User login
CVS in an Adult Patient
We present a 26‐year‐old white male with a chief complaint of nausea and vomiting. The patient described prodromal nausea followed by intractable vomiting for 2 days. Over the past 2 years he has experienced similar episodes occurring every 3 to 6 months. He has been hospitalized 5 times for this problem with no diagnosis given. There are no obvious precipitants. The symptoms consistently last 2 to 3 days and resolve with supportive care including intravenous fluids and antiemetics. The patient enjoys good health between the periods of sickness. He has never experienced coffee‐ground emesis or hematemesis. His past medical history is significant for attention deficit disorder and cholecystectomy. He takes no prescription medications. Social history is remarkable for tobacco abuse, binge drinking on weekends, and daily marijuana use. He is unemployed. His family history is unremarkable.
Physical examination at the time of admission was notable for tachycardia, orthostatic hypotension, and hypoactive bowel sounds. Otherwise physical examination was normal.
Diagnostic testing done on admission was notable for white blood cell count of 25,000, hemoglobin of 17.3, blood urea nitrogen 18, creatinine 1.4, aspartate aminotransferase (AST) 64, and alanine aminotransferase (ALT) 55. Pancreatic enzymes and acute abdominal series were normal.
The patient was admitted to the hospital with the presumptive diagnosis of viral gastroenteritis. Initial therapy included intravenous fluids and promethazine. Throughout hospital day 1, he remained nauseated and had multiple bouts of emesis. Records from the patient's hospitalization 5 months ago were obtained and reviewed. During this previous hospitalization, computed tomography (CT) scans of the abdomen and esophagogastroduodenoscopy (EGD) were performed, both of which were negative. Upon review of this recent workup, the diagnosis of cyclic vomiting syndrome (CVS) was entertained and the patient received a therapeutic trial of subcutaneous sumatriptan. His symptoms abated dramatically. Subsequently, he was able to keep oral liquids down and his orthostatic hypotension resolved. On hospital day 2, his white blood cell count normalized without intervention. Blood, urine, and stool cultures remained negative, and workup for acute intermittent porphyria was negative. Upon discharge from the hospital he was counseled to discontinue all marijuana use and was scheduled for follow‐up in the residents' clinic. He failed to keep this appointment. After being lost to follow‐up for 17 months, he presented to the emergency department with nausea and vomiting. As before, his symptoms promptly improved with sumatriptan.
Discussion
CVS, initially described in 1861 as a pediatric illness, is being increasingly recognized in adults.1 It has been estimated that up to 1.6% of children experience symptoms consistent with this disorder, but the prevalence in adults is unknown.2 The essential features of CVS, as noted in our patient, are multiple discrete episodes of nausea and vomiting lasting less than 1 week with absence of nausea and vomiting between episodes. The presentation of adults with CVS often differs from the pediatric form in that adults have longer, less frequent episodes, and the triggers are less evident.3
The etiology and pathogenesis of CVS remain unknown. A variety of physical and psychological stresses, including infection, overexertion, and emotional distress, have been noted to precipitate episodes.4 CVS has variably been associated with autonomic, mitochondrial, and endocrine disorders. The most prevalent theory in the literature, however, is that CVS and migraine headaches are different presentations of the same diathesis.5 Patients with both are noted to have similar patterns of symptoms and positive family history of migraines. The progression from CVS to migraines is noted frequently in individual patients. As many as 82% of the 214 children in a case series of CVS were noted to have a family history of migraines or to have or subsequently develop migraines.6 In addition, electroencephalogram findings and adrenergic autonomic abnormalities are similar in both sets of patients.3 In 1 case series of 17 patients with CVS, patients noted the possible association of episodes with menses (in 57% of women of reproductive age), and the improvement of symptoms with sleep (in 24%), clinical factors common in patients with migraines.3
CVS is one of the functional gastrointestinal disorders for which the diagnosis is clinical, with criteria based upon the consensus of expert opinion in the Rome III Criteria for Functional Gastrointestinal (GI) Disorders.7 At least 3 months, with onset at least 6 months previously of:
-
Stereotypical episodes of vomiting regarding onset (acute) and duration (less than 1 week);
-
3 or more discrete episodes in the prior year; and
-
Absence of nausea and vomiting between episodes.
Supportive criteria: History of migraine headaches or family history of migraine headaches.7
Making the diagnosis of CVS requires the exclusion of other disorders associated with recurrent vomiting. Examples include gastric outlet or small bowel obstruction, gastroparesis, vestibular neuritis, elevated intracranial pressure, inborn errors of metabolism, dysautonomia, porphyria, and alterations in the hypothalamic pituitary adrenal axis. The other functional nausea and vomiting disorders described in Rome III, specifically chronic idiopathic nausea and functional vomiting, also need to be considered.7 Many drugs can cause nausea and vomiting, and chronic marijuana use has been associated with cyclical hyperemesis.8 Our patient meets the diagnostic criteria for CVS, but his frequent marijuana use would preclude a diagnosis of functional vomiting, which by definition requires an absence of chronic cannabinoid use.
Determining which tests and procedures should be performed in the initial evaluation is based on clinical judgment, but commonly includes complete metabolic profile, urinalysis, upper GI series, EGD, neurological imaging, acute abdominal series, and CT of the abdomen and pelvis. In addition, pertinent metabolic screening including serum lactate, cortisol, pyruvate, ammonia, creatinine phosphokinase, carnitine, urinary organic acids, and porphobilinogen may be considered.5
Evidence‐based treatment of CVS is limited by the lack of controlled trials. Acutely, patients often require hospitalization and symptom management with aggressive hydration, antiemetics, and sometimes even sedative agents. Empiric abortive treatment with antimigraine mediations (sumitriptan, prochlorperazine, tricyclic antidepressants, and ketorolac) has been effective in case reports.911 Patients in whom a history of chronic cannabinoid use is elicited should be counseled that cessation may lead to an improvement in symptoms.
Just as with migraines, patients who experience frequent episodes of cyclic vomiting can benefit from prophylactic medications. Tricyclic antidepressants (TCAs) have been reported to be effective as prophylactic agents in children with CVS.12 An open‐label treatment group of 17 adult patients with CVS noted that 17% of patients had a complete remission with TCA therapy and almost 60% had a partial response.3 More recently, a retrospective case series of patients who had failed TCAs as maintenance therapy reported that 15 out of the 20 patients studied had improvement in the frequency of their vomiting episodes with the newer antiepileptic drugs zonisamide and levetiracem. However, moderate or severe side effects were reported in 45%.13
Conclusions
In summary, although CVS is still an uncommon diagnosis, it is being made more frequently in adults. Although recognition is increasing, there remains a significant delay between onset of symptoms and diagnosis in adults.4 CVS is a diagnosis of exclusion and should be considered when initial evaluation for recurrent nausea and vomiting are unrevealing. A wide range of medications show benefit for both abortive and prophylactic therapy. Increasing awareness of this disorder can lead to a reduction in invasive and costly diagnostic workups.
- .Evrose de la digestion, caracteriseo par des crises periodiques de vomissements et une profonde modification de l'assimilation.Gazette Medicale de Paris1861:312. [French]
- ,.Cyclical vomiting syndrome in children: a population‐based study.J Pediatr Gastroenterol Nutr.1995;21(4):454–458.
- ,.Cyclic vomiting syndrome in adults: clinical features and response to tricyclic antidepressants.Am J Gastroenterol.1999;94(10):2855–2860.
- ,,,.Cyclic vomiting syndrome in 41 adults: the illness, the patients, and problems of management.BMC Med.2005;3:20.
- ,,.Consensus statement—2nd International Scientific Symposium on CVS. The Faculty of The 2nd International Scientific Symposium on Cyclic Vomiting Syndrome.Dig Dis Sci.1999;44(8 suppl):9S–11S.
- ,,,,.Is cyclic vomiting syndrome related to migraine?J Pediatr.1999;134(5):567–572.
- ,,, et al.Functional gastroduodenal disorders.Gastroenterology.2006;130:1466–1479.
- ,,,.Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis use.Gut.2004;53:1566–1570.
- ,,.Sumatriptan in the treatment of cyclic vomiting.Ann Pharmacother.1995;29(10):997–999.
- .Recurrent vomiting in adults. A syndrome?Med J Aust.1983;1(7):329–331.
- ,,,,.Cyclic vomiting: association with multiple homeostatic abnormalities and response to ketorolac.Am J Gastroenterol.1996;91(10):2228–2232.
- .Effective prophylactic therapy for cyclic vomiting syndrome in children using amitriptyline or cyproheptadine.Pediatrics.1997;100(6):977–981.
- ,,,.Zonisamide or levetiracetam for adults with cyclic vomiting syndrome: a case series.Clin Gastroenterol Hepatol.2007;5(1):44–48.
We present a 26‐year‐old white male with a chief complaint of nausea and vomiting. The patient described prodromal nausea followed by intractable vomiting for 2 days. Over the past 2 years he has experienced similar episodes occurring every 3 to 6 months. He has been hospitalized 5 times for this problem with no diagnosis given. There are no obvious precipitants. The symptoms consistently last 2 to 3 days and resolve with supportive care including intravenous fluids and antiemetics. The patient enjoys good health between the periods of sickness. He has never experienced coffee‐ground emesis or hematemesis. His past medical history is significant for attention deficit disorder and cholecystectomy. He takes no prescription medications. Social history is remarkable for tobacco abuse, binge drinking on weekends, and daily marijuana use. He is unemployed. His family history is unremarkable.
Physical examination at the time of admission was notable for tachycardia, orthostatic hypotension, and hypoactive bowel sounds. Otherwise physical examination was normal.
Diagnostic testing done on admission was notable for white blood cell count of 25,000, hemoglobin of 17.3, blood urea nitrogen 18, creatinine 1.4, aspartate aminotransferase (AST) 64, and alanine aminotransferase (ALT) 55. Pancreatic enzymes and acute abdominal series were normal.
The patient was admitted to the hospital with the presumptive diagnosis of viral gastroenteritis. Initial therapy included intravenous fluids and promethazine. Throughout hospital day 1, he remained nauseated and had multiple bouts of emesis. Records from the patient's hospitalization 5 months ago were obtained and reviewed. During this previous hospitalization, computed tomography (CT) scans of the abdomen and esophagogastroduodenoscopy (EGD) were performed, both of which were negative. Upon review of this recent workup, the diagnosis of cyclic vomiting syndrome (CVS) was entertained and the patient received a therapeutic trial of subcutaneous sumatriptan. His symptoms abated dramatically. Subsequently, he was able to keep oral liquids down and his orthostatic hypotension resolved. On hospital day 2, his white blood cell count normalized without intervention. Blood, urine, and stool cultures remained negative, and workup for acute intermittent porphyria was negative. Upon discharge from the hospital he was counseled to discontinue all marijuana use and was scheduled for follow‐up in the residents' clinic. He failed to keep this appointment. After being lost to follow‐up for 17 months, he presented to the emergency department with nausea and vomiting. As before, his symptoms promptly improved with sumatriptan.
Discussion
CVS, initially described in 1861 as a pediatric illness, is being increasingly recognized in adults.1 It has been estimated that up to 1.6% of children experience symptoms consistent with this disorder, but the prevalence in adults is unknown.2 The essential features of CVS, as noted in our patient, are multiple discrete episodes of nausea and vomiting lasting less than 1 week with absence of nausea and vomiting between episodes. The presentation of adults with CVS often differs from the pediatric form in that adults have longer, less frequent episodes, and the triggers are less evident.3
The etiology and pathogenesis of CVS remain unknown. A variety of physical and psychological stresses, including infection, overexertion, and emotional distress, have been noted to precipitate episodes.4 CVS has variably been associated with autonomic, mitochondrial, and endocrine disorders. The most prevalent theory in the literature, however, is that CVS and migraine headaches are different presentations of the same diathesis.5 Patients with both are noted to have similar patterns of symptoms and positive family history of migraines. The progression from CVS to migraines is noted frequently in individual patients. As many as 82% of the 214 children in a case series of CVS were noted to have a family history of migraines or to have or subsequently develop migraines.6 In addition, electroencephalogram findings and adrenergic autonomic abnormalities are similar in both sets of patients.3 In 1 case series of 17 patients with CVS, patients noted the possible association of episodes with menses (in 57% of women of reproductive age), and the improvement of symptoms with sleep (in 24%), clinical factors common in patients with migraines.3
CVS is one of the functional gastrointestinal disorders for which the diagnosis is clinical, with criteria based upon the consensus of expert opinion in the Rome III Criteria for Functional Gastrointestinal (GI) Disorders.7 At least 3 months, with onset at least 6 months previously of:
-
Stereotypical episodes of vomiting regarding onset (acute) and duration (less than 1 week);
-
3 or more discrete episodes in the prior year; and
-
Absence of nausea and vomiting between episodes.
Supportive criteria: History of migraine headaches or family history of migraine headaches.7
Making the diagnosis of CVS requires the exclusion of other disorders associated with recurrent vomiting. Examples include gastric outlet or small bowel obstruction, gastroparesis, vestibular neuritis, elevated intracranial pressure, inborn errors of metabolism, dysautonomia, porphyria, and alterations in the hypothalamic pituitary adrenal axis. The other functional nausea and vomiting disorders described in Rome III, specifically chronic idiopathic nausea and functional vomiting, also need to be considered.7 Many drugs can cause nausea and vomiting, and chronic marijuana use has been associated with cyclical hyperemesis.8 Our patient meets the diagnostic criteria for CVS, but his frequent marijuana use would preclude a diagnosis of functional vomiting, which by definition requires an absence of chronic cannabinoid use.
Determining which tests and procedures should be performed in the initial evaluation is based on clinical judgment, but commonly includes complete metabolic profile, urinalysis, upper GI series, EGD, neurological imaging, acute abdominal series, and CT of the abdomen and pelvis. In addition, pertinent metabolic screening including serum lactate, cortisol, pyruvate, ammonia, creatinine phosphokinase, carnitine, urinary organic acids, and porphobilinogen may be considered.5
Evidence‐based treatment of CVS is limited by the lack of controlled trials. Acutely, patients often require hospitalization and symptom management with aggressive hydration, antiemetics, and sometimes even sedative agents. Empiric abortive treatment with antimigraine mediations (sumitriptan, prochlorperazine, tricyclic antidepressants, and ketorolac) has been effective in case reports.911 Patients in whom a history of chronic cannabinoid use is elicited should be counseled that cessation may lead to an improvement in symptoms.
Just as with migraines, patients who experience frequent episodes of cyclic vomiting can benefit from prophylactic medications. Tricyclic antidepressants (TCAs) have been reported to be effective as prophylactic agents in children with CVS.12 An open‐label treatment group of 17 adult patients with CVS noted that 17% of patients had a complete remission with TCA therapy and almost 60% had a partial response.3 More recently, a retrospective case series of patients who had failed TCAs as maintenance therapy reported that 15 out of the 20 patients studied had improvement in the frequency of their vomiting episodes with the newer antiepileptic drugs zonisamide and levetiracem. However, moderate or severe side effects were reported in 45%.13
Conclusions
In summary, although CVS is still an uncommon diagnosis, it is being made more frequently in adults. Although recognition is increasing, there remains a significant delay between onset of symptoms and diagnosis in adults.4 CVS is a diagnosis of exclusion and should be considered when initial evaluation for recurrent nausea and vomiting are unrevealing. A wide range of medications show benefit for both abortive and prophylactic therapy. Increasing awareness of this disorder can lead to a reduction in invasive and costly diagnostic workups.
We present a 26‐year‐old white male with a chief complaint of nausea and vomiting. The patient described prodromal nausea followed by intractable vomiting for 2 days. Over the past 2 years he has experienced similar episodes occurring every 3 to 6 months. He has been hospitalized 5 times for this problem with no diagnosis given. There are no obvious precipitants. The symptoms consistently last 2 to 3 days and resolve with supportive care including intravenous fluids and antiemetics. The patient enjoys good health between the periods of sickness. He has never experienced coffee‐ground emesis or hematemesis. His past medical history is significant for attention deficit disorder and cholecystectomy. He takes no prescription medications. Social history is remarkable for tobacco abuse, binge drinking on weekends, and daily marijuana use. He is unemployed. His family history is unremarkable.
Physical examination at the time of admission was notable for tachycardia, orthostatic hypotension, and hypoactive bowel sounds. Otherwise physical examination was normal.
Diagnostic testing done on admission was notable for white blood cell count of 25,000, hemoglobin of 17.3, blood urea nitrogen 18, creatinine 1.4, aspartate aminotransferase (AST) 64, and alanine aminotransferase (ALT) 55. Pancreatic enzymes and acute abdominal series were normal.
The patient was admitted to the hospital with the presumptive diagnosis of viral gastroenteritis. Initial therapy included intravenous fluids and promethazine. Throughout hospital day 1, he remained nauseated and had multiple bouts of emesis. Records from the patient's hospitalization 5 months ago were obtained and reviewed. During this previous hospitalization, computed tomography (CT) scans of the abdomen and esophagogastroduodenoscopy (EGD) were performed, both of which were negative. Upon review of this recent workup, the diagnosis of cyclic vomiting syndrome (CVS) was entertained and the patient received a therapeutic trial of subcutaneous sumatriptan. His symptoms abated dramatically. Subsequently, he was able to keep oral liquids down and his orthostatic hypotension resolved. On hospital day 2, his white blood cell count normalized without intervention. Blood, urine, and stool cultures remained negative, and workup for acute intermittent porphyria was negative. Upon discharge from the hospital he was counseled to discontinue all marijuana use and was scheduled for follow‐up in the residents' clinic. He failed to keep this appointment. After being lost to follow‐up for 17 months, he presented to the emergency department with nausea and vomiting. As before, his symptoms promptly improved with sumatriptan.
Discussion
CVS, initially described in 1861 as a pediatric illness, is being increasingly recognized in adults.1 It has been estimated that up to 1.6% of children experience symptoms consistent with this disorder, but the prevalence in adults is unknown.2 The essential features of CVS, as noted in our patient, are multiple discrete episodes of nausea and vomiting lasting less than 1 week with absence of nausea and vomiting between episodes. The presentation of adults with CVS often differs from the pediatric form in that adults have longer, less frequent episodes, and the triggers are less evident.3
The etiology and pathogenesis of CVS remain unknown. A variety of physical and psychological stresses, including infection, overexertion, and emotional distress, have been noted to precipitate episodes.4 CVS has variably been associated with autonomic, mitochondrial, and endocrine disorders. The most prevalent theory in the literature, however, is that CVS and migraine headaches are different presentations of the same diathesis.5 Patients with both are noted to have similar patterns of symptoms and positive family history of migraines. The progression from CVS to migraines is noted frequently in individual patients. As many as 82% of the 214 children in a case series of CVS were noted to have a family history of migraines or to have or subsequently develop migraines.6 In addition, electroencephalogram findings and adrenergic autonomic abnormalities are similar in both sets of patients.3 In 1 case series of 17 patients with CVS, patients noted the possible association of episodes with menses (in 57% of women of reproductive age), and the improvement of symptoms with sleep (in 24%), clinical factors common in patients with migraines.3
CVS is one of the functional gastrointestinal disorders for which the diagnosis is clinical, with criteria based upon the consensus of expert opinion in the Rome III Criteria for Functional Gastrointestinal (GI) Disorders.7 At least 3 months, with onset at least 6 months previously of:
-
Stereotypical episodes of vomiting regarding onset (acute) and duration (less than 1 week);
-
3 or more discrete episodes in the prior year; and
-
Absence of nausea and vomiting between episodes.
Supportive criteria: History of migraine headaches or family history of migraine headaches.7
Making the diagnosis of CVS requires the exclusion of other disorders associated with recurrent vomiting. Examples include gastric outlet or small bowel obstruction, gastroparesis, vestibular neuritis, elevated intracranial pressure, inborn errors of metabolism, dysautonomia, porphyria, and alterations in the hypothalamic pituitary adrenal axis. The other functional nausea and vomiting disorders described in Rome III, specifically chronic idiopathic nausea and functional vomiting, also need to be considered.7 Many drugs can cause nausea and vomiting, and chronic marijuana use has been associated with cyclical hyperemesis.8 Our patient meets the diagnostic criteria for CVS, but his frequent marijuana use would preclude a diagnosis of functional vomiting, which by definition requires an absence of chronic cannabinoid use.
Determining which tests and procedures should be performed in the initial evaluation is based on clinical judgment, but commonly includes complete metabolic profile, urinalysis, upper GI series, EGD, neurological imaging, acute abdominal series, and CT of the abdomen and pelvis. In addition, pertinent metabolic screening including serum lactate, cortisol, pyruvate, ammonia, creatinine phosphokinase, carnitine, urinary organic acids, and porphobilinogen may be considered.5
Evidence‐based treatment of CVS is limited by the lack of controlled trials. Acutely, patients often require hospitalization and symptom management with aggressive hydration, antiemetics, and sometimes even sedative agents. Empiric abortive treatment with antimigraine mediations (sumitriptan, prochlorperazine, tricyclic antidepressants, and ketorolac) has been effective in case reports.911 Patients in whom a history of chronic cannabinoid use is elicited should be counseled that cessation may lead to an improvement in symptoms.
Just as with migraines, patients who experience frequent episodes of cyclic vomiting can benefit from prophylactic medications. Tricyclic antidepressants (TCAs) have been reported to be effective as prophylactic agents in children with CVS.12 An open‐label treatment group of 17 adult patients with CVS noted that 17% of patients had a complete remission with TCA therapy and almost 60% had a partial response.3 More recently, a retrospective case series of patients who had failed TCAs as maintenance therapy reported that 15 out of the 20 patients studied had improvement in the frequency of their vomiting episodes with the newer antiepileptic drugs zonisamide and levetiracem. However, moderate or severe side effects were reported in 45%.13
Conclusions
In summary, although CVS is still an uncommon diagnosis, it is being made more frequently in adults. Although recognition is increasing, there remains a significant delay between onset of symptoms and diagnosis in adults.4 CVS is a diagnosis of exclusion and should be considered when initial evaluation for recurrent nausea and vomiting are unrevealing. A wide range of medications show benefit for both abortive and prophylactic therapy. Increasing awareness of this disorder can lead to a reduction in invasive and costly diagnostic workups.
- .Evrose de la digestion, caracteriseo par des crises periodiques de vomissements et une profonde modification de l'assimilation.Gazette Medicale de Paris1861:312. [French]
- ,.Cyclical vomiting syndrome in children: a population‐based study.J Pediatr Gastroenterol Nutr.1995;21(4):454–458.
- ,.Cyclic vomiting syndrome in adults: clinical features and response to tricyclic antidepressants.Am J Gastroenterol.1999;94(10):2855–2860.
- ,,,.Cyclic vomiting syndrome in 41 adults: the illness, the patients, and problems of management.BMC Med.2005;3:20.
- ,,.Consensus statement—2nd International Scientific Symposium on CVS. The Faculty of The 2nd International Scientific Symposium on Cyclic Vomiting Syndrome.Dig Dis Sci.1999;44(8 suppl):9S–11S.
- ,,,,.Is cyclic vomiting syndrome related to migraine?J Pediatr.1999;134(5):567–572.
- ,,, et al.Functional gastroduodenal disorders.Gastroenterology.2006;130:1466–1479.
- ,,,.Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis use.Gut.2004;53:1566–1570.
- ,,.Sumatriptan in the treatment of cyclic vomiting.Ann Pharmacother.1995;29(10):997–999.
- .Recurrent vomiting in adults. A syndrome?Med J Aust.1983;1(7):329–331.
- ,,,,.Cyclic vomiting: association with multiple homeostatic abnormalities and response to ketorolac.Am J Gastroenterol.1996;91(10):2228–2232.
- .Effective prophylactic therapy for cyclic vomiting syndrome in children using amitriptyline or cyproheptadine.Pediatrics.1997;100(6):977–981.
- ,,,.Zonisamide or levetiracetam for adults with cyclic vomiting syndrome: a case series.Clin Gastroenterol Hepatol.2007;5(1):44–48.
- .Evrose de la digestion, caracteriseo par des crises periodiques de vomissements et une profonde modification de l'assimilation.Gazette Medicale de Paris1861:312. [French]
- ,.Cyclical vomiting syndrome in children: a population‐based study.J Pediatr Gastroenterol Nutr.1995;21(4):454–458.
- ,.Cyclic vomiting syndrome in adults: clinical features and response to tricyclic antidepressants.Am J Gastroenterol.1999;94(10):2855–2860.
- ,,,.Cyclic vomiting syndrome in 41 adults: the illness, the patients, and problems of management.BMC Med.2005;3:20.
- ,,.Consensus statement—2nd International Scientific Symposium on CVS. The Faculty of The 2nd International Scientific Symposium on Cyclic Vomiting Syndrome.Dig Dis Sci.1999;44(8 suppl):9S–11S.
- ,,,,.Is cyclic vomiting syndrome related to migraine?J Pediatr.1999;134(5):567–572.
- ,,, et al.Functional gastroduodenal disorders.Gastroenterology.2006;130:1466–1479.
- ,,,.Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis use.Gut.2004;53:1566–1570.
- ,,.Sumatriptan in the treatment of cyclic vomiting.Ann Pharmacother.1995;29(10):997–999.
- .Recurrent vomiting in adults. A syndrome?Med J Aust.1983;1(7):329–331.
- ,,,,.Cyclic vomiting: association with multiple homeostatic abnormalities and response to ketorolac.Am J Gastroenterol.1996;91(10):2228–2232.
- .Effective prophylactic therapy for cyclic vomiting syndrome in children using amitriptyline or cyproheptadine.Pediatrics.1997;100(6):977–981.
- ,,,.Zonisamide or levetiracetam for adults with cyclic vomiting syndrome: a case series.Clin Gastroenterol Hepatol.2007;5(1):44–48.
Syncope from an Unusual Cause: Glossopharyngeal Neuralgia
Hyperpigmented Eruption on Chest
INH-Associated Hepatotoxicity
Reconstruction of a Chronic Monteggia Fracture With Associated Radioulnar Synostosis
Combined Medial and Lateral Condyle Elbow Fractures in a 3-Year-Old Boy
Fatal HIT
Heparin induced thrombocytopenia (HIT) is a significant, potentially life‐threatening immune‐mediated adverse event that occurs several days after commencement of therapy with unfractionated or low‐molecular weight heparin. There are several potential sequelae of HIT, the most frequent of these is thrombosis, including but not limited to deep venous thrombosis (DVT), pulmonary embolism (PE), myocardial infarction, limb arterial occlusion, and disseminated intravascular coagulation. The prothrombotic state induced by HIT can be very significant, generating a thrombosis risk 30 times that of the general population and a mortality risk of 17% to 30% in those patients who develop thrombosis.1, 2
Case Report
A 51‐year‐old female was transferred to our institution for further management of a prothrombotic state. Six days prior to transfer, she presented to an outside hospital with significant edema and discomfort of her left lower extremity. She was found to have bilateral pulmonary emboli and a left lower extremity DVT. She was anticoagulated with unfractionated heparin and transitioned to coumadin. Upon preparation for discharge she developed drastically increased edema of her left lower extremity. Coumadin was discontinued and she was transferred to our institution for alternate anticoagulation and potential interventional vascular treatments.
On examination, the patient reported pain in her legs bilaterally but was in no distress. She had marked edema of the left lower extremity with tender erythematous skin over the anterior thigh with mild cyanosis and pallor of the left toes. Pulses were not palpable but could be identified by handheld Doppler scan. Urgent bilateral lower extremity venous and arterial duplex studies were completed, revealing extensive thrombosis involving the entire deep and superficial venous system on the left and the superficial femoral, popliteal, and peroneal veins on the right.
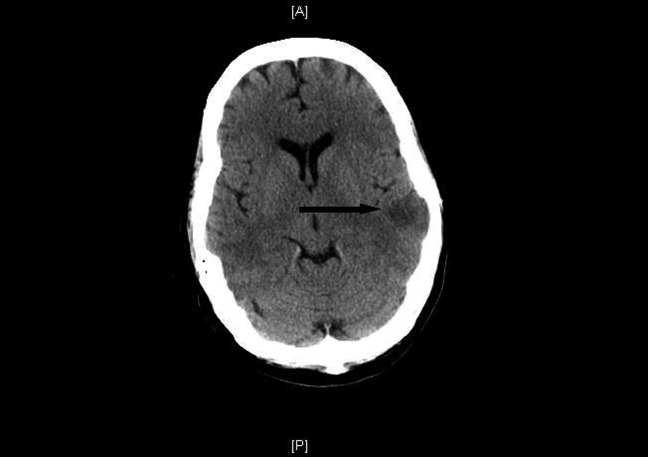
She was treated with an argatroban drip and a complete thrombophilia evaluation commenced. The following day she was mildly obtunded and slow to mentate. A noninfused computed tomography (CT) scan of the head revealed multiple acute left middle cerebral artery ischemic infarctions (Figure 1). CT scans of the chest, abdomen, and pelvis were done to assess for further thrombosis; bilateral renal infarcts were discovered.
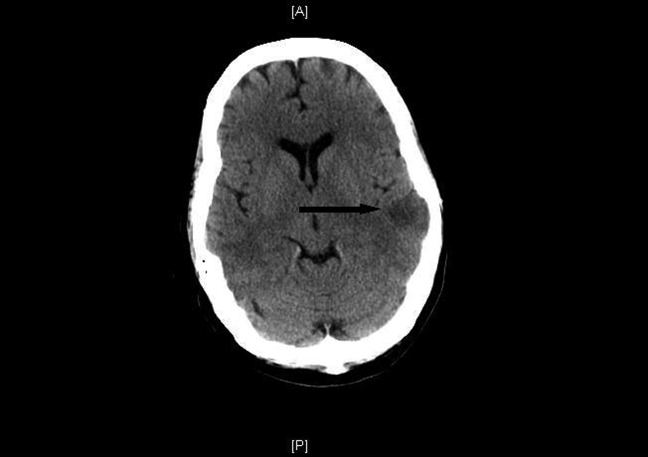
The hypercoagulable workup revealed prothrombin and Factor V Leiden gene mutations and anticardiolipin immunoglobulin (Ig)G, IgA and IgM that were all negative; however, heparin‐dependent antibody platelet factor 4 (PF4) enzyme‐linked immunosorbent assay (ELISA) was positive. Her preheparin platelet count was 149,000/L, 185,000/L at the time of her transfer and thrombosis extension, and 117,000/L at its nadir, 11 days after initial heparin exposure.
Despite lower extremity thrombectomy, right common femoral endarterectomy, and therapeutic anticoagulation, the patient continued to develop massive thrombosis and she expired. The patient underwent autopsy, which confirmed her extensive thrombosis and cited multisystem organ failure as the cause of death. Additionally, this examination revealed an occult high‐grade cervical cancer with lymphatic invasion.
Discussion
This case is an example of multiorgan failure as a result of the prothrombotic state induced by HIT. The thrombocytopenia of HIT is defined as either a platelet count of less than 150,000/L or a decrease of greater than 50% from baseline.3, 4 Despite the eventual confirmation of the diagnosis by PF4 ELISA (sensitivity 80%‐90%), the patient was not thrombocytopenic by definition at the time of extension of her thrombosis.5
Greinacher et al.3 retrospectively evaluated 408 patients with thrombosis associated with HIT and found that at the time of their thrombosis 40.2% became thrombocytopenic (>50% decrease in their platelet count) 1 or more days prior to their initial thrombosis, 26% became thrombocytopenic on the day of their initial thrombosis, and 33.5% had thrombosis that preceded their thrombocytopenia with a 3‐day median delay between thrombosis and thrombocytopenia. Our patient fell in the latter category, developing her thrombocytopenia 5 days after the extension of her thrombosis. The time course of this presentation places emphasis on the need for clinicians to be aware of this pattern and to have a suspicion for HIT in patients on heparin who develop thrombosis regardless of their platelet count at the time of the thrombotic event.
In addition, our patient had the occult diagnosis of cervical cancer. In a retrospective review, Opatrny and Warner6 found that thrombotic complications associated with HIT, venous thrombosis, and PE specifically, occurred more frequently in patients with malignancy than those without malignant disease. They evaluated 64 patients with the diagnosis of HIT, made by heparin‐PF4 ELISA, and discovered the incidence of thrombosis to be 73% in the patients with malignancy compared to 30% in the patients without malignancy. However, since our patient's cancer diagnosis was unknown at the time of the case events, it could not be considered.
There have been rare case reports published describing patients who develop thrombosis secondary to heparin‐dependent antibodies (HDA) without meeting the above definition of thrombocytopenia in HIT. Bream‐Rouwenhorst and Hobbs7 recently reported a similar case in which a 35‐year‐old woman with bilateral lower extremity arterial thrombosis had additional thrombotic events after reexposure to heparin; the patient had a positive heparin‐PF4 ELISA with a platelet count that remained consistently above 200,000/L and never fell below 75% of her baseline. They cite only 22 additional cases of patients with HDA without thrombocytopenia reported in the literature since 1965 and suggest that the term heparin‐associated thrombosis without HIT may be a more appropriate terminology to describe similar cases.
Conclusions
Early recognition and initiation of alternate anticoagulation are essential to the effective management of HIT and prevention of its sequelae. The possible diagnosis of HIT is important for clinicians to keep in mind for all patients that are receiving any form of heparin, not only those patients who present with thrombocytopenia but also those with otherwise unexplainable thrombosis regardless of the platelet count.
- ,,, et. al.The incidence of heparin‐induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study.Blood.2003;101(8):2955–2959.
- ,.Heparin‐induced thrombocytopenia, a prothrombotic disease.Hematol Oncol Clin North Am.2007;21:65–88.
- ,,,,,.Clinical features of heparin‐induced thrombocytopenia including risk factors for thrombosis: a retrospective analysis of 408 patients.Thromb Haemost.2005;94(1):132–135.
- ,,,.An improved definition of immune heparin‐induced thrombocytopenia in postoperative orthopedic patients.Arch Intern Med.2003;163:2518–2524.
- .Pathophysiology and laboratory testing of heparin‐induced thrombocytopenia.Semin Hematol.1998;35(suppl 5):3–8.
- ,.Risk of thrombosis in patients with malignancy and heparin‐induced thrombocytopenia.Am J Hematol.2004;76:240–244.
- ,.Heparin‐dependent antibodies and thrombosis with out heparin‐induced thrombocytopenia.Pharmacotherapy.2008;28(11):1401–1407.
Heparin induced thrombocytopenia (HIT) is a significant, potentially life‐threatening immune‐mediated adverse event that occurs several days after commencement of therapy with unfractionated or low‐molecular weight heparin. There are several potential sequelae of HIT, the most frequent of these is thrombosis, including but not limited to deep venous thrombosis (DVT), pulmonary embolism (PE), myocardial infarction, limb arterial occlusion, and disseminated intravascular coagulation. The prothrombotic state induced by HIT can be very significant, generating a thrombosis risk 30 times that of the general population and a mortality risk of 17% to 30% in those patients who develop thrombosis.1, 2
Case Report
A 51‐year‐old female was transferred to our institution for further management of a prothrombotic state. Six days prior to transfer, she presented to an outside hospital with significant edema and discomfort of her left lower extremity. She was found to have bilateral pulmonary emboli and a left lower extremity DVT. She was anticoagulated with unfractionated heparin and transitioned to coumadin. Upon preparation for discharge she developed drastically increased edema of her left lower extremity. Coumadin was discontinued and she was transferred to our institution for alternate anticoagulation and potential interventional vascular treatments.
On examination, the patient reported pain in her legs bilaterally but was in no distress. She had marked edema of the left lower extremity with tender erythematous skin over the anterior thigh with mild cyanosis and pallor of the left toes. Pulses were not palpable but could be identified by handheld Doppler scan. Urgent bilateral lower extremity venous and arterial duplex studies were completed, revealing extensive thrombosis involving the entire deep and superficial venous system on the left and the superficial femoral, popliteal, and peroneal veins on the right.
She was treated with an argatroban drip and a complete thrombophilia evaluation commenced. The following day she was mildly obtunded and slow to mentate. A noninfused computed tomography (CT) scan of the head revealed multiple acute left middle cerebral artery ischemic infarctions (Figure 1). CT scans of the chest, abdomen, and pelvis were done to assess for further thrombosis; bilateral renal infarcts were discovered.
The hypercoagulable workup revealed prothrombin and Factor V Leiden gene mutations and anticardiolipin immunoglobulin (Ig)G, IgA and IgM that were all negative; however, heparin‐dependent antibody platelet factor 4 (PF4) enzyme‐linked immunosorbent assay (ELISA) was positive. Her preheparin platelet count was 149,000/L, 185,000/L at the time of her transfer and thrombosis extension, and 117,000/L at its nadir, 11 days after initial heparin exposure.
Despite lower extremity thrombectomy, right common femoral endarterectomy, and therapeutic anticoagulation, the patient continued to develop massive thrombosis and she expired. The patient underwent autopsy, which confirmed her extensive thrombosis and cited multisystem organ failure as the cause of death. Additionally, this examination revealed an occult high‐grade cervical cancer with lymphatic invasion.
Discussion
This case is an example of multiorgan failure as a result of the prothrombotic state induced by HIT. The thrombocytopenia of HIT is defined as either a platelet count of less than 150,000/L or a decrease of greater than 50% from baseline.3, 4 Despite the eventual confirmation of the diagnosis by PF4 ELISA (sensitivity 80%‐90%), the patient was not thrombocytopenic by definition at the time of extension of her thrombosis.5
Greinacher et al.3 retrospectively evaluated 408 patients with thrombosis associated with HIT and found that at the time of their thrombosis 40.2% became thrombocytopenic (>50% decrease in their platelet count) 1 or more days prior to their initial thrombosis, 26% became thrombocytopenic on the day of their initial thrombosis, and 33.5% had thrombosis that preceded their thrombocytopenia with a 3‐day median delay between thrombosis and thrombocytopenia. Our patient fell in the latter category, developing her thrombocytopenia 5 days after the extension of her thrombosis. The time course of this presentation places emphasis on the need for clinicians to be aware of this pattern and to have a suspicion for HIT in patients on heparin who develop thrombosis regardless of their platelet count at the time of the thrombotic event.
In addition, our patient had the occult diagnosis of cervical cancer. In a retrospective review, Opatrny and Warner6 found that thrombotic complications associated with HIT, venous thrombosis, and PE specifically, occurred more frequently in patients with malignancy than those without malignant disease. They evaluated 64 patients with the diagnosis of HIT, made by heparin‐PF4 ELISA, and discovered the incidence of thrombosis to be 73% in the patients with malignancy compared to 30% in the patients without malignancy. However, since our patient's cancer diagnosis was unknown at the time of the case events, it could not be considered.
There have been rare case reports published describing patients who develop thrombosis secondary to heparin‐dependent antibodies (HDA) without meeting the above definition of thrombocytopenia in HIT. Bream‐Rouwenhorst and Hobbs7 recently reported a similar case in which a 35‐year‐old woman with bilateral lower extremity arterial thrombosis had additional thrombotic events after reexposure to heparin; the patient had a positive heparin‐PF4 ELISA with a platelet count that remained consistently above 200,000/L and never fell below 75% of her baseline. They cite only 22 additional cases of patients with HDA without thrombocytopenia reported in the literature since 1965 and suggest that the term heparin‐associated thrombosis without HIT may be a more appropriate terminology to describe similar cases.
Conclusions
Early recognition and initiation of alternate anticoagulation are essential to the effective management of HIT and prevention of its sequelae. The possible diagnosis of HIT is important for clinicians to keep in mind for all patients that are receiving any form of heparin, not only those patients who present with thrombocytopenia but also those with otherwise unexplainable thrombosis regardless of the platelet count.
Heparin induced thrombocytopenia (HIT) is a significant, potentially life‐threatening immune‐mediated adverse event that occurs several days after commencement of therapy with unfractionated or low‐molecular weight heparin. There are several potential sequelae of HIT, the most frequent of these is thrombosis, including but not limited to deep venous thrombosis (DVT), pulmonary embolism (PE), myocardial infarction, limb arterial occlusion, and disseminated intravascular coagulation. The prothrombotic state induced by HIT can be very significant, generating a thrombosis risk 30 times that of the general population and a mortality risk of 17% to 30% in those patients who develop thrombosis.1, 2
Case Report
A 51‐year‐old female was transferred to our institution for further management of a prothrombotic state. Six days prior to transfer, she presented to an outside hospital with significant edema and discomfort of her left lower extremity. She was found to have bilateral pulmonary emboli and a left lower extremity DVT. She was anticoagulated with unfractionated heparin and transitioned to coumadin. Upon preparation for discharge she developed drastically increased edema of her left lower extremity. Coumadin was discontinued and she was transferred to our institution for alternate anticoagulation and potential interventional vascular treatments.
On examination, the patient reported pain in her legs bilaterally but was in no distress. She had marked edema of the left lower extremity with tender erythematous skin over the anterior thigh with mild cyanosis and pallor of the left toes. Pulses were not palpable but could be identified by handheld Doppler scan. Urgent bilateral lower extremity venous and arterial duplex studies were completed, revealing extensive thrombosis involving the entire deep and superficial venous system on the left and the superficial femoral, popliteal, and peroneal veins on the right.
She was treated with an argatroban drip and a complete thrombophilia evaluation commenced. The following day she was mildly obtunded and slow to mentate. A noninfused computed tomography (CT) scan of the head revealed multiple acute left middle cerebral artery ischemic infarctions (Figure 1). CT scans of the chest, abdomen, and pelvis were done to assess for further thrombosis; bilateral renal infarcts were discovered.
The hypercoagulable workup revealed prothrombin and Factor V Leiden gene mutations and anticardiolipin immunoglobulin (Ig)G, IgA and IgM that were all negative; however, heparin‐dependent antibody platelet factor 4 (PF4) enzyme‐linked immunosorbent assay (ELISA) was positive. Her preheparin platelet count was 149,000/L, 185,000/L at the time of her transfer and thrombosis extension, and 117,000/L at its nadir, 11 days after initial heparin exposure.
Despite lower extremity thrombectomy, right common femoral endarterectomy, and therapeutic anticoagulation, the patient continued to develop massive thrombosis and she expired. The patient underwent autopsy, which confirmed her extensive thrombosis and cited multisystem organ failure as the cause of death. Additionally, this examination revealed an occult high‐grade cervical cancer with lymphatic invasion.
Discussion
This case is an example of multiorgan failure as a result of the prothrombotic state induced by HIT. The thrombocytopenia of HIT is defined as either a platelet count of less than 150,000/L or a decrease of greater than 50% from baseline.3, 4 Despite the eventual confirmation of the diagnosis by PF4 ELISA (sensitivity 80%‐90%), the patient was not thrombocytopenic by definition at the time of extension of her thrombosis.5
Greinacher et al.3 retrospectively evaluated 408 patients with thrombosis associated with HIT and found that at the time of their thrombosis 40.2% became thrombocytopenic (>50% decrease in their platelet count) 1 or more days prior to their initial thrombosis, 26% became thrombocytopenic on the day of their initial thrombosis, and 33.5% had thrombosis that preceded their thrombocytopenia with a 3‐day median delay between thrombosis and thrombocytopenia. Our patient fell in the latter category, developing her thrombocytopenia 5 days after the extension of her thrombosis. The time course of this presentation places emphasis on the need for clinicians to be aware of this pattern and to have a suspicion for HIT in patients on heparin who develop thrombosis regardless of their platelet count at the time of the thrombotic event.
In addition, our patient had the occult diagnosis of cervical cancer. In a retrospective review, Opatrny and Warner6 found that thrombotic complications associated with HIT, venous thrombosis, and PE specifically, occurred more frequently in patients with malignancy than those without malignant disease. They evaluated 64 patients with the diagnosis of HIT, made by heparin‐PF4 ELISA, and discovered the incidence of thrombosis to be 73% in the patients with malignancy compared to 30% in the patients without malignancy. However, since our patient's cancer diagnosis was unknown at the time of the case events, it could not be considered.
There have been rare case reports published describing patients who develop thrombosis secondary to heparin‐dependent antibodies (HDA) without meeting the above definition of thrombocytopenia in HIT. Bream‐Rouwenhorst and Hobbs7 recently reported a similar case in which a 35‐year‐old woman with bilateral lower extremity arterial thrombosis had additional thrombotic events after reexposure to heparin; the patient had a positive heparin‐PF4 ELISA with a platelet count that remained consistently above 200,000/L and never fell below 75% of her baseline. They cite only 22 additional cases of patients with HDA without thrombocytopenia reported in the literature since 1965 and suggest that the term heparin‐associated thrombosis without HIT may be a more appropriate terminology to describe similar cases.
Conclusions
Early recognition and initiation of alternate anticoagulation are essential to the effective management of HIT and prevention of its sequelae. The possible diagnosis of HIT is important for clinicians to keep in mind for all patients that are receiving any form of heparin, not only those patients who present with thrombocytopenia but also those with otherwise unexplainable thrombosis regardless of the platelet count.
- ,,, et. al.The incidence of heparin‐induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study.Blood.2003;101(8):2955–2959.
- ,.Heparin‐induced thrombocytopenia, a prothrombotic disease.Hematol Oncol Clin North Am.2007;21:65–88.
- ,,,,,.Clinical features of heparin‐induced thrombocytopenia including risk factors for thrombosis: a retrospective analysis of 408 patients.Thromb Haemost.2005;94(1):132–135.
- ,,,.An improved definition of immune heparin‐induced thrombocytopenia in postoperative orthopedic patients.Arch Intern Med.2003;163:2518–2524.
- .Pathophysiology and laboratory testing of heparin‐induced thrombocytopenia.Semin Hematol.1998;35(suppl 5):3–8.
- ,.Risk of thrombosis in patients with malignancy and heparin‐induced thrombocytopenia.Am J Hematol.2004;76:240–244.
- ,.Heparin‐dependent antibodies and thrombosis with out heparin‐induced thrombocytopenia.Pharmacotherapy.2008;28(11):1401–1407.
- ,,, et. al.The incidence of heparin‐induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study.Blood.2003;101(8):2955–2959.
- ,.Heparin‐induced thrombocytopenia, a prothrombotic disease.Hematol Oncol Clin North Am.2007;21:65–88.
- ,,,,,.Clinical features of heparin‐induced thrombocytopenia including risk factors for thrombosis: a retrospective analysis of 408 patients.Thromb Haemost.2005;94(1):132–135.
- ,,,.An improved definition of immune heparin‐induced thrombocytopenia in postoperative orthopedic patients.Arch Intern Med.2003;163:2518–2524.
- .Pathophysiology and laboratory testing of heparin‐induced thrombocytopenia.Semin Hematol.1998;35(suppl 5):3–8.
- ,.Risk of thrombosis in patients with malignancy and heparin‐induced thrombocytopenia.Am J Hematol.2004;76:240–244.
- ,.Heparin‐dependent antibodies and thrombosis with out heparin‐induced thrombocytopenia.Pharmacotherapy.2008;28(11):1401–1407.
ESIR and Peripheral Insulin Resistance
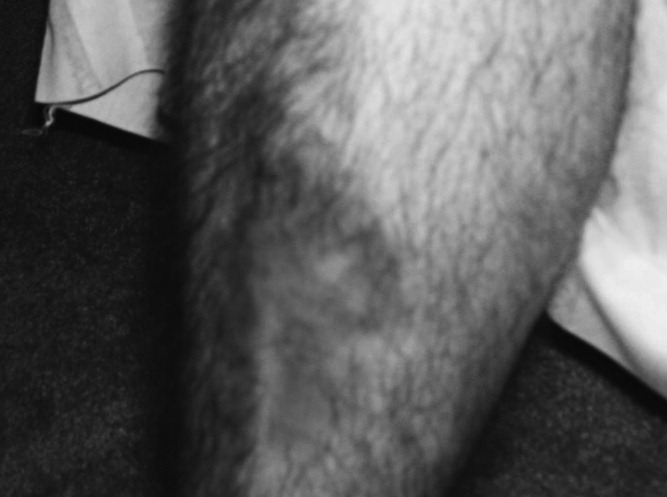
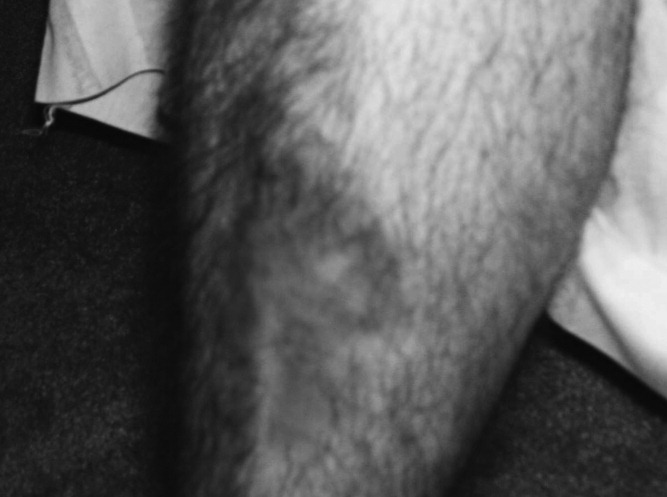
A 34‐year‐old man was admitted for evaluation of elevated blood glucose despite extremely high subcutaneous (SQ) insulin requirements. He had a 12‐year history of Type 2 diabetes mellitus (T2DM) without episodes of ketoacidosis, managed initially with oral medications (metformin with various sulfonylureas and thiazolidinediones). Three months prior to admission, he was transitioned to SQ insulin and thereafter his requirements escalated rapidly. By the time of his admission, his blood glucose measurements were consistently above 300 mg/dL despite injecting more than 4100 units of insulin daily. His regimen included 300 units of insulin glargine (Lantus) 2 times per day (BID) and 1.75 mL of Humilin U‐500 Insulin (875 units) 4 times per day (QID). Past medical history included metabolic syndrome, nonalcoholic steatohepatitis, and diabetic neuropathy. Physical exam was remarkable for centripetal obesity (body mass index [BMI] = 38.9 kg/m2), acanthosis nigricans, and necrobiosis lipoidica diabeticorum (NLD) (Figure 1).
We undertook an investigation to characterize this extreme insulin resistance. After 24 hours without insulin supplementation, and 12 hours of nothing by mouth (NPO), his blood glucose level was 280 mg/dL and his serum insulin was 133.5 IU/mL. We injected 12 units of insulin Aspart and subsequently measured his serum glucose and insulin once more. His blood glucose level had risen to 289 mg/dL and his serum insulin fell to 110.7 IU/mL. We then transitioned the patient to intravenous (IV) insulin. After a series of boluses totaling 400 units, his blood glucose normalized (90 mg/dL) and was maintained in normal range on a rate of 48 units per hour. Over 24 hours, we had infused over 1400 units.
During this time, we also drew several labs. Serum antiinsulin antibodies were undetectable (ARUP Laboratories, Salt Lake City, UT). A full rheumatologic workup was negative for systemic lupus erythematosus (SLE), rheumatoid factor, Sjgren's syndrome (SS)‐A and SS‐B. Androgen levels were normal, as were 24‐hour urine collections for cortisol and metanephrines. The patient was discharged on a regimen of U‐500 without glargine.
By 5 months after discharge, his blood glucose remained uncontrolled despite increasing doses of U‐500 (with or without metformin and thiazolidinediones). The patient was offered a gastric bypass operation. Now, 4 months postoperative, his blood glucose is controlled, no greater than 90 mg/dL in the morning and 125 mg/dL in the evening. He is off insulin, taking 30 mg pioglitazone (Actos) daily and 500 mg metformin 3 times per day (TID).
Discussion
Extreme insulin resistance (EIR), defined by daily insulin requirements in excess of 200 U, is a rare and frustrating condition.1 Rarer still is extreme subcutaneous insulin resistance (ESIR). A systematic Medline review revealed only 29 reported cases of ESIR, all of which involved patients that maintained IV sensitivity to insulin. Classic diagnostic criteria for ESIR include preserved sensitivity to IV insulin, failure to increase serum insulin with subcutaneous injection, and insulin degrading activity of subcutaneous tissue.2, 3 However, there are, at present, no laboratory tests that can test the final criterion. Indeed, very few of the published reports of ESIR satisfy it, with most studies considering as diagnostic of ESIR the constellation of EIR with failure to raise serum insulin after injection and preserved intravenous insulin sensitivity.
As was evident in the high doses of IV insulin required for blood glucose normalization, our patient also had a proven receptor‐level peripheral resistance. Beyond the common, multifactorial insulin resistance of T2DM, the published reports of patients with extreme peripheral resistance are of 2 types: (A) genetic (eg, Leprechaunism) and (B) acquired autoimmune (Table 1).4 This patient fits neither category. Patients with Type A are very sick, with a syndromic disease that sharply curtails their life expectancy. Patients with Type B acquire antibodies directed against their insulin receptors and are almost invariably elderly African‐American women with severe rheumatological disease, namely SLE. We could not test our patient for an insulin‐receptor antibody secondary to prohibitive cost. This is probably moot, given that his autoimmune workup was negative and, as above, patients with such antibodies are vastly different compared to our patients.
| Class of Insulin Resistance | Mechanism | Incidence | Treatment |
|---|---|---|---|
| |||
| Type 2 diabetes mellitus | Multifactorial | 3% of total population | Many |
| Type A receptor‐level insulin resistance | Congenital receptor defect | 86 cases | U‐500, insulin‐like growth factor‐1 |
| Type B receptor‐level insulin resistance | Antiinsulin receptor antibody | 50 cases | U‐500, immune modulation |
| Subcutaneous insulin resistance | Unknown; SQ protease? | 30 cases | U‐500, intraperitoneal insulin delivery, other |
Based on SQ insulin requirements, our patient had EIR. As his insulin levels failed to rise following an insulin injection, his EIR is thus subcutaneous in nature. However, among patients with this condition his failure to respond to IV insulin is unique. He does not fit criteria for types A or B insulin resistance; his condition is likely also due to an extreme version of the more common, multifactorial peripheral insulin resistance. This is supported by his successful response to the gastric bypass operation.5
The standard treatments for ESIR include: (1) concentrated regular insulin (U‐500) and (2) implantable intraperitoneal delivery; our patient received the former.6 U‐500 use in EIR has been shown to be more cost‐effective.1 Several reports have suggested success with protease inhibitors (aprotinin, nafamostat ointment), plasmapheresis, and intravenous immunoglobulin for extreme SQ resistance. Our case also represents the first treated successfully with a gastric bypass operation.
CONCLUSIONS
EIR can present a significant challenge for both the patient and hospitalist. The approach to this condition should begin with the determination of 24‐hour IV insulin requirement utilizing an insulin drip; serum insulin antibody evaluation; and endocrinology consultation. Our case also highlights a few important points about the broader management of diabetes mellitus. First, there are dermatological manifestations of diabetes that serve as potential markers for disease (namely acanthosis nigricans and NLD). Second, for patients with extreme insulin requirements, an extensive workup should be initiated and the patient should be transitioned to a concentrated regular insulin or intraperitoneal delivery. Third, our experience suggests a role for other measures such as gastric bypass that ought to be studied further.
- ,,.The use of U‐500 in patients with extreme insulin resistance.Diabetes Care.2005;28:1240–1244.
- ,.Impaired absorption of insulin as a cause insulin resistance.Diabetes.1975;24:443.
- ,,.Insulin resistance caused by massive degradation of subcutaneous insulin.Diabetes.1979;28:640–645.
- ,,, et al.Clinical course of genetic diseases of the insulin receptor: a 30‐year prospective.Medicine.2004;83:209–222.
- ,,, et al.Who would have thought it? An operation proves to be the most effective therapy for adult‐onset diabetes mellitus.Ann Surg.1995;222:339–352.
- ,,,,,.Extreme subcutaneous insulin resistance: a misunderstood syndrome.Diabetes Metab.2003;29:539–546.
A 34‐year‐old man was admitted for evaluation of elevated blood glucose despite extremely high subcutaneous (SQ) insulin requirements. He had a 12‐year history of Type 2 diabetes mellitus (T2DM) without episodes of ketoacidosis, managed initially with oral medications (metformin with various sulfonylureas and thiazolidinediones). Three months prior to admission, he was transitioned to SQ insulin and thereafter his requirements escalated rapidly. By the time of his admission, his blood glucose measurements were consistently above 300 mg/dL despite injecting more than 4100 units of insulin daily. His regimen included 300 units of insulin glargine (Lantus) 2 times per day (BID) and 1.75 mL of Humilin U‐500 Insulin (875 units) 4 times per day (QID). Past medical history included metabolic syndrome, nonalcoholic steatohepatitis, and diabetic neuropathy. Physical exam was remarkable for centripetal obesity (body mass index [BMI] = 38.9 kg/m2), acanthosis nigricans, and necrobiosis lipoidica diabeticorum (NLD) (Figure 1).
We undertook an investigation to characterize this extreme insulin resistance. After 24 hours without insulin supplementation, and 12 hours of nothing by mouth (NPO), his blood glucose level was 280 mg/dL and his serum insulin was 133.5 IU/mL. We injected 12 units of insulin Aspart and subsequently measured his serum glucose and insulin once more. His blood glucose level had risen to 289 mg/dL and his serum insulin fell to 110.7 IU/mL. We then transitioned the patient to intravenous (IV) insulin. After a series of boluses totaling 400 units, his blood glucose normalized (90 mg/dL) and was maintained in normal range on a rate of 48 units per hour. Over 24 hours, we had infused over 1400 units.
During this time, we also drew several labs. Serum antiinsulin antibodies were undetectable (ARUP Laboratories, Salt Lake City, UT). A full rheumatologic workup was negative for systemic lupus erythematosus (SLE), rheumatoid factor, Sjgren's syndrome (SS)‐A and SS‐B. Androgen levels were normal, as were 24‐hour urine collections for cortisol and metanephrines. The patient was discharged on a regimen of U‐500 without glargine.
By 5 months after discharge, his blood glucose remained uncontrolled despite increasing doses of U‐500 (with or without metformin and thiazolidinediones). The patient was offered a gastric bypass operation. Now, 4 months postoperative, his blood glucose is controlled, no greater than 90 mg/dL in the morning and 125 mg/dL in the evening. He is off insulin, taking 30 mg pioglitazone (Actos) daily and 500 mg metformin 3 times per day (TID).
Discussion
Extreme insulin resistance (EIR), defined by daily insulin requirements in excess of 200 U, is a rare and frustrating condition.1 Rarer still is extreme subcutaneous insulin resistance (ESIR). A systematic Medline review revealed only 29 reported cases of ESIR, all of which involved patients that maintained IV sensitivity to insulin. Classic diagnostic criteria for ESIR include preserved sensitivity to IV insulin, failure to increase serum insulin with subcutaneous injection, and insulin degrading activity of subcutaneous tissue.2, 3 However, there are, at present, no laboratory tests that can test the final criterion. Indeed, very few of the published reports of ESIR satisfy it, with most studies considering as diagnostic of ESIR the constellation of EIR with failure to raise serum insulin after injection and preserved intravenous insulin sensitivity.
As was evident in the high doses of IV insulin required for blood glucose normalization, our patient also had a proven receptor‐level peripheral resistance. Beyond the common, multifactorial insulin resistance of T2DM, the published reports of patients with extreme peripheral resistance are of 2 types: (A) genetic (eg, Leprechaunism) and (B) acquired autoimmune (Table 1).4 This patient fits neither category. Patients with Type A are very sick, with a syndromic disease that sharply curtails their life expectancy. Patients with Type B acquire antibodies directed against their insulin receptors and are almost invariably elderly African‐American women with severe rheumatological disease, namely SLE. We could not test our patient for an insulin‐receptor antibody secondary to prohibitive cost. This is probably moot, given that his autoimmune workup was negative and, as above, patients with such antibodies are vastly different compared to our patients.
| Class of Insulin Resistance | Mechanism | Incidence | Treatment |
|---|---|---|---|
| |||
| Type 2 diabetes mellitus | Multifactorial | 3% of total population | Many |
| Type A receptor‐level insulin resistance | Congenital receptor defect | 86 cases | U‐500, insulin‐like growth factor‐1 |
| Type B receptor‐level insulin resistance | Antiinsulin receptor antibody | 50 cases | U‐500, immune modulation |
| Subcutaneous insulin resistance | Unknown; SQ protease? | 30 cases | U‐500, intraperitoneal insulin delivery, other |
Based on SQ insulin requirements, our patient had EIR. As his insulin levels failed to rise following an insulin injection, his EIR is thus subcutaneous in nature. However, among patients with this condition his failure to respond to IV insulin is unique. He does not fit criteria for types A or B insulin resistance; his condition is likely also due to an extreme version of the more common, multifactorial peripheral insulin resistance. This is supported by his successful response to the gastric bypass operation.5
The standard treatments for ESIR include: (1) concentrated regular insulin (U‐500) and (2) implantable intraperitoneal delivery; our patient received the former.6 U‐500 use in EIR has been shown to be more cost‐effective.1 Several reports have suggested success with protease inhibitors (aprotinin, nafamostat ointment), plasmapheresis, and intravenous immunoglobulin for extreme SQ resistance. Our case also represents the first treated successfully with a gastric bypass operation.
CONCLUSIONS
EIR can present a significant challenge for both the patient and hospitalist. The approach to this condition should begin with the determination of 24‐hour IV insulin requirement utilizing an insulin drip; serum insulin antibody evaluation; and endocrinology consultation. Our case also highlights a few important points about the broader management of diabetes mellitus. First, there are dermatological manifestations of diabetes that serve as potential markers for disease (namely acanthosis nigricans and NLD). Second, for patients with extreme insulin requirements, an extensive workup should be initiated and the patient should be transitioned to a concentrated regular insulin or intraperitoneal delivery. Third, our experience suggests a role for other measures such as gastric bypass that ought to be studied further.
A 34‐year‐old man was admitted for evaluation of elevated blood glucose despite extremely high subcutaneous (SQ) insulin requirements. He had a 12‐year history of Type 2 diabetes mellitus (T2DM) without episodes of ketoacidosis, managed initially with oral medications (metformin with various sulfonylureas and thiazolidinediones). Three months prior to admission, he was transitioned to SQ insulin and thereafter his requirements escalated rapidly. By the time of his admission, his blood glucose measurements were consistently above 300 mg/dL despite injecting more than 4100 units of insulin daily. His regimen included 300 units of insulin glargine (Lantus) 2 times per day (BID) and 1.75 mL of Humilin U‐500 Insulin (875 units) 4 times per day (QID). Past medical history included metabolic syndrome, nonalcoholic steatohepatitis, and diabetic neuropathy. Physical exam was remarkable for centripetal obesity (body mass index [BMI] = 38.9 kg/m2), acanthosis nigricans, and necrobiosis lipoidica diabeticorum (NLD) (Figure 1).
We undertook an investigation to characterize this extreme insulin resistance. After 24 hours without insulin supplementation, and 12 hours of nothing by mouth (NPO), his blood glucose level was 280 mg/dL and his serum insulin was 133.5 IU/mL. We injected 12 units of insulin Aspart and subsequently measured his serum glucose and insulin once more. His blood glucose level had risen to 289 mg/dL and his serum insulin fell to 110.7 IU/mL. We then transitioned the patient to intravenous (IV) insulin. After a series of boluses totaling 400 units, his blood glucose normalized (90 mg/dL) and was maintained in normal range on a rate of 48 units per hour. Over 24 hours, we had infused over 1400 units.
During this time, we also drew several labs. Serum antiinsulin antibodies were undetectable (ARUP Laboratories, Salt Lake City, UT). A full rheumatologic workup was negative for systemic lupus erythematosus (SLE), rheumatoid factor, Sjgren's syndrome (SS)‐A and SS‐B. Androgen levels were normal, as were 24‐hour urine collections for cortisol and metanephrines. The patient was discharged on a regimen of U‐500 without glargine.
By 5 months after discharge, his blood glucose remained uncontrolled despite increasing doses of U‐500 (with or without metformin and thiazolidinediones). The patient was offered a gastric bypass operation. Now, 4 months postoperative, his blood glucose is controlled, no greater than 90 mg/dL in the morning and 125 mg/dL in the evening. He is off insulin, taking 30 mg pioglitazone (Actos) daily and 500 mg metformin 3 times per day (TID).
Discussion
Extreme insulin resistance (EIR), defined by daily insulin requirements in excess of 200 U, is a rare and frustrating condition.1 Rarer still is extreme subcutaneous insulin resistance (ESIR). A systematic Medline review revealed only 29 reported cases of ESIR, all of which involved patients that maintained IV sensitivity to insulin. Classic diagnostic criteria for ESIR include preserved sensitivity to IV insulin, failure to increase serum insulin with subcutaneous injection, and insulin degrading activity of subcutaneous tissue.2, 3 However, there are, at present, no laboratory tests that can test the final criterion. Indeed, very few of the published reports of ESIR satisfy it, with most studies considering as diagnostic of ESIR the constellation of EIR with failure to raise serum insulin after injection and preserved intravenous insulin sensitivity.
As was evident in the high doses of IV insulin required for blood glucose normalization, our patient also had a proven receptor‐level peripheral resistance. Beyond the common, multifactorial insulin resistance of T2DM, the published reports of patients with extreme peripheral resistance are of 2 types: (A) genetic (eg, Leprechaunism) and (B) acquired autoimmune (Table 1).4 This patient fits neither category. Patients with Type A are very sick, with a syndromic disease that sharply curtails their life expectancy. Patients with Type B acquire antibodies directed against their insulin receptors and are almost invariably elderly African‐American women with severe rheumatological disease, namely SLE. We could not test our patient for an insulin‐receptor antibody secondary to prohibitive cost. This is probably moot, given that his autoimmune workup was negative and, as above, patients with such antibodies are vastly different compared to our patients.
| Class of Insulin Resistance | Mechanism | Incidence | Treatment |
|---|---|---|---|
| |||
| Type 2 diabetes mellitus | Multifactorial | 3% of total population | Many |
| Type A receptor‐level insulin resistance | Congenital receptor defect | 86 cases | U‐500, insulin‐like growth factor‐1 |
| Type B receptor‐level insulin resistance | Antiinsulin receptor antibody | 50 cases | U‐500, immune modulation |
| Subcutaneous insulin resistance | Unknown; SQ protease? | 30 cases | U‐500, intraperitoneal insulin delivery, other |
Based on SQ insulin requirements, our patient had EIR. As his insulin levels failed to rise following an insulin injection, his EIR is thus subcutaneous in nature. However, among patients with this condition his failure to respond to IV insulin is unique. He does not fit criteria for types A or B insulin resistance; his condition is likely also due to an extreme version of the more common, multifactorial peripheral insulin resistance. This is supported by his successful response to the gastric bypass operation.5
The standard treatments for ESIR include: (1) concentrated regular insulin (U‐500) and (2) implantable intraperitoneal delivery; our patient received the former.6 U‐500 use in EIR has been shown to be more cost‐effective.1 Several reports have suggested success with protease inhibitors (aprotinin, nafamostat ointment), plasmapheresis, and intravenous immunoglobulin for extreme SQ resistance. Our case also represents the first treated successfully with a gastric bypass operation.
CONCLUSIONS
EIR can present a significant challenge for both the patient and hospitalist. The approach to this condition should begin with the determination of 24‐hour IV insulin requirement utilizing an insulin drip; serum insulin antibody evaluation; and endocrinology consultation. Our case also highlights a few important points about the broader management of diabetes mellitus. First, there are dermatological manifestations of diabetes that serve as potential markers for disease (namely acanthosis nigricans and NLD). Second, for patients with extreme insulin requirements, an extensive workup should be initiated and the patient should be transitioned to a concentrated regular insulin or intraperitoneal delivery. Third, our experience suggests a role for other measures such as gastric bypass that ought to be studied further.
- ,,.The use of U‐500 in patients with extreme insulin resistance.Diabetes Care.2005;28:1240–1244.
- ,.Impaired absorption of insulin as a cause insulin resistance.Diabetes.1975;24:443.
- ,,.Insulin resistance caused by massive degradation of subcutaneous insulin.Diabetes.1979;28:640–645.
- ,,, et al.Clinical course of genetic diseases of the insulin receptor: a 30‐year prospective.Medicine.2004;83:209–222.
- ,,, et al.Who would have thought it? An operation proves to be the most effective therapy for adult‐onset diabetes mellitus.Ann Surg.1995;222:339–352.
- ,,,,,.Extreme subcutaneous insulin resistance: a misunderstood syndrome.Diabetes Metab.2003;29:539–546.
- ,,.The use of U‐500 in patients with extreme insulin resistance.Diabetes Care.2005;28:1240–1244.
- ,.Impaired absorption of insulin as a cause insulin resistance.Diabetes.1975;24:443.
- ,,.Insulin resistance caused by massive degradation of subcutaneous insulin.Diabetes.1979;28:640–645.
- ,,, et al.Clinical course of genetic diseases of the insulin receptor: a 30‐year prospective.Medicine.2004;83:209–222.
- ,,, et al.Who would have thought it? An operation proves to be the most effective therapy for adult‐onset diabetes mellitus.Ann Surg.1995;222:339–352.
- ,,,,,.Extreme subcutaneous insulin resistance: a misunderstood syndrome.Diabetes Metab.2003;29:539–546.
Hyponatremia: SIADH or CSW?
An 83‐year‐old man admitted for weakness, lethargy, and mental status changes was found to have human immunodeficiency virus (HIV) disease and cryptococcal meningitis. His hospital course was complicated by worsening hyponatremia (sodium 136 mEq/L). By hospital day 6, the patient's serum sodium had declined to 127 mEq/L from his admission level of 133 mEq/L. The initial impression was that the patient had syndrome of inappropriate antidiuretic hormone (SIADH) and fluid restriction to less than 1500 mL per day was initiated. By hospital day 11, serum sodium continued to decline, to 123 mEq/L, despite fluid restriction.
The past medical history was remarkable for coronary artery disease, hypertension, hyperlipidemia, and anemia, but by self‐report he had not been taking any medications. His review of systems was positive for intermittent bouts of diarrhea.
Vital signs on day 11 included a temperature of 37.3C, blood pressure (BP) of 105/55 mm Hg, and pulse of 90 beats per minute. The BP on admission had been 145/86 mm Hg but had steadily declined with fluid restriction. On physical examination, he appeared thin and cachetic with no evidence of jugular venous distention, rales, or peripheral edema to suggest volume overload. He had been receiving 2 to 4 L of isotonic saline daily for 5 days before the fluid restriction was initiated. The urine output continuously exceeded his intake by at least 500 mL per day throughout his hospital course. His only inpatient medications were amphotericin B and flucytosine. For nutritional supplementation, he was receiving a high‐calorie supplement with free‐water flushes via a nasogastric tube.
Laboratory results revealed a serum sodium concentration of 123 mEq/L, serum potassium of 4.4 mEq/L, serum creatinine of 0.6 mg/dL, urine sodium of 139 mEq/L, serum osmolality of 272 mOsm/kg, and urine osmolality of 598 mOsm/kg (see Table 1). Urinalysis revealed a specific gravity of 1.030. A random serum cortisol level was 11.1 g/dL. A thyroid‐stimulating hormone (TSH) level was 1.32 IU/mL. Brain natriuretic peptide (BNP) was elevated, at 686 pg/mL. A fractional excretion of uric acid was also elevated, at 83.8%.
| Parameters | Day 1 | Fluid Restriction Initiated: Day 6 | Day 8 | Fluid Resuscitation Initiated: Day 11 | Day 13 | Day 15 | Day 26 | Day 37* | Day 40 | Day 44 |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Na (mEq/L) | 133 | 127 | 126 | 123 | 131 | 119 | 140 | 131 | 132 | 135 |
| K (mEq/L) | 4.2 | 4.2 | ||||||||
| BUN (mg/dL) | 39 | 36 | ||||||||
| Cr (mg/dL) | 1.1 | 0.9 | ||||||||
| UNa (mEq/L) | 139 | 86 | 154 | 138 | ||||||
| UOsm (mOsm/kg) | 598 | 362 | 376 | |||||||
| SOsm (mOsm/kg) | 272 | 273 | 279 | |||||||
| BNP (pg/mL) | 686 | 900 | 222 | |||||||
| SUA (mg/dL) | 1.7 | 2.6 | 1.6 | |||||||
| UUA (mg/dL) | 38 | 11 | ||||||||
| FEUA (%) | 83.82 | 28.21 | ||||||||
| FENa (%) | 3.94 | 7.33 | ||||||||
| BP (mm Hg) | 147 | 136 | 122 | 105 | 101 | 90 | 125 | 132 | 140 | |
| Total input (mL) | 700 | NR | 1285 | 3320 | NR | 3040 | 4030 | 4240 | 3120 | 1900 |
| Urine output (mL) | 500 | NR | 2400 | 6501 | NR | 3150 | 3380 | 2950 | 1900 | 950 |
The clinical assessment was volume depletion given the high urine specific gravity, decreasing BP, and a negative fluid balance. The hyponatremia was determined to be due to sodium loss rather than dilution from inappropriate antidiuretic hormone secretion. Intravenous fluid (IVF) hydration with isotonic saline was initiated with a goal to keep the patient in positive fluid balance. The serum sodium level gradually improved to 140 mEq/L over the next 10 days. Attempts to decrease the rate of IVF resulted in a fall in serum sodium and improved when isotonic saline was increased. Eventually, the patient was placed on fludrocortisone, which normalized his urine output and serum sodium.
The response to the treatment regimen supported our diagnosis of cerebral salt wasting (CSW). The patient's serum sodium concentration upon discharge was 135 mEq/L.
Discussion
Our case illustrates the diagnostic challenge presented to physicians when they manage hyponatremia in the setting of a central nervous system (CNS) event. Hyponatremia (sodium 136 mEq/L) has been associated with confusion, lethargy, seizures, coma, and even death.1 Hyponatremia has been reported to occur in up to 30% of the patients with subarachnoid hemorrhage.2, 3
SIADH is frequently the cause of hyponatremia in a patient with a concurrent intracranial process. However, CSW is an important diagnosis to consider and differentiate from SIADH. In a retrospective review of 316 patients with subarachnoid hemorrhage and hyponatremia, 69% were determined to be due to SIADH while 6.5% were from CSW.4 Both CSW and SIADH have been reported to occur in the setting of head trauma, intracranial or metastatic neoplasm, carcinomatous or infectious meningitis, subarachnoid hemorrhage, and CNS surgery. Cryptococcal meningitis as an etiology of CSW has not been previously reported.
The main differentiating feature between SIADH and CSW is that CSW is a dysfunction of renal sodium absorption whereas in SIADH renal sodium handling is intact. This also leads to a difference in the extracellular volume status. SIADH is associated with an increased to normal volume status whereas CSW is a volume‐depleted state. Our patient exhibited a low serum osmolality and a high urine osmolality in the context of hyponatremia, which is present in both CSW and SIADH. However, the clinical course and presentation suggested volume loss, specifically the diarrhea, high urine specific gravity, declining BP, and a negative fluid balance. Some other features that are helpful in determining the volume status may include orthostatic changes, tachycardia, and skin turgor.
Our patient had a low serum uric acid, which is also present in both SIADH and CSW. The key difference between the 2 is that while uric acid will improve with resolution of hyponatremia in SIADH, it will remain low in CSW, as in our patient's uric acid levels, which remained low after normalization of the serum sodium.
Finally, the hyponatremia improved with isotonic fluid repletion, which would not occur in SIADH. The majority of the CSW patients will respond to volume repletion alone, as CSW is a transient condition that will usually resolve in 3 to 4 weeks.3 However, a few patients may require fludrocortisone, as was needed in our patient.
The renal wasting of sodium in CSW is poorly understood. Some postulated mechanisms cite the disruption of sympathetic neural input to the kidney and natriuresis induced by natriuretic peptides. Natriuretic peptides, in particular BNP, have been reported to be elevated in patients with CSW.5, 6 Natriuretic peptides cause salt wasting by inhibition of sodium reabsorption in renal tubule and intramedullary collecting.5, 6 Renin and aldosterone release can also inhibited by the natriuretic peptides. BNP levels were elevated in our patient despite volume loss and no signs of congestive heart failure. Cardiac congestion is a possible etiology for the elevated BNP levels, which peaked to 900 pg/mL on hospital day 37. However, 3 days later the BNP levels declined to 222 pg/mL despite the fact that he was continually in positive fluid balance, suggesting that the BNP elevation was due to CSW and not heart failure.
Conclusions
Our case illustrates the diagnostic and management challenge of hyponatremia in the setting of a CNS event. Both SIADH and CSW are possible etiologies but it is important to make a differentiation. Levels of natriuretic peptides and changes in fractional excretion of uric acid may help differentiate between the 2 conditions.6 The key difference mechanistically is that CSW is due to sodium‐handling deficits, whereas in SIADH sodium‐handling is intact. It is essential to establish volume status since SIADH is a euvolemic to mildly hypervolemic state vs. CSW, which is a volume‐depleted state.7
CSW is well recognized in the neurosurgical arena. The hospitalist will encounter neurosurgical patients with increasing frequency, and thus having an understanding of this disorder, including its diagnosis and treatment, is key.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- .Hyponatremia in acute brain disease: the cerebral salt wasting syndrome.Eur J Intern Med.2002;13(1):9–14.
- .Cerebral salt wasting syndrome: a review.Neurosurgery.1996;38(1):152–160.
- ,,, et al.The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage.Clin Endocrinol (Oxf).2006;64(3):250–254.
- ,,.Brain natriuretic peptide and cerebral vasospasm in subarachnoid hemorrhage. Clinical and TCD correlations.Stroke.2000;31(1):118–122.
- ,,, et al.Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage.Lancet.1997;349(9047):245–249.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83(5):905–908.
An 83‐year‐old man admitted for weakness, lethargy, and mental status changes was found to have human immunodeficiency virus (HIV) disease and cryptococcal meningitis. His hospital course was complicated by worsening hyponatremia (sodium 136 mEq/L). By hospital day 6, the patient's serum sodium had declined to 127 mEq/L from his admission level of 133 mEq/L. The initial impression was that the patient had syndrome of inappropriate antidiuretic hormone (SIADH) and fluid restriction to less than 1500 mL per day was initiated. By hospital day 11, serum sodium continued to decline, to 123 mEq/L, despite fluid restriction.
The past medical history was remarkable for coronary artery disease, hypertension, hyperlipidemia, and anemia, but by self‐report he had not been taking any medications. His review of systems was positive for intermittent bouts of diarrhea.
Vital signs on day 11 included a temperature of 37.3C, blood pressure (BP) of 105/55 mm Hg, and pulse of 90 beats per minute. The BP on admission had been 145/86 mm Hg but had steadily declined with fluid restriction. On physical examination, he appeared thin and cachetic with no evidence of jugular venous distention, rales, or peripheral edema to suggest volume overload. He had been receiving 2 to 4 L of isotonic saline daily for 5 days before the fluid restriction was initiated. The urine output continuously exceeded his intake by at least 500 mL per day throughout his hospital course. His only inpatient medications were amphotericin B and flucytosine. For nutritional supplementation, he was receiving a high‐calorie supplement with free‐water flushes via a nasogastric tube.
Laboratory results revealed a serum sodium concentration of 123 mEq/L, serum potassium of 4.4 mEq/L, serum creatinine of 0.6 mg/dL, urine sodium of 139 mEq/L, serum osmolality of 272 mOsm/kg, and urine osmolality of 598 mOsm/kg (see Table 1). Urinalysis revealed a specific gravity of 1.030. A random serum cortisol level was 11.1 g/dL. A thyroid‐stimulating hormone (TSH) level was 1.32 IU/mL. Brain natriuretic peptide (BNP) was elevated, at 686 pg/mL. A fractional excretion of uric acid was also elevated, at 83.8%.
| Parameters | Day 1 | Fluid Restriction Initiated: Day 6 | Day 8 | Fluid Resuscitation Initiated: Day 11 | Day 13 | Day 15 | Day 26 | Day 37* | Day 40 | Day 44 |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Na (mEq/L) | 133 | 127 | 126 | 123 | 131 | 119 | 140 | 131 | 132 | 135 |
| K (mEq/L) | 4.2 | 4.2 | ||||||||
| BUN (mg/dL) | 39 | 36 | ||||||||
| Cr (mg/dL) | 1.1 | 0.9 | ||||||||
| UNa (mEq/L) | 139 | 86 | 154 | 138 | ||||||
| UOsm (mOsm/kg) | 598 | 362 | 376 | |||||||
| SOsm (mOsm/kg) | 272 | 273 | 279 | |||||||
| BNP (pg/mL) | 686 | 900 | 222 | |||||||
| SUA (mg/dL) | 1.7 | 2.6 | 1.6 | |||||||
| UUA (mg/dL) | 38 | 11 | ||||||||
| FEUA (%) | 83.82 | 28.21 | ||||||||
| FENa (%) | 3.94 | 7.33 | ||||||||
| BP (mm Hg) | 147 | 136 | 122 | 105 | 101 | 90 | 125 | 132 | 140 | |
| Total input (mL) | 700 | NR | 1285 | 3320 | NR | 3040 | 4030 | 4240 | 3120 | 1900 |
| Urine output (mL) | 500 | NR | 2400 | 6501 | NR | 3150 | 3380 | 2950 | 1900 | 950 |
The clinical assessment was volume depletion given the high urine specific gravity, decreasing BP, and a negative fluid balance. The hyponatremia was determined to be due to sodium loss rather than dilution from inappropriate antidiuretic hormone secretion. Intravenous fluid (IVF) hydration with isotonic saline was initiated with a goal to keep the patient in positive fluid balance. The serum sodium level gradually improved to 140 mEq/L over the next 10 days. Attempts to decrease the rate of IVF resulted in a fall in serum sodium and improved when isotonic saline was increased. Eventually, the patient was placed on fludrocortisone, which normalized his urine output and serum sodium.
The response to the treatment regimen supported our diagnosis of cerebral salt wasting (CSW). The patient's serum sodium concentration upon discharge was 135 mEq/L.
Discussion
Our case illustrates the diagnostic challenge presented to physicians when they manage hyponatremia in the setting of a central nervous system (CNS) event. Hyponatremia (sodium 136 mEq/L) has been associated with confusion, lethargy, seizures, coma, and even death.1 Hyponatremia has been reported to occur in up to 30% of the patients with subarachnoid hemorrhage.2, 3
SIADH is frequently the cause of hyponatremia in a patient with a concurrent intracranial process. However, CSW is an important diagnosis to consider and differentiate from SIADH. In a retrospective review of 316 patients with subarachnoid hemorrhage and hyponatremia, 69% were determined to be due to SIADH while 6.5% were from CSW.4 Both CSW and SIADH have been reported to occur in the setting of head trauma, intracranial or metastatic neoplasm, carcinomatous or infectious meningitis, subarachnoid hemorrhage, and CNS surgery. Cryptococcal meningitis as an etiology of CSW has not been previously reported.
The main differentiating feature between SIADH and CSW is that CSW is a dysfunction of renal sodium absorption whereas in SIADH renal sodium handling is intact. This also leads to a difference in the extracellular volume status. SIADH is associated with an increased to normal volume status whereas CSW is a volume‐depleted state. Our patient exhibited a low serum osmolality and a high urine osmolality in the context of hyponatremia, which is present in both CSW and SIADH. However, the clinical course and presentation suggested volume loss, specifically the diarrhea, high urine specific gravity, declining BP, and a negative fluid balance. Some other features that are helpful in determining the volume status may include orthostatic changes, tachycardia, and skin turgor.
Our patient had a low serum uric acid, which is also present in both SIADH and CSW. The key difference between the 2 is that while uric acid will improve with resolution of hyponatremia in SIADH, it will remain low in CSW, as in our patient's uric acid levels, which remained low after normalization of the serum sodium.
Finally, the hyponatremia improved with isotonic fluid repletion, which would not occur in SIADH. The majority of the CSW patients will respond to volume repletion alone, as CSW is a transient condition that will usually resolve in 3 to 4 weeks.3 However, a few patients may require fludrocortisone, as was needed in our patient.
The renal wasting of sodium in CSW is poorly understood. Some postulated mechanisms cite the disruption of sympathetic neural input to the kidney and natriuresis induced by natriuretic peptides. Natriuretic peptides, in particular BNP, have been reported to be elevated in patients with CSW.5, 6 Natriuretic peptides cause salt wasting by inhibition of sodium reabsorption in renal tubule and intramedullary collecting.5, 6 Renin and aldosterone release can also inhibited by the natriuretic peptides. BNP levels were elevated in our patient despite volume loss and no signs of congestive heart failure. Cardiac congestion is a possible etiology for the elevated BNP levels, which peaked to 900 pg/mL on hospital day 37. However, 3 days later the BNP levels declined to 222 pg/mL despite the fact that he was continually in positive fluid balance, suggesting that the BNP elevation was due to CSW and not heart failure.
Conclusions
Our case illustrates the diagnostic and management challenge of hyponatremia in the setting of a CNS event. Both SIADH and CSW are possible etiologies but it is important to make a differentiation. Levels of natriuretic peptides and changes in fractional excretion of uric acid may help differentiate between the 2 conditions.6 The key difference mechanistically is that CSW is due to sodium‐handling deficits, whereas in SIADH sodium‐handling is intact. It is essential to establish volume status since SIADH is a euvolemic to mildly hypervolemic state vs. CSW, which is a volume‐depleted state.7
CSW is well recognized in the neurosurgical arena. The hospitalist will encounter neurosurgical patients with increasing frequency, and thus having an understanding of this disorder, including its diagnosis and treatment, is key.
An 83‐year‐old man admitted for weakness, lethargy, and mental status changes was found to have human immunodeficiency virus (HIV) disease and cryptococcal meningitis. His hospital course was complicated by worsening hyponatremia (sodium 136 mEq/L). By hospital day 6, the patient's serum sodium had declined to 127 mEq/L from his admission level of 133 mEq/L. The initial impression was that the patient had syndrome of inappropriate antidiuretic hormone (SIADH) and fluid restriction to less than 1500 mL per day was initiated. By hospital day 11, serum sodium continued to decline, to 123 mEq/L, despite fluid restriction.
The past medical history was remarkable for coronary artery disease, hypertension, hyperlipidemia, and anemia, but by self‐report he had not been taking any medications. His review of systems was positive for intermittent bouts of diarrhea.
Vital signs on day 11 included a temperature of 37.3C, blood pressure (BP) of 105/55 mm Hg, and pulse of 90 beats per minute. The BP on admission had been 145/86 mm Hg but had steadily declined with fluid restriction. On physical examination, he appeared thin and cachetic with no evidence of jugular venous distention, rales, or peripheral edema to suggest volume overload. He had been receiving 2 to 4 L of isotonic saline daily for 5 days before the fluid restriction was initiated. The urine output continuously exceeded his intake by at least 500 mL per day throughout his hospital course. His only inpatient medications were amphotericin B and flucytosine. For nutritional supplementation, he was receiving a high‐calorie supplement with free‐water flushes via a nasogastric tube.
Laboratory results revealed a serum sodium concentration of 123 mEq/L, serum potassium of 4.4 mEq/L, serum creatinine of 0.6 mg/dL, urine sodium of 139 mEq/L, serum osmolality of 272 mOsm/kg, and urine osmolality of 598 mOsm/kg (see Table 1). Urinalysis revealed a specific gravity of 1.030. A random serum cortisol level was 11.1 g/dL. A thyroid‐stimulating hormone (TSH) level was 1.32 IU/mL. Brain natriuretic peptide (BNP) was elevated, at 686 pg/mL. A fractional excretion of uric acid was also elevated, at 83.8%.
| Parameters | Day 1 | Fluid Restriction Initiated: Day 6 | Day 8 | Fluid Resuscitation Initiated: Day 11 | Day 13 | Day 15 | Day 26 | Day 37* | Day 40 | Day 44 |
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
| Na (mEq/L) | 133 | 127 | 126 | 123 | 131 | 119 | 140 | 131 | 132 | 135 |
| K (mEq/L) | 4.2 | 4.2 | ||||||||
| BUN (mg/dL) | 39 | 36 | ||||||||
| Cr (mg/dL) | 1.1 | 0.9 | ||||||||
| UNa (mEq/L) | 139 | 86 | 154 | 138 | ||||||
| UOsm (mOsm/kg) | 598 | 362 | 376 | |||||||
| SOsm (mOsm/kg) | 272 | 273 | 279 | |||||||
| BNP (pg/mL) | 686 | 900 | 222 | |||||||
| SUA (mg/dL) | 1.7 | 2.6 | 1.6 | |||||||
| UUA (mg/dL) | 38 | 11 | ||||||||
| FEUA (%) | 83.82 | 28.21 | ||||||||
| FENa (%) | 3.94 | 7.33 | ||||||||
| BP (mm Hg) | 147 | 136 | 122 | 105 | 101 | 90 | 125 | 132 | 140 | |
| Total input (mL) | 700 | NR | 1285 | 3320 | NR | 3040 | 4030 | 4240 | 3120 | 1900 |
| Urine output (mL) | 500 | NR | 2400 | 6501 | NR | 3150 | 3380 | 2950 | 1900 | 950 |
The clinical assessment was volume depletion given the high urine specific gravity, decreasing BP, and a negative fluid balance. The hyponatremia was determined to be due to sodium loss rather than dilution from inappropriate antidiuretic hormone secretion. Intravenous fluid (IVF) hydration with isotonic saline was initiated with a goal to keep the patient in positive fluid balance. The serum sodium level gradually improved to 140 mEq/L over the next 10 days. Attempts to decrease the rate of IVF resulted in a fall in serum sodium and improved when isotonic saline was increased. Eventually, the patient was placed on fludrocortisone, which normalized his urine output and serum sodium.
The response to the treatment regimen supported our diagnosis of cerebral salt wasting (CSW). The patient's serum sodium concentration upon discharge was 135 mEq/L.
Discussion
Our case illustrates the diagnostic challenge presented to physicians when they manage hyponatremia in the setting of a central nervous system (CNS) event. Hyponatremia (sodium 136 mEq/L) has been associated with confusion, lethargy, seizures, coma, and even death.1 Hyponatremia has been reported to occur in up to 30% of the patients with subarachnoid hemorrhage.2, 3
SIADH is frequently the cause of hyponatremia in a patient with a concurrent intracranial process. However, CSW is an important diagnosis to consider and differentiate from SIADH. In a retrospective review of 316 patients with subarachnoid hemorrhage and hyponatremia, 69% were determined to be due to SIADH while 6.5% were from CSW.4 Both CSW and SIADH have been reported to occur in the setting of head trauma, intracranial or metastatic neoplasm, carcinomatous or infectious meningitis, subarachnoid hemorrhage, and CNS surgery. Cryptococcal meningitis as an etiology of CSW has not been previously reported.
The main differentiating feature between SIADH and CSW is that CSW is a dysfunction of renal sodium absorption whereas in SIADH renal sodium handling is intact. This also leads to a difference in the extracellular volume status. SIADH is associated with an increased to normal volume status whereas CSW is a volume‐depleted state. Our patient exhibited a low serum osmolality and a high urine osmolality in the context of hyponatremia, which is present in both CSW and SIADH. However, the clinical course and presentation suggested volume loss, specifically the diarrhea, high urine specific gravity, declining BP, and a negative fluid balance. Some other features that are helpful in determining the volume status may include orthostatic changes, tachycardia, and skin turgor.
Our patient had a low serum uric acid, which is also present in both SIADH and CSW. The key difference between the 2 is that while uric acid will improve with resolution of hyponatremia in SIADH, it will remain low in CSW, as in our patient's uric acid levels, which remained low after normalization of the serum sodium.
Finally, the hyponatremia improved with isotonic fluid repletion, which would not occur in SIADH. The majority of the CSW patients will respond to volume repletion alone, as CSW is a transient condition that will usually resolve in 3 to 4 weeks.3 However, a few patients may require fludrocortisone, as was needed in our patient.
The renal wasting of sodium in CSW is poorly understood. Some postulated mechanisms cite the disruption of sympathetic neural input to the kidney and natriuresis induced by natriuretic peptides. Natriuretic peptides, in particular BNP, have been reported to be elevated in patients with CSW.5, 6 Natriuretic peptides cause salt wasting by inhibition of sodium reabsorption in renal tubule and intramedullary collecting.5, 6 Renin and aldosterone release can also inhibited by the natriuretic peptides. BNP levels were elevated in our patient despite volume loss and no signs of congestive heart failure. Cardiac congestion is a possible etiology for the elevated BNP levels, which peaked to 900 pg/mL on hospital day 37. However, 3 days later the BNP levels declined to 222 pg/mL despite the fact that he was continually in positive fluid balance, suggesting that the BNP elevation was due to CSW and not heart failure.
Conclusions
Our case illustrates the diagnostic and management challenge of hyponatremia in the setting of a CNS event. Both SIADH and CSW are possible etiologies but it is important to make a differentiation. Levels of natriuretic peptides and changes in fractional excretion of uric acid may help differentiate between the 2 conditions.6 The key difference mechanistically is that CSW is due to sodium‐handling deficits, whereas in SIADH sodium‐handling is intact. It is essential to establish volume status since SIADH is a euvolemic to mildly hypervolemic state vs. CSW, which is a volume‐depleted state.7
CSW is well recognized in the neurosurgical arena. The hospitalist will encounter neurosurgical patients with increasing frequency, and thus having an understanding of this disorder, including its diagnosis and treatment, is key.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- .Hyponatremia in acute brain disease: the cerebral salt wasting syndrome.Eur J Intern Med.2002;13(1):9–14.
- .Cerebral salt wasting syndrome: a review.Neurosurgery.1996;38(1):152–160.
- ,,, et al.The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage.Clin Endocrinol (Oxf).2006;64(3):250–254.
- ,,.Brain natriuretic peptide and cerebral vasospasm in subarachnoid hemorrhage. Clinical and TCD correlations.Stroke.2000;31(1):118–122.
- ,,, et al.Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage.Lancet.1997;349(9047):245–249.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83(5):905–908.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- .Hyponatremia in acute brain disease: the cerebral salt wasting syndrome.Eur J Intern Med.2002;13(1):9–14.
- .Cerebral salt wasting syndrome: a review.Neurosurgery.1996;38(1):152–160.
- ,,, et al.The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage.Clin Endocrinol (Oxf).2006;64(3):250–254.
- ,,.Brain natriuretic peptide and cerebral vasospasm in subarachnoid hemorrhage. Clinical and TCD correlations.Stroke.2000;31(1):118–122.
- ,,, et al.Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage.Lancet.1997;349(9047):245–249.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83(5):905–908.
Cefepime: Underrecognized Cause of NCSE
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.