User login
Primary Localized Cutaneous Nodular Amyloidosis of the Thighs
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
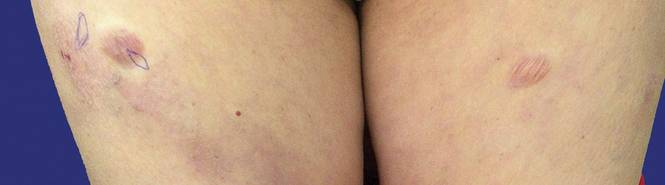
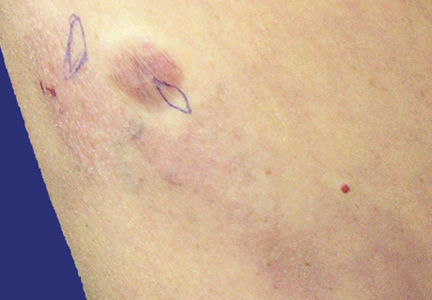
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
Histopathologic Findings
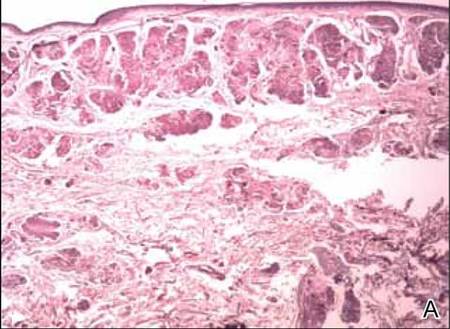
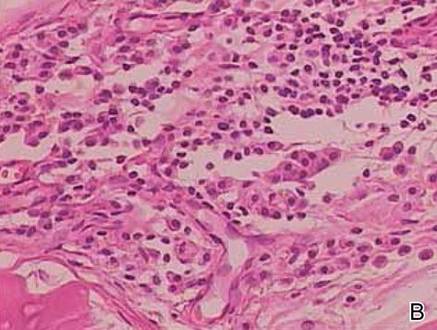
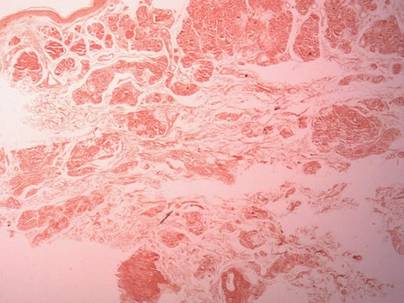
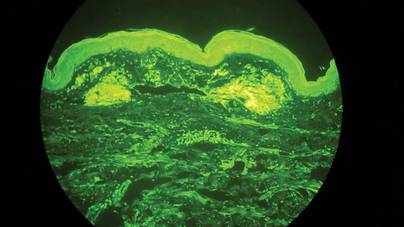
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
|
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
Histopathologic Findings
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
|
|
|
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
Case Report
Clinical Findings
A 65-year-old woman presented with multiple asymptomatic discrete nodules and atrophic plaques on the thighs of 4 years’ duration. The lesions had started as 2 small, asymptomatic, madder red plaques symmetrically located on the anterior aspect of each thigh that had gradually increased in number and size, particularly on the right thigh. Two years later, 2 new atrophic plaques appeared on the anterior aspect of each. The lesions developed slowly but never remitted and had been misdiagnosed as primary macular atrophy of skin by several outpatient clinics. The patient’s general health was good and her personal and family history was unremarkable.
Physical examination revealed multiple madder red plaques and nodules of various shapes and sizes (ie, 1–3 cm in diameter) on the anterior aspect of the right thigh. The lesions were slightly elevated with a waxy surface, firm, and painless to palpation. One similar lesion was noted on the anterior aspect of the left thigh. Two 2-cm, brown-red, atrophic plaques also were noted in a symmetrical distribution on the anterior aspect of each thigh. The plaque surfaces were slightly crinkly and shiny, and anetodermalike lesions produced a buttonhole sign identical to a neurofibroma on palpation (Figure 1).
Histopathologic Findings
Two biopsy specimens were taken from a nodule and an atrophic plaque on the right thigh. Microscopic examination revealed deposition of homogeneous eosinophilic material in the reticular dermis and subcutis as well as around the fine vessels (Figure 2A). There was mild cellular infiltration of lymphocytes, plasma cells, and giant cells in the dermis, especially adjacent to deposits and around the vessels (Figure 2B). The homogeneous material appeared salmon pink on Congo red staining and bright green by thioflavin T staining using a fluorescent microscope (Figures 3 and 4). These results suggested the characteristic features of cutaneous nodular amyloidosis.
Figure 2. Histopathologically, homogeneous eosinophilic material deposited in the reticular dermis and subcutis was noted (A)(H&E, original magnification ×25). Microscopic examination showed lymphocytes and plasma cells infiltrated in the dermis, especially adjacent to deposits and around the vessels (B)(H&E, original magnification ×200). |
|
|
|
|
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Serum protein electrophoresis was normal and no Bence Jones proteins were detected. Serum IgA, IgG, and IgM levels showed no abnormalities. Electrocardiogram, chest radiography, and abdominal ultrasound were normal.
A diagnosis of primary localized cutaneous nodular amyloidosis (PLCNA) was made based on clinical, histopathologic, and laboratory findings. Although surgical excision in stages was proposed, the patient refused treatment because the lesions were asymptomatic. There was no obvious progression of the skin lesions and no abnormal systemic findings during 2.5 years’ follow-up.
Comment
Amyloidosis is a spectrum of diseases consisting of deposition of amyloid proteins in various tissues. Clinically, amyloidosis is divided into both primary and secondary forms of systemic amyloidosis, hemodialysis-associated amyloidosis, heredofamilial amyloidosis, and cutaneous amyloidosis. Primary cutaneous amyloidosis is localized to the skin without other organ involvement and does not occur in systemic amyloidosis. Secondary cutaneous involvement in systemic amyloidosis is rare. Most cases of primary localized cutaneous amyloidosis (PLCA) are sporadic but approximately 10% of cases may be familial.1 There are 3 main forms of localized cutaneous amyloidosis: macular, lichen, and nodular amyloidosis. Nodular cutaneous amyloidosis is the rarest form of PLCA.
Nodular amyloidosis was first described by Gottron in 1950.2 Its cutaneous lesions may present as single or multiple nodules, occasionally with overlying atrophic plaques. The lesions consist of firm, smooth-surfaced, waxy or rubbery, pink to tan papules, plaques, or nodules measuring up to several centimeters. On some lesions, surface telangiectasia may be seen. Bullous-appearing and anetodermalike lesions have been reported.3 The acral region is the most common location, followed by the legs, head, trunk, arms, and genitalia, respectively.4 In some cases the lesions can spontaneously improve over time. In our patient, the lesions were composed of both multiple nodules and atrophic plaques, which is uncommon.
The pathogenesis of amyloid deposition is still unknown. Cutaneous macular and lichen amyloidosis may originate from degenerated keratinocyte intermediate filaments. Nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.5 Some components of amyloid in some cases of PLCNA may consist of κ and λ immunoglobulin light chains, with most reported cases being of the l subtype.6 The results of one study indicated that β2-microglobulin was another major component of amyloid fibrils and that β2-microglobulin was partly subjected to the modification of advanced glycation end product in PLCNA.7
The histopathologic examination of PLCNA is characterized by large deposits of amorphous, sometimes fissured, pale, eosinophilic material in the papillary dermis, reticular dermis, and subcutaneous fat. The overlying epidermis may exhibit flattened rete ridges. Amyloid may occur within vessel walls and adnexal structures, sometimes in a ring surrounding individual fat cells. Clinically, the lesions of PLCNA may be indistinguishable from nodular deposits of amyloid occurring in primary systemic amyloidosis or myeloma-associated amyloidosis. Histopathologically, PLCNA usually has a variable infiltrate of plasma cells and lymphocytes at the periphery or within the amyloid deposits,6 but no single stain is highly sensitive and specific. Congo red–stained deposits showed salmon pink amorphous material or apple green birefringence with polarizing microscopy.8 Amyloid derived from immunoglobulin light chains, including cutaneous nodular amyloid, also stain positive for anti-human λ light chain antibody on immunohistochemistry. Additionally, amyloid can stain positively with methyl violet and crystal violet Gram stains, Picrosirius red, thioflavin T, Dylon, and periodic acid–Schiff stains.
Clinically, some PLCNA lesions can be removed via surgical excision or laser if they are cosmetically disfiguring or symptomatic. Other methods have been attempted to improve the appearance of the lesions, such as intralesional corticosteroids, cryotherapy, and dermabrasion,9 but they usually are not helpful and have a high rate of recurrence. Although PLCNA often is a benign cutaneous disorder and some cases of PLCNA could be reactive diseases rather than neoplastic ones, some patients may develop underlying systemic amyloidosis or even paraproteinemia.10 Northcutt and Vanover11 indicated that systemic amyloidosis may be expected in less than 15% of 47 patients with localized cutaneous amyloidosis during follow-up by reviewing most of the related literature. Some cases may be associated with Sjögren syndrome (SJS), CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia) syndrome, dermatomyositis, and diabetes mellitus.12-14 Polyclonal immunoglobulin amyloid has been reported only in PLCNA with SJS, which may be due to the fact that a certain population of SJS develops polyclonal B-cell proliferation and hyperglobulinemia.12 Woollons and Black15 estimated the rate of progression of PLCNA to systemic amyloidosis to be only 7%, which is much lower than the rate in the literature by a large clinical follow-up study on PLCNA.16 However, all patients with PLCNA should have a systemic evaluation and should be advised to undergo long-term clinical follow-up to help prevent progression to systemic amyloidosis or plasma cell dyscrasia.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
1. Sakuma TH, Hans-Filho G, Arita K, et al. Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol. 2009;145:695-699.
2. Rodermund OE. On amyloidosis cutis nodularis atrophicans (Gottron 1950). at the same time a contribution to the classification of amyloidosis [in German]. Arch Klin Exp Dermatol. 1967;230:153-171.
3. Chapel TA, Birmingham DJ, Malinowski YE. Nodular primary localized cutaneous amyloidosis. Arch Dermatol. 1977;113:1248-1249.
4. Criado PR, Silva CS, Vasconcellos C, et al. Extensive nodular cutaneous amyloidosis: an unusual presentation. J Eur Acad Dermatol Venereol. 2005;19:481-483.
5. Touart DM, Sau P. Cutaneous deposition diseases. part I [published correction appears in J Am Acad Dermatol. 1998;39:1042]. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-174.
6. Borrowman TA, Lutz ME, Walsh JS. Cutaneous nodular amyloidosis masquerading as a foot callus. J Am Acad Dermatol. 2003;49:307-310.
7. Fujimoto N, Yajima M, Ohnishi Y, et al. Advanced glycation end product-modified beta2-microglobulin is a component of amyloid fibrils of primary localized cutaneous nodular amyloidosis. J Invest Dermatol. 2002;118:479-484.
8. Clement CG, Truong LD. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol. 2014;45:1766-1772.
9. Lien MH, Railan D, Nelson BR. The efficacy of dermabrasion in the treatment of nodular amyloidosis. J Am Acad Dermatol. 1997;36(2, pt 2):315-316.
10. Taylor SC, Baker E, Grossman ME. Nodular vulvar amyloid as a presentation of systemic amyloidosis. J Am Acad Dermatol. 1991;24:139.
11. Northcutt AD, Vanover MJ. Nodular cutaneous amyloidosis involving the vulva. case report and literature review. Arch Dermatol. 1985;121:518-521.
12. Yoneyama K, Tochigi N, Oikawa A, et al. Primary localized cutaneous nodular amyloidosis in a patient with Sjögren’s syndrome: a review of the literature. J Dermatol. 2005;32:120-123.
13. Summers EM, Kendrick CG. Primary localized cutaneous nodular amyloidosis and CREST syndrome: a case report and review of the literature. Cutis. 2008;82:55-59.
14. Taniguchi Y, Horino T, Terada Y. Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol. 2009;36:1088-1089.
15. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145:105-109.
16. Brownstein MH, Helwig EB. The cutaneous amyloidoses. I. localized forms. Arch Dermatol. 1970;102:8-19.
Practice Points
- The cutaneous lesions of primary localized cutaneous nodular amyloidosis (PLCNA) may present as single or multiple nodules, occasionally with overlying atrophic plaques and some with surface telangiectasia.
- Primary localized cutaneous nodular amyloidosis may represent a localized plasma cell dyscrasia that can be associated with a monoclonal gammopathy or multiple myeloma.
- Although PLCNA often is a benign cutaneous disorder, some patients can develop underlying systemic amyloidosis or even paraproteinemia.
Burning pain from chest to back • allodynia and hyperesthesia • extreme sensitivity at the left T5 dermatome • Dx?
THE CASE
A 27-year-old woman in the 21st week of her first pregnancy came to our clinic complaining of a constant burning pain that spread around her left chest wall to her back. She graded the pain as a 10 on a 0 to 10 visual analog scale. The pain, which began 3 months earlier, became worse when she took a deep breath, ate, or walked, but was alleviated by applying warm compresses. Our patient hadn’t slept well since the pain began. Her medical history was noteworthy for chickenpox at age 5.
During the physical examination, palpating her left upper abdominal quadrant and left lower chest wall elicited tenderness. We noted allodynia and hyperesthesia in these regions, and the left T5 dermatome revealed extreme sensitivity.
THE DIAGNOSIS
We decided to test for antibodies to the varicella-zoster virus (VZV) based on the location of the pain along a dermatome. A serum anti-VZV immunoglobulin G (IgG) level was high at 1.9. Since our patient hadn’t been vaccinated against VZV, her high IgG level may have been the result of reactivation of the virus. Based on this test result and our patient’s history and physical exam findings (ie, neuropathic pain along a dermatome without a typical herpes zoster rash), we diagnosed zoster sine herpete (ZSH).
DISCUSSION
One million new cases of herpes zoster (shingles) are diagnosed in the United States each year, with a rate of 3 to 4 cases per 1000 people.1 One in 3 patients develops postherpetic neuralgia, depending on age and immunocompetence.1
In ZSH, the neuropathic pain of herpes zoster occurs without the typical zoster rash.2 Since the rash is absent, the diagnosis is often missed. The incidence of ZSH is unknown.
Although many pregnant women suffer from thoracic and/or abdominal neuropathic pain, there are no reports in the literature that describe ZSH in pregnant women.3
The appropriate diagnostic tests for ZSH are polymerase chain reaction for VZV DNA and anti-VZV IgG.2,4-7 A definitive diagnosis can be reached by identifying herpes zoster DNA in cerebrospinal fluid (CSF) and organism-specific immunoglobulins. However, a high titer of serum IgG antibodies or a positive IgM antibodies test typically provides a high degree of certainty for the diagnosis.8 For our patient, we decided not to test her CSF because we felt that her clinical course and positive IgG test were sufficient to establish the diagnosis.
The differential diagnosis of radicular pain during pregnancy includes cutaneous nerve entrapment. The expanding uterus could increase pressure on cutaneous nerves in the abdominal wall and cause pain. Although nerve entrapment would be expected to cause impingement and sometimes hypoesthesia, ZSH usually causes allodynia and hyperesthesia, as was the case in our patient.3
Pregnancy affects choice of treatment
Treatments for ZSH include acyclovir and local anesthesia.8 A single injection of lidocaine (8 cc) may completely eliminate the ZSH pain by affecting the nerve action potential.9 Corticosteroids are used to suppress inflammation and decrease erythema, swelling, warmth at the site, and local tenderness.
Our patient. We decided to treat our patient with only a nerve block because the potential adverse effects of acyclovir in the second trimester of pregnancy are unclear.10 She received 1 cc of betamethasone acetate (3 mg) and betamethasone sodium phosphate (3 mg) and 8 cc of 2% lidocaine. The patient reported immediate pain relief, which lasted until delivery.
THE TAKEAWAY
ZSH is characterized by neuropathic pain along a dermatome that’s associated with herpes zoster and is not accompanied by the characteristic rash. Many pregnant women suffer from thoracic and abdominal wall neuropathic pain. Neuropathic radicular pain in the absence of a rash should raise suspicion of ZSH. Considering this syndrome at an early stage can avert unnecessary testing and reduce the patient’s pain.
1. Cohen JI. Clinical practice: Herpes zoster. N Engl J Med. 2013;369:255-263.
2. Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med. 2007;74:489-504.
3. Peleg R, Gohar J, Koretz M, et al. Abdominal wall pain in pregnant women caused by thoracic lateral cutaneous nerve entrapment. Eur J Obstet Gynecol Reprod Biol. 1997;74:169-171.
4. Gilden DH, Wright RR, Schneck SA, et al. Zoster sine herpete, a clinical variant. Ann Neurol. 1994;35:530-533.
5. Amlie-Lefond C, Mackin GA, Ferguson M, et al. Another case of virologically confirmed zoster sine herpete, with electrophysiologic correlation. J Neurovirol. 1996;2:136-138.
6. Blumenthal DT, Shacham-Shmueli E, Bokstein F, et al. Zoster sine herpete: virologic verification by detection of anti-VZV IgG antibody in CSF. Neurology. 2011;76:484-485.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;2:418-421.
8. Kennedy PG. Zoster sine herpete: it would be rash to ignore it. Neurology. 2011;76:416-417.
9. Baranowski AP, De Courcey J, Bonello E. A trial of intravenous lidocaine on the pain and allodynia of postherpetic neuralgia. J Pain Symptom Manage. 1999;17:429-433.
10. Stone KM, Reiff-Eldridge R, White AD, et al. Pregnancy outcomes following systemic prenatal acyclovir exposure: Conclusions from the international acyclovir pregnancy registry, 1984-1999. Birth Defects Res A Clin Mol Teratol. 2004;70:201-207.
THE CASE
A 27-year-old woman in the 21st week of her first pregnancy came to our clinic complaining of a constant burning pain that spread around her left chest wall to her back. She graded the pain as a 10 on a 0 to 10 visual analog scale. The pain, which began 3 months earlier, became worse when she took a deep breath, ate, or walked, but was alleviated by applying warm compresses. Our patient hadn’t slept well since the pain began. Her medical history was noteworthy for chickenpox at age 5.
During the physical examination, palpating her left upper abdominal quadrant and left lower chest wall elicited tenderness. We noted allodynia and hyperesthesia in these regions, and the left T5 dermatome revealed extreme sensitivity.
THE DIAGNOSIS
We decided to test for antibodies to the varicella-zoster virus (VZV) based on the location of the pain along a dermatome. A serum anti-VZV immunoglobulin G (IgG) level was high at 1.9. Since our patient hadn’t been vaccinated against VZV, her high IgG level may have been the result of reactivation of the virus. Based on this test result and our patient’s history and physical exam findings (ie, neuropathic pain along a dermatome without a typical herpes zoster rash), we diagnosed zoster sine herpete (ZSH).
DISCUSSION
One million new cases of herpes zoster (shingles) are diagnosed in the United States each year, with a rate of 3 to 4 cases per 1000 people.1 One in 3 patients develops postherpetic neuralgia, depending on age and immunocompetence.1
In ZSH, the neuropathic pain of herpes zoster occurs without the typical zoster rash.2 Since the rash is absent, the diagnosis is often missed. The incidence of ZSH is unknown.
Although many pregnant women suffer from thoracic and/or abdominal neuropathic pain, there are no reports in the literature that describe ZSH in pregnant women.3
The appropriate diagnostic tests for ZSH are polymerase chain reaction for VZV DNA and anti-VZV IgG.2,4-7 A definitive diagnosis can be reached by identifying herpes zoster DNA in cerebrospinal fluid (CSF) and organism-specific immunoglobulins. However, a high titer of serum IgG antibodies or a positive IgM antibodies test typically provides a high degree of certainty for the diagnosis.8 For our patient, we decided not to test her CSF because we felt that her clinical course and positive IgG test were sufficient to establish the diagnosis.
The differential diagnosis of radicular pain during pregnancy includes cutaneous nerve entrapment. The expanding uterus could increase pressure on cutaneous nerves in the abdominal wall and cause pain. Although nerve entrapment would be expected to cause impingement and sometimes hypoesthesia, ZSH usually causes allodynia and hyperesthesia, as was the case in our patient.3
Pregnancy affects choice of treatment
Treatments for ZSH include acyclovir and local anesthesia.8 A single injection of lidocaine (8 cc) may completely eliminate the ZSH pain by affecting the nerve action potential.9 Corticosteroids are used to suppress inflammation and decrease erythema, swelling, warmth at the site, and local tenderness.
Our patient. We decided to treat our patient with only a nerve block because the potential adverse effects of acyclovir in the second trimester of pregnancy are unclear.10 She received 1 cc of betamethasone acetate (3 mg) and betamethasone sodium phosphate (3 mg) and 8 cc of 2% lidocaine. The patient reported immediate pain relief, which lasted until delivery.
THE TAKEAWAY
ZSH is characterized by neuropathic pain along a dermatome that’s associated with herpes zoster and is not accompanied by the characteristic rash. Many pregnant women suffer from thoracic and abdominal wall neuropathic pain. Neuropathic radicular pain in the absence of a rash should raise suspicion of ZSH. Considering this syndrome at an early stage can avert unnecessary testing and reduce the patient’s pain.
THE CASE
A 27-year-old woman in the 21st week of her first pregnancy came to our clinic complaining of a constant burning pain that spread around her left chest wall to her back. She graded the pain as a 10 on a 0 to 10 visual analog scale. The pain, which began 3 months earlier, became worse when she took a deep breath, ate, or walked, but was alleviated by applying warm compresses. Our patient hadn’t slept well since the pain began. Her medical history was noteworthy for chickenpox at age 5.
During the physical examination, palpating her left upper abdominal quadrant and left lower chest wall elicited tenderness. We noted allodynia and hyperesthesia in these regions, and the left T5 dermatome revealed extreme sensitivity.
THE DIAGNOSIS
We decided to test for antibodies to the varicella-zoster virus (VZV) based on the location of the pain along a dermatome. A serum anti-VZV immunoglobulin G (IgG) level was high at 1.9. Since our patient hadn’t been vaccinated against VZV, her high IgG level may have been the result of reactivation of the virus. Based on this test result and our patient’s history and physical exam findings (ie, neuropathic pain along a dermatome without a typical herpes zoster rash), we diagnosed zoster sine herpete (ZSH).
DISCUSSION
One million new cases of herpes zoster (shingles) are diagnosed in the United States each year, with a rate of 3 to 4 cases per 1000 people.1 One in 3 patients develops postherpetic neuralgia, depending on age and immunocompetence.1
In ZSH, the neuropathic pain of herpes zoster occurs without the typical zoster rash.2 Since the rash is absent, the diagnosis is often missed. The incidence of ZSH is unknown.
Although many pregnant women suffer from thoracic and/or abdominal neuropathic pain, there are no reports in the literature that describe ZSH in pregnant women.3
The appropriate diagnostic tests for ZSH are polymerase chain reaction for VZV DNA and anti-VZV IgG.2,4-7 A definitive diagnosis can be reached by identifying herpes zoster DNA in cerebrospinal fluid (CSF) and organism-specific immunoglobulins. However, a high titer of serum IgG antibodies or a positive IgM antibodies test typically provides a high degree of certainty for the diagnosis.8 For our patient, we decided not to test her CSF because we felt that her clinical course and positive IgG test were sufficient to establish the diagnosis.
The differential diagnosis of radicular pain during pregnancy includes cutaneous nerve entrapment. The expanding uterus could increase pressure on cutaneous nerves in the abdominal wall and cause pain. Although nerve entrapment would be expected to cause impingement and sometimes hypoesthesia, ZSH usually causes allodynia and hyperesthesia, as was the case in our patient.3
Pregnancy affects choice of treatment
Treatments for ZSH include acyclovir and local anesthesia.8 A single injection of lidocaine (8 cc) may completely eliminate the ZSH pain by affecting the nerve action potential.9 Corticosteroids are used to suppress inflammation and decrease erythema, swelling, warmth at the site, and local tenderness.
Our patient. We decided to treat our patient with only a nerve block because the potential adverse effects of acyclovir in the second trimester of pregnancy are unclear.10 She received 1 cc of betamethasone acetate (3 mg) and betamethasone sodium phosphate (3 mg) and 8 cc of 2% lidocaine. The patient reported immediate pain relief, which lasted until delivery.
THE TAKEAWAY
ZSH is characterized by neuropathic pain along a dermatome that’s associated with herpes zoster and is not accompanied by the characteristic rash. Many pregnant women suffer from thoracic and abdominal wall neuropathic pain. Neuropathic radicular pain in the absence of a rash should raise suspicion of ZSH. Considering this syndrome at an early stage can avert unnecessary testing and reduce the patient’s pain.
1. Cohen JI. Clinical practice: Herpes zoster. N Engl J Med. 2013;369:255-263.
2. Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med. 2007;74:489-504.
3. Peleg R, Gohar J, Koretz M, et al. Abdominal wall pain in pregnant women caused by thoracic lateral cutaneous nerve entrapment. Eur J Obstet Gynecol Reprod Biol. 1997;74:169-171.
4. Gilden DH, Wright RR, Schneck SA, et al. Zoster sine herpete, a clinical variant. Ann Neurol. 1994;35:530-533.
5. Amlie-Lefond C, Mackin GA, Ferguson M, et al. Another case of virologically confirmed zoster sine herpete, with electrophysiologic correlation. J Neurovirol. 1996;2:136-138.
6. Blumenthal DT, Shacham-Shmueli E, Bokstein F, et al. Zoster sine herpete: virologic verification by detection of anti-VZV IgG antibody in CSF. Neurology. 2011;76:484-485.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;2:418-421.
8. Kennedy PG. Zoster sine herpete: it would be rash to ignore it. Neurology. 2011;76:416-417.
9. Baranowski AP, De Courcey J, Bonello E. A trial of intravenous lidocaine on the pain and allodynia of postherpetic neuralgia. J Pain Symptom Manage. 1999;17:429-433.
10. Stone KM, Reiff-Eldridge R, White AD, et al. Pregnancy outcomes following systemic prenatal acyclovir exposure: Conclusions from the international acyclovir pregnancy registry, 1984-1999. Birth Defects Res A Clin Mol Teratol. 2004;70:201-207.
1. Cohen JI. Clinical practice: Herpes zoster. N Engl J Med. 2013;369:255-263.
2. Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med. 2007;74:489-504.
3. Peleg R, Gohar J, Koretz M, et al. Abdominal wall pain in pregnant women caused by thoracic lateral cutaneous nerve entrapment. Eur J Obstet Gynecol Reprod Biol. 1997;74:169-171.
4. Gilden DH, Wright RR, Schneck SA, et al. Zoster sine herpete, a clinical variant. Ann Neurol. 1994;35:530-533.
5. Amlie-Lefond C, Mackin GA, Ferguson M, et al. Another case of virologically confirmed zoster sine herpete, with electrophysiologic correlation. J Neurovirol. 1996;2:136-138.
6. Blumenthal DT, Shacham-Shmueli E, Bokstein F, et al. Zoster sine herpete: virologic verification by detection of anti-VZV IgG antibody in CSF. Neurology. 2011;76:484-485.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;2:418-421.
8. Kennedy PG. Zoster sine herpete: it would be rash to ignore it. Neurology. 2011;76:416-417.
9. Baranowski AP, De Courcey J, Bonello E. A trial of intravenous lidocaine on the pain and allodynia of postherpetic neuralgia. J Pain Symptom Manage. 1999;17:429-433.
10. Stone KM, Reiff-Eldridge R, White AD, et al. Pregnancy outcomes following systemic prenatal acyclovir exposure: Conclusions from the international acyclovir pregnancy registry, 1984-1999. Birth Defects Res A Clin Mol Teratol. 2004;70:201-207.
Bifrontal headache • blurred vision • vomiting • Dx?
THE CASE
A 55-year-old woman presented to the emergency department (ED) with a bifrontal headache that she’d had for one day. She also had blurred vision and was vomiting shortly before coming to the hospital. The patient had no history of hypertension, migraine headaches, seizure disorder, autoimmune disorders, or cerebrovascular disease.
Her vital signs, including a blood pressure of 114/63 mm Hg, were normal, but a physical examination revealed subjective vision loss. She was only able to see objects moving on a horizontal plane. Her finger-to-nose exam, pupillary reflexes, and extra-ocular movements were normal, but peripheral vision was limited on her left side. No other neurologic deficits were noted.
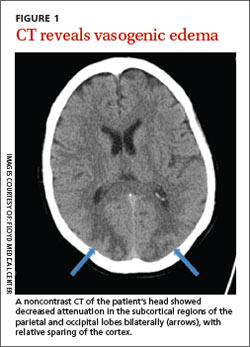
The patient was admitted to the hospital and most of her laboratory work-up was normal, including a basic metabolic panel, complete blood count, coagulation studies, brain natriuretic peptide test, and cardiac enzymes. Her white blood cell count was 19,700/mcL, but no source of infection was found. A computed tomography (CT) scan of her head without contrast showed low-density, patchy areas in the subcortical regions of the parietal and occipital lobes bilaterally (FIGURE 1, arrows), with relative sparing of the cortex.
THE DIAGNOSIS
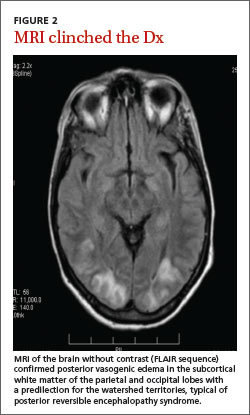
Based on our patient’s presentation and radiologic findings, we made a diagnosis of posterior reversible encephalopathy syndrome (PRES). However, because we could not rule out an ischemic cerebrovascular event at the time of presentation, we started the patient on aspirin and clopidogrel 75 mg to prevent possible future ischemic events. The next day, we ordered magnetic resonance imaging (MRI) of the head and neck, which documented the edema and confirmed the diagnosis of PRES (FIGURE 2).
DISCUSSION
PRES is a neurotoxic state associated with a unique pattern of brain vasogenic edema seen on CT or MRI. The edema is often widespread but is predominantly found in the parietal and occipital regions.1 PRES is seen in patients with a variety of conditions, including hypertension and bone marrow or organ transplantation, as well as in those receiving immunosuppressive or cytotoxic medications.1 Patients with PRES typically present with headaches and seizures.2 Visual abnormalities (most commonly cortical blindness), occur in 15% to 20% of patients with PRES.2-4
Hinchey et al3 first described reversible posterior leukoencephalopathy syndrome (which later became known as PRES) in 1996. Most of the 15 patients included in this original report had a history of hypertension or immunosuppression. These cases were associated with cerebral edema in portions of the posterior cerebral white matter. It is thought that hypertension alters the blood-brain barrier and causes the acute changes that occur in PRES.3
Besides hypertension and immunosuppression, the risk factors most commonly associated with PRES include preeclampsia/eclampsia; sepsis, particularly due to grampositive organisms; Wegener’s granulomatosis, scleroderma, and polyarteritis nodosa; cancer chemotherapy; bone marrow or stem cell transplantation; and renal disease.1,4-6
Although a clear cause of PRES has not yet been established, researchers have proposed 2 theories. The first postulates that a sudden increase in systemic blood pressure causes vasoconstriction, which leads to ischemia and edema.1-4,7,8 However, several studies have also described cases of PRES in patients with mild elevations in blood pressure,1,5-7 and mild edema has been observed even in normotensive patients1,5 (as was the case with our patient).
The second theory links PRES to the loss of brain autoregulation, a function that maintains steady blood flow when blood pressure fluctuates.6 A loss of this regulatory mechanism causes endothelial dysfunction, capillary leakage, and disruption in the blood-brain barrier.1,2,4,6-8 These changes then lead to cerebral vasodilatation and edema.2 Immunotherapy has also been associated with increased endothelial dysfunction.2
The evidence on the link between the severity of PRES and clinical outcomes is conflicting.
One study that followed 113 PRES patients over 6 years did not find an association between the severity of clinical presentation and the extent of vasogenic edema found on imaging studies.5 Of these 113 patients, 69 had PRES primarily due to hypertension, and 21 were receiving cytotoxic medications.5 In contrast, a larger retrospective study that followed patients with PRES for 12 years found that severe cases, which included patients with severe cerebral edema and altered mental status, had poor outcomes.4 Small studies have reported that 14% of patients with PRES develop cerebral hemorrhage.8
When to suspect this condition. PRES should be part of the differential diagnosis for any patient who presents with headache and vision loss. It is important to distinguish PRES from an acute cerebrovascular accident (CVA) because the 2 conditions are managed differently.2 In addition, PRES lesions can be misdiagnosed as tumors, especially in a patient with a history of malignant disease in whom the condition appears after chemotherapy.9
Treatment targets the underlying causes
Treatment options for PRES are limited. Hypertension in a patient with PRES requires prompt intervention to avoid progression of the disease.2 The use of intravenous (IV) calcium-channel blockers or IV beta-blockers for these patients is common.2,8
Patients with seizures should be treated with anticonvulsant medication, but longterm antiepileptic treatment usually is not required.2 Patients who take immunosuppressant or cytotoxic drugs should stop them indefinitely upon presenting with PRES.2
For a pregnant woman with preeclampsia/eclampsia, delivery of the placenta, which is considered to be the cause of PRES in these cases, is curative.1 However, women can develop PRES several weeks after delivery.1
In most cases, the symptoms associated with PRES will resolve once treatment is initiated, and neurologic recovery can be expected within 2 weeks.2
Our patient regained her sight the following morning and was discharged home 2 days after admission. Her blood pressure remained normal. She returned to the hospital unresponsive the day after she had been discharged. Family members stated that she had taken 15 packets of an aspirin/caffeine combination to control a new headache.
Her blood pressure was elevated at 159/79 mm Hg. A CT of the brain showed a hemorrhagic stroke within the left occipital lobe and posterior parietal lobe with a midline shift of 8 mm. We don’t know if the aspirin use contributed to the hemorrhagic event or if it was a sequela of PRES.
The patient died 4 days later.
THE TAKEAWAY
PRES is a neurotoxic condition that causes headache, seizures, and vision loss. Most patients will present with elevated blood pressure and imaging studies will reveal a specific pattern of vasogenic edema that is predominately found in the parietal and occipital regions.
Treating the hypertension may result in a more favorable recovery. Normotensive patients are harder to treat because there is no specific therapy for PRES. Follow-up imaging may help to assess the resolution of the syndrome.
1. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29:1036-1042.
2. Stott VL, Hurrell MA, Anderson TJ. Reversible posterior leukoencephalopathy syndrome: a misnomer reviewed. Intern Med J. 2005;35:83-90.
3. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
4. Liman TG, Bohner G, Endres M, et al. Discharge status and in-hospital mortality in posterior reversible encephalopathy syndrome. Acta Neurol Scand. 2014;130:34-39.
5. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85:427-432.
6. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29:1043-1049.
7. Ay H, Buonanno FS, Schaefer PW, et al. Posterior leukoencephalopathy without severe hypertension: utility of diffusion-weighted MRI. Neurology. 1998;51:1369-1376.
8. Legriel S, Schraub O, Azoulay E, et al; Critically III Posterior Reversible Encephalopathy Syndrome Study Group (CYPRESS). Determinants of recovery from severe posterior reversible encephalopathy syndrome. PLoS ONE. 2012;7:e44534.
9. Morina D, Ntoulias G, Maslehaty H, et al. Posterior reversible encephalopathy syndrome mimicking cerebral metastasis: contraindication for biopsy. Clin Pract. 2014;4:632.
THE CASE
A 55-year-old woman presented to the emergency department (ED) with a bifrontal headache that she’d had for one day. She also had blurred vision and was vomiting shortly before coming to the hospital. The patient had no history of hypertension, migraine headaches, seizure disorder, autoimmune disorders, or cerebrovascular disease.
Her vital signs, including a blood pressure of 114/63 mm Hg, were normal, but a physical examination revealed subjective vision loss. She was only able to see objects moving on a horizontal plane. Her finger-to-nose exam, pupillary reflexes, and extra-ocular movements were normal, but peripheral vision was limited on her left side. No other neurologic deficits were noted.
The patient was admitted to the hospital and most of her laboratory work-up was normal, including a basic metabolic panel, complete blood count, coagulation studies, brain natriuretic peptide test, and cardiac enzymes. Her white blood cell count was 19,700/mcL, but no source of infection was found. A computed tomography (CT) scan of her head without contrast showed low-density, patchy areas in the subcortical regions of the parietal and occipital lobes bilaterally (FIGURE 1, arrows), with relative sparing of the cortex.
THE DIAGNOSIS
Based on our patient’s presentation and radiologic findings, we made a diagnosis of posterior reversible encephalopathy syndrome (PRES). However, because we could not rule out an ischemic cerebrovascular event at the time of presentation, we started the patient on aspirin and clopidogrel 75 mg to prevent possible future ischemic events. The next day, we ordered magnetic resonance imaging (MRI) of the head and neck, which documented the edema and confirmed the diagnosis of PRES (FIGURE 2).
DISCUSSION
PRES is a neurotoxic state associated with a unique pattern of brain vasogenic edema seen on CT or MRI. The edema is often widespread but is predominantly found in the parietal and occipital regions.1 PRES is seen in patients with a variety of conditions, including hypertension and bone marrow or organ transplantation, as well as in those receiving immunosuppressive or cytotoxic medications.1 Patients with PRES typically present with headaches and seizures.2 Visual abnormalities (most commonly cortical blindness), occur in 15% to 20% of patients with PRES.2-4
Hinchey et al3 first described reversible posterior leukoencephalopathy syndrome (which later became known as PRES) in 1996. Most of the 15 patients included in this original report had a history of hypertension or immunosuppression. These cases were associated with cerebral edema in portions of the posterior cerebral white matter. It is thought that hypertension alters the blood-brain barrier and causes the acute changes that occur in PRES.3
Besides hypertension and immunosuppression, the risk factors most commonly associated with PRES include preeclampsia/eclampsia; sepsis, particularly due to grampositive organisms; Wegener’s granulomatosis, scleroderma, and polyarteritis nodosa; cancer chemotherapy; bone marrow or stem cell transplantation; and renal disease.1,4-6
Although a clear cause of PRES has not yet been established, researchers have proposed 2 theories. The first postulates that a sudden increase in systemic blood pressure causes vasoconstriction, which leads to ischemia and edema.1-4,7,8 However, several studies have also described cases of PRES in patients with mild elevations in blood pressure,1,5-7 and mild edema has been observed even in normotensive patients1,5 (as was the case with our patient).
The second theory links PRES to the loss of brain autoregulation, a function that maintains steady blood flow when blood pressure fluctuates.6 A loss of this regulatory mechanism causes endothelial dysfunction, capillary leakage, and disruption in the blood-brain barrier.1,2,4,6-8 These changes then lead to cerebral vasodilatation and edema.2 Immunotherapy has also been associated with increased endothelial dysfunction.2
The evidence on the link between the severity of PRES and clinical outcomes is conflicting.
One study that followed 113 PRES patients over 6 years did not find an association between the severity of clinical presentation and the extent of vasogenic edema found on imaging studies.5 Of these 113 patients, 69 had PRES primarily due to hypertension, and 21 were receiving cytotoxic medications.5 In contrast, a larger retrospective study that followed patients with PRES for 12 years found that severe cases, which included patients with severe cerebral edema and altered mental status, had poor outcomes.4 Small studies have reported that 14% of patients with PRES develop cerebral hemorrhage.8
When to suspect this condition. PRES should be part of the differential diagnosis for any patient who presents with headache and vision loss. It is important to distinguish PRES from an acute cerebrovascular accident (CVA) because the 2 conditions are managed differently.2 In addition, PRES lesions can be misdiagnosed as tumors, especially in a patient with a history of malignant disease in whom the condition appears after chemotherapy.9
Treatment targets the underlying causes
Treatment options for PRES are limited. Hypertension in a patient with PRES requires prompt intervention to avoid progression of the disease.2 The use of intravenous (IV) calcium-channel blockers or IV beta-blockers for these patients is common.2,8
Patients with seizures should be treated with anticonvulsant medication, but longterm antiepileptic treatment usually is not required.2 Patients who take immunosuppressant or cytotoxic drugs should stop them indefinitely upon presenting with PRES.2
For a pregnant woman with preeclampsia/eclampsia, delivery of the placenta, which is considered to be the cause of PRES in these cases, is curative.1 However, women can develop PRES several weeks after delivery.1
In most cases, the symptoms associated with PRES will resolve once treatment is initiated, and neurologic recovery can be expected within 2 weeks.2
Our patient regained her sight the following morning and was discharged home 2 days after admission. Her blood pressure remained normal. She returned to the hospital unresponsive the day after she had been discharged. Family members stated that she had taken 15 packets of an aspirin/caffeine combination to control a new headache.
Her blood pressure was elevated at 159/79 mm Hg. A CT of the brain showed a hemorrhagic stroke within the left occipital lobe and posterior parietal lobe with a midline shift of 8 mm. We don’t know if the aspirin use contributed to the hemorrhagic event or if it was a sequela of PRES.
The patient died 4 days later.
THE TAKEAWAY
PRES is a neurotoxic condition that causes headache, seizures, and vision loss. Most patients will present with elevated blood pressure and imaging studies will reveal a specific pattern of vasogenic edema that is predominately found in the parietal and occipital regions.
Treating the hypertension may result in a more favorable recovery. Normotensive patients are harder to treat because there is no specific therapy for PRES. Follow-up imaging may help to assess the resolution of the syndrome.
THE CASE
A 55-year-old woman presented to the emergency department (ED) with a bifrontal headache that she’d had for one day. She also had blurred vision and was vomiting shortly before coming to the hospital. The patient had no history of hypertension, migraine headaches, seizure disorder, autoimmune disorders, or cerebrovascular disease.
Her vital signs, including a blood pressure of 114/63 mm Hg, were normal, but a physical examination revealed subjective vision loss. She was only able to see objects moving on a horizontal plane. Her finger-to-nose exam, pupillary reflexes, and extra-ocular movements were normal, but peripheral vision was limited on her left side. No other neurologic deficits were noted.
The patient was admitted to the hospital and most of her laboratory work-up was normal, including a basic metabolic panel, complete blood count, coagulation studies, brain natriuretic peptide test, and cardiac enzymes. Her white blood cell count was 19,700/mcL, but no source of infection was found. A computed tomography (CT) scan of her head without contrast showed low-density, patchy areas in the subcortical regions of the parietal and occipital lobes bilaterally (FIGURE 1, arrows), with relative sparing of the cortex.
THE DIAGNOSIS
Based on our patient’s presentation and radiologic findings, we made a diagnosis of posterior reversible encephalopathy syndrome (PRES). However, because we could not rule out an ischemic cerebrovascular event at the time of presentation, we started the patient on aspirin and clopidogrel 75 mg to prevent possible future ischemic events. The next day, we ordered magnetic resonance imaging (MRI) of the head and neck, which documented the edema and confirmed the diagnosis of PRES (FIGURE 2).
DISCUSSION
PRES is a neurotoxic state associated with a unique pattern of brain vasogenic edema seen on CT or MRI. The edema is often widespread but is predominantly found in the parietal and occipital regions.1 PRES is seen in patients with a variety of conditions, including hypertension and bone marrow or organ transplantation, as well as in those receiving immunosuppressive or cytotoxic medications.1 Patients with PRES typically present with headaches and seizures.2 Visual abnormalities (most commonly cortical blindness), occur in 15% to 20% of patients with PRES.2-4
Hinchey et al3 first described reversible posterior leukoencephalopathy syndrome (which later became known as PRES) in 1996. Most of the 15 patients included in this original report had a history of hypertension or immunosuppression. These cases were associated with cerebral edema in portions of the posterior cerebral white matter. It is thought that hypertension alters the blood-brain barrier and causes the acute changes that occur in PRES.3
Besides hypertension and immunosuppression, the risk factors most commonly associated with PRES include preeclampsia/eclampsia; sepsis, particularly due to grampositive organisms; Wegener’s granulomatosis, scleroderma, and polyarteritis nodosa; cancer chemotherapy; bone marrow or stem cell transplantation; and renal disease.1,4-6
Although a clear cause of PRES has not yet been established, researchers have proposed 2 theories. The first postulates that a sudden increase in systemic blood pressure causes vasoconstriction, which leads to ischemia and edema.1-4,7,8 However, several studies have also described cases of PRES in patients with mild elevations in blood pressure,1,5-7 and mild edema has been observed even in normotensive patients1,5 (as was the case with our patient).
The second theory links PRES to the loss of brain autoregulation, a function that maintains steady blood flow when blood pressure fluctuates.6 A loss of this regulatory mechanism causes endothelial dysfunction, capillary leakage, and disruption in the blood-brain barrier.1,2,4,6-8 These changes then lead to cerebral vasodilatation and edema.2 Immunotherapy has also been associated with increased endothelial dysfunction.2
The evidence on the link between the severity of PRES and clinical outcomes is conflicting.
One study that followed 113 PRES patients over 6 years did not find an association between the severity of clinical presentation and the extent of vasogenic edema found on imaging studies.5 Of these 113 patients, 69 had PRES primarily due to hypertension, and 21 were receiving cytotoxic medications.5 In contrast, a larger retrospective study that followed patients with PRES for 12 years found that severe cases, which included patients with severe cerebral edema and altered mental status, had poor outcomes.4 Small studies have reported that 14% of patients with PRES develop cerebral hemorrhage.8
When to suspect this condition. PRES should be part of the differential diagnosis for any patient who presents with headache and vision loss. It is important to distinguish PRES from an acute cerebrovascular accident (CVA) because the 2 conditions are managed differently.2 In addition, PRES lesions can be misdiagnosed as tumors, especially in a patient with a history of malignant disease in whom the condition appears after chemotherapy.9
Treatment targets the underlying causes
Treatment options for PRES are limited. Hypertension in a patient with PRES requires prompt intervention to avoid progression of the disease.2 The use of intravenous (IV) calcium-channel blockers or IV beta-blockers for these patients is common.2,8
Patients with seizures should be treated with anticonvulsant medication, but longterm antiepileptic treatment usually is not required.2 Patients who take immunosuppressant or cytotoxic drugs should stop them indefinitely upon presenting with PRES.2
For a pregnant woman with preeclampsia/eclampsia, delivery of the placenta, which is considered to be the cause of PRES in these cases, is curative.1 However, women can develop PRES several weeks after delivery.1
In most cases, the symptoms associated with PRES will resolve once treatment is initiated, and neurologic recovery can be expected within 2 weeks.2
Our patient regained her sight the following morning and was discharged home 2 days after admission. Her blood pressure remained normal. She returned to the hospital unresponsive the day after she had been discharged. Family members stated that she had taken 15 packets of an aspirin/caffeine combination to control a new headache.
Her blood pressure was elevated at 159/79 mm Hg. A CT of the brain showed a hemorrhagic stroke within the left occipital lobe and posterior parietal lobe with a midline shift of 8 mm. We don’t know if the aspirin use contributed to the hemorrhagic event or if it was a sequela of PRES.
The patient died 4 days later.
THE TAKEAWAY
PRES is a neurotoxic condition that causes headache, seizures, and vision loss. Most patients will present with elevated blood pressure and imaging studies will reveal a specific pattern of vasogenic edema that is predominately found in the parietal and occipital regions.
Treating the hypertension may result in a more favorable recovery. Normotensive patients are harder to treat because there is no specific therapy for PRES. Follow-up imaging may help to assess the resolution of the syndrome.
1. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29:1036-1042.
2. Stott VL, Hurrell MA, Anderson TJ. Reversible posterior leukoencephalopathy syndrome: a misnomer reviewed. Intern Med J. 2005;35:83-90.
3. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
4. Liman TG, Bohner G, Endres M, et al. Discharge status and in-hospital mortality in posterior reversible encephalopathy syndrome. Acta Neurol Scand. 2014;130:34-39.
5. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85:427-432.
6. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29:1043-1049.
7. Ay H, Buonanno FS, Schaefer PW, et al. Posterior leukoencephalopathy without severe hypertension: utility of diffusion-weighted MRI. Neurology. 1998;51:1369-1376.
8. Legriel S, Schraub O, Azoulay E, et al; Critically III Posterior Reversible Encephalopathy Syndrome Study Group (CYPRESS). Determinants of recovery from severe posterior reversible encephalopathy syndrome. PLoS ONE. 2012;7:e44534.
9. Morina D, Ntoulias G, Maslehaty H, et al. Posterior reversible encephalopathy syndrome mimicking cerebral metastasis: contraindication for biopsy. Clin Pract. 2014;4:632.
1. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29:1036-1042.
2. Stott VL, Hurrell MA, Anderson TJ. Reversible posterior leukoencephalopathy syndrome: a misnomer reviewed. Intern Med J. 2005;35:83-90.
3. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
4. Liman TG, Bohner G, Endres M, et al. Discharge status and in-hospital mortality in posterior reversible encephalopathy syndrome. Acta Neurol Scand. 2014;130:34-39.
5. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85:427-432.
6. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29:1043-1049.
7. Ay H, Buonanno FS, Schaefer PW, et al. Posterior leukoencephalopathy without severe hypertension: utility of diffusion-weighted MRI. Neurology. 1998;51:1369-1376.
8. Legriel S, Schraub O, Azoulay E, et al; Critically III Posterior Reversible Encephalopathy Syndrome Study Group (CYPRESS). Determinants of recovery from severe posterior reversible encephalopathy syndrome. PLoS ONE. 2012;7:e44534.
9. Morina D, Ntoulias G, Maslehaty H, et al. Posterior reversible encephalopathy syndrome mimicking cerebral metastasis: contraindication for biopsy. Clin Pract. 2014;4:632.

Case Report: High-Pressure Injection Hand Injury
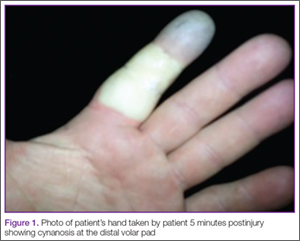
A 42-year-old healthy man presented to the ED 3 hours after sustaining an injury to the tip of his left index finger from a pressurized washer. He stated that while at work, he had touched the jet nozzle of the washer to “test” its pressure, and had experienced immediate pain in his entire finger as well as blanching in his mid-finger. He took a picture of his finger with his cell phone approximately 5 minutes postinjury, which showed cynanosis at the distal volar pad (Figure 1).
On presentation to the ED, physical examination revealed only an innocuous 1-mm puncture wound to the middle of the volar pad of the distal phalanx of his left index finger, with mild tenderness along the length of the finger but no swelling. The rest of the finger and hand appeared intact with normal color and sensation.
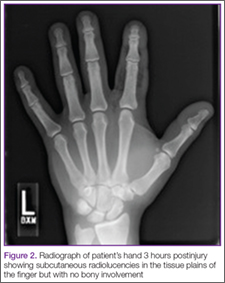
Three weeks after discharge, however, the patient developed pain, diffuse swelling, and purulent drainage from the same injured finger and presented again to the ED. He was immediately taken to the operating room where a broad dissection was performed and drains were placed. Two months later, he reported a complete resolution of the infection and was still working on regaining full functioning of his finger by attending physical therapy.
High-Pressure Injection injuries
Diagnosis
At the time of presentation, most high-pressure injection injuries to the hand appear innocuous but frequently result in severe sequelae, including functional disability and amputation. High-pressure injuries occur when soft tissues are placed in contact or near the opening of a high-pressure device or malfunctioning equipment (eg, pinhole rupture in a hydraulic hose).
While the literature reports that 100 psi or greater is required to break the skin,1 patients presenting to the ED typically report operating machinery shooting between 2,000 and 12,000 psi.2,3 Case-report reviews of patients with high-pressure injection injuries by Schoo et al4 and Hogan and Ruland5 found this type of injury most often occurred in the nondominant hand of male laborers—primarily in the index finger—with 30% to 48% of such injuries resulting in amputation of the digit.
Mechanical and Chemical Injuries
High-pressure injection injuries can be both mechanical as well as chemical. A mechanical high-pressure injury depends upon the magnitude of the injection force, with higher pressures associated with increased dysfunction and incidence of amputation. A chemical-related injury depends upon the duration of exposure and the volume of the material injected.
A high-pressure injury results from a tearing of the soft tissues, with shearing and dissection within and among the tissue plains down the finger and into the palm and proximal tissues.4,6 In this type of injury, there is often direct injury to the neurovascular bundles.
Along with the mechanical aspect of injury, a concurrent chemical injury depends upon the type of material injected (ie, gas or liquid), cytotoxicity, concentration, and inflammatory properties.6 Hogan and Ruland5 described the incidence of amputation associated with injected materials to be greater than 40% with diesel fuel, paint thinner, oil, and paint; 20% to 40% with undercoating, hydraulic fluid, and grease; and 0% with air and water. Oil-based paint injections were associated with a 58% amputation rate comapred to only 6% for latex paints.5
In general, evaluation of high-pressure-related injuries begins with a thorough history to identify the injected material and its attributes, pressure of injection, mechanism of exposure, time elapsed since the event, and the patient’s tetanus immunization status. Interventions include radiographs to evaluate for proximal spread of injected material; tetanus immunization, if required; prophylactic antibiotic therapy7; analgesia; and often emergency surgical consultation. The use of corticosteroid therapy has not been shown to impact the amputation rate or the incidence of infection.5
Type of Injury and Treatment
Wong et al8 developed the following guidelines categorizing the type and recommended treatment of mild, moderate, and severe high-pressure injection injuries:
Mild Injuries. In mild high-pressure injection injuries, patients tend to seek immediate treatment. These types of injuries typically involve oil or water, with a relatively low-pressure exposure, and preserved circulation in the injured area.8 Treatment includes conservative management with an option for surgery.5,8
Moderate Injuries. In moderate injection injuries, there is more significant tissue damage from any of the aforementioned materials, but with preserved neurovascular structures. Treatment often includes debridement in addition to antibiotic therapy.
Severe Injuries. These types of injection injuries often involve paints and solvents, high-pressure exposure, and usually present with an abnormal neurovasculature. The best chance for salvage injuries involves rapid surgical intervention for debridement and reconstruction.9,10
Surgical Evaluation and Debridement
Despite optimal care, amputation and dysfunction rates with high-risk injection injuries (due to higher psi, organic solvents, and/or delayed definitive care) have been reported from 48% to 80%.10-13 Immediate surgical evaluation is often recommended when injection injuries are encountered.10,13 Patients who receive immediate surgical debridement (ie, within 6 hours from injury), specifically those injected with organic solvents, have a lower amputation rate than those who do not have immediate surgical intervention.13
In some reports, more than 80% of those injected with organic solvents who did not have immediate washout and debridement required amputation.4 Other effects of postponed debridement include delayed return to work time and decreased functionality of the hand.13,14
Conclusion
Although high-pressure injection injuries often appear mild at initial presentation since the immediate symptoms of the neurovascular injury often resolves, patients often experience severe sequelae including infection, disability, or amputation. The severity of the injury not only depends upon the psi, but also the type and amount of chemical injected. Because of the high morbidity, it is imperative that emergency physicians are aware of and identify these types of injuries and their sequelae.10,11,13,14
Dr Wilson is a third year resident in the department of emergency medicine at the Alpert Medical School, Brown University, Providence, Rhode Island. Dr Hack is the director for the division of medical toxicology at Brown University and the director of the educational program in medical toxicology. He is an associate professor at Warren Alpert Medical School; and an attending physician in the department of emergency medicine at Brown University, Rhode Island Hospital, Miriam Hospital, Providence.
Disclosure: The authors report no conflicts of interest.
- Scott AR. Occupational high-pressure injection injuries: pathogenesis and prevention. J Soc Occup Med. 1983;33(2):56-59.
- Pappou IP, Deal DN. High-pressure injection injuries. J Hand Surg Am. 2012;37(11):2404-2407.
- Soyuncu S, Bektas F, Dinc S. High-pressure air injection injury to the upper extremity. J Emerg Med. 2013;45(1):96-98.
- Schoo MJ, Scott FA, Boswick JA Jr. High-pressure injection injuries of the hand. J Trauma. 1980;20(3):229-238.
- Hogan CJ, Ruland RT. High-pressure injection injuries to the upper extremity: a review of the literature. J Orthop Trauma. 2006;20(7):503-511.
- Rosenwasser MP, Wei DH. High-pressure injection injuries to the hand. J Am Acad Orthop Surg. 2014;22(1):38-45.
- Mirzayan R, Schnall SB, Chon JH, Holtom PD, Pazakis MJ, Stevanovic MV. Culture results and amputation rates in high-pressure paint gun injuries of the hand. Orthopedics. 2001;24(6):587-589.
- Wong TC, Ip FK, Wu WC. High-pressure injection injuries of the hand in a Chinese population. J Hand Surg Br. 2005;30(6):588-592.
- Pinto MR, Turkula-Pinto LD, Cooney WP, Wood MB, Dobyns JH: High-pressure injection injuries of the hand: review of 25 patients managed by open wound technique. J Hand Surg Am. 1993;18(1):125-130.
- Kaufman, HD. High pressure injection injuries, the problems, pathogenesis and management. Hand. 1970;2(1);63-73.
- Vasilevski D, Noorbergen M, Depierreux M, Lafontaine M. High-pressure injection injuries to the hand. Am J Emerg Med. 2000;18(7):820-824.
- Bekler H, Gokce A, Beyzadeoglu T, Parmaksizoglu F. The surgical treatment and outcomes of high pressure injection injuries of the hand. J Hand Surg Eur Vol. 2007;32(4):394-399.
- Amsdell SL, Hammert WC. High-pressure injection injuries in the hand: current treatment concepts. Plast Reconstr Surg. 2013;132(4):586e-591e.
- Hart RG, Smith GD, Haq A. Prevention of high-pressure injection injuries to the hand. Am J Emerg Med. 2005;24(1):73-76.
A 42-year-old healthy man presented to the ED 3 hours after sustaining an injury to the tip of his left index finger from a pressurized washer. He stated that while at work, he had touched the jet nozzle of the washer to “test” its pressure, and had experienced immediate pain in his entire finger as well as blanching in his mid-finger. He took a picture of his finger with his cell phone approximately 5 minutes postinjury, which showed cynanosis at the distal volar pad (Figure 1).
On presentation to the ED, physical examination revealed only an innocuous 1-mm puncture wound to the middle of the volar pad of the distal phalanx of his left index finger, with mild tenderness along the length of the finger but no swelling. The rest of the finger and hand appeared intact with normal color and sensation.
Three weeks after discharge, however, the patient developed pain, diffuse swelling, and purulent drainage from the same injured finger and presented again to the ED. He was immediately taken to the operating room where a broad dissection was performed and drains were placed. Two months later, he reported a complete resolution of the infection and was still working on regaining full functioning of his finger by attending physical therapy.
High-Pressure Injection injuries
Diagnosis
At the time of presentation, most high-pressure injection injuries to the hand appear innocuous but frequently result in severe sequelae, including functional disability and amputation. High-pressure injuries occur when soft tissues are placed in contact or near the opening of a high-pressure device or malfunctioning equipment (eg, pinhole rupture in a hydraulic hose).
While the literature reports that 100 psi or greater is required to break the skin,1 patients presenting to the ED typically report operating machinery shooting between 2,000 and 12,000 psi.2,3 Case-report reviews of patients with high-pressure injection injuries by Schoo et al4 and Hogan and Ruland5 found this type of injury most often occurred in the nondominant hand of male laborers—primarily in the index finger—with 30% to 48% of such injuries resulting in amputation of the digit.
Mechanical and Chemical Injuries
High-pressure injection injuries can be both mechanical as well as chemical. A mechanical high-pressure injury depends upon the magnitude of the injection force, with higher pressures associated with increased dysfunction and incidence of amputation. A chemical-related injury depends upon the duration of exposure and the volume of the material injected.
A high-pressure injury results from a tearing of the soft tissues, with shearing and dissection within and among the tissue plains down the finger and into the palm and proximal tissues.4,6 In this type of injury, there is often direct injury to the neurovascular bundles.
Along with the mechanical aspect of injury, a concurrent chemical injury depends upon the type of material injected (ie, gas or liquid), cytotoxicity, concentration, and inflammatory properties.6 Hogan and Ruland5 described the incidence of amputation associated with injected materials to be greater than 40% with diesel fuel, paint thinner, oil, and paint; 20% to 40% with undercoating, hydraulic fluid, and grease; and 0% with air and water. Oil-based paint injections were associated with a 58% amputation rate comapred to only 6% for latex paints.5
In general, evaluation of high-pressure-related injuries begins with a thorough history to identify the injected material and its attributes, pressure of injection, mechanism of exposure, time elapsed since the event, and the patient’s tetanus immunization status. Interventions include radiographs to evaluate for proximal spread of injected material; tetanus immunization, if required; prophylactic antibiotic therapy7; analgesia; and often emergency surgical consultation. The use of corticosteroid therapy has not been shown to impact the amputation rate or the incidence of infection.5
Type of Injury and Treatment
Wong et al8 developed the following guidelines categorizing the type and recommended treatment of mild, moderate, and severe high-pressure injection injuries:
Mild Injuries. In mild high-pressure injection injuries, patients tend to seek immediate treatment. These types of injuries typically involve oil or water, with a relatively low-pressure exposure, and preserved circulation in the injured area.8 Treatment includes conservative management with an option for surgery.5,8
Moderate Injuries. In moderate injection injuries, there is more significant tissue damage from any of the aforementioned materials, but with preserved neurovascular structures. Treatment often includes debridement in addition to antibiotic therapy.
Severe Injuries. These types of injection injuries often involve paints and solvents, high-pressure exposure, and usually present with an abnormal neurovasculature. The best chance for salvage injuries involves rapid surgical intervention for debridement and reconstruction.9,10
Surgical Evaluation and Debridement
Despite optimal care, amputation and dysfunction rates with high-risk injection injuries (due to higher psi, organic solvents, and/or delayed definitive care) have been reported from 48% to 80%.10-13 Immediate surgical evaluation is often recommended when injection injuries are encountered.10,13 Patients who receive immediate surgical debridement (ie, within 6 hours from injury), specifically those injected with organic solvents, have a lower amputation rate than those who do not have immediate surgical intervention.13
In some reports, more than 80% of those injected with organic solvents who did not have immediate washout and debridement required amputation.4 Other effects of postponed debridement include delayed return to work time and decreased functionality of the hand.13,14
Conclusion
Although high-pressure injection injuries often appear mild at initial presentation since the immediate symptoms of the neurovascular injury often resolves, patients often experience severe sequelae including infection, disability, or amputation. The severity of the injury not only depends upon the psi, but also the type and amount of chemical injected. Because of the high morbidity, it is imperative that emergency physicians are aware of and identify these types of injuries and their sequelae.10,11,13,14
Dr Wilson is a third year resident in the department of emergency medicine at the Alpert Medical School, Brown University, Providence, Rhode Island. Dr Hack is the director for the division of medical toxicology at Brown University and the director of the educational program in medical toxicology. He is an associate professor at Warren Alpert Medical School; and an attending physician in the department of emergency medicine at Brown University, Rhode Island Hospital, Miriam Hospital, Providence.
Disclosure: The authors report no conflicts of interest.
A 42-year-old healthy man presented to the ED 3 hours after sustaining an injury to the tip of his left index finger from a pressurized washer. He stated that while at work, he had touched the jet nozzle of the washer to “test” its pressure, and had experienced immediate pain in his entire finger as well as blanching in his mid-finger. He took a picture of his finger with his cell phone approximately 5 minutes postinjury, which showed cynanosis at the distal volar pad (Figure 1).
On presentation to the ED, physical examination revealed only an innocuous 1-mm puncture wound to the middle of the volar pad of the distal phalanx of his left index finger, with mild tenderness along the length of the finger but no swelling. The rest of the finger and hand appeared intact with normal color and sensation.
Three weeks after discharge, however, the patient developed pain, diffuse swelling, and purulent drainage from the same injured finger and presented again to the ED. He was immediately taken to the operating room where a broad dissection was performed and drains were placed. Two months later, he reported a complete resolution of the infection and was still working on regaining full functioning of his finger by attending physical therapy.
High-Pressure Injection injuries
Diagnosis
At the time of presentation, most high-pressure injection injuries to the hand appear innocuous but frequently result in severe sequelae, including functional disability and amputation. High-pressure injuries occur when soft tissues are placed in contact or near the opening of a high-pressure device or malfunctioning equipment (eg, pinhole rupture in a hydraulic hose).
While the literature reports that 100 psi or greater is required to break the skin,1 patients presenting to the ED typically report operating machinery shooting between 2,000 and 12,000 psi.2,3 Case-report reviews of patients with high-pressure injection injuries by Schoo et al4 and Hogan and Ruland5 found this type of injury most often occurred in the nondominant hand of male laborers—primarily in the index finger—with 30% to 48% of such injuries resulting in amputation of the digit.
Mechanical and Chemical Injuries
High-pressure injection injuries can be both mechanical as well as chemical. A mechanical high-pressure injury depends upon the magnitude of the injection force, with higher pressures associated with increased dysfunction and incidence of amputation. A chemical-related injury depends upon the duration of exposure and the volume of the material injected.
A high-pressure injury results from a tearing of the soft tissues, with shearing and dissection within and among the tissue plains down the finger and into the palm and proximal tissues.4,6 In this type of injury, there is often direct injury to the neurovascular bundles.
Along with the mechanical aspect of injury, a concurrent chemical injury depends upon the type of material injected (ie, gas or liquid), cytotoxicity, concentration, and inflammatory properties.6 Hogan and Ruland5 described the incidence of amputation associated with injected materials to be greater than 40% with diesel fuel, paint thinner, oil, and paint; 20% to 40% with undercoating, hydraulic fluid, and grease; and 0% with air and water. Oil-based paint injections were associated with a 58% amputation rate comapred to only 6% for latex paints.5
In general, evaluation of high-pressure-related injuries begins with a thorough history to identify the injected material and its attributes, pressure of injection, mechanism of exposure, time elapsed since the event, and the patient’s tetanus immunization status. Interventions include radiographs to evaluate for proximal spread of injected material; tetanus immunization, if required; prophylactic antibiotic therapy7; analgesia; and often emergency surgical consultation. The use of corticosteroid therapy has not been shown to impact the amputation rate or the incidence of infection.5
Type of Injury and Treatment
Wong et al8 developed the following guidelines categorizing the type and recommended treatment of mild, moderate, and severe high-pressure injection injuries:
Mild Injuries. In mild high-pressure injection injuries, patients tend to seek immediate treatment. These types of injuries typically involve oil or water, with a relatively low-pressure exposure, and preserved circulation in the injured area.8 Treatment includes conservative management with an option for surgery.5,8
Moderate Injuries. In moderate injection injuries, there is more significant tissue damage from any of the aforementioned materials, but with preserved neurovascular structures. Treatment often includes debridement in addition to antibiotic therapy.
Severe Injuries. These types of injection injuries often involve paints and solvents, high-pressure exposure, and usually present with an abnormal neurovasculature. The best chance for salvage injuries involves rapid surgical intervention for debridement and reconstruction.9,10
Surgical Evaluation and Debridement
Despite optimal care, amputation and dysfunction rates with high-risk injection injuries (due to higher psi, organic solvents, and/or delayed definitive care) have been reported from 48% to 80%.10-13 Immediate surgical evaluation is often recommended when injection injuries are encountered.10,13 Patients who receive immediate surgical debridement (ie, within 6 hours from injury), specifically those injected with organic solvents, have a lower amputation rate than those who do not have immediate surgical intervention.13
In some reports, more than 80% of those injected with organic solvents who did not have immediate washout and debridement required amputation.4 Other effects of postponed debridement include delayed return to work time and decreased functionality of the hand.13,14
Conclusion
Although high-pressure injection injuries often appear mild at initial presentation since the immediate symptoms of the neurovascular injury often resolves, patients often experience severe sequelae including infection, disability, or amputation. The severity of the injury not only depends upon the psi, but also the type and amount of chemical injected. Because of the high morbidity, it is imperative that emergency physicians are aware of and identify these types of injuries and their sequelae.10,11,13,14
Dr Wilson is a third year resident in the department of emergency medicine at the Alpert Medical School, Brown University, Providence, Rhode Island. Dr Hack is the director for the division of medical toxicology at Brown University and the director of the educational program in medical toxicology. He is an associate professor at Warren Alpert Medical School; and an attending physician in the department of emergency medicine at Brown University, Rhode Island Hospital, Miriam Hospital, Providence.
Disclosure: The authors report no conflicts of interest.
- Scott AR. Occupational high-pressure injection injuries: pathogenesis and prevention. J Soc Occup Med. 1983;33(2):56-59.
- Pappou IP, Deal DN. High-pressure injection injuries. J Hand Surg Am. 2012;37(11):2404-2407.
- Soyuncu S, Bektas F, Dinc S. High-pressure air injection injury to the upper extremity. J Emerg Med. 2013;45(1):96-98.
- Schoo MJ, Scott FA, Boswick JA Jr. High-pressure injection injuries of the hand. J Trauma. 1980;20(3):229-238.
- Hogan CJ, Ruland RT. High-pressure injection injuries to the upper extremity: a review of the literature. J Orthop Trauma. 2006;20(7):503-511.
- Rosenwasser MP, Wei DH. High-pressure injection injuries to the hand. J Am Acad Orthop Surg. 2014;22(1):38-45.
- Mirzayan R, Schnall SB, Chon JH, Holtom PD, Pazakis MJ, Stevanovic MV. Culture results and amputation rates in high-pressure paint gun injuries of the hand. Orthopedics. 2001;24(6):587-589.
- Wong TC, Ip FK, Wu WC. High-pressure injection injuries of the hand in a Chinese population. J Hand Surg Br. 2005;30(6):588-592.
- Pinto MR, Turkula-Pinto LD, Cooney WP, Wood MB, Dobyns JH: High-pressure injection injuries of the hand: review of 25 patients managed by open wound technique. J Hand Surg Am. 1993;18(1):125-130.
- Kaufman, HD. High pressure injection injuries, the problems, pathogenesis and management. Hand. 1970;2(1);63-73.
- Vasilevski D, Noorbergen M, Depierreux M, Lafontaine M. High-pressure injection injuries to the hand. Am J Emerg Med. 2000;18(7):820-824.
- Bekler H, Gokce A, Beyzadeoglu T, Parmaksizoglu F. The surgical treatment and outcomes of high pressure injection injuries of the hand. J Hand Surg Eur Vol. 2007;32(4):394-399.
- Amsdell SL, Hammert WC. High-pressure injection injuries in the hand: current treatment concepts. Plast Reconstr Surg. 2013;132(4):586e-591e.
- Hart RG, Smith GD, Haq A. Prevention of high-pressure injection injuries to the hand. Am J Emerg Med. 2005;24(1):73-76.
- Scott AR. Occupational high-pressure injection injuries: pathogenesis and prevention. J Soc Occup Med. 1983;33(2):56-59.
- Pappou IP, Deal DN. High-pressure injection injuries. J Hand Surg Am. 2012;37(11):2404-2407.
- Soyuncu S, Bektas F, Dinc S. High-pressure air injection injury to the upper extremity. J Emerg Med. 2013;45(1):96-98.
- Schoo MJ, Scott FA, Boswick JA Jr. High-pressure injection injuries of the hand. J Trauma. 1980;20(3):229-238.
- Hogan CJ, Ruland RT. High-pressure injection injuries to the upper extremity: a review of the literature. J Orthop Trauma. 2006;20(7):503-511.
- Rosenwasser MP, Wei DH. High-pressure injection injuries to the hand. J Am Acad Orthop Surg. 2014;22(1):38-45.
- Mirzayan R, Schnall SB, Chon JH, Holtom PD, Pazakis MJ, Stevanovic MV. Culture results and amputation rates in high-pressure paint gun injuries of the hand. Orthopedics. 2001;24(6):587-589.
- Wong TC, Ip FK, Wu WC. High-pressure injection injuries of the hand in a Chinese population. J Hand Surg Br. 2005;30(6):588-592.
- Pinto MR, Turkula-Pinto LD, Cooney WP, Wood MB, Dobyns JH: High-pressure injection injuries of the hand: review of 25 patients managed by open wound technique. J Hand Surg Am. 1993;18(1):125-130.
- Kaufman, HD. High pressure injection injuries, the problems, pathogenesis and management. Hand. 1970;2(1);63-73.
- Vasilevski D, Noorbergen M, Depierreux M, Lafontaine M. High-pressure injection injuries to the hand. Am J Emerg Med. 2000;18(7):820-824.
- Bekler H, Gokce A, Beyzadeoglu T, Parmaksizoglu F. The surgical treatment and outcomes of high pressure injection injuries of the hand. J Hand Surg Eur Vol. 2007;32(4):394-399.
- Amsdell SL, Hammert WC. High-pressure injection injuries in the hand: current treatment concepts. Plast Reconstr Surg. 2013;132(4):586e-591e.
- Hart RG, Smith GD, Haq A. Prevention of high-pressure injection injuries to the hand. Am J Emerg Med. 2005;24(1):73-76.
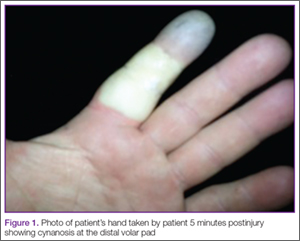
Case Studies in Toxicology: Babies and Booze—Pediatric Considerations in the Management of Ethanol Intoxication
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.

Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
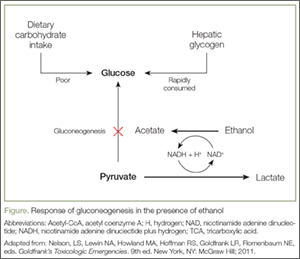
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.
Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.
Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.

Knee Extensor Mechanism Reconstruction With Complete Extensor Allograft After Failure of Patellar Tendon Repair
The extensor mechanism of the knee comprises the quadriceps tendon, the patella, and the patellar tendon. The extensor mechanism may be damaged by injury to these structures, with consequences such as the inability to actively extend the knee and hemarthrosis.1,2 Disruption of this mechanism is rare, and the most common injury pattern is an eccentric contraction of the quadriceps tendon on a flexed knee causing a tendon (quadriceps or patellar) rupture or a patella fracture.1,2
Patellar tendon ruptures are more common in persons younger than 40 years.1 Treatment is surgical, regardless of age and physical activity. In the acute setting, repair can be end-to-end suture or transosseous tunnel insertion. End-to-end suturing is difficult in chronic patellar tendon ruptures because of patella alta secondary to quadriceps contraction.3 Treatment options for chronic ruptures may involve transpatellar traction4 or tendon reinforcement with fascia lata, a semitendinosus band, or synthetic materials.3-5 Alternatively, tendon autograft and allografts have also been recommended, especially in extreme situations.1,6 Furthermore, animal experiments have shown that a compact platelet-rich fibrin scaffold (CPFS) has the potential to accelerate healing of patellar tendon defects and to act as a bioscaffold for graft augmentation.7
We describe the case of a 30-year-old man who underwent extensor mechanism reconstruction with cadaveric tendon–patellar tendon–bone allograft for failure of an infected primary end-to-end repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 30-year-old healthy man landed on an empty glass fish tank, resulting in a traumatic right-knee arthrotomy. On initial evaluation, the patient had a negative straight-leg-raise test and impaired knee extension. The patient was taken urgently to the operating room for irrigation and débridement and concurrent repair of the patellar tendon laceration. Antibiotic prophylaxis with 2 g of intravenous (IV) cefazolin was given in the emergency room.
Intraoperatively, after visualizing the patellar tendon laceration and excluding any associated chondral lesions, we proceeded with extensive débridement and irrigation using 9 L of normal saline pulse lavage. After we achieved a clean site, we proceeded to repair the patellar tendon using No. 2 FiberWire sutures (Arthrex, Naples, Florida) with a classic Krackow repair8 consisting of 2 sutures run in a 4-row fashion through the patella and the patellar tendon. The suture was securely tightened and then tested for stability to at least 90° of knee flexion. The retinaculum was repaired using No. 0 Vicryl sutures (Ethicon, Somerville, New Jersey). After wound closure and dressing, the patient was placed in a hinged knee brace locked in extension at all times after surgery. Antibiotic treatment with IV cefazolin was administered for 48 hours.
Postoperative management consisted of weight-bearing as tolerated on the operative limb and appropriate deep venous thrombosis prophylaxis. The patient followed up in clinic 2 weeks and 4 weeks after surgery. At 4 weeks, the patient was noted to have a secondary wound infection with superficial dehiscence and serosanguineous drainage. No wound opening was noticed, and local wound care was performed with a 1-week course of oral cephalexin. The patient was scheduled to follow up a few weeks later but did not follow up for a year.
At 1-year follow-up, the patient reported that he had had a steady progression of his knee range of motion (ROM) with decreased pain. However, over time, the patient noted subjective instability of the knee, with frequent falls occurring close to his 1-year follow-up. Examination of his knee showed that his active ROM ranged from 20° in extension to 120° in flexion, with a weak extensor mechanism. Passively, his knee could be brought to full extension. His incision was well healed, but it had an area of bogginess in the middle. Radiographs showed patella alta on the affected knee, with a lengthening of the patellar tendon of 7.70 cm on the right compared with 5.18 cm on the left. Magnetic resonance imaging (MRI) showed moderate-to-severe patellar tendinosis with small fluid pockets around the surgical material and evidence of acute patellar enthesopathy. The laboratory values showed a white blood cell count of 7580/μL (normal, 4500-11,000/μL), an erythrocyte sedimentation rate of 2 mm/h (normal, 1-15 mm/h), and a C-reactive protein level of 1.93 mg/dL (normal, 0.00-0.29 mg/dL). Based on the clinical examination and imaging findings, there was a concern for a possible chronic deep-tissue infection, in addition to failure of the primary patellar tendon repair. Operative versus nonoperative management options were discussed with the patient, and he elected to undergo surgery.
During surgery, the patellar laxity was confirmed, and the patellar tendon was noticed to be chronically thickened and surrounded by unhealthy tissue. Initially, an extensive soft-tissue débridement was performed, and all patellar tendon loculations visualized on the preoperative MRI were drained; a solid purulent-like fluid was expressed. Unfortunately, the extensive and required débridement did not allow the preservation of the patellar tendon. Appropriate cultures were taken and sent for immediate Gram-stain analysis, which returned negative. Tissue samples from the patellar tendon were also sent to the pathology department for analysis. Intraoperatively, the infrapatellar defect was filled temporarily with a tobramycin cement spacer mixed with 2 g of vancomycin in a manner similar to that of the Masquelet technique used for infected long-bone nonunions with bone loss.9,10 This technique is a 2-stage procedure that promotes the formation of a biologic membrane that allows bone healing in the reconstruction of long-bone defects. The first stage consists of a radical débridement with soft-tissue repair by flaps when needed, with the insertion of a polymethylmethacrylate cement spacer into the bone defect. The second stage is usually performed 6 to 8 weeks later, with removal of the spacer and preservation of the induced membrane, which is filled with iliac crest bone autograft augmented (if necessary) with demineralized allograft.
The incision was closed primarily, and after surgery, the patient was allowed to bear weight as tolerated in a hinged knee brace locked in extension. Final laboratory analysis from cultures and tissue samples revealed acute and chronic inflammation with more than 20 neutrophils per high-powered field. No organisms grew from aerobic, anaerobic, fungal, or mycobacterial cultures. The infectious disease service was consulted and recommended oral cephalexin.
Because all cultures were negative, all laboratory examinations did not indicate any residual infections, and no bony involvement was noticed intraoperatively or in the preoperative knee MRI, we decided to proceed with the second stage of the Masquelet technique after 2 weeks. The patient returned to the operating room for final reconstruction of his patellar tendon using a custom-ordered cadaveric tendon–patellar tendon–bone allograft, the length of which was determined by measuring the contralateral patellar tendon, ie, 5.18 cm (Figure 1A). The previous anterior knee incision was reopened and extended distally past the tibial tuberosity and proximally toward the quadriceps tendon. The antibiotic spacer was removed. We proceeded with a repeat irrigation and débridement and the allograft transfer. The selected allograft was customized by reducing the tibial bone component to an approximately 1×2-cm bone block and by reducing the allograft patellar thickness with an oscillating saw, leaving an approximately 2-mm thick patellar bone graft attached to the patellar tendon. In a similar technique using an oscillating saw, we shaved off the anterior cortex of the patient’s patella to accommodate, in a sandwich fashion, the patellar allograft. Proximally, the quadriceps tendon insertion was split longitudinally and partially separated from the superior pole of the patellar tendon to allow seating and fixation of the modified quadriceps allograft tendon component.
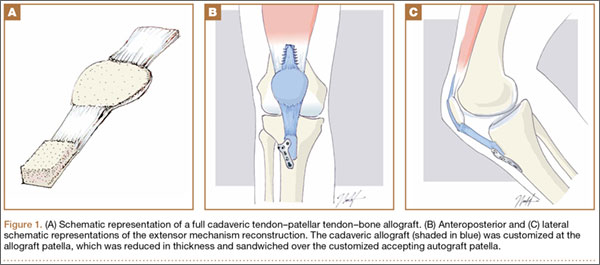
We proceeded with the fixation of the allograft first distally on the patella. The anterior cortex of the tibial tuberosity was resected to allow the perfect seating of the bone block allograft. The graft was secured with a 4.0-mm fully threaded cancellous lag screw and reinforced with a 2.4-mm, 3-hole T-volar buttress plate (Synthes, Paoli, Pennsylvania). The plate was contoured to better fit the patient’s tibia. We sutured the patellar allograft tendon to the patella using two No. 2-0 FiberWire sutures in Krackow suture technique8 (Figures 1B, 1C). We obtained good fixation of the patellar tendon, and the distance between the patellar insertion and the inferior patellar pole was the same as before surgery: 5.57 cm and comparable to the contralateral side (Figures 2A-2C). The patellar allograft and autograft sandwich were secured with additional No. 2-0 FiberWire sutures, and the quadriceps allograft and autograft were secured with the cross-stitch technique with the same material. Fine suturing of the quadriceps tendon was done with No. 0 Vicryl sutures. After the fixation was completed, we tested the stability of the reconstruction and found good flexion up to 120°.
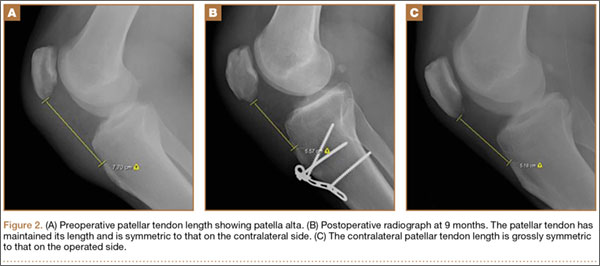
The postoperative protocol consisted of weight-bearing as tolerated in full extension and passive knee ROM, using a continuous passive ROM machine from 0° to 45° for the first 4 weeks, followed by active ROM, increased as tolerated, during the next 8 weeks.
The patient was seen in clinic 3 and 9 months after surgery. At the 3-month follow-up appointment, the patient’s examination showed knee ROM from 0° extension to 130° of flexion, no secondary infection signs, and radiographic evidence of a well-healing patellar allograft with symmetric patellar tendon length to the contralateral side. At 9-month follow-up, the patient’s active ROM was from 0° extension to 140° flexion (Figures 3A, 3B), and he had returned to his preinjury level of functioning.
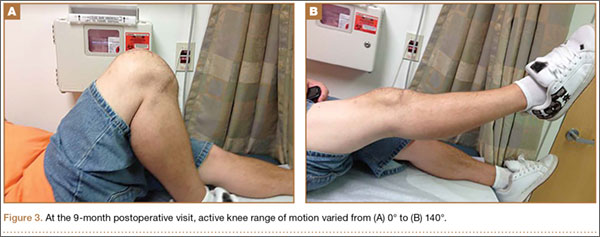
Discussion
This case report describes the successful reconstruction of a patellar tendon defect with cadaveric tendon–patellar tendon–bone allograft. Extensor mechanism injuries are uncommon in general, and the incidence of patellar tendon injury is higher in men than in women.2 Patellar tendon tears occur frequently in active patients younger than 40 years, usually as a result of sudden quadriceps contraction with the knee slightly flexed.1 Treatment of patellar tendon injury is surgical, and functional outcomes for patients with this injury are equivalent to those of patients with quadriceps tendon injuries or patellar fractures.2 Acute patellar tendon tears can be repaired by end-to-end suturing or transosseous tunnel insertion in the tibia or patella.1 Reinforcement is often added between the patella and tibial tuberosity, using a semitendinosus band or wire.1 End-to-end suture is performed using a thick resorbable suture. It is important to avoid patella alta during suturing, comparing the position of the patella with the contralateral patella with the knee in 45° of flexion. In proximal avulsion, the tendon is anchored to the bone by 2 thick nonresorbable sutures through 2 parallel bone tunnels to the proximal pole of the patella. Distal avulsion is rare in adults, but it can be managed by using staples or suture anchors.1
End-to-end suturing of chronic patellar tendon defects is difficult more than 45 days after injury primarily because of difficulties in correcting patella alta secondary to the upward force exerted by the quadriceps tendon.1,3 Extreme situations similar to the case we present warrant Achilles or patellar tendon allograft for reconstruction of the extensor mechanism.1,3,6,9
Extensor mechanism allograft also provides an effective remedy for severe quadriceps deficiency caused by loss of the patella, patellar tendon, and quadriceps tendon in total knee arthroplasty.10 However, in such cases, late failure is common, and major quadriceps deficiency occurs after removal of the allograft material.10 To improve outcome, a novel technique using the medial gastrocnemius muscle transferred to the muscular portion of the vastus medialis and lateralis flaps provides a secure and strong closure of the anterior knee, thereby restoring the extensor mechanism of the knee.10
Patellar tendon reconstruction with allograft tissue has been successfully used, especially in cases related to chronic patellar tendon ruptures11 and total knee arthroplasty.6,12-14 Crossett and colleagues12 showed that, at 2-year follow-up, the average knee score for pain, ROM, and stability had improved from 26 points (range, 6-39 points) before surgery to 81 points (range, 40-92 points). The average knee score for function had also improved: 14 points (range, 0-35 points) before surgery to 53 points (range, 30-90 points).12 Primary repair may succeed in early intervention, but in an established rupture, allograft reconstruction is often necessary. Achilles tendon is the preferred allograft, with the calcaneus fragment embedded into the proximal tibia as a new tubercle and the tendon sutured into the remaining extensor mechanism.1,11 The repair is further protected using a cable loop from the superior pole of the patella to a drill hole in the upper tibia.9 Techniques have also been described involving passage of the proximal aspect of the allograft tendon through patellar bone tunnels and suture fixation to the native quadriceps tendon.11,15 However, in our technique, we shaved off the anterior cortex of the patient’s patella to allow a sandwich-type over-position of the allograft to secure fixation to the patella.
Another alternative to allograft reconstruction involves biocompatible scaffolds. Such scaffolds incorporate the use of platelets in a fibrin framework. A CPFS, produced from blood and calcium gluconate to improve healing of patellar tendon defects, has been described in animal studies.7 In the rabbit model, CPFS acts as a provisional bioscaffold that can accelerate healing of an injured patellar tendon repair, potentially secondary to several growth factors derived from platelets.7 Platelets are biocompatible sources of growth factors, and CPFS can act as a scaffold to restore the mechanical integrity of injured soft tissue.7,16 In addition, CPFS can act to lower donor-site morbidity associated with harvesting tissue autograft.7 However, to our knowledge, such scaffolds have not been used in human trials. The LARS biocompatible ligament (Corin Group PLC, Cirencester, United Kingdom), currently not approved by the US Food and Drug Administration, is used for reconstructions of isolated or multiple knee ligament injuries.17 This graft requires the presence of healthy tissue with good blood supply from which new tendon or ligament can grow in. Sometimes it is also used for extensor mechanism reconstruction after radical tumor resection around the knee; however, good results are achieved in only 59% of cases,18 and to our knowledge, only 1 case of primary repair of a patellar tendon rupture has been published.19
Techniques involving the use of tendon–patellar tendon–bone graft with fixation via the sandwich-type over-position of the allograft for chronic patellar tendon rupture have not been described in the literature. In our patient, given the extensive patellar tendon lesion and inflammation with chronic tissue degeneration, there was no option but to use allograft. To improve the patient’s outcome, we chose the strongest possible allograft, tendon–patellar tendon–bone graft.
Conclusion
Revision patellar tendon reconstruction is a challenging, but necessary, procedure to restore the extensor mechanism of the knee, especially in young, active individuals. Various options to reconstruct the tissue defects are available. Our patient was successfully treated with a tendon–patellar tendon–bone allograft reconstruction.
1. Saragaglia D, Pison A, Rubens-Duval B. Acute and old ruptures of the extensor apparatus of the knee in adults (excluding knee replacement). Orthop Traumatol Surg Res. 2013;99(1 suppl):S67-S76.
2. Tejwani NC, Lekic N, Bechtel C, Montero N, Egol KA. Outcomes after knee joint extensor mechanism disruptions: is it better to fracture the patella or rupture the tendon? J Orthop Trauma. 2012;26(11):648-651.
3. Ecker ML, Lotke PA, Glazer RM. Late reconstruction of the patellar tendon. J Bone Joint Surg Am. 1979;61(6):884-886.
4. Siwek CW, Rao JP. Ruptures of the extensor mechanism of the knee joint. J Bone Joint Surg Am. 1981;63(6):932-937.
5. Levy M, Goldstein J, Rosner M. A method of repair for quadriceps tendon or patellar ligament (tendon) ruptures without cast immobilization. Preliminary report. Clin Orthop Relat Res. 1987;218:297-301.
6. Burks RT, Edelson RH. Allograft reconstruction of the patellar ligament. A case report. J Bone Joint Surg Am. 1994;76(7):1077-1079.
7. Matsunaga D, Akizuki S, Takizawa T, Omae S, Kato H. Compact platelet-rich fibrin scaffold to improve healing of patellar tendon defects and for medial collateral ligament reconstruction. Knee. 2013;20(6):545-550.
8. Krackow KA, Thomas SC, Jones LC. Ligament-tendon fixation: analysis of a new stitch and comparison with standard techniques. Orthopedics. 1988;11(6):909-917.
9. Brooks P. Extensor mechanism ruptures. Orthopedics. 2009;32(9):683-684.
10. Whiteside LA. Surgical technique: muscle transfer restores extensor function after failed patella-patellar tendon allograft. Clin Orthop Relat Res. 2014;472(1):218-226.
11. Farmer K, Cosgarea AJ. Procedure 25. Acute and chronic patellar tendon ruptures. In: Miller MD, Cole BJ, Cosgarea AJ, Sekiya JK, eds. Operative Techniques: Sports Knee Surgery. Philadelphia, PA: Saunders (Elsevier); 2008:397-417.
12. Crossett LS, Sinha RK, Sechriest VF, Rubash HE. Reconstruction of a ruptured patellar tendon with achilles tendon allograft following total knee arthroplasty. J Bone Joint Surg Am. 2002;84(8):1354-1361.
13. Lahav A, Burks RT, Scholl MD. Allograft reconstruction of the patellar tendon: 12-year follow-up. Am J Orthop. 2004;33(12):623-624.
14. Yoo JH, Chang JD, Seo YJ, Baek SW. Reconstruction of a patellar tendon with Achilles tendon allograft for severe patellar infera--a case report. Knee. 2011;18(5):350-353.
15. Saldua NS, Mazurek MT. Procedure 37. Quadriceps and patellar tendon repair. In: Reider B, Terry MA, Provencher MT, eds. Operative Techniques: Sports Medicine Surgery. Philadelphia, PA: Saunders (Elsevier); 2010:623-640.
16. Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91(1):4-15.
17. Ibrahim SAR, Ahmad FHF, Salah M, Al Misfer ARK, Ghaffer SA, Khirat S. Surgical management of traumatic knee dislocation. Arthroscopy. 2008;24(2):178-187.
18. Dominkus M, Sabeti M, Toma C, Abdolvahab F, Trieb K, Kotz RI. Reconstructing the extensor apparatus with a new polyester ligament. Clin Orthop Relat Res. 2006;453:328-334.
19. Naim S, Gougoulias N, Griffiths D. Patellar tendon reconstruction using LARS ligament: surgical technique and case report. Strategies Trauma Limb Reconstr. 2011;6(1):39-41.
The extensor mechanism of the knee comprises the quadriceps tendon, the patella, and the patellar tendon. The extensor mechanism may be damaged by injury to these structures, with consequences such as the inability to actively extend the knee and hemarthrosis.1,2 Disruption of this mechanism is rare, and the most common injury pattern is an eccentric contraction of the quadriceps tendon on a flexed knee causing a tendon (quadriceps or patellar) rupture or a patella fracture.1,2
Patellar tendon ruptures are more common in persons younger than 40 years.1 Treatment is surgical, regardless of age and physical activity. In the acute setting, repair can be end-to-end suture or transosseous tunnel insertion. End-to-end suturing is difficult in chronic patellar tendon ruptures because of patella alta secondary to quadriceps contraction.3 Treatment options for chronic ruptures may involve transpatellar traction4 or tendon reinforcement with fascia lata, a semitendinosus band, or synthetic materials.3-5 Alternatively, tendon autograft and allografts have also been recommended, especially in extreme situations.1,6 Furthermore, animal experiments have shown that a compact platelet-rich fibrin scaffold (CPFS) has the potential to accelerate healing of patellar tendon defects and to act as a bioscaffold for graft augmentation.7
We describe the case of a 30-year-old man who underwent extensor mechanism reconstruction with cadaveric tendon–patellar tendon–bone allograft for failure of an infected primary end-to-end repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 30-year-old healthy man landed on an empty glass fish tank, resulting in a traumatic right-knee arthrotomy. On initial evaluation, the patient had a negative straight-leg-raise test and impaired knee extension. The patient was taken urgently to the operating room for irrigation and débridement and concurrent repair of the patellar tendon laceration. Antibiotic prophylaxis with 2 g of intravenous (IV) cefazolin was given in the emergency room.
Intraoperatively, after visualizing the patellar tendon laceration and excluding any associated chondral lesions, we proceeded with extensive débridement and irrigation using 9 L of normal saline pulse lavage. After we achieved a clean site, we proceeded to repair the patellar tendon using No. 2 FiberWire sutures (Arthrex, Naples, Florida) with a classic Krackow repair8 consisting of 2 sutures run in a 4-row fashion through the patella and the patellar tendon. The suture was securely tightened and then tested for stability to at least 90° of knee flexion. The retinaculum was repaired using No. 0 Vicryl sutures (Ethicon, Somerville, New Jersey). After wound closure and dressing, the patient was placed in a hinged knee brace locked in extension at all times after surgery. Antibiotic treatment with IV cefazolin was administered for 48 hours.
Postoperative management consisted of weight-bearing as tolerated on the operative limb and appropriate deep venous thrombosis prophylaxis. The patient followed up in clinic 2 weeks and 4 weeks after surgery. At 4 weeks, the patient was noted to have a secondary wound infection with superficial dehiscence and serosanguineous drainage. No wound opening was noticed, and local wound care was performed with a 1-week course of oral cephalexin. The patient was scheduled to follow up a few weeks later but did not follow up for a year.
At 1-year follow-up, the patient reported that he had had a steady progression of his knee range of motion (ROM) with decreased pain. However, over time, the patient noted subjective instability of the knee, with frequent falls occurring close to his 1-year follow-up. Examination of his knee showed that his active ROM ranged from 20° in extension to 120° in flexion, with a weak extensor mechanism. Passively, his knee could be brought to full extension. His incision was well healed, but it had an area of bogginess in the middle. Radiographs showed patella alta on the affected knee, with a lengthening of the patellar tendon of 7.70 cm on the right compared with 5.18 cm on the left. Magnetic resonance imaging (MRI) showed moderate-to-severe patellar tendinosis with small fluid pockets around the surgical material and evidence of acute patellar enthesopathy. The laboratory values showed a white blood cell count of 7580/μL (normal, 4500-11,000/μL), an erythrocyte sedimentation rate of 2 mm/h (normal, 1-15 mm/h), and a C-reactive protein level of 1.93 mg/dL (normal, 0.00-0.29 mg/dL). Based on the clinical examination and imaging findings, there was a concern for a possible chronic deep-tissue infection, in addition to failure of the primary patellar tendon repair. Operative versus nonoperative management options were discussed with the patient, and he elected to undergo surgery.
During surgery, the patellar laxity was confirmed, and the patellar tendon was noticed to be chronically thickened and surrounded by unhealthy tissue. Initially, an extensive soft-tissue débridement was performed, and all patellar tendon loculations visualized on the preoperative MRI were drained; a solid purulent-like fluid was expressed. Unfortunately, the extensive and required débridement did not allow the preservation of the patellar tendon. Appropriate cultures were taken and sent for immediate Gram-stain analysis, which returned negative. Tissue samples from the patellar tendon were also sent to the pathology department for analysis. Intraoperatively, the infrapatellar defect was filled temporarily with a tobramycin cement spacer mixed with 2 g of vancomycin in a manner similar to that of the Masquelet technique used for infected long-bone nonunions with bone loss.9,10 This technique is a 2-stage procedure that promotes the formation of a biologic membrane that allows bone healing in the reconstruction of long-bone defects. The first stage consists of a radical débridement with soft-tissue repair by flaps when needed, with the insertion of a polymethylmethacrylate cement spacer into the bone defect. The second stage is usually performed 6 to 8 weeks later, with removal of the spacer and preservation of the induced membrane, which is filled with iliac crest bone autograft augmented (if necessary) with demineralized allograft.
The incision was closed primarily, and after surgery, the patient was allowed to bear weight as tolerated in a hinged knee brace locked in extension. Final laboratory analysis from cultures and tissue samples revealed acute and chronic inflammation with more than 20 neutrophils per high-powered field. No organisms grew from aerobic, anaerobic, fungal, or mycobacterial cultures. The infectious disease service was consulted and recommended oral cephalexin.
Because all cultures were negative, all laboratory examinations did not indicate any residual infections, and no bony involvement was noticed intraoperatively or in the preoperative knee MRI, we decided to proceed with the second stage of the Masquelet technique after 2 weeks. The patient returned to the operating room for final reconstruction of his patellar tendon using a custom-ordered cadaveric tendon–patellar tendon–bone allograft, the length of which was determined by measuring the contralateral patellar tendon, ie, 5.18 cm (Figure 1A). The previous anterior knee incision was reopened and extended distally past the tibial tuberosity and proximally toward the quadriceps tendon. The antibiotic spacer was removed. We proceeded with a repeat irrigation and débridement and the allograft transfer. The selected allograft was customized by reducing the tibial bone component to an approximately 1×2-cm bone block and by reducing the allograft patellar thickness with an oscillating saw, leaving an approximately 2-mm thick patellar bone graft attached to the patellar tendon. In a similar technique using an oscillating saw, we shaved off the anterior cortex of the patient’s patella to accommodate, in a sandwich fashion, the patellar allograft. Proximally, the quadriceps tendon insertion was split longitudinally and partially separated from the superior pole of the patellar tendon to allow seating and fixation of the modified quadriceps allograft tendon component.
We proceeded with the fixation of the allograft first distally on the patella. The anterior cortex of the tibial tuberosity was resected to allow the perfect seating of the bone block allograft. The graft was secured with a 4.0-mm fully threaded cancellous lag screw and reinforced with a 2.4-mm, 3-hole T-volar buttress plate (Synthes, Paoli, Pennsylvania). The plate was contoured to better fit the patient’s tibia. We sutured the patellar allograft tendon to the patella using two No. 2-0 FiberWire sutures in Krackow suture technique8 (Figures 1B, 1C). We obtained good fixation of the patellar tendon, and the distance between the patellar insertion and the inferior patellar pole was the same as before surgery: 5.57 cm and comparable to the contralateral side (Figures 2A-2C). The patellar allograft and autograft sandwich were secured with additional No. 2-0 FiberWire sutures, and the quadriceps allograft and autograft were secured with the cross-stitch technique with the same material. Fine suturing of the quadriceps tendon was done with No. 0 Vicryl sutures. After the fixation was completed, we tested the stability of the reconstruction and found good flexion up to 120°.
The postoperative protocol consisted of weight-bearing as tolerated in full extension and passive knee ROM, using a continuous passive ROM machine from 0° to 45° for the first 4 weeks, followed by active ROM, increased as tolerated, during the next 8 weeks.
The patient was seen in clinic 3 and 9 months after surgery. At the 3-month follow-up appointment, the patient’s examination showed knee ROM from 0° extension to 130° of flexion, no secondary infection signs, and radiographic evidence of a well-healing patellar allograft with symmetric patellar tendon length to the contralateral side. At 9-month follow-up, the patient’s active ROM was from 0° extension to 140° flexion (Figures 3A, 3B), and he had returned to his preinjury level of functioning.
Discussion
This case report describes the successful reconstruction of a patellar tendon defect with cadaveric tendon–patellar tendon–bone allograft. Extensor mechanism injuries are uncommon in general, and the incidence of patellar tendon injury is higher in men than in women.2 Patellar tendon tears occur frequently in active patients younger than 40 years, usually as a result of sudden quadriceps contraction with the knee slightly flexed.1 Treatment of patellar tendon injury is surgical, and functional outcomes for patients with this injury are equivalent to those of patients with quadriceps tendon injuries or patellar fractures.2 Acute patellar tendon tears can be repaired by end-to-end suturing or transosseous tunnel insertion in the tibia or patella.1 Reinforcement is often added between the patella and tibial tuberosity, using a semitendinosus band or wire.1 End-to-end suture is performed using a thick resorbable suture. It is important to avoid patella alta during suturing, comparing the position of the patella with the contralateral patella with the knee in 45° of flexion. In proximal avulsion, the tendon is anchored to the bone by 2 thick nonresorbable sutures through 2 parallel bone tunnels to the proximal pole of the patella. Distal avulsion is rare in adults, but it can be managed by using staples or suture anchors.1
End-to-end suturing of chronic patellar tendon defects is difficult more than 45 days after injury primarily because of difficulties in correcting patella alta secondary to the upward force exerted by the quadriceps tendon.1,3 Extreme situations similar to the case we present warrant Achilles or patellar tendon allograft for reconstruction of the extensor mechanism.1,3,6,9
Extensor mechanism allograft also provides an effective remedy for severe quadriceps deficiency caused by loss of the patella, patellar tendon, and quadriceps tendon in total knee arthroplasty.10 However, in such cases, late failure is common, and major quadriceps deficiency occurs after removal of the allograft material.10 To improve outcome, a novel technique using the medial gastrocnemius muscle transferred to the muscular portion of the vastus medialis and lateralis flaps provides a secure and strong closure of the anterior knee, thereby restoring the extensor mechanism of the knee.10
Patellar tendon reconstruction with allograft tissue has been successfully used, especially in cases related to chronic patellar tendon ruptures11 and total knee arthroplasty.6,12-14 Crossett and colleagues12 showed that, at 2-year follow-up, the average knee score for pain, ROM, and stability had improved from 26 points (range, 6-39 points) before surgery to 81 points (range, 40-92 points). The average knee score for function had also improved: 14 points (range, 0-35 points) before surgery to 53 points (range, 30-90 points).12 Primary repair may succeed in early intervention, but in an established rupture, allograft reconstruction is often necessary. Achilles tendon is the preferred allograft, with the calcaneus fragment embedded into the proximal tibia as a new tubercle and the tendon sutured into the remaining extensor mechanism.1,11 The repair is further protected using a cable loop from the superior pole of the patella to a drill hole in the upper tibia.9 Techniques have also been described involving passage of the proximal aspect of the allograft tendon through patellar bone tunnels and suture fixation to the native quadriceps tendon.11,15 However, in our technique, we shaved off the anterior cortex of the patient’s patella to allow a sandwich-type over-position of the allograft to secure fixation to the patella.
Another alternative to allograft reconstruction involves biocompatible scaffolds. Such scaffolds incorporate the use of platelets in a fibrin framework. A CPFS, produced from blood and calcium gluconate to improve healing of patellar tendon defects, has been described in animal studies.7 In the rabbit model, CPFS acts as a provisional bioscaffold that can accelerate healing of an injured patellar tendon repair, potentially secondary to several growth factors derived from platelets.7 Platelets are biocompatible sources of growth factors, and CPFS can act as a scaffold to restore the mechanical integrity of injured soft tissue.7,16 In addition, CPFS can act to lower donor-site morbidity associated with harvesting tissue autograft.7 However, to our knowledge, such scaffolds have not been used in human trials. The LARS biocompatible ligament (Corin Group PLC, Cirencester, United Kingdom), currently not approved by the US Food and Drug Administration, is used for reconstructions of isolated or multiple knee ligament injuries.17 This graft requires the presence of healthy tissue with good blood supply from which new tendon or ligament can grow in. Sometimes it is also used for extensor mechanism reconstruction after radical tumor resection around the knee; however, good results are achieved in only 59% of cases,18 and to our knowledge, only 1 case of primary repair of a patellar tendon rupture has been published.19
Techniques involving the use of tendon–patellar tendon–bone graft with fixation via the sandwich-type over-position of the allograft for chronic patellar tendon rupture have not been described in the literature. In our patient, given the extensive patellar tendon lesion and inflammation with chronic tissue degeneration, there was no option but to use allograft. To improve the patient’s outcome, we chose the strongest possible allograft, tendon–patellar tendon–bone graft.
Conclusion
Revision patellar tendon reconstruction is a challenging, but necessary, procedure to restore the extensor mechanism of the knee, especially in young, active individuals. Various options to reconstruct the tissue defects are available. Our patient was successfully treated with a tendon–patellar tendon–bone allograft reconstruction.
The extensor mechanism of the knee comprises the quadriceps tendon, the patella, and the patellar tendon. The extensor mechanism may be damaged by injury to these structures, with consequences such as the inability to actively extend the knee and hemarthrosis.1,2 Disruption of this mechanism is rare, and the most common injury pattern is an eccentric contraction of the quadriceps tendon on a flexed knee causing a tendon (quadriceps or patellar) rupture or a patella fracture.1,2
Patellar tendon ruptures are more common in persons younger than 40 years.1 Treatment is surgical, regardless of age and physical activity. In the acute setting, repair can be end-to-end suture or transosseous tunnel insertion. End-to-end suturing is difficult in chronic patellar tendon ruptures because of patella alta secondary to quadriceps contraction.3 Treatment options for chronic ruptures may involve transpatellar traction4 or tendon reinforcement with fascia lata, a semitendinosus band, or synthetic materials.3-5 Alternatively, tendon autograft and allografts have also been recommended, especially in extreme situations.1,6 Furthermore, animal experiments have shown that a compact platelet-rich fibrin scaffold (CPFS) has the potential to accelerate healing of patellar tendon defects and to act as a bioscaffold for graft augmentation.7
We describe the case of a 30-year-old man who underwent extensor mechanism reconstruction with cadaveric tendon–patellar tendon–bone allograft for failure of an infected primary end-to-end repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 30-year-old healthy man landed on an empty glass fish tank, resulting in a traumatic right-knee arthrotomy. On initial evaluation, the patient had a negative straight-leg-raise test and impaired knee extension. The patient was taken urgently to the operating room for irrigation and débridement and concurrent repair of the patellar tendon laceration. Antibiotic prophylaxis with 2 g of intravenous (IV) cefazolin was given in the emergency room.
Intraoperatively, after visualizing the patellar tendon laceration and excluding any associated chondral lesions, we proceeded with extensive débridement and irrigation using 9 L of normal saline pulse lavage. After we achieved a clean site, we proceeded to repair the patellar tendon using No. 2 FiberWire sutures (Arthrex, Naples, Florida) with a classic Krackow repair8 consisting of 2 sutures run in a 4-row fashion through the patella and the patellar tendon. The suture was securely tightened and then tested for stability to at least 90° of knee flexion. The retinaculum was repaired using No. 0 Vicryl sutures (Ethicon, Somerville, New Jersey). After wound closure and dressing, the patient was placed in a hinged knee brace locked in extension at all times after surgery. Antibiotic treatment with IV cefazolin was administered for 48 hours.
Postoperative management consisted of weight-bearing as tolerated on the operative limb and appropriate deep venous thrombosis prophylaxis. The patient followed up in clinic 2 weeks and 4 weeks after surgery. At 4 weeks, the patient was noted to have a secondary wound infection with superficial dehiscence and serosanguineous drainage. No wound opening was noticed, and local wound care was performed with a 1-week course of oral cephalexin. The patient was scheduled to follow up a few weeks later but did not follow up for a year.
At 1-year follow-up, the patient reported that he had had a steady progression of his knee range of motion (ROM) with decreased pain. However, over time, the patient noted subjective instability of the knee, with frequent falls occurring close to his 1-year follow-up. Examination of his knee showed that his active ROM ranged from 20° in extension to 120° in flexion, with a weak extensor mechanism. Passively, his knee could be brought to full extension. His incision was well healed, but it had an area of bogginess in the middle. Radiographs showed patella alta on the affected knee, with a lengthening of the patellar tendon of 7.70 cm on the right compared with 5.18 cm on the left. Magnetic resonance imaging (MRI) showed moderate-to-severe patellar tendinosis with small fluid pockets around the surgical material and evidence of acute patellar enthesopathy. The laboratory values showed a white blood cell count of 7580/μL (normal, 4500-11,000/μL), an erythrocyte sedimentation rate of 2 mm/h (normal, 1-15 mm/h), and a C-reactive protein level of 1.93 mg/dL (normal, 0.00-0.29 mg/dL). Based on the clinical examination and imaging findings, there was a concern for a possible chronic deep-tissue infection, in addition to failure of the primary patellar tendon repair. Operative versus nonoperative management options were discussed with the patient, and he elected to undergo surgery.
During surgery, the patellar laxity was confirmed, and the patellar tendon was noticed to be chronically thickened and surrounded by unhealthy tissue. Initially, an extensive soft-tissue débridement was performed, and all patellar tendon loculations visualized on the preoperative MRI were drained; a solid purulent-like fluid was expressed. Unfortunately, the extensive and required débridement did not allow the preservation of the patellar tendon. Appropriate cultures were taken and sent for immediate Gram-stain analysis, which returned negative. Tissue samples from the patellar tendon were also sent to the pathology department for analysis. Intraoperatively, the infrapatellar defect was filled temporarily with a tobramycin cement spacer mixed with 2 g of vancomycin in a manner similar to that of the Masquelet technique used for infected long-bone nonunions with bone loss.9,10 This technique is a 2-stage procedure that promotes the formation of a biologic membrane that allows bone healing in the reconstruction of long-bone defects. The first stage consists of a radical débridement with soft-tissue repair by flaps when needed, with the insertion of a polymethylmethacrylate cement spacer into the bone defect. The second stage is usually performed 6 to 8 weeks later, with removal of the spacer and preservation of the induced membrane, which is filled with iliac crest bone autograft augmented (if necessary) with demineralized allograft.
The incision was closed primarily, and after surgery, the patient was allowed to bear weight as tolerated in a hinged knee brace locked in extension. Final laboratory analysis from cultures and tissue samples revealed acute and chronic inflammation with more than 20 neutrophils per high-powered field. No organisms grew from aerobic, anaerobic, fungal, or mycobacterial cultures. The infectious disease service was consulted and recommended oral cephalexin.
Because all cultures were negative, all laboratory examinations did not indicate any residual infections, and no bony involvement was noticed intraoperatively or in the preoperative knee MRI, we decided to proceed with the second stage of the Masquelet technique after 2 weeks. The patient returned to the operating room for final reconstruction of his patellar tendon using a custom-ordered cadaveric tendon–patellar tendon–bone allograft, the length of which was determined by measuring the contralateral patellar tendon, ie, 5.18 cm (Figure 1A). The previous anterior knee incision was reopened and extended distally past the tibial tuberosity and proximally toward the quadriceps tendon. The antibiotic spacer was removed. We proceeded with a repeat irrigation and débridement and the allograft transfer. The selected allograft was customized by reducing the tibial bone component to an approximately 1×2-cm bone block and by reducing the allograft patellar thickness with an oscillating saw, leaving an approximately 2-mm thick patellar bone graft attached to the patellar tendon. In a similar technique using an oscillating saw, we shaved off the anterior cortex of the patient’s patella to accommodate, in a sandwich fashion, the patellar allograft. Proximally, the quadriceps tendon insertion was split longitudinally and partially separated from the superior pole of the patellar tendon to allow seating and fixation of the modified quadriceps allograft tendon component.
We proceeded with the fixation of the allograft first distally on the patella. The anterior cortex of the tibial tuberosity was resected to allow the perfect seating of the bone block allograft. The graft was secured with a 4.0-mm fully threaded cancellous lag screw and reinforced with a 2.4-mm, 3-hole T-volar buttress plate (Synthes, Paoli, Pennsylvania). The plate was contoured to better fit the patient’s tibia. We sutured the patellar allograft tendon to the patella using two No. 2-0 FiberWire sutures in Krackow suture technique8 (Figures 1B, 1C). We obtained good fixation of the patellar tendon, and the distance between the patellar insertion and the inferior patellar pole was the same as before surgery: 5.57 cm and comparable to the contralateral side (Figures 2A-2C). The patellar allograft and autograft sandwich were secured with additional No. 2-0 FiberWire sutures, and the quadriceps allograft and autograft were secured with the cross-stitch technique with the same material. Fine suturing of the quadriceps tendon was done with No. 0 Vicryl sutures. After the fixation was completed, we tested the stability of the reconstruction and found good flexion up to 120°.
The postoperative protocol consisted of weight-bearing as tolerated in full extension and passive knee ROM, using a continuous passive ROM machine from 0° to 45° for the first 4 weeks, followed by active ROM, increased as tolerated, during the next 8 weeks.
The patient was seen in clinic 3 and 9 months after surgery. At the 3-month follow-up appointment, the patient’s examination showed knee ROM from 0° extension to 130° of flexion, no secondary infection signs, and radiographic evidence of a well-healing patellar allograft with symmetric patellar tendon length to the contralateral side. At 9-month follow-up, the patient’s active ROM was from 0° extension to 140° flexion (Figures 3A, 3B), and he had returned to his preinjury level of functioning.
Discussion
This case report describes the successful reconstruction of a patellar tendon defect with cadaveric tendon–patellar tendon–bone allograft. Extensor mechanism injuries are uncommon in general, and the incidence of patellar tendon injury is higher in men than in women.2 Patellar tendon tears occur frequently in active patients younger than 40 years, usually as a result of sudden quadriceps contraction with the knee slightly flexed.1 Treatment of patellar tendon injury is surgical, and functional outcomes for patients with this injury are equivalent to those of patients with quadriceps tendon injuries or patellar fractures.2 Acute patellar tendon tears can be repaired by end-to-end suturing or transosseous tunnel insertion in the tibia or patella.1 Reinforcement is often added between the patella and tibial tuberosity, using a semitendinosus band or wire.1 End-to-end suture is performed using a thick resorbable suture. It is important to avoid patella alta during suturing, comparing the position of the patella with the contralateral patella with the knee in 45° of flexion. In proximal avulsion, the tendon is anchored to the bone by 2 thick nonresorbable sutures through 2 parallel bone tunnels to the proximal pole of the patella. Distal avulsion is rare in adults, but it can be managed by using staples or suture anchors.1
End-to-end suturing of chronic patellar tendon defects is difficult more than 45 days after injury primarily because of difficulties in correcting patella alta secondary to the upward force exerted by the quadriceps tendon.1,3 Extreme situations similar to the case we present warrant Achilles or patellar tendon allograft for reconstruction of the extensor mechanism.1,3,6,9
Extensor mechanism allograft also provides an effective remedy for severe quadriceps deficiency caused by loss of the patella, patellar tendon, and quadriceps tendon in total knee arthroplasty.10 However, in such cases, late failure is common, and major quadriceps deficiency occurs after removal of the allograft material.10 To improve outcome, a novel technique using the medial gastrocnemius muscle transferred to the muscular portion of the vastus medialis and lateralis flaps provides a secure and strong closure of the anterior knee, thereby restoring the extensor mechanism of the knee.10
Patellar tendon reconstruction with allograft tissue has been successfully used, especially in cases related to chronic patellar tendon ruptures11 and total knee arthroplasty.6,12-14 Crossett and colleagues12 showed that, at 2-year follow-up, the average knee score for pain, ROM, and stability had improved from 26 points (range, 6-39 points) before surgery to 81 points (range, 40-92 points). The average knee score for function had also improved: 14 points (range, 0-35 points) before surgery to 53 points (range, 30-90 points).12 Primary repair may succeed in early intervention, but in an established rupture, allograft reconstruction is often necessary. Achilles tendon is the preferred allograft, with the calcaneus fragment embedded into the proximal tibia as a new tubercle and the tendon sutured into the remaining extensor mechanism.1,11 The repair is further protected using a cable loop from the superior pole of the patella to a drill hole in the upper tibia.9 Techniques have also been described involving passage of the proximal aspect of the allograft tendon through patellar bone tunnels and suture fixation to the native quadriceps tendon.11,15 However, in our technique, we shaved off the anterior cortex of the patient’s patella to allow a sandwich-type over-position of the allograft to secure fixation to the patella.
Another alternative to allograft reconstruction involves biocompatible scaffolds. Such scaffolds incorporate the use of platelets in a fibrin framework. A CPFS, produced from blood and calcium gluconate to improve healing of patellar tendon defects, has been described in animal studies.7 In the rabbit model, CPFS acts as a provisional bioscaffold that can accelerate healing of an injured patellar tendon repair, potentially secondary to several growth factors derived from platelets.7 Platelets are biocompatible sources of growth factors, and CPFS can act as a scaffold to restore the mechanical integrity of injured soft tissue.7,16 In addition, CPFS can act to lower donor-site morbidity associated with harvesting tissue autograft.7 However, to our knowledge, such scaffolds have not been used in human trials. The LARS biocompatible ligament (Corin Group PLC, Cirencester, United Kingdom), currently not approved by the US Food and Drug Administration, is used for reconstructions of isolated or multiple knee ligament injuries.17 This graft requires the presence of healthy tissue with good blood supply from which new tendon or ligament can grow in. Sometimes it is also used for extensor mechanism reconstruction after radical tumor resection around the knee; however, good results are achieved in only 59% of cases,18 and to our knowledge, only 1 case of primary repair of a patellar tendon rupture has been published.19
Techniques involving the use of tendon–patellar tendon–bone graft with fixation via the sandwich-type over-position of the allograft for chronic patellar tendon rupture have not been described in the literature. In our patient, given the extensive patellar tendon lesion and inflammation with chronic tissue degeneration, there was no option but to use allograft. To improve the patient’s outcome, we chose the strongest possible allograft, tendon–patellar tendon–bone graft.
Conclusion
Revision patellar tendon reconstruction is a challenging, but necessary, procedure to restore the extensor mechanism of the knee, especially in young, active individuals. Various options to reconstruct the tissue defects are available. Our patient was successfully treated with a tendon–patellar tendon–bone allograft reconstruction.
1. Saragaglia D, Pison A, Rubens-Duval B. Acute and old ruptures of the extensor apparatus of the knee in adults (excluding knee replacement). Orthop Traumatol Surg Res. 2013;99(1 suppl):S67-S76.
2. Tejwani NC, Lekic N, Bechtel C, Montero N, Egol KA. Outcomes after knee joint extensor mechanism disruptions: is it better to fracture the patella or rupture the tendon? J Orthop Trauma. 2012;26(11):648-651.
3. Ecker ML, Lotke PA, Glazer RM. Late reconstruction of the patellar tendon. J Bone Joint Surg Am. 1979;61(6):884-886.
4. Siwek CW, Rao JP. Ruptures of the extensor mechanism of the knee joint. J Bone Joint Surg Am. 1981;63(6):932-937.
5. Levy M, Goldstein J, Rosner M. A method of repair for quadriceps tendon or patellar ligament (tendon) ruptures without cast immobilization. Preliminary report. Clin Orthop Relat Res. 1987;218:297-301.
6. Burks RT, Edelson RH. Allograft reconstruction of the patellar ligament. A case report. J Bone Joint Surg Am. 1994;76(7):1077-1079.
7. Matsunaga D, Akizuki S, Takizawa T, Omae S, Kato H. Compact platelet-rich fibrin scaffold to improve healing of patellar tendon defects and for medial collateral ligament reconstruction. Knee. 2013;20(6):545-550.
8. Krackow KA, Thomas SC, Jones LC. Ligament-tendon fixation: analysis of a new stitch and comparison with standard techniques. Orthopedics. 1988;11(6):909-917.
9. Brooks P. Extensor mechanism ruptures. Orthopedics. 2009;32(9):683-684.
10. Whiteside LA. Surgical technique: muscle transfer restores extensor function after failed patella-patellar tendon allograft. Clin Orthop Relat Res. 2014;472(1):218-226.
11. Farmer K, Cosgarea AJ. Procedure 25. Acute and chronic patellar tendon ruptures. In: Miller MD, Cole BJ, Cosgarea AJ, Sekiya JK, eds. Operative Techniques: Sports Knee Surgery. Philadelphia, PA: Saunders (Elsevier); 2008:397-417.
12. Crossett LS, Sinha RK, Sechriest VF, Rubash HE. Reconstruction of a ruptured patellar tendon with achilles tendon allograft following total knee arthroplasty. J Bone Joint Surg Am. 2002;84(8):1354-1361.
13. Lahav A, Burks RT, Scholl MD. Allograft reconstruction of the patellar tendon: 12-year follow-up. Am J Orthop. 2004;33(12):623-624.
14. Yoo JH, Chang JD, Seo YJ, Baek SW. Reconstruction of a patellar tendon with Achilles tendon allograft for severe patellar infera--a case report. Knee. 2011;18(5):350-353.
15. Saldua NS, Mazurek MT. Procedure 37. Quadriceps and patellar tendon repair. In: Reider B, Terry MA, Provencher MT, eds. Operative Techniques: Sports Medicine Surgery. Philadelphia, PA: Saunders (Elsevier); 2010:623-640.
16. Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91(1):4-15.
17. Ibrahim SAR, Ahmad FHF, Salah M, Al Misfer ARK, Ghaffer SA, Khirat S. Surgical management of traumatic knee dislocation. Arthroscopy. 2008;24(2):178-187.
18. Dominkus M, Sabeti M, Toma C, Abdolvahab F, Trieb K, Kotz RI. Reconstructing the extensor apparatus with a new polyester ligament. Clin Orthop Relat Res. 2006;453:328-334.
19. Naim S, Gougoulias N, Griffiths D. Patellar tendon reconstruction using LARS ligament: surgical technique and case report. Strategies Trauma Limb Reconstr. 2011;6(1):39-41.
1. Saragaglia D, Pison A, Rubens-Duval B. Acute and old ruptures of the extensor apparatus of the knee in adults (excluding knee replacement). Orthop Traumatol Surg Res. 2013;99(1 suppl):S67-S76.
2. Tejwani NC, Lekic N, Bechtel C, Montero N, Egol KA. Outcomes after knee joint extensor mechanism disruptions: is it better to fracture the patella or rupture the tendon? J Orthop Trauma. 2012;26(11):648-651.
3. Ecker ML, Lotke PA, Glazer RM. Late reconstruction of the patellar tendon. J Bone Joint Surg Am. 1979;61(6):884-886.
4. Siwek CW, Rao JP. Ruptures of the extensor mechanism of the knee joint. J Bone Joint Surg Am. 1981;63(6):932-937.
5. Levy M, Goldstein J, Rosner M. A method of repair for quadriceps tendon or patellar ligament (tendon) ruptures without cast immobilization. Preliminary report. Clin Orthop Relat Res. 1987;218:297-301.
6. Burks RT, Edelson RH. Allograft reconstruction of the patellar ligament. A case report. J Bone Joint Surg Am. 1994;76(7):1077-1079.
7. Matsunaga D, Akizuki S, Takizawa T, Omae S, Kato H. Compact platelet-rich fibrin scaffold to improve healing of patellar tendon defects and for medial collateral ligament reconstruction. Knee. 2013;20(6):545-550.
8. Krackow KA, Thomas SC, Jones LC. Ligament-tendon fixation: analysis of a new stitch and comparison with standard techniques. Orthopedics. 1988;11(6):909-917.
9. Brooks P. Extensor mechanism ruptures. Orthopedics. 2009;32(9):683-684.
10. Whiteside LA. Surgical technique: muscle transfer restores extensor function after failed patella-patellar tendon allograft. Clin Orthop Relat Res. 2014;472(1):218-226.
11. Farmer K, Cosgarea AJ. Procedure 25. Acute and chronic patellar tendon ruptures. In: Miller MD, Cole BJ, Cosgarea AJ, Sekiya JK, eds. Operative Techniques: Sports Knee Surgery. Philadelphia, PA: Saunders (Elsevier); 2008:397-417.
12. Crossett LS, Sinha RK, Sechriest VF, Rubash HE. Reconstruction of a ruptured patellar tendon with achilles tendon allograft following total knee arthroplasty. J Bone Joint Surg Am. 2002;84(8):1354-1361.
13. Lahav A, Burks RT, Scholl MD. Allograft reconstruction of the patellar tendon: 12-year follow-up. Am J Orthop. 2004;33(12):623-624.
14. Yoo JH, Chang JD, Seo YJ, Baek SW. Reconstruction of a patellar tendon with Achilles tendon allograft for severe patellar infera--a case report. Knee. 2011;18(5):350-353.
15. Saldua NS, Mazurek MT. Procedure 37. Quadriceps and patellar tendon repair. In: Reider B, Terry MA, Provencher MT, eds. Operative Techniques: Sports Medicine Surgery. Philadelphia, PA: Saunders (Elsevier); 2010:623-640.
16. Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91(1):4-15.
17. Ibrahim SAR, Ahmad FHF, Salah M, Al Misfer ARK, Ghaffer SA, Khirat S. Surgical management of traumatic knee dislocation. Arthroscopy. 2008;24(2):178-187.
18. Dominkus M, Sabeti M, Toma C, Abdolvahab F, Trieb K, Kotz RI. Reconstructing the extensor apparatus with a new polyester ligament. Clin Orthop Relat Res. 2006;453:328-334.
19. Naim S, Gougoulias N, Griffiths D. Patellar tendon reconstruction using LARS ligament: surgical technique and case report. Strategies Trauma Limb Reconstr. 2011;6(1):39-41.
Intra-Articular Dislocation of the Patella With Associated Hoffa Fracture in a Skeletally Immature Patient
In 1887, Midelfart1 first reported on an intra-articular dislocation of the patella, and since then approximately 50 cases have been reported in the worldwide literature.2 Also known as an inferior patellar dislocation, these rare traumatic events occur when the patella dislocates intra-articularly. Because the patella commonly rotates about its horizontal axis, the articular surface is facing proximally or distally. The patella becomes lodged within the trochlea and locks the knee joint. Most cases described in the literature involved adolescent boys, with the patella difficult to reduce. Most patients required open reduction, while those who underwent successful closed reduction often needed general anesthesia.3
Similarly, coronal shear fractures of the femoral condyle (ie, Hoffa fractures) are an uncommon fracture pattern typically seen in adults. These fractures are even more infrequent in skeletally immature patients, with fewer than 5 cases documented in the literature.4-7 In our case report, we present a 14-year-old boy with a coronal shear fracture of the femoral condyle associated with an intra-articular patellar dislocation. To our knowledge, this constellation of injuries has not been reported. Additionally, closed reduction of the patella was successful after intra-articular lidocaine injection, without the need for sedation or general anesthesia. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy presented to our institution after sustaining a direct blow to his left knee. The injury occurred as he jumped and landed on a flexed knee while playing with friends. The patient was unable to ambulate after the injury, and his left knee was locked in a slightly flexed position. Examination in the emergency department showed the knee to be held in approximately 60º of flexion, with an obvious bony prominence noted anteriorly over the femoral condyles. The patient was unable to perform a straight leg raise or any active range of motion (ROM) at the knee. Radiographs performed with the knee maintained in flexion confirmed that the patella was displaced into the knee joint and was rotated with the articular surface facing distally. Also noted was a coronal shear fracture of the lateral femoral condyle (Figures 1A, 1B).
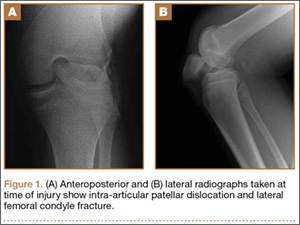
The patient received pain medication and an intra-articular lidocaine injection prior to a reduction attempt by the orthopedic resident. With the patient supine, the hip was gently flexed to relax the quadriceps muscle. As the knee was flexed up to 110º, the prominent patella was gripped between the thumb and fingers to gently free and elevate the patella out of the intercondylar notch.
After reduction, an immediate return of normal patellar contour and patellofemoral tracking was observed as the knee was gently extended. There was no obvious defect to the patellar or quadriceps tendons, and the patient was able to perform a straight-leg raise, confirming the integrity of the extensor mechanism. Radiographs performed after the reduction confirmed relocation of the patella in correct anatomic position, as well as a lateral femoral condyle fracture (Figures 2A, 2B). Magnetic resonance imaging (MRI) of the knee confirmed no full-thickness quadriceps or patellar tendon tear. A computed tomography (CT) scan of the knee showed a comminuted fracture of the lateral femoral condyle in the coronal plane, as well as multiple bone fragments within the joint (Figures 3A, 3B). The patient was placed in a bulky soft dressing and underwent open reduction and internal fixation of the fracture.
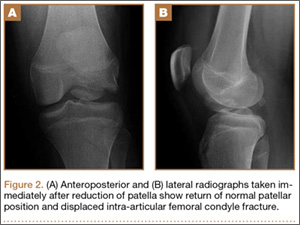
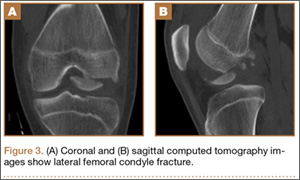
A 10-cm incision was made over the anterior aspect of the knee, and after dissection to the level of the retinaculum, a lateral parapatellar arthrotomy was performed. The patella was retracted medially to identify and free the fracture fragments. The fracture fragments were provisionally reduced and stabilized with three 0.065-in Kirschner wires. An area of osteochondral impaction proximal to the fracture was elevated and allograft bone was incorporated below the articular surface (Figures 4A, 4B). Rigid fixation of the fracture was achieved using 3 screws (2 Bio-Compression Screws [Arthrex Inc., Naples, Florida] and 1 Synthes cannulated screw [Synthes, West Chester, Pennsylvania]). The screws were placed in posteroanterior (PA) direction and inserted into the weight-bearing articular surface of the femoral condyle (Figures 4C, 4D). The screws were countersunk, and stable fixation with compression of the fracture was achieved. Reduction and screw position were verified with fluoroscopic views. The wound was closed in layers, and the patient was discharged home the next day.
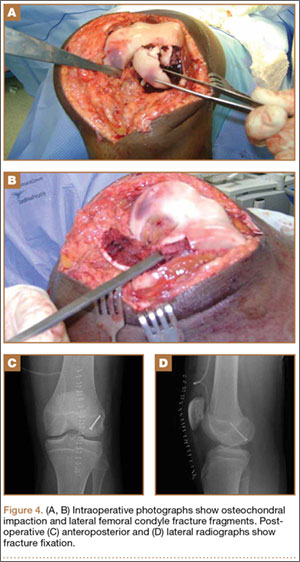
Postoperatively, the patient was non-weight-bearing on the affected limb with a hinged-knee brace to allow for knee ROM exercises immediately. He was also given a continuous passive motion device to maintain knee motion. At the 6-week mark, the patient’s fracture alignment appeared to be well-maintained and showed interval healing. Clinically, the patient was noted to have limited knee ROM. The decision was made to take the patient to the operating room primarily for a manipulation under anesthesia and resection of scar tissue from postoperative arthrofibrosis. Arthroscopic screw removal was also planned as a secondary procedure at the same time in order to prevent the possibility of chondral injury from screw migration. During the procedure, the patient was noted to have improved ROM from 5º to 85º premanipulation to 5º to 110º postoperatively. At 3 months after the initial injury, the patient was allowed to begin progressive weight-bearing on the left knee. At most recent follow-up, after 12 months, the patient was able to ambulate and bear weight on the left leg without pain. Plain radiographs show a well-healed fracture with no evidence of collapse of the femoral condyle (Figures 5A, 5B). His active ROM of the left knee was 5º to 110º without pain (Figures 5C, 5D).
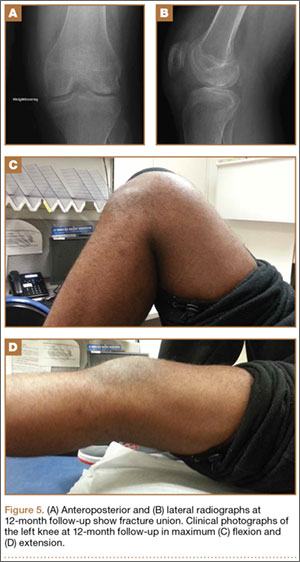
Discussion
In the vast majority of patellar dislocations, the patella dislocates laterally over the trochlear groove. Inferior, or intra-articular, dislocations of the patella are rare. The mechanism of injury is usually a blow onto the patella with a flexed knee. The 2 groups commonly involved are adolescent boys and the elderly.8,9 In young men, it is thought that lax patellar attachments place adolescents at higher risk for this type of injury.10-12 While patella fractures and frank extensor mechanism ruptures are uncommon in this age group, the same mechanism of injury can lead to stripping of the deep fibers of the patellar tendon from the superior pole of the patella.3,13 The intact superficial fibers of the tendon allow the patella to hinge and displace into the joint.14
Inferior dislocations of the patella are classified into 2 types based on the orientation of the articular surface and the presence of osteophytes.15 Type I inferior dislocations occur after a direct blow to a flexed knee forces the superior pole of the patella into the intercondylar notch. Type II dislocations are caused by osteophytes on the superior pole of the patella that become wedged in the intercondylar notch and dislocate the patella inferiorly. In type I dislocations, the patella is rotated in the horizontal plane and the articular surface often faces inferiorly, but type II dislocations do not involve rotation of the articular surface. Type II injuries are seen more commonly in the elderly.
Our patient was able to tolerate a closed reduction of the patella after an intra-articular lidocaine injection, and a successful reduction was achieved without great difficulty. However, the majority of reports describe the need for an open reduction of inferior patellar dislocations.3,8 When closed reductions were a success, they were performed under general anesthesia or conscious sedation.3 It is thought that the difficulty of reduction results from the tension of the quadriceps muscle pulling the patella superiorly into intercondylar notch.11,16 However, successful closed reduction may be more likely in patients with less patellar rotation and entrapment within the intercondylar notch, as well as in patients whose knee is near full extension at presentation.17-19 Successful closed reduction is also seen in elderly patients, where dislocation is generally caused by less forceful impact and held by osteophytes. In these patients, the knee is commonly held in extension.12,15,20-22
The fracture pattern seen in this case also shows a rare fracture in skeletally immature patients, with only a few case reports in the literature. Isolated coronal plane femur fractures account for 0.65% of all femur fractures and are usually seen in adults after high-energy trauma.23 In the skeletally immature, the fracture can occur with lower-energy mechanisms. The typical mechanism is thought to be a shearing force to the femur caused by an axial load to the knee in 90° or more of flexion.4,24 A CT scan is recommended for better identification of the fracture and to plan treatment.25,26 Because of their intra-articular nature and tenuous blood supply, Hoffa fractures tend to do poorly with nonoperative treatment and are prone to displacement and nonunion.27,28 The goal of operative treatment is to obtain anatomic reduction and rigid fixation. While operative fixation techniques are varied, screw fixation with multiple smaller diameter screws has equal pullout strength compared to larger screws and may minimize damage to the articular cartilage.29-31 By preserving blood supply to the fracture, and allowing for early active mobilization, operative treatment generally provides good long-term functional outcomes in these fracture patterns.24
Conclusion
We describe a case in which the patella of an adolescent boy dislocated inferiorly into the knee joint, with an associated coronal shear fracture of the lateral femoral condyle. To our knowledge, this constellation of injuries has not been reported. For this uncommon injury pattern, we recommend a sequential treatment algorithm to minimize morbidity. We recommend first attempting a closed reduction of the patella with adequate pain control to avoid the morbidity associated with general anesthesia. After a successful reduction, an advanced imaging study (eg, MRI) is advisable to assess for concomitant soft-tissue injuries and preoperative planning, if necessary. The mechanism of injury and force required to cause a patellar dislocation of this nature leaves a high likelihood of other injuries. When a fracture is noted on plain radiographs after reduction, a CT scan can provide important information for planning surgical fixation of the fracture. Even in a skeletally immature patient, the principle of direct reduction and stable interfragmentary fixation of an articular fracture is critical for long-term function, even after a significant trauma to the knee.
1. Midelfart V. En sjelden luxation of patella. Norsk Magazin for Laegevidenskaben. 1887;4:588.
2. Kramer DE, Simoni MK. Horizontal intra-articular patellar dislocation resulting in quadriceps avulsion and medial patellofemoral ligament tear: a case report. J Pediatr Orthop B. 2013;22(4):329-332.
3. van den Broek TA, Moll PJ. Horizontal rotation of the patella. A case report with review of the literature. Acta Orthop Scand. 1985;56(5):436-438.
4. Flanagin BA, Cruz AI, Medvecky MJ. Hoffa fracture in a 14-year-old. Orthopedics. 2011;34(2):138.
5. Strauss E, Nelson JM, Abdelwahab IF. Fracture of the lateral femoral condyle. A case report. Bull Hosp Jt Dis Orthop Inst. 1984;44(1):86-90.
6. Biau DJ, Schranz PJ. Transverse Hoffa’s or deep osteochondral fracture? An unusual fracture of the lateral femoral condyle in a child. Injury. 2005;36(7):862-865.
7. McDonough PW, Bernstein RM. Nonunion of a Hoffa fracture in a child. J Orthop Trauma. 2000;14(7):519-521.
8. Brady TA, Russell D. Interarticular horizontal dislocation of the patella. A case report. J Bone Joint Surg Am. 1965;47(7):1393-1396.
9. Yuguero M, Gonzalez JA, Carma A, Huguet J. Intra-articular patellar dislocation. Orthopedics. 2003;26(5):517-518.
10. Frangakis EK. Intra-articular dislocation of the patella. A case report. J Bone Joint Surg Am. 1974;56(2):423-424.
11. Nanda R, Yadav RS, Thakur M. Intra-articular dislocation of the patella. J Trauma. 2000;48(1):159-160.
12. Choudhary RK, Tice JW. Intra-articular dislocation of the patella with incomplete rotation--two case reports and a review of the literature. Knee. 2004;11(2):125-127.
13. Chatziantoniou I, Diakos G, Pantelelli M. Horizontal dislocation of the patella. Case report. EEXOT. 2008;59(2):112-114.
14. McHugh G, Ryan E, Cleary M, Kenny P, O’Flanagan S, Keogh P. Intra-articular dislocation of the patella. Case Rep Orthop. 2013;2013:535803.
15. Bankes MJ, Eastwood DM. Inferior dislocation of the patella in the degenerate knee. Injury. 2002;33(6):528-529.
16. Theodorides A, Guo S, Case R. Intra-articular dislocation of the patella: A case report and review of the literature. Injury Extra. 2010;41(10):103-105.
17. Dimentberg RA. Intra-articular dislocation of the patella: case report and literature review. Clin J Sport Med. 1997;7(2):126-128.
18. Morin WD, Steadman JR. Case report of a successful closed reduction without anesthesia. Clin Orthop. 1993(297):179-181.
19. Murakami Y. Intra-articular dislocation of the patella. A case report. Clin Orthop. 1982;171:137-139.
20. Joshi RP. Inferior dislocation of the patella. Injury. 1997;28(5-6):389-390.
21. Garner JP, Pike JM, George CD. Intra-articular dislocation of the patella: two cases and literature review. J Trauma. 1999;47(4):780-783.
22. McCarthy TA, Quinn B, Pegum JM. Inferior dislocation of the patella: an unusual cause of a locked knee. Ir J Med Sci. 2001;170(3):209-210.
23. Manfredini M, Gildone A, Ferrante R, Bernasconi S, Massari L. Unicondylar femoral fractures: therapeutic strategy and long-term results. A review of 23 patients. Acta Orthop Belg. 2001;67(2):132-138.
24. Holmes SM, Bomback D, Baumgaertner MR. Coronal fractures of the femoral condyle: a brief report of five cases. J Orthop Trauma. 2004;18(5):316-319.
25. Nork SE, Segina DN, Aflatoon K, et al. The association between supracondylar-intercondylar distal femoral fractures and coronal plane fractures. J Bone Joint Surg Am. 2005;87(3):564-569.
26. Allmann KH, Altehoefer C, Wildanger G, et al. Hoffa fracture--a radiologic diagnostic approach. J Belge Radiol. 1996;79(5):201-202.
27. Oztürk A, Ozkan Y, Ozdemir RM. Nonunion of a Hoffa fracture in an adult. Chir Organi Mov. 2009;93(3):183-185.
28. Lewis SL, Pozo JL, Muirhead-Allwood WF. Coronal fractures of the lateral femoral condyle. J Bone Joint Surg Br. 1989;71(1):118-120.
29. Arastu MH, Kokke MC, Duffy PJ, Korley RE, Buckley RE. Coronal plane partial articular fractures of the distal femoral condyle: current concepts in management. Bone Joint J. 2013;95-B(9):1165-1171.
30. Westmoreland GL, McLaurin TM, Hutton WC. Screw pullout strength: a biomechanical comparison of large-fragment and small-fragment fixation in the tibial plateau. J Orthop Trauma. 2002;16(3):178-181.
31. Jarit GJ, Kummer FJ, Gibber MJ, Egol KA. A mechanical evaluation of two fixation methods using cancellous screws for coronal fractures of the lateral condyle of the distal femur (OTA type 33B). J Orthop Trauma. 2006;20(4):273-276.
In 1887, Midelfart1 first reported on an intra-articular dislocation of the patella, and since then approximately 50 cases have been reported in the worldwide literature.2 Also known as an inferior patellar dislocation, these rare traumatic events occur when the patella dislocates intra-articularly. Because the patella commonly rotates about its horizontal axis, the articular surface is facing proximally or distally. The patella becomes lodged within the trochlea and locks the knee joint. Most cases described in the literature involved adolescent boys, with the patella difficult to reduce. Most patients required open reduction, while those who underwent successful closed reduction often needed general anesthesia.3
Similarly, coronal shear fractures of the femoral condyle (ie, Hoffa fractures) are an uncommon fracture pattern typically seen in adults. These fractures are even more infrequent in skeletally immature patients, with fewer than 5 cases documented in the literature.4-7 In our case report, we present a 14-year-old boy with a coronal shear fracture of the femoral condyle associated with an intra-articular patellar dislocation. To our knowledge, this constellation of injuries has not been reported. Additionally, closed reduction of the patella was successful after intra-articular lidocaine injection, without the need for sedation or general anesthesia. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy presented to our institution after sustaining a direct blow to his left knee. The injury occurred as he jumped and landed on a flexed knee while playing with friends. The patient was unable to ambulate after the injury, and his left knee was locked in a slightly flexed position. Examination in the emergency department showed the knee to be held in approximately 60º of flexion, with an obvious bony prominence noted anteriorly over the femoral condyles. The patient was unable to perform a straight leg raise or any active range of motion (ROM) at the knee. Radiographs performed with the knee maintained in flexion confirmed that the patella was displaced into the knee joint and was rotated with the articular surface facing distally. Also noted was a coronal shear fracture of the lateral femoral condyle (Figures 1A, 1B).
The patient received pain medication and an intra-articular lidocaine injection prior to a reduction attempt by the orthopedic resident. With the patient supine, the hip was gently flexed to relax the quadriceps muscle. As the knee was flexed up to 110º, the prominent patella was gripped between the thumb and fingers to gently free and elevate the patella out of the intercondylar notch.
After reduction, an immediate return of normal patellar contour and patellofemoral tracking was observed as the knee was gently extended. There was no obvious defect to the patellar or quadriceps tendons, and the patient was able to perform a straight-leg raise, confirming the integrity of the extensor mechanism. Radiographs performed after the reduction confirmed relocation of the patella in correct anatomic position, as well as a lateral femoral condyle fracture (Figures 2A, 2B). Magnetic resonance imaging (MRI) of the knee confirmed no full-thickness quadriceps or patellar tendon tear. A computed tomography (CT) scan of the knee showed a comminuted fracture of the lateral femoral condyle in the coronal plane, as well as multiple bone fragments within the joint (Figures 3A, 3B). The patient was placed in a bulky soft dressing and underwent open reduction and internal fixation of the fracture.
A 10-cm incision was made over the anterior aspect of the knee, and after dissection to the level of the retinaculum, a lateral parapatellar arthrotomy was performed. The patella was retracted medially to identify and free the fracture fragments. The fracture fragments were provisionally reduced and stabilized with three 0.065-in Kirschner wires. An area of osteochondral impaction proximal to the fracture was elevated and allograft bone was incorporated below the articular surface (Figures 4A, 4B). Rigid fixation of the fracture was achieved using 3 screws (2 Bio-Compression Screws [Arthrex Inc., Naples, Florida] and 1 Synthes cannulated screw [Synthes, West Chester, Pennsylvania]). The screws were placed in posteroanterior (PA) direction and inserted into the weight-bearing articular surface of the femoral condyle (Figures 4C, 4D). The screws were countersunk, and stable fixation with compression of the fracture was achieved. Reduction and screw position were verified with fluoroscopic views. The wound was closed in layers, and the patient was discharged home the next day.
Postoperatively, the patient was non-weight-bearing on the affected limb with a hinged-knee brace to allow for knee ROM exercises immediately. He was also given a continuous passive motion device to maintain knee motion. At the 6-week mark, the patient’s fracture alignment appeared to be well-maintained and showed interval healing. Clinically, the patient was noted to have limited knee ROM. The decision was made to take the patient to the operating room primarily for a manipulation under anesthesia and resection of scar tissue from postoperative arthrofibrosis. Arthroscopic screw removal was also planned as a secondary procedure at the same time in order to prevent the possibility of chondral injury from screw migration. During the procedure, the patient was noted to have improved ROM from 5º to 85º premanipulation to 5º to 110º postoperatively. At 3 months after the initial injury, the patient was allowed to begin progressive weight-bearing on the left knee. At most recent follow-up, after 12 months, the patient was able to ambulate and bear weight on the left leg without pain. Plain radiographs show a well-healed fracture with no evidence of collapse of the femoral condyle (Figures 5A, 5B). His active ROM of the left knee was 5º to 110º without pain (Figures 5C, 5D).
Discussion
In the vast majority of patellar dislocations, the patella dislocates laterally over the trochlear groove. Inferior, or intra-articular, dislocations of the patella are rare. The mechanism of injury is usually a blow onto the patella with a flexed knee. The 2 groups commonly involved are adolescent boys and the elderly.8,9 In young men, it is thought that lax patellar attachments place adolescents at higher risk for this type of injury.10-12 While patella fractures and frank extensor mechanism ruptures are uncommon in this age group, the same mechanism of injury can lead to stripping of the deep fibers of the patellar tendon from the superior pole of the patella.3,13 The intact superficial fibers of the tendon allow the patella to hinge and displace into the joint.14
Inferior dislocations of the patella are classified into 2 types based on the orientation of the articular surface and the presence of osteophytes.15 Type I inferior dislocations occur after a direct blow to a flexed knee forces the superior pole of the patella into the intercondylar notch. Type II dislocations are caused by osteophytes on the superior pole of the patella that become wedged in the intercondylar notch and dislocate the patella inferiorly. In type I dislocations, the patella is rotated in the horizontal plane and the articular surface often faces inferiorly, but type II dislocations do not involve rotation of the articular surface. Type II injuries are seen more commonly in the elderly.
Our patient was able to tolerate a closed reduction of the patella after an intra-articular lidocaine injection, and a successful reduction was achieved without great difficulty. However, the majority of reports describe the need for an open reduction of inferior patellar dislocations.3,8 When closed reductions were a success, they were performed under general anesthesia or conscious sedation.3 It is thought that the difficulty of reduction results from the tension of the quadriceps muscle pulling the patella superiorly into intercondylar notch.11,16 However, successful closed reduction may be more likely in patients with less patellar rotation and entrapment within the intercondylar notch, as well as in patients whose knee is near full extension at presentation.17-19 Successful closed reduction is also seen in elderly patients, where dislocation is generally caused by less forceful impact and held by osteophytes. In these patients, the knee is commonly held in extension.12,15,20-22
The fracture pattern seen in this case also shows a rare fracture in skeletally immature patients, with only a few case reports in the literature. Isolated coronal plane femur fractures account for 0.65% of all femur fractures and are usually seen in adults after high-energy trauma.23 In the skeletally immature, the fracture can occur with lower-energy mechanisms. The typical mechanism is thought to be a shearing force to the femur caused by an axial load to the knee in 90° or more of flexion.4,24 A CT scan is recommended for better identification of the fracture and to plan treatment.25,26 Because of their intra-articular nature and tenuous blood supply, Hoffa fractures tend to do poorly with nonoperative treatment and are prone to displacement and nonunion.27,28 The goal of operative treatment is to obtain anatomic reduction and rigid fixation. While operative fixation techniques are varied, screw fixation with multiple smaller diameter screws has equal pullout strength compared to larger screws and may minimize damage to the articular cartilage.29-31 By preserving blood supply to the fracture, and allowing for early active mobilization, operative treatment generally provides good long-term functional outcomes in these fracture patterns.24
Conclusion
We describe a case in which the patella of an adolescent boy dislocated inferiorly into the knee joint, with an associated coronal shear fracture of the lateral femoral condyle. To our knowledge, this constellation of injuries has not been reported. For this uncommon injury pattern, we recommend a sequential treatment algorithm to minimize morbidity. We recommend first attempting a closed reduction of the patella with adequate pain control to avoid the morbidity associated with general anesthesia. After a successful reduction, an advanced imaging study (eg, MRI) is advisable to assess for concomitant soft-tissue injuries and preoperative planning, if necessary. The mechanism of injury and force required to cause a patellar dislocation of this nature leaves a high likelihood of other injuries. When a fracture is noted on plain radiographs after reduction, a CT scan can provide important information for planning surgical fixation of the fracture. Even in a skeletally immature patient, the principle of direct reduction and stable interfragmentary fixation of an articular fracture is critical for long-term function, even after a significant trauma to the knee.
In 1887, Midelfart1 first reported on an intra-articular dislocation of the patella, and since then approximately 50 cases have been reported in the worldwide literature.2 Also known as an inferior patellar dislocation, these rare traumatic events occur when the patella dislocates intra-articularly. Because the patella commonly rotates about its horizontal axis, the articular surface is facing proximally or distally. The patella becomes lodged within the trochlea and locks the knee joint. Most cases described in the literature involved adolescent boys, with the patella difficult to reduce. Most patients required open reduction, while those who underwent successful closed reduction often needed general anesthesia.3
Similarly, coronal shear fractures of the femoral condyle (ie, Hoffa fractures) are an uncommon fracture pattern typically seen in adults. These fractures are even more infrequent in skeletally immature patients, with fewer than 5 cases documented in the literature.4-7 In our case report, we present a 14-year-old boy with a coronal shear fracture of the femoral condyle associated with an intra-articular patellar dislocation. To our knowledge, this constellation of injuries has not been reported. Additionally, closed reduction of the patella was successful after intra-articular lidocaine injection, without the need for sedation or general anesthesia. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy presented to our institution after sustaining a direct blow to his left knee. The injury occurred as he jumped and landed on a flexed knee while playing with friends. The patient was unable to ambulate after the injury, and his left knee was locked in a slightly flexed position. Examination in the emergency department showed the knee to be held in approximately 60º of flexion, with an obvious bony prominence noted anteriorly over the femoral condyles. The patient was unable to perform a straight leg raise or any active range of motion (ROM) at the knee. Radiographs performed with the knee maintained in flexion confirmed that the patella was displaced into the knee joint and was rotated with the articular surface facing distally. Also noted was a coronal shear fracture of the lateral femoral condyle (Figures 1A, 1B).
The patient received pain medication and an intra-articular lidocaine injection prior to a reduction attempt by the orthopedic resident. With the patient supine, the hip was gently flexed to relax the quadriceps muscle. As the knee was flexed up to 110º, the prominent patella was gripped between the thumb and fingers to gently free and elevate the patella out of the intercondylar notch.
After reduction, an immediate return of normal patellar contour and patellofemoral tracking was observed as the knee was gently extended. There was no obvious defect to the patellar or quadriceps tendons, and the patient was able to perform a straight-leg raise, confirming the integrity of the extensor mechanism. Radiographs performed after the reduction confirmed relocation of the patella in correct anatomic position, as well as a lateral femoral condyle fracture (Figures 2A, 2B). Magnetic resonance imaging (MRI) of the knee confirmed no full-thickness quadriceps or patellar tendon tear. A computed tomography (CT) scan of the knee showed a comminuted fracture of the lateral femoral condyle in the coronal plane, as well as multiple bone fragments within the joint (Figures 3A, 3B). The patient was placed in a bulky soft dressing and underwent open reduction and internal fixation of the fracture.
A 10-cm incision was made over the anterior aspect of the knee, and after dissection to the level of the retinaculum, a lateral parapatellar arthrotomy was performed. The patella was retracted medially to identify and free the fracture fragments. The fracture fragments were provisionally reduced and stabilized with three 0.065-in Kirschner wires. An area of osteochondral impaction proximal to the fracture was elevated and allograft bone was incorporated below the articular surface (Figures 4A, 4B). Rigid fixation of the fracture was achieved using 3 screws (2 Bio-Compression Screws [Arthrex Inc., Naples, Florida] and 1 Synthes cannulated screw [Synthes, West Chester, Pennsylvania]). The screws were placed in posteroanterior (PA) direction and inserted into the weight-bearing articular surface of the femoral condyle (Figures 4C, 4D). The screws were countersunk, and stable fixation with compression of the fracture was achieved. Reduction and screw position were verified with fluoroscopic views. The wound was closed in layers, and the patient was discharged home the next day.
Postoperatively, the patient was non-weight-bearing on the affected limb with a hinged-knee brace to allow for knee ROM exercises immediately. He was also given a continuous passive motion device to maintain knee motion. At the 6-week mark, the patient’s fracture alignment appeared to be well-maintained and showed interval healing. Clinically, the patient was noted to have limited knee ROM. The decision was made to take the patient to the operating room primarily for a manipulation under anesthesia and resection of scar tissue from postoperative arthrofibrosis. Arthroscopic screw removal was also planned as a secondary procedure at the same time in order to prevent the possibility of chondral injury from screw migration. During the procedure, the patient was noted to have improved ROM from 5º to 85º premanipulation to 5º to 110º postoperatively. At 3 months after the initial injury, the patient was allowed to begin progressive weight-bearing on the left knee. At most recent follow-up, after 12 months, the patient was able to ambulate and bear weight on the left leg without pain. Plain radiographs show a well-healed fracture with no evidence of collapse of the femoral condyle (Figures 5A, 5B). His active ROM of the left knee was 5º to 110º without pain (Figures 5C, 5D).
Discussion
In the vast majority of patellar dislocations, the patella dislocates laterally over the trochlear groove. Inferior, or intra-articular, dislocations of the patella are rare. The mechanism of injury is usually a blow onto the patella with a flexed knee. The 2 groups commonly involved are adolescent boys and the elderly.8,9 In young men, it is thought that lax patellar attachments place adolescents at higher risk for this type of injury.10-12 While patella fractures and frank extensor mechanism ruptures are uncommon in this age group, the same mechanism of injury can lead to stripping of the deep fibers of the patellar tendon from the superior pole of the patella.3,13 The intact superficial fibers of the tendon allow the patella to hinge and displace into the joint.14
Inferior dislocations of the patella are classified into 2 types based on the orientation of the articular surface and the presence of osteophytes.15 Type I inferior dislocations occur after a direct blow to a flexed knee forces the superior pole of the patella into the intercondylar notch. Type II dislocations are caused by osteophytes on the superior pole of the patella that become wedged in the intercondylar notch and dislocate the patella inferiorly. In type I dislocations, the patella is rotated in the horizontal plane and the articular surface often faces inferiorly, but type II dislocations do not involve rotation of the articular surface. Type II injuries are seen more commonly in the elderly.
Our patient was able to tolerate a closed reduction of the patella after an intra-articular lidocaine injection, and a successful reduction was achieved without great difficulty. However, the majority of reports describe the need for an open reduction of inferior patellar dislocations.3,8 When closed reductions were a success, they were performed under general anesthesia or conscious sedation.3 It is thought that the difficulty of reduction results from the tension of the quadriceps muscle pulling the patella superiorly into intercondylar notch.11,16 However, successful closed reduction may be more likely in patients with less patellar rotation and entrapment within the intercondylar notch, as well as in patients whose knee is near full extension at presentation.17-19 Successful closed reduction is also seen in elderly patients, where dislocation is generally caused by less forceful impact and held by osteophytes. In these patients, the knee is commonly held in extension.12,15,20-22
The fracture pattern seen in this case also shows a rare fracture in skeletally immature patients, with only a few case reports in the literature. Isolated coronal plane femur fractures account for 0.65% of all femur fractures and are usually seen in adults after high-energy trauma.23 In the skeletally immature, the fracture can occur with lower-energy mechanisms. The typical mechanism is thought to be a shearing force to the femur caused by an axial load to the knee in 90° or more of flexion.4,24 A CT scan is recommended for better identification of the fracture and to plan treatment.25,26 Because of their intra-articular nature and tenuous blood supply, Hoffa fractures tend to do poorly with nonoperative treatment and are prone to displacement and nonunion.27,28 The goal of operative treatment is to obtain anatomic reduction and rigid fixation. While operative fixation techniques are varied, screw fixation with multiple smaller diameter screws has equal pullout strength compared to larger screws and may minimize damage to the articular cartilage.29-31 By preserving blood supply to the fracture, and allowing for early active mobilization, operative treatment generally provides good long-term functional outcomes in these fracture patterns.24
Conclusion
We describe a case in which the patella of an adolescent boy dislocated inferiorly into the knee joint, with an associated coronal shear fracture of the lateral femoral condyle. To our knowledge, this constellation of injuries has not been reported. For this uncommon injury pattern, we recommend a sequential treatment algorithm to minimize morbidity. We recommend first attempting a closed reduction of the patella with adequate pain control to avoid the morbidity associated with general anesthesia. After a successful reduction, an advanced imaging study (eg, MRI) is advisable to assess for concomitant soft-tissue injuries and preoperative planning, if necessary. The mechanism of injury and force required to cause a patellar dislocation of this nature leaves a high likelihood of other injuries. When a fracture is noted on plain radiographs after reduction, a CT scan can provide important information for planning surgical fixation of the fracture. Even in a skeletally immature patient, the principle of direct reduction and stable interfragmentary fixation of an articular fracture is critical for long-term function, even after a significant trauma to the knee.
1. Midelfart V. En sjelden luxation of patella. Norsk Magazin for Laegevidenskaben. 1887;4:588.
2. Kramer DE, Simoni MK. Horizontal intra-articular patellar dislocation resulting in quadriceps avulsion and medial patellofemoral ligament tear: a case report. J Pediatr Orthop B. 2013;22(4):329-332.
3. van den Broek TA, Moll PJ. Horizontal rotation of the patella. A case report with review of the literature. Acta Orthop Scand. 1985;56(5):436-438.
4. Flanagin BA, Cruz AI, Medvecky MJ. Hoffa fracture in a 14-year-old. Orthopedics. 2011;34(2):138.
5. Strauss E, Nelson JM, Abdelwahab IF. Fracture of the lateral femoral condyle. A case report. Bull Hosp Jt Dis Orthop Inst. 1984;44(1):86-90.
6. Biau DJ, Schranz PJ. Transverse Hoffa’s or deep osteochondral fracture? An unusual fracture of the lateral femoral condyle in a child. Injury. 2005;36(7):862-865.
7. McDonough PW, Bernstein RM. Nonunion of a Hoffa fracture in a child. J Orthop Trauma. 2000;14(7):519-521.
8. Brady TA, Russell D. Interarticular horizontal dislocation of the patella. A case report. J Bone Joint Surg Am. 1965;47(7):1393-1396.
9. Yuguero M, Gonzalez JA, Carma A, Huguet J. Intra-articular patellar dislocation. Orthopedics. 2003;26(5):517-518.
10. Frangakis EK. Intra-articular dislocation of the patella. A case report. J Bone Joint Surg Am. 1974;56(2):423-424.
11. Nanda R, Yadav RS, Thakur M. Intra-articular dislocation of the patella. J Trauma. 2000;48(1):159-160.
12. Choudhary RK, Tice JW. Intra-articular dislocation of the patella with incomplete rotation--two case reports and a review of the literature. Knee. 2004;11(2):125-127.
13. Chatziantoniou I, Diakos G, Pantelelli M. Horizontal dislocation of the patella. Case report. EEXOT. 2008;59(2):112-114.
14. McHugh G, Ryan E, Cleary M, Kenny P, O’Flanagan S, Keogh P. Intra-articular dislocation of the patella. Case Rep Orthop. 2013;2013:535803.
15. Bankes MJ, Eastwood DM. Inferior dislocation of the patella in the degenerate knee. Injury. 2002;33(6):528-529.
16. Theodorides A, Guo S, Case R. Intra-articular dislocation of the patella: A case report and review of the literature. Injury Extra. 2010;41(10):103-105.
17. Dimentberg RA. Intra-articular dislocation of the patella: case report and literature review. Clin J Sport Med. 1997;7(2):126-128.
18. Morin WD, Steadman JR. Case report of a successful closed reduction without anesthesia. Clin Orthop. 1993(297):179-181.
19. Murakami Y. Intra-articular dislocation of the patella. A case report. Clin Orthop. 1982;171:137-139.
20. Joshi RP. Inferior dislocation of the patella. Injury. 1997;28(5-6):389-390.
21. Garner JP, Pike JM, George CD. Intra-articular dislocation of the patella: two cases and literature review. J Trauma. 1999;47(4):780-783.
22. McCarthy TA, Quinn B, Pegum JM. Inferior dislocation of the patella: an unusual cause of a locked knee. Ir J Med Sci. 2001;170(3):209-210.
23. Manfredini M, Gildone A, Ferrante R, Bernasconi S, Massari L. Unicondylar femoral fractures: therapeutic strategy and long-term results. A review of 23 patients. Acta Orthop Belg. 2001;67(2):132-138.
24. Holmes SM, Bomback D, Baumgaertner MR. Coronal fractures of the femoral condyle: a brief report of five cases. J Orthop Trauma. 2004;18(5):316-319.
25. Nork SE, Segina DN, Aflatoon K, et al. The association between supracondylar-intercondylar distal femoral fractures and coronal plane fractures. J Bone Joint Surg Am. 2005;87(3):564-569.
26. Allmann KH, Altehoefer C, Wildanger G, et al. Hoffa fracture--a radiologic diagnostic approach. J Belge Radiol. 1996;79(5):201-202.
27. Oztürk A, Ozkan Y, Ozdemir RM. Nonunion of a Hoffa fracture in an adult. Chir Organi Mov. 2009;93(3):183-185.
28. Lewis SL, Pozo JL, Muirhead-Allwood WF. Coronal fractures of the lateral femoral condyle. J Bone Joint Surg Br. 1989;71(1):118-120.
29. Arastu MH, Kokke MC, Duffy PJ, Korley RE, Buckley RE. Coronal plane partial articular fractures of the distal femoral condyle: current concepts in management. Bone Joint J. 2013;95-B(9):1165-1171.
30. Westmoreland GL, McLaurin TM, Hutton WC. Screw pullout strength: a biomechanical comparison of large-fragment and small-fragment fixation in the tibial plateau. J Orthop Trauma. 2002;16(3):178-181.
31. Jarit GJ, Kummer FJ, Gibber MJ, Egol KA. A mechanical evaluation of two fixation methods using cancellous screws for coronal fractures of the lateral condyle of the distal femur (OTA type 33B). J Orthop Trauma. 2006;20(4):273-276.
1. Midelfart V. En sjelden luxation of patella. Norsk Magazin for Laegevidenskaben. 1887;4:588.
2. Kramer DE, Simoni MK. Horizontal intra-articular patellar dislocation resulting in quadriceps avulsion and medial patellofemoral ligament tear: a case report. J Pediatr Orthop B. 2013;22(4):329-332.
3. van den Broek TA, Moll PJ. Horizontal rotation of the patella. A case report with review of the literature. Acta Orthop Scand. 1985;56(5):436-438.
4. Flanagin BA, Cruz AI, Medvecky MJ. Hoffa fracture in a 14-year-old. Orthopedics. 2011;34(2):138.
5. Strauss E, Nelson JM, Abdelwahab IF. Fracture of the lateral femoral condyle. A case report. Bull Hosp Jt Dis Orthop Inst. 1984;44(1):86-90.
6. Biau DJ, Schranz PJ. Transverse Hoffa’s or deep osteochondral fracture? An unusual fracture of the lateral femoral condyle in a child. Injury. 2005;36(7):862-865.
7. McDonough PW, Bernstein RM. Nonunion of a Hoffa fracture in a child. J Orthop Trauma. 2000;14(7):519-521.
8. Brady TA, Russell D. Interarticular horizontal dislocation of the patella. A case report. J Bone Joint Surg Am. 1965;47(7):1393-1396.
9. Yuguero M, Gonzalez JA, Carma A, Huguet J. Intra-articular patellar dislocation. Orthopedics. 2003;26(5):517-518.
10. Frangakis EK. Intra-articular dislocation of the patella. A case report. J Bone Joint Surg Am. 1974;56(2):423-424.
11. Nanda R, Yadav RS, Thakur M. Intra-articular dislocation of the patella. J Trauma. 2000;48(1):159-160.
12. Choudhary RK, Tice JW. Intra-articular dislocation of the patella with incomplete rotation--two case reports and a review of the literature. Knee. 2004;11(2):125-127.
13. Chatziantoniou I, Diakos G, Pantelelli M. Horizontal dislocation of the patella. Case report. EEXOT. 2008;59(2):112-114.
14. McHugh G, Ryan E, Cleary M, Kenny P, O’Flanagan S, Keogh P. Intra-articular dislocation of the patella. Case Rep Orthop. 2013;2013:535803.
15. Bankes MJ, Eastwood DM. Inferior dislocation of the patella in the degenerate knee. Injury. 2002;33(6):528-529.
16. Theodorides A, Guo S, Case R. Intra-articular dislocation of the patella: A case report and review of the literature. Injury Extra. 2010;41(10):103-105.
17. Dimentberg RA. Intra-articular dislocation of the patella: case report and literature review. Clin J Sport Med. 1997;7(2):126-128.
18. Morin WD, Steadman JR. Case report of a successful closed reduction without anesthesia. Clin Orthop. 1993(297):179-181.
19. Murakami Y. Intra-articular dislocation of the patella. A case report. Clin Orthop. 1982;171:137-139.
20. Joshi RP. Inferior dislocation of the patella. Injury. 1997;28(5-6):389-390.
21. Garner JP, Pike JM, George CD. Intra-articular dislocation of the patella: two cases and literature review. J Trauma. 1999;47(4):780-783.
22. McCarthy TA, Quinn B, Pegum JM. Inferior dislocation of the patella: an unusual cause of a locked knee. Ir J Med Sci. 2001;170(3):209-210.
23. Manfredini M, Gildone A, Ferrante R, Bernasconi S, Massari L. Unicondylar femoral fractures: therapeutic strategy and long-term results. A review of 23 patients. Acta Orthop Belg. 2001;67(2):132-138.
24. Holmes SM, Bomback D, Baumgaertner MR. Coronal fractures of the femoral condyle: a brief report of five cases. J Orthop Trauma. 2004;18(5):316-319.
25. Nork SE, Segina DN, Aflatoon K, et al. The association between supracondylar-intercondylar distal femoral fractures and coronal plane fractures. J Bone Joint Surg Am. 2005;87(3):564-569.
26. Allmann KH, Altehoefer C, Wildanger G, et al. Hoffa fracture--a radiologic diagnostic approach. J Belge Radiol. 1996;79(5):201-202.
27. Oztürk A, Ozkan Y, Ozdemir RM. Nonunion of a Hoffa fracture in an adult. Chir Organi Mov. 2009;93(3):183-185.
28. Lewis SL, Pozo JL, Muirhead-Allwood WF. Coronal fractures of the lateral femoral condyle. J Bone Joint Surg Br. 1989;71(1):118-120.
29. Arastu MH, Kokke MC, Duffy PJ, Korley RE, Buckley RE. Coronal plane partial articular fractures of the distal femoral condyle: current concepts in management. Bone Joint J. 2013;95-B(9):1165-1171.
30. Westmoreland GL, McLaurin TM, Hutton WC. Screw pullout strength: a biomechanical comparison of large-fragment and small-fragment fixation in the tibial plateau. J Orthop Trauma. 2002;16(3):178-181.
31. Jarit GJ, Kummer FJ, Gibber MJ, Egol KA. A mechanical evaluation of two fixation methods using cancellous screws for coronal fractures of the lateral condyle of the distal femur (OTA type 33B). J Orthop Trauma. 2006;20(4):273-276.
Primary Apocrine Adenocarcinoma of the Axilla
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
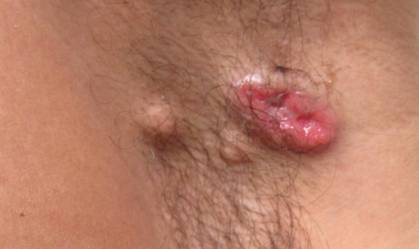
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
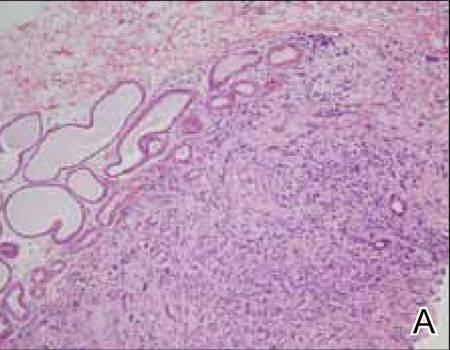
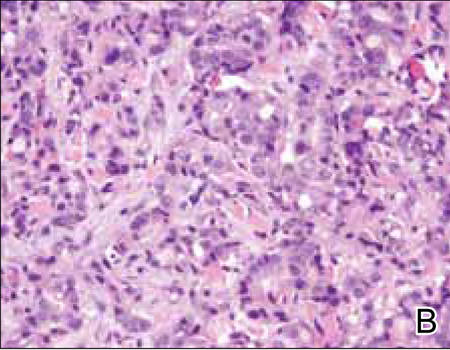
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
|
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
|
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
Practice Points
- Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy with metastatic potential.
It arises in areas of high apocrine gland density including the axillae and anogenital region. - Apocrine adenocarcinoma must be differentiated from various infections and cutaneous metastases from internal malignancies.
- Primary apocrine differentiation with invasion to adjacent tissue is a key histopathologic feature of AA.
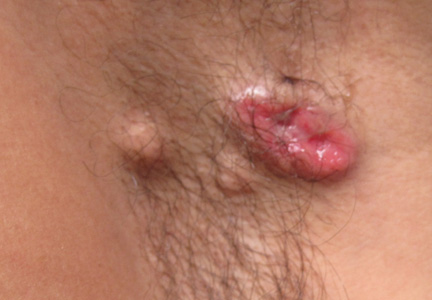
Significant response to lacosamide in a patient with severe chemotherapy-induced peripheral neuropathy
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Young Man With Headache, Confusion, and Hearing Loss
A 25-year-old male was found wandering naked in his room with the shower running after failing to come to work on a Monday morning. When found, he was able to talk and follow some commands but was confused about what was happening. He had experienced a right periorbital headache with nausea and vomiting for several days before admission. A computed tomography (CT) scan of the head at an outside hospital was negative.
Related: Pain, Anxiety, and Dementia: A Catastrophic Outcome
The patient had no history of tick or insect bites, skin rash, chest pain, shortness of breath, trauma, or illicit drug or alcohol use. He smoked a half pack of cigarettes per day. The patient had spent time in the military in the Middle East and North Africa 3 years earlier and had 3 tattoos. Over the past few months, he had been noted to be more aggressive, including having gotten into a bar fight. His past medical history was significant only for documented hearing loss in the right ear per reports from the air base.
On examination, the patient’s temperature was 97.3°F, heart rate 47 bpm, respirations 20 breathes/min, and blood pressure 97/60 mm Hg. His neck was supple, and the remainder of the general examination was normal. The neurologic examination revealed the patient to be awake and alert but apathetic, irritable, and refusing to talk after a few minutes. He was slow to respond, spoke loudly, and had a poor attention span. The patient was disoriented to time and place and remembered 0/5 objects at 5 minutes.
Blood tests revealed 12,500/μL white blood cell (WBC) count (increased mononuclear cells, 13.7%); hemoglobin, 14.6 g/dL; platelet count 194,000/μL; and hematocrit, 43.6%. Electrolytes, general chemistries, vitamin B12, thyroid-stimulating hormone, copper, erythrocyte sedimentation rate, and urinalysis were all normal. The CT head scan was normal, and the urine drug screen and alcohol levels were negative.
The initial audiologic evaluation revealed absent acoustic reflexes bilaterally at 500 Hz, 1 KHz, 2 KHz, and 4 KHz. The brain stem auditory evoked potentials showed no replicable waveforms from the right ear and a wave I present in the left ear with no other replicable waveforms.
A very broad differential diagnosis was considered at this point (Table 1). Lumbar puncture was performed with an opening pressure of 17.5 cm H2O. There were 10 WBC/μL with 12% segmented polymorphonuclear cells and 83% lymphocytes, 30 red blood cells/μL, glucose of 71 mg/dL, and protein of 221 mg/dL. The Venereal Disease Research Laboratory Test was nonreactive; cryptococcal antigen, acid-fast stain, and bacterial and fungal cultures were negative. The electroencephalogram (EEG) showed mild diffuse slowing with frontal intermittent rhythmic delta activity. The magnetic resonance imaging (MRI) was significant for leptomeningeal and pachymeningeal enhancement with a small area of restricted diffusion in the splenium of the corpus callosum (Figure 1). Other cerebral spinal fluid and serum studies were negative or nonreactive (Table 2).

The patient completed a 2-week course of ceftriaxone 1 gram q 12 hours, vancomycin 1,000 mg q 12 hours, acyclovir 700 mg q 8 hours, and doxycycline 100 mg bid without any notable clinical change. A repeat lumbar puncture was acellular and had a protein of 254 mg/dL.
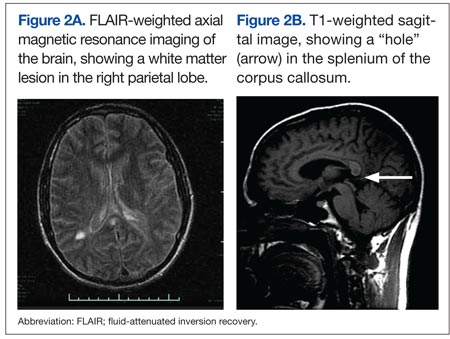
Two days later he worsened, becoming more withdrawn, unable to speak, irritable, and unwilling to be examined. He refused to get out of bed even with his family members present. A repeat MRI at this time showed continued meningeal enhancement, enlargement of the previously seen corpus callosal lesion, a new white matter lesion in the right parietal region, and a dark hole in the corpus callosum on the sagittal T1 image (Figures 2A and 2B). Audiometric testing showed profound hearing loss at low and high frequencies, with severe loss at middle frequencies in both ears.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Diagnosis
At this point, a diagnosis must explain encephalitis/encephalopathy, hearing loss, and MRI findings of meningeal enhancement and lesions in the corpus callosum and right parietal white matter. The main differential at this point included acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS), infection, and vasculitis/vasculopathy (especially primary central nervous system vasculitis). Acute disseminated encephalomyelitis usually has large, asymmetric lesions in the subcortical white matter and gray white junction, with corpus callosal lesions being unusual. Meningeal enhancement is very rare, and hearing loss would be unusual as well.1
Encephalopathy would be unusual in MS and if seen is usually associated with large confluent lesions (the Marburg variant). Meningeal enhancement would be rare on MRI, and the location of the corpus callosal lesion as shown on the T1- sagittal MRI would be atypical for both MS and ADEM (Figure 2B). Hearing loss has been described in MS, with a 4% to 5% incidence, often as the first manifestation and usually with full recovery.2 With the extensive evaluation and treatment in this case, infection was unlikely at this point. Primary central nervous system vasculitis remained a definite possibility and could explain most of the findings. However, there have been no reports of hearing loss or corpus callosal lesions in the literature with this latter condition.
The presence of encephalopathy and hearing loss, in addition to the location of the corpus callosal lesion as demonstrated on the sagittal T1-weighted MRI (Figure 2B) suggested the need for an ophthalmologic consult with dilated retinoscopy and fluorescein angiography (Figure 3). A retinal examination showed branch retinal artery occlusions with cotton wool spots (infarctions). Fluorescein angiography showed branch retinal artery occlusions and arteriolar wall hyperfluoresence in one area. This demonstrated the final feature of the triad of encephalopathy, hearing loss, and branch retinal artery occlusions, confirming the diagnosis of Susac syndrome (SS).
Discussion and Literature Review
The patient was treated with a 3-day course of IV methylprednisolone 1 g daily for 3 days, followed by oral prednisone 60 mg daily for 1 week, followed by a slow taper thereafter. Both his cognition and behavior improved by the second day of treatment, and this continued during his hospital stay. After a short stay in the rehabilitation unit, he was transferred to a facility closer to his home. Mental status improved almost to baseline, but he got minimal if any improvement in his hearing function. Despite the branch retinal occlusions, he had no noticeable deficit in his visual function.
John O. Susac, MD, first described 2 women with a triad of encephalopathy, hearing loss, and branch retinal artery occlusions as a syndrome that subsequently was named after him.3 The syndrome most frequently affects women aged 20 to 40 years. Headaches consistent with migraines occur at onset in a majority of patients.4 Encephalopathy may be acute or subacute and mild to severe. Symptoms can include mood changes, personality change, bizarre behavior, hallucinations, memory and cognitive difficulties, ataxia, seizures, corticospinal tract signs, and myoclonus.5,6 The retinopathy may cause scintillating scotomata or segmental loss of vision but may also be asymptomatic due to occlusions in very distal, branch arteries. Hearing loss may be acute and severe or insidious and mild. Audiometry shows low-to-mid frequency hearing loss.
Related: Infliximab-Induced Complications
Hearing loss is usually permanent and, if severe, may require a cochlear implant.7 The disease course is variable and unpredictable. It may be monophasic, lasting under 2 years. This is often the case if encephalopathy occurs in the first 2 years. Susac syndrome can also have a polycyclic course, with remissions lasting up to 18 years. A chronic continuous course has also been described.8 All 3 components of the triad are not always present, and those without encephalopathy are more likely to have a polycyclic or chronic continuous course. The differential diagnosis is broad, as in the present case.
A cerebral spinal fluid evaluation often shows an elevated protein of 100 to 3,000 mg/dL, a mild lymphocytic pleocytosis (5-30 cells/mm), and oligoclonal bands may be present. Antiendothelial antibodies are present in the serum but not specific (also seen in systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and juvenile dermatomyositis).8 The EEG usually shows diffuse slowing. The MRI is almost always abnormal, and studies have shown virtually 100% have corpus callosal lesions. These occur in the central region of the corpus callosum, consistent with infarction. Demyelinating lesions, with MS or ADEM, on the other hand, tend to occur on the inferior surface of the corpus callosum. If SS is suspected, a sagittal fluid- attenuated inversion recovery (FLAIR) MRI should be obtained to look for these changes. About one-third or more of MRIs show leptomeningeal enhancement, and other lesions can be found scattered throughout the white matter, cerebellum, brain stem, and gray matter.9
Because relatively few cases have been described, SS etiology remains obscure at this time. The disease has an affinity for small precapillary arterioles, of > 100 μm in diameter. The pathology shows necrosis and inflammatory changes of the endothelial cells, making them the primary site of the immune attack. This immune-mediated injury leads to narrowing and occlusion of the microvasculature, with resulting ischemia of the brain, retina, and cochlea. This pathology is very similar to that of juvenile dermatomyositis, which involves muscle, skin, and the gastrointestinal tract.10
Treatment approaches are based on treatments for juvenile dermatomyositis. It is suggested that the patient be given pulse methylprednisolone therapy of 1 g per day for 3 days followed by prednisone 60 mg to 80 mg per day for 4 weeks. Newer recommendations suggest giving IV immunoglobulin in the first week as well, followed by additional courses every month for 6 months. Cyclophosphamide or mycophenolate mofetil should be considered for long-term treatment with consideration of etanercept, cyclosporine, or rituximab in those who fail to respond.10 Aggressive treatment is suggested, because this is a self-limiting disorder, but the deficits tend to be permanent.
Related: Rituximab and Primary Sjögren Syndrome
This patient was atypical, because SS primarily affects young females. Review of the literature indicates that men account for about 25% of patients.8 The presentation, however, was not unusual and demonstrated the difficulty in making this diagnosis. In this patient with encephalopathy, the unusual feature was hearing loss, but it must be kept in mind that both hearing loss and visual changes can be difficult to identify in a confused patient. Brain stem auditory evoked potentials may be helpful in investigating hearing loss in noncooperative patients. An MRI may show centrally located corpus callosal lesions. If SS is suspected, sagittal FLAIR images, which often are not routinely done, should be obtained.
The most helpful evaluation is a dilated direct retinoscopy, which will usually show the branch retinal artery occlusions, and if not, fluorescein angiography will usually show a change. The presence of Gass plaques, yellow-white retinal arterial wall plaques from lipid deposition into the damaged arterial wall, with hyperfluoresence on fluorescein angiography is considered pathognomonic of SS.8 Establishing the diagnosis of SS as soon as possible is critical, because early treatment may lessen the degree of permanent disability.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Eckstein C, Saidha S, Levy M. A differential diagnosis of central nervous system demyelination: beyond multiple sclerosis. J Neurol. 2012;259(5):801-816.
2. Hellmann MA, Steiner I, Mosberg-Galili R. Sudden sensorineural hearing loss in multiple sclerosis: clinical course and possible pathogenesis. Acta Neurol Scand. 2011;124(4):245-249.
3. Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology. 1979;29(3):313-316.
4. Papo T, Biousse V, Lehoang P, et al. Susac syndrome. Medicine (Baltimore). 1998;77(1):3-11.
5. Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44(4):591-593.
6. Hahn JS, Lannin WC, Sarwal MM. Microangiopathy of brain, retina, and inner ear (Susac’s syndrome) in an adolescent female presenting as acute disseminated encephalomyelitis. Pediatrics. 2004;114(1):276-281.
7. Roeser MM, Driscoll CL, Shallop JK, Gifford RH, Kasperbauer JL, Gluth MB. Susac syndrome—a report of cochlear implantation and review of otologic manifestations in twenty-three patients. Otol Neurotol. 2009;30(1):34-40.
8. Bitra RK, Eggenberger E. Review of Susac syndrome. Curr Opin Ophthalmol. 2011;22(6):472-476.
9. Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susac’s syndrome. Neurology. 2003;61(12): 1783-1787.
10. Rennebohm RM, Susac JO. Treatment of Susac’s syndrome. J Neurol Sci. 2007;257(1-2):215-220.
A 25-year-old male was found wandering naked in his room with the shower running after failing to come to work on a Monday morning. When found, he was able to talk and follow some commands but was confused about what was happening. He had experienced a right periorbital headache with nausea and vomiting for several days before admission. A computed tomography (CT) scan of the head at an outside hospital was negative.
Related: Pain, Anxiety, and Dementia: A Catastrophic Outcome
The patient had no history of tick or insect bites, skin rash, chest pain, shortness of breath, trauma, or illicit drug or alcohol use. He smoked a half pack of cigarettes per day. The patient had spent time in the military in the Middle East and North Africa 3 years earlier and had 3 tattoos. Over the past few months, he had been noted to be more aggressive, including having gotten into a bar fight. His past medical history was significant only for documented hearing loss in the right ear per reports from the air base.
On examination, the patient’s temperature was 97.3°F, heart rate 47 bpm, respirations 20 breathes/min, and blood pressure 97/60 mm Hg. His neck was supple, and the remainder of the general examination was normal. The neurologic examination revealed the patient to be awake and alert but apathetic, irritable, and refusing to talk after a few minutes. He was slow to respond, spoke loudly, and had a poor attention span. The patient was disoriented to time and place and remembered 0/5 objects at 5 minutes.
Blood tests revealed 12,500/μL white blood cell (WBC) count (increased mononuclear cells, 13.7%); hemoglobin, 14.6 g/dL; platelet count 194,000/μL; and hematocrit, 43.6%. Electrolytes, general chemistries, vitamin B12, thyroid-stimulating hormone, copper, erythrocyte sedimentation rate, and urinalysis were all normal. The CT head scan was normal, and the urine drug screen and alcohol levels were negative.
The initial audiologic evaluation revealed absent acoustic reflexes bilaterally at 500 Hz, 1 KHz, 2 KHz, and 4 KHz. The brain stem auditory evoked potentials showed no replicable waveforms from the right ear and a wave I present in the left ear with no other replicable waveforms.
A very broad differential diagnosis was considered at this point (Table 1). Lumbar puncture was performed with an opening pressure of 17.5 cm H2O. There were 10 WBC/μL with 12% segmented polymorphonuclear cells and 83% lymphocytes, 30 red blood cells/μL, glucose of 71 mg/dL, and protein of 221 mg/dL. The Venereal Disease Research Laboratory Test was nonreactive; cryptococcal antigen, acid-fast stain, and bacterial and fungal cultures were negative. The electroencephalogram (EEG) showed mild diffuse slowing with frontal intermittent rhythmic delta activity. The magnetic resonance imaging (MRI) was significant for leptomeningeal and pachymeningeal enhancement with a small area of restricted diffusion in the splenium of the corpus callosum (Figure 1). Other cerebral spinal fluid and serum studies were negative or nonreactive (Table 2).
The patient completed a 2-week course of ceftriaxone 1 gram q 12 hours, vancomycin 1,000 mg q 12 hours, acyclovir 700 mg q 8 hours, and doxycycline 100 mg bid without any notable clinical change. A repeat lumbar puncture was acellular and had a protein of 254 mg/dL.
Two days later he worsened, becoming more withdrawn, unable to speak, irritable, and unwilling to be examined. He refused to get out of bed even with his family members present. A repeat MRI at this time showed continued meningeal enhancement, enlargement of the previously seen corpus callosal lesion, a new white matter lesion in the right parietal region, and a dark hole in the corpus callosum on the sagittal T1 image (Figures 2A and 2B). Audiometric testing showed profound hearing loss at low and high frequencies, with severe loss at middle frequencies in both ears.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Diagnosis
At this point, a diagnosis must explain encephalitis/encephalopathy, hearing loss, and MRI findings of meningeal enhancement and lesions in the corpus callosum and right parietal white matter. The main differential at this point included acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS), infection, and vasculitis/vasculopathy (especially primary central nervous system vasculitis). Acute disseminated encephalomyelitis usually has large, asymmetric lesions in the subcortical white matter and gray white junction, with corpus callosal lesions being unusual. Meningeal enhancement is very rare, and hearing loss would be unusual as well.1
Encephalopathy would be unusual in MS and if seen is usually associated with large confluent lesions (the Marburg variant). Meningeal enhancement would be rare on MRI, and the location of the corpus callosal lesion as shown on the T1- sagittal MRI would be atypical for both MS and ADEM (Figure 2B). Hearing loss has been described in MS, with a 4% to 5% incidence, often as the first manifestation and usually with full recovery.2 With the extensive evaluation and treatment in this case, infection was unlikely at this point. Primary central nervous system vasculitis remained a definite possibility and could explain most of the findings. However, there have been no reports of hearing loss or corpus callosal lesions in the literature with this latter condition.
The presence of encephalopathy and hearing loss, in addition to the location of the corpus callosal lesion as demonstrated on the sagittal T1-weighted MRI (Figure 2B) suggested the need for an ophthalmologic consult with dilated retinoscopy and fluorescein angiography (Figure 3). A retinal examination showed branch retinal artery occlusions with cotton wool spots (infarctions). Fluorescein angiography showed branch retinal artery occlusions and arteriolar wall hyperfluoresence in one area. This demonstrated the final feature of the triad of encephalopathy, hearing loss, and branch retinal artery occlusions, confirming the diagnosis of Susac syndrome (SS).
Discussion and Literature Review
The patient was treated with a 3-day course of IV methylprednisolone 1 g daily for 3 days, followed by oral prednisone 60 mg daily for 1 week, followed by a slow taper thereafter. Both his cognition and behavior improved by the second day of treatment, and this continued during his hospital stay. After a short stay in the rehabilitation unit, he was transferred to a facility closer to his home. Mental status improved almost to baseline, but he got minimal if any improvement in his hearing function. Despite the branch retinal occlusions, he had no noticeable deficit in his visual function.
John O. Susac, MD, first described 2 women with a triad of encephalopathy, hearing loss, and branch retinal artery occlusions as a syndrome that subsequently was named after him.3 The syndrome most frequently affects women aged 20 to 40 years. Headaches consistent with migraines occur at onset in a majority of patients.4 Encephalopathy may be acute or subacute and mild to severe. Symptoms can include mood changes, personality change, bizarre behavior, hallucinations, memory and cognitive difficulties, ataxia, seizures, corticospinal tract signs, and myoclonus.5,6 The retinopathy may cause scintillating scotomata or segmental loss of vision but may also be asymptomatic due to occlusions in very distal, branch arteries. Hearing loss may be acute and severe or insidious and mild. Audiometry shows low-to-mid frequency hearing loss.
Related: Infliximab-Induced Complications
Hearing loss is usually permanent and, if severe, may require a cochlear implant.7 The disease course is variable and unpredictable. It may be monophasic, lasting under 2 years. This is often the case if encephalopathy occurs in the first 2 years. Susac syndrome can also have a polycyclic course, with remissions lasting up to 18 years. A chronic continuous course has also been described.8 All 3 components of the triad are not always present, and those without encephalopathy are more likely to have a polycyclic or chronic continuous course. The differential diagnosis is broad, as in the present case.
A cerebral spinal fluid evaluation often shows an elevated protein of 100 to 3,000 mg/dL, a mild lymphocytic pleocytosis (5-30 cells/mm), and oligoclonal bands may be present. Antiendothelial antibodies are present in the serum but not specific (also seen in systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and juvenile dermatomyositis).8 The EEG usually shows diffuse slowing. The MRI is almost always abnormal, and studies have shown virtually 100% have corpus callosal lesions. These occur in the central region of the corpus callosum, consistent with infarction. Demyelinating lesions, with MS or ADEM, on the other hand, tend to occur on the inferior surface of the corpus callosum. If SS is suspected, a sagittal fluid- attenuated inversion recovery (FLAIR) MRI should be obtained to look for these changes. About one-third or more of MRIs show leptomeningeal enhancement, and other lesions can be found scattered throughout the white matter, cerebellum, brain stem, and gray matter.9
Because relatively few cases have been described, SS etiology remains obscure at this time. The disease has an affinity for small precapillary arterioles, of > 100 μm in diameter. The pathology shows necrosis and inflammatory changes of the endothelial cells, making them the primary site of the immune attack. This immune-mediated injury leads to narrowing and occlusion of the microvasculature, with resulting ischemia of the brain, retina, and cochlea. This pathology is very similar to that of juvenile dermatomyositis, which involves muscle, skin, and the gastrointestinal tract.10
Treatment approaches are based on treatments for juvenile dermatomyositis. It is suggested that the patient be given pulse methylprednisolone therapy of 1 g per day for 3 days followed by prednisone 60 mg to 80 mg per day for 4 weeks. Newer recommendations suggest giving IV immunoglobulin in the first week as well, followed by additional courses every month for 6 months. Cyclophosphamide or mycophenolate mofetil should be considered for long-term treatment with consideration of etanercept, cyclosporine, or rituximab in those who fail to respond.10 Aggressive treatment is suggested, because this is a self-limiting disorder, but the deficits tend to be permanent.
Related: Rituximab and Primary Sjögren Syndrome
This patient was atypical, because SS primarily affects young females. Review of the literature indicates that men account for about 25% of patients.8 The presentation, however, was not unusual and demonstrated the difficulty in making this diagnosis. In this patient with encephalopathy, the unusual feature was hearing loss, but it must be kept in mind that both hearing loss and visual changes can be difficult to identify in a confused patient. Brain stem auditory evoked potentials may be helpful in investigating hearing loss in noncooperative patients. An MRI may show centrally located corpus callosal lesions. If SS is suspected, sagittal FLAIR images, which often are not routinely done, should be obtained.
The most helpful evaluation is a dilated direct retinoscopy, which will usually show the branch retinal artery occlusions, and if not, fluorescein angiography will usually show a change. The presence of Gass plaques, yellow-white retinal arterial wall plaques from lipid deposition into the damaged arterial wall, with hyperfluoresence on fluorescein angiography is considered pathognomonic of SS.8 Establishing the diagnosis of SS as soon as possible is critical, because early treatment may lessen the degree of permanent disability.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
A 25-year-old male was found wandering naked in his room with the shower running after failing to come to work on a Monday morning. When found, he was able to talk and follow some commands but was confused about what was happening. He had experienced a right periorbital headache with nausea and vomiting for several days before admission. A computed tomography (CT) scan of the head at an outside hospital was negative.
Related: Pain, Anxiety, and Dementia: A Catastrophic Outcome
The patient had no history of tick or insect bites, skin rash, chest pain, shortness of breath, trauma, or illicit drug or alcohol use. He smoked a half pack of cigarettes per day. The patient had spent time in the military in the Middle East and North Africa 3 years earlier and had 3 tattoos. Over the past few months, he had been noted to be more aggressive, including having gotten into a bar fight. His past medical history was significant only for documented hearing loss in the right ear per reports from the air base.
On examination, the patient’s temperature was 97.3°F, heart rate 47 bpm, respirations 20 breathes/min, and blood pressure 97/60 mm Hg. His neck was supple, and the remainder of the general examination was normal. The neurologic examination revealed the patient to be awake and alert but apathetic, irritable, and refusing to talk after a few minutes. He was slow to respond, spoke loudly, and had a poor attention span. The patient was disoriented to time and place and remembered 0/5 objects at 5 minutes.
Blood tests revealed 12,500/μL white blood cell (WBC) count (increased mononuclear cells, 13.7%); hemoglobin, 14.6 g/dL; platelet count 194,000/μL; and hematocrit, 43.6%. Electrolytes, general chemistries, vitamin B12, thyroid-stimulating hormone, copper, erythrocyte sedimentation rate, and urinalysis were all normal. The CT head scan was normal, and the urine drug screen and alcohol levels were negative.
The initial audiologic evaluation revealed absent acoustic reflexes bilaterally at 500 Hz, 1 KHz, 2 KHz, and 4 KHz. The brain stem auditory evoked potentials showed no replicable waveforms from the right ear and a wave I present in the left ear with no other replicable waveforms.
A very broad differential diagnosis was considered at this point (Table 1). Lumbar puncture was performed with an opening pressure of 17.5 cm H2O. There were 10 WBC/μL with 12% segmented polymorphonuclear cells and 83% lymphocytes, 30 red blood cells/μL, glucose of 71 mg/dL, and protein of 221 mg/dL. The Venereal Disease Research Laboratory Test was nonreactive; cryptococcal antigen, acid-fast stain, and bacterial and fungal cultures were negative. The electroencephalogram (EEG) showed mild diffuse slowing with frontal intermittent rhythmic delta activity. The magnetic resonance imaging (MRI) was significant for leptomeningeal and pachymeningeal enhancement with a small area of restricted diffusion in the splenium of the corpus callosum (Figure 1). Other cerebral spinal fluid and serum studies were negative or nonreactive (Table 2).
The patient completed a 2-week course of ceftriaxone 1 gram q 12 hours, vancomycin 1,000 mg q 12 hours, acyclovir 700 mg q 8 hours, and doxycycline 100 mg bid without any notable clinical change. A repeat lumbar puncture was acellular and had a protein of 254 mg/dL.
Two days later he worsened, becoming more withdrawn, unable to speak, irritable, and unwilling to be examined. He refused to get out of bed even with his family members present. A repeat MRI at this time showed continued meningeal enhancement, enlargement of the previously seen corpus callosal lesion, a new white matter lesion in the right parietal region, and a dark hole in the corpus callosum on the sagittal T1 image (Figures 2A and 2B). Audiometric testing showed profound hearing loss at low and high frequencies, with severe loss at middle frequencies in both ears.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Diagnosis
At this point, a diagnosis must explain encephalitis/encephalopathy, hearing loss, and MRI findings of meningeal enhancement and lesions in the corpus callosum and right parietal white matter. The main differential at this point included acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS), infection, and vasculitis/vasculopathy (especially primary central nervous system vasculitis). Acute disseminated encephalomyelitis usually has large, asymmetric lesions in the subcortical white matter and gray white junction, with corpus callosal lesions being unusual. Meningeal enhancement is very rare, and hearing loss would be unusual as well.1
Encephalopathy would be unusual in MS and if seen is usually associated with large confluent lesions (the Marburg variant). Meningeal enhancement would be rare on MRI, and the location of the corpus callosal lesion as shown on the T1- sagittal MRI would be atypical for both MS and ADEM (Figure 2B). Hearing loss has been described in MS, with a 4% to 5% incidence, often as the first manifestation and usually with full recovery.2 With the extensive evaluation and treatment in this case, infection was unlikely at this point. Primary central nervous system vasculitis remained a definite possibility and could explain most of the findings. However, there have been no reports of hearing loss or corpus callosal lesions in the literature with this latter condition.
The presence of encephalopathy and hearing loss, in addition to the location of the corpus callosal lesion as demonstrated on the sagittal T1-weighted MRI (Figure 2B) suggested the need for an ophthalmologic consult with dilated retinoscopy and fluorescein angiography (Figure 3). A retinal examination showed branch retinal artery occlusions with cotton wool spots (infarctions). Fluorescein angiography showed branch retinal artery occlusions and arteriolar wall hyperfluoresence in one area. This demonstrated the final feature of the triad of encephalopathy, hearing loss, and branch retinal artery occlusions, confirming the diagnosis of Susac syndrome (SS).
Discussion and Literature Review
The patient was treated with a 3-day course of IV methylprednisolone 1 g daily for 3 days, followed by oral prednisone 60 mg daily for 1 week, followed by a slow taper thereafter. Both his cognition and behavior improved by the second day of treatment, and this continued during his hospital stay. After a short stay in the rehabilitation unit, he was transferred to a facility closer to his home. Mental status improved almost to baseline, but he got minimal if any improvement in his hearing function. Despite the branch retinal occlusions, he had no noticeable deficit in his visual function.
John O. Susac, MD, first described 2 women with a triad of encephalopathy, hearing loss, and branch retinal artery occlusions as a syndrome that subsequently was named after him.3 The syndrome most frequently affects women aged 20 to 40 years. Headaches consistent with migraines occur at onset in a majority of patients.4 Encephalopathy may be acute or subacute and mild to severe. Symptoms can include mood changes, personality change, bizarre behavior, hallucinations, memory and cognitive difficulties, ataxia, seizures, corticospinal tract signs, and myoclonus.5,6 The retinopathy may cause scintillating scotomata or segmental loss of vision but may also be asymptomatic due to occlusions in very distal, branch arteries. Hearing loss may be acute and severe or insidious and mild. Audiometry shows low-to-mid frequency hearing loss.
Related: Infliximab-Induced Complications
Hearing loss is usually permanent and, if severe, may require a cochlear implant.7 The disease course is variable and unpredictable. It may be monophasic, lasting under 2 years. This is often the case if encephalopathy occurs in the first 2 years. Susac syndrome can also have a polycyclic course, with remissions lasting up to 18 years. A chronic continuous course has also been described.8 All 3 components of the triad are not always present, and those without encephalopathy are more likely to have a polycyclic or chronic continuous course. The differential diagnosis is broad, as in the present case.
A cerebral spinal fluid evaluation often shows an elevated protein of 100 to 3,000 mg/dL, a mild lymphocytic pleocytosis (5-30 cells/mm), and oligoclonal bands may be present. Antiendothelial antibodies are present in the serum but not specific (also seen in systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and juvenile dermatomyositis).8 The EEG usually shows diffuse slowing. The MRI is almost always abnormal, and studies have shown virtually 100% have corpus callosal lesions. These occur in the central region of the corpus callosum, consistent with infarction. Demyelinating lesions, with MS or ADEM, on the other hand, tend to occur on the inferior surface of the corpus callosum. If SS is suspected, a sagittal fluid- attenuated inversion recovery (FLAIR) MRI should be obtained to look for these changes. About one-third or more of MRIs show leptomeningeal enhancement, and other lesions can be found scattered throughout the white matter, cerebellum, brain stem, and gray matter.9
Because relatively few cases have been described, SS etiology remains obscure at this time. The disease has an affinity for small precapillary arterioles, of > 100 μm in diameter. The pathology shows necrosis and inflammatory changes of the endothelial cells, making them the primary site of the immune attack. This immune-mediated injury leads to narrowing and occlusion of the microvasculature, with resulting ischemia of the brain, retina, and cochlea. This pathology is very similar to that of juvenile dermatomyositis, which involves muscle, skin, and the gastrointestinal tract.10
Treatment approaches are based on treatments for juvenile dermatomyositis. It is suggested that the patient be given pulse methylprednisolone therapy of 1 g per day for 3 days followed by prednisone 60 mg to 80 mg per day for 4 weeks. Newer recommendations suggest giving IV immunoglobulin in the first week as well, followed by additional courses every month for 6 months. Cyclophosphamide or mycophenolate mofetil should be considered for long-term treatment with consideration of etanercept, cyclosporine, or rituximab in those who fail to respond.10 Aggressive treatment is suggested, because this is a self-limiting disorder, but the deficits tend to be permanent.
Related: Rituximab and Primary Sjögren Syndrome
This patient was atypical, because SS primarily affects young females. Review of the literature indicates that men account for about 25% of patients.8 The presentation, however, was not unusual and demonstrated the difficulty in making this diagnosis. In this patient with encephalopathy, the unusual feature was hearing loss, but it must be kept in mind that both hearing loss and visual changes can be difficult to identify in a confused patient. Brain stem auditory evoked potentials may be helpful in investigating hearing loss in noncooperative patients. An MRI may show centrally located corpus callosal lesions. If SS is suspected, sagittal FLAIR images, which often are not routinely done, should be obtained.
The most helpful evaluation is a dilated direct retinoscopy, which will usually show the branch retinal artery occlusions, and if not, fluorescein angiography will usually show a change. The presence of Gass plaques, yellow-white retinal arterial wall plaques from lipid deposition into the damaged arterial wall, with hyperfluoresence on fluorescein angiography is considered pathognomonic of SS.8 Establishing the diagnosis of SS as soon as possible is critical, because early treatment may lessen the degree of permanent disability.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Eckstein C, Saidha S, Levy M. A differential diagnosis of central nervous system demyelination: beyond multiple sclerosis. J Neurol. 2012;259(5):801-816.
2. Hellmann MA, Steiner I, Mosberg-Galili R. Sudden sensorineural hearing loss in multiple sclerosis: clinical course and possible pathogenesis. Acta Neurol Scand. 2011;124(4):245-249.
3. Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology. 1979;29(3):313-316.
4. Papo T, Biousse V, Lehoang P, et al. Susac syndrome. Medicine (Baltimore). 1998;77(1):3-11.
5. Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44(4):591-593.
6. Hahn JS, Lannin WC, Sarwal MM. Microangiopathy of brain, retina, and inner ear (Susac’s syndrome) in an adolescent female presenting as acute disseminated encephalomyelitis. Pediatrics. 2004;114(1):276-281.
7. Roeser MM, Driscoll CL, Shallop JK, Gifford RH, Kasperbauer JL, Gluth MB. Susac syndrome—a report of cochlear implantation and review of otologic manifestations in twenty-three patients. Otol Neurotol. 2009;30(1):34-40.
8. Bitra RK, Eggenberger E. Review of Susac syndrome. Curr Opin Ophthalmol. 2011;22(6):472-476.
9. Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susac’s syndrome. Neurology. 2003;61(12): 1783-1787.
10. Rennebohm RM, Susac JO. Treatment of Susac’s syndrome. J Neurol Sci. 2007;257(1-2):215-220.
1. Eckstein C, Saidha S, Levy M. A differential diagnosis of central nervous system demyelination: beyond multiple sclerosis. J Neurol. 2012;259(5):801-816.
2. Hellmann MA, Steiner I, Mosberg-Galili R. Sudden sensorineural hearing loss in multiple sclerosis: clinical course and possible pathogenesis. Acta Neurol Scand. 2011;124(4):245-249.
3. Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology. 1979;29(3):313-316.
4. Papo T, Biousse V, Lehoang P, et al. Susac syndrome. Medicine (Baltimore). 1998;77(1):3-11.
5. Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44(4):591-593.
6. Hahn JS, Lannin WC, Sarwal MM. Microangiopathy of brain, retina, and inner ear (Susac’s syndrome) in an adolescent female presenting as acute disseminated encephalomyelitis. Pediatrics. 2004;114(1):276-281.
7. Roeser MM, Driscoll CL, Shallop JK, Gifford RH, Kasperbauer JL, Gluth MB. Susac syndrome—a report of cochlear implantation and review of otologic manifestations in twenty-three patients. Otol Neurotol. 2009;30(1):34-40.
8. Bitra RK, Eggenberger E. Review of Susac syndrome. Curr Opin Ophthalmol. 2011;22(6):472-476.
9. Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susac’s syndrome. Neurology. 2003;61(12): 1783-1787.
10. Rennebohm RM, Susac JO. Treatment of Susac’s syndrome. J Neurol Sci. 2007;257(1-2):215-220.