse perspiration, tachycardia, and hypertension.3
Diagnosis of Pink Disease
The differential diagnosis of palmoplantar rash is broad and includes rickettsial disease; syphilis; scabies; toxic shock syndrome; infective endocarditis; meningococcal infection; hand-foot-and-mouth disease; dermatophytosis; and palmoplantar keratodermas. The involvement of the hands and feet in our patient, along with hyperhidrosis, tachycardia, and paresthesia, led us to believe that her condition was a variation of pink disease. The patient’s age at presentation (18 years) was unique, as it is atypical for pink disease. Although the polyarthropathy was attributed to Campylobacter, it is important to note that high levels of mercury exposure also have been associated with polyarthritis,8 polyneuropathy,4 and neuromuscular abnormalities on electromyography.4 Therefore, it is possible that the presence of these symptoms in our patient was either secondary to or compounded by mercury exposure.
Mercury Poisoning
Diagnosis of mercury poisoning can be made by assessing blood, urine, hair, or nail concentrations. However, as mercury deposits in multiple organs, individual concentrations do not correlate with total-body mercury levels.1 Currently, no universal diagnostic criteria for mercury toxicity exist, though a provocation test with the chelating agent 2,
Elemental mercury, as found in some thermometers, dental amalgams, and electrical appliances (eg, certain switches, fluorescent light bulbs), can be converted to inorganic mercury in the body.9 Elemental mercury is vaporized at room temperature; the predominant route of exposure is by subsequent inhalation and lung absorbtion.10 Cutaneous absorption of high concentrations of elementary mercury in either liquid or vapor form may occur, though the rate is slow and absorption is poor. In cases of accidental exposure, contaminated clothing should be removed and immediately decontaminated or disposed. Exposed skin should be washed with a mild soap and water and rinsed thoroughly.10
The treatment of inorganic mercury poisoning is accomplished with the chelating agents succimer, dimercaptopropanesulfonate, dimercaprol, or D-penicillamine.1 In symptomatic cases with high clinical suspicion, the first dose of chelation treatment should be initiated early without delay for laboratory confirmation, as treatment efficacy decreases with an increased interim between exposure and onset of chelation.11 Combination chelation therapy also may be used in treatment. Plasma exchange or hemodialysis are treatment options for extreme, life-threatening cases.1
Conclusion
Mercury exposure should be included in the differential diagnosis of patients presenting with a rash on the palms and soles, especially in young patients with systemic symptoms. A high level of suspicion and a thorough history can prevent a delay in treatment and an unnecessarily extensive and expensive workup. An emphasis on early diagnosis and treatment is important for optimal outcomes and can prevent the severe and potentially devastating consequences of mercury toxicity.
Mercury poisoning affects multiple body systems, leading to variable clinical presentations. Mercury intoxication at low levels frequently presents with weakness, fatigue, weight loss, and abdominal pain. At higher levels of mercury intoxication, tremors and neurologic dysfunction are more prevalent.1 Dermatologic manifestations of mercury exposure vary and include pink disease (acrodynia), mercury exanthem, contact dermatitis, and cutaneous granulomas. Untreated mercury poisoning may result in severe complications, including renal tubular necrosis, pneumonitis, persistent neurologic dysfunction, and fatality in some cases.1,2
Pink disease is a rare disease that typically arises in infants and young children from chronic mercury exposure.3 We report a unique presentation of pink disease occurring in an 18-year-old woman following mercury exposure.
Case Report
An 18-year-old woman who was previously healthy presented to the hospital for evaluation of body aches and back pain. She reported a transient rash on the torso 2 weeks prior, but at the current presentation, only the distal upper and lower extremities were involved. A review of systems revealed myalgia, most severe in the lower back; muscle spasms; stiffness in the fingers; abdominal pain; constipation; paresthesia in the hands and feet; hyperhidrosis; and generalized weakness.
Vitals on admission revealed tachycardia (112 beats per minute). Physical examination revealed the patient was pale and fatigued; she appeared to be in pain, with observable facial grimacing and muscle spasms in the legs. She had poorly demarcated pink macules and papules scattered on the left palm (Figure 1), right forearm, right wrist, and dorsal aspects of the feet including the soles. A few pinpoint pustules were present on the left fifth digit.
An extensive workup was initiated to rule out infectious, autoimmune, or toxic etiologies. Two 4-mm punch biopsies of the left palm were performed for hematoxylin and eosin staining and tissue culture. Findings on hematoxylin and eosin stain were nonspecific, showing acanthosis, orthokeratosis, and a mild interface and perivascular lymphocytic infiltrate (Figure 2); superficial bacterial colonization was present, but the tissue culture was negative.
Laboratory studies showed mild transaminitis, and stool was positive for Campylobacter antigen. Electromyography showed myokymia (fascicular muscle contractions). A heavy metal serum panel and urine screen were positive for elevated mercury levels, with a serum mercury level of 23 µg/L (reference range, 0.0–14.9 µg/L) and a urine mercury level of 76 µg/L (reference range, 0–19 µg/L).
Upon further questioning, it was discovered that the patient’s brother and neighbor found a glass bottle containing mercury in their house 10 days prior. They played with the mercury beads with their hands, throwing them around the room and spilling them around the house, which led to mercury exposure in multiple individuals, including our patient. Of note, her brother and neighbor also were hospitalized at the same time as our patient with similar symptoms.
A diagnosis of mercury poisoning was made along with a component of postinfectious reactive arthropathy due to Campylobacter. The myokymia and skin eruption were believed to be secondary to mercury poisoning. The patient was started on ciprofloxacin (750 mg twice daily), intravenous immunoglobulin for Campylobacter, a 2-week treatment regimen with the chelating agent succimer (500 mg twice daily) for mercury poisoning, and a 3-day regimen of pulse intravenous steroids (intravenous methylprednisolone 500 mg once daily) to reduce inflammation. Repeat mercury levels showed a downward trend, and the rash improved with time. All family members were advised to undergo testing for mercury exposure.
Comment
Manifestations of Mercury Poisoning
Dermatologic manifestations of mercury exposure are varied. The most common—allergic contact dermatitis—presents after repeat systemic or topical exposure.4 Mercury exanthem is an acute systemic contact dermatitis most commonly triggered by mercury vapor inhalation. It manifests as an erythematous maculopapular eruption predominantly involving the flexural areas and the anterior thighs in a V-shaped distribution.5 Purpura may be seen in severe cases. Cutaneous granulomas after direct injection of mercury also have been reported as well as cutaneous hyperpigmentation after chronic mercury absorption.6
Presentation of Pink Disease
Pink disease occurs in children after chronic mercury exposure. It was a common pediatric disorder in the 19th century due to the presence of mercury in certain anthelmintics and teething powders.7 However, prevalence drastically decreased after the removal of mercury from these products.3 Although pink disease classically was associated with mercury ingestion, cases also occurred secondary to external application of mercury.7 Additionally, in 1988 a case was reported in a 14-month-old girl after inhalation of mercury vapor from a spilled bottle of mercury.3
Pink disease begins with pink discoloration of the fingertips, nose, and toes, and later progresses to involvement of the hands and feet. Erythema, edema, and desquamation of the hands and feet are seen, along with irritability and autonomic dysfunction that manifests as profuse perspiration, tachycardia, and hypertension.3
Diagnosis of Pink Disease
The differential diagnosis of palmoplantar rash is broad and includes rickettsial disease; syphilis; scabies; toxic shock syndrome; infective endocarditis; meningococcal infection; hand-foot-and-mouth disease; dermatophytosis; and palmoplantar keratodermas. The involvement of the hands and feet in our patient, along with hyperhidrosis, tachycardia, and paresthesia, led us to believe that her condition was a variation of pink disease. The patient’s age at presentation (18 years) was unique, as it is atypical for pink disease. Although the polyarthropathy was attributed to Campylobacter, it is important to note that high levels of mercury exposure also have been associated with polyarthritis,8 polyneuropathy,4 and neuromuscular abnormalities on electromyography.4 Therefore, it is possible that the presence of these symptoms in our patient was either secondary to or compounded by mercury exposure.
Mercury Poisoning
Diagnosis of mercury poisoning can be made by assessing blood, urine, hair, or nail concentrations. However, as mercury deposits in multiple organs, individual concentrations do not correlate with total-body mercury levels.1 Currently, no universal diagnostic criteria for mercury toxicity exist, though a provocation test with the chelating agent 2,
Elemental mercury, as found in some thermometers, dental amalgams, and electrical appliances (eg, certain switches, fluorescent light bulbs), can be converted to inorganic mercury in the body.9 Elemental mercury is vaporized at room temperature; the predominant route of exposure is by subsequent inhalation and lung absorbtion.10 Cutaneous absorption of high concentrations of elementary mercury in either liquid or vapor form may occur, though the rate is slow and absorption is poor. In cases of accidental exposure, contaminated clothing should be removed and immediately decontaminated or disposed. Exposed skin should be washed with a mild soap and water and rinsed thoroughly.10
The treatment of inorganic mercury poisoning is accomplished with the chelating agents succimer, dimercaptopropanesulfonate, dimercaprol, or D-penicillamine.1 In symptomatic cases with high clinical suspicion, the first dose of chelation treatment should be initiated early without delay for laboratory confirmation, as treatment efficacy decreases with an increased interim between exposure and onset of chelation.11 Combination chelation therapy also may be used in treatment. Plasma exchange or hemodialysis are treatment options for extreme, life-threatening cases.1
Conclusion
Mercury exposure should be included in the differential diagnosis of patients presenting with a rash on the palms and soles, especially in young patients with systemic symptoms. A high level of suspicion and a thorough history can prevent a delay in treatment and an unnecessarily extensive and expensive workup. An emphasis on early diagnosis and treatment is important for optimal outcomes and can prevent the severe and potentially devastating consequences of mercury toxicity.
- Bernhoft RA. Mercury toxicity and treatment: a review of the literature. J Environ Public Health. 2012;2012:460508.
- Kamensky OL, Horton D, Kingsley DP, et al. A case of accidental mercury intoxication. J Emerg Med. 2019;56:275-278.
- Dinehart SM, Dillard R, Raimer SS, et al. Cutaneous manifestations of acrodynia (pink disease). Arch Dermatol. 1988;124:107-109.
- Malek A, Aouad K, El Khoury R, et al. Chronic mercury intoxication masquerading as systemic disease: a case report and review of the literature. Eur J Case Rep Intern Med. 2017;4:000632.
- Nakayama H, Niki F, Shono M, et al. Mercury exanthem. Contact Dermatitis. 1983;9:411-417.
- Boyd AS, Seger D, Vannucci S, et al. Mercury exposure and cutaneous disease. J Am Acad Dermatol. 2000;43:81-90.
- Warkany J. Acrodynia—postmortem of a disease. Am J Dis Child. 1966;112:147-156.
- Karatas¸ GK, Tosun AK, Karacehennem E, et al. Mercury poisoning: an unusual cause of polyarthritis. Clin Rheumatol. 2002;21:73-75.
- Mercury Factsheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/biomonitoring/Mercury_FactSheet.html. Reviewed April 7, 2017. Accessed October 21, 2020.
- Medical management guidelines for mercury. Agency for Toxic Substances & Disease Registry website. https://www.atsdr.cdc .gov/MMG/MMG.asp?id=106&tid=24. Update October 21, 2014. Accessed September 11, 2020.
- Kosnett MJ. The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol. 2013;9:347-354.
- Bernhoft RA. Mercury toxicity and treatment: a review of the literature. J Environ Public Health. 2012;2012:460508.
- Kamensky OL, Horton D, Kingsley DP, et al. A case of accidental mercury intoxication. J Emerg Med. 2019;56:275-278.
- Dinehart SM, Dillard R, Raimer SS, et al. Cutaneous manifestations of acrodynia (pink disease). Arch Dermatol. 1988;124:107-109.
- Malek A, Aouad K, El Khoury R, et al. Chronic mercury intoxication masquerading as systemic disease: a case report and review of the literature. Eur J Case Rep Intern Med. 2017;4:000632.
- Nakayama H, Niki F, Shono M, et al. Mercury exanthem. Contact Dermatitis. 1983;9:411-417.
- Boyd AS, Seger D, Vannucci S, et al. Mercury exposure and cutaneous disease. J Am Acad Dermatol. 2000;43:81-90.
- Warkany J. Acrodynia—postmortem of a disease. Am J Dis Child. 1966;112:147-156.
- Karatas¸ GK, Tosun AK, Karacehennem E, et al. Mercury poisoning: an unusual cause of polyarthritis. Clin Rheumatol. 2002;21:73-75.
- Mercury Factsheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/biomonitoring/Mercury_FactSheet.html. Reviewed April 7, 2017. Accessed October 21, 2020.
- Medical management guidelines for mercury. Agency for Toxic Substances & Disease Registry website. https://www.atsdr.cdc .gov/MMG/MMG.asp?id=106&tid=24. Update October 21, 2014. Accessed September 11, 2020.
- Kosnett MJ. The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol. 2013;9:347-354.
Practice Points
- The dermatologic and histologic presentation of mercury exposure may be nonspecific, requiring a high degree of clinical suspicion to make a diagnosis.
- Mercury exposure should be included in the differential diagnosis in patients presenting with a rash of the palms and soles, especially in young patients with systemic symptoms.
An Unusual Skin Infection With Achromobacter xylosoxidans
Case Report
A 50-year-old woman presented with a sore, tender, red lump on the right superior buttock of 5 months’ duration. Five months prior to presentation the patient used this area to attach the infusion set for an insulin pump, which was left in place for 7 days as opposed to the 2 or 3 days recommended by the device manufacturer. A firm, slightly tender lump formed, similar to prior scars that had developed from use of the insulin pump. However, the lump began to grow and get softer. It was intermittently warm and red. Although the area was sore and tender, she never had any major pain. She also denied any fever, malaise, or other systemic symptoms.
The patient indicated a medical history of type 1 diabetes mellitus diagnosed at 9 years of age; hypertension; asthma; gastroesophageal reflux disease; allergic rhinitis; migraine headaches; depression; hidradenitis suppurativa that resolved after surgical excision; and recurrent vaginal yeast infections, especially when taking antibiotics. She had a surgical history of hidradenitis suppurativa excision at the inguinal folds, bilateral carpal tunnel release, tubal ligation, abdominoplasty, and cholecystectomy. The patient’s current medications included insulin aspart, mometasone furoate, inhaled fluticasone, pantoprazole, cetirizine, spironolactone, duloxetine, sumatriptan, fluconazole, topiramate, and enalapril.
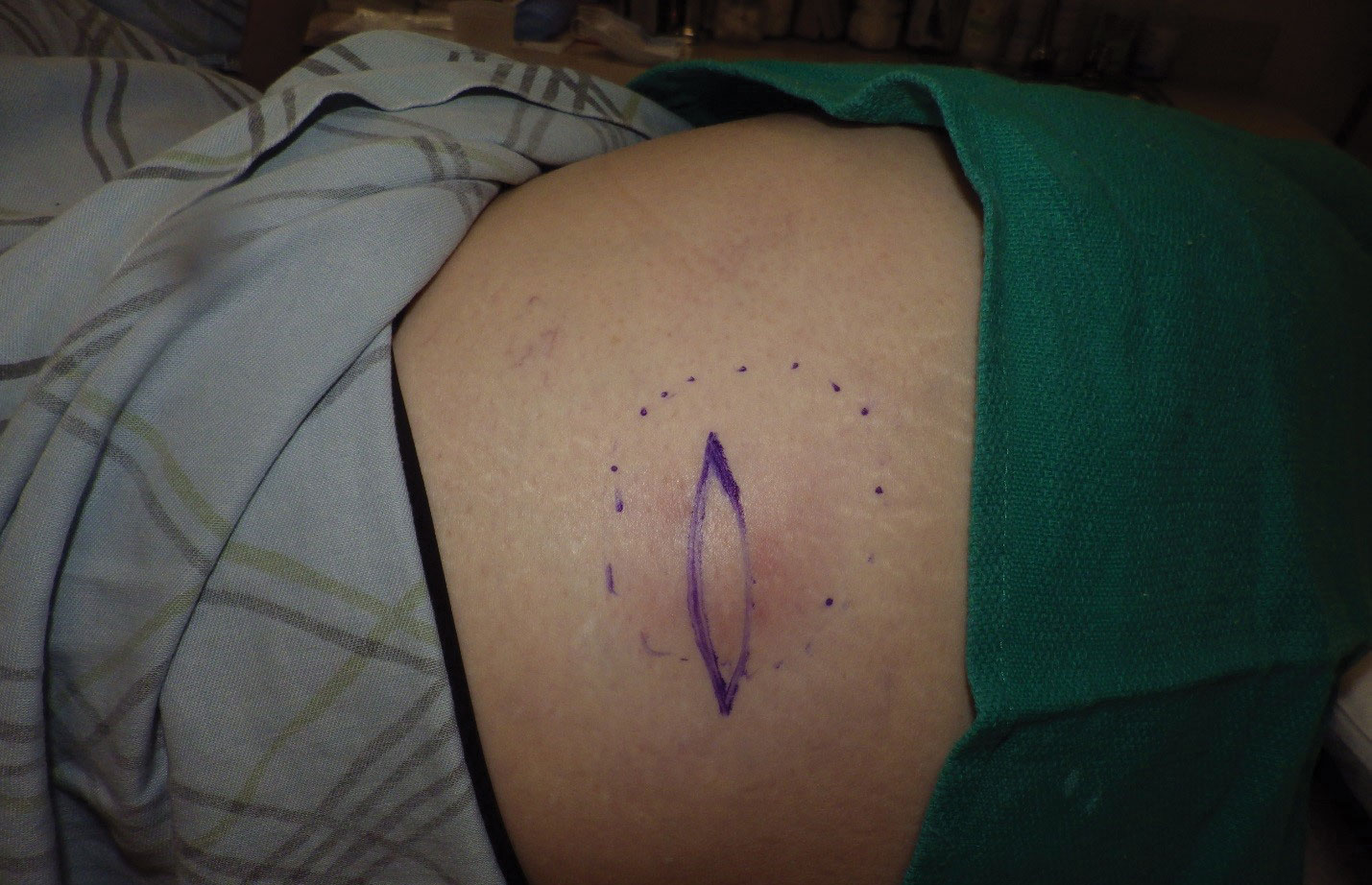
Physical examination revealed normal vital signs and the patient was afebrile. She had no swollen or tender lymph nodes. There was a 5.5×7.0-cm, soft, tender, erythematous subcutaneous mass with no visible punctum or overlying epidermal change on the right superior buttock (Figure 1). Based on the history and physical examination, the differential diagnosis included subcutaneous fat necrosis, epidermal inclusion cyst, and an abscess.
The patient was scheduled for excision of the mass the day after presenting to the clinic. During excision, 10 mL of thick purulent liquid was drained. A sample of the liquid was sent for Gram stain, aerobic and anaerobic culture, and antibiotic sensitivities. Necrotic-appearing adipose and fibrotic tissues were dissected and extirpated through an elliptical incision and submitted for pathologic evaluation.
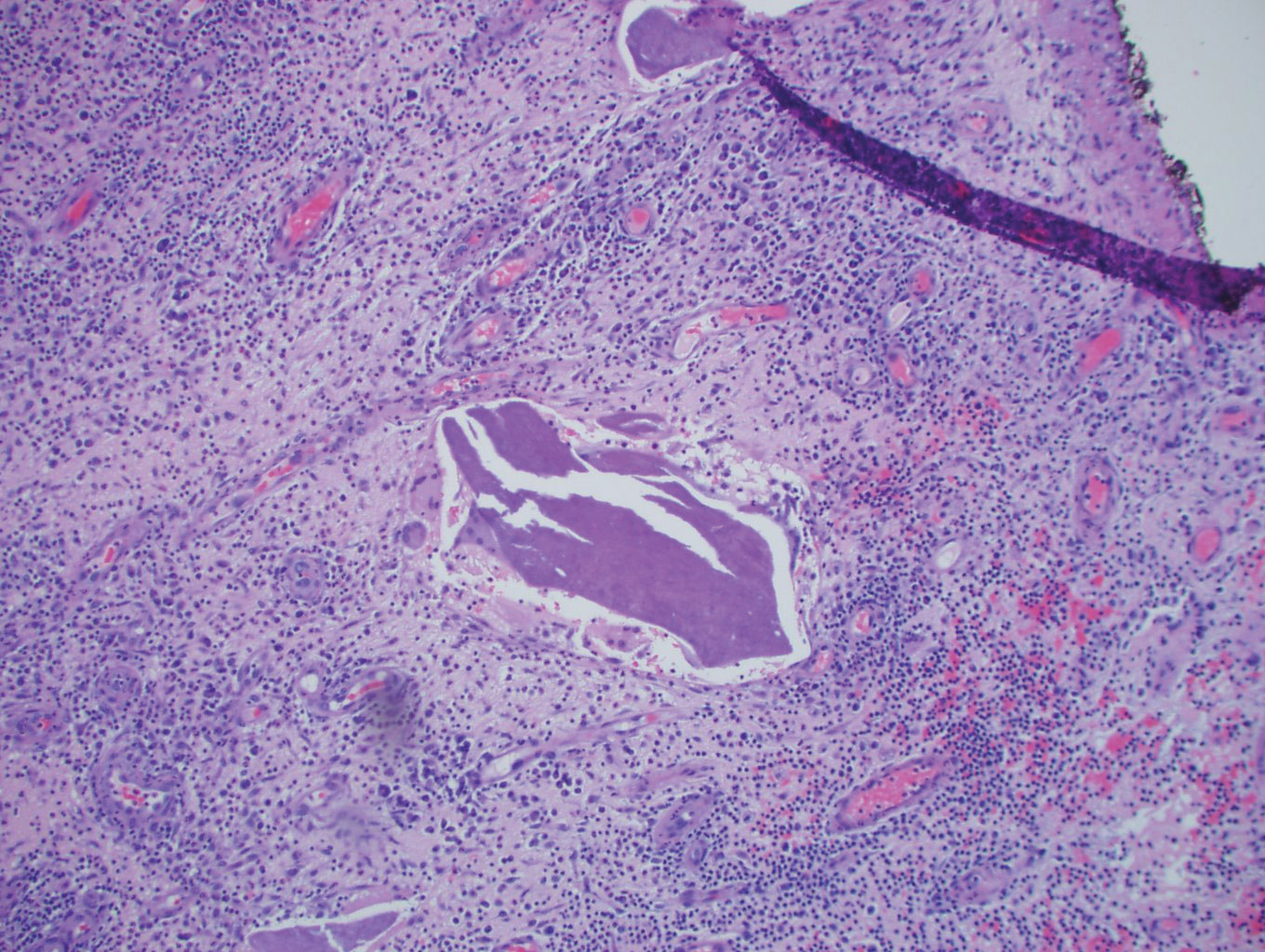
Histopathology showed a subcutaneous defect with palisaded granulomatous inflammation and sclerosis (Figure 2). There was no detection of microorganisms with Grocott-Gomori methenamine-silver, tissue Gram, or acid-fast stains. There was a focus of acellular material embedded within the inflammation (Figure 3). The Gram stain of the purulent material showed few white blood cells and rare gram-negative bacilli. Culture grew moderate Achromobacter xylosoxidans resistant to cefepime, cefotaxime, and gentamicin. The culture was susceptible to ceftazidime, imipenem, levofloxacin, piperacillin, and trimethoprim-sulfamethoxazole (TMP-SMX).

The patient was prescribed oral TMP-SMX (160 mg of TMP and 800 mg of SMX) twice daily for 10 days. The patient tolerated the procedure and the subsequent antibiotics well. The patient had normal levels of IgA, IgG, and IgM, as well as a negative screening test for human immunodeficiency virus. She healed well from the surgical procedure and has had no recurrence of symptoms.
Comment
Achromobacter xylosoxidans is a nonfermentative, non–spore-forming, motile, gram-negative, aerobic, catalase-positive and oxidase-positive flagellate bacterium. It is an emerging pathogen that was first isolated in 1971 from patients with chronic otitis media.1 Since its recognition, it has been documented to cause a variety of infections, including pneumonia, meningitis, osteomyelitis, endocarditis, and bacteremia, as well as abdominal, urinary tract, ocular, and skin and soft tissue infections.2,3 Those affected usually are immunocompromised, have hematologic disorders, or have indwelling catheters.4 Strains of A xylosoxidans have shown resistance to multiple antibiotics including penicillins, cephalosporins, carbapenems, aminoglycosides, macrolides, fluoroquinolones, and TMP-SMX. Achromobacter xylosoxidans has been documented to form biofilms on plastics, including on contact lenses, urinary and intravenous catheters, and reusable tissue dispensers treated with disinfectant solution.4-6 One study demonstrated that A xylosoxidans is even capable of biodegradation of plastic, using the plastic as its sole source of carbon.7
Our case illustrates an indolent infection with A xylosoxidans forming a granulomatous abscess at the site of an insulin pump that was left in place for 7 days in an immunocompetent patient. Although infections with A xylosoxidans in patients with urinary or intravenous catheters have been reported,4 our case is unique, as the insulin pump was the source of such an infection. It is possible that the subcutaneous focus of acellular material described on the pathology report represented a partially biodegraded piece of the insulin pump catheter that broke off and was serving as a nidus of infection for A xylosoxidans. Although multidrug resistance is common, the culture grown from our patient was susceptible to TMP-SMX, among other antibiotics. Our patient was treated successfully with surgical excision, drainage, and a 10-day course of TMP-SMX.
Conclusion
Health care providers should recognize A xylosoxidans as an emerging pathogen that is capable of forming biofilms on “disinfected” surfaces and medical products, especially plastics. Achromobacter xylosoxidans may be resistant to multiple antibiotics and can cause infections with various presentations.
- Yabuuchi E, Oyama A. Achromobacter xylosoxidans n. sp. from human ear discharge. Jpn J Microbiol. 1971;15:477-481.
- Rodrigues CG, Rays J, Kanegae MY. Native-valve endocarditis caused by Achromobacter xylosoxidans: a case report and review of literature. Autops Case Rep. 2017;7:50-55.
- Tena D, Martínez NM, Losa C, et al. Skin and soft tissue infection caused by Achromobacter xylosoxidans: report of 14 cases. Scand J Infect Dis. 2014;46:130-135.
- Pérez Barragán E, Sandino Pérez J, Corbella L, et al. Achromobacter xylosoxidans bacteremia: clinical and microbiological features in a 10-year case series. Rev Esp Quimioter. 2018;31:268-273.
- Konstantinović N, Ćirković I, Đukić S, et al. Biofilm formation of Achromobacter xylosoxidans on contact lens. Acta Microbiol Immunol Hung. 2017;64:293-300.
- Günther F, Merle U, Frank U, et al. Pseudobacteremia outbreak of biofilm-forming Achromobacter xylosoxidans—environmental transmission. BMC Infect Dis. 2016;16:584.
- Kowalczyk A, Chyc M, Ryszka P, et al. Achromobacter xylosoxidans as a new microorganism strain colonizing high-density polyethylene as a key step to its biodegradation. Environ Sci Pollut Res Int. 2016;23:11349-11356.
Case Report
A 50-year-old woman presented with a sore, tender, red lump on the right superior buttock of 5 months’ duration. Five months prior to presentation the patient used this area to attach the infusion set for an insulin pump, which was left in place for 7 days as opposed to the 2 or 3 days recommended by the device manufacturer. A firm, slightly tender lump formed, similar to prior scars that had developed from use of the insulin pump. However, the lump began to grow and get softer. It was intermittently warm and red. Although the area was sore and tender, she never had any major pain. She also denied any fever, malaise, or other systemic symptoms.
The patient indicated a medical history of type 1 diabetes mellitus diagnosed at 9 years of age; hypertension; asthma; gastroesophageal reflux disease; allergic rhinitis; migraine headaches; depression; hidradenitis suppurativa that resolved after surgical excision; and recurrent vaginal yeast infections, especially when taking antibiotics. She had a surgical history of hidradenitis suppurativa excision at the inguinal folds, bilateral carpal tunnel release, tubal ligation, abdominoplasty, and cholecystectomy. The patient’s current medications included insulin aspart, mometasone furoate, inhaled fluticasone, pantoprazole, cetirizine, spironolactone, duloxetine, sumatriptan, fluconazole, topiramate, and enalapril.
Physical examination revealed normal vital signs and the patient was afebrile. She had no swollen or tender lymph nodes. There was a 5.5×7.0-cm, soft, tender, erythematous subcutaneous mass with no visible punctum or overlying epidermal change on the right superior buttock (Figure 1). Based on the history and physical examination, the differential diagnosis included subcutaneous fat necrosis, epidermal inclusion cyst, and an abscess.
The patient was scheduled for excision of the mass the day after presenting to the clinic. During excision, 10 mL of thick purulent liquid was drained. A sample of the liquid was sent for Gram stain, aerobic and anaerobic culture, and antibiotic sensitivities. Necrotic-appearing adipose and fibrotic tissues were dissected and extirpated through an elliptical incision and submitted for pathologic evaluation.
Histopathology showed a subcutaneous defect with palisaded granulomatous inflammation and sclerosis (Figure 2). There was no detection of microorganisms with Grocott-Gomori methenamine-silver, tissue Gram, or acid-fast stains. There was a focus of acellular material embedded within the inflammation (Figure 3). The Gram stain of the purulent material showed few white blood cells and rare gram-negative bacilli. Culture grew moderate Achromobacter xylosoxidans resistant to cefepime, cefotaxime, and gentamicin. The culture was susceptible to ceftazidime, imipenem, levofloxacin, piperacillin, and trimethoprim-sulfamethoxazole (TMP-SMX).
The patient was prescribed oral TMP-SMX (160 mg of TMP and 800 mg of SMX) twice daily for 10 days. The patient tolerated the procedure and the subsequent antibiotics well. The patient had normal levels of IgA, IgG, and IgM, as well as a negative screening test for human immunodeficiency virus. She healed well from the surgical procedure and has had no recurrence of symptoms.
Comment
Achromobacter xylosoxidans is a nonfermentative, non–spore-forming, motile, gram-negative, aerobic, catalase-positive and oxidase-positive flagellate bacterium. It is an emerging pathogen that was first isolated in 1971 from patients with chronic otitis media.1 Since its recognition, it has been documented to cause a variety of infections, including pneumonia, meningitis, osteomyelitis, endocarditis, and bacteremia, as well as abdominal, urinary tract, ocular, and skin and soft tissue infections.2,3 Those affected usually are immunocompromised, have hematologic disorders, or have indwelling catheters.4 Strains of A xylosoxidans have shown resistance to multiple antibiotics including penicillins, cephalosporins, carbapenems, aminoglycosides, macrolides, fluoroquinolones, and TMP-SMX. Achromobacter xylosoxidans has been documented to form biofilms on plastics, including on contact lenses, urinary and intravenous catheters, and reusable tissue dispensers treated with disinfectant solution.4-6 One study demonstrated that A xylosoxidans is even capable of biodegradation of plastic, using the plastic as its sole source of carbon.7
Our case illustrates an indolent infection with A xylosoxidans forming a granulomatous abscess at the site of an insulin pump that was left in place for 7 days in an immunocompetent patient. Although infections with A xylosoxidans in patients with urinary or intravenous catheters have been reported,4 our case is unique, as the insulin pump was the source of such an infection. It is possible that the subcutaneous focus of acellular material described on the pathology report represented a partially biodegraded piece of the insulin pump catheter that broke off and was serving as a nidus of infection for A xylosoxidans. Although multidrug resistance is common, the culture grown from our patient was susceptible to TMP-SMX, among other antibiotics. Our patient was treated successfully with surgical excision, drainage, and a 10-day course of TMP-SMX.
Conclusion
Health care providers should recognize A xylosoxidans as an emerging pathogen that is capable of forming biofilms on “disinfected” surfaces and medical products, especially plastics. Achromobacter xylosoxidans may be resistant to multiple antibiotics and can cause infections with various presentations.
Case Report
A 50-year-old woman presented with a sore, tender, red lump on the right superior buttock of 5 months’ duration. Five months prior to presentation the patient used this area to attach the infusion set for an insulin pump, which was left in place for 7 days as opposed to the 2 or 3 days recommended by the device manufacturer. A firm, slightly tender lump formed, similar to prior scars that had developed from use of the insulin pump. However, the lump began to grow and get softer. It was intermittently warm and red. Although the area was sore and tender, she never had any major pain. She also denied any fever, malaise, or other systemic symptoms.
The patient indicated a medical history of type 1 diabetes mellitus diagnosed at 9 years of age; hypertension; asthma; gastroesophageal reflux disease; allergic rhinitis; migraine headaches; depression; hidradenitis suppurativa that resolved after surgical excision; and recurrent vaginal yeast infections, especially when taking antibiotics. She had a surgical history of hidradenitis suppurativa excision at the inguinal folds, bilateral carpal tunnel release, tubal ligation, abdominoplasty, and cholecystectomy. The patient’s current medications included insulin aspart, mometasone furoate, inhaled fluticasone, pantoprazole, cetirizine, spironolactone, duloxetine, sumatriptan, fluconazole, topiramate, and enalapril.
Physical examination revealed normal vital signs and the patient was afebrile. She had no swollen or tender lymph nodes. There was a 5.5×7.0-cm, soft, tender, erythematous subcutaneous mass with no visible punctum or overlying epidermal change on the right superior buttock (Figure 1). Based on the history and physical examination, the differential diagnosis included subcutaneous fat necrosis, epidermal inclusion cyst, and an abscess.
The patient was scheduled for excision of the mass the day after presenting to the clinic. During excision, 10 mL of thick purulent liquid was drained. A sample of the liquid was sent for Gram stain, aerobic and anaerobic culture, and antibiotic sensitivities. Necrotic-appearing adipose and fibrotic tissues were dissected and extirpated through an elliptical incision and submitted for pathologic evaluation.
Histopathology showed a subcutaneous defect with palisaded granulomatous inflammation and sclerosis (Figure 2). There was no detection of microorganisms with Grocott-Gomori methenamine-silver, tissue Gram, or acid-fast stains. There was a focus of acellular material embedded within the inflammation (Figure 3). The Gram stain of the purulent material showed few white blood cells and rare gram-negative bacilli. Culture grew moderate Achromobacter xylosoxidans resistant to cefepime, cefotaxime, and gentamicin. The culture was susceptible to ceftazidime, imipenem, levofloxacin, piperacillin, and trimethoprim-sulfamethoxazole (TMP-SMX).
The patient was prescribed oral TMP-SMX (160 mg of TMP and 800 mg of SMX) twice daily for 10 days. The patient tolerated the procedure and the subsequent antibiotics well. The patient had normal levels of IgA, IgG, and IgM, as well as a negative screening test for human immunodeficiency virus. She healed well from the surgical procedure and has had no recurrence of symptoms.
Comment
Achromobacter xylosoxidans is a nonfermentative, non–spore-forming, motile, gram-negative, aerobic, catalase-positive and oxidase-positive flagellate bacterium. It is an emerging pathogen that was first isolated in 1971 from patients with chronic otitis media.1 Since its recognition, it has been documented to cause a variety of infections, including pneumonia, meningitis, osteomyelitis, endocarditis, and bacteremia, as well as abdominal, urinary tract, ocular, and skin and soft tissue infections.2,3 Those affected usually are immunocompromised, have hematologic disorders, or have indwelling catheters.4 Strains of A xylosoxidans have shown resistance to multiple antibiotics including penicillins, cephalosporins, carbapenems, aminoglycosides, macrolides, fluoroquinolones, and TMP-SMX. Achromobacter xylosoxidans has been documented to form biofilms on plastics, including on contact lenses, urinary and intravenous catheters, and reusable tissue dispensers treated with disinfectant solution.4-6 One study demonstrated that A xylosoxidans is even capable of biodegradation of plastic, using the plastic as its sole source of carbon.7
Our case illustrates an indolent infection with A xylosoxidans forming a granulomatous abscess at the site of an insulin pump that was left in place for 7 days in an immunocompetent patient. Although infections with A xylosoxidans in patients with urinary or intravenous catheters have been reported,4 our case is unique, as the insulin pump was the source of such an infection. It is possible that the subcutaneous focus of acellular material described on the pathology report represented a partially biodegraded piece of the insulin pump catheter that broke off and was serving as a nidus of infection for A xylosoxidans. Although multidrug resistance is common, the culture grown from our patient was susceptible to TMP-SMX, among other antibiotics. Our patient was treated successfully with surgical excision, drainage, and a 10-day course of TMP-SMX.
Conclusion
Health care providers should recognize A xylosoxidans as an emerging pathogen that is capable of forming biofilms on “disinfected” surfaces and medical products, especially plastics. Achromobacter xylosoxidans may be resistant to multiple antibiotics and can cause infections with various presentations.
- Yabuuchi E, Oyama A. Achromobacter xylosoxidans n. sp. from human ear discharge. Jpn J Microbiol. 1971;15:477-481.
- Rodrigues CG, Rays J, Kanegae MY. Native-valve endocarditis caused by Achromobacter xylosoxidans: a case report and review of literature. Autops Case Rep. 2017;7:50-55.
- Tena D, Martínez NM, Losa C, et al. Skin and soft tissue infection caused by Achromobacter xylosoxidans: report of 14 cases. Scand J Infect Dis. 2014;46:130-135.
- Pérez Barragán E, Sandino Pérez J, Corbella L, et al. Achromobacter xylosoxidans bacteremia: clinical and microbiological features in a 10-year case series. Rev Esp Quimioter. 2018;31:268-273.
- Konstantinović N, Ćirković I, Đukić S, et al. Biofilm formation of Achromobacter xylosoxidans on contact lens. Acta Microbiol Immunol Hung. 2017;64:293-300.
- Günther F, Merle U, Frank U, et al. Pseudobacteremia outbreak of biofilm-forming Achromobacter xylosoxidans—environmental transmission. BMC Infect Dis. 2016;16:584.
- Kowalczyk A, Chyc M, Ryszka P, et al. Achromobacter xylosoxidans as a new microorganism strain colonizing high-density polyethylene as a key step to its biodegradation. Environ Sci Pollut Res Int. 2016;23:11349-11356.
- Yabuuchi E, Oyama A. Achromobacter xylosoxidans n. sp. from human ear discharge. Jpn J Microbiol. 1971;15:477-481.
- Rodrigues CG, Rays J, Kanegae MY. Native-valve endocarditis caused by Achromobacter xylosoxidans: a case report and review of literature. Autops Case Rep. 2017;7:50-55.
- Tena D, Martínez NM, Losa C, et al. Skin and soft tissue infection caused by Achromobacter xylosoxidans: report of 14 cases. Scand J Infect Dis. 2014;46:130-135.
- Pérez Barragán E, Sandino Pérez J, Corbella L, et al. Achromobacter xylosoxidans bacteremia: clinical and microbiological features in a 10-year case series. Rev Esp Quimioter. 2018;31:268-273.
- Konstantinović N, Ćirković I, Đukić S, et al. Biofilm formation of Achromobacter xylosoxidans on contact lens. Acta Microbiol Immunol Hung. 2017;64:293-300.
- Günther F, Merle U, Frank U, et al. Pseudobacteremia outbreak of biofilm-forming Achromobacter xylosoxidans—environmental transmission. BMC Infect Dis. 2016;16:584.
- Kowalczyk A, Chyc M, Ryszka P, et al. Achromobacter xylosoxidans as a new microorganism strain colonizing high-density polyethylene as a key step to its biodegradation. Environ Sci Pollut Res Int. 2016;23:11349-11356.
Practice Points
- Achromobacter xylosoxidans is an emerging pathogen primarily in the immunocompromised patient.
- Achromobacter xylosoxidans can form biofilms on plastics treated with disinfectant solution, including medical products.
- Strains of A xylosoxidans have shown multiantibiotic resistance.

Cutaneous Leishmaniasis Successfully Treated With Miltefosine
Leishmaniasis is a neglected parasitic disease with an estimated annual incidence of 1.3 million cases, the majority of which manifest as cutaneous leishmaniasis.1 The cutaneous and mucosal forms demonstrate substantial global burden with morbidity and socioeconomic repercussions, while the visceral form is responsible for up to 30,000 deaths annually.2 Despite increasing prevalence in the United States, awareness and diagnosis remain relatively low.3 We describe 2 cases of cutaneous leishmaniasis in New England, United States, in travelers returning from Central America, both successfully treated with miltefosine. We also review prevention, diagnosis, and treatment options.
Case Reports
Patient 1
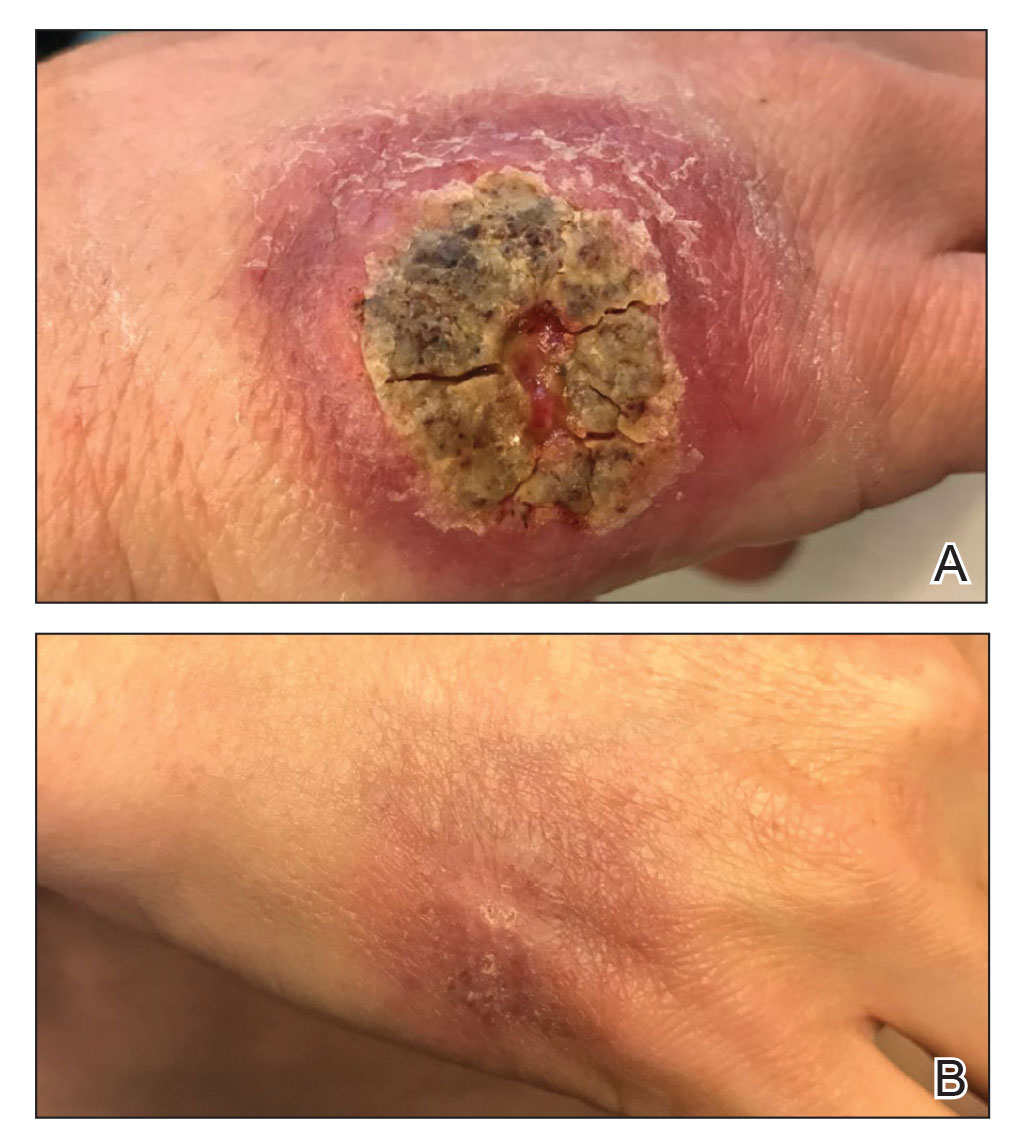
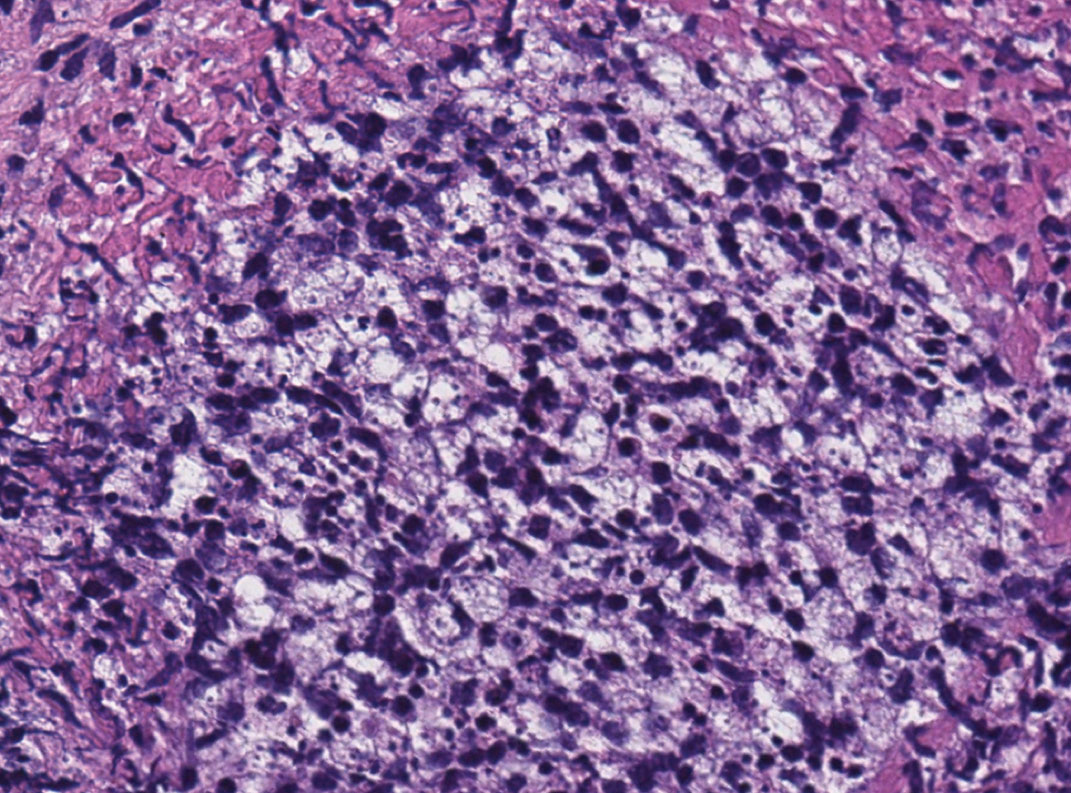
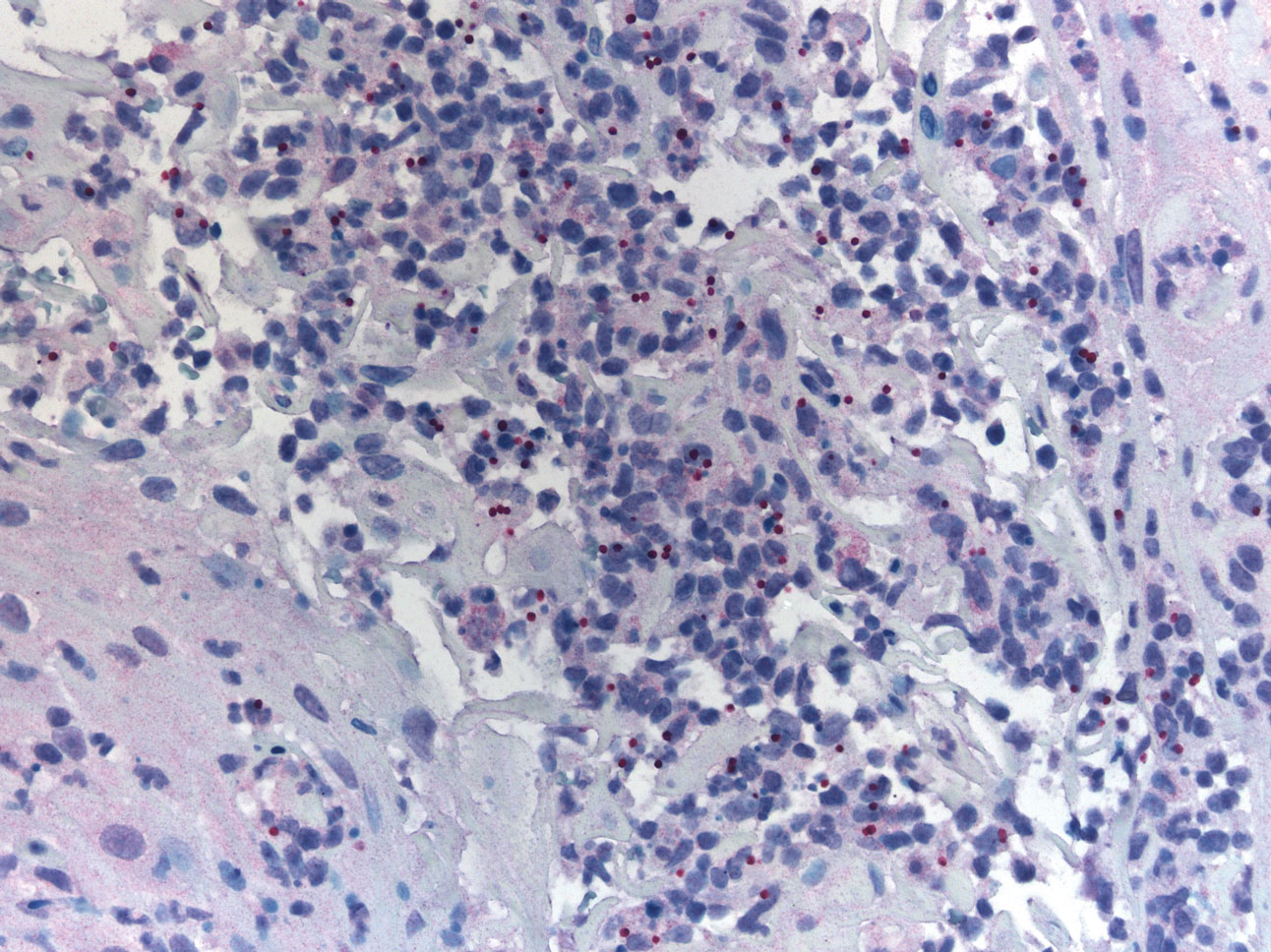
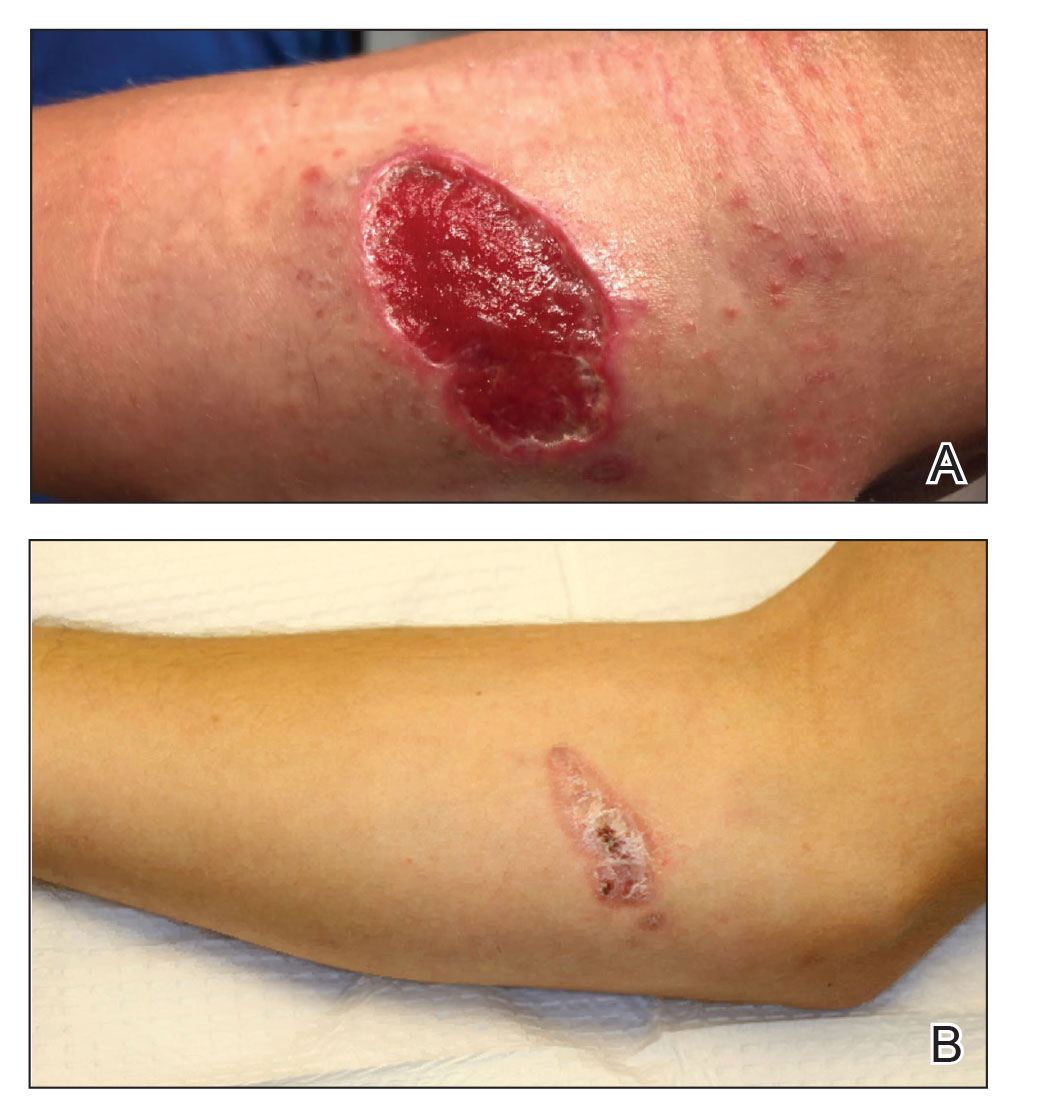
A 47-year-old woman presented with an enlarging, 2-cm, erythematous, ulcerated nodule on the right dorsal hand of 2 weeks’ duration with accompanying right epitrochlear lymphadenopathy (Figure 1A). She noticed the lesion 10 weeks after returning from Panama, where she had been photographing the jungle. Prior to the initial presentation to dermatology, salicylic acid wart remover, intramuscular ceftriaxone, and oral trimethoprim had failed to alleviate the lesion. Her laboratory results were notable for an elevated C-reactive protein level of 5.4 mg/L (reference range, ≤4.9 mg/L). A punch biopsy demonstrated pseudoepitheliomatous hyperplasia with diffuse dermal lymphohistiocytic inflammation and small intracytoplasmic structures within histiocytes consistent with leishmaniasis (Figure 2). Immunohistochemistry was consistent with leishmaniasis (Figure 3), and polymerase chain reaction performed by the Centers for Disease Control and Prevention (CDC) identified the pathogen as Leishmania braziliensis.
Patient 2
An 18-year-old man presented with an enlarging, well-delineated, tender ulcer of 6 weeks’ duration measuring 2.5×2 cm with an erythematous and edematous border on the right medial forearm with associated epitrochlear lymphadenopathy (Figure 4). Nine weeks prior to initial presentation, he had returned from a 3-month outdoor adventure trip to the Florida Keys, Costa Rica, and Panama. He had used bug repellent intermittently, slept under a bug net, and did not recall any trauma or bite at the ulcer site. Biopsy and tissue culture were obtained, and histopathology demonstrated an ulcer with a dense dermal lymphogranulomatous infiltrate and intracytoplasmic organisms consistent with leishmaniasis. Polymerase chain reaction by the CDC identified the pathogen as Leishmania panamensis.
Treatment
Both patients were prescribed oral miltefosine 50 mg twice daily for 28 days. Patient 1 initiated treatment 1 month after lesion onset, and patient 2 initiated treatment 2.5 months after initial presentation. Both patients had noticeable clinical improvement within 21 days of starting treatment, with lesions diminishing in size and lymphadenopathy resolving. Within 2 months of treatment, patient 1’s ulcer completely resolved with only postinflammatory hyperpigmentation (Figure 1B), while patient 2’s ulcer was noticeably smaller and shallower compared with its peak size of 4.2×2.4 cm (Figure 4B). Miltefosine was well tolerated by both patients; emesis resolved with ondansetron in patient 1 and spontaneously in patient 2, who had asymptomatic temporary hyperkalemia of 5.2 mmol/L (reference range, 3.5–5.0 mmol/L).
Comment
Epidemiology and Prevention
Risk factors for leishmaniasis include weak immunity, poverty, poor housing, poor sanitation, malnutrition, urbanization, climate change, and human migration.4 Our patients were most directly affected by travel to locations where leishmaniasis is endemic. Despite an increasing prevalence of endemic leishmaniasis and new animal hosts in the southern United States, most patients diagnosed in the United States are infected abroad by Leishmania mexicana and L braziliensis, both cutaneous New World species.3 Our patients were infected by species within the New World subgenus Viannia that have potential for mucocutaneous spread.4
Because there is no chemoprophylaxis or acquired active immunity such as vaccines that can mitigate the risk for leishmaniasis, public health efforts focus on preventive measures. Although difficult to achieve, avoidance of the phlebotomine sand fly species that transmit the obligate intracellular Leishmania parasite is a most effective measure.4 Travelers entering geographic regions with higher risk for leishmaniasis should be aware of the inherent risk and determine which methods of prevention, such as N,N-diethyl-meta-toluamide (DEET) insecticides or permethrin-treated protective clothing, are most feasible. Although higher concentrations of DEET provide longer protection, the effectiveness tends to plateau at approximately 50%.5
Presentation and Prognosis
For patients who develop leishmaniasis, the disease course and prognosis depend greatly on the species and manifestation. The most common form of leishmaniasis is localized cutaneous leishmaniasis, which has an annual incidence of up to 1 million cases. It initially presents as macules, usually at the site of inoculation within several months to years of infection.6 The macules expand into papules and plaques that reach maximum size over at least 1 week4 and then progress into crusted ulcers up to 5 cm in diameter with raised edges. Although usually painless and self-limited, these lesions can take years to spontaneously heal, with the risk for atrophic scarring and altered pigmentation. Lymphatic involvement manifests as lymphadenitis or regional lymphadenopathy and is common with lesions caused by the subgenus Viannia.6
Leishmania braziliensis and L panamensis, the species that infected our patients, can uniquely cause cutaneous leishmaniasis that metastasizes into mucocutaneous leishmaniasis, which always affects the nasal mucosa. Risk factors for transformation include a primary lesion site above the waist, multiple or large primary lesions, and delayed healing of primary cutaneous leishmaniasis. Mucocutaneous leishmaniasis can result in notable morbidity and even mortality from invasion and destruction of nasal and oropharyngeal mucosa, as well as intercurrent pneumonia, especially if treatment is insufficient or delayed.4
Diagnosis
Prompt treatment relies on accurate and timely diagnosis, which is complicated by the relative unfamiliarity with leishmaniasis in the United States. The differential diagnosis for cutaneous leishmaniasis is broad, including deep fungal infection, Mycobacterium infection, cutaneous granulomatous conditions, nonmelanoma cutaneous neoplasms, and trauma. Taking a thorough patient history, including potential exposures and travels; having high clinical suspicion; and being aware of classic presentation allows for identification of leishmaniasis and subsequent stratification by manifestation.7
Diagnosis is made by detecting Leishmania organisms or DNA using light microscopy and staining to visualize the kinetoplast in an amastigote, molecular methods, or specialized culturing.7 The CDC is a valuable diagnostic partner for confirmation and speciation. Specific instructions for specimen collection and transportation can be found by contacting the CDC or reading their guide.8 To provide prompt care and reassurance to patients, it is important to be aware of the coordination effort that may be needed to send samples, receive results, and otherwise correspond with a separate institution.
Treatment
Treatment of cutaneous leishmaniasis is indicated to decrease the risk for mucosal dissemination and clinical reactivation of lesions, accelerate healing of lesions, decrease local morbidity caused by large or persistent lesions, and decrease the reservoir of infection in places where infected humans serve as reservoir hosts. Oral treatments include ketoconazole, itraconazole, and fluconazole, recommended at doses ranging from 200 to 600 mg daily for at least 28 days. For severe, refractory, or visceral leishmaniasis, parenteral choices include
Miltefosine is becoming a more common treatment of leishmaniasis because of its oral route, tolerability in nonpregnant patients, and commercial availability. It was approved by the US Food and Drug Administration in 2014 for cutaneous leishmaniasis due to L braziliensis, L panamensis, and Leishmania guyanensis; mucosal leishmaniasis due to L braziliensis; and visceral leishmaniasis due to Leishmania donovani in patients at least 12 years of age. For cutaneous leishmaniasis, the standard dosage of 50 mg twice daily (for patients weighing 30–44 kg) or 3 times daily (for patients weighing 45 kg or more) for 28 consecutive days has cure rates of 48% to 85% by 6 months after therapy ends. Cure is defined as epithelialization of lesions, no enlargement greater than 50% in lesions, no appearance of new lesions, and/or negative parasitology. The antileishmanial mechanism of action is unknown and likely involves interaction with lipids, inhibition of cytochrome c oxidase, and apoptosislike cell death. Miltefosine is contraindicated in pregnancy. The most common adverse reactions in patients include nausea (35.9%–41.7%), motion sickness (29.2%), headache (28.1%), and emesis (4.5%–27.5%). With the exception of headache, these adverse reactions can decrease with administration of food, fluids, and antiemetics. Potentially more serious but rarer adverse reactions include elevated serum creatinine (5%–25%) and transaminases (5%). Although our patients had mild hyperkalemia, it is not an established adverse reaction. However, renal injury has been reported.10
Conclusion
Cutaneous leishmaniasis is increasing in prevalence in the United States due to increased foreign travel. Providers should be familiar with the cutaneous presentation of leishmaniasis, even in areas of low prevalence, to limit the risk for mucocutaneous dissemination from infection with the subgenus Viannia. Prompt treatment is vital to ensuring the best prognosis, and first-line treatment with miltefosine should be strongly considered given its efficacy and tolerability.
- Babuadze G, Alvar J, Argaw D, et al. Epidemiology of visceral leishmaniasis in Georgia. PLoS Negl Trop Dis. 2014;8:e2725.
- Leishmaniasis. World Health Organization website. https://www.afro.who.int/health-topics/Leishmaniasis. Accessed September 15, 2020.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039.
- Leishmaniasis. World Health Organization website. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. Update March 2, 2020. Accessed September 15, 2020.
- Centers for Disease Control and Prevention. Guidelines for DEET insect repellent use. https://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 20, 2020.
- Buescher MD, Rutledge LC, Wirtz RA, et al. The dose-persistence relationship of DEET against Aedes aegypti. Mosq News. 1983;43:364-366.
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:e202-e264.
- US Department of Health and Human Services. Practical guide for specimen collection and reference diagnosis of leishmaniasis. Centers for Disease Control and Prevention website. https://www.cdc.gov/parasites/leishmaniasis/resources/pdf/cdc_diagnosis_guide_leishmaniasis_2016.pdf. Accessed September 15, 2020.
- Visceral leishmaniasis. Drugs for Neglected Diseases Initiative website. https://www.dndi.org/diseases-projects/leishmaniasis/. Accessed September 15, 2020.
- Impavido Medication Guide. Food and Drug Administration Web site. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204684s000lbl.pdf. Revised March 2014. Accessed May 18, 2020.
Leishmaniasis is a neglected parasitic disease with an estimated annual incidence of 1.3 million cases, the majority of which manifest as cutaneous leishmaniasis.1 The cutaneous and mucosal forms demonstrate substantial global burden with morbidity and socioeconomic repercussions, while the visceral form is responsible for up to 30,000 deaths annually.2 Despite increasing prevalence in the United States, awareness and diagnosis remain relatively low.3 We describe 2 cases of cutaneous leishmaniasis in New England, United States, in travelers returning from Central America, both successfully treated with miltefosine. We also review prevention, diagnosis, and treatment options.
Case Reports
Patient 1
A 47-year-old woman presented with an enlarging, 2-cm, erythematous, ulcerated nodule on the right dorsal hand of 2 weeks’ duration with accompanying right epitrochlear lymphadenopathy (Figure 1A). She noticed the lesion 10 weeks after returning from Panama, where she had been photographing the jungle. Prior to the initial presentation to dermatology, salicylic acid wart remover, intramuscular ceftriaxone, and oral trimethoprim had failed to alleviate the lesion. Her laboratory results were notable for an elevated C-reactive protein level of 5.4 mg/L (reference range, ≤4.9 mg/L). A punch biopsy demonstrated pseudoepitheliomatous hyperplasia with diffuse dermal lymphohistiocytic inflammation and small intracytoplasmic structures within histiocytes consistent with leishmaniasis (Figure 2). Immunohistochemistry was consistent with leishmaniasis (Figure 3), and polymerase chain reaction performed by the Centers for Disease Control and Prevention (CDC) identified the pathogen as Leishmania braziliensis.
Patient 2
An 18-year-old man presented with an enlarging, well-delineated, tender ulcer of 6 weeks’ duration measuring 2.5×2 cm with an erythematous and edematous border on the right medial forearm with associated epitrochlear lymphadenopathy (Figure 4). Nine weeks prior to initial presentation, he had returned from a 3-month outdoor adventure trip to the Florida Keys, Costa Rica, and Panama. He had used bug repellent intermittently, slept under a bug net, and did not recall any trauma or bite at the ulcer site. Biopsy and tissue culture were obtained, and histopathology demonstrated an ulcer with a dense dermal lymphogranulomatous infiltrate and intracytoplasmic organisms consistent with leishmaniasis. Polymerase chain reaction by the CDC identified the pathogen as Leishmania panamensis.
Treatment
Both patients were prescribed oral miltefosine 50 mg twice daily for 28 days. Patient 1 initiated treatment 1 month after lesion onset, and patient 2 initiated treatment 2.5 months after initial presentation. Both patients had noticeable clinical improvement within 21 days of starting treatment, with lesions diminishing in size and lymphadenopathy resolving. Within 2 months of treatment, patient 1’s ulcer completely resolved with only postinflammatory hyperpigmentation (Figure 1B), while patient 2’s ulcer was noticeably smaller and shallower compared with its peak size of 4.2×2.4 cm (Figure 4B). Miltefosine was well tolerated by both patients; emesis resolved with ondansetron in patient 1 and spontaneously in patient 2, who had asymptomatic temporary hyperkalemia of 5.2 mmol/L (reference range, 3.5–5.0 mmol/L).
Comment
Epidemiology and Prevention
Risk factors for leishmaniasis include weak immunity, poverty, poor housing, poor sanitation, malnutrition, urbanization, climate change, and human migration.4 Our patients were most directly affected by travel to locations where leishmaniasis is endemic. Despite an increasing prevalence of endemic leishmaniasis and new animal hosts in the southern United States, most patients diagnosed in the United States are infected abroad by Leishmania mexicana and L braziliensis, both cutaneous New World species.3 Our patients were infected by species within the New World subgenus Viannia that have potential for mucocutaneous spread.4
Because there is no chemoprophylaxis or acquired active immunity such as vaccines that can mitigate the risk for leishmaniasis, public health efforts focus on preventive measures. Although difficult to achieve, avoidance of the phlebotomine sand fly species that transmit the obligate intracellular Leishmania parasite is a most effective measure.4 Travelers entering geographic regions with higher risk for leishmaniasis should be aware of the inherent risk and determine which methods of prevention, such as N,N-diethyl-meta-toluamide (DEET) insecticides or permethrin-treated protective clothing, are most feasible. Although higher concentrations of DEET provide longer protection, the effectiveness tends to plateau at approximately 50%.5
Presentation and Prognosis
For patients who develop leishmaniasis, the disease course and prognosis depend greatly on the species and manifestation. The most common form of leishmaniasis is localized cutaneous leishmaniasis, which has an annual incidence of up to 1 million cases. It initially presents as macules, usually at the site of inoculation within several months to years of infection.6 The macules expand into papules and plaques that reach maximum size over at least 1 week4 and then progress into crusted ulcers up to 5 cm in diameter with raised edges. Although usually painless and self-limited, these lesions can take years to spontaneously heal, with the risk for atrophic scarring and altered pigmentation. Lymphatic involvement manifests as lymphadenitis or regional lymphadenopathy and is common with lesions caused by the subgenus Viannia.6
Leishmania braziliensis and L panamensis, the species that infected our patients, can uniquely cause cutaneous leishmaniasis that metastasizes into mucocutaneous leishmaniasis, which always affects the nasal mucosa. Risk factors for transformation include a primary lesion site above the waist, multiple or large primary lesions, and delayed healing of primary cutaneous leishmaniasis. Mucocutaneous leishmaniasis can result in notable morbidity and even mortality from invasion and destruction of nasal and oropharyngeal mucosa, as well as intercurrent pneumonia, especially if treatment is insufficient or delayed.4
Diagnosis
Prompt treatment relies on accurate and timely diagnosis, which is complicated by the relative unfamiliarity with leishmaniasis in the United States. The differential diagnosis for cutaneous leishmaniasis is broad, including deep fungal infection, Mycobacterium infection, cutaneous granulomatous conditions, nonmelanoma cutaneous neoplasms, and trauma. Taking a thorough patient history, including potential exposures and travels; having high clinical suspicion; and being aware of classic presentation allows for identification of leishmaniasis and subsequent stratification by manifestation.7
Diagnosis is made by detecting Leishmania organisms or DNA using light microscopy and staining to visualize the kinetoplast in an amastigote, molecular methods, or specialized culturing.7 The CDC is a valuable diagnostic partner for confirmation and speciation. Specific instructions for specimen collection and transportation can be found by contacting the CDC or reading their guide.8 To provide prompt care and reassurance to patients, it is important to be aware of the coordination effort that may be needed to send samples, receive results, and otherwise correspond with a separate institution.
Treatment
Treatment of cutaneous leishmaniasis is indicated to decrease the risk for mucosal dissemination and clinical reactivation of lesions, accelerate healing of lesions, decrease local morbidity caused by large or persistent lesions, and decrease the reservoir of infection in places where infected humans serve as reservoir hosts. Oral treatments include ketoconazole, itraconazole, and fluconazole, recommended at doses ranging from 200 to 600 mg daily for at least 28 days. For severe, refractory, or visceral leishmaniasis, parenteral choices include
Miltefosine is becoming a more common treatment of leishmaniasis because of its oral route, tolerability in nonpregnant patients, and commercial availability. It was approved by the US Food and Drug Administration in 2014 for cutaneous leishmaniasis due to L braziliensis, L panamensis, and Leishmania guyanensis; mucosal leishmaniasis due to L braziliensis; and visceral leishmaniasis due to Leishmania donovani in patients at least 12 years of age. For cutaneous leishmaniasis, the standard dosage of 50 mg twice daily (for patients weighing 30–44 kg) or 3 times daily (for patients weighing 45 kg or more) for 28 consecutive days has cure rates of 48% to 85% by 6 months after therapy ends. Cure is defined as epithelialization of lesions, no enlargement greater than 50% in lesions, no appearance of new lesions, and/or negative parasitology. The antileishmanial mechanism of action is unknown and likely involves interaction with lipids, inhibition of cytochrome c oxidase, and apoptosislike cell death. Miltefosine is contraindicated in pregnancy. The most common adverse reactions in patients include nausea (35.9%–41.7%), motion sickness (29.2%), headache (28.1%), and emesis (4.5%–27.5%). With the exception of headache, these adverse reactions can decrease with administration of food, fluids, and antiemetics. Potentially more serious but rarer adverse reactions include elevated serum creatinine (5%–25%) and transaminases (5%). Although our patients had mild hyperkalemia, it is not an established adverse reaction. However, renal injury has been reported.10
Conclusion
Cutaneous leishmaniasis is increasing in prevalence in the United States due to increased foreign travel. Providers should be familiar with the cutaneous presentation of leishmaniasis, even in areas of low prevalence, to limit the risk for mucocutaneous dissemination from infection with the subgenus Viannia. Prompt treatment is vital to ensuring the best prognosis, and first-line treatment with miltefosine should be strongly considered given its efficacy and tolerability.
Leishmaniasis is a neglected parasitic disease with an estimated annual incidence of 1.3 million cases, the majority of which manifest as cutaneous leishmaniasis.1 The cutaneous and mucosal forms demonstrate substantial global burden with morbidity and socioeconomic repercussions, while the visceral form is responsible for up to 30,000 deaths annually.2 Despite increasing prevalence in the United States, awareness and diagnosis remain relatively low.3 We describe 2 cases of cutaneous leishmaniasis in New England, United States, in travelers returning from Central America, both successfully treated with miltefosine. We also review prevention, diagnosis, and treatment options.
Case Reports
Patient 1
A 47-year-old woman presented with an enlarging, 2-cm, erythematous, ulcerated nodule on the right dorsal hand of 2 weeks’ duration with accompanying right epitrochlear lymphadenopathy (Figure 1A). She noticed the lesion 10 weeks after returning from Panama, where she had been photographing the jungle. Prior to the initial presentation to dermatology, salicylic acid wart remover, intramuscular ceftriaxone, and oral trimethoprim had failed to alleviate the lesion. Her laboratory results were notable for an elevated C-reactive protein level of 5.4 mg/L (reference range, ≤4.9 mg/L). A punch biopsy demonstrated pseudoepitheliomatous hyperplasia with diffuse dermal lymphohistiocytic inflammation and small intracytoplasmic structures within histiocytes consistent with leishmaniasis (Figure 2). Immunohistochemistry was consistent with leishmaniasis (Figure 3), and polymerase chain reaction performed by the Centers for Disease Control and Prevention (CDC) identified the pathogen as Leishmania braziliensis.
Patient 2
An 18-year-old man presented with an enlarging, well-delineated, tender ulcer of 6 weeks’ duration measuring 2.5×2 cm with an erythematous and edematous border on the right medial forearm with associated epitrochlear lymphadenopathy (Figure 4). Nine weeks prior to initial presentation, he had returned from a 3-month outdoor adventure trip to the Florida Keys, Costa Rica, and Panama. He had used bug repellent intermittently, slept under a bug net, and did not recall any trauma or bite at the ulcer site. Biopsy and tissue culture were obtained, and histopathology demonstrated an ulcer with a dense dermal lymphogranulomatous infiltrate and intracytoplasmic organisms consistent with leishmaniasis. Polymerase chain reaction by the CDC identified the pathogen as Leishmania panamensis.
Treatment
Both patients were prescribed oral miltefosine 50 mg twice daily for 28 days. Patient 1 initiated treatment 1 month after lesion onset, and patient 2 initiated treatment 2.5 months after initial presentation. Both patients had noticeable clinical improvement within 21 days of starting treatment, with lesions diminishing in size and lymphadenopathy resolving. Within 2 months of treatment, patient 1’s ulcer completely resolved with only postinflammatory hyperpigmentation (Figure 1B), while patient 2’s ulcer was noticeably smaller and shallower compared with its peak size of 4.2×2.4 cm (Figure 4B). Miltefosine was well tolerated by both patients; emesis resolved with ondansetron in patient 1 and spontaneously in patient 2, who had asymptomatic temporary hyperkalemia of 5.2 mmol/L (reference range, 3.5–5.0 mmol/L).
Comment
Epidemiology and Prevention
Risk factors for leishmaniasis include weak immunity, poverty, poor housing, poor sanitation, malnutrition, urbanization, climate change, and human migration.4 Our patients were most directly affected by travel to locations where leishmaniasis is endemic. Despite an increasing prevalence of endemic leishmaniasis and new animal hosts in the southern United States, most patients diagnosed in the United States are infected abroad by Leishmania mexicana and L braziliensis, both cutaneous New World species.3 Our patients were infected by species within the New World subgenus Viannia that have potential for mucocutaneous spread.4
Because there is no chemoprophylaxis or acquired active immunity such as vaccines that can mitigate the risk for leishmaniasis, public health efforts focus on preventive measures. Although difficult to achieve, avoidance of the phlebotomine sand fly species that transmit the obligate intracellular Leishmania parasite is a most effective measure.4 Travelers entering geographic regions with higher risk for leishmaniasis should be aware of the inherent risk and determine which methods of prevention, such as N,N-diethyl-meta-toluamide (DEET) insecticides or permethrin-treated protective clothing, are most feasible. Although higher concentrations of DEET provide longer protection, the effectiveness tends to plateau at approximately 50%.5
Presentation and Prognosis
For patients who develop leishmaniasis, the disease course and prognosis depend greatly on the species and manifestation. The most common form of leishmaniasis is localized cutaneous leishmaniasis, which has an annual incidence of up to 1 million cases. It initially presents as macules, usually at the site of inoculation within several months to years of infection.6 The macules expand into papules and plaques that reach maximum size over at least 1 week4 and then progress into crusted ulcers up to 5 cm in diameter with raised edges. Although usually painless and self-limited, these lesions can take years to spontaneously heal, with the risk for atrophic scarring and altered pigmentation. Lymphatic involvement manifests as lymphadenitis or regional lymphadenopathy and is common with lesions caused by the subgenus Viannia.6
Leishmania braziliensis and L panamensis, the species that infected our patients, can uniquely cause cutaneous leishmaniasis that metastasizes into mucocutaneous leishmaniasis, which always affects the nasal mucosa. Risk factors for transformation include a primary lesion site above the waist, multiple or large primary lesions, and delayed healing of primary cutaneous leishmaniasis. Mucocutaneous leishmaniasis can result in notable morbidity and even mortality from invasion and destruction of nasal and oropharyngeal mucosa, as well as intercurrent pneumonia, especially if treatment is insufficient or delayed.4
Diagnosis
Prompt treatment relies on accurate and timely diagnosis, which is complicated by the relative unfamiliarity with leishmaniasis in the United States. The differential diagnosis for cutaneous leishmaniasis is broad, including deep fungal infection, Mycobacterium infection, cutaneous granulomatous conditions, nonmelanoma cutaneous neoplasms, and trauma. Taking a thorough patient history, including potential exposures and travels; having high clinical suspicion; and being aware of classic presentation allows for identification of leishmaniasis and subsequent stratification by manifestation.7
Diagnosis is made by detecting Leishmania organisms or DNA using light microscopy and staining to visualize the kinetoplast in an amastigote, molecular methods, or specialized culturing.7 The CDC is a valuable diagnostic partner for confirmation and speciation. Specific instructions for specimen collection and transportation can be found by contacting the CDC or reading their guide.8 To provide prompt care and reassurance to patients, it is important to be aware of the coordination effort that may be needed to send samples, receive results, and otherwise correspond with a separate institution.
Treatment
Treatment of cutaneous leishmaniasis is indicated to decrease the risk for mucosal dissemination and clinical reactivation of lesions, accelerate healing of lesions, decrease local morbidity caused by large or persistent lesions, and decrease the reservoir of infection in places where infected humans serve as reservoir hosts. Oral treatments include ketoconazole, itraconazole, and fluconazole, recommended at doses ranging from 200 to 600 mg daily for at least 28 days. For severe, refractory, or visceral leishmaniasis, parenteral choices include
Miltefosine is becoming a more common treatment of leishmaniasis because of its oral route, tolerability in nonpregnant patients, and commercial availability. It was approved by the US Food and Drug Administration in 2014 for cutaneous leishmaniasis due to L braziliensis, L panamensis, and Leishmania guyanensis; mucosal leishmaniasis due to L braziliensis; and visceral leishmaniasis due to Leishmania donovani in patients at least 12 years of age. For cutaneous leishmaniasis, the standard dosage of 50 mg twice daily (for patients weighing 30–44 kg) or 3 times daily (for patients weighing 45 kg or more) for 28 consecutive days has cure rates of 48% to 85% by 6 months after therapy ends. Cure is defined as epithelialization of lesions, no enlargement greater than 50% in lesions, no appearance of new lesions, and/or negative parasitology. The antileishmanial mechanism of action is unknown and likely involves interaction with lipids, inhibition of cytochrome c oxidase, and apoptosislike cell death. Miltefosine is contraindicated in pregnancy. The most common adverse reactions in patients include nausea (35.9%–41.7%), motion sickness (29.2%), headache (28.1%), and emesis (4.5%–27.5%). With the exception of headache, these adverse reactions can decrease with administration of food, fluids, and antiemetics. Potentially more serious but rarer adverse reactions include elevated serum creatinine (5%–25%) and transaminases (5%). Although our patients had mild hyperkalemia, it is not an established adverse reaction. However, renal injury has been reported.10
Conclusion
Cutaneous leishmaniasis is increasing in prevalence in the United States due to increased foreign travel. Providers should be familiar with the cutaneous presentation of leishmaniasis, even in areas of low prevalence, to limit the risk for mucocutaneous dissemination from infection with the subgenus Viannia. Prompt treatment is vital to ensuring the best prognosis, and first-line treatment with miltefosine should be strongly considered given its efficacy and tolerability.
- Babuadze G, Alvar J, Argaw D, et al. Epidemiology of visceral leishmaniasis in Georgia. PLoS Negl Trop Dis. 2014;8:e2725.
- Leishmaniasis. World Health Organization website. https://www.afro.who.int/health-topics/Leishmaniasis. Accessed September 15, 2020.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039.
- Leishmaniasis. World Health Organization website. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. Update March 2, 2020. Accessed September 15, 2020.
- Centers for Disease Control and Prevention. Guidelines for DEET insect repellent use. https://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 20, 2020.
- Buescher MD, Rutledge LC, Wirtz RA, et al. The dose-persistence relationship of DEET against Aedes aegypti. Mosq News. 1983;43:364-366.
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:e202-e264.
- US Department of Health and Human Services. Practical guide for specimen collection and reference diagnosis of leishmaniasis. Centers for Disease Control and Prevention website. https://www.cdc.gov/parasites/leishmaniasis/resources/pdf/cdc_diagnosis_guide_leishmaniasis_2016.pdf. Accessed September 15, 2020.
- Visceral leishmaniasis. Drugs for Neglected Diseases Initiative website. https://www.dndi.org/diseases-projects/leishmaniasis/. Accessed September 15, 2020.
- Impavido Medication Guide. Food and Drug Administration Web site. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204684s000lbl.pdf. Revised March 2014. Accessed May 18, 2020.
- Babuadze G, Alvar J, Argaw D, et al. Epidemiology of visceral leishmaniasis in Georgia. PLoS Negl Trop Dis. 2014;8:e2725.
- Leishmaniasis. World Health Organization website. https://www.afro.who.int/health-topics/Leishmaniasis. Accessed September 15, 2020.
- McIlwee BE, Weis SE, Hosler GA. Incidence of endemic human cutaneous leishmaniasis in the United States. JAMA Dermatol. 2018;154:1032-1039.
- Leishmaniasis. World Health Organization website. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. Update March 2, 2020. Accessed September 15, 2020.
- Centers for Disease Control and Prevention. Guidelines for DEET insect repellent use. https://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 20, 2020.
- Buescher MD, Rutledge LC, Wirtz RA, et al. The dose-persistence relationship of DEET against Aedes aegypti. Mosq News. 1983;43:364-366.
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:e202-e264.
- US Department of Health and Human Services. Practical guide for specimen collection and reference diagnosis of leishmaniasis. Centers for Disease Control and Prevention website. https://www.cdc.gov/parasites/leishmaniasis/resources/pdf/cdc_diagnosis_guide_leishmaniasis_2016.pdf. Accessed September 15, 2020.
- Visceral leishmaniasis. Drugs for Neglected Diseases Initiative website. https://www.dndi.org/diseases-projects/leishmaniasis/. Accessed September 15, 2020.
- Impavido Medication Guide. Food and Drug Administration Web site. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204684s000lbl.pdf. Revised March 2014. Accessed May 18, 2020.
Practice Points
- Avoiding phlebotomine sand fly vector bites is the most effective way to prevent leishmaniasis.
- Prompt diagnosis and treatment of cutaneous leishmaniasis caused by Leishmania species that have potential for mucocutaneous spread are key to limiting morbidity and mortality.
- Partnering with the Centers for Disease Control and Prevention is critical for timely diagnosis.
- Miltefosine should be considered as a first-line agent for cutaneous leishmaniasis given its efficacy, tolerability, and ease of administration.
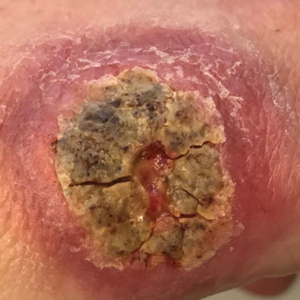
28-year-old woman • weakness • anxiety • altered mental status • Dx?
THE CASE
A 28-year-old woman with an extensive psychiatric history—including generalized anxiety disorder, panic disorder, and recent postpartum depression—presented with a chief complaint of right leg weakness. She stated this weakness had begun 4 days earlier. It occurred episodically and was preceded by tingling and cramping sensations. Each episode lasted a couple of minutes and spontaneously resolved. Associated with it, she experienced slurred speech and altered mentation. There was no loss of consciousness and no pain. A panic attack usually followed, consisting of feelings of impending doom, rapid breathing, palpitations, and nausea.
She had 3 prior diagnostic evaluations for this same chief complaint, twice in an emergency department (ED) and once with her primary care physician. These evaluations included lab work and extensive head imaging, which demonstrated no acute intracranial pathology. At each previous presentation, the diagnosis was an exacerbation of her anxiety disorder, and she was treated with lorazepam.
At the current presentation, her vital signs were stable. Examination revealed a notably anxious patient. She repeatedly expressed concern that she might have a brain tumor or some other deadly disease, as she had a family history of brain cancer. Her physical exam was entirely normal, including normal strength, sensation, and reflexes in all extremities.
Further head imaging (computed tomography, CT angiography, and magnetic resonance imaging of the brain) failed to reveal an etiology of her symptoms. With no clear organic cause, her medical providers again suspected an anxiety or panic episode. She was given reassurance, and an outpatient neurology consult was arranged.
THE DIAGNOSIS
One week later, at her outpatient neurology appointment, an electroencephalogram (EEG) was performed. Following photic stimulation, the EEG showed multiple right- and left-hemisphere foci of cortical hyperexcitability including a subtle sharp component (see FIGURE). Immediately following the longest of these episodes, the patient expressed a sense of anxiety and an altered sensorium similar to her prior presentations.

The EEG findings, in addition to the postictal anxiety symptoms and clinical history, were all important components that led the treating neurologist to the diagnosis of localization-related (focal) epilepsy.1 The patient was started on oxcarbazepine, a first-line anti-epileptic medication used in the treatment of focal epilepsy.2 She is being followed by a neurologist regularly and after optimizing her anti-epileptic medication, is no longer having seizures.
DISCUSSION
The difficulty of this case stems from the atypical presentation of the patient’s seizures. The key step to the correct diagnosis was a neurological consultation and an ensuing EEG. However, the patient received a vast spectrum of care, including multiple work-ups, prior to a conclusive diagnosis—which highlights an important issue health care providers must address.
Continue to: The role of bias
The role of bias. From the patient’s initial visits to the ED to her hospital admission, there was a prominent affixation, known as the anchoring bias,3 by the clinicians providing her care: All were focused heavily on her psychiatric features. Conversely, the evaluation for patients with suspected psychiatric diagnoses should focus on successfully ruling out major organic etiology with a broad differential diagnosis. It is crucial for providers to take a step back and make a conscious attempt to avoid fixation on a particular diagnosis, especially when it is psychiatric in nature. This allows the provider to actively consider alternative explanations for a patient presentation and work through a more encompassing differential.
The distinguishing symptoms. There is a common association between comorbid mood disorders (eg, depression, anxiety) and epilepsy.4 Another clue is ictal anxiety or nervousness, which is commonly observed in patients with partial seizures (and occurred with our patient).
These ictal episodes can be difficult to identify within the context of an isolated psychiatric diagnosis.5 The distinction can be clarified by the presence of associated somatic symptoms, which in this case included unilateral cramping, paresthesia, and weakness. These symptoms should clue in a practitioner to the possibility of underlying neurologic pathology, which should prompt the ordering of either an EEG or, at minimum, a neurological consultation.
THE TAKEAWAY
This case report shows how anchoring bias can lead to a delay in diagnosis and treatment. Avoidance of this type of bias requires heightened cognitive awareness by medical providers. A more system-based approach is to have structured diagnostic assessments,6 such as conducting a thorough neurological exam for patients with somatic symptoms and exacerbating comorbid psychiatric conditions.
It may also help to review cases like this with colleagues from diverse disciplinary backgrounds, highlighting thought processes and sharing uncertainty.3 These processes may shed light on confounding diagnoses that might be playing a role in a patient’s presentation and ultimately aid in the decision-making process.
CORRESPONDENCE
Paimon Ameli, DO, Naval Medical Center San Diego, 34800 Bob Wilson Drive, San Diego, CA 92134; paimonm@gmail.com
1. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522-530.
2. Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000-1015.
3. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
4. Jackson MJ, Turkington D. Depression and anxiety in epilepsy. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i45-i47.
5. López-Gómez M, Espinola M, Ramirez-Bermudez J, et al. Clinical presentation of anxiety among patients with epilepsy. Neuropsychiatr Dis Treat. 2008;4:1235-1239.
6. Etchells E. Anchoring bias with critical implications. Published June 2015. Patient Safety Network. https://psnet.ahrq.gov/web-mm/anchoring-bias-critical-implications. Accessed September 29, 2020.
THE CASE
A 28-year-old woman with an extensive psychiatric history—including generalized anxiety disorder, panic disorder, and recent postpartum depression—presented with a chief complaint of right leg weakness. She stated this weakness had begun 4 days earlier. It occurred episodically and was preceded by tingling and cramping sensations. Each episode lasted a couple of minutes and spontaneously resolved. Associated with it, she experienced slurred speech and altered mentation. There was no loss of consciousness and no pain. A panic attack usually followed, consisting of feelings of impending doom, rapid breathing, palpitations, and nausea.
She had 3 prior diagnostic evaluations for this same chief complaint, twice in an emergency department (ED) and once with her primary care physician. These evaluations included lab work and extensive head imaging, which demonstrated no acute intracranial pathology. At each previous presentation, the diagnosis was an exacerbation of her anxiety disorder, and she was treated with lorazepam.
At the current presentation, her vital signs were stable. Examination revealed a notably anxious patient. She repeatedly expressed concern that she might have a brain tumor or some other deadly disease, as she had a family history of brain cancer. Her physical exam was entirely normal, including normal strength, sensation, and reflexes in all extremities.
Further head imaging (computed tomography, CT angiography, and magnetic resonance imaging of the brain) failed to reveal an etiology of her symptoms. With no clear organic cause, her medical providers again suspected an anxiety or panic episode. She was given reassurance, and an outpatient neurology consult was arranged.
THE DIAGNOSIS
One week later, at her outpatient neurology appointment, an electroencephalogram (EEG) was performed. Following photic stimulation, the EEG showed multiple right- and left-hemisphere foci of cortical hyperexcitability including a subtle sharp component (see FIGURE). Immediately following the longest of these episodes, the patient expressed a sense of anxiety and an altered sensorium similar to her prior presentations.
The EEG findings, in addition to the postictal anxiety symptoms and clinical history, were all important components that led the treating neurologist to the diagnosis of localization-related (focal) epilepsy.1 The patient was started on oxcarbazepine, a first-line anti-epileptic medication used in the treatment of focal epilepsy.2 She is being followed by a neurologist regularly and after optimizing her anti-epileptic medication, is no longer having seizures.
DISCUSSION
The difficulty of this case stems from the atypical presentation of the patient’s seizures. The key step to the correct diagnosis was a neurological consultation and an ensuing EEG. However, the patient received a vast spectrum of care, including multiple work-ups, prior to a conclusive diagnosis—which highlights an important issue health care providers must address.
Continue to: The role of bias
The role of bias. From the patient’s initial visits to the ED to her hospital admission, there was a prominent affixation, known as the anchoring bias,3 by the clinicians providing her care: All were focused heavily on her psychiatric features. Conversely, the evaluation for patients with suspected psychiatric diagnoses should focus on successfully ruling out major organic etiology with a broad differential diagnosis. It is crucial for providers to take a step back and make a conscious attempt to avoid fixation on a particular diagnosis, especially when it is psychiatric in nature. This allows the provider to actively consider alternative explanations for a patient presentation and work through a more encompassing differential.
The distinguishing symptoms. There is a common association between comorbid mood disorders (eg, depression, anxiety) and epilepsy.4 Another clue is ictal anxiety or nervousness, which is commonly observed in patients with partial seizures (and occurred with our patient).
These ictal episodes can be difficult to identify within the context of an isolated psychiatric diagnosis.5 The distinction can be clarified by the presence of associated somatic symptoms, which in this case included unilateral cramping, paresthesia, and weakness. These symptoms should clue in a practitioner to the possibility of underlying neurologic pathology, which should prompt the ordering of either an EEG or, at minimum, a neurological consultation.
THE TAKEAWAY
This case report shows how anchoring bias can lead to a delay in diagnosis and treatment. Avoidance of this type of bias requires heightened cognitive awareness by medical providers. A more system-based approach is to have structured diagnostic assessments,6 such as conducting a thorough neurological exam for patients with somatic symptoms and exacerbating comorbid psychiatric conditions.
It may also help to review cases like this with colleagues from diverse disciplinary backgrounds, highlighting thought processes and sharing uncertainty.3 These processes may shed light on confounding diagnoses that might be playing a role in a patient’s presentation and ultimately aid in the decision-making process.
CORRESPONDENCE
Paimon Ameli, DO, Naval Medical Center San Diego, 34800 Bob Wilson Drive, San Diego, CA 92134; paimonm@gmail.com
THE CASE
A 28-year-old woman with an extensive psychiatric history—including generalized anxiety disorder, panic disorder, and recent postpartum depression—presented with a chief complaint of right leg weakness. She stated this weakness had begun 4 days earlier. It occurred episodically and was preceded by tingling and cramping sensations. Each episode lasted a couple of minutes and spontaneously resolved. Associated with it, she experienced slurred speech and altered mentation. There was no loss of consciousness and no pain. A panic attack usually followed, consisting of feelings of impending doom, rapid breathing, palpitations, and nausea.
She had 3 prior diagnostic evaluations for this same chief complaint, twice in an emergency department (ED) and once with her primary care physician. These evaluations included lab work and extensive head imaging, which demonstrated no acute intracranial pathology. At each previous presentation, the diagnosis was an exacerbation of her anxiety disorder, and she was treated with lorazepam.
At the current presentation, her vital signs were stable. Examination revealed a notably anxious patient. She repeatedly expressed concern that she might have a brain tumor or some other deadly disease, as she had a family history of brain cancer. Her physical exam was entirely normal, including normal strength, sensation, and reflexes in all extremities.
Further head imaging (computed tomography, CT angiography, and magnetic resonance imaging of the brain) failed to reveal an etiology of her symptoms. With no clear organic cause, her medical providers again suspected an anxiety or panic episode. She was given reassurance, and an outpatient neurology consult was arranged.
THE DIAGNOSIS
One week later, at her outpatient neurology appointment, an electroencephalogram (EEG) was performed. Following photic stimulation, the EEG showed multiple right- and left-hemisphere foci of cortical hyperexcitability including a subtle sharp component (see FIGURE). Immediately following the longest of these episodes, the patient expressed a sense of anxiety and an altered sensorium similar to her prior presentations.
The EEG findings, in addition to the postictal anxiety symptoms and clinical history, were all important components that led the treating neurologist to the diagnosis of localization-related (focal) epilepsy.1 The patient was started on oxcarbazepine, a first-line anti-epileptic medication used in the treatment of focal epilepsy.2 She is being followed by a neurologist regularly and after optimizing her anti-epileptic medication, is no longer having seizures.
DISCUSSION
The difficulty of this case stems from the atypical presentation of the patient’s seizures. The key step to the correct diagnosis was a neurological consultation and an ensuing EEG. However, the patient received a vast spectrum of care, including multiple work-ups, prior to a conclusive diagnosis—which highlights an important issue health care providers must address.
Continue to: The role of bias
The role of bias. From the patient’s initial visits to the ED to her hospital admission, there was a prominent affixation, known as the anchoring bias,3 by the clinicians providing her care: All were focused heavily on her psychiatric features. Conversely, the evaluation for patients with suspected psychiatric diagnoses should focus on successfully ruling out major organic etiology with a broad differential diagnosis. It is crucial for providers to take a step back and make a conscious attempt to avoid fixation on a particular diagnosis, especially when it is psychiatric in nature. This allows the provider to actively consider alternative explanations for a patient presentation and work through a more encompassing differential.
The distinguishing symptoms. There is a common association between comorbid mood disorders (eg, depression, anxiety) and epilepsy.4 Another clue is ictal anxiety or nervousness, which is commonly observed in patients with partial seizures (and occurred with our patient).
These ictal episodes can be difficult to identify within the context of an isolated psychiatric diagnosis.5 The distinction can be clarified by the presence of associated somatic symptoms, which in this case included unilateral cramping, paresthesia, and weakness. These symptoms should clue in a practitioner to the possibility of underlying neurologic pathology, which should prompt the ordering of either an EEG or, at minimum, a neurological consultation.
THE TAKEAWAY
This case report shows how anchoring bias can lead to a delay in diagnosis and treatment. Avoidance of this type of bias requires heightened cognitive awareness by medical providers. A more system-based approach is to have structured diagnostic assessments,6 such as conducting a thorough neurological exam for patients with somatic symptoms and exacerbating comorbid psychiatric conditions.
It may also help to review cases like this with colleagues from diverse disciplinary backgrounds, highlighting thought processes and sharing uncertainty.3 These processes may shed light on confounding diagnoses that might be playing a role in a patient’s presentation and ultimately aid in the decision-making process.
CORRESPONDENCE
Paimon Ameli, DO, Naval Medical Center San Diego, 34800 Bob Wilson Drive, San Diego, CA 92134; paimonm@gmail.com
1. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522-530.
2. Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000-1015.
3. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
4. Jackson MJ, Turkington D. Depression and anxiety in epilepsy. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i45-i47.
5. López-Gómez M, Espinola M, Ramirez-Bermudez J, et al. Clinical presentation of anxiety among patients with epilepsy. Neuropsychiatr Dis Treat. 2008;4:1235-1239.
6. Etchells E. Anchoring bias with critical implications. Published June 2015. Patient Safety Network. https://psnet.ahrq.gov/web-mm/anchoring-bias-critical-implications. Accessed September 29, 2020.
1. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522-530.
2. Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000-1015.
3. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
4. Jackson MJ, Turkington D. Depression and anxiety in epilepsy. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i45-i47.
5. López-Gómez M, Espinola M, Ramirez-Bermudez J, et al. Clinical presentation of anxiety among patients with epilepsy. Neuropsychiatr Dis Treat. 2008;4:1235-1239.
6. Etchells E. Anchoring bias with critical implications. Published June 2015. Patient Safety Network. https://psnet.ahrq.gov/web-mm/anchoring-bias-critical-implications. Accessed September 29, 2020.

Cutaneous T-cell Lymphoma and Concomitant Atopic Dermatitis Responding to Dupilumab
Patients with cutaneous T-cell lymphoma (CTCL) often are diagnosed with atopic dermatitis (AD) or psoriasis before receiving their CTCL diagnosis. The effects of new biologic therapies for AD such as dupilumab, an IL-4/IL-13 antagonist, on CTCL are unknown. Dupilumab may be beneficial in CTCL given that helper T cell (TH2) cytokines are increased in advanced CTCL.1 We present a patient with definitive CTCL and concomitant AD who was safely treated with dupilumab and experienced improvement in both CTCL and AD.
Case Report
A 68-year-old man presented with increased itching from AD and a new rash on the arms, neck, chest, back, and lower extremities (Figures 1A and 2A). He had a medical history of AD and CTCL diagnosed by biopsy and peripheral blood flow cytometry (stage IVA1 [T4N0M0B2]) that was being treated with comprehensive multimodality therapy consisting of bexarotene 375 mg daily, interferon alfa-2b 3 mIU 3 times weekly, interferon gamma-1b 2 mIU 3 times weekly, total skin electron beam therapy followed by narrowband UVB twice weekly, and extracorporeal photopheresis every 4 weeks, which resulted in a partial clinical response for 6 months. A biopsy performed at the current presentation showed focal spongiosis and features of lichen simplex chronicus with no evidence suggestive of CTCL. Peripheral blood flow cytometry showed stable B1-staged disease burden (CD4/CD8, 2.6:1); CD4+/CD7−, 12% [91/µL]; CD4+/CD26−, 21% [155/µL]). Treatment with potent and superpotent topical steroids was attempted for more than 6 months and was unsuccessful in relieving the symptoms.
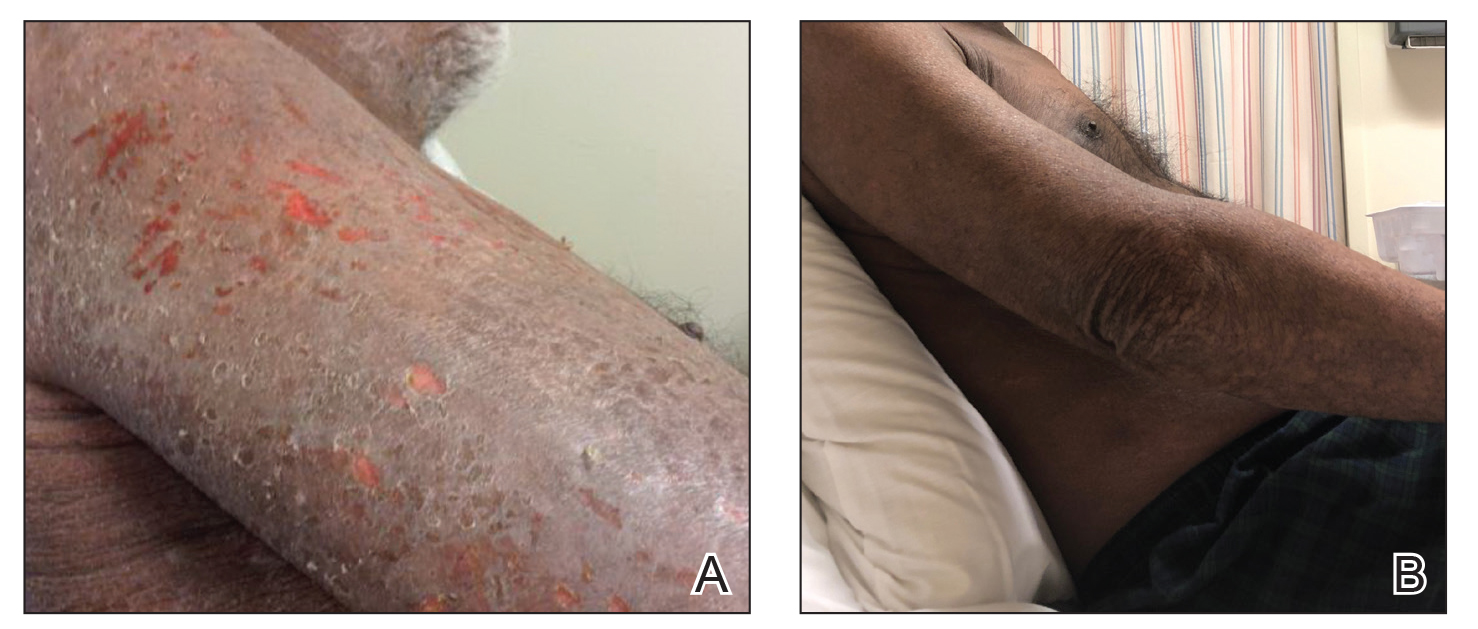
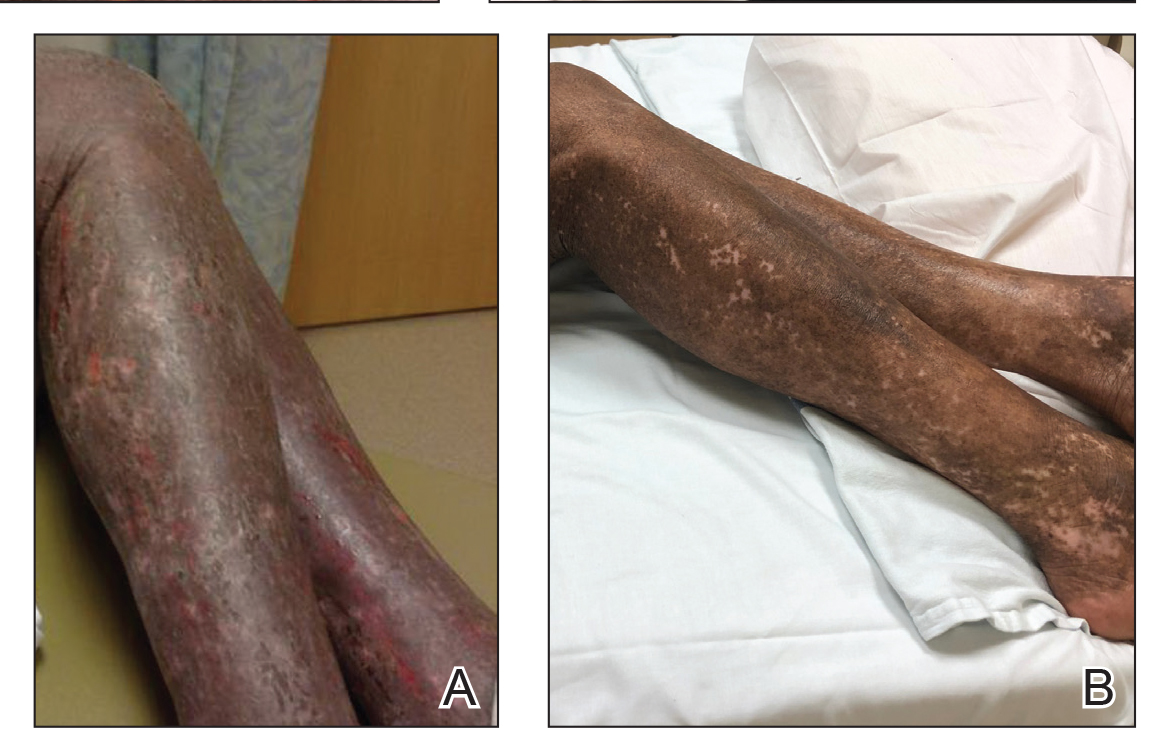
Given the recalcitrant nature of the patient’s rash and itching, dupilumab was added to his CTCL regimen. Prior to initiating dupilumab, the patient reported a numeric rating scale itch intensity of 7 out of 10. After 4 weeks of treatment with dupilumab, the patient reported a numeric rating scale itch intensity of 1. Over a 3-month period, the patient’s rash improved dramatically (Figures 1B and 2B), making it possible to decrease CTCL treatments—bexarotene decreased to 300 mg, interferon alfa-2b to 3 mIU twice weekly, interferon gamma-1b to 2 mIU twice weekly, extracorporeal photopheresis every 5 weeks, and narrowband UVB was discontinued completely. A comparison of the patient’s flow cytometry analysis from before treatment to 3 months after dupilumab showed an overall slight reduction in CTCL B1 blood involvement and normalization of the patient’s absolute eosinophil count and serum lactate dehydrogenase level. The patient tolerated the treatment well without any adverse events and has maintained clinical response for 6 months.
Comment
Cutaneous T-cell lymphomas represent a heterogeneous group of T-cell lymphoproliferative disorders involving the skin.2 The definitive diagnosis of CTCL is challenging, as the clinical and pathologic features often are nonspecific in early disease. Frequently, undiagnosed patients are treated empirically with immunosuppressive agents. Tumor necrosis factor inhibitors and cyclosporine are both associated with progression or worsening of undiagnosed CTCL.3,4 Dupilumab was the first US Food and Drug Administration–approved biologic for the treatment of moderate to severe AD. Cutaneous T-cell lymphoma has immunologic features, such as TH2 skewing, that overlap with AD; however, the effects of dupilumab in CTCL are not yet known.5,6 Our group has seen patients initially thought to have AD who received dupilumab without improvement and were subsequently diagnosed with CTCL, suggesting dupilumab did not affect CTCL tumor cells. Given these findings, there was concern that dupilumab might exacerbate undiagnosed CTCL. Our patient with definitive, severe, refractory CTCL noted marked improvement in both AD and underlying CTCL with the addition of dupilumab. No other treatments were added. The response was so dramatic that we were able to wean the doses and frequencies of several CTCL treatments. Our findings suggest that dupilumab may be beneficial in a certain subset of CTCL patients with a history of AD or known concomitant AD. Prospective studies are needed to fully investigate dupilumab safety and efficacy in CTCL and whether it has any primary effects on tumor burden in addition to benefit for itch and skin symptom relief.
- Guenova E, Watanabe R, Teague JE, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013;19:3755-3763.
- Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:151-165.
- Martinez-Escala ME, Posligua AL, Wickless H, et al. Progression of undiagnosed cutaneous lymphoma after anti-tumor necrosis factor-alpha therapy. J Am Acad Dermatol. 2018;78:1068-1076.
- Pielop JA, Jones D, Duvic M. Transient CD30+ nodal transformation of cutaneous T-cell lymphoma associated with cyclosporine treatment. Int J Dermatol. 2001;40:505-511.
- Saulite I, Hoetzenecker W, Weidinger S, et al. Sézary syndrome and atopic dermatitis: comparison of immunological aspects and targets [published online May 17, 2016]. BioMed Res Int. doi:10.1155/2016/9717530.
- Sigurdsson V, Toonstra J, Bihari IC, et al. Interleukin 4 and interferon-gamma expression of the dermal infiltrate in patients with erythroderma and mycosis fungoides. an immuno-histochemical study. J Cutan Pathol. 2000;27:429-435.
Patients with cutaneous T-cell lymphoma (CTCL) often are diagnosed with atopic dermatitis (AD) or psoriasis before receiving their CTCL diagnosis. The effects of new biologic therapies for AD such as dupilumab, an IL-4/IL-13 antagonist, on CTCL are unknown. Dupilumab may be beneficial in CTCL given that helper T cell (TH2) cytokines are increased in advanced CTCL.1 We present a patient with definitive CTCL and concomitant AD who was safely treated with dupilumab and experienced improvement in both CTCL and AD.
Case Report
A 68-year-old man presented with increased itching from AD and a new rash on the arms, neck, chest, back, and lower extremities (Figures 1A and 2A). He had a medical history of AD and CTCL diagnosed by biopsy and peripheral blood flow cytometry (stage IVA1 [T4N0M0B2]) that was being treated with comprehensive multimodality therapy consisting of bexarotene 375 mg daily, interferon alfa-2b 3 mIU 3 times weekly, interferon gamma-1b 2 mIU 3 times weekly, total skin electron beam therapy followed by narrowband UVB twice weekly, and extracorporeal photopheresis every 4 weeks, which resulted in a partial clinical response for 6 months. A biopsy performed at the current presentation showed focal spongiosis and features of lichen simplex chronicus with no evidence suggestive of CTCL. Peripheral blood flow cytometry showed stable B1-staged disease burden (CD4/CD8, 2.6:1); CD4+/CD7−, 12% [91/µL]; CD4+/CD26−, 21% [155/µL]). Treatment with potent and superpotent topical steroids was attempted for more than 6 months and was unsuccessful in relieving the symptoms.
Given the recalcitrant nature of the patient’s rash and itching, dupilumab was added to his CTCL regimen. Prior to initiating dupilumab, the patient reported a numeric rating scale itch intensity of 7 out of 10. After 4 weeks of treatment with dupilumab, the patient reported a numeric rating scale itch intensity of 1. Over a 3-month period, the patient’s rash improved dramatically (Figures 1B and 2B), making it possible to decrease CTCL treatments—bexarotene decreased to 300 mg, interferon alfa-2b to 3 mIU twice weekly, interferon gamma-1b to 2 mIU twice weekly, extracorporeal photopheresis every 5 weeks, and narrowband UVB was discontinued completely. A comparison of the patient’s flow cytometry analysis from before treatment to 3 months after dupilumab showed an overall slight reduction in CTCL B1 blood involvement and normalization of the patient’s absolute eosinophil count and serum lactate dehydrogenase level. The patient tolerated the treatment well without any adverse events and has maintained clinical response for 6 months.
Comment
Cutaneous T-cell lymphomas represent a heterogeneous group of T-cell lymphoproliferative disorders involving the skin.2 The definitive diagnosis of CTCL is challenging, as the clinical and pathologic features often are nonspecific in early disease. Frequently, undiagnosed patients are treated empirically with immunosuppressive agents. Tumor necrosis factor inhibitors and cyclosporine are both associated with progression or worsening of undiagnosed CTCL.3,4 Dupilumab was the first US Food and Drug Administration–approved biologic for the treatment of moderate to severe AD. Cutaneous T-cell lymphoma has immunologic features, such as TH2 skewing, that overlap with AD; however, the effects of dupilumab in CTCL are not yet known.5,6 Our group has seen patients initially thought to have AD who received dupilumab without improvement and were subsequently diagnosed with CTCL, suggesting dupilumab did not affect CTCL tumor cells. Given these findings, there was concern that dupilumab might exacerbate undiagnosed CTCL. Our patient with definitive, severe, refractory CTCL noted marked improvement in both AD and underlying CTCL with the addition of dupilumab. No other treatments were added. The response was so dramatic that we were able to wean the doses and frequencies of several CTCL treatments. Our findings suggest that dupilumab may be beneficial in a certain subset of CTCL patients with a history of AD or known concomitant AD. Prospective studies are needed to fully investigate dupilumab safety and efficacy in CTCL and whether it has any primary effects on tumor burden in addition to benefit for itch and skin symptom relief.
Patients with cutaneous T-cell lymphoma (CTCL) often are diagnosed with atopic dermatitis (AD) or psoriasis before receiving their CTCL diagnosis. The effects of new biologic therapies for AD such as dupilumab, an IL-4/IL-13 antagonist, on CTCL are unknown. Dupilumab may be beneficial in CTCL given that helper T cell (TH2) cytokines are increased in advanced CTCL.1 We present a patient with definitive CTCL and concomitant AD who was safely treated with dupilumab and experienced improvement in both CTCL and AD.
Case Report
A 68-year-old man presented with increased itching from AD and a new rash on the arms, neck, chest, back, and lower extremities (Figures 1A and 2A). He had a medical history of AD and CTCL diagnosed by biopsy and peripheral blood flow cytometry (stage IVA1 [T4N0M0B2]) that was being treated with comprehensive multimodality therapy consisting of bexarotene 375 mg daily, interferon alfa-2b 3 mIU 3 times weekly, interferon gamma-1b 2 mIU 3 times weekly, total skin electron beam therapy followed by narrowband UVB twice weekly, and extracorporeal photopheresis every 4 weeks, which resulted in a partial clinical response for 6 months. A biopsy performed at the current presentation showed focal spongiosis and features of lichen simplex chronicus with no evidence suggestive of CTCL. Peripheral blood flow cytometry showed stable B1-staged disease burden (CD4/CD8, 2.6:1); CD4+/CD7−, 12% [91/µL]; CD4+/CD26−, 21% [155/µL]). Treatment with potent and superpotent topical steroids was attempted for more than 6 months and was unsuccessful in relieving the symptoms.
Given the recalcitrant nature of the patient’s rash and itching, dupilumab was added to his CTCL regimen. Prior to initiating dupilumab, the patient reported a numeric rating scale itch intensity of 7 out of 10. After 4 weeks of treatment with dupilumab, the patient reported a numeric rating scale itch intensity of 1. Over a 3-month period, the patient’s rash improved dramatically (Figures 1B and 2B), making it possible to decrease CTCL treatments—bexarotene decreased to 300 mg, interferon alfa-2b to 3 mIU twice weekly, interferon gamma-1b to 2 mIU twice weekly, extracorporeal photopheresis every 5 weeks, and narrowband UVB was discontinued completely. A comparison of the patient’s flow cytometry analysis from before treatment to 3 months after dupilumab showed an overall slight reduction in CTCL B1 blood involvement and normalization of the patient’s absolute eosinophil count and serum lactate dehydrogenase level. The patient tolerated the treatment well without any adverse events and has maintained clinical response for 6 months.
Comment
Cutaneous T-cell lymphomas represent a heterogeneous group of T-cell lymphoproliferative disorders involving the skin.2 The definitive diagnosis of CTCL is challenging, as the clinical and pathologic features often are nonspecific in early disease. Frequently, undiagnosed patients are treated empirically with immunosuppressive agents. Tumor necrosis factor inhibitors and cyclosporine are both associated with progression or worsening of undiagnosed CTCL.3,4 Dupilumab was the first US Food and Drug Administration–approved biologic for the treatment of moderate to severe AD. Cutaneous T-cell lymphoma has immunologic features, such as TH2 skewing, that overlap with AD; however, the effects of dupilumab in CTCL are not yet known.5,6 Our group has seen patients initially thought to have AD who received dupilumab without improvement and were subsequently diagnosed with CTCL, suggesting dupilumab did not affect CTCL tumor cells. Given these findings, there was concern that dupilumab might exacerbate undiagnosed CTCL. Our patient with definitive, severe, refractory CTCL noted marked improvement in both AD and underlying CTCL with the addition of dupilumab. No other treatments were added. The response was so dramatic that we were able to wean the doses and frequencies of several CTCL treatments. Our findings suggest that dupilumab may be beneficial in a certain subset of CTCL patients with a history of AD or known concomitant AD. Prospective studies are needed to fully investigate dupilumab safety and efficacy in CTCL and whether it has any primary effects on tumor burden in addition to benefit for itch and skin symptom relief.
- Guenova E, Watanabe R, Teague JE, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013;19:3755-3763.
- Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:151-165.
- Martinez-Escala ME, Posligua AL, Wickless H, et al. Progression of undiagnosed cutaneous lymphoma after anti-tumor necrosis factor-alpha therapy. J Am Acad Dermatol. 2018;78:1068-1076.
- Pielop JA, Jones D, Duvic M. Transient CD30+ nodal transformation of cutaneous T-cell lymphoma associated with cyclosporine treatment. Int J Dermatol. 2001;40:505-511.
- Saulite I, Hoetzenecker W, Weidinger S, et al. Sézary syndrome and atopic dermatitis: comparison of immunological aspects and targets [published online May 17, 2016]. BioMed Res Int. doi:10.1155/2016/9717530.
- Sigurdsson V, Toonstra J, Bihari IC, et al. Interleukin 4 and interferon-gamma expression of the dermal infiltrate in patients with erythroderma and mycosis fungoides. an immuno-histochemical study. J Cutan Pathol. 2000;27:429-435.
- Guenova E, Watanabe R, Teague JE, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013;19:3755-3763.
- Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:151-165.
- Martinez-Escala ME, Posligua AL, Wickless H, et al. Progression of undiagnosed cutaneous lymphoma after anti-tumor necrosis factor-alpha therapy. J Am Acad Dermatol. 2018;78:1068-1076.
- Pielop JA, Jones D, Duvic M. Transient CD30+ nodal transformation of cutaneous T-cell lymphoma associated with cyclosporine treatment. Int J Dermatol. 2001;40:505-511.
- Saulite I, Hoetzenecker W, Weidinger S, et al. Sézary syndrome and atopic dermatitis: comparison of immunological aspects and targets [published online May 17, 2016]. BioMed Res Int. doi:10.1155/2016/9717530.
- Sigurdsson V, Toonstra J, Bihari IC, et al. Interleukin 4 and interferon-gamma expression of the dermal infiltrate in patients with erythroderma and mycosis fungoides. an immuno-histochemical study. J Cutan Pathol. 2000;27:429-435.
Practice Points
- The diagnosis of cutaneous T-cell lymphoma (CTCL), particularly early-stage disease, remains challenging and often requires a combination of serial clinical evaluations as well as laboratory diagnostic examinations.
- Dupilumab and its effect on helper T cell (TH2) skewing may play a role in the future management of CTCL.
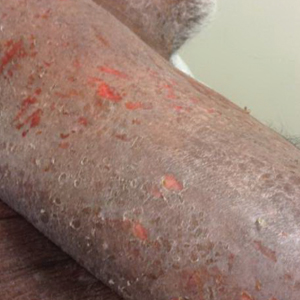
4-year-old girl • limited movement & diffuse pain in both arms • pronated hands • Dx?
THE CASE
A 4-year-old girl was triaged to the Pediatric Emergency Department (PED) Fast Track, complaining of pain and limited movement in both arms. For an unknown reason, she had attempted to lift a heavy, 3-person sofa several hours earlier.
Her prior medical history included left nursemaid elbow (NME) at both 15 months and 33 months of age. Neither event had a known mechanism of injury. In both episodes, it was noted in the medical record that the child was not using her arm, “was holding it funny,” and was complaining of pain. Each time, she presented about 24 hours after symptom onset.
During the physical exam in the PED, the patient showed no signs of acute distress. She held both arms close to her body, with a slight flexion at the elbows, and her hands were pronated. She could not pinpoint the location of her discomfort and described diffuse pain in her forearms, elbows, and upper arms. Examination revealed no localized pain or tenderness in her hands, wrists, or clavicles. Radial pulses were easily palpated, and capillary refill was less than 2 seconds. There was no swelling or bruising. The rest of her physical exam was normal.
DIAGNOSIS
The patient was given a diagnosis of self-inflicted bilateral
DISCUSSION
BNME is an uncommon diagnosis; a literature review of reported cases indicates none were self-inflicted.1-4 However, NME is a common injury and is easily reduced. The classic mechanism of injury for NME involves the elbow in extension, while the forearm is pronated, and a sudden brisk axial traction is applied. This combination of motions causes the annular ligament to slip over the head of the radius and become displaced downward into the radiohumeral joint, where it becomes entrapped. In this case, the patient apparently exerted enough longitudinal traction while trying to lift the couch to produce the injury.
NME occurs most commonly in the left arm of girls between the ages of 4 months and 7 years and peaks at around the age of 2 years.5 A 2014 study by Irie et al6 corroborated the findings on left-side predominance and increased incidence with age, noting that frequency of injury peaked at 6 months in those younger than 1 year of age and at 2 years for those 1 year or older. However, the researchers found no significant sex difference.6
NME is radiographically indistinguishable from a healthy elbow.7 To prevent unnecessary expense and radiation exposure in young children, prereduction radiographs should only be used to rule out the possibility of fracture or other injury.7 Krul et al8 recommend restricting x-ray use to cases with an unclear history or those that are due to trauma other than an arm pull.
Continue to: Methods of reduction
Methods of reduction. Once NME is diagnosed, there are 2 methods of reduction: hyper-pronation and supination-flexion. Reduction is best performed with the child sitting in the parent’s lap with the injured arm facing the examiner.
Success rates for both methods of NME reduction are statistically similar; however, first-attempt success rates are significantly higher with the hyper-pronation method than with supination-flexion.9 Furthermore, physicians have deemed the hyper-pronation method significantly easier to perform than supination-flexion.9 A Cochrane review by Krul et al10 concluded that the hyper-pronation method may result in lower failure rates than supination-flexion, but due to limited evidence, the researchers were unable to draw any conclusions on other outcomes, such as pain. Green et al11 noted that hyper-pronation is perceived by parents of children with NME as being less painful. For these reasons, hyper-pronation should be utilized as the first method of reduction, followed by supination-flexion if the former does not work.12
Additional management. In a limited study of 50 children with pulled-elbow injuries, ultrasound revealed that 78% had an intact yet interposed radial annular ligament and 22% had a tear in the radial annular ligament.13 The authors propose that if, after appropriate reduction methods are attempted, no pop is felt, or there is no prompt clinical improvement, and ultrasound is not available to assess the integrity of the annular ligament, the child should be placed in a splint for 7 days
Our patient returned to the PED 3 days later, complaining of pain and an inability to move her left arm after her older sibling pulled her by her outstretched arms. She was once again diagnosed with NME, the injury was reduced, and she was using the arm within minutes. She has not presented to either the PED or the pediatric clinic with a similar complaint since. Discarding outliers, NME recurrence rates fall within a range of 23.7% to 32.9%.14,15
THE TAKEAWAY
Pre-reduction x-rays are not warranted in cases of NME unless there is suspicion for fracture or another injury. The 2 reduction methods, hyper-pronation and supination-flexion, are easily mastered. Any reduction should be quick, easy, and as painless as possible. Hyper-pronation should be utilized first, as this maneuver seems to be the more successful and is perceived by parents as being less painful. However, it is always most helpful to be proficient in both methods. If, after appropriate attempts at reduction, the child has not regained the use of the arm, 7 days of splinting is recommended, along with an orthopedic referral.
CORRESPONDENCE
Robert N. Anderson, DNP, APRN, (F)NP-C, ENP-BC, Vanderbilt Health, 512 Autumn Springs Court, Suite 100 C, Franklin, TN 37067; bob.anderson@vumc.org
1. Quan L, Marcuse EK. The epidemiology and treatment of radial head subluxation. Am J Dis Child. 1985;139:1194-1197.
2. Michaels MG. A case of bilateral nursemaid’s elbow. Pediatr Emerg Care. 1989;5:226-227.
3. Meiner EV, Sama AE, Lee DC, et al. Bilateral nursemaid’s elbow. Am J Emerg Med. 2004;6:502-503.
4. Wang YX, Zhang G, Song B, et al. Radial head subluxation in pediatric clinics and emergency departments in China. Chin J Traum. 2019;22:340-344.
5. Schunk JE. Radial head subluxation: epidemiology and treatment of 87 episodes. Ann Emerg Med. 1990;19:1019-1023.
6. Irie T, Sono T, Hayama Y, et al. Investigation on 2331 cases of pulled elbow over the last 10 years. Pediatr Rep. 2014;6:5090. doi: 10.4081/pr.2014.5090
7. Eismann EA, Cosco ED, Wall EJ. Absence of radiographic abnormalities in nursemaid’s elbows. J Pediatr Orthop. 2014;34:426-431.
8. Krul M, van der Wouden JC, Koes BW, et al. Nursemaid’s Elbow: its diagnostic clues and preferred means of reduction. J Fam Pract. 2010:59:E5-E7.
9. Bek D, Yildiz C, Köse O, et al. Pronation versus supination maneuvers for the reduction of ‘pulled elbow’: a randomized clinical trial. Eur J Emerg Med. 2009;16:135-138.
10. Krul M, van der Wouden JC, Kruithof EJ, et al. Manipulative interventions for reducing pulled elbow in young children. Cochrane Database Syst Rev. 2017.
11. Green DA, Linares MYR, Garcia Peña BM, et al. Randomized comparison of pain perception during radial head subluxation reduction using supination-flexion of forced pronation. Pediatr Emerg Care. 2006;22:235-238.
12. García-Mata S, Hidalgo-Ovejero A. Efficacy of reduction maneuvers for “pulled elbow” in children: a prospective study of 115 cases. J Pediatr Orthop. 2014;34:432-436.
13. Diab HS, Hamed MMS, Allam Y. Obscure pathology of pulled elbow: dynamic high-resolution ultrasound-assisted classification. J Child Orthop. 2010;4:539-543.
14. Teach SJ, Schutzman SA. Prospective study of recurrent radial head subluxation. Arch Pediatr Adolesc Med. 1996;150:164-166.
15. Macias CG, Bothner J, Wiebe R. Comparison of supination/flexion to hyperpronation in the reduction of radial head subluxation. Pediatrics. 1998;102:E10. doi: 10.1542/peds.102.1.e10.
THE CASE
A 4-year-old girl was triaged to the Pediatric Emergency Department (PED) Fast Track, complaining of pain and limited movement in both arms. For an unknown reason, she had attempted to lift a heavy, 3-person sofa several hours earlier.
Her prior medical history included left nursemaid elbow (NME) at both 15 months and 33 months of age. Neither event had a known mechanism of injury. In both episodes, it was noted in the medical record that the child was not using her arm, “was holding it funny,” and was complaining of pain. Each time, she presented about 24 hours after symptom onset.
During the physical exam in the PED, the patient showed no signs of acute distress. She held both arms close to her body, with a slight flexion at the elbows, and her hands were pronated. She could not pinpoint the location of her discomfort and described diffuse pain in her forearms, elbows, and upper arms. Examination revealed no localized pain or tenderness in her hands, wrists, or clavicles. Radial pulses were easily palpated, and capillary refill was less than 2 seconds. There was no swelling or bruising. The rest of her physical exam was normal.
DIAGNOSIS
The patient was given a diagnosis of self-inflicted bilateral
DISCUSSION
BNME is an uncommon diagnosis; a literature review of reported cases indicates none were self-inflicted.1-4 However, NME is a common injury and is easily reduced. The classic mechanism of injury for NME involves the elbow in extension, while the forearm is pronated, and a sudden brisk axial traction is applied. This combination of motions causes the annular ligament to slip over the head of the radius and become displaced downward into the radiohumeral joint, where it becomes entrapped. In this case, the patient apparently exerted enough longitudinal traction while trying to lift the couch to produce the injury.
NME occurs most commonly in the left arm of girls between the ages of 4 months and 7 years and peaks at around the age of 2 years.5 A 2014 study by Irie et al6 corroborated the findings on left-side predominance and increased incidence with age, noting that frequency of injury peaked at 6 months in those younger than 1 year of age and at 2 years for those 1 year or older. However, the researchers found no significant sex difference.6
NME is radiographically indistinguishable from a healthy elbow.7 To prevent unnecessary expense and radiation exposure in young children, prereduction radiographs should only be used to rule out the possibility of fracture or other injury.7 Krul et al8 recommend restricting x-ray use to cases with an unclear history or those that are due to trauma other than an arm pull.
Continue to: Methods of reduction
Methods of reduction. Once NME is diagnosed, there are 2 methods of reduction: hyper-pronation and supination-flexion. Reduction is best performed with the child sitting in the parent’s lap with the injured arm facing the examiner.
Success rates for both methods of NME reduction are statistically similar; however, first-attempt success rates are significantly higher with the hyper-pronation method than with supination-flexion.9 Furthermore, physicians have deemed the hyper-pronation method significantly easier to perform than supination-flexion.9 A Cochrane review by Krul et al10 concluded that the hyper-pronation method may result in lower failure rates than supination-flexion, but due to limited evidence, the researchers were unable to draw any conclusions on other outcomes, such as pain. Green et al11 noted that hyper-pronation is perceived by parents of children with NME as being less painful. For these reasons, hyper-pronation should be utilized as the first method of reduction, followed by supination-flexion if the former does not work.12
Additional management. In a limited study of 50 children with pulled-elbow injuries, ultrasound revealed that 78% had an intact yet interposed radial annular ligament and 22% had a tear in the radial annular ligament.13 The authors propose that if, after appropriate reduction methods are attempted, no pop is felt, or there is no prompt clinical improvement, and ultrasound is not available to assess the integrity of the annular ligament, the child should be placed in a splint for 7 days
Our patient returned to the PED 3 days later, complaining of pain and an inability to move her left arm after her older sibling pulled her by her outstretched arms. She was once again diagnosed with NME, the injury was reduced, and she was using the arm within minutes. She has not presented to either the PED or the pediatric clinic with a similar complaint since. Discarding outliers, NME recurrence rates fall within a range of 23.7% to 32.9%.14,15
THE TAKEAWAY
Pre-reduction x-rays are not warranted in cases of NME unless there is suspicion for fracture or another injury. The 2 reduction methods, hyper-pronation and supination-flexion, are easily mastered. Any reduction should be quick, easy, and as painless as possible. Hyper-pronation should be utilized first, as this maneuver seems to be the more successful and is perceived by parents as being less painful. However, it is always most helpful to be proficient in both methods. If, after appropriate attempts at reduction, the child has not regained the use of the arm, 7 days of splinting is recommended, along with an orthopedic referral.
CORRESPONDENCE
Robert N. Anderson, DNP, APRN, (F)NP-C, ENP-BC, Vanderbilt Health, 512 Autumn Springs Court, Suite 100 C, Franklin, TN 37067; bob.anderson@vumc.org
THE CASE
A 4-year-old girl was triaged to the Pediatric Emergency Department (PED) Fast Track, complaining of pain and limited movement in both arms. For an unknown reason, she had attempted to lift a heavy, 3-person sofa several hours earlier.
Her prior medical history included left nursemaid elbow (NME) at both 15 months and 33 months of age. Neither event had a known mechanism of injury. In both episodes, it was noted in the medical record that the child was not using her arm, “was holding it funny,” and was complaining of pain. Each time, she presented about 24 hours after symptom onset.
During the physical exam in the PED, the patient showed no signs of acute distress. She held both arms close to her body, with a slight flexion at the elbows, and her hands were pronated. She could not pinpoint the location of her discomfort and described diffuse pain in her forearms, elbows, and upper arms. Examination revealed no localized pain or tenderness in her hands, wrists, or clavicles. Radial pulses were easily palpated, and capillary refill was less than 2 seconds. There was no swelling or bruising. The rest of her physical exam was normal.
DIAGNOSIS
The patient was given a diagnosis of self-inflicted bilateral
DISCUSSION
BNME is an uncommon diagnosis; a literature review of reported cases indicates none were self-inflicted.1-4 However, NME is a common injury and is easily reduced. The classic mechanism of injury for NME involves the elbow in extension, while the forearm is pronated, and a sudden brisk axial traction is applied. This combination of motions causes the annular ligament to slip over the head of the radius and become displaced downward into the radiohumeral joint, where it becomes entrapped. In this case, the patient apparently exerted enough longitudinal traction while trying to lift the couch to produce the injury.
NME occurs most commonly in the left arm of girls between the ages of 4 months and 7 years and peaks at around the age of 2 years.5 A 2014 study by Irie et al6 corroborated the findings on left-side predominance and increased incidence with age, noting that frequency of injury peaked at 6 months in those younger than 1 year of age and at 2 years for those 1 year or older. However, the researchers found no significant sex difference.6
NME is radiographically indistinguishable from a healthy elbow.7 To prevent unnecessary expense and radiation exposure in young children, prereduction radiographs should only be used to rule out the possibility of fracture or other injury.7 Krul et al8 recommend restricting x-ray use to cases with an unclear history or those that are due to trauma other than an arm pull.
Continue to: Methods of reduction
Methods of reduction. Once NME is diagnosed, there are 2 methods of reduction: hyper-pronation and supination-flexion. Reduction is best performed with the child sitting in the parent’s lap with the injured arm facing the examiner.
Success rates for both methods of NME reduction are statistically similar; however, first-attempt success rates are significantly higher with the hyper-pronation method than with supination-flexion.9 Furthermore, physicians have deemed the hyper-pronation method significantly easier to perform than supination-flexion.9 A Cochrane review by Krul et al10 concluded that the hyper-pronation method may result in lower failure rates than supination-flexion, but due to limited evidence, the researchers were unable to draw any conclusions on other outcomes, such as pain. Green et al11 noted that hyper-pronation is perceived by parents of children with NME as being less painful. For these reasons, hyper-pronation should be utilized as the first method of reduction, followed by supination-flexion if the former does not work.12
Additional management. In a limited study of 50 children with pulled-elbow injuries, ultrasound revealed that 78% had an intact yet interposed radial annular ligament and 22% had a tear in the radial annular ligament.13 The authors propose that if, after appropriate reduction methods are attempted, no pop is felt, or there is no prompt clinical improvement, and ultrasound is not available to assess the integrity of the annular ligament, the child should be placed in a splint for 7 days
Our patient returned to the PED 3 days later, complaining of pain and an inability to move her left arm after her older sibling pulled her by her outstretched arms. She was once again diagnosed with NME, the injury was reduced, and she was using the arm within minutes. She has not presented to either the PED or the pediatric clinic with a similar complaint since. Discarding outliers, NME recurrence rates fall within a range of 23.7% to 32.9%.14,15
THE TAKEAWAY
Pre-reduction x-rays are not warranted in cases of NME unless there is suspicion for fracture or another injury. The 2 reduction methods, hyper-pronation and supination-flexion, are easily mastered. Any reduction should be quick, easy, and as painless as possible. Hyper-pronation should be utilized first, as this maneuver seems to be the more successful and is perceived by parents as being less painful. However, it is always most helpful to be proficient in both methods. If, after appropriate attempts at reduction, the child has not regained the use of the arm, 7 days of splinting is recommended, along with an orthopedic referral.
CORRESPONDENCE
Robert N. Anderson, DNP, APRN, (F)NP-C, ENP-BC, Vanderbilt Health, 512 Autumn Springs Court, Suite 100 C, Franklin, TN 37067; bob.anderson@vumc.org
1. Quan L, Marcuse EK. The epidemiology and treatment of radial head subluxation. Am J Dis Child. 1985;139:1194-1197.
2. Michaels MG. A case of bilateral nursemaid’s elbow. Pediatr Emerg Care. 1989;5:226-227.
3. Meiner EV, Sama AE, Lee DC, et al. Bilateral nursemaid’s elbow. Am J Emerg Med. 2004;6:502-503.
4. Wang YX, Zhang G, Song B, et al. Radial head subluxation in pediatric clinics and emergency departments in China. Chin J Traum. 2019;22:340-344.
5. Schunk JE. Radial head subluxation: epidemiology and treatment of 87 episodes. Ann Emerg Med. 1990;19:1019-1023.
6. Irie T, Sono T, Hayama Y, et al. Investigation on 2331 cases of pulled elbow over the last 10 years. Pediatr Rep. 2014;6:5090. doi: 10.4081/pr.2014.5090
7. Eismann EA, Cosco ED, Wall EJ. Absence of radiographic abnormalities in nursemaid’s elbows. J Pediatr Orthop. 2014;34:426-431.
8. Krul M, van der Wouden JC, Koes BW, et al. Nursemaid’s Elbow: its diagnostic clues and preferred means of reduction. J Fam Pract. 2010:59:E5-E7.
9. Bek D, Yildiz C, Köse O, et al. Pronation versus supination maneuvers for the reduction of ‘pulled elbow’: a randomized clinical trial. Eur J Emerg Med. 2009;16:135-138.
10. Krul M, van der Wouden JC, Kruithof EJ, et al. Manipulative interventions for reducing pulled elbow in young children. Cochrane Database Syst Rev. 2017.
11. Green DA, Linares MYR, Garcia Peña BM, et al. Randomized comparison of pain perception during radial head subluxation reduction using supination-flexion of forced pronation. Pediatr Emerg Care. 2006;22:235-238.
12. García-Mata S, Hidalgo-Ovejero A. Efficacy of reduction maneuvers for “pulled elbow” in children: a prospective study of 115 cases. J Pediatr Orthop. 2014;34:432-436.
13. Diab HS, Hamed MMS, Allam Y. Obscure pathology of pulled elbow: dynamic high-resolution ultrasound-assisted classification. J Child Orthop. 2010;4:539-543.
14. Teach SJ, Schutzman SA. Prospective study of recurrent radial head subluxation. Arch Pediatr Adolesc Med. 1996;150:164-166.
15. Macias CG, Bothner J, Wiebe R. Comparison of supination/flexion to hyperpronation in the reduction of radial head subluxation. Pediatrics. 1998;102:E10. doi: 10.1542/peds.102.1.e10.
1. Quan L, Marcuse EK. The epidemiology and treatment of radial head subluxation. Am J Dis Child. 1985;139:1194-1197.
2. Michaels MG. A case of bilateral nursemaid’s elbow. Pediatr Emerg Care. 1989;5:226-227.
3. Meiner EV, Sama AE, Lee DC, et al. Bilateral nursemaid’s elbow. Am J Emerg Med. 2004;6:502-503.
4. Wang YX, Zhang G, Song B, et al. Radial head subluxation in pediatric clinics and emergency departments in China. Chin J Traum. 2019;22:340-344.
5. Schunk JE. Radial head subluxation: epidemiology and treatment of 87 episodes. Ann Emerg Med. 1990;19:1019-1023.
6. Irie T, Sono T, Hayama Y, et al. Investigation on 2331 cases of pulled elbow over the last 10 years. Pediatr Rep. 2014;6:5090. doi: 10.4081/pr.2014.5090
7. Eismann EA, Cosco ED, Wall EJ. Absence of radiographic abnormalities in nursemaid’s elbows. J Pediatr Orthop. 2014;34:426-431.
8. Krul M, van der Wouden JC, Koes BW, et al. Nursemaid’s Elbow: its diagnostic clues and preferred means of reduction. J Fam Pract. 2010:59:E5-E7.
9. Bek D, Yildiz C, Köse O, et al. Pronation versus supination maneuvers for the reduction of ‘pulled elbow’: a randomized clinical trial. Eur J Emerg Med. 2009;16:135-138.
10. Krul M, van der Wouden JC, Kruithof EJ, et al. Manipulative interventions for reducing pulled elbow in young children. Cochrane Database Syst Rev. 2017.
11. Green DA, Linares MYR, Garcia Peña BM, et al. Randomized comparison of pain perception during radial head subluxation reduction using supination-flexion of forced pronation. Pediatr Emerg Care. 2006;22:235-238.
12. García-Mata S, Hidalgo-Ovejero A. Efficacy of reduction maneuvers for “pulled elbow” in children: a prospective study of 115 cases. J Pediatr Orthop. 2014;34:432-436.
13. Diab HS, Hamed MMS, Allam Y. Obscure pathology of pulled elbow: dynamic high-resolution ultrasound-assisted classification. J Child Orthop. 2010;4:539-543.
14. Teach SJ, Schutzman SA. Prospective study of recurrent radial head subluxation. Arch Pediatr Adolesc Med. 1996;150:164-166.
15. Macias CG, Bothner J, Wiebe R. Comparison of supination/flexion to hyperpronation in the reduction of radial head subluxation. Pediatrics. 1998;102:E10. doi: 10.1542/peds.102.1.e10.

45-year-old man • fever • generalized rash • recent history of calcaneal osteomyelitis • Dx?
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).

Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).

Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The

Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; w.jacob.cobb@gmail.com
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).
Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).
Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The
Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; w.jacob.cobb@gmail.com
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).
Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).
Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The
Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; w.jacob.cobb@gmail.com
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.