User login
Molluscum Contagiosum Superimposed on Lymphangioma Circumscriptum
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
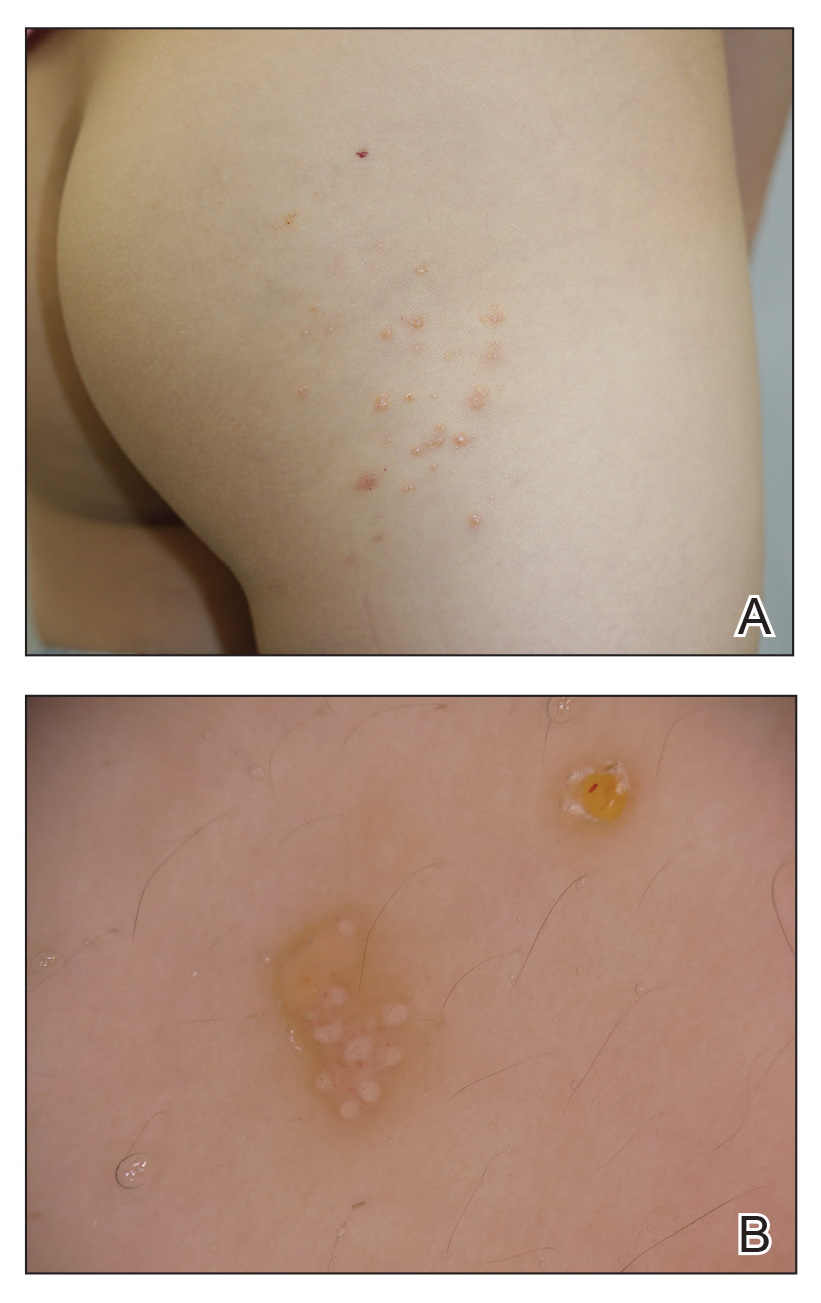
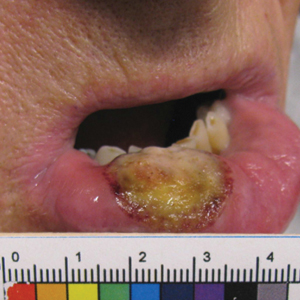
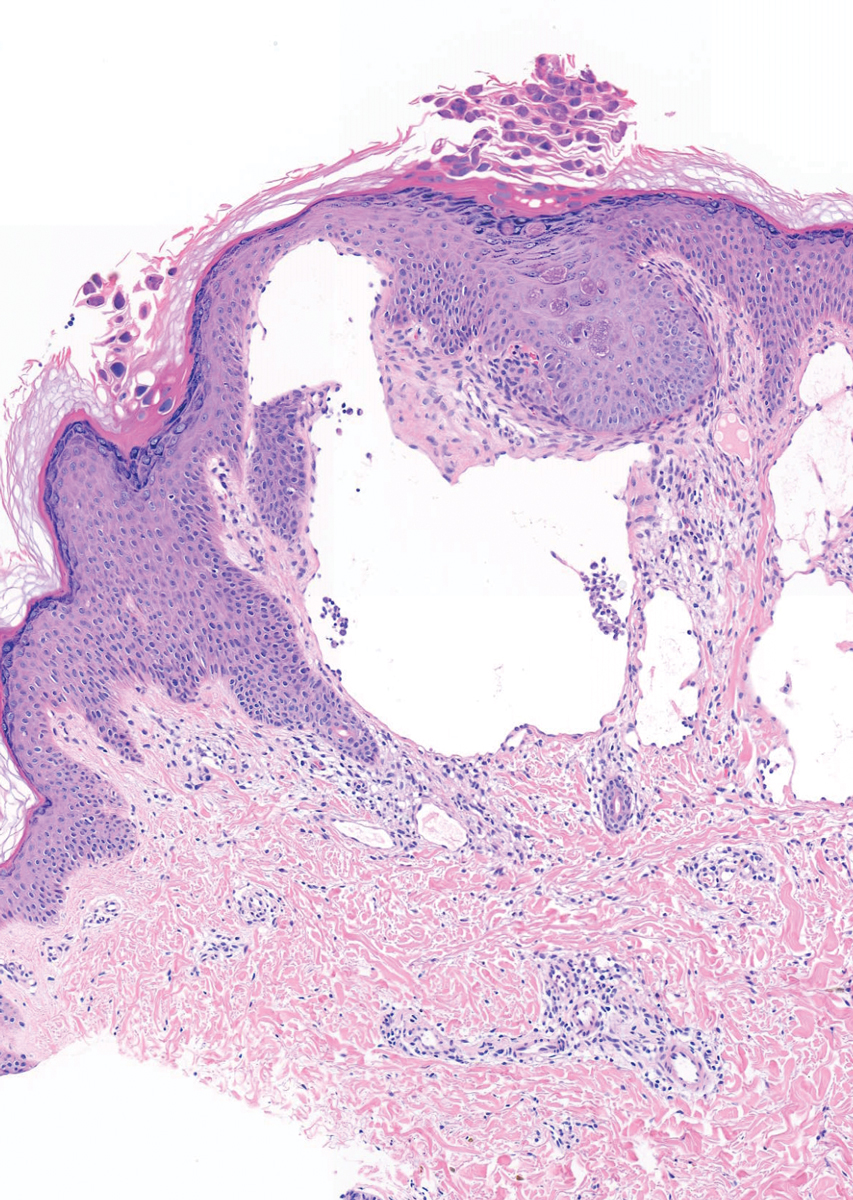
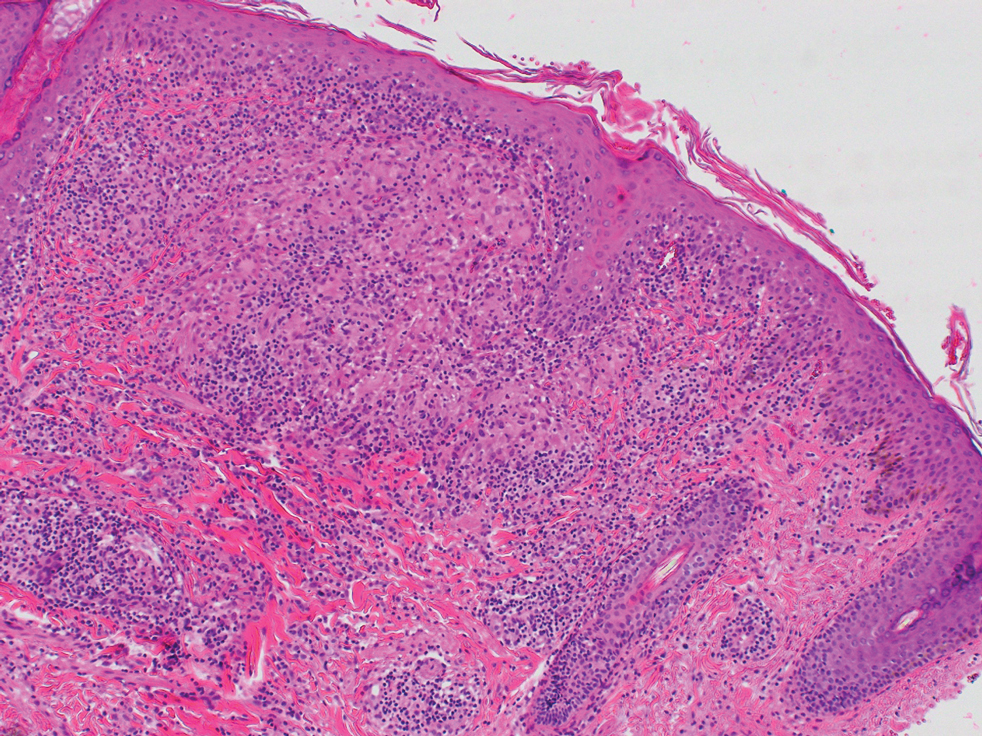
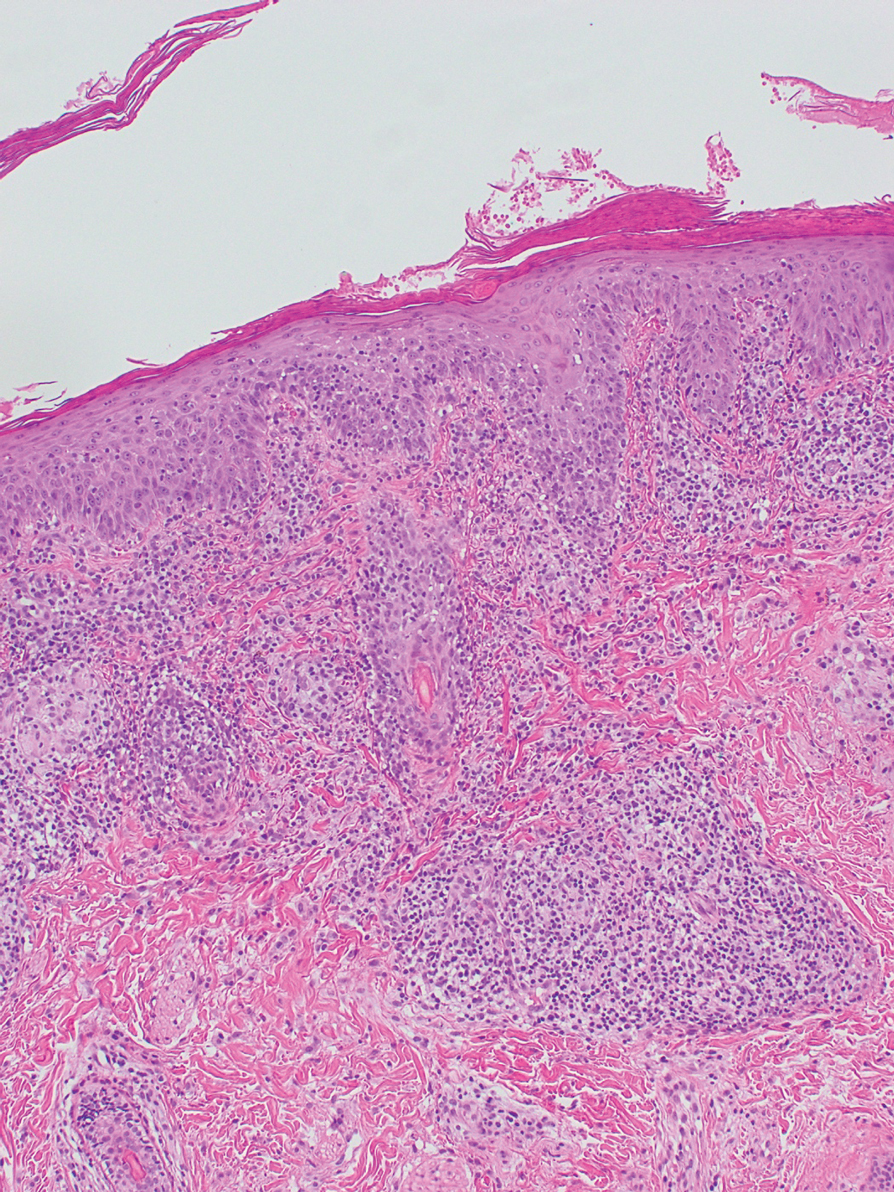
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
Practice Points
- Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system that can be misdiagnosed as molluscum contagiosum (MC).
- Secondary infection of LC is common, with Staphylococcus aureus being the most common entity, but MC virus also can be secondarily infected.
Enoxaparin-Induced Hemorrhagic Bullae at Sites of Trauma and Endothelial Pathology
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
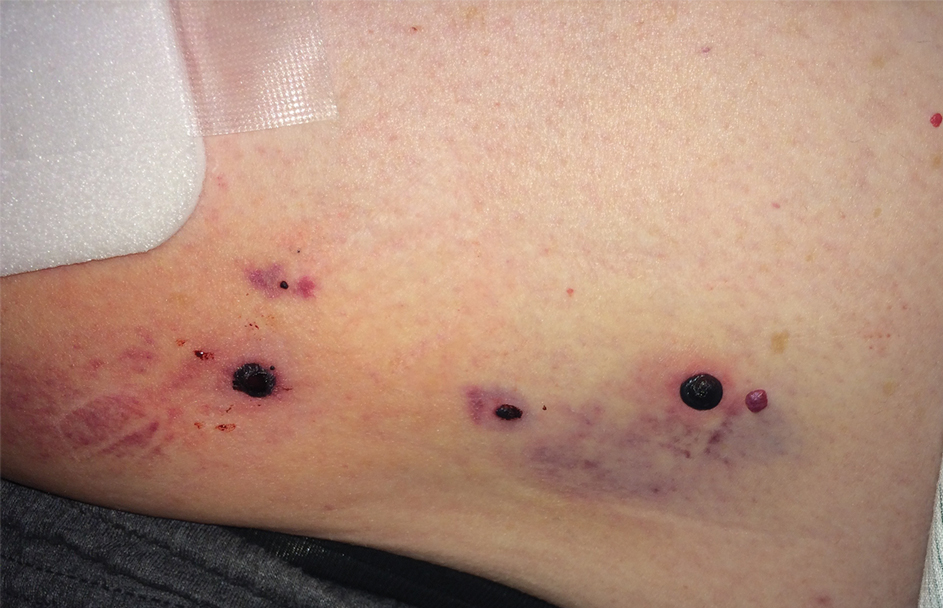
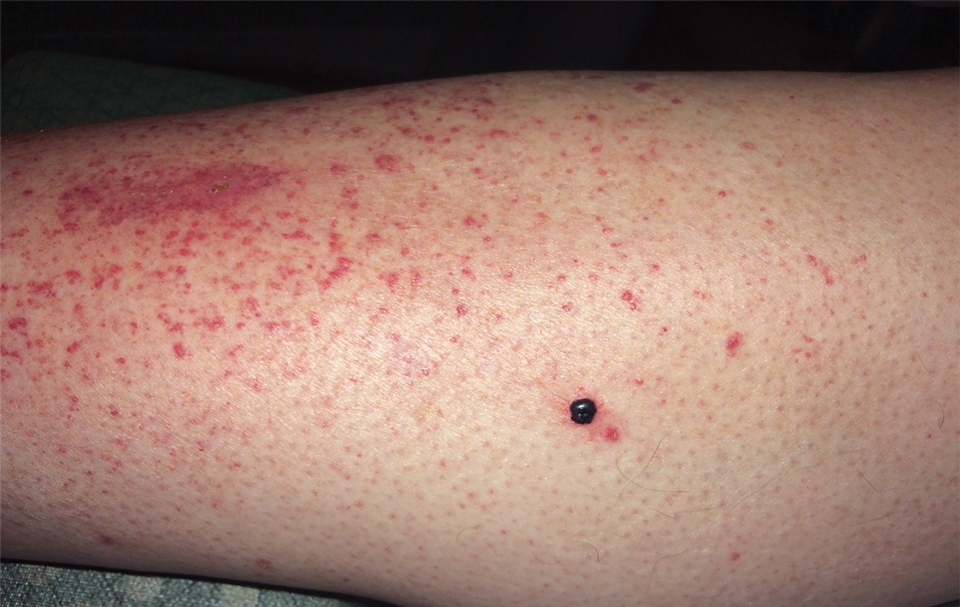
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
Granulomatous Facial Dermatoses
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
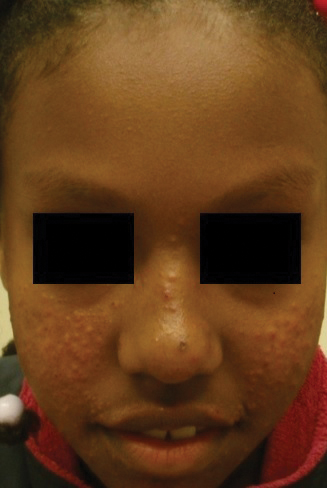
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
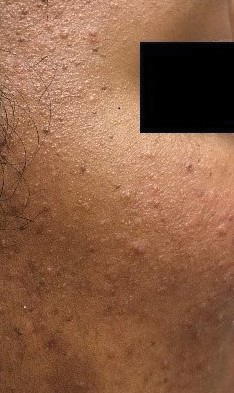
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
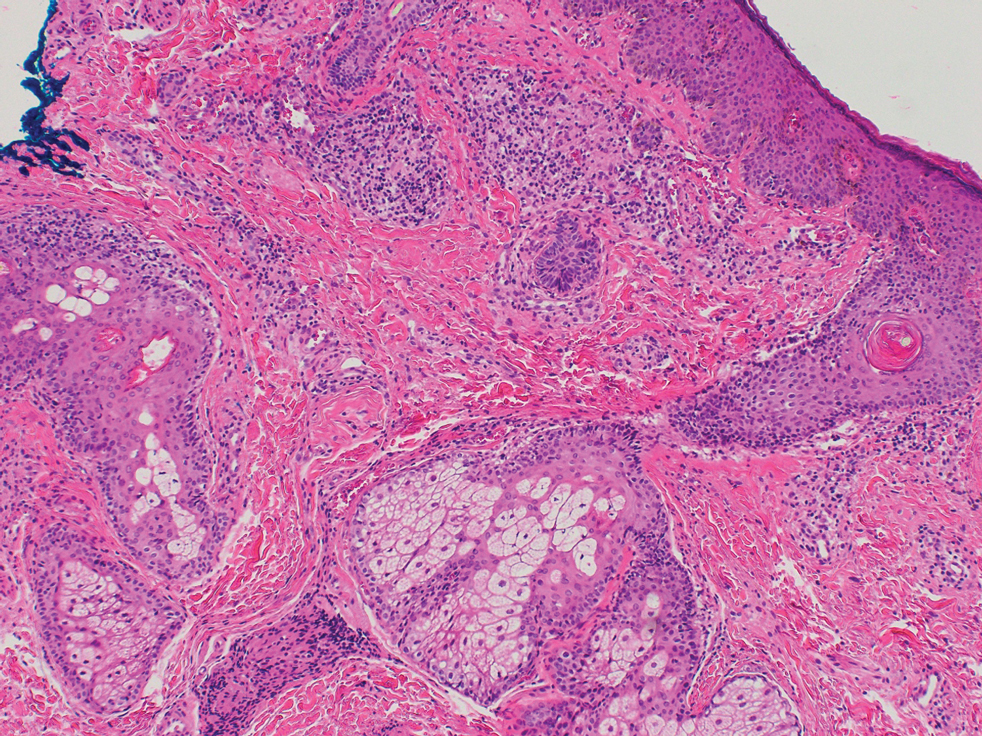
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Practice Points
- Dermatologists should be aware that noninfectious granulomatous dermatosis of the face can be caused by granulomatous periorificial dermatitis, granulomatous rosacea, lupus miliaris disseminatus faciei, and papular sarcoidosis.
- These conditions lie on a spectrum, suggested by their historical description and clinical and histological features.
- Because their clinical courses can vary considerably from patient to patient, a thorough effort should be made to differentiate these conditions.
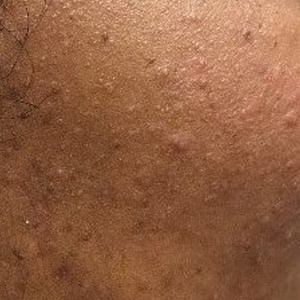
Atypical Presentation of Pityriasis Rubra Pilaris: Challenges in Diagnosis and Management
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare inflammatory dermatosis of unknown etiology characterized by erythematosquamous salmon-colored plaques with well-demarcated islands of unaffected skin and hyperkeratotic follicles.1 In the United States, an incidence of 1 in 3500to 5000 patients presenting to dermatology clinics has been reported.2 Pityriasis rubra pilaris has several subtypes and variability in presentation that can make accurate and timely diagnosis challenging.3-5 Herein, we present a case of PRP with complex diagnostic and therapeutic challenges.
A 22-year-old woman presented with symmetrical, well-demarcated, hyperkeratotic, erythematous plaques with a carnauba wax–like appearance on the palms (Figure 1), soles, elbows, and trunk covering approximately 5% of the body surface area. Two weeks prior to presentation, she experienced an upper respiratory tract infection without any treatment and subsequently developed redness on the palms, which became very hard and scaly. The redness then spread to the elbows, soles, and trunk. She reported itching as well as pain in areas of fissuring. Hand mobility became restricted due to thick scale.
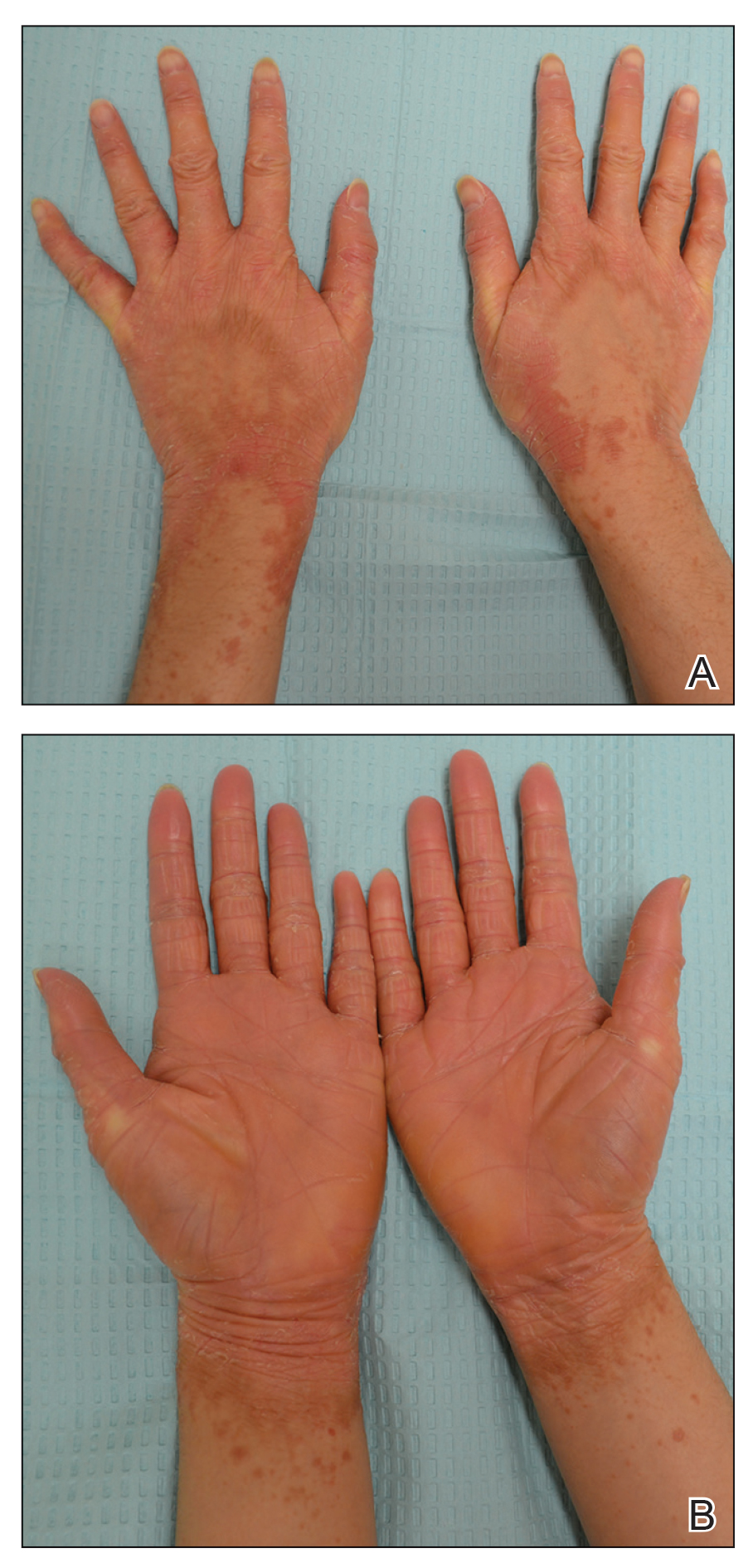
The patient’s medical history was notable for suspected psoriasis 9 years prior, but there were no records or biopsy reports that could be obtained to confirm the diagnosis. She also reported a similar skin condition in her father, which also was diagnosed as psoriasis, but this diagnosis could not be verified.
Although the morphology of the lesions was most consistent with localized PRP, atypical psoriasis, palmoplantar keratoderma (PPK), and erythroderma progressive symmetrica (EPS) also were considered given the personal and family history of suspected psoriasis. A biopsy could not be obtained due to an insurance issue. She was started on clobetasol cream 0.05% and ointment. At 2-week follow-up, her condition remained unchanged. Empiric systemic treatment was discussed, which would potentially work for diagnoses of both PRP and psoriasis. Due to the history of psoriasis and level of discomfort, cyclosporine 300 mg once daily was started to gain rapid control of the disease. Methotrexate also was considered due to its efficacy and economic considerations but was not selected due to patient concerns about the medication.
After 10 weeks of cyclosporine treatment, our patient showed some improvement of the skin with decreased scale and flattening of plaques but not complete resolution. At this point, a biopsy was able to be obtained with prior authorization. A 4-mm punch biopsy of the right flank demonstrated a psoriasiform and papillated epidermis with multifocally capped, compact parakeratosis and minimal lymphocytic infiltrate consistent with PRP. Although EPS also was on the histologic differential, clinical history was more consistent with a diagnosis of PRP. There was some minimal improvement with cyclosporine, but with the diagnosis of PRP confirmed, a systemic retinoid became the treatment of choice. Although acitretin is the preferred treatment for PRP, given that pregnancy would be contraindicated during and for 3 years following acitretin therapy, a trial of isotretinoin 40 mg once daily was started due to its shorter half-life compared to acitretin and was continued for 3 months (Figure 2).6,7
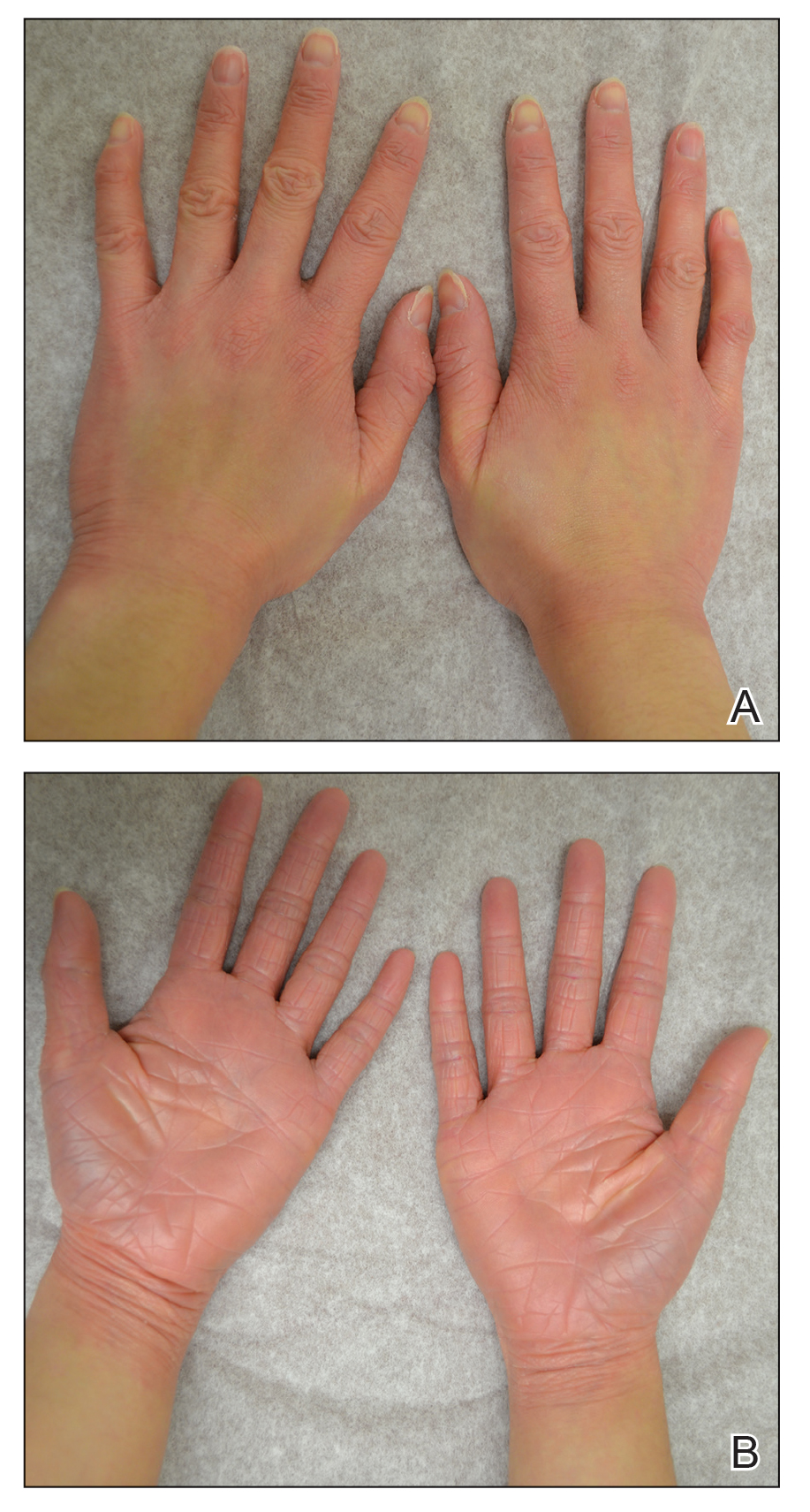
The diagnosis of PRP often can be challenging given the variety of clinical presentations. This case was an atypical presentation of PRP with several learning points, as our patient’s condition did not fit perfectly into any of the 6 types of PRP. The age of onset was atypical at 22 years old. Pityriasis rubra pilaris typically presents with a bimodal age distribution, appearing either in the first decade or the fifth to sixth decades of life.3,8 Her clinical presentation was atypical for adult-onset types I and II, which typically present with cephalocaudal progression or ichthyosiform dermatitis, respectively. Her presentation also was atypical for juvenile onset in types III, IV, and V, which tend to present in younger children and with different physical examination findings.3,8
The morphology of our patient’s lesions also was atypical for PRP, PPK, EPS, and psoriasis. The clinical presentation had features of these entities with erythema, fissuring, xerosis, carnauba wax–like appearance, symmetric scale, and well-demarcated plaques. Although these findings are not mutually exclusive, their combined presentation is atypical. Coupled with the ambiguous family history of similar skin disease in the patient’s father, the discussion of genodermatoses, particularly PPK, further confounded the diagnosis.4,9 When evaluating for PRP, especially with any family history of skin conditions, genodermatoses should be considered. Furthermore, our patient’s remote and unverifiable history of psoriasis serves as a cautionary reminder that prior diagnoses and medical history always should be reasonably scrutinized. Additionally, a drug-induced PRP eruption also should be considered. Although our patient received no medical treatment for the upper respiratory tract infection prior to the onset of PRP, there have been several reports of drug-induced PRP.10-12
The therapeutic challenge in this case is one that often is encountered in clinical practice. The health care system often may pose a barrier to diagnosis by inhibiting particular services required for adequate patient care. For our patient, diagnosis was delayed by several weeks due to difficulties obtaining a diagnostic skin biopsy. When faced with challenges from health care infrastructure, creativity with treatment options, such as finding an empiric treatment option (cyclosporine in this case), must be considered.
Systemic retinoids have been found to be efficacious treatment options for PRP, but when dealing with a woman of reproductive age, reproductive preferences must be discussed before identifying an appropriate treatment regimen.1,13-15 The half-life of acitretin compared to isotretinoin is 2 days vs 22 hours.6,16 With alcohol consumption, acitretin can be metabolized to etretinate, which has a half-life of 120 days.17 In our patient, isotretinoin was a more manageable option to allow for greater reproductive freedom upon treatment completion.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris: a review of diagnosis and treatment. Am J Clin Dermatol. 2010;11:157-170.
- Shenefelt PD. Pityriasis rubra pilaris. Medscape website. Updated September 11, 2020. Accessed September 28, 2021. https://reference.medscape.com/article/1107742-overview
- Griffiths WA. Pityriasis rubra pilaris. Clin Exp Dermatol. 1980;5:105-112.
- Itin PH, Lautenschlager S. Palmoplantar keratoderma and associated syndromes. Semin Dermatol. 1995;14:152-161.
- Guidelines of care for psoriasis. Committee on Guidelines of Care. Task Force on Psoriasis. J Am Acad Dermatol. 1993;28:632-637.
- Larsen FG, Jakobsen P, Eriksen H, et al. The pharmacokinetics of acitretin and its 13-cis-metabolite in psoriatic patients. J Clin Pharmacol. 1991;31:477-483.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Sørensen KB, Thestrup-Pedersen K. Pityriasis rubra pilaris: a retrospective analysis of 43 patients. Acta Derm Venereol. 1999;79:405-406.
- Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol. 1994;131:1-14.
- Cutaneous reactions to labetalol. Br Med J. 1978;1:987.
- Plana A, Carrascosa JM, Vilavella M. Pityriasis rubra pilaris‐like reaction induced by imatinib. Clin Exp Dermatol. 2013;38:520-522.
- Gajinov ZT, Matc´ MB, Duran VD, et al. Drug-related pityriasis rubra pilaris with acantholysis. Vojnosanit Pregl. 2013;70:871-873.
- Clayton BD, Jorizzo JL, Hitchcock MG, et al. Adult pityriasis rubra pilaris: a 10-year case series. J Am Acad Dermatol. 1997;36:959-964.
- Cohen PR, Prystowsky JH. Pityriasis rubra pilaris: a review of diagnosis and treatment. J Am Acad Dermatol. 1989;20:801-807.
- Dicken CH. Isotretinoin treatment of pityriasis rubra pilaris. J Am Acad Dermatol. 1987;16(2 pt 1):297-301.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Grønhøj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare inflammatory dermatosis of unknown etiology characterized by erythematosquamous salmon-colored plaques with well-demarcated islands of unaffected skin and hyperkeratotic follicles.1 In the United States, an incidence of 1 in 3500to 5000 patients presenting to dermatology clinics has been reported.2 Pityriasis rubra pilaris has several subtypes and variability in presentation that can make accurate and timely diagnosis challenging.3-5 Herein, we present a case of PRP with complex diagnostic and therapeutic challenges.
A 22-year-old woman presented with symmetrical, well-demarcated, hyperkeratotic, erythematous plaques with a carnauba wax–like appearance on the palms (Figure 1), soles, elbows, and trunk covering approximately 5% of the body surface area. Two weeks prior to presentation, she experienced an upper respiratory tract infection without any treatment and subsequently developed redness on the palms, which became very hard and scaly. The redness then spread to the elbows, soles, and trunk. She reported itching as well as pain in areas of fissuring. Hand mobility became restricted due to thick scale.
The patient’s medical history was notable for suspected psoriasis 9 years prior, but there were no records or biopsy reports that could be obtained to confirm the diagnosis. She also reported a similar skin condition in her father, which also was diagnosed as psoriasis, but this diagnosis could not be verified.
Although the morphology of the lesions was most consistent with localized PRP, atypical psoriasis, palmoplantar keratoderma (PPK), and erythroderma progressive symmetrica (EPS) also were considered given the personal and family history of suspected psoriasis. A biopsy could not be obtained due to an insurance issue. She was started on clobetasol cream 0.05% and ointment. At 2-week follow-up, her condition remained unchanged. Empiric systemic treatment was discussed, which would potentially work for diagnoses of both PRP and psoriasis. Due to the history of psoriasis and level of discomfort, cyclosporine 300 mg once daily was started to gain rapid control of the disease. Methotrexate also was considered due to its efficacy and economic considerations but was not selected due to patient concerns about the medication.
After 10 weeks of cyclosporine treatment, our patient showed some improvement of the skin with decreased scale and flattening of plaques but not complete resolution. At this point, a biopsy was able to be obtained with prior authorization. A 4-mm punch biopsy of the right flank demonstrated a psoriasiform and papillated epidermis with multifocally capped, compact parakeratosis and minimal lymphocytic infiltrate consistent with PRP. Although EPS also was on the histologic differential, clinical history was more consistent with a diagnosis of PRP. There was some minimal improvement with cyclosporine, but with the diagnosis of PRP confirmed, a systemic retinoid became the treatment of choice. Although acitretin is the preferred treatment for PRP, given that pregnancy would be contraindicated during and for 3 years following acitretin therapy, a trial of isotretinoin 40 mg once daily was started due to its shorter half-life compared to acitretin and was continued for 3 months (Figure 2).6,7
The diagnosis of PRP often can be challenging given the variety of clinical presentations. This case was an atypical presentation of PRP with several learning points, as our patient’s condition did not fit perfectly into any of the 6 types of PRP. The age of onset was atypical at 22 years old. Pityriasis rubra pilaris typically presents with a bimodal age distribution, appearing either in the first decade or the fifth to sixth decades of life.3,8 Her clinical presentation was atypical for adult-onset types I and II, which typically present with cephalocaudal progression or ichthyosiform dermatitis, respectively. Her presentation also was atypical for juvenile onset in types III, IV, and V, which tend to present in younger children and with different physical examination findings.3,8
The morphology of our patient’s lesions also was atypical for PRP, PPK, EPS, and psoriasis. The clinical presentation had features of these entities with erythema, fissuring, xerosis, carnauba wax–like appearance, symmetric scale, and well-demarcated plaques. Although these findings are not mutually exclusive, their combined presentation is atypical. Coupled with the ambiguous family history of similar skin disease in the patient’s father, the discussion of genodermatoses, particularly PPK, further confounded the diagnosis.4,9 When evaluating for PRP, especially with any family history of skin conditions, genodermatoses should be considered. Furthermore, our patient’s remote and unverifiable history of psoriasis serves as a cautionary reminder that prior diagnoses and medical history always should be reasonably scrutinized. Additionally, a drug-induced PRP eruption also should be considered. Although our patient received no medical treatment for the upper respiratory tract infection prior to the onset of PRP, there have been several reports of drug-induced PRP.10-12
The therapeutic challenge in this case is one that often is encountered in clinical practice. The health care system often may pose a barrier to diagnosis by inhibiting particular services required for adequate patient care. For our patient, diagnosis was delayed by several weeks due to difficulties obtaining a diagnostic skin biopsy. When faced with challenges from health care infrastructure, creativity with treatment options, such as finding an empiric treatment option (cyclosporine in this case), must be considered.
Systemic retinoids have been found to be efficacious treatment options for PRP, but when dealing with a woman of reproductive age, reproductive preferences must be discussed before identifying an appropriate treatment regimen.1,13-15 The half-life of acitretin compared to isotretinoin is 2 days vs 22 hours.6,16 With alcohol consumption, acitretin can be metabolized to etretinate, which has a half-life of 120 days.17 In our patient, isotretinoin was a more manageable option to allow for greater reproductive freedom upon treatment completion.
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare inflammatory dermatosis of unknown etiology characterized by erythematosquamous salmon-colored plaques with well-demarcated islands of unaffected skin and hyperkeratotic follicles.1 In the United States, an incidence of 1 in 3500to 5000 patients presenting to dermatology clinics has been reported.2 Pityriasis rubra pilaris has several subtypes and variability in presentation that can make accurate and timely diagnosis challenging.3-5 Herein, we present a case of PRP with complex diagnostic and therapeutic challenges.
A 22-year-old woman presented with symmetrical, well-demarcated, hyperkeratotic, erythematous plaques with a carnauba wax–like appearance on the palms (Figure 1), soles, elbows, and trunk covering approximately 5% of the body surface area. Two weeks prior to presentation, she experienced an upper respiratory tract infection without any treatment and subsequently developed redness on the palms, which became very hard and scaly. The redness then spread to the elbows, soles, and trunk. She reported itching as well as pain in areas of fissuring. Hand mobility became restricted due to thick scale.
The patient’s medical history was notable for suspected psoriasis 9 years prior, but there were no records or biopsy reports that could be obtained to confirm the diagnosis. She also reported a similar skin condition in her father, which also was diagnosed as psoriasis, but this diagnosis could not be verified.
Although the morphology of the lesions was most consistent with localized PRP, atypical psoriasis, palmoplantar keratoderma (PPK), and erythroderma progressive symmetrica (EPS) also were considered given the personal and family history of suspected psoriasis. A biopsy could not be obtained due to an insurance issue. She was started on clobetasol cream 0.05% and ointment. At 2-week follow-up, her condition remained unchanged. Empiric systemic treatment was discussed, which would potentially work for diagnoses of both PRP and psoriasis. Due to the history of psoriasis and level of discomfort, cyclosporine 300 mg once daily was started to gain rapid control of the disease. Methotrexate also was considered due to its efficacy and economic considerations but was not selected due to patient concerns about the medication.
After 10 weeks of cyclosporine treatment, our patient showed some improvement of the skin with decreased scale and flattening of plaques but not complete resolution. At this point, a biopsy was able to be obtained with prior authorization. A 4-mm punch biopsy of the right flank demonstrated a psoriasiform and papillated epidermis with multifocally capped, compact parakeratosis and minimal lymphocytic infiltrate consistent with PRP. Although EPS also was on the histologic differential, clinical history was more consistent with a diagnosis of PRP. There was some minimal improvement with cyclosporine, but with the diagnosis of PRP confirmed, a systemic retinoid became the treatment of choice. Although acitretin is the preferred treatment for PRP, given that pregnancy would be contraindicated during and for 3 years following acitretin therapy, a trial of isotretinoin 40 mg once daily was started due to its shorter half-life compared to acitretin and was continued for 3 months (Figure 2).6,7
The diagnosis of PRP often can be challenging given the variety of clinical presentations. This case was an atypical presentation of PRP with several learning points, as our patient’s condition did not fit perfectly into any of the 6 types of PRP. The age of onset was atypical at 22 years old. Pityriasis rubra pilaris typically presents with a bimodal age distribution, appearing either in the first decade or the fifth to sixth decades of life.3,8 Her clinical presentation was atypical for adult-onset types I and II, which typically present with cephalocaudal progression or ichthyosiform dermatitis, respectively. Her presentation also was atypical for juvenile onset in types III, IV, and V, which tend to present in younger children and with different physical examination findings.3,8
The morphology of our patient’s lesions also was atypical for PRP, PPK, EPS, and psoriasis. The clinical presentation had features of these entities with erythema, fissuring, xerosis, carnauba wax–like appearance, symmetric scale, and well-demarcated plaques. Although these findings are not mutually exclusive, their combined presentation is atypical. Coupled with the ambiguous family history of similar skin disease in the patient’s father, the discussion of genodermatoses, particularly PPK, further confounded the diagnosis.4,9 When evaluating for PRP, especially with any family history of skin conditions, genodermatoses should be considered. Furthermore, our patient’s remote and unverifiable history of psoriasis serves as a cautionary reminder that prior diagnoses and medical history always should be reasonably scrutinized. Additionally, a drug-induced PRP eruption also should be considered. Although our patient received no medical treatment for the upper respiratory tract infection prior to the onset of PRP, there have been several reports of drug-induced PRP.10-12
The therapeutic challenge in this case is one that often is encountered in clinical practice. The health care system often may pose a barrier to diagnosis by inhibiting particular services required for adequate patient care. For our patient, diagnosis was delayed by several weeks due to difficulties obtaining a diagnostic skin biopsy. When faced with challenges from health care infrastructure, creativity with treatment options, such as finding an empiric treatment option (cyclosporine in this case), must be considered.
Systemic retinoids have been found to be efficacious treatment options for PRP, but when dealing with a woman of reproductive age, reproductive preferences must be discussed before identifying an appropriate treatment regimen.1,13-15 The half-life of acitretin compared to isotretinoin is 2 days vs 22 hours.6,16 With alcohol consumption, acitretin can be metabolized to etretinate, which has a half-life of 120 days.17 In our patient, isotretinoin was a more manageable option to allow for greater reproductive freedom upon treatment completion.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris: a review of diagnosis and treatment. Am J Clin Dermatol. 2010;11:157-170.
- Shenefelt PD. Pityriasis rubra pilaris. Medscape website. Updated September 11, 2020. Accessed September 28, 2021. https://reference.medscape.com/article/1107742-overview
- Griffiths WA. Pityriasis rubra pilaris. Clin Exp Dermatol. 1980;5:105-112.
- Itin PH, Lautenschlager S. Palmoplantar keratoderma and associated syndromes. Semin Dermatol. 1995;14:152-161.
- Guidelines of care for psoriasis. Committee on Guidelines of Care. Task Force on Psoriasis. J Am Acad Dermatol. 1993;28:632-637.
- Larsen FG, Jakobsen P, Eriksen H, et al. The pharmacokinetics of acitretin and its 13-cis-metabolite in psoriatic patients. J Clin Pharmacol. 1991;31:477-483.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Sørensen KB, Thestrup-Pedersen K. Pityriasis rubra pilaris: a retrospective analysis of 43 patients. Acta Derm Venereol. 1999;79:405-406.
- Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol. 1994;131:1-14.
- Cutaneous reactions to labetalol. Br Med J. 1978;1:987.
- Plana A, Carrascosa JM, Vilavella M. Pityriasis rubra pilaris‐like reaction induced by imatinib. Clin Exp Dermatol. 2013;38:520-522.
- Gajinov ZT, Matc´ MB, Duran VD, et al. Drug-related pityriasis rubra pilaris with acantholysis. Vojnosanit Pregl. 2013;70:871-873.
- Clayton BD, Jorizzo JL, Hitchcock MG, et al. Adult pityriasis rubra pilaris: a 10-year case series. J Am Acad Dermatol. 1997;36:959-964.
- Cohen PR, Prystowsky JH. Pityriasis rubra pilaris: a review of diagnosis and treatment. J Am Acad Dermatol. 1989;20:801-807.
- Dicken CH. Isotretinoin treatment of pityriasis rubra pilaris. J Am Acad Dermatol. 1987;16(2 pt 1):297-301.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Grønhøj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris: a review of diagnosis and treatment. Am J Clin Dermatol. 2010;11:157-170.
- Shenefelt PD. Pityriasis rubra pilaris. Medscape website. Updated September 11, 2020. Accessed September 28, 2021. https://reference.medscape.com/article/1107742-overview
- Griffiths WA. Pityriasis rubra pilaris. Clin Exp Dermatol. 1980;5:105-112.
- Itin PH, Lautenschlager S. Palmoplantar keratoderma and associated syndromes. Semin Dermatol. 1995;14:152-161.
- Guidelines of care for psoriasis. Committee on Guidelines of Care. Task Force on Psoriasis. J Am Acad Dermatol. 1993;28:632-637.
- Larsen FG, Jakobsen P, Eriksen H, et al. The pharmacokinetics of acitretin and its 13-cis-metabolite in psoriatic patients. J Clin Pharmacol. 1991;31:477-483.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Sørensen KB, Thestrup-Pedersen K. Pityriasis rubra pilaris: a retrospective analysis of 43 patients. Acta Derm Venereol. 1999;79:405-406.
- Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol. 1994;131:1-14.
- Cutaneous reactions to labetalol. Br Med J. 1978;1:987.
- Plana A, Carrascosa JM, Vilavella M. Pityriasis rubra pilaris‐like reaction induced by imatinib. Clin Exp Dermatol. 2013;38:520-522.
- Gajinov ZT, Matc´ MB, Duran VD, et al. Drug-related pityriasis rubra pilaris with acantholysis. Vojnosanit Pregl. 2013;70:871-873.
- Clayton BD, Jorizzo JL, Hitchcock MG, et al. Adult pityriasis rubra pilaris: a 10-year case series. J Am Acad Dermatol. 1997;36:959-964.
- Cohen PR, Prystowsky JH. Pityriasis rubra pilaris: a review of diagnosis and treatment. J Am Acad Dermatol. 1989;20:801-807.
- Dicken CH. Isotretinoin treatment of pityriasis rubra pilaris. J Am Acad Dermatol. 1987;16(2 pt 1):297-301.
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169.
- Grønhøj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
Practice Points
- Pityriasis rubra pilaris (PRP) is a rare inflammatory dermatosis of unknown etiology characterized by erythematosquamous salmon-colored plaques with well-demarcated islands of unaffected skin and hyperkeratotic follicles.
- The diagnosis of PRP often can be challenging given the variety of clinical presentations.
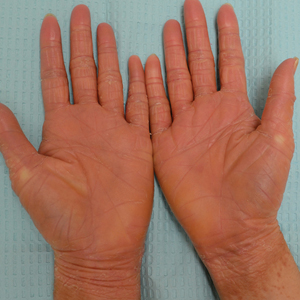
Paraneoplastic Signs in Bladder Transitional Cell Carcinoma: An Unusual Presentation
To the Editor:
A 40-year-old Somalian man presented to the dermatology clinic with lesions on the eyelids, tongue, lips, and hands of 8 years’ duration. He was a former refugee who had faced considerable stigma from his community due to his appearance. A review of systems was remarkable for decreased appetite but no weight loss. He reported no abdominal distention, early satiety, or urinary symptoms, and he had no personal history of diabetes mellitus or obesity. Physical examination demonstrated hyperpigmented velvety plaques in all skin folds and on the genitalia. Massive papillomatosis of the eyelid margins, tongue, and lips also was noted (Figure 1A). Flesh-colored papules also were scattered across the face. Punctate, flesh-colored papules were present on the volar and palmar hands (Figure 2A). Histopathology demonstrated pronounced papillomatous epidermal hyperplasia with negative human papillomavirus (HPV) type 16 and HPV-18 DNA studies. Given the appearance of malignant acanthosis nigricans with oral and conjunctival features, cutaneous papillomatosis, and tripe palms, concern for underlying malignancy was high. Malignancy workup, including upper and lower endoscopy as well as serial computed tomography scans of the chest, abdomen, and pelvis, was unrevealing.
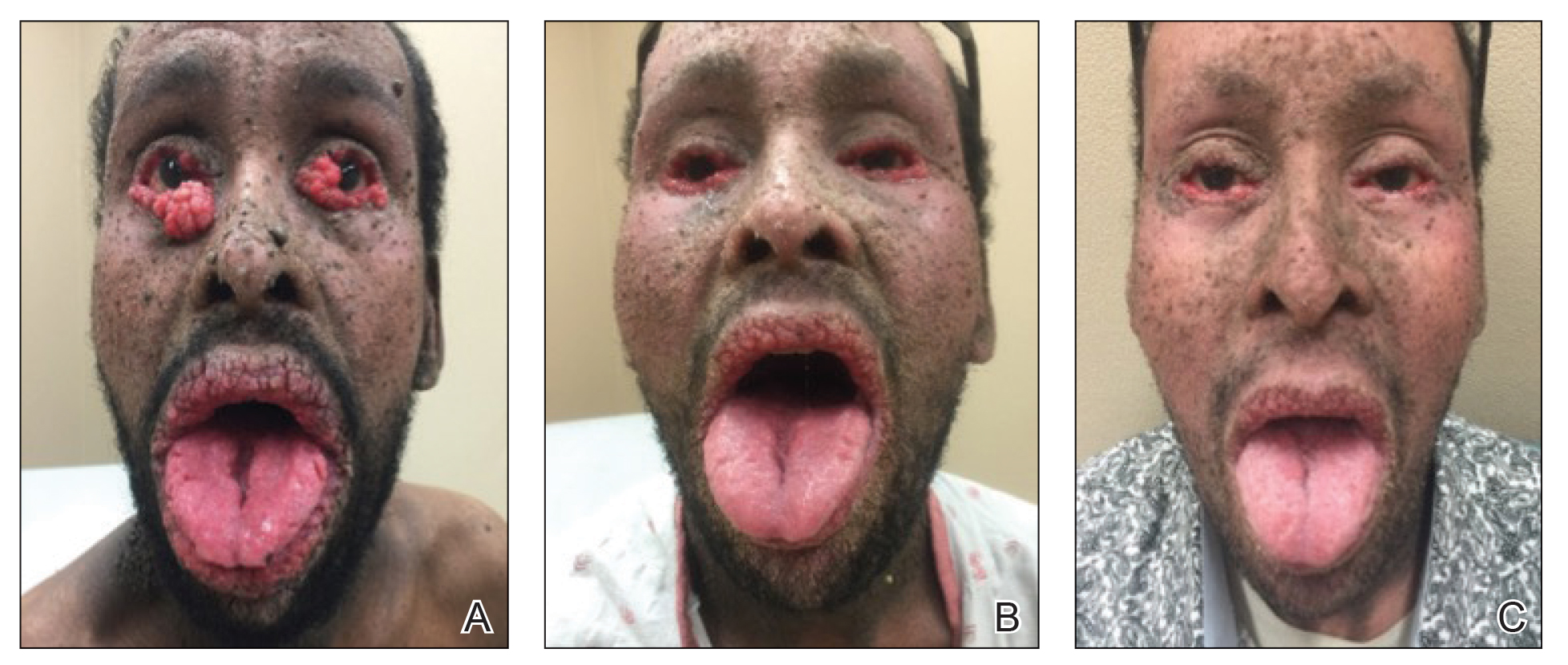
Laboratory investigation revealed a positive Schistosoma IgG antibody (0.38 geometric mean egg count) and peripheral eosinophilia (1.09 ×103/μL), which normalized after praziquantel therapy. With no malignancy identified over the preceding 6-month period, treatment with acitretin 50 mg daily was initiated based on limited literature support.1-3 Treatment led to reduction in the size and number of papillomas (Figure 1B) and tripe palms (Figure 2B) with increased mobility of hands, lips, and tongue. The patient underwent oculoplastic surgery to reduce the papilloma burden along the eyelid margins. Subsequent cystoscopy 9 months after the initial presentation revealed low-grade transitional cell carcinoma of the bladder. Intraoperative mitomycin C led to tumor shrinkage and, with continued treatment with daily acitretin, dramatic improvement of all cutaneous and mucosal symptoms (Figure 1C and Figure 2C). To date, his cutaneous symptoms have resolved.
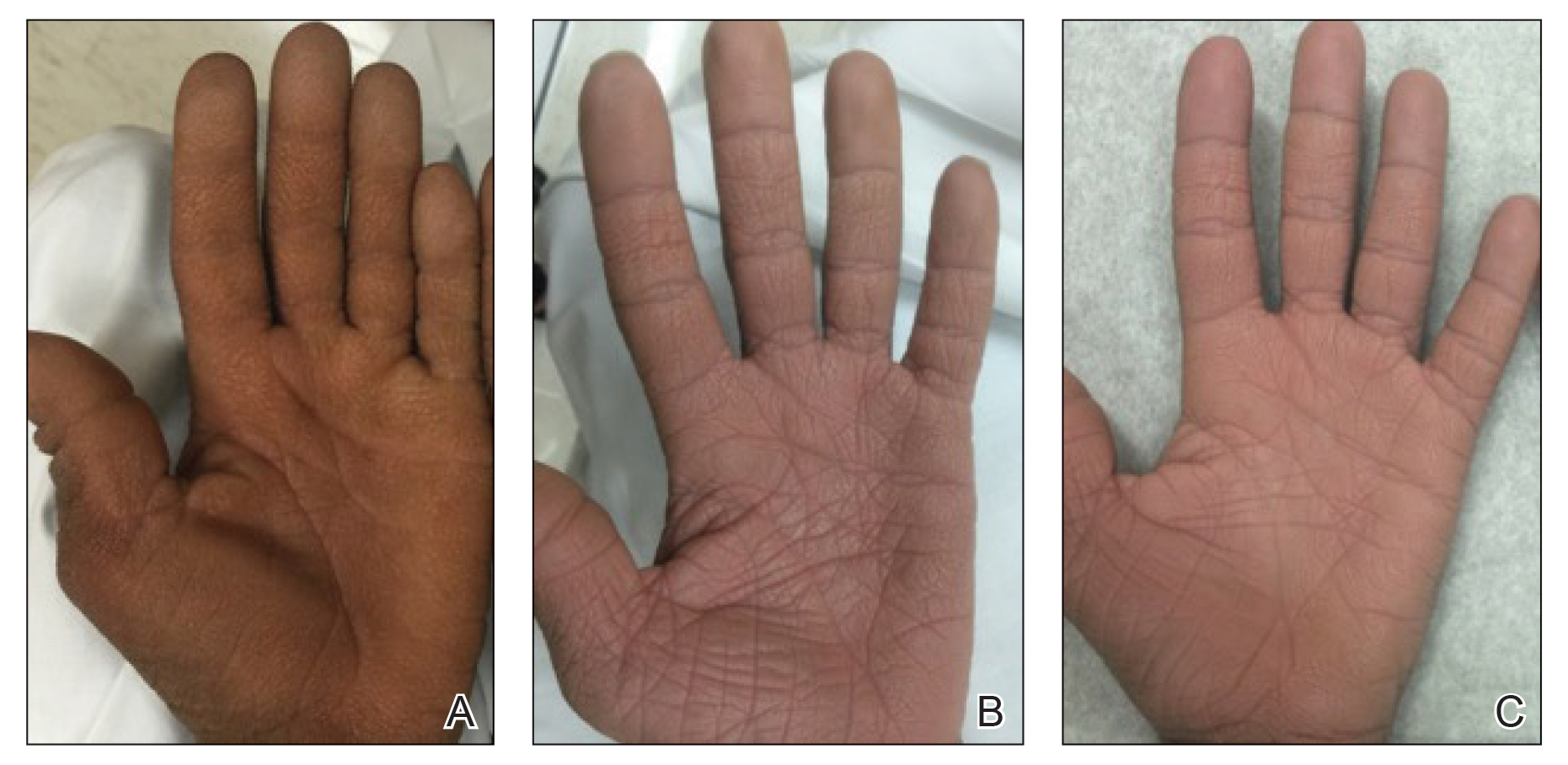
This case demonstrated a unique presentation of multiple paraneoplastic signs in bladder transitional cell carcinoma. The presence of malignant acanthosis nigricans (including oral and conjunctival involvement), cutaneous papillomatosis, and tripe palms have been individually documented in various types of gastric malignancies.4 Acanthosis nigricans often is secondary to diabetes and obesity, presenting with diffuse, thickened, velvety plaques in the flexural areas. Malignant acanthosis nigricans is a rare, rapidly progressive condition that often presents over a period of weeks to months; it almost always is associated with internal malignancies. It often has more extensive involvement, extending beyond the flexural areas, than typical acanthosis nigricans.4 Oral involvement can be either hypertrophic or papillomatous; papillomatosis of the oral mucosa was reported in over 40% of malignant acanthosis nigricans cases (N=200).5 Cases with conjunctival involvement are less common.6 Although malignant acanthosis nigricans often is codiagnosed with a malignancy, it can precede the cancer diagnosis in some cases.7,8 A majority of cases are associated with adenocarcinomas of the gastrointestinal tract.4 Progressive mucocutaneous papillomatosis also is a rare paraneoplastic condition that most commonly is associated with gastric adenocarcinomas. Progressive mucocutaneous papillomatosis often presents rapidly as verrucous growths on cutaneous surfaces (including the hands and face) but also can affect mucosal surfaces such as the mouth and conjunctiva.9-11 Tripe palms are characterized by exaggerated dermatoglyphics with diffuse palmar ridging and hyperkeratosis. Tripe palms most often are associated with pulmonary malignancies. When tripe palms are present with malignant acanthosis nigricans, they reflect up to a one-third incidence of gastrointestinal malignancy.12,13
Despite the individual presentation of these paraneoplastic signs in a variety of malignancies, synchronous presentation is rare. A brief literature review only identified 6 cases of concurrent acanthosis nigricans, tripe palms, and progressive mucocutaneous papillomatosis with an underlying gastrointestinal malignancy.1,11,14-17 Two additional reports described tripe palms with oral acanthosis nigricans and progressive mucocutaneous papillomatosis in metastatic gastric adenocarcinoma and renal urothelial carcinoma.2,18 An additional case of all 3 paraneoplastic conditions was reported in the setting of metastatic cervical cancer (HPV positive).19 Per a recent case report and literature review,20 there have only been 8 cases of acanthosis nigricans reported in bladder transitional cell carcinoma,20-27 half of which have included oral malignant acanthosis nigricans.20-23 Only one report of concurrent cutaneous and oral malignant acanthosis nigricans and triple palms in the setting of bladder cancer has been reported.20 Given the extensive conjunctival involvement and cutaneous papillomatosis in our patient, ours is a rarely reported case of concurrent malignant mucocutaneous acanthosis nigricans, tripe palms, and progressive papillomatosis in transitional cell bladder carcinoma. We believe it is imperative to consider the role of this malignancy as a cause of these paraneoplastic conditions.
Although these paraneoplastic conditions rarely co-occur, our case further offers a common molecular pathway for these conditions.28 In these paraneoplastic conditions, the stimulating factor is thought to be tumor growth factor α, which is structurally related to epidermal growth factor (EGF). Epidermal growth factor receptors (EGFRs) are found in the basal layer of the epidermis, where activation stimulates keratinocyte growth and leads to the cutaneous manifestation of symptoms.28 Fibroblast growth factor receptor 3 mutations are found in most noninvasive transitional cell tumors of the bladder.29 The fibroblast growth factor pathway is distinctly different from the tumor growth factor α and EGF pathways.30 However, this association with transitional cell carcinoma suggests that fibroblast growth factor receptor 3 also may be implicated in these paraneoplastic conditions.
Our patient responded well to treatment with acitretin 50 mg daily. The mechanism of action of retinoids involves inducing mitotic activity and desmosomal shedding.31 Retinoids downregulate EGFR expression and activation in EGF-stimulated cells.32 We hypothesize that these oral retinoids decreased the growth stimulus and thereby improved cutaneous signs in the setting of our patient’s transitional cell cancer. Although definitive therapy is malignancy management, our case highlights the utility of adjunctive measures such as oral retinoids and surgical debulking. While previous cases have reported use of retinoids at a lower dosage than used in this case, oral lesions often have only been mildly improved with little impact on other cutaneous symptoms.1,2 In one case of malignant acanthosis nigricans and oral papillomatosis, isotretinoin 25 mg once every 2 to 3 days led to a moderate decrease in hyperkeratosis and papillomas, but the patient was lost to follow-up.3 Our case highlights the use of higher daily doses of oral retinoids for over 9 months, resulting in marked improvement in both the mucosal and cutaneous symptoms of acanthosis nigricans, progressive mucocutaneous papillomatosis, and tripe palms. Therefore, oral acitretin should be considered as adjuvant therapy for these paraneoplastic conditions.
By reporting this case, we hope to demonstrate the importance of considering other forms of malignancies in the presence of paraneoplastic conditions. Although gastric malignancies more commonly are associated with these conditions, bladder carcinomas also can present with cutaneous manifestations. The presence of these paraneoplastic conditions alone or together rarely is reported in urologic cancers and generally is considered to be an indicator of poor prognosis. Paraneoplastic conditions often develop rapidly and occur in very advanced malignancies.4 The disfiguring presentation in our case also had unusual diagnostic challenges. The presence of these conditions for 8 years and nonmetastatic advanced malignancy suggest a more indolent process and that these signs are not always an indicator of poor prognosis. Future patients with these paraneoplastic conditions may benefit from both a thorough malignancy screen, including cystoscopy, and high daily doses of oral retinoids.
- Stawczyk-Macieja M, Szczerkowska-Dobosz A, Nowicki R, et al. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma. Postepy Dermatol Alergol. 2014;31:56-58.
- Lee HC, Ker KJ, Chong W-S. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Swineford SL, Drucker CR. Palliative treatment of paraneoplastic acanthosis nigricans and oral florid papillomatosis with retinoids. J Drugs Dermatol. 2010;9:1151-1153.
- Wick MR, Patterson JW. Cutaneous paraneoplastic syndromes [published online January 31, 2019]. Semin Diagn Pathol. 2019;36:211-228.
- Tyler MT, Ficarra G, Silverman S, et al. Malignant acanthosis nigricans with florid papillary oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:445-449.
- Zhang X, Liu R, Liu Y, et al. Malignant acanthosis nigricans: a case report. BMC Ophthalmology. 2020;20:1-4.
- Curth HO. Dermatoses and malignant internal tumours. Arch Dermatol Syphil. 1955;71:95-107.
- Krawczyk M, Mykala-Cies´la J, Kolodziej-Jaskula A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
- Singhi MK, Gupta LK, Bansal M, et al. Florid cutaneous papillomatosis with adenocarcinoma of stomach in a 35 year old male. Indian J Dermatol Venereol Leprol. 2005;71:195-196.
- Klieb HB, Avon SL, Gilbert J, et al. Florid cutaneous and mucosal papillomatosis: mucocutaneous markers of an underlying gastric malignancy. J Clin Oncol. 2013;31:E218-E219.
- Yang YH, Zhang RZ, Kang DH, et al. Three paraneoplastic signs in the same patient with gastric adenocarcinoma. Dermatol Online J. 2013;19:18966.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Chantarojanasiri T, Buranathawornsom A, Sirinawasatien A. Diffuse esophageal squamous papillomatosis: a rare disease associated with acanthosis nigricans and tripe palms. Case Rep Gastroenterol. 2020;14:702-706.
- Muhammad R, Iftikhar N, Sarfraz T, et al. Malignant acanthosis nigricans: an indicator of internal malignancy. J Coll Physicians Surg Pak. 2019;29:888-890.
- Brinca A, Cardoso JC, Brites MM, et al. Florid cutaneous papillomatosis and acanthosis nigricans maligna revealing gastric adenocarcinoma. An Bras Dermatol. 2011;86:573-577.
- Vilas-Sueiro A, Suárez-Amor O, Monteagudo B, et al. Malignant acanthosis nigricans, florid cutaneous and mucosal papillomatosis, and tripe palms in a man with gastric adenocarcinoma. Actas Dermosifiliogr. 2015;106:438-439.
- Paravina M, Ljubisavljevic´ D. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma—a case report. Serbian J Dermatology Venereol. 2015;7:5-14.
- Kleikamp S, Böhm M, Frosch P, et al. Acanthosis nigricans, papillomatosis mucosae and “tripe” palms in a patient with metastasized gastric carcinoma [in German]. Dtsch Med Wochenschr. 2006;131:1209-1213.
- Mikhail GR, Fachnie DM, Drukker BH, et al. Generalized malignant acanthosis nigricans. Arch Dermatol. 1979;115:201-202.
- Zhang R, Jiang M, Lei W, et al. Malignant acanthosis nigricans with recurrent bladder cancer: a case report and review of literature. Onco Targets Ther. 2021;14:951.
- Olek-Hrab K, Silny W, Zaba R, et al. Co-occurrence of acanthosis nigricans and bladder adenocarcinoma-case report. Contemp Oncol (Pozn). 2013;17:327-330.
- Canjuga I, Mravak-Stipetic´ M, Kopic´V, et al. Oral acanthosis nigricans: case report and comparison with literature reports. Acta Dermatovenerol Croat. 2008;16:91-95.
- Cairo F, Rubino I, Rotundo R, et al. Oral acanthosis nigricans as a marker of internal malignancy. a case report. J Periodontol. 2001;72:1271-1275.
- Möhrenschlager M, Vocks E, Wessner DB, et al. 2001;165:1629-1630.
- Singh GK, Sen D, Mulajker DS, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Indian J Dermatol. 2011;56:722-725.
- Gohji K, Hasunuma Y, Gotoh A, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Int J Dermatol. 1994;33:433-435.
- Pinto WBVR, Badia BML, Souza PVS, et al. Paraneoplastic motor neuronopathy and malignant acanthosis nigricans. Arq Neuropsiquiatr. 2019;77:527.
- Koyama S, Ikeda K, Sato M, et al. Transforming growth factor–alpha (TGF-alpha)-producing gastric carcinoma with acanthosis nigricans: an endocrine effect of TGF alpha in the pathogenesis of cutaneous paraneoplastic syndrome and epithelial hyperplasia of the esophagus. J Gastroenterol. 1997;32:71-77.
- Billerey C, Chopin D, Aubriot-Lorton MH, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955-1959.
- Lee C-J, Lee M-H, Cho Y-Y. Fibroblast and epidermal growth factors utilize different signaling pathways to induce anchorage-independent cell transformation in JB6 Cl41 mouse skin epidermal cells. J Cancer Prev. 2014;19:199-208.
- Darmstadt GL, Yokel BK, Horn TD. Treatment of acanthosis nigricans with tretinoin. Arch Dermatol. 1991;127:1139-1140.
- Sah JF, Eckert RL, Chandraratna RA, et al. Retinoids suppress epidermal growth factor–associated cell proliferation by inhibiting epidermal growth factor receptor–dependent ERK1/2 activation. J Biol Chem. 2002;277:9728-9735.
To the Editor:
A 40-year-old Somalian man presented to the dermatology clinic with lesions on the eyelids, tongue, lips, and hands of 8 years’ duration. He was a former refugee who had faced considerable stigma from his community due to his appearance. A review of systems was remarkable for decreased appetite but no weight loss. He reported no abdominal distention, early satiety, or urinary symptoms, and he had no personal history of diabetes mellitus or obesity. Physical examination demonstrated hyperpigmented velvety plaques in all skin folds and on the genitalia. Massive papillomatosis of the eyelid margins, tongue, and lips also was noted (Figure 1A). Flesh-colored papules also were scattered across the face. Punctate, flesh-colored papules were present on the volar and palmar hands (Figure 2A). Histopathology demonstrated pronounced papillomatous epidermal hyperplasia with negative human papillomavirus (HPV) type 16 and HPV-18 DNA studies. Given the appearance of malignant acanthosis nigricans with oral and conjunctival features, cutaneous papillomatosis, and tripe palms, concern for underlying malignancy was high. Malignancy workup, including upper and lower endoscopy as well as serial computed tomography scans of the chest, abdomen, and pelvis, was unrevealing.
Laboratory investigation revealed a positive Schistosoma IgG antibody (0.38 geometric mean egg count) and peripheral eosinophilia (1.09 ×103/μL), which normalized after praziquantel therapy. With no malignancy identified over the preceding 6-month period, treatment with acitretin 50 mg daily was initiated based on limited literature support.1-3 Treatment led to reduction in the size and number of papillomas (Figure 1B) and tripe palms (Figure 2B) with increased mobility of hands, lips, and tongue. The patient underwent oculoplastic surgery to reduce the papilloma burden along the eyelid margins. Subsequent cystoscopy 9 months after the initial presentation revealed low-grade transitional cell carcinoma of the bladder. Intraoperative mitomycin C led to tumor shrinkage and, with continued treatment with daily acitretin, dramatic improvement of all cutaneous and mucosal symptoms (Figure 1C and Figure 2C). To date, his cutaneous symptoms have resolved.
This case demonstrated a unique presentation of multiple paraneoplastic signs in bladder transitional cell carcinoma. The presence of malignant acanthosis nigricans (including oral and conjunctival involvement), cutaneous papillomatosis, and tripe palms have been individually documented in various types of gastric malignancies.4 Acanthosis nigricans often is secondary to diabetes and obesity, presenting with diffuse, thickened, velvety plaques in the flexural areas. Malignant acanthosis nigricans is a rare, rapidly progressive condition that often presents over a period of weeks to months; it almost always is associated with internal malignancies. It often has more extensive involvement, extending beyond the flexural areas, than typical acanthosis nigricans.4 Oral involvement can be either hypertrophic or papillomatous; papillomatosis of the oral mucosa was reported in over 40% of malignant acanthosis nigricans cases (N=200).5 Cases with conjunctival involvement are less common.6 Although malignant acanthosis nigricans often is codiagnosed with a malignancy, it can precede the cancer diagnosis in some cases.7,8 A majority of cases are associated with adenocarcinomas of the gastrointestinal tract.4 Progressive mucocutaneous papillomatosis also is a rare paraneoplastic condition that most commonly is associated with gastric adenocarcinomas. Progressive mucocutaneous papillomatosis often presents rapidly as verrucous growths on cutaneous surfaces (including the hands and face) but also can affect mucosal surfaces such as the mouth and conjunctiva.9-11 Tripe palms are characterized by exaggerated dermatoglyphics with diffuse palmar ridging and hyperkeratosis. Tripe palms most often are associated with pulmonary malignancies. When tripe palms are present with malignant acanthosis nigricans, they reflect up to a one-third incidence of gastrointestinal malignancy.12,13
Despite the individual presentation of these paraneoplastic signs in a variety of malignancies, synchronous presentation is rare. A brief literature review only identified 6 cases of concurrent acanthosis nigricans, tripe palms, and progressive mucocutaneous papillomatosis with an underlying gastrointestinal malignancy.1,11,14-17 Two additional reports described tripe palms with oral acanthosis nigricans and progressive mucocutaneous papillomatosis in metastatic gastric adenocarcinoma and renal urothelial carcinoma.2,18 An additional case of all 3 paraneoplastic conditions was reported in the setting of metastatic cervical cancer (HPV positive).19 Per a recent case report and literature review,20 there have only been 8 cases of acanthosis nigricans reported in bladder transitional cell carcinoma,20-27 half of which have included oral malignant acanthosis nigricans.20-23 Only one report of concurrent cutaneous and oral malignant acanthosis nigricans and triple palms in the setting of bladder cancer has been reported.20 Given the extensive conjunctival involvement and cutaneous papillomatosis in our patient, ours is a rarely reported case of concurrent malignant mucocutaneous acanthosis nigricans, tripe palms, and progressive papillomatosis in transitional cell bladder carcinoma. We believe it is imperative to consider the role of this malignancy as a cause of these paraneoplastic conditions.
Although these paraneoplastic conditions rarely co-occur, our case further offers a common molecular pathway for these conditions.28 In these paraneoplastic conditions, the stimulating factor is thought to be tumor growth factor α, which is structurally related to epidermal growth factor (EGF). Epidermal growth factor receptors (EGFRs) are found in the basal layer of the epidermis, where activation stimulates keratinocyte growth and leads to the cutaneous manifestation of symptoms.28 Fibroblast growth factor receptor 3 mutations are found in most noninvasive transitional cell tumors of the bladder.29 The fibroblast growth factor pathway is distinctly different from the tumor growth factor α and EGF pathways.30 However, this association with transitional cell carcinoma suggests that fibroblast growth factor receptor 3 also may be implicated in these paraneoplastic conditions.
Our patient responded well to treatment with acitretin 50 mg daily. The mechanism of action of retinoids involves inducing mitotic activity and desmosomal shedding.31 Retinoids downregulate EGFR expression and activation in EGF-stimulated cells.32 We hypothesize that these oral retinoids decreased the growth stimulus and thereby improved cutaneous signs in the setting of our patient’s transitional cell cancer. Although definitive therapy is malignancy management, our case highlights the utility of adjunctive measures such as oral retinoids and surgical debulking. While previous cases have reported use of retinoids at a lower dosage than used in this case, oral lesions often have only been mildly improved with little impact on other cutaneous symptoms.1,2 In one case of malignant acanthosis nigricans and oral papillomatosis, isotretinoin 25 mg once every 2 to 3 days led to a moderate decrease in hyperkeratosis and papillomas, but the patient was lost to follow-up.3 Our case highlights the use of higher daily doses of oral retinoids for over 9 months, resulting in marked improvement in both the mucosal and cutaneous symptoms of acanthosis nigricans, progressive mucocutaneous papillomatosis, and tripe palms. Therefore, oral acitretin should be considered as adjuvant therapy for these paraneoplastic conditions.
By reporting this case, we hope to demonstrate the importance of considering other forms of malignancies in the presence of paraneoplastic conditions. Although gastric malignancies more commonly are associated with these conditions, bladder carcinomas also can present with cutaneous manifestations. The presence of these paraneoplastic conditions alone or together rarely is reported in urologic cancers and generally is considered to be an indicator of poor prognosis. Paraneoplastic conditions often develop rapidly and occur in very advanced malignancies.4 The disfiguring presentation in our case also had unusual diagnostic challenges. The presence of these conditions for 8 years and nonmetastatic advanced malignancy suggest a more indolent process and that these signs are not always an indicator of poor prognosis. Future patients with these paraneoplastic conditions may benefit from both a thorough malignancy screen, including cystoscopy, and high daily doses of oral retinoids.
To the Editor:
A 40-year-old Somalian man presented to the dermatology clinic with lesions on the eyelids, tongue, lips, and hands of 8 years’ duration. He was a former refugee who had faced considerable stigma from his community due to his appearance. A review of systems was remarkable for decreased appetite but no weight loss. He reported no abdominal distention, early satiety, or urinary symptoms, and he had no personal history of diabetes mellitus or obesity. Physical examination demonstrated hyperpigmented velvety plaques in all skin folds and on the genitalia. Massive papillomatosis of the eyelid margins, tongue, and lips also was noted (Figure 1A). Flesh-colored papules also were scattered across the face. Punctate, flesh-colored papules were present on the volar and palmar hands (Figure 2A). Histopathology demonstrated pronounced papillomatous epidermal hyperplasia with negative human papillomavirus (HPV) type 16 and HPV-18 DNA studies. Given the appearance of malignant acanthosis nigricans with oral and conjunctival features, cutaneous papillomatosis, and tripe palms, concern for underlying malignancy was high. Malignancy workup, including upper and lower endoscopy as well as serial computed tomography scans of the chest, abdomen, and pelvis, was unrevealing.
Laboratory investigation revealed a positive Schistosoma IgG antibody (0.38 geometric mean egg count) and peripheral eosinophilia (1.09 ×103/μL), which normalized after praziquantel therapy. With no malignancy identified over the preceding 6-month period, treatment with acitretin 50 mg daily was initiated based on limited literature support.1-3 Treatment led to reduction in the size and number of papillomas (Figure 1B) and tripe palms (Figure 2B) with increased mobility of hands, lips, and tongue. The patient underwent oculoplastic surgery to reduce the papilloma burden along the eyelid margins. Subsequent cystoscopy 9 months after the initial presentation revealed low-grade transitional cell carcinoma of the bladder. Intraoperative mitomycin C led to tumor shrinkage and, with continued treatment with daily acitretin, dramatic improvement of all cutaneous and mucosal symptoms (Figure 1C and Figure 2C). To date, his cutaneous symptoms have resolved.
This case demonstrated a unique presentation of multiple paraneoplastic signs in bladder transitional cell carcinoma. The presence of malignant acanthosis nigricans (including oral and conjunctival involvement), cutaneous papillomatosis, and tripe palms have been individually documented in various types of gastric malignancies.4 Acanthosis nigricans often is secondary to diabetes and obesity, presenting with diffuse, thickened, velvety plaques in the flexural areas. Malignant acanthosis nigricans is a rare, rapidly progressive condition that often presents over a period of weeks to months; it almost always is associated with internal malignancies. It often has more extensive involvement, extending beyond the flexural areas, than typical acanthosis nigricans.4 Oral involvement can be either hypertrophic or papillomatous; papillomatosis of the oral mucosa was reported in over 40% of malignant acanthosis nigricans cases (N=200).5 Cases with conjunctival involvement are less common.6 Although malignant acanthosis nigricans often is codiagnosed with a malignancy, it can precede the cancer diagnosis in some cases.7,8 A majority of cases are associated with adenocarcinomas of the gastrointestinal tract.4 Progressive mucocutaneous papillomatosis also is a rare paraneoplastic condition that most commonly is associated with gastric adenocarcinomas. Progressive mucocutaneous papillomatosis often presents rapidly as verrucous growths on cutaneous surfaces (including the hands and face) but also can affect mucosal surfaces such as the mouth and conjunctiva.9-11 Tripe palms are characterized by exaggerated dermatoglyphics with diffuse palmar ridging and hyperkeratosis. Tripe palms most often are associated with pulmonary malignancies. When tripe palms are present with malignant acanthosis nigricans, they reflect up to a one-third incidence of gastrointestinal malignancy.12,13
Despite the individual presentation of these paraneoplastic signs in a variety of malignancies, synchronous presentation is rare. A brief literature review only identified 6 cases of concurrent acanthosis nigricans, tripe palms, and progressive mucocutaneous papillomatosis with an underlying gastrointestinal malignancy.1,11,14-17 Two additional reports described tripe palms with oral acanthosis nigricans and progressive mucocutaneous papillomatosis in metastatic gastric adenocarcinoma and renal urothelial carcinoma.2,18 An additional case of all 3 paraneoplastic conditions was reported in the setting of metastatic cervical cancer (HPV positive).19 Per a recent case report and literature review,20 there have only been 8 cases of acanthosis nigricans reported in bladder transitional cell carcinoma,20-27 half of which have included oral malignant acanthosis nigricans.20-23 Only one report of concurrent cutaneous and oral malignant acanthosis nigricans and triple palms in the setting of bladder cancer has been reported.20 Given the extensive conjunctival involvement and cutaneous papillomatosis in our patient, ours is a rarely reported case of concurrent malignant mucocutaneous acanthosis nigricans, tripe palms, and progressive papillomatosis in transitional cell bladder carcinoma. We believe it is imperative to consider the role of this malignancy as a cause of these paraneoplastic conditions.
Although these paraneoplastic conditions rarely co-occur, our case further offers a common molecular pathway for these conditions.28 In these paraneoplastic conditions, the stimulating factor is thought to be tumor growth factor α, which is structurally related to epidermal growth factor (EGF). Epidermal growth factor receptors (EGFRs) are found in the basal layer of the epidermis, where activation stimulates keratinocyte growth and leads to the cutaneous manifestation of symptoms.28 Fibroblast growth factor receptor 3 mutations are found in most noninvasive transitional cell tumors of the bladder.29 The fibroblast growth factor pathway is distinctly different from the tumor growth factor α and EGF pathways.30 However, this association with transitional cell carcinoma suggests that fibroblast growth factor receptor 3 also may be implicated in these paraneoplastic conditions.
Our patient responded well to treatment with acitretin 50 mg daily. The mechanism of action of retinoids involves inducing mitotic activity and desmosomal shedding.31 Retinoids downregulate EGFR expression and activation in EGF-stimulated cells.32 We hypothesize that these oral retinoids decreased the growth stimulus and thereby improved cutaneous signs in the setting of our patient’s transitional cell cancer. Although definitive therapy is malignancy management, our case highlights the utility of adjunctive measures such as oral retinoids and surgical debulking. While previous cases have reported use of retinoids at a lower dosage than used in this case, oral lesions often have only been mildly improved with little impact on other cutaneous symptoms.1,2 In one case of malignant acanthosis nigricans and oral papillomatosis, isotretinoin 25 mg once every 2 to 3 days led to a moderate decrease in hyperkeratosis and papillomas, but the patient was lost to follow-up.3 Our case highlights the use of higher daily doses of oral retinoids for over 9 months, resulting in marked improvement in both the mucosal and cutaneous symptoms of acanthosis nigricans, progressive mucocutaneous papillomatosis, and tripe palms. Therefore, oral acitretin should be considered as adjuvant therapy for these paraneoplastic conditions.
By reporting this case, we hope to demonstrate the importance of considering other forms of malignancies in the presence of paraneoplastic conditions. Although gastric malignancies more commonly are associated with these conditions, bladder carcinomas also can present with cutaneous manifestations. The presence of these paraneoplastic conditions alone or together rarely is reported in urologic cancers and generally is considered to be an indicator of poor prognosis. Paraneoplastic conditions often develop rapidly and occur in very advanced malignancies.4 The disfiguring presentation in our case also had unusual diagnostic challenges. The presence of these conditions for 8 years and nonmetastatic advanced malignancy suggest a more indolent process and that these signs are not always an indicator of poor prognosis. Future patients with these paraneoplastic conditions may benefit from both a thorough malignancy screen, including cystoscopy, and high daily doses of oral retinoids.
- Stawczyk-Macieja M, Szczerkowska-Dobosz A, Nowicki R, et al. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma. Postepy Dermatol Alergol. 2014;31:56-58.
- Lee HC, Ker KJ, Chong W-S. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Swineford SL, Drucker CR. Palliative treatment of paraneoplastic acanthosis nigricans and oral florid papillomatosis with retinoids. J Drugs Dermatol. 2010;9:1151-1153.
- Wick MR, Patterson JW. Cutaneous paraneoplastic syndromes [published online January 31, 2019]. Semin Diagn Pathol. 2019;36:211-228.
- Tyler MT, Ficarra G, Silverman S, et al. Malignant acanthosis nigricans with florid papillary oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:445-449.
- Zhang X, Liu R, Liu Y, et al. Malignant acanthosis nigricans: a case report. BMC Ophthalmology. 2020;20:1-4.
- Curth HO. Dermatoses and malignant internal tumours. Arch Dermatol Syphil. 1955;71:95-107.
- Krawczyk M, Mykala-Cies´la J, Kolodziej-Jaskula A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
- Singhi MK, Gupta LK, Bansal M, et al. Florid cutaneous papillomatosis with adenocarcinoma of stomach in a 35 year old male. Indian J Dermatol Venereol Leprol. 2005;71:195-196.
- Klieb HB, Avon SL, Gilbert J, et al. Florid cutaneous and mucosal papillomatosis: mucocutaneous markers of an underlying gastric malignancy. J Clin Oncol. 2013;31:E218-E219.
- Yang YH, Zhang RZ, Kang DH, et al. Three paraneoplastic signs in the same patient with gastric adenocarcinoma. Dermatol Online J. 2013;19:18966.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Chantarojanasiri T, Buranathawornsom A, Sirinawasatien A. Diffuse esophageal squamous papillomatosis: a rare disease associated with acanthosis nigricans and tripe palms. Case Rep Gastroenterol. 2020;14:702-706.
- Muhammad R, Iftikhar N, Sarfraz T, et al. Malignant acanthosis nigricans: an indicator of internal malignancy. J Coll Physicians Surg Pak. 2019;29:888-890.
- Brinca A, Cardoso JC, Brites MM, et al. Florid cutaneous papillomatosis and acanthosis nigricans maligna revealing gastric adenocarcinoma. An Bras Dermatol. 2011;86:573-577.
- Vilas-Sueiro A, Suárez-Amor O, Monteagudo B, et al. Malignant acanthosis nigricans, florid cutaneous and mucosal papillomatosis, and tripe palms in a man with gastric adenocarcinoma. Actas Dermosifiliogr. 2015;106:438-439.
- Paravina M, Ljubisavljevic´ D. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma—a case report. Serbian J Dermatology Venereol. 2015;7:5-14.
- Kleikamp S, Böhm M, Frosch P, et al. Acanthosis nigricans, papillomatosis mucosae and “tripe” palms in a patient with metastasized gastric carcinoma [in German]. Dtsch Med Wochenschr. 2006;131:1209-1213.
- Mikhail GR, Fachnie DM, Drukker BH, et al. Generalized malignant acanthosis nigricans. Arch Dermatol. 1979;115:201-202.
- Zhang R, Jiang M, Lei W, et al. Malignant acanthosis nigricans with recurrent bladder cancer: a case report and review of literature. Onco Targets Ther. 2021;14:951.
- Olek-Hrab K, Silny W, Zaba R, et al. Co-occurrence of acanthosis nigricans and bladder adenocarcinoma-case report. Contemp Oncol (Pozn). 2013;17:327-330.
- Canjuga I, Mravak-Stipetic´ M, Kopic´V, et al. Oral acanthosis nigricans: case report and comparison with literature reports. Acta Dermatovenerol Croat. 2008;16:91-95.
- Cairo F, Rubino I, Rotundo R, et al. Oral acanthosis nigricans as a marker of internal malignancy. a case report. J Periodontol. 2001;72:1271-1275.
- Möhrenschlager M, Vocks E, Wessner DB, et al. 2001;165:1629-1630.
- Singh GK, Sen D, Mulajker DS, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Indian J Dermatol. 2011;56:722-725.
- Gohji K, Hasunuma Y, Gotoh A, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Int J Dermatol. 1994;33:433-435.
- Pinto WBVR, Badia BML, Souza PVS, et al. Paraneoplastic motor neuronopathy and malignant acanthosis nigricans. Arq Neuropsiquiatr. 2019;77:527.
- Koyama S, Ikeda K, Sato M, et al. Transforming growth factor–alpha (TGF-alpha)-producing gastric carcinoma with acanthosis nigricans: an endocrine effect of TGF alpha in the pathogenesis of cutaneous paraneoplastic syndrome and epithelial hyperplasia of the esophagus. J Gastroenterol. 1997;32:71-77.
- Billerey C, Chopin D, Aubriot-Lorton MH, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955-1959.
- Lee C-J, Lee M-H, Cho Y-Y. Fibroblast and epidermal growth factors utilize different signaling pathways to induce anchorage-independent cell transformation in JB6 Cl41 mouse skin epidermal cells. J Cancer Prev. 2014;19:199-208.
- Darmstadt GL, Yokel BK, Horn TD. Treatment of acanthosis nigricans with tretinoin. Arch Dermatol. 1991;127:1139-1140.
- Sah JF, Eckert RL, Chandraratna RA, et al. Retinoids suppress epidermal growth factor–associated cell proliferation by inhibiting epidermal growth factor receptor–dependent ERK1/2 activation. J Biol Chem. 2002;277:9728-9735.
- Stawczyk-Macieja M, Szczerkowska-Dobosz A, Nowicki R, et al. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma. Postepy Dermatol Alergol. 2014;31:56-58.
- Lee HC, Ker KJ, Chong W-S. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Swineford SL, Drucker CR. Palliative treatment of paraneoplastic acanthosis nigricans and oral florid papillomatosis with retinoids. J Drugs Dermatol. 2010;9:1151-1153.
- Wick MR, Patterson JW. Cutaneous paraneoplastic syndromes [published online January 31, 2019]. Semin Diagn Pathol. 2019;36:211-228.
- Tyler MT, Ficarra G, Silverman S, et al. Malignant acanthosis nigricans with florid papillary oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:445-449.
- Zhang X, Liu R, Liu Y, et al. Malignant acanthosis nigricans: a case report. BMC Ophthalmology. 2020;20:1-4.
- Curth HO. Dermatoses and malignant internal tumours. Arch Dermatol Syphil. 1955;71:95-107.
- Krawczyk M, Mykala-Cies´la J, Kolodziej-Jaskula A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
- Singhi MK, Gupta LK, Bansal M, et al. Florid cutaneous papillomatosis with adenocarcinoma of stomach in a 35 year old male. Indian J Dermatol Venereol Leprol. 2005;71:195-196.
- Klieb HB, Avon SL, Gilbert J, et al. Florid cutaneous and mucosal papillomatosis: mucocutaneous markers of an underlying gastric malignancy. J Clin Oncol. 2013;31:E218-E219.
- Yang YH, Zhang RZ, Kang DH, et al. Three paraneoplastic signs in the same patient with gastric adenocarcinoma. Dermatol Online J. 2013;19:18966.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Chantarojanasiri T, Buranathawornsom A, Sirinawasatien A. Diffuse esophageal squamous papillomatosis: a rare disease associated with acanthosis nigricans and tripe palms. Case Rep Gastroenterol. 2020;14:702-706.
- Muhammad R, Iftikhar N, Sarfraz T, et al. Malignant acanthosis nigricans: an indicator of internal malignancy. J Coll Physicians Surg Pak. 2019;29:888-890.
- Brinca A, Cardoso JC, Brites MM, et al. Florid cutaneous papillomatosis and acanthosis nigricans maligna revealing gastric adenocarcinoma. An Bras Dermatol. 2011;86:573-577.
- Vilas-Sueiro A, Suárez-Amor O, Monteagudo B, et al. Malignant acanthosis nigricans, florid cutaneous and mucosal papillomatosis, and tripe palms in a man with gastric adenocarcinoma. Actas Dermosifiliogr. 2015;106:438-439.
- Paravina M, Ljubisavljevic´ D. Malignant acanthosis nigricans, florid cutaneous papillomatosis and tripe palms syndrome associated with gastric adenocarcinoma—a case report. Serbian J Dermatology Venereol. 2015;7:5-14.
- Kleikamp S, Böhm M, Frosch P, et al. Acanthosis nigricans, papillomatosis mucosae and “tripe” palms in a patient with metastasized gastric carcinoma [in German]. Dtsch Med Wochenschr. 2006;131:1209-1213.
- Mikhail GR, Fachnie DM, Drukker BH, et al. Generalized malignant acanthosis nigricans. Arch Dermatol. 1979;115:201-202.
- Zhang R, Jiang M, Lei W, et al. Malignant acanthosis nigricans with recurrent bladder cancer: a case report and review of literature. Onco Targets Ther. 2021;14:951.
- Olek-Hrab K, Silny W, Zaba R, et al. Co-occurrence of acanthosis nigricans and bladder adenocarcinoma-case report. Contemp Oncol (Pozn). 2013;17:327-330.
- Canjuga I, Mravak-Stipetic´ M, Kopic´V, et al. Oral acanthosis nigricans: case report and comparison with literature reports. Acta Dermatovenerol Croat. 2008;16:91-95.
- Cairo F, Rubino I, Rotundo R, et al. Oral acanthosis nigricans as a marker of internal malignancy. a case report. J Periodontol. 2001;72:1271-1275.
- Möhrenschlager M, Vocks E, Wessner DB, et al. 2001;165:1629-1630.
- Singh GK, Sen D, Mulajker DS, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Indian J Dermatol. 2011;56:722-725.
- Gohji K, Hasunuma Y, Gotoh A, et al. Acanthosis nigricans associated with transitional cell carcinoma of the urinary bladder. Int J Dermatol. 1994;33:433-435.
- Pinto WBVR, Badia BML, Souza PVS, et al. Paraneoplastic motor neuronopathy and malignant acanthosis nigricans. Arq Neuropsiquiatr. 2019;77:527.
- Koyama S, Ikeda K, Sato M, et al. Transforming growth factor–alpha (TGF-alpha)-producing gastric carcinoma with acanthosis nigricans: an endocrine effect of TGF alpha in the pathogenesis of cutaneous paraneoplastic syndrome and epithelial hyperplasia of the esophagus. J Gastroenterol. 1997;32:71-77.
- Billerey C, Chopin D, Aubriot-Lorton MH, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955-1959.
- Lee C-J, Lee M-H, Cho Y-Y. Fibroblast and epidermal growth factors utilize different signaling pathways to induce anchorage-independent cell transformation in JB6 Cl41 mouse skin epidermal cells. J Cancer Prev. 2014;19:199-208.
- Darmstadt GL, Yokel BK, Horn TD. Treatment of acanthosis nigricans with tretinoin. Arch Dermatol. 1991;127:1139-1140.
- Sah JF, Eckert RL, Chandraratna RA, et al. Retinoids suppress epidermal growth factor–associated cell proliferation by inhibiting epidermal growth factor receptor–dependent ERK1/2 activation. J Biol Chem. 2002;277:9728-9735.
Practice Points
- Paraneoplastic conditions may present secondary to urologic malignancy. Providers should perform thorough malignancy screening, including urologic cystoscopy, in patients presenting with paraneoplastic signs and no identified malignancy.
- Oral retinoids, such as acitretin, may be used as an adjuvant treatment to treat paraneoplastic cutaneous symptoms. The definitive treatment is malignancy management.
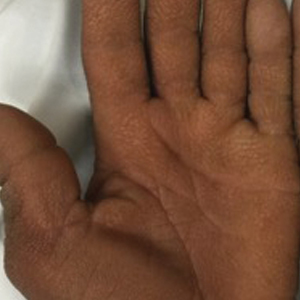
Kikuchi-Fujimoto Disease in an Adolescent Boy
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.

Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
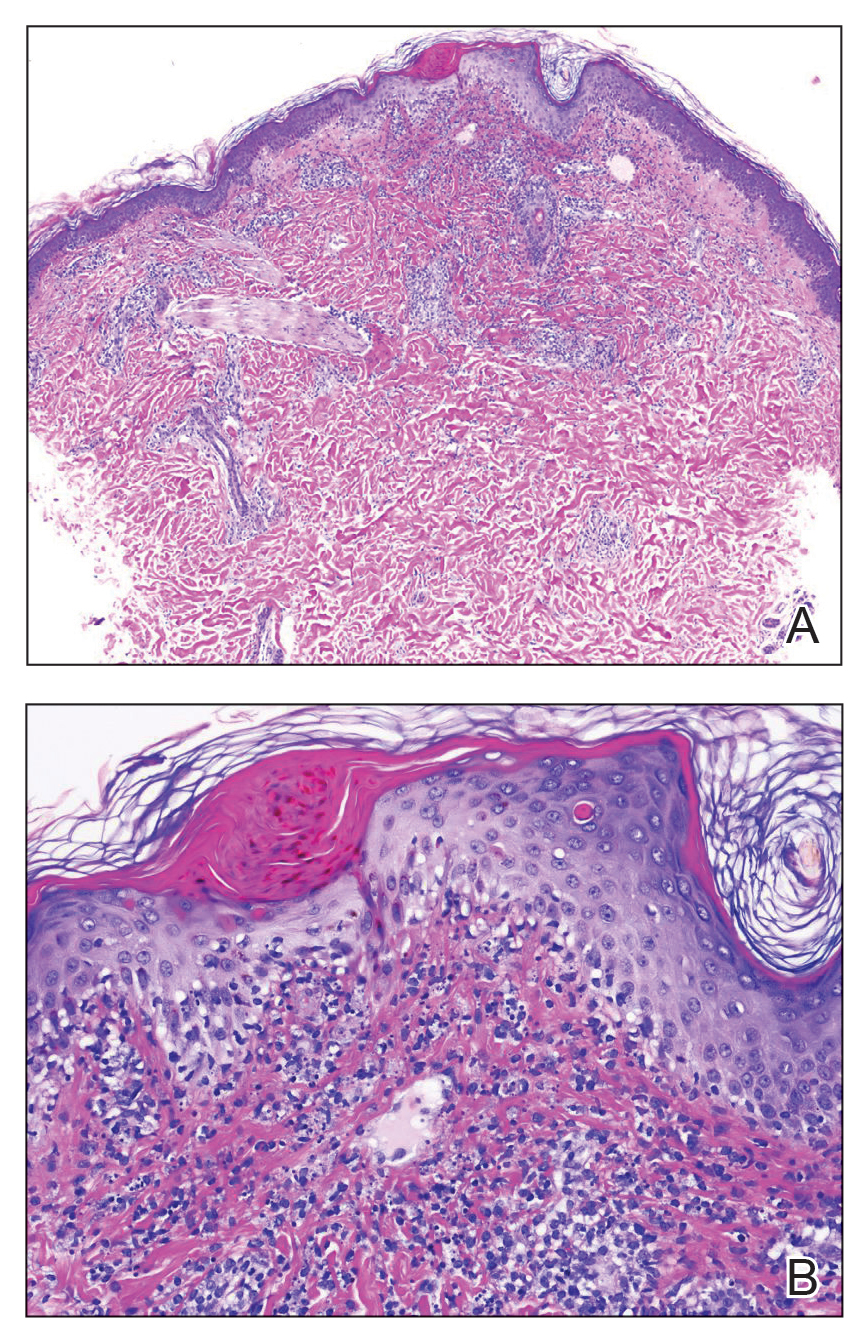
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.

Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
Practice Points
- Kikuchi-Fujimoto disease is an uncommon, self-limited condition characterized by benign lymphadenopathy and variable systemic symptoms.
- Definitive diagnosis is made by excisional lymph node biopsy.
- Treatment options include oral corticosteroids, nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.
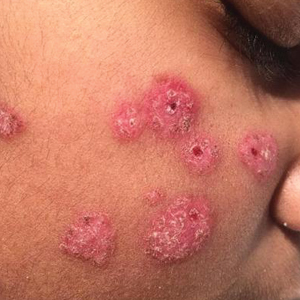
Botulinum Toxin for the Treatment of Intractable Raynaud Phenomenon
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
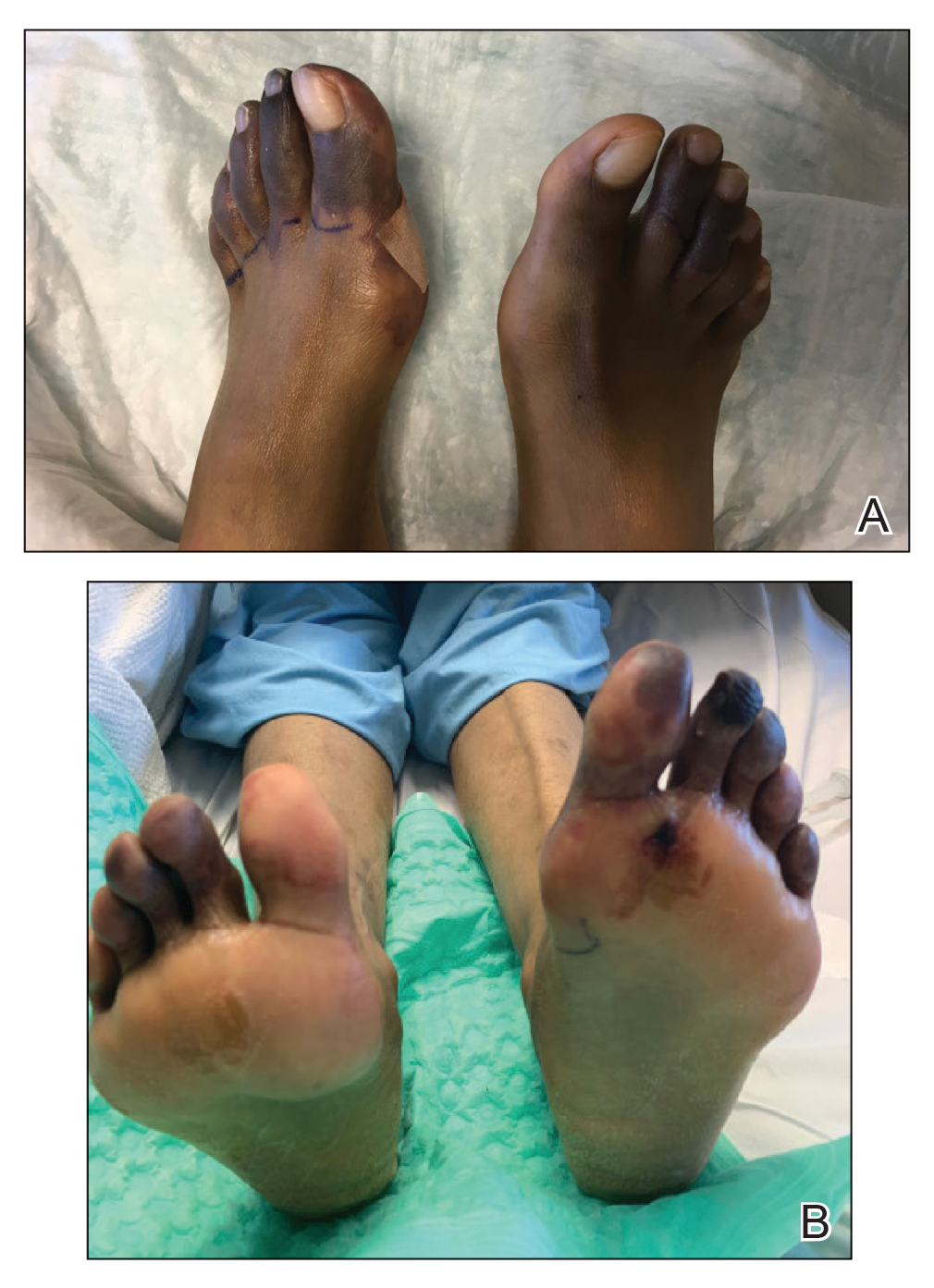
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
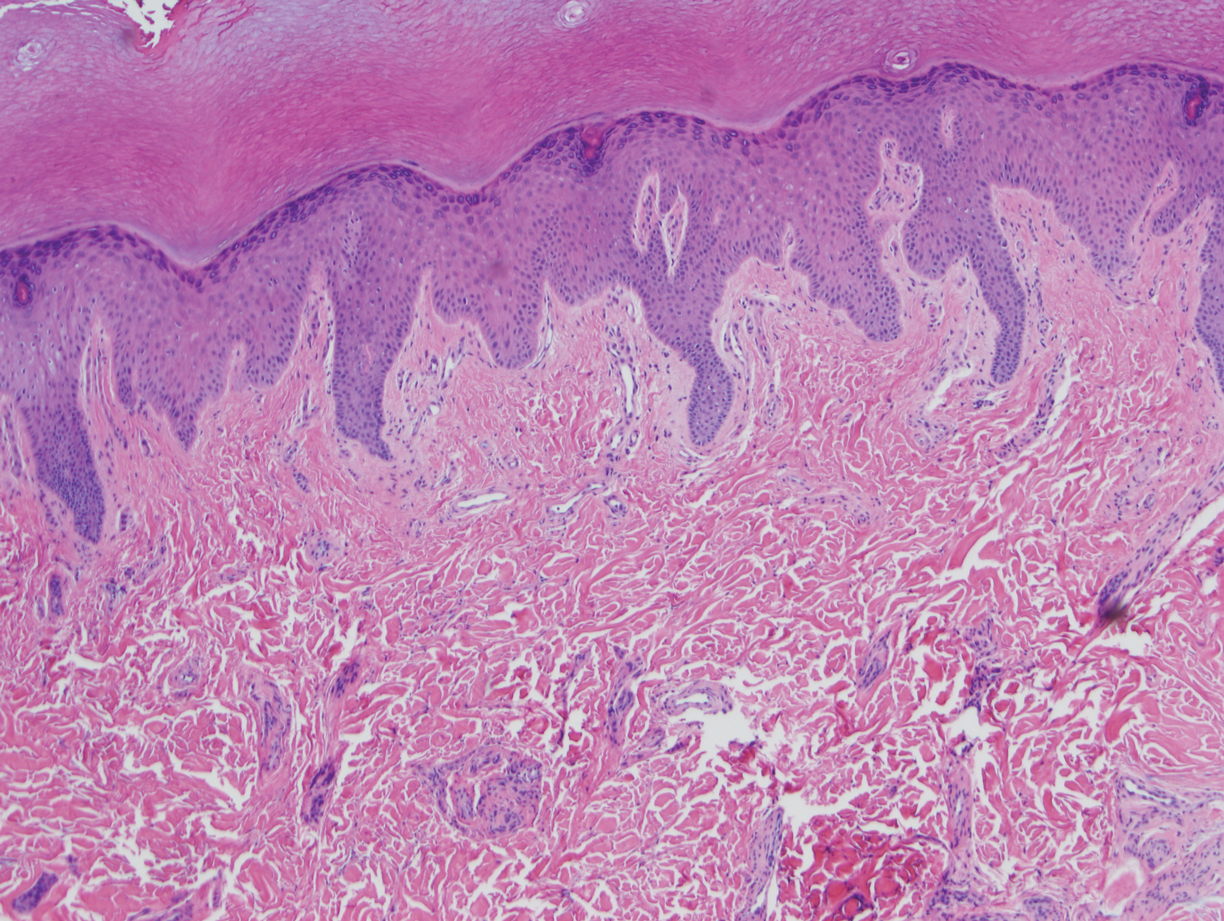
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
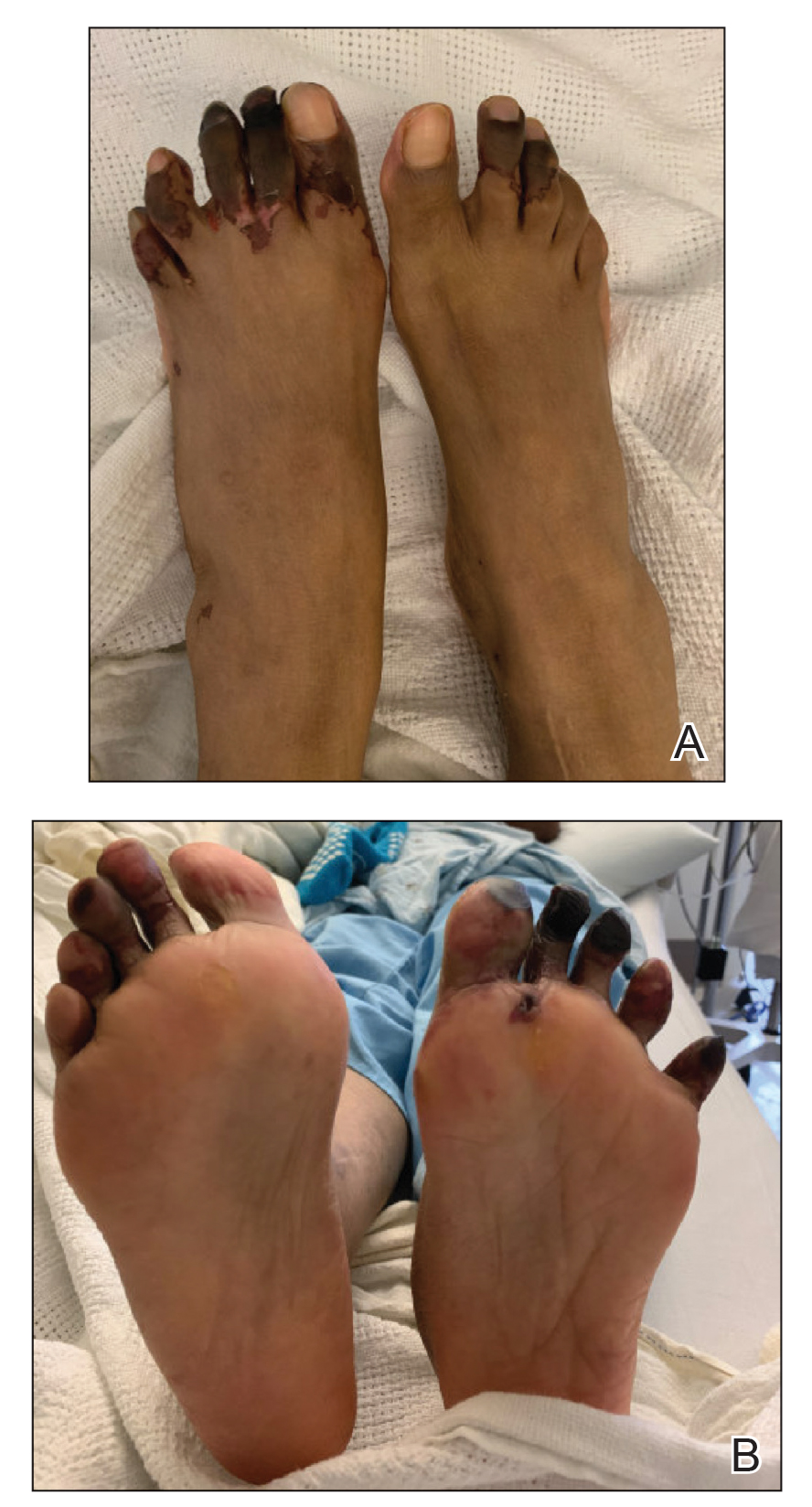
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
Practice Points
- Raynaud phenomenon (RP) is a vascular disorder characterized by episodic vasospasms of the digits often due to cold temperature or stress.
- OnabotulinumtoxinA has been implemented as a treatment of intractable RP after failure with traditional treatments, such as calcium channel blockers, angiotensin receptor blockers, prostaglandins, endothelin receptor blockers, and phosphodiesterase 5 inhibitors.
- A standard technique of delivery of onabotulinumtoxinA involves injection of 5 U/mL into the medial and lateral aspects of each finger at its base (near the metacarpal head) for a total of 50 U per hand or foot.
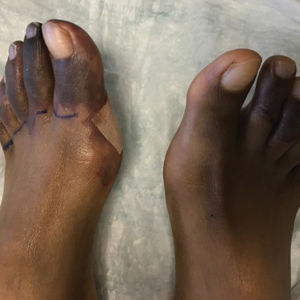
Flagellate Shiitake Mushroom Reaction With Histologic Features of Acute Generalized Exanthematous Pustulosis
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.

A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.

The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
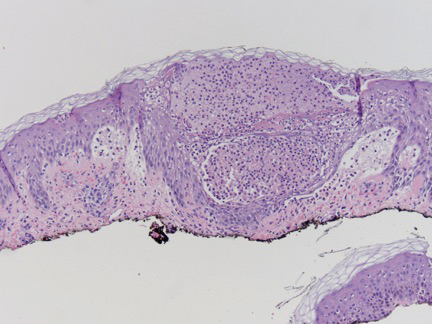
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.
A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.
The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.
A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.
The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
Practice Points
- Ingestion of shiitake mushrooms and bleomycin is associated with flagellate dermatitis.
- Acute generalized exanthematous pustulosis (AGEP) is a rare condition associated with certain drug ingestion.
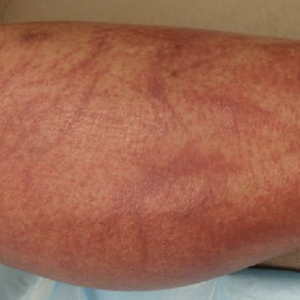
Overlapping Phenotypic Features of PTEN Hamartoma Tumor Syndrome and Birt-Hogg-Dubé Syndrome
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.

Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).

Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.

The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10

Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.

Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
PRACTICE POINTS
- PTEN hamartoma tumor syndrome (PHTS) represents a spectrum of disorders caused by autosomal-dominant germline mutations in PTEN.
- Our patient presented with phenotypic features of PHTS and Birt-Hogg-Dubé syndrome. Given that both syndromes cause alterations in mammalian target of rapamycin signaling, overlapping phenotypic features may be seen.
- Recognizing overlapping phenotypic features of these syndromes will allow for timely diagnosis and surveillance for malignancy.
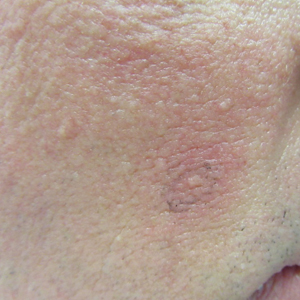
A Severe Presentation of Plasma Cell Cheilitis
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
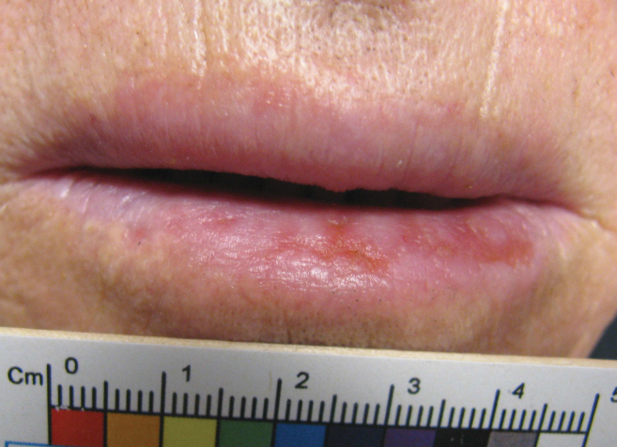
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
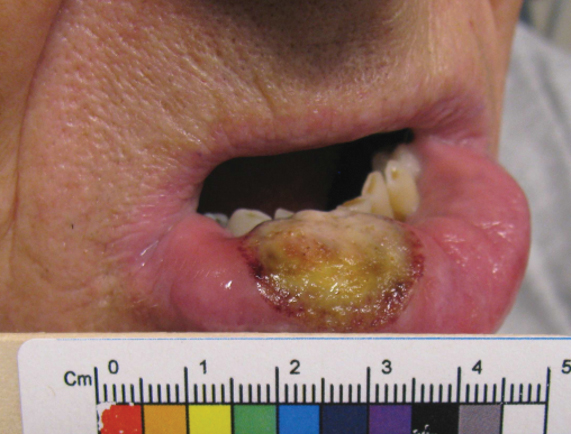
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
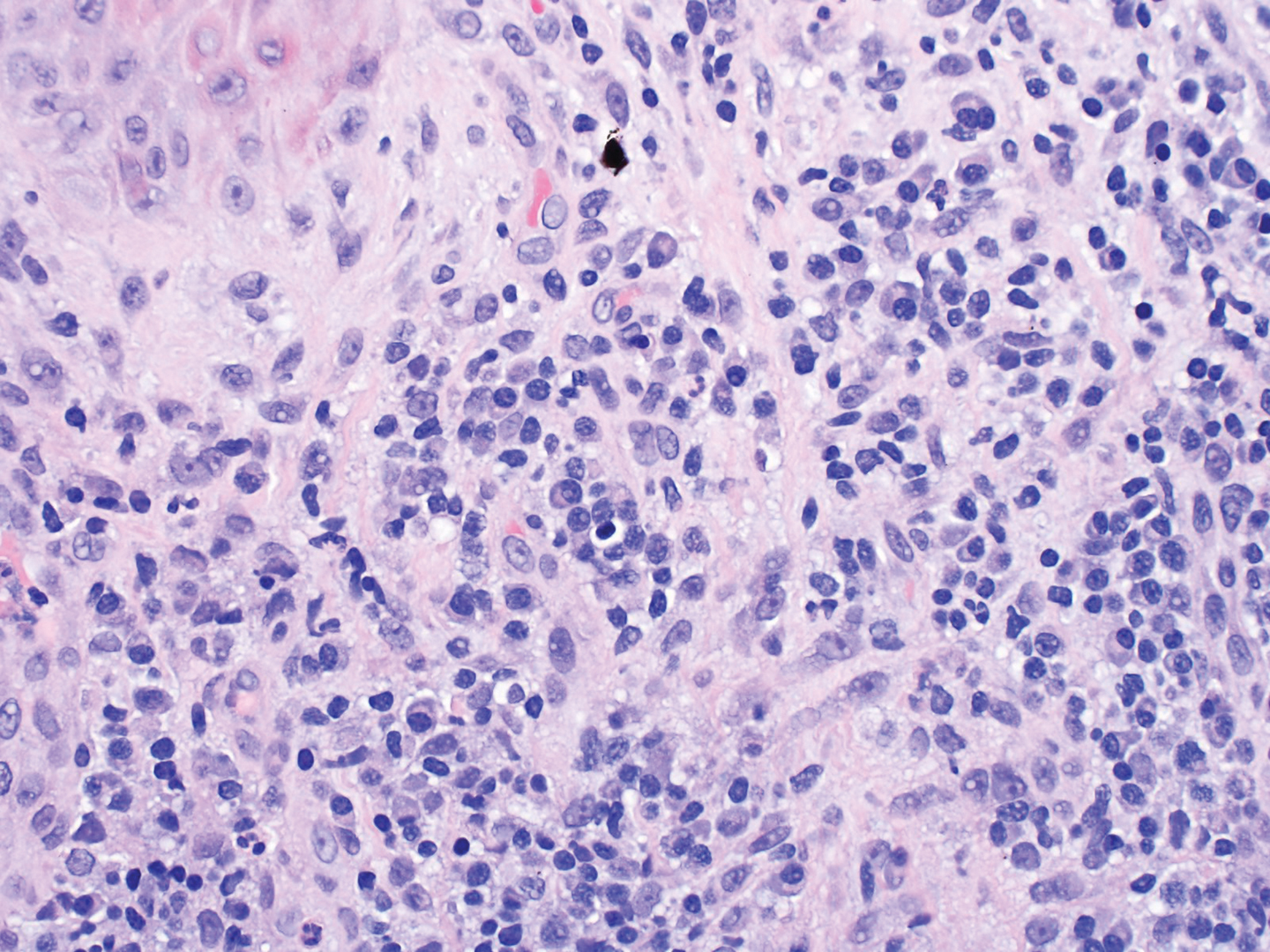
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
PRACTICE POINTS
- Plasma cell cheilitis (PCC) is a benign condition that affects the lower lip in older individuals, presenting as a nonspecific, red-brown patch or plaque that can progress slowly to erosions and edema.
- Our patient with PCC experienced full resolution of symptoms with application of a class I topical corticosteroid.