User login
Autosomal-Dominant Familial Angiolipomatosis
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
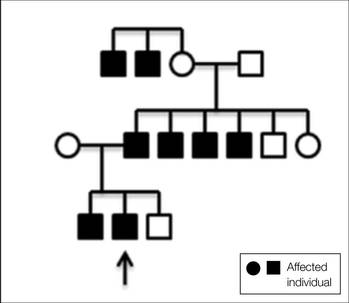
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
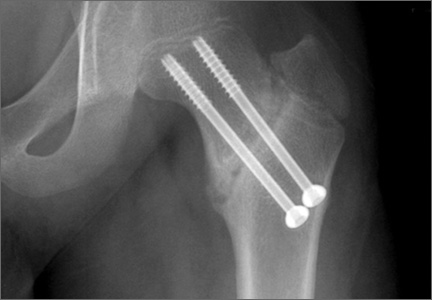
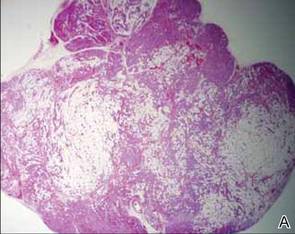
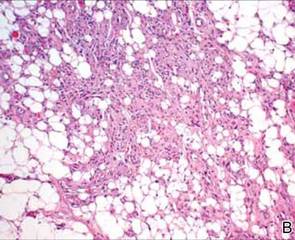
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
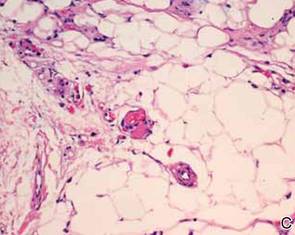
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.
Easy bruising • low platelet count • recent cold-like illness • Dx?
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
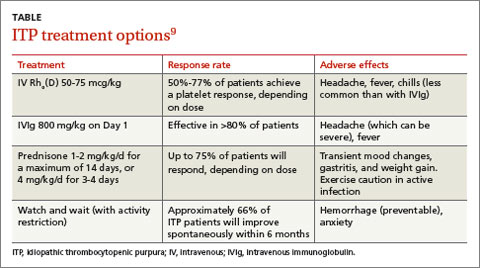
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
Hyperthyroidism • myalgia • rapidly progressing paralysis • Dx?
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
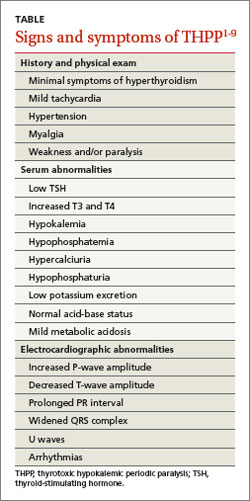
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.

Case Report: Conus Medullaris Syndrome From Spinal Metastasis
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.

Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
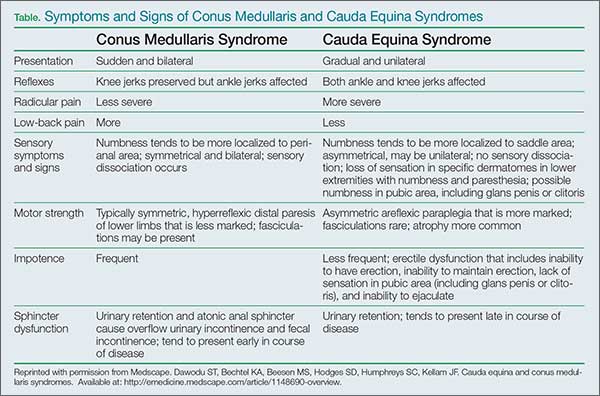
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.
Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.
Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.

Case Report: Rapidly Ascending Weakness in a 22-year-old Man
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
Case Studies in Toxicology: You Can’t See Dragonfly or Hear NBOMe, but They Can Still Hurt You
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
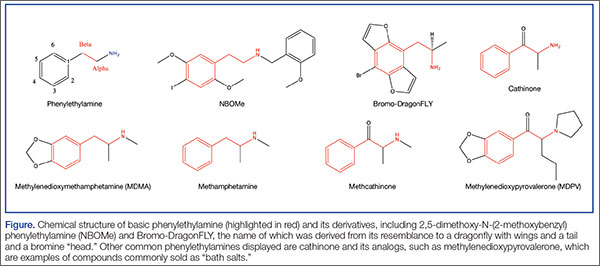
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.

Unusual Form and Location of a Tumor: Multiosseous Ewing Sarcoma in the Foot
Ewing sarcomas are characterized as primitive malignant round cell tumors.1 These tumors are diagnosed by neuroectodermal differentiation and by their common histologic and immunohistochemical properties.2 Ewing sarcoma is the second most common malignant bone tumor in adolescents and young adults. It is the fourth most common primary malignant tumor, accounting for about 9% of all malignant tumors of bone. The most common primary bone tumors are multiple myeloma, osteosarcoma, and chondrosarcoma.3
The diaphyses of long bones (eg, femur, tibia, humerus) and flat bones (eg, pelvis, scapula) are the most commonly involved sites. Involvement of bones in the hands and feet is uncommon (3%-5% of reported cases).4 The foot bones most commonly involved include the calcaneus and the metatarsals, in the series by Casadei and colleagues.5
About 90% of Ewing sarcoma cases present before age 20 years (mean age, 13 years).6 Typical presentation is that of localized pain at the involved site. Some patients have systemic symptoms, such as fever, malaise, weight loss, leukocytosis, and increased erythrocyte sedimentation rate (ESR) mimicking infection. Radiographically, Ewing sarcoma appears as a permeative destructive bone lesion with a moth-eaten appearance (almost 76% of cases).7 This is usually associated with lamellated periosteal new bone formation or an “onion skin” appearance. Less commonly, a sunburst configuration with an associated soft-tissue mass can be seen. Computed tomography (CT) and magnetic resonance imaging (MRI) show the osseous extent of the tumor and the presence or absence of the soft-tissue component of the tumor. Radionuclide bone scans show increased technetium-99m methylene diphosphonate accumulation and are typically hot.6
Histopathologically, the tumor is composed of small, uniformly sized cells characterized by an almost clear eosinophilic cytoplasm and very little intercellular matrix. There are lobules and strands divided by prominent septa. Macroscopically, appearance can range from a soft, fleshy solid mass to an almost liquid form, as the lesion does not produce any matrix. At time of surgery, the tumor may have a liquefied component and the appearance of pus.6 Prognostic factors are tumor site in foot and treatment according to the series by Casadei and colleagues.5 Patients with large central tumors, especially in the pelvis, have worse outcomes than patients with distal tumors.8
In this article, we report a case of multifocal Ewing sarcoma involving multiple bones in the foot. Given the multifocal nature of the disease confined to the foot, the initial impression was that of osteomyelitis. We describe the histologic, radiologic, and diagnostic features of the tumor and outline treatment and prognosis. To our knowledge, this is the first report of multifocal Ewing sarcoma involving multiple bones in the foot. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old man visited our clinic with the chief complaints of left foot pain and swelling. The pain started 10 months earlier and was followed by swelling. Complaints started after a minor local trauma. The man sought outside medical attention 8 months after pain onset. At his first visit at another institution, an initial radiograph was reported as normal, and all laboratory measures, including complete blood cell count (CBC) differential, ESR, and C-reactive protein (CRP) level, were within normal limits. Under the erroneous diagnosis of infection, the patient was treated with cloxacillin 500 mg 4 times a day for 4 weeks.
The patient’s pain had started 10 months before presentation (2 months after antibiotic therapy was initiated) (Figure 1). Physical examination at our institution revealed a palpable mass on the dorsum of the left foot. Anteroposterior and lateral plain radiographs showed a permeative lytic lesion with cortical destruction in the left calcaneus, navicular, cuboid, and cuneiform bones and in all metatarsal bones except the first (Figure 2). A soft-tissue mass around the involved bones was noted as well. The talus was not involved (Figure 3).
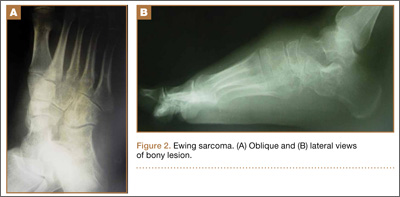
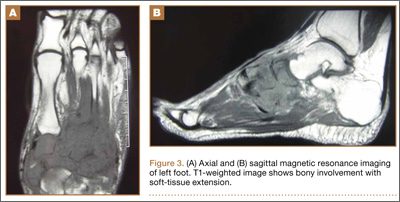
CT showed permeative destruction of left foot bones, including the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first. Invasion through the overlying cortex of the involved bones indicated aggressive biological activity of the tumor (Figure 4). MRI showed a destructive bony lesion of the mentioned bones associated with the soft-tissue mass (Figure 3).
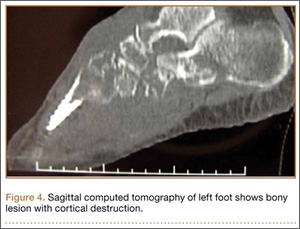
Bone scan showed increased uptake in the involved areas (Figure 5). Chest plain radiographs and CT showed no distant metastasis. An incisional biopsy was performed, and histopathology showed a malignant small round cell tumor, identified as Ewing sarcoma (Figure 6). An immunohistochemistry study demonstrated positive CD99 and negative cytokeratin, leukocyte common antigen, desmin, and synaptophysin.
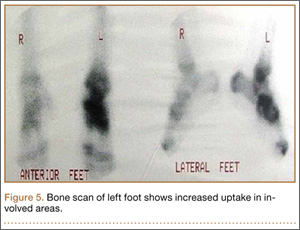
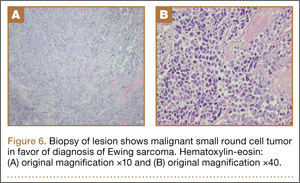
The patient was started on 4 cycles of adjuvant chemotherapy. Cycles 1 and 3 involved cyclophosphamide 2 g, vincristine 2 g, and doxorubicin 50 mg; cycles 2 and 4 involved ifosfamide 3.5 g and etoposide 200 mg. Tumor shrinkage occurred after chemotherapy. Clinical response to preoperative chemotherapy was documented by a decrease in tumor size at follow-ups. The patient underwent below-knee amputation.
Postoperative histopathology confirmed the diagnosis of Ewing sarcoma of the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first (Figure 7). At 2-year follow-up, the patient had no evidence of local recurrence or distant metastasis based on chest CT and clinical examination of the affected site.
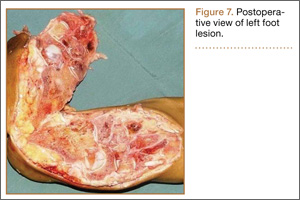
Discussion
Ewing sarcoma is the prototype of round small cell malignancies that arise from the long bones and the flat bones. It seldom involves the hands or feet. To our knowledge, this is the first report of Ewing sarcoma of the foot with multiple-bone involvement. Our literature review found a case of Ewing sarcoma of the first phalanx of the third toe, the second metatarsal bone, the cuneiform, the cuboid, and the talus, with lesser soft-tissue extension compared with our patient’s case.9
As this foot tumor is rare, there are few reports on its clinical aspects, appropriate treatment, and long-term outcome. For treatment of nonmetastatic Ewing sarcoma, limb-salvage surgery is advised if response has been good and there is a reasonable expectation of negative margins and good functional results.
Radiation and surgery may be part of the overall treatment plan. Radiation without surgery has a unique role in pelvic Ewing sarcoma, in contrast to extremity Ewing sarcoma. In our opinion, margins and histologic necrosis in the resection specimen are examined after surgery, and, if the margins are widely negative and histologic response is good, no further local control is advised. If the margin is positive, postoperative radiation therapy is recommended.1 Amputation has gradually become a (rare) choice in the treatment of extremity sarcomas.9 In our patient’s case, surgery was preferred over radiation therapy after chemotherapy because of the low risk of local side effects and the expected high efficacy. In addition, radiation at such high doses for Ewing sarcoma in the foot causes functional impairment. Because of the multiple-bone involvement, a salvage procedure was not possible for our patient. Given the calcaneal involvement, however, below-knee amputation was considered safer than ankle disarticulation.
Multiple-bone involvement occurs in the advanced stage of Ewing sarcoma, usually after visceral and pulmonary metastases are detected.9 The case reported by Rammal and colleagues9 had both multiple-bone involvement in the foot and pulmonary metastasis. The authors indicated that hematogenous spread of the tumor was discerned because the lesions were noncontiguous.9 Our patient had no distant metastases. We think his tumor originated in a tarsal or midtarsal bone and extended to adjacent bones. Therefore, it probably spread through its capsular and ligamentous attachment among tarsal and midtarsal bones, as the involvement was contiguous rather than distinct.
Average delay from symptom onset to diagnosis was reported to be 34 weeks.3 Average physician delay from initial visit to correct diagnosis was reported to be 19 weeks.3 Patients may have erythema, fever, and swelling, suggestive of osteomyelitis.3 Laboratory results may show increased white blood cell count and elevated ESR and CRP level.3 In addition, needle biopsy of the tumor may reveal an appearance grossly similar to that of pus.3 Therefore, physicians may send all the tissue out for microbiological analysis (according to the erroneous diagnosis of infection) and none out for pathologic analysis. The situation can be further complicated when Ewing sarcoma occurs in the foot, an uncommon site. In this special case, multiple-bone involvement can present a misleading clinical picture of infection.10 In other words, infection is one of the best choices in the differential diagnosis.7 Also to be considered are multicentric giant cell tumor, fibrosarcoma,11 and osteosarcoma.12
1. Herring JA. Malignant tumors of bone. In: Herring JA, ed. Tachdjian’s Pediatric Orthopaedics. Philadelphia, PA: Saunders Elsevier; 2008:2324-2327.
2. Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am J Pathol. 1987;127(3):507-518.
3. Canale ST, Beaty JH. Malignant tumors of bone. In: Canale ST, ed. Campbell’s Operative Orthopaedics. Philadelphia, PA: Mosby Elsevier; 2008:910-913.
4. Unni KK. Ewing sarcoma. In: Unni KK, ed. Dahlin’s Bone Tumor: General Aspects and Data on 11087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:121-142.
5. Casadei R, Magnani M, Biagini R, Mercuri M. Prognostic factors in Ewing’s sarcoma of the foot. Clin Orthop. 2004;(420):230-238.
6. Greenspan A, Jundt G, Remagen W. Bone-forming (osteogenic) lesions. In: Greenspan A, Jundt G, Remagen W, eds. Differential Diagnosis in Orthopaedic Oncology. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:114.
7. Metcalfe JE, Grimer RJ. Ewing’s sarcoma of the foot masquerading as osteomyelitis. Foot Ankle Surg. 2004;10(1):29-33.
8. Hoffmann C, Ahrens S, Dunst J, et al. Pelvis Ewing sarcoma: a retrospective analysis of 241 cases. Cancer. 1999;85(4):869-877.
9. Rammal H, Ghanem I, Torbey PH, Dagher F, Kharrat K. Multifocal Ewing sarcoma of the foot. J Pediatr Hematol Oncol. 2008;30(4):298-300.
10. Ledermann HP, Morrison WB, Schweitzer ME. MR image analysis of pedal osteomyelitis: distribution, patterns of spread, and frequency of associated ulceration and septic arthritis. Radiology. 2002;223(3):747-755.
11. Dhillon MS, Prabhudev Prasad AP, Virk MS, Aggarwal S. Multicentric giant cell tumor involving the same foot: a case report and review of literature. Indian J Orthop. 2007;41(2):154-157.
12. Baraga JJ, Amarami KK, Swee RG, Wold L, Unni KK. Radiographic features of Ewing’s sarcoma of the bones of the hand and feet. Skeletal Radiol. 2001;30(3):121-126.
Ewing sarcomas are characterized as primitive malignant round cell tumors.1 These tumors are diagnosed by neuroectodermal differentiation and by their common histologic and immunohistochemical properties.2 Ewing sarcoma is the second most common malignant bone tumor in adolescents and young adults. It is the fourth most common primary malignant tumor, accounting for about 9% of all malignant tumors of bone. The most common primary bone tumors are multiple myeloma, osteosarcoma, and chondrosarcoma.3
The diaphyses of long bones (eg, femur, tibia, humerus) and flat bones (eg, pelvis, scapula) are the most commonly involved sites. Involvement of bones in the hands and feet is uncommon (3%-5% of reported cases).4 The foot bones most commonly involved include the calcaneus and the metatarsals, in the series by Casadei and colleagues.5
About 90% of Ewing sarcoma cases present before age 20 years (mean age, 13 years).6 Typical presentation is that of localized pain at the involved site. Some patients have systemic symptoms, such as fever, malaise, weight loss, leukocytosis, and increased erythrocyte sedimentation rate (ESR) mimicking infection. Radiographically, Ewing sarcoma appears as a permeative destructive bone lesion with a moth-eaten appearance (almost 76% of cases).7 This is usually associated with lamellated periosteal new bone formation or an “onion skin” appearance. Less commonly, a sunburst configuration with an associated soft-tissue mass can be seen. Computed tomography (CT) and magnetic resonance imaging (MRI) show the osseous extent of the tumor and the presence or absence of the soft-tissue component of the tumor. Radionuclide bone scans show increased technetium-99m methylene diphosphonate accumulation and are typically hot.6
Histopathologically, the tumor is composed of small, uniformly sized cells characterized by an almost clear eosinophilic cytoplasm and very little intercellular matrix. There are lobules and strands divided by prominent septa. Macroscopically, appearance can range from a soft, fleshy solid mass to an almost liquid form, as the lesion does not produce any matrix. At time of surgery, the tumor may have a liquefied component and the appearance of pus.6 Prognostic factors are tumor site in foot and treatment according to the series by Casadei and colleagues.5 Patients with large central tumors, especially in the pelvis, have worse outcomes than patients with distal tumors.8
In this article, we report a case of multifocal Ewing sarcoma involving multiple bones in the foot. Given the multifocal nature of the disease confined to the foot, the initial impression was that of osteomyelitis. We describe the histologic, radiologic, and diagnostic features of the tumor and outline treatment and prognosis. To our knowledge, this is the first report of multifocal Ewing sarcoma involving multiple bones in the foot. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old man visited our clinic with the chief complaints of left foot pain and swelling. The pain started 10 months earlier and was followed by swelling. Complaints started after a minor local trauma. The man sought outside medical attention 8 months after pain onset. At his first visit at another institution, an initial radiograph was reported as normal, and all laboratory measures, including complete blood cell count (CBC) differential, ESR, and C-reactive protein (CRP) level, were within normal limits. Under the erroneous diagnosis of infection, the patient was treated with cloxacillin 500 mg 4 times a day for 4 weeks.
The patient’s pain had started 10 months before presentation (2 months after antibiotic therapy was initiated) (Figure 1). Physical examination at our institution revealed a palpable mass on the dorsum of the left foot. Anteroposterior and lateral plain radiographs showed a permeative lytic lesion with cortical destruction in the left calcaneus, navicular, cuboid, and cuneiform bones and in all metatarsal bones except the first (Figure 2). A soft-tissue mass around the involved bones was noted as well. The talus was not involved (Figure 3).
CT showed permeative destruction of left foot bones, including the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first. Invasion through the overlying cortex of the involved bones indicated aggressive biological activity of the tumor (Figure 4). MRI showed a destructive bony lesion of the mentioned bones associated with the soft-tissue mass (Figure 3).
Bone scan showed increased uptake in the involved areas (Figure 5). Chest plain radiographs and CT showed no distant metastasis. An incisional biopsy was performed, and histopathology showed a malignant small round cell tumor, identified as Ewing sarcoma (Figure 6). An immunohistochemistry study demonstrated positive CD99 and negative cytokeratin, leukocyte common antigen, desmin, and synaptophysin.
The patient was started on 4 cycles of adjuvant chemotherapy. Cycles 1 and 3 involved cyclophosphamide 2 g, vincristine 2 g, and doxorubicin 50 mg; cycles 2 and 4 involved ifosfamide 3.5 g and etoposide 200 mg. Tumor shrinkage occurred after chemotherapy. Clinical response to preoperative chemotherapy was documented by a decrease in tumor size at follow-ups. The patient underwent below-knee amputation.
Postoperative histopathology confirmed the diagnosis of Ewing sarcoma of the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first (Figure 7). At 2-year follow-up, the patient had no evidence of local recurrence or distant metastasis based on chest CT and clinical examination of the affected site.
Discussion
Ewing sarcoma is the prototype of round small cell malignancies that arise from the long bones and the flat bones. It seldom involves the hands or feet. To our knowledge, this is the first report of Ewing sarcoma of the foot with multiple-bone involvement. Our literature review found a case of Ewing sarcoma of the first phalanx of the third toe, the second metatarsal bone, the cuneiform, the cuboid, and the talus, with lesser soft-tissue extension compared with our patient’s case.9
As this foot tumor is rare, there are few reports on its clinical aspects, appropriate treatment, and long-term outcome. For treatment of nonmetastatic Ewing sarcoma, limb-salvage surgery is advised if response has been good and there is a reasonable expectation of negative margins and good functional results.
Radiation and surgery may be part of the overall treatment plan. Radiation without surgery has a unique role in pelvic Ewing sarcoma, in contrast to extremity Ewing sarcoma. In our opinion, margins and histologic necrosis in the resection specimen are examined after surgery, and, if the margins are widely negative and histologic response is good, no further local control is advised. If the margin is positive, postoperative radiation therapy is recommended.1 Amputation has gradually become a (rare) choice in the treatment of extremity sarcomas.9 In our patient’s case, surgery was preferred over radiation therapy after chemotherapy because of the low risk of local side effects and the expected high efficacy. In addition, radiation at such high doses for Ewing sarcoma in the foot causes functional impairment. Because of the multiple-bone involvement, a salvage procedure was not possible for our patient. Given the calcaneal involvement, however, below-knee amputation was considered safer than ankle disarticulation.
Multiple-bone involvement occurs in the advanced stage of Ewing sarcoma, usually after visceral and pulmonary metastases are detected.9 The case reported by Rammal and colleagues9 had both multiple-bone involvement in the foot and pulmonary metastasis. The authors indicated that hematogenous spread of the tumor was discerned because the lesions were noncontiguous.9 Our patient had no distant metastases. We think his tumor originated in a tarsal or midtarsal bone and extended to adjacent bones. Therefore, it probably spread through its capsular and ligamentous attachment among tarsal and midtarsal bones, as the involvement was contiguous rather than distinct.
Average delay from symptom onset to diagnosis was reported to be 34 weeks.3 Average physician delay from initial visit to correct diagnosis was reported to be 19 weeks.3 Patients may have erythema, fever, and swelling, suggestive of osteomyelitis.3 Laboratory results may show increased white blood cell count and elevated ESR and CRP level.3 In addition, needle biopsy of the tumor may reveal an appearance grossly similar to that of pus.3 Therefore, physicians may send all the tissue out for microbiological analysis (according to the erroneous diagnosis of infection) and none out for pathologic analysis. The situation can be further complicated when Ewing sarcoma occurs in the foot, an uncommon site. In this special case, multiple-bone involvement can present a misleading clinical picture of infection.10 In other words, infection is one of the best choices in the differential diagnosis.7 Also to be considered are multicentric giant cell tumor, fibrosarcoma,11 and osteosarcoma.12
Ewing sarcomas are characterized as primitive malignant round cell tumors.1 These tumors are diagnosed by neuroectodermal differentiation and by their common histologic and immunohistochemical properties.2 Ewing sarcoma is the second most common malignant bone tumor in adolescents and young adults. It is the fourth most common primary malignant tumor, accounting for about 9% of all malignant tumors of bone. The most common primary bone tumors are multiple myeloma, osteosarcoma, and chondrosarcoma.3
The diaphyses of long bones (eg, femur, tibia, humerus) and flat bones (eg, pelvis, scapula) are the most commonly involved sites. Involvement of bones in the hands and feet is uncommon (3%-5% of reported cases).4 The foot bones most commonly involved include the calcaneus and the metatarsals, in the series by Casadei and colleagues.5
About 90% of Ewing sarcoma cases present before age 20 years (mean age, 13 years).6 Typical presentation is that of localized pain at the involved site. Some patients have systemic symptoms, such as fever, malaise, weight loss, leukocytosis, and increased erythrocyte sedimentation rate (ESR) mimicking infection. Radiographically, Ewing sarcoma appears as a permeative destructive bone lesion with a moth-eaten appearance (almost 76% of cases).7 This is usually associated with lamellated periosteal new bone formation or an “onion skin” appearance. Less commonly, a sunburst configuration with an associated soft-tissue mass can be seen. Computed tomography (CT) and magnetic resonance imaging (MRI) show the osseous extent of the tumor and the presence or absence of the soft-tissue component of the tumor. Radionuclide bone scans show increased technetium-99m methylene diphosphonate accumulation and are typically hot.6
Histopathologically, the tumor is composed of small, uniformly sized cells characterized by an almost clear eosinophilic cytoplasm and very little intercellular matrix. There are lobules and strands divided by prominent septa. Macroscopically, appearance can range from a soft, fleshy solid mass to an almost liquid form, as the lesion does not produce any matrix. At time of surgery, the tumor may have a liquefied component and the appearance of pus.6 Prognostic factors are tumor site in foot and treatment according to the series by Casadei and colleagues.5 Patients with large central tumors, especially in the pelvis, have worse outcomes than patients with distal tumors.8
In this article, we report a case of multifocal Ewing sarcoma involving multiple bones in the foot. Given the multifocal nature of the disease confined to the foot, the initial impression was that of osteomyelitis. We describe the histologic, radiologic, and diagnostic features of the tumor and outline treatment and prognosis. To our knowledge, this is the first report of multifocal Ewing sarcoma involving multiple bones in the foot. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old man visited our clinic with the chief complaints of left foot pain and swelling. The pain started 10 months earlier and was followed by swelling. Complaints started after a minor local trauma. The man sought outside medical attention 8 months after pain onset. At his first visit at another institution, an initial radiograph was reported as normal, and all laboratory measures, including complete blood cell count (CBC) differential, ESR, and C-reactive protein (CRP) level, were within normal limits. Under the erroneous diagnosis of infection, the patient was treated with cloxacillin 500 mg 4 times a day for 4 weeks.
The patient’s pain had started 10 months before presentation (2 months after antibiotic therapy was initiated) (Figure 1). Physical examination at our institution revealed a palpable mass on the dorsum of the left foot. Anteroposterior and lateral plain radiographs showed a permeative lytic lesion with cortical destruction in the left calcaneus, navicular, cuboid, and cuneiform bones and in all metatarsal bones except the first (Figure 2). A soft-tissue mass around the involved bones was noted as well. The talus was not involved (Figure 3).
CT showed permeative destruction of left foot bones, including the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first. Invasion through the overlying cortex of the involved bones indicated aggressive biological activity of the tumor (Figure 4). MRI showed a destructive bony lesion of the mentioned bones associated with the soft-tissue mass (Figure 3).
Bone scan showed increased uptake in the involved areas (Figure 5). Chest plain radiographs and CT showed no distant metastasis. An incisional biopsy was performed, and histopathology showed a malignant small round cell tumor, identified as Ewing sarcoma (Figure 6). An immunohistochemistry study demonstrated positive CD99 and negative cytokeratin, leukocyte common antigen, desmin, and synaptophysin.
The patient was started on 4 cycles of adjuvant chemotherapy. Cycles 1 and 3 involved cyclophosphamide 2 g, vincristine 2 g, and doxorubicin 50 mg; cycles 2 and 4 involved ifosfamide 3.5 g and etoposide 200 mg. Tumor shrinkage occurred after chemotherapy. Clinical response to preoperative chemotherapy was documented by a decrease in tumor size at follow-ups. The patient underwent below-knee amputation.
Postoperative histopathology confirmed the diagnosis of Ewing sarcoma of the calcaneus, navicular, cuboid, and cuneiform bones and all metatarsal bones except the first (Figure 7). At 2-year follow-up, the patient had no evidence of local recurrence or distant metastasis based on chest CT and clinical examination of the affected site.
Discussion
Ewing sarcoma is the prototype of round small cell malignancies that arise from the long bones and the flat bones. It seldom involves the hands or feet. To our knowledge, this is the first report of Ewing sarcoma of the foot with multiple-bone involvement. Our literature review found a case of Ewing sarcoma of the first phalanx of the third toe, the second metatarsal bone, the cuneiform, the cuboid, and the talus, with lesser soft-tissue extension compared with our patient’s case.9
As this foot tumor is rare, there are few reports on its clinical aspects, appropriate treatment, and long-term outcome. For treatment of nonmetastatic Ewing sarcoma, limb-salvage surgery is advised if response has been good and there is a reasonable expectation of negative margins and good functional results.
Radiation and surgery may be part of the overall treatment plan. Radiation without surgery has a unique role in pelvic Ewing sarcoma, in contrast to extremity Ewing sarcoma. In our opinion, margins and histologic necrosis in the resection specimen are examined after surgery, and, if the margins are widely negative and histologic response is good, no further local control is advised. If the margin is positive, postoperative radiation therapy is recommended.1 Amputation has gradually become a (rare) choice in the treatment of extremity sarcomas.9 In our patient’s case, surgery was preferred over radiation therapy after chemotherapy because of the low risk of local side effects and the expected high efficacy. In addition, radiation at such high doses for Ewing sarcoma in the foot causes functional impairment. Because of the multiple-bone involvement, a salvage procedure was not possible for our patient. Given the calcaneal involvement, however, below-knee amputation was considered safer than ankle disarticulation.
Multiple-bone involvement occurs in the advanced stage of Ewing sarcoma, usually after visceral and pulmonary metastases are detected.9 The case reported by Rammal and colleagues9 had both multiple-bone involvement in the foot and pulmonary metastasis. The authors indicated that hematogenous spread of the tumor was discerned because the lesions were noncontiguous.9 Our patient had no distant metastases. We think his tumor originated in a tarsal or midtarsal bone and extended to adjacent bones. Therefore, it probably spread through its capsular and ligamentous attachment among tarsal and midtarsal bones, as the involvement was contiguous rather than distinct.
Average delay from symptom onset to diagnosis was reported to be 34 weeks.3 Average physician delay from initial visit to correct diagnosis was reported to be 19 weeks.3 Patients may have erythema, fever, and swelling, suggestive of osteomyelitis.3 Laboratory results may show increased white blood cell count and elevated ESR and CRP level.3 In addition, needle biopsy of the tumor may reveal an appearance grossly similar to that of pus.3 Therefore, physicians may send all the tissue out for microbiological analysis (according to the erroneous diagnosis of infection) and none out for pathologic analysis. The situation can be further complicated when Ewing sarcoma occurs in the foot, an uncommon site. In this special case, multiple-bone involvement can present a misleading clinical picture of infection.10 In other words, infection is one of the best choices in the differential diagnosis.7 Also to be considered are multicentric giant cell tumor, fibrosarcoma,11 and osteosarcoma.12
1. Herring JA. Malignant tumors of bone. In: Herring JA, ed. Tachdjian’s Pediatric Orthopaedics. Philadelphia, PA: Saunders Elsevier; 2008:2324-2327.
2. Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am J Pathol. 1987;127(3):507-518.
3. Canale ST, Beaty JH. Malignant tumors of bone. In: Canale ST, ed. Campbell’s Operative Orthopaedics. Philadelphia, PA: Mosby Elsevier; 2008:910-913.
4. Unni KK. Ewing sarcoma. In: Unni KK, ed. Dahlin’s Bone Tumor: General Aspects and Data on 11087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:121-142.
5. Casadei R, Magnani M, Biagini R, Mercuri M. Prognostic factors in Ewing’s sarcoma of the foot. Clin Orthop. 2004;(420):230-238.
6. Greenspan A, Jundt G, Remagen W. Bone-forming (osteogenic) lesions. In: Greenspan A, Jundt G, Remagen W, eds. Differential Diagnosis in Orthopaedic Oncology. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:114.
7. Metcalfe JE, Grimer RJ. Ewing’s sarcoma of the foot masquerading as osteomyelitis. Foot Ankle Surg. 2004;10(1):29-33.
8. Hoffmann C, Ahrens S, Dunst J, et al. Pelvis Ewing sarcoma: a retrospective analysis of 241 cases. Cancer. 1999;85(4):869-877.
9. Rammal H, Ghanem I, Torbey PH, Dagher F, Kharrat K. Multifocal Ewing sarcoma of the foot. J Pediatr Hematol Oncol. 2008;30(4):298-300.
10. Ledermann HP, Morrison WB, Schweitzer ME. MR image analysis of pedal osteomyelitis: distribution, patterns of spread, and frequency of associated ulceration and septic arthritis. Radiology. 2002;223(3):747-755.
11. Dhillon MS, Prabhudev Prasad AP, Virk MS, Aggarwal S. Multicentric giant cell tumor involving the same foot: a case report and review of literature. Indian J Orthop. 2007;41(2):154-157.
12. Baraga JJ, Amarami KK, Swee RG, Wold L, Unni KK. Radiographic features of Ewing’s sarcoma of the bones of the hand and feet. Skeletal Radiol. 2001;30(3):121-126.
1. Herring JA. Malignant tumors of bone. In: Herring JA, ed. Tachdjian’s Pediatric Orthopaedics. Philadelphia, PA: Saunders Elsevier; 2008:2324-2327.
2. Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am J Pathol. 1987;127(3):507-518.
3. Canale ST, Beaty JH. Malignant tumors of bone. In: Canale ST, ed. Campbell’s Operative Orthopaedics. Philadelphia, PA: Mosby Elsevier; 2008:910-913.
4. Unni KK. Ewing sarcoma. In: Unni KK, ed. Dahlin’s Bone Tumor: General Aspects and Data on 11087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:121-142.
5. Casadei R, Magnani M, Biagini R, Mercuri M. Prognostic factors in Ewing’s sarcoma of the foot. Clin Orthop. 2004;(420):230-238.
6. Greenspan A, Jundt G, Remagen W. Bone-forming (osteogenic) lesions. In: Greenspan A, Jundt G, Remagen W, eds. Differential Diagnosis in Orthopaedic Oncology. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:114.
7. Metcalfe JE, Grimer RJ. Ewing’s sarcoma of the foot masquerading as osteomyelitis. Foot Ankle Surg. 2004;10(1):29-33.
8. Hoffmann C, Ahrens S, Dunst J, et al. Pelvis Ewing sarcoma: a retrospective analysis of 241 cases. Cancer. 1999;85(4):869-877.
9. Rammal H, Ghanem I, Torbey PH, Dagher F, Kharrat K. Multifocal Ewing sarcoma of the foot. J Pediatr Hematol Oncol. 2008;30(4):298-300.
10. Ledermann HP, Morrison WB, Schweitzer ME. MR image analysis of pedal osteomyelitis: distribution, patterns of spread, and frequency of associated ulceration and septic arthritis. Radiology. 2002;223(3):747-755.
11. Dhillon MS, Prabhudev Prasad AP, Virk MS, Aggarwal S. Multicentric giant cell tumor involving the same foot: a case report and review of literature. Indian J Orthop. 2007;41(2):154-157.
12. Baraga JJ, Amarami KK, Swee RG, Wold L, Unni KK. Radiographic features of Ewing’s sarcoma of the bones of the hand and feet. Skeletal Radiol. 2001;30(3):121-126.
Spontaneous, Chronic Expanding Posterior Thigh Hematoma Mimicking Soft-Tissue Sarcoma in a Morbidly Obese Pregnant Woman
Soft-tissue sarcomas are quite rare, with an annual incidence of 20 to 30 per 1,000,000 persons in the United States.1 Because of their heterogeneous presentation, they remain a diagnostic challenge and are often initially confused for more common, benign disorders.2 Chronic expanding hematoma, first described by Friedlander and colleagues3 in 1968, is a rare entity that is particularly difficult to distinguish from soft-tissue malignancy.3-5 Chronic expanding hematoma is defined as a hematoma that gradually expands over 1 month or longer, is absent of neoplastic change on histologic sections, and does not occur in the setting of coagulopathy.6
Typically associated with remote trauma, these lesions often present as a slowly growing mass on the anterior or lateral thigh, calf, or buttock.3-4,7-9 They have been reported to persist as long as 46 years, with sizes ranging from 3 to 55 cm in maximum diameter.7 On imaging, they have a cystic appearance with a dense fibrous capsule.7-8 Most cases resolve uneventfully after drainage or marginal excision, although some cases require repeated intervention.7 This case report describes a morbidly obese patient with a chronic expanding hematoma in the distal posterior thigh whose definitive treatment was delayed 6 months because of her pregnancy status and inability to lie prone for open biopsy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old morbidly obese woman, who was pregnant at 12 weeks gestation, was seen in an orthopedic oncology clinic with a 1-month history of a slowly growing, painful posterior thigh mass. She had no history of cancer or bleeding disorder, and denied a history of trauma or constitutional symptoms consistent with malignancy. Coagulation studies were normal. Magnetic resonance imaging (MRI) obtained 2 weeks prior in the emergency room showed a cystic lesion with mass-like components in the posterior compartment of the distal right thigh, measuring 17 cm longitudinally. The lesion was located adjacent to, but not involving, the sciatic nerve and femoral vasculature. On initial examination, the large soft-tissue mass was evident and moderately painful to palpation; no skin changes were noted, and the patient had a normal sensorimotor examination. Fine-needle aspiration was performed, which resulted in amorphous debris consistent with hematoma.
Repeat MRI 2 months later showed increased size of the lesion (9.5×10.5 cm axial, 22.0 cm craniocaudal). Although most findings of a more extensive imaging protocol, including precontrast and postcontrast sequences, were consistent with hematoma, the lesion also had several characteristics that indicated soft-tissue sarcoma. Specifically, findings suggestive of chronic hematoma included the hyperintense short tau inversion recovery (STIR) T1/T2 signal of the cystic component consistent with proteinaceous fluid and the low STIR TI/T2 signal of the periphery consistent with a rim of hemosiderin (Figure 1). Additionally, the cystic component of the lesion had multiple fine septations that are atypical for a hematoma (Figure 1), and several lymph nodes greater than 1.7 cm in short axis were noted in the anterior thigh and hemipelvis that were suspicious of metastatic lymphadenopathy. The encapsulated appearance of the lesion with a sharply defined margin and short transition zone were also reassuring findings for a benign lesion (Figures 1, 2A, 2B). However, several findings were identified that suggested soft-tissue sarcoma, including a nodular soft-tissue component on the medial wall of the lesion that had heterogeneous enhancement with contrast (Figure 2B). We, therefore, proceeded with ultrasound-guided core needle biopsy of the mass and cytologic sampling of the fluid components, which were again consistent with hematoma; no evidence of internal vascular flow was noted on Doppler ultrasound. Ultrasound-guided right inguinal lymph node biopsy was also performed and was negative for malignancy. Because of her large body habitus and pregnancy status, it was agreed that open biopsy should be delayed until after delivery to avoid placing the patient in a prone position.


The patient visited the emergency room several times during the following months because of intermittent exacerbations of her lower extremity pain, swelling, and occasional paresthesias. About 6 months after initial presentation, repeat MRI again showed increased size of the mass (13.5×13.5 cm axial, 28 cm craniocaudal). There was also increased displacement of the adjacent neurovascular structures but no evidence of deep vein thrombosis. Because of concerns about the increased symptomatology of her thigh mass and possible sampling error of the previous biopsies, an elective cesarean section was performed at 35 weeks gestation. One week later, after clearance by her obstetrician, we proceeded with open biopsy of the mass in prone position. Initial sampling was negative for malignancy on frozen section; then, we expressed 1.75 L of brown fluid and solidified blood products, irrigated copiously, and placed a surgical drain. The permanent histologic specimens were again consistent with hematoma, and microbial cultures were negative. A week later, the patient accidentally removed her drain, and she presented with a fever (101°F) on postoperative day (POD) 15. Computed tomography showed reaccumulation of fluid; duplex ultrasound was negative. She was placed on cephalexin and underwent ultrasound-guided replacement of the drain with removal of an additional 750 mL fluid on POD 20. She drained an additional 150 to 200 mL/d for 1 month, with marked improvement in her leg swelling and knee range of motion. The drainage decreased during the next 3 weeks, and the drain was removed on POD 75.
Discussion
The presence of a hematoma in the extremities is usually a straightforward diagnosis. However, the unusual circumstances of this case highlight all the indications for investigation for possible soft-tissue sarcoma when a patient presents with what appears to be a benign condition.
Hematomas are rare in the absence of trauma or coagulopathy, with chronic expansion of hematomas rarer still.4,7,10-11 The patient had no evidence of coagulopathy because of her ability to have an uncomplicated pregnancy and elective cesarean section. She denied a history of trauma, and the location of her hematoma at the posterior distal thigh is an uncommon site of injury. In this setting, fine-needle aspiration and serial imaging to assess for progressive increase in lesion size were indicated to rule out malignancy.2
MRI is the gold-standard imaging modality for distinguishing soft-tissue masses from hematomas.5,12-14 Unlike the typical appearance of a hematoma, sarcomas of the soft-tissue extremities are often complex cystic lesions with multiple septations, internal soft-tissue components, and relatively ill-defined margins.15-17 However, as a hematoma becomes chronic, it can develop a fibrinous capsule, and the contents can manifest an atypical, heterogeneous appearance from scattered, progressive accumulation of blood products that is essentially indistinguishable from sarcomas on imaging.5
Because of the expansion of the hematoma and the atypical appearance of the mass on imaging, repeated core biopsy and, eventually, open biopsy were indicated, despite a preliminary negative diagnosis based on fine-needle aspiration. This resulted from the possibility of sampling error that is particularly relevant to cystic sarcomas, because only portions of the mass may be composed of malignant cells.2 An unusual aspect of this case is the regional lymphadenopathy noted on MRI, because regional lymphatic spread is a known mechanism of metastasis in soft-tissue sarcomas.18 However, the inguinal biopsies showed a chronic inflammatory infiltrate and were negative for malignancy, and enlarged nodes were not seen on imaging several months later. It is possible that the lymphadenopathy resulted from an unrelated process; alternatively, it may have been secondary to impaired lymphatic drainage because of mass effect from the hematoma, which also caused temporary lower extremity swelling.
The distal posterior thigh is an unreported location for a chronic expanding hematoma. Our patient developed slowly progressive lower-limb swelling and, eventually, paresthesias because of displacement of the neurovasculature, an unusual sequela that was recently reported in a similar case of an acute spontaneous hematoma in a patient on warfarin.19 Rupture of a Baker cyst is a possible inciting factor in our patient, although the proximal location of the lesion and the clearly defined tissue plane on MRI between the hematoma and the popliteal region make this unlikely. Finally, the patient’s lesion showed no evidence of vascular flow on Doppler ultrasonography, although giant hematomas secondary to popliteal aneurysm rupture have been reported.20-22
Conclusion
This case highlights the features of a chronic expanding hematoma that can suggest soft-tissue sarcoma and shows the recommended diagnostic steps to differentiate the 2 conditions. This case also describes an unreported location for a chronic expanding hematoma with resulting progressive neurovascular displacement caused by mass effect. We recommend careful monitoring of patients with similarly expansile lesions in this region for signs of neurovascular compromise.
1. O’Sullivan B, Pisters PW. Staging and prognostic factor evaluation in soft tissue sarcoma. Surg Oncol Clin N Am. 2003;12(2):333-353.
2. Rougraff B. The diagnosis and management of soft tissue sarcomas of the extremities in the adult. Curr Probl Cancer. 1999;23(1):1-50.
3. Friedlander HL, Bump RG. Chronic expanding hematoma of the calf. A case report. J Bone Joint Surg Am. 1968;50(6):1237-1241.
4. Liu CW, Kuo CL, Tsai TY, Lin LC, Wu CC. Massive gluteal mass mimicking sarcoma: chronic expanding hematoma. Formosan J Musculoskeletal Disord. 2011;2(3):106-108.
5. Taieb S, Penel N, Vanseymortier L, Ceugnart L. Soft tissue sarcomas or intramuscular haematomas? Eur J Radiol. 2009;72(1):44-49.
6. Reid JD, Kommareddi S, Lankerani M, Park MC. Chronic expanding hematomas. A clinicopathologic entity. JAMA. 1980;244(21):2441-2442.
7. Okada K, Sugiyama T, Kato H, Tani T. Chronic expanding hematoma mimicking soft tissue neoplasm. J Clin Oncol. 2001;19(11):2971-2972.
8. Negoro K, Uchida K, Yayama T, Kokubo Y, Baba H. Chronic expanding hematoma of the thigh. Joint Bone Spine. 2012;79(2):192-194.
9. Goddard MS, Vakil JJ, McCarthy EF, Khanuja HS. Chronic expanding hematoma of the lateral thigh and massive bony destruction after a failed total hip arthroplasty. J Arthroplasty. 2011;26(2):338.e13-.e15.
10. Radford DM, Schuh ME, Nambisan RN, Karakousis CP. Pseudo-tumor of the calf. Eur J Surg Oncol. 1993;19(3):300-301.
11. Mann HA, Hilton A, Goddard NJ, Smith MA, Holloway B, Lee CA. Synovial sarcoma mimicking haemophilic pseudotumour. Sarcoma. 2006;2006:27212.
12. Kransdorf MJ, Murphey MD. Radiologic evaluation of soft-tissue masses: a current perspective. AJR Am J Roentgenol. 2000;175(3):575-587.
13. Vanel D, Verstraete KL, Shapeero LG. Primary tumors of the musculoskeletal system. Radiol Clin North Am. 1997;35(1):213-237.
14. Siegel MJ. Magnetic resonance imaging of musculoskeletal soft tissue masses. Radiol Clin North Am. 2001;39(4):701-720.
15. O’Connor EE, Dixon LB, Peabody T, Stacy GS. MRI of cystic and soft-tissue masses of the shoulder joint. AJR Am J Roentgenol. 2004;183(1):39-47.
16. Bermejo A, De Bustamante TD, Martinez A, Carrera R, Zabia E, Manjon P. MR imaging in the evaluation of cystic-appearing soft-tissue masses of the extremities. Radiographics. 2013;33(3):833-855.
17. Morrison C, Wakely PE Jr, Ashman CJ, Lemley D, Theil K. Cystic synovial sarcoma. Ann Diagn Pathol. 2001;5(1):48-56.
18. Eilber FC, Rosen G, Nelson SD, et al. High-grade extremity soft tissue sarcomas: factors predictive of local recurrence and its effect on morbidity and mortality. Ann Surg. 2003;237(2):218-226.
19. Kuo CH. Peripheral neuropathy and lower limb swelling caused by a giant popliteal fossa hematoma. Neurol Sci. 2012;33(2):475-476.
20. Reijnen MM, de Rhoter W, Zeebregts CJ. Treatment of a symptomatic popliteal pseudoaneurysm using a stent-graft and ultrasound-guided evacuation of the haematoma. Emerg Radiol. 2009;16(2):167-169.
21. Rossi FH, Veith FJ, Lipsitz EC, Izukawa NM, Oliveira LA, Silva DG. Giant femoropopliteal artery aneurysm and vein rupture. Vascular. 2004;12(4):263-265.
22. Lamoca LM, Alerany MB, Hernando LL. Endovascular therapy for a ruptured popliteal aneurysm. Catheter Cardiovasc Interv. 2010;75(3):427-429.
Soft-tissue sarcomas are quite rare, with an annual incidence of 20 to 30 per 1,000,000 persons in the United States.1 Because of their heterogeneous presentation, they remain a diagnostic challenge and are often initially confused for more common, benign disorders.2 Chronic expanding hematoma, first described by Friedlander and colleagues3 in 1968, is a rare entity that is particularly difficult to distinguish from soft-tissue malignancy.3-5 Chronic expanding hematoma is defined as a hematoma that gradually expands over 1 month or longer, is absent of neoplastic change on histologic sections, and does not occur in the setting of coagulopathy.6
Typically associated with remote trauma, these lesions often present as a slowly growing mass on the anterior or lateral thigh, calf, or buttock.3-4,7-9 They have been reported to persist as long as 46 years, with sizes ranging from 3 to 55 cm in maximum diameter.7 On imaging, they have a cystic appearance with a dense fibrous capsule.7-8 Most cases resolve uneventfully after drainage or marginal excision, although some cases require repeated intervention.7 This case report describes a morbidly obese patient with a chronic expanding hematoma in the distal posterior thigh whose definitive treatment was delayed 6 months because of her pregnancy status and inability to lie prone for open biopsy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old morbidly obese woman, who was pregnant at 12 weeks gestation, was seen in an orthopedic oncology clinic with a 1-month history of a slowly growing, painful posterior thigh mass. She had no history of cancer or bleeding disorder, and denied a history of trauma or constitutional symptoms consistent with malignancy. Coagulation studies were normal. Magnetic resonance imaging (MRI) obtained 2 weeks prior in the emergency room showed a cystic lesion with mass-like components in the posterior compartment of the distal right thigh, measuring 17 cm longitudinally. The lesion was located adjacent to, but not involving, the sciatic nerve and femoral vasculature. On initial examination, the large soft-tissue mass was evident and moderately painful to palpation; no skin changes were noted, and the patient had a normal sensorimotor examination. Fine-needle aspiration was performed, which resulted in amorphous debris consistent with hematoma.
Repeat MRI 2 months later showed increased size of the lesion (9.5×10.5 cm axial, 22.0 cm craniocaudal). Although most findings of a more extensive imaging protocol, including precontrast and postcontrast sequences, were consistent with hematoma, the lesion also had several characteristics that indicated soft-tissue sarcoma. Specifically, findings suggestive of chronic hematoma included the hyperintense short tau inversion recovery (STIR) T1/T2 signal of the cystic component consistent with proteinaceous fluid and the low STIR TI/T2 signal of the periphery consistent with a rim of hemosiderin (Figure 1). Additionally, the cystic component of the lesion had multiple fine septations that are atypical for a hematoma (Figure 1), and several lymph nodes greater than 1.7 cm in short axis were noted in the anterior thigh and hemipelvis that were suspicious of metastatic lymphadenopathy. The encapsulated appearance of the lesion with a sharply defined margin and short transition zone were also reassuring findings for a benign lesion (Figures 1, 2A, 2B). However, several findings were identified that suggested soft-tissue sarcoma, including a nodular soft-tissue component on the medial wall of the lesion that had heterogeneous enhancement with contrast (Figure 2B). We, therefore, proceeded with ultrasound-guided core needle biopsy of the mass and cytologic sampling of the fluid components, which were again consistent with hematoma; no evidence of internal vascular flow was noted on Doppler ultrasound. Ultrasound-guided right inguinal lymph node biopsy was also performed and was negative for malignancy. Because of her large body habitus and pregnancy status, it was agreed that open biopsy should be delayed until after delivery to avoid placing the patient in a prone position.
The patient visited the emergency room several times during the following months because of intermittent exacerbations of her lower extremity pain, swelling, and occasional paresthesias. About 6 months after initial presentation, repeat MRI again showed increased size of the mass (13.5×13.5 cm axial, 28 cm craniocaudal). There was also increased displacement of the adjacent neurovascular structures but no evidence of deep vein thrombosis. Because of concerns about the increased symptomatology of her thigh mass and possible sampling error of the previous biopsies, an elective cesarean section was performed at 35 weeks gestation. One week later, after clearance by her obstetrician, we proceeded with open biopsy of the mass in prone position. Initial sampling was negative for malignancy on frozen section; then, we expressed 1.75 L of brown fluid and solidified blood products, irrigated copiously, and placed a surgical drain. The permanent histologic specimens were again consistent with hematoma, and microbial cultures were negative. A week later, the patient accidentally removed her drain, and she presented with a fever (101°F) on postoperative day (POD) 15. Computed tomography showed reaccumulation of fluid; duplex ultrasound was negative. She was placed on cephalexin and underwent ultrasound-guided replacement of the drain with removal of an additional 750 mL fluid on POD 20. She drained an additional 150 to 200 mL/d for 1 month, with marked improvement in her leg swelling and knee range of motion. The drainage decreased during the next 3 weeks, and the drain was removed on POD 75.
Discussion
The presence of a hematoma in the extremities is usually a straightforward diagnosis. However, the unusual circumstances of this case highlight all the indications for investigation for possible soft-tissue sarcoma when a patient presents with what appears to be a benign condition.
Hematomas are rare in the absence of trauma or coagulopathy, with chronic expansion of hematomas rarer still.4,7,10-11 The patient had no evidence of coagulopathy because of her ability to have an uncomplicated pregnancy and elective cesarean section. She denied a history of trauma, and the location of her hematoma at the posterior distal thigh is an uncommon site of injury. In this setting, fine-needle aspiration and serial imaging to assess for progressive increase in lesion size were indicated to rule out malignancy.2
MRI is the gold-standard imaging modality for distinguishing soft-tissue masses from hematomas.5,12-14 Unlike the typical appearance of a hematoma, sarcomas of the soft-tissue extremities are often complex cystic lesions with multiple septations, internal soft-tissue components, and relatively ill-defined margins.15-17 However, as a hematoma becomes chronic, it can develop a fibrinous capsule, and the contents can manifest an atypical, heterogeneous appearance from scattered, progressive accumulation of blood products that is essentially indistinguishable from sarcomas on imaging.5
Because of the expansion of the hematoma and the atypical appearance of the mass on imaging, repeated core biopsy and, eventually, open biopsy were indicated, despite a preliminary negative diagnosis based on fine-needle aspiration. This resulted from the possibility of sampling error that is particularly relevant to cystic sarcomas, because only portions of the mass may be composed of malignant cells.2 An unusual aspect of this case is the regional lymphadenopathy noted on MRI, because regional lymphatic spread is a known mechanism of metastasis in soft-tissue sarcomas.18 However, the inguinal biopsies showed a chronic inflammatory infiltrate and were negative for malignancy, and enlarged nodes were not seen on imaging several months later. It is possible that the lymphadenopathy resulted from an unrelated process; alternatively, it may have been secondary to impaired lymphatic drainage because of mass effect from the hematoma, which also caused temporary lower extremity swelling.
The distal posterior thigh is an unreported location for a chronic expanding hematoma. Our patient developed slowly progressive lower-limb swelling and, eventually, paresthesias because of displacement of the neurovasculature, an unusual sequela that was recently reported in a similar case of an acute spontaneous hematoma in a patient on warfarin.19 Rupture of a Baker cyst is a possible inciting factor in our patient, although the proximal location of the lesion and the clearly defined tissue plane on MRI between the hematoma and the popliteal region make this unlikely. Finally, the patient’s lesion showed no evidence of vascular flow on Doppler ultrasonography, although giant hematomas secondary to popliteal aneurysm rupture have been reported.20-22
Conclusion
This case highlights the features of a chronic expanding hematoma that can suggest soft-tissue sarcoma and shows the recommended diagnostic steps to differentiate the 2 conditions. This case also describes an unreported location for a chronic expanding hematoma with resulting progressive neurovascular displacement caused by mass effect. We recommend careful monitoring of patients with similarly expansile lesions in this region for signs of neurovascular compromise.
Soft-tissue sarcomas are quite rare, with an annual incidence of 20 to 30 per 1,000,000 persons in the United States.1 Because of their heterogeneous presentation, they remain a diagnostic challenge and are often initially confused for more common, benign disorders.2 Chronic expanding hematoma, first described by Friedlander and colleagues3 in 1968, is a rare entity that is particularly difficult to distinguish from soft-tissue malignancy.3-5 Chronic expanding hematoma is defined as a hematoma that gradually expands over 1 month or longer, is absent of neoplastic change on histologic sections, and does not occur in the setting of coagulopathy.6
Typically associated with remote trauma, these lesions often present as a slowly growing mass on the anterior or lateral thigh, calf, or buttock.3-4,7-9 They have been reported to persist as long as 46 years, with sizes ranging from 3 to 55 cm in maximum diameter.7 On imaging, they have a cystic appearance with a dense fibrous capsule.7-8 Most cases resolve uneventfully after drainage or marginal excision, although some cases require repeated intervention.7 This case report describes a morbidly obese patient with a chronic expanding hematoma in the distal posterior thigh whose definitive treatment was delayed 6 months because of her pregnancy status and inability to lie prone for open biopsy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old morbidly obese woman, who was pregnant at 12 weeks gestation, was seen in an orthopedic oncology clinic with a 1-month history of a slowly growing, painful posterior thigh mass. She had no history of cancer or bleeding disorder, and denied a history of trauma or constitutional symptoms consistent with malignancy. Coagulation studies were normal. Magnetic resonance imaging (MRI) obtained 2 weeks prior in the emergency room showed a cystic lesion with mass-like components in the posterior compartment of the distal right thigh, measuring 17 cm longitudinally. The lesion was located adjacent to, but not involving, the sciatic nerve and femoral vasculature. On initial examination, the large soft-tissue mass was evident and moderately painful to palpation; no skin changes were noted, and the patient had a normal sensorimotor examination. Fine-needle aspiration was performed, which resulted in amorphous debris consistent with hematoma.
Repeat MRI 2 months later showed increased size of the lesion (9.5×10.5 cm axial, 22.0 cm craniocaudal). Although most findings of a more extensive imaging protocol, including precontrast and postcontrast sequences, were consistent with hematoma, the lesion also had several characteristics that indicated soft-tissue sarcoma. Specifically, findings suggestive of chronic hematoma included the hyperintense short tau inversion recovery (STIR) T1/T2 signal of the cystic component consistent with proteinaceous fluid and the low STIR TI/T2 signal of the periphery consistent with a rim of hemosiderin (Figure 1). Additionally, the cystic component of the lesion had multiple fine septations that are atypical for a hematoma (Figure 1), and several lymph nodes greater than 1.7 cm in short axis were noted in the anterior thigh and hemipelvis that were suspicious of metastatic lymphadenopathy. The encapsulated appearance of the lesion with a sharply defined margin and short transition zone were also reassuring findings for a benign lesion (Figures 1, 2A, 2B). However, several findings were identified that suggested soft-tissue sarcoma, including a nodular soft-tissue component on the medial wall of the lesion that had heterogeneous enhancement with contrast (Figure 2B). We, therefore, proceeded with ultrasound-guided core needle biopsy of the mass and cytologic sampling of the fluid components, which were again consistent with hematoma; no evidence of internal vascular flow was noted on Doppler ultrasound. Ultrasound-guided right inguinal lymph node biopsy was also performed and was negative for malignancy. Because of her large body habitus and pregnancy status, it was agreed that open biopsy should be delayed until after delivery to avoid placing the patient in a prone position.
The patient visited the emergency room several times during the following months because of intermittent exacerbations of her lower extremity pain, swelling, and occasional paresthesias. About 6 months after initial presentation, repeat MRI again showed increased size of the mass (13.5×13.5 cm axial, 28 cm craniocaudal). There was also increased displacement of the adjacent neurovascular structures but no evidence of deep vein thrombosis. Because of concerns about the increased symptomatology of her thigh mass and possible sampling error of the previous biopsies, an elective cesarean section was performed at 35 weeks gestation. One week later, after clearance by her obstetrician, we proceeded with open biopsy of the mass in prone position. Initial sampling was negative for malignancy on frozen section; then, we expressed 1.75 L of brown fluid and solidified blood products, irrigated copiously, and placed a surgical drain. The permanent histologic specimens were again consistent with hematoma, and microbial cultures were negative. A week later, the patient accidentally removed her drain, and she presented with a fever (101°F) on postoperative day (POD) 15. Computed tomography showed reaccumulation of fluid; duplex ultrasound was negative. She was placed on cephalexin and underwent ultrasound-guided replacement of the drain with removal of an additional 750 mL fluid on POD 20. She drained an additional 150 to 200 mL/d for 1 month, with marked improvement in her leg swelling and knee range of motion. The drainage decreased during the next 3 weeks, and the drain was removed on POD 75.
Discussion
The presence of a hematoma in the extremities is usually a straightforward diagnosis. However, the unusual circumstances of this case highlight all the indications for investigation for possible soft-tissue sarcoma when a patient presents with what appears to be a benign condition.
Hematomas are rare in the absence of trauma or coagulopathy, with chronic expansion of hematomas rarer still.4,7,10-11 The patient had no evidence of coagulopathy because of her ability to have an uncomplicated pregnancy and elective cesarean section. She denied a history of trauma, and the location of her hematoma at the posterior distal thigh is an uncommon site of injury. In this setting, fine-needle aspiration and serial imaging to assess for progressive increase in lesion size were indicated to rule out malignancy.2
MRI is the gold-standard imaging modality for distinguishing soft-tissue masses from hematomas.5,12-14 Unlike the typical appearance of a hematoma, sarcomas of the soft-tissue extremities are often complex cystic lesions with multiple septations, internal soft-tissue components, and relatively ill-defined margins.15-17 However, as a hematoma becomes chronic, it can develop a fibrinous capsule, and the contents can manifest an atypical, heterogeneous appearance from scattered, progressive accumulation of blood products that is essentially indistinguishable from sarcomas on imaging.5
Because of the expansion of the hematoma and the atypical appearance of the mass on imaging, repeated core biopsy and, eventually, open biopsy were indicated, despite a preliminary negative diagnosis based on fine-needle aspiration. This resulted from the possibility of sampling error that is particularly relevant to cystic sarcomas, because only portions of the mass may be composed of malignant cells.2 An unusual aspect of this case is the regional lymphadenopathy noted on MRI, because regional lymphatic spread is a known mechanism of metastasis in soft-tissue sarcomas.18 However, the inguinal biopsies showed a chronic inflammatory infiltrate and were negative for malignancy, and enlarged nodes were not seen on imaging several months later. It is possible that the lymphadenopathy resulted from an unrelated process; alternatively, it may have been secondary to impaired lymphatic drainage because of mass effect from the hematoma, which also caused temporary lower extremity swelling.
The distal posterior thigh is an unreported location for a chronic expanding hematoma. Our patient developed slowly progressive lower-limb swelling and, eventually, paresthesias because of displacement of the neurovasculature, an unusual sequela that was recently reported in a similar case of an acute spontaneous hematoma in a patient on warfarin.19 Rupture of a Baker cyst is a possible inciting factor in our patient, although the proximal location of the lesion and the clearly defined tissue plane on MRI between the hematoma and the popliteal region make this unlikely. Finally, the patient’s lesion showed no evidence of vascular flow on Doppler ultrasonography, although giant hematomas secondary to popliteal aneurysm rupture have been reported.20-22
Conclusion
This case highlights the features of a chronic expanding hematoma that can suggest soft-tissue sarcoma and shows the recommended diagnostic steps to differentiate the 2 conditions. This case also describes an unreported location for a chronic expanding hematoma with resulting progressive neurovascular displacement caused by mass effect. We recommend careful monitoring of patients with similarly expansile lesions in this region for signs of neurovascular compromise.
1. O’Sullivan B, Pisters PW. Staging and prognostic factor evaluation in soft tissue sarcoma. Surg Oncol Clin N Am. 2003;12(2):333-353.
2. Rougraff B. The diagnosis and management of soft tissue sarcomas of the extremities in the adult. Curr Probl Cancer. 1999;23(1):1-50.
3. Friedlander HL, Bump RG. Chronic expanding hematoma of the calf. A case report. J Bone Joint Surg Am. 1968;50(6):1237-1241.
4. Liu CW, Kuo CL, Tsai TY, Lin LC, Wu CC. Massive gluteal mass mimicking sarcoma: chronic expanding hematoma. Formosan J Musculoskeletal Disord. 2011;2(3):106-108.
5. Taieb S, Penel N, Vanseymortier L, Ceugnart L. Soft tissue sarcomas or intramuscular haematomas? Eur J Radiol. 2009;72(1):44-49.
6. Reid JD, Kommareddi S, Lankerani M, Park MC. Chronic expanding hematomas. A clinicopathologic entity. JAMA. 1980;244(21):2441-2442.
7. Okada K, Sugiyama T, Kato H, Tani T. Chronic expanding hematoma mimicking soft tissue neoplasm. J Clin Oncol. 2001;19(11):2971-2972.
8. Negoro K, Uchida K, Yayama T, Kokubo Y, Baba H. Chronic expanding hematoma of the thigh. Joint Bone Spine. 2012;79(2):192-194.
9. Goddard MS, Vakil JJ, McCarthy EF, Khanuja HS. Chronic expanding hematoma of the lateral thigh and massive bony destruction after a failed total hip arthroplasty. J Arthroplasty. 2011;26(2):338.e13-.e15.
10. Radford DM, Schuh ME, Nambisan RN, Karakousis CP. Pseudo-tumor of the calf. Eur J Surg Oncol. 1993;19(3):300-301.
11. Mann HA, Hilton A, Goddard NJ, Smith MA, Holloway B, Lee CA. Synovial sarcoma mimicking haemophilic pseudotumour. Sarcoma. 2006;2006:27212.
12. Kransdorf MJ, Murphey MD. Radiologic evaluation of soft-tissue masses: a current perspective. AJR Am J Roentgenol. 2000;175(3):575-587.
13. Vanel D, Verstraete KL, Shapeero LG. Primary tumors of the musculoskeletal system. Radiol Clin North Am. 1997;35(1):213-237.
14. Siegel MJ. Magnetic resonance imaging of musculoskeletal soft tissue masses. Radiol Clin North Am. 2001;39(4):701-720.
15. O’Connor EE, Dixon LB, Peabody T, Stacy GS. MRI of cystic and soft-tissue masses of the shoulder joint. AJR Am J Roentgenol. 2004;183(1):39-47.
16. Bermejo A, De Bustamante TD, Martinez A, Carrera R, Zabia E, Manjon P. MR imaging in the evaluation of cystic-appearing soft-tissue masses of the extremities. Radiographics. 2013;33(3):833-855.
17. Morrison C, Wakely PE Jr, Ashman CJ, Lemley D, Theil K. Cystic synovial sarcoma. Ann Diagn Pathol. 2001;5(1):48-56.
18. Eilber FC, Rosen G, Nelson SD, et al. High-grade extremity soft tissue sarcomas: factors predictive of local recurrence and its effect on morbidity and mortality. Ann Surg. 2003;237(2):218-226.
19. Kuo CH. Peripheral neuropathy and lower limb swelling caused by a giant popliteal fossa hematoma. Neurol Sci. 2012;33(2):475-476.
20. Reijnen MM, de Rhoter W, Zeebregts CJ. Treatment of a symptomatic popliteal pseudoaneurysm using a stent-graft and ultrasound-guided evacuation of the haematoma. Emerg Radiol. 2009;16(2):167-169.
21. Rossi FH, Veith FJ, Lipsitz EC, Izukawa NM, Oliveira LA, Silva DG. Giant femoropopliteal artery aneurysm and vein rupture. Vascular. 2004;12(4):263-265.
22. Lamoca LM, Alerany MB, Hernando LL. Endovascular therapy for a ruptured popliteal aneurysm. Catheter Cardiovasc Interv. 2010;75(3):427-429.
1. O’Sullivan B, Pisters PW. Staging and prognostic factor evaluation in soft tissue sarcoma. Surg Oncol Clin N Am. 2003;12(2):333-353.
2. Rougraff B. The diagnosis and management of soft tissue sarcomas of the extremities in the adult. Curr Probl Cancer. 1999;23(1):1-50.
3. Friedlander HL, Bump RG. Chronic expanding hematoma of the calf. A case report. J Bone Joint Surg Am. 1968;50(6):1237-1241.
4. Liu CW, Kuo CL, Tsai TY, Lin LC, Wu CC. Massive gluteal mass mimicking sarcoma: chronic expanding hematoma. Formosan J Musculoskeletal Disord. 2011;2(3):106-108.
5. Taieb S, Penel N, Vanseymortier L, Ceugnart L. Soft tissue sarcomas or intramuscular haematomas? Eur J Radiol. 2009;72(1):44-49.
6. Reid JD, Kommareddi S, Lankerani M, Park MC. Chronic expanding hematomas. A clinicopathologic entity. JAMA. 1980;244(21):2441-2442.
7. Okada K, Sugiyama T, Kato H, Tani T. Chronic expanding hematoma mimicking soft tissue neoplasm. J Clin Oncol. 2001;19(11):2971-2972.
8. Negoro K, Uchida K, Yayama T, Kokubo Y, Baba H. Chronic expanding hematoma of the thigh. Joint Bone Spine. 2012;79(2):192-194.
9. Goddard MS, Vakil JJ, McCarthy EF, Khanuja HS. Chronic expanding hematoma of the lateral thigh and massive bony destruction after a failed total hip arthroplasty. J Arthroplasty. 2011;26(2):338.e13-.e15.
10. Radford DM, Schuh ME, Nambisan RN, Karakousis CP. Pseudo-tumor of the calf. Eur J Surg Oncol. 1993;19(3):300-301.
11. Mann HA, Hilton A, Goddard NJ, Smith MA, Holloway B, Lee CA. Synovial sarcoma mimicking haemophilic pseudotumour. Sarcoma. 2006;2006:27212.
12. Kransdorf MJ, Murphey MD. Radiologic evaluation of soft-tissue masses: a current perspective. AJR Am J Roentgenol. 2000;175(3):575-587.
13. Vanel D, Verstraete KL, Shapeero LG. Primary tumors of the musculoskeletal system. Radiol Clin North Am. 1997;35(1):213-237.
14. Siegel MJ. Magnetic resonance imaging of musculoskeletal soft tissue masses. Radiol Clin North Am. 2001;39(4):701-720.
15. O’Connor EE, Dixon LB, Peabody T, Stacy GS. MRI of cystic and soft-tissue masses of the shoulder joint. AJR Am J Roentgenol. 2004;183(1):39-47.
16. Bermejo A, De Bustamante TD, Martinez A, Carrera R, Zabia E, Manjon P. MR imaging in the evaluation of cystic-appearing soft-tissue masses of the extremities. Radiographics. 2013;33(3):833-855.
17. Morrison C, Wakely PE Jr, Ashman CJ, Lemley D, Theil K. Cystic synovial sarcoma. Ann Diagn Pathol. 2001;5(1):48-56.
18. Eilber FC, Rosen G, Nelson SD, et al. High-grade extremity soft tissue sarcomas: factors predictive of local recurrence and its effect on morbidity and mortality. Ann Surg. 2003;237(2):218-226.
19. Kuo CH. Peripheral neuropathy and lower limb swelling caused by a giant popliteal fossa hematoma. Neurol Sci. 2012;33(2):475-476.
20. Reijnen MM, de Rhoter W, Zeebregts CJ. Treatment of a symptomatic popliteal pseudoaneurysm using a stent-graft and ultrasound-guided evacuation of the haematoma. Emerg Radiol. 2009;16(2):167-169.
21. Rossi FH, Veith FJ, Lipsitz EC, Izukawa NM, Oliveira LA, Silva DG. Giant femoropopliteal artery aneurysm and vein rupture. Vascular. 2004;12(4):263-265.
22. Lamoca LM, Alerany MB, Hernando LL. Endovascular therapy for a ruptured popliteal aneurysm. Catheter Cardiovasc Interv. 2010;75(3):427-429.
Severe Neurologic Manifestations of Fat Embolism Syndrome in a Polytrauma Patient
Fat embolism syndrome (FES) was first described by Von Bergmann in 1873 in a patient with a fractured femur.1 While fat within the circulation (fat embolism) is relatively common following long-bone fracture, the clinical pattern of symptoms that make up FES is less so, occurring in 1% to 3% of isolated long-bone fractures and 5% to 10% of patients with multiple skeletal trauma.1 A variety of clinical, laboratory, and imaging criteria has been described, classically by Gurd in 1970 (Table).1-6 Most commonly, however, it is a diagnosis of exclusion when the classic triad of respiratory difficulty, neurologic abnormalities, and a characteristic petechial rash are present in the appropriate clinical setting.6
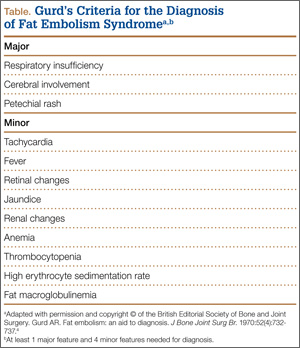
The neurologic sequelae of this syndrome can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of these symptoms usually occurs between 24 hours and 48 hours (mean, 40 hours) after trauma.1 While these neurologic manifestations occur in up to 86% of patients with FES, it is rare for them to be present without the pulmonary symptoms of dyspnea, hypoxemia, and tachypnea, which are the most common presenting symptoms of the disease.1-6 In this case report, we describe severe, rapid-onset neurologic manifestations, without the typical pulmonary involvement, as the primary clinical presentation of FES in a polytrauma patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A previously healthy 50-year-old man presented to the emergency room in transfer from an outside hospital after a rollover motor vehicle collision in which he was ejected approximately 50 feet. Injuries included a right proximal humerus fracture/dislocation (Figure 1), right ulnar styloid fracture, L1 compression fracture, and multiple rib fractures. On admission, the patient had an ethanol level of 969 mg/L (.097%) and a urine drug screen positive only for opioids, presumably because of pain medication given that day. He denied a history of alcohol abuse and reported consuming 2 to 3 beers per week. The patient was awake, alert, and oriented with a Glasgow Coma Scale (GCS) of 15. He was tachycardic (heart rate, 126), tachypneic (respiratory rate, 24), and febrile (temperature, 38.6°C [101.5°F]), and his white blood cell count was elevated at 29.5×109/L. On examination, his right arm was found to be neurovascularly intact; it was placed in a sling with a forearm splint, and the patient was admitted to the intermediate special care unit on spine precautions with a plan for right shoulder hemiarthroplasty the following day.
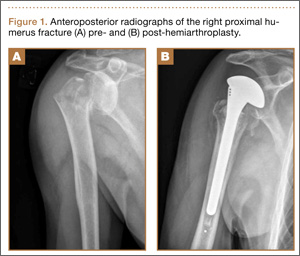
Overnight the patient’s mental status began to deteriorate, and approximately 10 hours after initial assessment, he was not answering questions but was able to respond to some commands. On hospital day 2, approximately 20 hours after initial assessment, the patient had a GCS of 8, was not responding to commands, and moved only in response to painful stimuli. The patient had been prescribed morphine by patient-controlled analgesia and had received intravenous hydromorphone on the day of admission, although the amount of medication delivered was not thought adequate to explain this deterioration. On the morning of hospital day 2, noncontrast brain computed tomography (CT) was normal with no evidence of intracranial hemorrhage or infarct. This was followed by brain magnetic resonance imaging (MRI), with the T2-weighted images showing numerous, small hyperintense lesions in subcortical and periventricular white matter, corpus callosum, basal ganglia, brain stem, and cerebellar hemispheres (Figure 2). The lesions also showed hyperintensity on diffusion-weighted MRI and were interpreted to be consistent with multiple, tiny infarcts (Figure 3). In addition, susceptibility-weighted sequences showed low signal in the same areas, suggesting multiple microhemorrhages, a pattern consistent with FES. Oxygen saturations remained 95% to 99%, and chest radiograph revealed clear lung fields without infiltrate. On hospital day 2, the patient was transferred to the intensive care unit and intubated for airway protection owing to an inability to clear secretions, although arterial blood gas levels remained normal. An echocardiogram revealed no right-to-left shunt, such as a patent foramen ovale (PFO); an electroencephalogram showed no seizure-like activity. No petechial rash was noted on skin examination. The patient was treated with supportive care. Right shoulder hemiarthroplasty was performed on hospital day 7 without complications (Figure 1). On hospital day 13, the patient was following commands and on day 14 he was extubated. His mental status continued to improve, and he was discharged to a rehabilitation facility after 36 days. On last follow-up, 6 months after initial injury, the patient was recovering well with no residual neurologic deficits and only minor limitation in range of motion of the right shoulder.
Discussion
This case presented an interesting diagnostic challenge regarding the patient’s rapid decline in mental status, with a differential diagnosis including diffuse axonal injury (DAI), anoxic brain injury, posttraumatic seizure, other intracranial pathology, such as stroke or hemorrhage, and FES. FES was diagnosed, when other possibilities were ruled out, given the characteristic findings on brain MRI described above in the context of multiple fractures.
Pathophysiology
Despite its recognition in 1873, there is no consensus on the pathophysiological mechanism that causes the clinical symptoms of FES. In the setting of trauma, there are 2 predominant theories. The mechanical theory postulates that fat globules enter the circulation through disrupted venules after the fracture of marrow-containing bones, passing to the arterial circulation through pulmonary vasculature, or paradoxically, by way of a right-to-left shunt, such as a PFO.1,3 The presence of fat in the heart, visualized as echogenic material in the right and left atria on transesophageal echocardiography, has been confirmed in multiple studies during orthopedic procedures, including total knee arthroplasty and femoral reaming.8,9 These fat particles can lodge as microembolisms in target organs such as the skin and brain. However, autopsy studies have shown a lack of correlation of the severity of symptoms and the quantity of intravascular fat.1 In addition, the typical 24- to 72-hour delay in the onset of symptoms after initial trauma would argue against a solely mechanical explanation.10
Alternatively or concomitantly, the biochemical theory proposes that embolized fat may be degraded to toxic intermediaries, such as free fatty acids and C-reactive protein, which cause end-organ damage.3 This has been shown in an animal model, in which intravascular injection of free fatty acids was associated with endothelial damage and increased capillary permeability in the lung, leading to acute respiratory distress syndrome (ARDS).11 The same mechanism could explain injury to other end organs and is consistent with the delay in onset of symptoms after acute injury. In our patient’s case, the absence of pulmonary involvement, lack of a right-to-left vascular shunt such as a PFO, and presence of a systemic inflammatory response on admission may implicate the production of toxic intermediaries from the metabolism of embolized fat as the source of this patient’s FES.
Clinical Presentation
The initial presentation of FES usually manifests as respiratory distress and hypoxia.10 Chest radiographs are often normal, as in our patient, but can show bilateral diffuse interstitial or alveolar infiltrates.2,6 CT more often has findings, including bilateral ground-glass opacities with interlobar septal thickening.12 A petechial rash can be found on the head, neck, anterior thorax, axillae, subconjunctiva, and oral mucous membranes, although it occurs in only 20% to 50% of cases.1,2,13 Neurologic sequelae are present in up to 80% of patients,7 with onset typically following pulmonary symptoms.1,10 These sequelae can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of symptoms generally occurs between 24 and 48 hours after trauma,1 although they have been reported as early as 12 hours.10 This case is an example of an atypical course, with the initial presentation of neurologic symptoms at approximately 14 hours after trauma with rapid progression to coma without classic pulmonary symptoms.
Diagnosis
Owing to the nonspecific clinical features of FES, a variety of clinical, laboratory, and imaging criteria has been described. Of these criteria, the most frequently referenced is by Gurd in 1970,4,5 who divided the features into major and minor, with 1 major and 4 minor features required to make the diagnosis (Table). In applying these criteria to our patient, we found that he exhibited the major criteria of cerebral involvement and minor criteria of tachycardia, fever, and thrombocytopenia. Respiratory insufficiency and petechial rash, as well as jaundice, renal changes, and anemia were negative features. Retinal changes, elevated erythrocyte sedimentation rate, and fat macroglobulinemia were not tested or examined. Although in our case the clinical and laboratory criteria for the diagnosis of FES as defined by Gurd were not met, the sensitivity of Gurd’s and other criteria is debated.10
Laboratory tests specific for the disease have not been developed. Although elevated serum levels of lipase, increased blood lipid levels, and fat globules in the urine, sputum, and blood have all been proposed, they are found in trauma patients with and without FES.2,5,6
The nonspecific nature of the signs and symptoms of FES and the lack of reliable laboratory tests for diagnosis of the syndrome highlight the importance of radiographic evaluation in patients with neurologic symptoms. Brain CT scans are usually negative,14 although, in some cases, they may show diffuse edema with scattered low attenuating areas and hemorrhage.15 MRI is more sensitive, and T2-weighted images typically reveal multiple small, nonconfluent hyperintense lesions, usually in the periventricular, subcortical, and deep white matter, sometimes referred to as the “starfield” pattern.14,16 The differential diagnosis for these findings is broad and, in addition to FES, includes DAI, vasogenic edema with microinfarcts, and demyelinating disease.14 Sensitivity and specificity may be increased with the addition of diffusion-weighted MRI, which shows scattered bright spots on a dark background in a similar “starfield” pattern as on T2-weighted images.15 Susceptibility-weighted MRI has recently been introduced as having utility in the diagnosis of FES, with areas of low-signal intensity indicating diffuse microhemorrhages.17 DAI can show a similar pattern; however, the autopsy-confirmed locations of the abnormalities are distinct, with those of FES being found in cerebral and cerebellar white matter and splenium of the corpus callosum and radiographic abnormalities of DAI being found in the gray-white matter junction, dorsolateral brainstem, and splenium of corpus callosum.17
Prevention and Treatment
Of primary importance in the prevention of FES is early stabilization of fractures. Several studies have shown a decreased incidence of FES when long-bone fractures are treated with immediate operative fixation.18,19 However, in the setting of polytrauma, the desire for early definitive treatment must be balanced against the risks for the exaggerated immune response from prolonged surgery.20 The timing of fracture fixation to prevent sequelae of the inflammatory response, such as ARDS and multiple organ dysfunction syndrome, is still debated. In a review article, Pape and colleagues20 suggest classifying the multiply injured patient as stable, borderline, unstable, and in extremis based on clinical and laboratory criteria. They recommend early definitive fixation for stable patients and those patients who are borderline or unstable and responsive to resuscitation, whereas damage-control orthopedics and staged fracture fixation should be considered in the other groups.
Several pharmacologic interventions have been described, although their effects are highly variable and none have clear indications.1-3,6 The most heavily researched is corticosteroids, with the proposed mechanisms of action including blunting of the inflammatory response, stabilizing the pulmonary capillary membrane to reduce interstitial edema, preventing activation of the complement system, and retarding platelet aggregation.21 A recent meta-analysis to assess this intervention examined 6 studies with a total of 386 patients with long-bone fractures who were randomized to treatment with corticosteroids or supportive care only.22 They found a reduced risk for FES in those patients who received corticosteroids, but there was no difference in mortality between groups. Given these results, the utility of corticosteroids is still debated.
Once FES has occurred, treatment options usually focus on supportive care, with most patients having a full recovery.1,3 No specific treatments are available, and symptomatic treatment is the suggested approach, including ensuring adequate oxygenation and ventilation and providing hemodynamic support and volume and blood-product resuscitation as needed.1-3,6
Conclusion
We have presented a case of FES unique in its rapid onset, an initial presentation with neurologic manifestations without typical pulmonary involvement, and the mechanism of end-organ damage without a right-to-left shunt. This case emphasizes the importance of considering FES in the patient with deteriorating mental status in the setting of multiple fractures, particularly in the absence of other characteristic clinical findings, such as pulmonary distress and the pathognomonic petechial rash. Brain MRI can play an important role in diagnosing those patients presenting with predominantly neurological symptoms. Early recognition of this condition allows for the anticipation of complications of the disease process, such as respiratory distress, and the potential need for mechanical ventilation and hemodynamic support.
1. Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics. 1996;19(1):41-49.
2. Levy D. The fat embolism syndrome. A review. Clin Orthop. 1990;261:281-286.
3. Mellor A, Soni N. Fat embolism. Anaesthesia. 2001;56(2):145-154.
4. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surg Br. 1970:52(4):732-737.
5. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
6. Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132(4):435-439.
7. Jacobson DM, Terrence CF, Reinmuth OM. The neurologic manifestations of fat embolism. Neurology. 1986;36(6):847-851.
8. Sulek CA, Davies LK, Enneking FK, Gearen PA, Lobato EB. Cerebral microembolism diagnosed by transcranial Doppler during total knee arthroplasty: correlation with transesophageal echocardiography. Anesthesiology. 1999;91(3):672-676.
9. Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41(Suppl 2):S90-S93.
10. Gupta B, D’souza N, Sawhney C, et al. Analyzing fat embolism syndrome in trauma patients at AIIMS Apex Trauma Center, New Delhi, India. J Emerg Trauma Shock. 2011;4(3):337–341.
11. King EG, Wagner WW Jr, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest. 1971:59(5):524-530.
12. Malagari K, Economopoulos N, Stoupis C, et al. High-resolution CT findings in mild pulmonary fat embolism. Chest. 2003:123(4):1196-1201.
13. King MB, Harmon KR. Unusual forms of pulmonary embolism. Clin Chest Med. 1994;15(3):561-580.
14. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.
15. Simon AD, Ulmer JL, Strottmann JM. Contrast-enhanced MR imaging of cerebral fat embolism: case report and review of the literature. AJNR Am J Neuroradiol. 2003;24(1):97-101.
16. Butteriss DJ, Mahad D, Soh C, Walls T, Weir D, Birchall D. Reversible cytotoxic cerebral edema in cerebral fat embolism. AJNR Am J Neuroradiol. 2006;27(3):620-623.
17. Zaitsu Y, Terae S, Kudo K, et al. Susceptibility-weighted imaging of cerebral fat embolism. J Comput Assist Tomogr. 2010;34(1):107-112.
18. Riska EB, Myllynen P. Fat embolism in patients with multiple injuries. J Trauma. 1982;22(11):891-894.
19. Svenningsen S, Nesse O, Finsen V, Hole A, Benum P. Prevention of fat embolism syndrome in patients with femoral fractures–immediate or delayed operative fixation? Ann Chir Gynaecol. 1987;76(3):163-166.
20. Pape HC, Tornetta P, Tarkin I, Tzioupis C, Sabeson V, Olson SA. Timing of fracture fixation in multitrauma patients: the role of early total care and damage control surgery. J Am Acad Orthop Surg. 2009;17(9):541-549.
21. Gosseling HR, Pellegrini VD Jr. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop. 1982;165:68-82.
22. Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009:52(5):386-393.
Fat embolism syndrome (FES) was first described by Von Bergmann in 1873 in a patient with a fractured femur.1 While fat within the circulation (fat embolism) is relatively common following long-bone fracture, the clinical pattern of symptoms that make up FES is less so, occurring in 1% to 3% of isolated long-bone fractures and 5% to 10% of patients with multiple skeletal trauma.1 A variety of clinical, laboratory, and imaging criteria has been described, classically by Gurd in 1970 (Table).1-6 Most commonly, however, it is a diagnosis of exclusion when the classic triad of respiratory difficulty, neurologic abnormalities, and a characteristic petechial rash are present in the appropriate clinical setting.6
The neurologic sequelae of this syndrome can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of these symptoms usually occurs between 24 hours and 48 hours (mean, 40 hours) after trauma.1 While these neurologic manifestations occur in up to 86% of patients with FES, it is rare for them to be present without the pulmonary symptoms of dyspnea, hypoxemia, and tachypnea, which are the most common presenting symptoms of the disease.1-6 In this case report, we describe severe, rapid-onset neurologic manifestations, without the typical pulmonary involvement, as the primary clinical presentation of FES in a polytrauma patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A previously healthy 50-year-old man presented to the emergency room in transfer from an outside hospital after a rollover motor vehicle collision in which he was ejected approximately 50 feet. Injuries included a right proximal humerus fracture/dislocation (Figure 1), right ulnar styloid fracture, L1 compression fracture, and multiple rib fractures. On admission, the patient had an ethanol level of 969 mg/L (.097%) and a urine drug screen positive only for opioids, presumably because of pain medication given that day. He denied a history of alcohol abuse and reported consuming 2 to 3 beers per week. The patient was awake, alert, and oriented with a Glasgow Coma Scale (GCS) of 15. He was tachycardic (heart rate, 126), tachypneic (respiratory rate, 24), and febrile (temperature, 38.6°C [101.5°F]), and his white blood cell count was elevated at 29.5×109/L. On examination, his right arm was found to be neurovascularly intact; it was placed in a sling with a forearm splint, and the patient was admitted to the intermediate special care unit on spine precautions with a plan for right shoulder hemiarthroplasty the following day.
Overnight the patient’s mental status began to deteriorate, and approximately 10 hours after initial assessment, he was not answering questions but was able to respond to some commands. On hospital day 2, approximately 20 hours after initial assessment, the patient had a GCS of 8, was not responding to commands, and moved only in response to painful stimuli. The patient had been prescribed morphine by patient-controlled analgesia and had received intravenous hydromorphone on the day of admission, although the amount of medication delivered was not thought adequate to explain this deterioration. On the morning of hospital day 2, noncontrast brain computed tomography (CT) was normal with no evidence of intracranial hemorrhage or infarct. This was followed by brain magnetic resonance imaging (MRI), with the T2-weighted images showing numerous, small hyperintense lesions in subcortical and periventricular white matter, corpus callosum, basal ganglia, brain stem, and cerebellar hemispheres (Figure 2). The lesions also showed hyperintensity on diffusion-weighted MRI and were interpreted to be consistent with multiple, tiny infarcts (Figure 3). In addition, susceptibility-weighted sequences showed low signal in the same areas, suggesting multiple microhemorrhages, a pattern consistent with FES. Oxygen saturations remained 95% to 99%, and chest radiograph revealed clear lung fields without infiltrate. On hospital day 2, the patient was transferred to the intensive care unit and intubated for airway protection owing to an inability to clear secretions, although arterial blood gas levels remained normal. An echocardiogram revealed no right-to-left shunt, such as a patent foramen ovale (PFO); an electroencephalogram showed no seizure-like activity. No petechial rash was noted on skin examination. The patient was treated with supportive care. Right shoulder hemiarthroplasty was performed on hospital day 7 without complications (Figure 1). On hospital day 13, the patient was following commands and on day 14 he was extubated. His mental status continued to improve, and he was discharged to a rehabilitation facility after 36 days. On last follow-up, 6 months after initial injury, the patient was recovering well with no residual neurologic deficits and only minor limitation in range of motion of the right shoulder.
Discussion
This case presented an interesting diagnostic challenge regarding the patient’s rapid decline in mental status, with a differential diagnosis including diffuse axonal injury (DAI), anoxic brain injury, posttraumatic seizure, other intracranial pathology, such as stroke or hemorrhage, and FES. FES was diagnosed, when other possibilities were ruled out, given the characteristic findings on brain MRI described above in the context of multiple fractures.
Pathophysiology
Despite its recognition in 1873, there is no consensus on the pathophysiological mechanism that causes the clinical symptoms of FES. In the setting of trauma, there are 2 predominant theories. The mechanical theory postulates that fat globules enter the circulation through disrupted venules after the fracture of marrow-containing bones, passing to the arterial circulation through pulmonary vasculature, or paradoxically, by way of a right-to-left shunt, such as a PFO.1,3 The presence of fat in the heart, visualized as echogenic material in the right and left atria on transesophageal echocardiography, has been confirmed in multiple studies during orthopedic procedures, including total knee arthroplasty and femoral reaming.8,9 These fat particles can lodge as microembolisms in target organs such as the skin and brain. However, autopsy studies have shown a lack of correlation of the severity of symptoms and the quantity of intravascular fat.1 In addition, the typical 24- to 72-hour delay in the onset of symptoms after initial trauma would argue against a solely mechanical explanation.10
Alternatively or concomitantly, the biochemical theory proposes that embolized fat may be degraded to toxic intermediaries, such as free fatty acids and C-reactive protein, which cause end-organ damage.3 This has been shown in an animal model, in which intravascular injection of free fatty acids was associated with endothelial damage and increased capillary permeability in the lung, leading to acute respiratory distress syndrome (ARDS).11 The same mechanism could explain injury to other end organs and is consistent with the delay in onset of symptoms after acute injury. In our patient’s case, the absence of pulmonary involvement, lack of a right-to-left vascular shunt such as a PFO, and presence of a systemic inflammatory response on admission may implicate the production of toxic intermediaries from the metabolism of embolized fat as the source of this patient’s FES.
Clinical Presentation
The initial presentation of FES usually manifests as respiratory distress and hypoxia.10 Chest radiographs are often normal, as in our patient, but can show bilateral diffuse interstitial or alveolar infiltrates.2,6 CT more often has findings, including bilateral ground-glass opacities with interlobar septal thickening.12 A petechial rash can be found on the head, neck, anterior thorax, axillae, subconjunctiva, and oral mucous membranes, although it occurs in only 20% to 50% of cases.1,2,13 Neurologic sequelae are present in up to 80% of patients,7 with onset typically following pulmonary symptoms.1,10 These sequelae can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of symptoms generally occurs between 24 and 48 hours after trauma,1 although they have been reported as early as 12 hours.10 This case is an example of an atypical course, with the initial presentation of neurologic symptoms at approximately 14 hours after trauma with rapid progression to coma without classic pulmonary symptoms.
Diagnosis
Owing to the nonspecific clinical features of FES, a variety of clinical, laboratory, and imaging criteria has been described. Of these criteria, the most frequently referenced is by Gurd in 1970,4,5 who divided the features into major and minor, with 1 major and 4 minor features required to make the diagnosis (Table). In applying these criteria to our patient, we found that he exhibited the major criteria of cerebral involvement and minor criteria of tachycardia, fever, and thrombocytopenia. Respiratory insufficiency and petechial rash, as well as jaundice, renal changes, and anemia were negative features. Retinal changes, elevated erythrocyte sedimentation rate, and fat macroglobulinemia were not tested or examined. Although in our case the clinical and laboratory criteria for the diagnosis of FES as defined by Gurd were not met, the sensitivity of Gurd’s and other criteria is debated.10
Laboratory tests specific for the disease have not been developed. Although elevated serum levels of lipase, increased blood lipid levels, and fat globules in the urine, sputum, and blood have all been proposed, they are found in trauma patients with and without FES.2,5,6
The nonspecific nature of the signs and symptoms of FES and the lack of reliable laboratory tests for diagnosis of the syndrome highlight the importance of radiographic evaluation in patients with neurologic symptoms. Brain CT scans are usually negative,14 although, in some cases, they may show diffuse edema with scattered low attenuating areas and hemorrhage.15 MRI is more sensitive, and T2-weighted images typically reveal multiple small, nonconfluent hyperintense lesions, usually in the periventricular, subcortical, and deep white matter, sometimes referred to as the “starfield” pattern.14,16 The differential diagnosis for these findings is broad and, in addition to FES, includes DAI, vasogenic edema with microinfarcts, and demyelinating disease.14 Sensitivity and specificity may be increased with the addition of diffusion-weighted MRI, which shows scattered bright spots on a dark background in a similar “starfield” pattern as on T2-weighted images.15 Susceptibility-weighted MRI has recently been introduced as having utility in the diagnosis of FES, with areas of low-signal intensity indicating diffuse microhemorrhages.17 DAI can show a similar pattern; however, the autopsy-confirmed locations of the abnormalities are distinct, with those of FES being found in cerebral and cerebellar white matter and splenium of the corpus callosum and radiographic abnormalities of DAI being found in the gray-white matter junction, dorsolateral brainstem, and splenium of corpus callosum.17
Prevention and Treatment
Of primary importance in the prevention of FES is early stabilization of fractures. Several studies have shown a decreased incidence of FES when long-bone fractures are treated with immediate operative fixation.18,19 However, in the setting of polytrauma, the desire for early definitive treatment must be balanced against the risks for the exaggerated immune response from prolonged surgery.20 The timing of fracture fixation to prevent sequelae of the inflammatory response, such as ARDS and multiple organ dysfunction syndrome, is still debated. In a review article, Pape and colleagues20 suggest classifying the multiply injured patient as stable, borderline, unstable, and in extremis based on clinical and laboratory criteria. They recommend early definitive fixation for stable patients and those patients who are borderline or unstable and responsive to resuscitation, whereas damage-control orthopedics and staged fracture fixation should be considered in the other groups.
Several pharmacologic interventions have been described, although their effects are highly variable and none have clear indications.1-3,6 The most heavily researched is corticosteroids, with the proposed mechanisms of action including blunting of the inflammatory response, stabilizing the pulmonary capillary membrane to reduce interstitial edema, preventing activation of the complement system, and retarding platelet aggregation.21 A recent meta-analysis to assess this intervention examined 6 studies with a total of 386 patients with long-bone fractures who were randomized to treatment with corticosteroids or supportive care only.22 They found a reduced risk for FES in those patients who received corticosteroids, but there was no difference in mortality between groups. Given these results, the utility of corticosteroids is still debated.
Once FES has occurred, treatment options usually focus on supportive care, with most patients having a full recovery.1,3 No specific treatments are available, and symptomatic treatment is the suggested approach, including ensuring adequate oxygenation and ventilation and providing hemodynamic support and volume and blood-product resuscitation as needed.1-3,6
Conclusion
We have presented a case of FES unique in its rapid onset, an initial presentation with neurologic manifestations without typical pulmonary involvement, and the mechanism of end-organ damage without a right-to-left shunt. This case emphasizes the importance of considering FES in the patient with deteriorating mental status in the setting of multiple fractures, particularly in the absence of other characteristic clinical findings, such as pulmonary distress and the pathognomonic petechial rash. Brain MRI can play an important role in diagnosing those patients presenting with predominantly neurological symptoms. Early recognition of this condition allows for the anticipation of complications of the disease process, such as respiratory distress, and the potential need for mechanical ventilation and hemodynamic support.
Fat embolism syndrome (FES) was first described by Von Bergmann in 1873 in a patient with a fractured femur.1 While fat within the circulation (fat embolism) is relatively common following long-bone fracture, the clinical pattern of symptoms that make up FES is less so, occurring in 1% to 3% of isolated long-bone fractures and 5% to 10% of patients with multiple skeletal trauma.1 A variety of clinical, laboratory, and imaging criteria has been described, classically by Gurd in 1970 (Table).1-6 Most commonly, however, it is a diagnosis of exclusion when the classic triad of respiratory difficulty, neurologic abnormalities, and a characteristic petechial rash are present in the appropriate clinical setting.6
The neurologic sequelae of this syndrome can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of these symptoms usually occurs between 24 hours and 48 hours (mean, 40 hours) after trauma.1 While these neurologic manifestations occur in up to 86% of patients with FES, it is rare for them to be present without the pulmonary symptoms of dyspnea, hypoxemia, and tachypnea, which are the most common presenting symptoms of the disease.1-6 In this case report, we describe severe, rapid-onset neurologic manifestations, without the typical pulmonary involvement, as the primary clinical presentation of FES in a polytrauma patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A previously healthy 50-year-old man presented to the emergency room in transfer from an outside hospital after a rollover motor vehicle collision in which he was ejected approximately 50 feet. Injuries included a right proximal humerus fracture/dislocation (Figure 1), right ulnar styloid fracture, L1 compression fracture, and multiple rib fractures. On admission, the patient had an ethanol level of 969 mg/L (.097%) and a urine drug screen positive only for opioids, presumably because of pain medication given that day. He denied a history of alcohol abuse and reported consuming 2 to 3 beers per week. The patient was awake, alert, and oriented with a Glasgow Coma Scale (GCS) of 15. He was tachycardic (heart rate, 126), tachypneic (respiratory rate, 24), and febrile (temperature, 38.6°C [101.5°F]), and his white blood cell count was elevated at 29.5×109/L. On examination, his right arm was found to be neurovascularly intact; it was placed in a sling with a forearm splint, and the patient was admitted to the intermediate special care unit on spine precautions with a plan for right shoulder hemiarthroplasty the following day.
Overnight the patient’s mental status began to deteriorate, and approximately 10 hours after initial assessment, he was not answering questions but was able to respond to some commands. On hospital day 2, approximately 20 hours after initial assessment, the patient had a GCS of 8, was not responding to commands, and moved only in response to painful stimuli. The patient had been prescribed morphine by patient-controlled analgesia and had received intravenous hydromorphone on the day of admission, although the amount of medication delivered was not thought adequate to explain this deterioration. On the morning of hospital day 2, noncontrast brain computed tomography (CT) was normal with no evidence of intracranial hemorrhage or infarct. This was followed by brain magnetic resonance imaging (MRI), with the T2-weighted images showing numerous, small hyperintense lesions in subcortical and periventricular white matter, corpus callosum, basal ganglia, brain stem, and cerebellar hemispheres (Figure 2). The lesions also showed hyperintensity on diffusion-weighted MRI and were interpreted to be consistent with multiple, tiny infarcts (Figure 3). In addition, susceptibility-weighted sequences showed low signal in the same areas, suggesting multiple microhemorrhages, a pattern consistent with FES. Oxygen saturations remained 95% to 99%, and chest radiograph revealed clear lung fields without infiltrate. On hospital day 2, the patient was transferred to the intensive care unit and intubated for airway protection owing to an inability to clear secretions, although arterial blood gas levels remained normal. An echocardiogram revealed no right-to-left shunt, such as a patent foramen ovale (PFO); an electroencephalogram showed no seizure-like activity. No petechial rash was noted on skin examination. The patient was treated with supportive care. Right shoulder hemiarthroplasty was performed on hospital day 7 without complications (Figure 1). On hospital day 13, the patient was following commands and on day 14 he was extubated. His mental status continued to improve, and he was discharged to a rehabilitation facility after 36 days. On last follow-up, 6 months after initial injury, the patient was recovering well with no residual neurologic deficits and only minor limitation in range of motion of the right shoulder.
Discussion
This case presented an interesting diagnostic challenge regarding the patient’s rapid decline in mental status, with a differential diagnosis including diffuse axonal injury (DAI), anoxic brain injury, posttraumatic seizure, other intracranial pathology, such as stroke or hemorrhage, and FES. FES was diagnosed, when other possibilities were ruled out, given the characteristic findings on brain MRI described above in the context of multiple fractures.
Pathophysiology
Despite its recognition in 1873, there is no consensus on the pathophysiological mechanism that causes the clinical symptoms of FES. In the setting of trauma, there are 2 predominant theories. The mechanical theory postulates that fat globules enter the circulation through disrupted venules after the fracture of marrow-containing bones, passing to the arterial circulation through pulmonary vasculature, or paradoxically, by way of a right-to-left shunt, such as a PFO.1,3 The presence of fat in the heart, visualized as echogenic material in the right and left atria on transesophageal echocardiography, has been confirmed in multiple studies during orthopedic procedures, including total knee arthroplasty and femoral reaming.8,9 These fat particles can lodge as microembolisms in target organs such as the skin and brain. However, autopsy studies have shown a lack of correlation of the severity of symptoms and the quantity of intravascular fat.1 In addition, the typical 24- to 72-hour delay in the onset of symptoms after initial trauma would argue against a solely mechanical explanation.10
Alternatively or concomitantly, the biochemical theory proposes that embolized fat may be degraded to toxic intermediaries, such as free fatty acids and C-reactive protein, which cause end-organ damage.3 This has been shown in an animal model, in which intravascular injection of free fatty acids was associated with endothelial damage and increased capillary permeability in the lung, leading to acute respiratory distress syndrome (ARDS).11 The same mechanism could explain injury to other end organs and is consistent with the delay in onset of symptoms after acute injury. In our patient’s case, the absence of pulmonary involvement, lack of a right-to-left vascular shunt such as a PFO, and presence of a systemic inflammatory response on admission may implicate the production of toxic intermediaries from the metabolism of embolized fat as the source of this patient’s FES.
Clinical Presentation
The initial presentation of FES usually manifests as respiratory distress and hypoxia.10 Chest radiographs are often normal, as in our patient, but can show bilateral diffuse interstitial or alveolar infiltrates.2,6 CT more often has findings, including bilateral ground-glass opacities with interlobar septal thickening.12 A petechial rash can be found on the head, neck, anterior thorax, axillae, subconjunctiva, and oral mucous membranes, although it occurs in only 20% to 50% of cases.1,2,13 Neurologic sequelae are present in up to 80% of patients,7 with onset typically following pulmonary symptoms.1,10 These sequelae can range from headache, confusion, and agitation to stupor, focal neurologic signs, and, less commonly, coma.7 Onset of symptoms generally occurs between 24 and 48 hours after trauma,1 although they have been reported as early as 12 hours.10 This case is an example of an atypical course, with the initial presentation of neurologic symptoms at approximately 14 hours after trauma with rapid progression to coma without classic pulmonary symptoms.
Diagnosis
Owing to the nonspecific clinical features of FES, a variety of clinical, laboratory, and imaging criteria has been described. Of these criteria, the most frequently referenced is by Gurd in 1970,4,5 who divided the features into major and minor, with 1 major and 4 minor features required to make the diagnosis (Table). In applying these criteria to our patient, we found that he exhibited the major criteria of cerebral involvement and minor criteria of tachycardia, fever, and thrombocytopenia. Respiratory insufficiency and petechial rash, as well as jaundice, renal changes, and anemia were negative features. Retinal changes, elevated erythrocyte sedimentation rate, and fat macroglobulinemia were not tested or examined. Although in our case the clinical and laboratory criteria for the diagnosis of FES as defined by Gurd were not met, the sensitivity of Gurd’s and other criteria is debated.10
Laboratory tests specific for the disease have not been developed. Although elevated serum levels of lipase, increased blood lipid levels, and fat globules in the urine, sputum, and blood have all been proposed, they are found in trauma patients with and without FES.2,5,6
The nonspecific nature of the signs and symptoms of FES and the lack of reliable laboratory tests for diagnosis of the syndrome highlight the importance of radiographic evaluation in patients with neurologic symptoms. Brain CT scans are usually negative,14 although, in some cases, they may show diffuse edema with scattered low attenuating areas and hemorrhage.15 MRI is more sensitive, and T2-weighted images typically reveal multiple small, nonconfluent hyperintense lesions, usually in the periventricular, subcortical, and deep white matter, sometimes referred to as the “starfield” pattern.14,16 The differential diagnosis for these findings is broad and, in addition to FES, includes DAI, vasogenic edema with microinfarcts, and demyelinating disease.14 Sensitivity and specificity may be increased with the addition of diffusion-weighted MRI, which shows scattered bright spots on a dark background in a similar “starfield” pattern as on T2-weighted images.15 Susceptibility-weighted MRI has recently been introduced as having utility in the diagnosis of FES, with areas of low-signal intensity indicating diffuse microhemorrhages.17 DAI can show a similar pattern; however, the autopsy-confirmed locations of the abnormalities are distinct, with those of FES being found in cerebral and cerebellar white matter and splenium of the corpus callosum and radiographic abnormalities of DAI being found in the gray-white matter junction, dorsolateral brainstem, and splenium of corpus callosum.17
Prevention and Treatment
Of primary importance in the prevention of FES is early stabilization of fractures. Several studies have shown a decreased incidence of FES when long-bone fractures are treated with immediate operative fixation.18,19 However, in the setting of polytrauma, the desire for early definitive treatment must be balanced against the risks for the exaggerated immune response from prolonged surgery.20 The timing of fracture fixation to prevent sequelae of the inflammatory response, such as ARDS and multiple organ dysfunction syndrome, is still debated. In a review article, Pape and colleagues20 suggest classifying the multiply injured patient as stable, borderline, unstable, and in extremis based on clinical and laboratory criteria. They recommend early definitive fixation for stable patients and those patients who are borderline or unstable and responsive to resuscitation, whereas damage-control orthopedics and staged fracture fixation should be considered in the other groups.
Several pharmacologic interventions have been described, although their effects are highly variable and none have clear indications.1-3,6 The most heavily researched is corticosteroids, with the proposed mechanisms of action including blunting of the inflammatory response, stabilizing the pulmonary capillary membrane to reduce interstitial edema, preventing activation of the complement system, and retarding platelet aggregation.21 A recent meta-analysis to assess this intervention examined 6 studies with a total of 386 patients with long-bone fractures who were randomized to treatment with corticosteroids or supportive care only.22 They found a reduced risk for FES in those patients who received corticosteroids, but there was no difference in mortality between groups. Given these results, the utility of corticosteroids is still debated.
Once FES has occurred, treatment options usually focus on supportive care, with most patients having a full recovery.1,3 No specific treatments are available, and symptomatic treatment is the suggested approach, including ensuring adequate oxygenation and ventilation and providing hemodynamic support and volume and blood-product resuscitation as needed.1-3,6
Conclusion
We have presented a case of FES unique in its rapid onset, an initial presentation with neurologic manifestations without typical pulmonary involvement, and the mechanism of end-organ damage without a right-to-left shunt. This case emphasizes the importance of considering FES in the patient with deteriorating mental status in the setting of multiple fractures, particularly in the absence of other characteristic clinical findings, such as pulmonary distress and the pathognomonic petechial rash. Brain MRI can play an important role in diagnosing those patients presenting with predominantly neurological symptoms. Early recognition of this condition allows for the anticipation of complications of the disease process, such as respiratory distress, and the potential need for mechanical ventilation and hemodynamic support.
1. Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics. 1996;19(1):41-49.
2. Levy D. The fat embolism syndrome. A review. Clin Orthop. 1990;261:281-286.
3. Mellor A, Soni N. Fat embolism. Anaesthesia. 2001;56(2):145-154.
4. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surg Br. 1970:52(4):732-737.
5. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
6. Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132(4):435-439.
7. Jacobson DM, Terrence CF, Reinmuth OM. The neurologic manifestations of fat embolism. Neurology. 1986;36(6):847-851.
8. Sulek CA, Davies LK, Enneking FK, Gearen PA, Lobato EB. Cerebral microembolism diagnosed by transcranial Doppler during total knee arthroplasty: correlation with transesophageal echocardiography. Anesthesiology. 1999;91(3):672-676.
9. Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41(Suppl 2):S90-S93.
10. Gupta B, D’souza N, Sawhney C, et al. Analyzing fat embolism syndrome in trauma patients at AIIMS Apex Trauma Center, New Delhi, India. J Emerg Trauma Shock. 2011;4(3):337–341.
11. King EG, Wagner WW Jr, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest. 1971:59(5):524-530.
12. Malagari K, Economopoulos N, Stoupis C, et al. High-resolution CT findings in mild pulmonary fat embolism. Chest. 2003:123(4):1196-1201.
13. King MB, Harmon KR. Unusual forms of pulmonary embolism. Clin Chest Med. 1994;15(3):561-580.
14. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.
15. Simon AD, Ulmer JL, Strottmann JM. Contrast-enhanced MR imaging of cerebral fat embolism: case report and review of the literature. AJNR Am J Neuroradiol. 2003;24(1):97-101.
16. Butteriss DJ, Mahad D, Soh C, Walls T, Weir D, Birchall D. Reversible cytotoxic cerebral edema in cerebral fat embolism. AJNR Am J Neuroradiol. 2006;27(3):620-623.
17. Zaitsu Y, Terae S, Kudo K, et al. Susceptibility-weighted imaging of cerebral fat embolism. J Comput Assist Tomogr. 2010;34(1):107-112.
18. Riska EB, Myllynen P. Fat embolism in patients with multiple injuries. J Trauma. 1982;22(11):891-894.
19. Svenningsen S, Nesse O, Finsen V, Hole A, Benum P. Prevention of fat embolism syndrome in patients with femoral fractures–immediate or delayed operative fixation? Ann Chir Gynaecol. 1987;76(3):163-166.
20. Pape HC, Tornetta P, Tarkin I, Tzioupis C, Sabeson V, Olson SA. Timing of fracture fixation in multitrauma patients: the role of early total care and damage control surgery. J Am Acad Orthop Surg. 2009;17(9):541-549.
21. Gosseling HR, Pellegrini VD Jr. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop. 1982;165:68-82.
22. Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009:52(5):386-393.
1. Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics. 1996;19(1):41-49.
2. Levy D. The fat embolism syndrome. A review. Clin Orthop. 1990;261:281-286.
3. Mellor A, Soni N. Fat embolism. Anaesthesia. 2001;56(2):145-154.
4. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surg Br. 1970:52(4):732-737.
5. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
6. Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132(4):435-439.
7. Jacobson DM, Terrence CF, Reinmuth OM. The neurologic manifestations of fat embolism. Neurology. 1986;36(6):847-851.
8. Sulek CA, Davies LK, Enneking FK, Gearen PA, Lobato EB. Cerebral microembolism diagnosed by transcranial Doppler during total knee arthroplasty: correlation with transesophageal echocardiography. Anesthesiology. 1999;91(3):672-676.
9. Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41(Suppl 2):S90-S93.
10. Gupta B, D’souza N, Sawhney C, et al. Analyzing fat embolism syndrome in trauma patients at AIIMS Apex Trauma Center, New Delhi, India. J Emerg Trauma Shock. 2011;4(3):337–341.
11. King EG, Wagner WW Jr, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest. 1971:59(5):524-530.
12. Malagari K, Economopoulos N, Stoupis C, et al. High-resolution CT findings in mild pulmonary fat embolism. Chest. 2003:123(4):1196-1201.
13. King MB, Harmon KR. Unusual forms of pulmonary embolism. Clin Chest Med. 1994;15(3):561-580.
14. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.
15. Simon AD, Ulmer JL, Strottmann JM. Contrast-enhanced MR imaging of cerebral fat embolism: case report and review of the literature. AJNR Am J Neuroradiol. 2003;24(1):97-101.
16. Butteriss DJ, Mahad D, Soh C, Walls T, Weir D, Birchall D. Reversible cytotoxic cerebral edema in cerebral fat embolism. AJNR Am J Neuroradiol. 2006;27(3):620-623.
17. Zaitsu Y, Terae S, Kudo K, et al. Susceptibility-weighted imaging of cerebral fat embolism. J Comput Assist Tomogr. 2010;34(1):107-112.
18. Riska EB, Myllynen P. Fat embolism in patients with multiple injuries. J Trauma. 1982;22(11):891-894.
19. Svenningsen S, Nesse O, Finsen V, Hole A, Benum P. Prevention of fat embolism syndrome in patients with femoral fractures–immediate or delayed operative fixation? Ann Chir Gynaecol. 1987;76(3):163-166.
20. Pape HC, Tornetta P, Tarkin I, Tzioupis C, Sabeson V, Olson SA. Timing of fracture fixation in multitrauma patients: the role of early total care and damage control surgery. J Am Acad Orthop Surg. 2009;17(9):541-549.
21. Gosseling HR, Pellegrini VD Jr. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop. 1982;165:68-82.
22. Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009:52(5):386-393.
Subtrochanteric Femur Fracture After Removal of Screws for Femoral Neck Fracture in a Child
Subtrochanteric fractures and other complications related to hardware removal in children with slipped capital femoral epiphysis (SCFE) have been well documented.1-3 Subtrochanteric fractures after cannulated screw fixation of femoral neck fractures in adults have also been well recognized,4 and there are several reports on the topic.4,5 However, there are no reports on subtrochanteric fractures after removal of the screws for femoral neck fractures in children.
In this article, we report the case of a child who sustained a subtrochanteric fracture after the screw removal and healing that followed a femoral neck fracture. The patient’s parent provided written informed consent for print and electronic publication of this case report. In addition, our institutional review board approved this case report.
Case Report
A 10-year-old boy was brought to our emergency department with the chief complaint of left hip pain after a car accident. Anteroposterior and axial lateral radiographs showed a displaced cervicotrochanteric femoral neck fracture (Figures 1A, 1B). The patient was admitted to the hospital and underwent closed reduction and internal fixation with two 3.5-mm cannulated titanium screws within 12 hours of arrival. The screws did not cross the physis to avoid iatrogenic injury of the capital femoral epiphysis (Figures 2A, 2B). The entry point was located at the lower level of the lesser trochanter. The lateral cortex was penetrated only once by the guide wire for the placement of each screw.
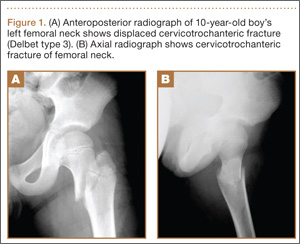
The patient was discharged to home care with a crutch and an ischial weight-bearing long leg brace for protection from unexpected external force. Two months after surgery, we allowed the patient to walk with the brace and without the crutch. Full-weight-bearing ambulation was allowed 3 months after surgery.
About 9 months after initial surgery, we removed 2 titanium screws, which were completely covered with growing new bone. The lateral cortex surrounding the screw heads was chiseled from the lower level of the lesser trochanter to remove the completely immersed screw heads (Figures 3A, 3B).
After screw removal, we recommended non-weight-bearing crutch-walking for 2 weeks followed by partial weight-bearing with crutch for another month. However, the patient started full weight-bearing 2 weeks after screw removal. One month after screw removal, he was brought to the emergency department with severe left hip pain after missing a step on a path. Anteroposterior and lateral radiographs showed an oblique subtrochanteric fracture at the empty screw holes (Figures 4A, 4B). A plate and 4 screws were placed to stabilize the subtrochanteric fracture, and a hip spica cast was applied and was to be worn for 3 weeks (Figures 5A, 5B).
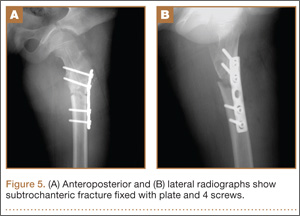
At final follow-up, 6 months after the second surgery, the fracture was healed, and there had been no complications, such as avascular necrosis of the femoral head and leg-length discrepancy (Figures 6A, 6B).
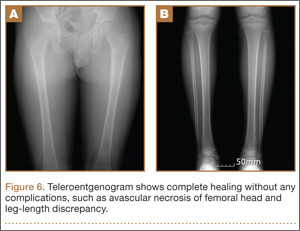
Discussion
Although in situ pinning of SCFE is a common procedure with good results, the rate of complications of hardware removal can be as high as 34%; these complications are well documented.5 Subtrochanteric fracture as a complication of proximal femoral neck pinning in adults is also well documented.4,5 However, there are no reports on subtrochanteric fractures after screw removal in the treatment of femoral neck fractures in children.
Brooks and colleagues6 emphasized the point that multiple passes weakened the lateral cortex, decreased the energy-absorbing capacity by 55.2%, and increased local stress. Even if a screw is placed in a relatively safe zone above the lesser trochanter, pie-crusting of the cortex can weaken it enough to predispose it to failure under a relatively normal load.7 We inserted 2 cannulated titanium screws without repositioning or multiple drilling, and the femoral neck fracture was united.
The common denominator for subtrochanteric fractures after screw or pin fixation of femoral neck fractures in adults seemed to be the entry point of the lateral cortex below the level of the most inferior edge of the lesser trochanter.4 The pin should have its entry site proximal to the level of the lesser trochnater. Paloski and colleagues7 and Canale and colleagues8 hypothesized that this screw acted as a stress riser to the normal bone, which underwent abnormal loads caused by the patient’s habitus and later mechanism of injury. In our patient’s case, the appropriate starting point for perpendicular penetration of the femoral neck fracture line was on the lateral femoral cortex at the level of the lesser trochanter. We thought this entry on the lateral cortex might predispose the patient to a subtrochanteric fracture. The starting point of the screw is considered the most important factor in preventing fracture after screw removal.
As titanium pins cause very tight bone ingrowth,9,10 the surface of titanium screws used for femoral neck fractures in children are smoothed to reduce turning force.1 The hexagonal sockets wore off rapidly and proved to be too weak to overcome the necessary torque for loosening the pin from the bone.
Lee and colleagues10 found that significantly more operative time was needed to remove titanium pins (vs steel pins) after 12 months or longer. When Asnis III pins (Howmedica, Rutherford, New Jersey) were used in the treatment of femoral neck fractures in aged patients, similar problems did not occur. One possible explanation is that bone density is higher in adolescents than in adults. In addition, more bone ingrowth and higher bone compression might occur in adolescent bones.1 Given the considerable disadvantages noted in their series, Ilchmann and Parsch1 concluded that use of cannulated titanium screws should be suspended and that stainless steel pins are safe to use in SCFE.
In our patient’s case, we also struggled to remove titanium screws. Subtrochanteric fractures can be complications after removal of screws for femoral neck fractures in children. If there are no specific screw-related symptoms, one should consider leaving the screw in place and avoiding screw removal.
1. Ilchmann T, Parsch K. Complications at screw removal in slipped capital femoral epiphysis treated by cannulated titanium screws. Arch Orthop Trauma Surg. 2006;126(6):359-363.
2. Raney EM, Freccero LA, Dolan DE, Lighter R, Fillman L, Chambers HG. Evidence-based analysis of removal of orthopaedic implants in the pediatric population. J Pediatr Orthop. 2008;28(7):701-704.
3. Karagkevrekis CB, Rahman H. Subtrochanteric femoral fracture following removal of screw for slipped capital femoral epiphysis. Injury. 2003;38(4):320-321.
4. Kloen P, Rubel IF, Lyden JP, Helfet DL. Subtrochanteric fracture after cannulated screw fixation of femoral neck fractures: a report of four cases. J Orthop Trauma. 2003;17(3):225-229.
5. Karr RK, Schwab JP. Subtrochanteric fracture as complication of proximal femoral pinning. Clin Orthop. 1985;(194):214-217.
6. Brooks DB, Burstein AH, Frankel VH. The biomechanics of torsional fractures. The stress concentration effect of a drill hole. J Bone Joint Surg Am. 1970;52(3):507-514.
7. Paloski M, Taylor BC, Willits M. Subtrochanteric femur fracture after slipped capital femoral epiphysis pinning: a novel treatment. Adv Orthop. 2011;2011:809136.
8. Canale ST, Casillas M, Banta JV. Displaced femoral neck fractures at the bone–screw interface after in situ fixation of slipped capital femoral epiphysis. J Pediatr Orthop. 1997;17(2):212-215.
9. Vresilovic EJ, Spindler KP, Robertson WW Jr, Davidson RS, Drummond DS. Failure of pin removal after in situ pinning of slipped capital femoral epiphysis: a comparison of different pin types. J Pediatr Orthop. 1990;10(6):764-768.
10. Lee TK, Haynes RJ, Longo JA, Chu JR. Pin removal in slipped capital femoral epiphysis: the unsuitability of titanium devices. J Pediatr Orthop. 1996;16(1):49-52.
Subtrochanteric fractures and other complications related to hardware removal in children with slipped capital femoral epiphysis (SCFE) have been well documented.1-3 Subtrochanteric fractures after cannulated screw fixation of femoral neck fractures in adults have also been well recognized,4 and there are several reports on the topic.4,5 However, there are no reports on subtrochanteric fractures after removal of the screws for femoral neck fractures in children.
In this article, we report the case of a child who sustained a subtrochanteric fracture after the screw removal and healing that followed a femoral neck fracture. The patient’s parent provided written informed consent for print and electronic publication of this case report. In addition, our institutional review board approved this case report.
Case Report
A 10-year-old boy was brought to our emergency department with the chief complaint of left hip pain after a car accident. Anteroposterior and axial lateral radiographs showed a displaced cervicotrochanteric femoral neck fracture (Figures 1A, 1B). The patient was admitted to the hospital and underwent closed reduction and internal fixation with two 3.5-mm cannulated titanium screws within 12 hours of arrival. The screws did not cross the physis to avoid iatrogenic injury of the capital femoral epiphysis (Figures 2A, 2B). The entry point was located at the lower level of the lesser trochanter. The lateral cortex was penetrated only once by the guide wire for the placement of each screw.
The patient was discharged to home care with a crutch and an ischial weight-bearing long leg brace for protection from unexpected external force. Two months after surgery, we allowed the patient to walk with the brace and without the crutch. Full-weight-bearing ambulation was allowed 3 months after surgery.
About 9 months after initial surgery, we removed 2 titanium screws, which were completely covered with growing new bone. The lateral cortex surrounding the screw heads was chiseled from the lower level of the lesser trochanter to remove the completely immersed screw heads (Figures 3A, 3B).
After screw removal, we recommended non-weight-bearing crutch-walking for 2 weeks followed by partial weight-bearing with crutch for another month. However, the patient started full weight-bearing 2 weeks after screw removal. One month after screw removal, he was brought to the emergency department with severe left hip pain after missing a step on a path. Anteroposterior and lateral radiographs showed an oblique subtrochanteric fracture at the empty screw holes (Figures 4A, 4B). A plate and 4 screws were placed to stabilize the subtrochanteric fracture, and a hip spica cast was applied and was to be worn for 3 weeks (Figures 5A, 5B).
At final follow-up, 6 months after the second surgery, the fracture was healed, and there had been no complications, such as avascular necrosis of the femoral head and leg-length discrepancy (Figures 6A, 6B).
Discussion
Although in situ pinning of SCFE is a common procedure with good results, the rate of complications of hardware removal can be as high as 34%; these complications are well documented.5 Subtrochanteric fracture as a complication of proximal femoral neck pinning in adults is also well documented.4,5 However, there are no reports on subtrochanteric fractures after screw removal in the treatment of femoral neck fractures in children.
Brooks and colleagues6 emphasized the point that multiple passes weakened the lateral cortex, decreased the energy-absorbing capacity by 55.2%, and increased local stress. Even if a screw is placed in a relatively safe zone above the lesser trochanter, pie-crusting of the cortex can weaken it enough to predispose it to failure under a relatively normal load.7 We inserted 2 cannulated titanium screws without repositioning or multiple drilling, and the femoral neck fracture was united.
The common denominator for subtrochanteric fractures after screw or pin fixation of femoral neck fractures in adults seemed to be the entry point of the lateral cortex below the level of the most inferior edge of the lesser trochanter.4 The pin should have its entry site proximal to the level of the lesser trochnater. Paloski and colleagues7 and Canale and colleagues8 hypothesized that this screw acted as a stress riser to the normal bone, which underwent abnormal loads caused by the patient’s habitus and later mechanism of injury. In our patient’s case, the appropriate starting point for perpendicular penetration of the femoral neck fracture line was on the lateral femoral cortex at the level of the lesser trochanter. We thought this entry on the lateral cortex might predispose the patient to a subtrochanteric fracture. The starting point of the screw is considered the most important factor in preventing fracture after screw removal.
As titanium pins cause very tight bone ingrowth,9,10 the surface of titanium screws used for femoral neck fractures in children are smoothed to reduce turning force.1 The hexagonal sockets wore off rapidly and proved to be too weak to overcome the necessary torque for loosening the pin from the bone.
Lee and colleagues10 found that significantly more operative time was needed to remove titanium pins (vs steel pins) after 12 months or longer. When Asnis III pins (Howmedica, Rutherford, New Jersey) were used in the treatment of femoral neck fractures in aged patients, similar problems did not occur. One possible explanation is that bone density is higher in adolescents than in adults. In addition, more bone ingrowth and higher bone compression might occur in adolescent bones.1 Given the considerable disadvantages noted in their series, Ilchmann and Parsch1 concluded that use of cannulated titanium screws should be suspended and that stainless steel pins are safe to use in SCFE.
In our patient’s case, we also struggled to remove titanium screws. Subtrochanteric fractures can be complications after removal of screws for femoral neck fractures in children. If there are no specific screw-related symptoms, one should consider leaving the screw in place and avoiding screw removal.
Subtrochanteric fractures and other complications related to hardware removal in children with slipped capital femoral epiphysis (SCFE) have been well documented.1-3 Subtrochanteric fractures after cannulated screw fixation of femoral neck fractures in adults have also been well recognized,4 and there are several reports on the topic.4,5 However, there are no reports on subtrochanteric fractures after removal of the screws for femoral neck fractures in children.
In this article, we report the case of a child who sustained a subtrochanteric fracture after the screw removal and healing that followed a femoral neck fracture. The patient’s parent provided written informed consent for print and electronic publication of this case report. In addition, our institutional review board approved this case report.
Case Report
A 10-year-old boy was brought to our emergency department with the chief complaint of left hip pain after a car accident. Anteroposterior and axial lateral radiographs showed a displaced cervicotrochanteric femoral neck fracture (Figures 1A, 1B). The patient was admitted to the hospital and underwent closed reduction and internal fixation with two 3.5-mm cannulated titanium screws within 12 hours of arrival. The screws did not cross the physis to avoid iatrogenic injury of the capital femoral epiphysis (Figures 2A, 2B). The entry point was located at the lower level of the lesser trochanter. The lateral cortex was penetrated only once by the guide wire for the placement of each screw.
The patient was discharged to home care with a crutch and an ischial weight-bearing long leg brace for protection from unexpected external force. Two months after surgery, we allowed the patient to walk with the brace and without the crutch. Full-weight-bearing ambulation was allowed 3 months after surgery.
About 9 months after initial surgery, we removed 2 titanium screws, which were completely covered with growing new bone. The lateral cortex surrounding the screw heads was chiseled from the lower level of the lesser trochanter to remove the completely immersed screw heads (Figures 3A, 3B).
After screw removal, we recommended non-weight-bearing crutch-walking for 2 weeks followed by partial weight-bearing with crutch for another month. However, the patient started full weight-bearing 2 weeks after screw removal. One month after screw removal, he was brought to the emergency department with severe left hip pain after missing a step on a path. Anteroposterior and lateral radiographs showed an oblique subtrochanteric fracture at the empty screw holes (Figures 4A, 4B). A plate and 4 screws were placed to stabilize the subtrochanteric fracture, and a hip spica cast was applied and was to be worn for 3 weeks (Figures 5A, 5B).
At final follow-up, 6 months after the second surgery, the fracture was healed, and there had been no complications, such as avascular necrosis of the femoral head and leg-length discrepancy (Figures 6A, 6B).
Discussion
Although in situ pinning of SCFE is a common procedure with good results, the rate of complications of hardware removal can be as high as 34%; these complications are well documented.5 Subtrochanteric fracture as a complication of proximal femoral neck pinning in adults is also well documented.4,5 However, there are no reports on subtrochanteric fractures after screw removal in the treatment of femoral neck fractures in children.
Brooks and colleagues6 emphasized the point that multiple passes weakened the lateral cortex, decreased the energy-absorbing capacity by 55.2%, and increased local stress. Even if a screw is placed in a relatively safe zone above the lesser trochanter, pie-crusting of the cortex can weaken it enough to predispose it to failure under a relatively normal load.7 We inserted 2 cannulated titanium screws without repositioning or multiple drilling, and the femoral neck fracture was united.
The common denominator for subtrochanteric fractures after screw or pin fixation of femoral neck fractures in adults seemed to be the entry point of the lateral cortex below the level of the most inferior edge of the lesser trochanter.4 The pin should have its entry site proximal to the level of the lesser trochnater. Paloski and colleagues7 and Canale and colleagues8 hypothesized that this screw acted as a stress riser to the normal bone, which underwent abnormal loads caused by the patient’s habitus and later mechanism of injury. In our patient’s case, the appropriate starting point for perpendicular penetration of the femoral neck fracture line was on the lateral femoral cortex at the level of the lesser trochanter. We thought this entry on the lateral cortex might predispose the patient to a subtrochanteric fracture. The starting point of the screw is considered the most important factor in preventing fracture after screw removal.
As titanium pins cause very tight bone ingrowth,9,10 the surface of titanium screws used for femoral neck fractures in children are smoothed to reduce turning force.1 The hexagonal sockets wore off rapidly and proved to be too weak to overcome the necessary torque for loosening the pin from the bone.
Lee and colleagues10 found that significantly more operative time was needed to remove titanium pins (vs steel pins) after 12 months or longer. When Asnis III pins (Howmedica, Rutherford, New Jersey) were used in the treatment of femoral neck fractures in aged patients, similar problems did not occur. One possible explanation is that bone density is higher in adolescents than in adults. In addition, more bone ingrowth and higher bone compression might occur in adolescent bones.1 Given the considerable disadvantages noted in their series, Ilchmann and Parsch1 concluded that use of cannulated titanium screws should be suspended and that stainless steel pins are safe to use in SCFE.
In our patient’s case, we also struggled to remove titanium screws. Subtrochanteric fractures can be complications after removal of screws for femoral neck fractures in children. If there are no specific screw-related symptoms, one should consider leaving the screw in place and avoiding screw removal.
1. Ilchmann T, Parsch K. Complications at screw removal in slipped capital femoral epiphysis treated by cannulated titanium screws. Arch Orthop Trauma Surg. 2006;126(6):359-363.
2. Raney EM, Freccero LA, Dolan DE, Lighter R, Fillman L, Chambers HG. Evidence-based analysis of removal of orthopaedic implants in the pediatric population. J Pediatr Orthop. 2008;28(7):701-704.
3. Karagkevrekis CB, Rahman H. Subtrochanteric femoral fracture following removal of screw for slipped capital femoral epiphysis. Injury. 2003;38(4):320-321.
4. Kloen P, Rubel IF, Lyden JP, Helfet DL. Subtrochanteric fracture after cannulated screw fixation of femoral neck fractures: a report of four cases. J Orthop Trauma. 2003;17(3):225-229.
5. Karr RK, Schwab JP. Subtrochanteric fracture as complication of proximal femoral pinning. Clin Orthop. 1985;(194):214-217.
6. Brooks DB, Burstein AH, Frankel VH. The biomechanics of torsional fractures. The stress concentration effect of a drill hole. J Bone Joint Surg Am. 1970;52(3):507-514.
7. Paloski M, Taylor BC, Willits M. Subtrochanteric femur fracture after slipped capital femoral epiphysis pinning: a novel treatment. Adv Orthop. 2011;2011:809136.
8. Canale ST, Casillas M, Banta JV. Displaced femoral neck fractures at the bone–screw interface after in situ fixation of slipped capital femoral epiphysis. J Pediatr Orthop. 1997;17(2):212-215.
9. Vresilovic EJ, Spindler KP, Robertson WW Jr, Davidson RS, Drummond DS. Failure of pin removal after in situ pinning of slipped capital femoral epiphysis: a comparison of different pin types. J Pediatr Orthop. 1990;10(6):764-768.
10. Lee TK, Haynes RJ, Longo JA, Chu JR. Pin removal in slipped capital femoral epiphysis: the unsuitability of titanium devices. J Pediatr Orthop. 1996;16(1):49-52.
1. Ilchmann T, Parsch K. Complications at screw removal in slipped capital femoral epiphysis treated by cannulated titanium screws. Arch Orthop Trauma Surg. 2006;126(6):359-363.
2. Raney EM, Freccero LA, Dolan DE, Lighter R, Fillman L, Chambers HG. Evidence-based analysis of removal of orthopaedic implants in the pediatric population. J Pediatr Orthop. 2008;28(7):701-704.
3. Karagkevrekis CB, Rahman H. Subtrochanteric femoral fracture following removal of screw for slipped capital femoral epiphysis. Injury. 2003;38(4):320-321.
4. Kloen P, Rubel IF, Lyden JP, Helfet DL. Subtrochanteric fracture after cannulated screw fixation of femoral neck fractures: a report of four cases. J Orthop Trauma. 2003;17(3):225-229.
5. Karr RK, Schwab JP. Subtrochanteric fracture as complication of proximal femoral pinning. Clin Orthop. 1985;(194):214-217.
6. Brooks DB, Burstein AH, Frankel VH. The biomechanics of torsional fractures. The stress concentration effect of a drill hole. J Bone Joint Surg Am. 1970;52(3):507-514.
7. Paloski M, Taylor BC, Willits M. Subtrochanteric femur fracture after slipped capital femoral epiphysis pinning: a novel treatment. Adv Orthop. 2011;2011:809136.
8. Canale ST, Casillas M, Banta JV. Displaced femoral neck fractures at the bone–screw interface after in situ fixation of slipped capital femoral epiphysis. J Pediatr Orthop. 1997;17(2):212-215.
9. Vresilovic EJ, Spindler KP, Robertson WW Jr, Davidson RS, Drummond DS. Failure of pin removal after in situ pinning of slipped capital femoral epiphysis: a comparison of different pin types. J Pediatr Orthop. 1990;10(6):764-768.
10. Lee TK, Haynes RJ, Longo JA, Chu JR. Pin removal in slipped capital femoral epiphysis: the unsuitability of titanium devices. J Pediatr Orthop. 1996;16(1):49-52.