User login
At-Home Treatment of Pigmented Lesions With a Zinc Chloride Preparation
To the Editor:
Zinc chloride originally was used by Dr. Frederic Mohs as an in vivo tissue fixative during the early phases of Mohs micrographic surgery.1 Although this technique has since been replaced with fresh frozen tissue fixation, zinc chloride still is found in topical preparations that are readily available to patients. Specifically, black salve describes variably composed topical preparations that share the common ingredients zinc chloride and Sanguinaria canadensis (bloodroot).2 Patients self-treat with these unregulated compounds, but the majority do not have their lesions evaluated by a clinician prior to use and are unaware of the potential risks.3-5 Products containing zinc chloride and S canadensis that are not marketed as black salve present a new problem for the dermatology community.
A 73-year-old man presented to our dermatology clinic for the focused evaluation of scaly lesions on the face and nose. At this visit, it was recommended he undergo a total-body skin examination for skin cancer screening given his age and substantial photodamage.
Physical examination revealed more than 20 superficial, 3- to 10-mm scars predominantly over the trunk. One scar over the left mid-back had a large, 1.2-cm peripheral rim of dark brown pigment that was clinically concerning for a melanocytic neoplasm. Shave removal of this lesion was performed. Histologic examination showed melanoma in situ with a central scar. The central scar spanned the depth of the dermis, and the melanocytic component was absent in this area, raising the question if prior biopsy or treatment had been performed on this lesion. During a discussion of the results with the patient, he was questioned about prior biopsy or treatment of this lesion. He reported prior use of a topical all-natural cream containing zinc chloride and S canadensis that he purchased online, which he had used to treat this lesion as well as numerous presumed moles.
The trend of at-home mole removal products containing the traditional ingredients in black salve seems to be one of rapidly shifting product availability as well as a departure from marketing items as black salve. Many prior black salve products are no longer available.4 The product that our patient used is a topical cream marketed as a treatment for moles and skin tags.6 Despite not being marketed as black salve, it does contain zinc chloride and S canadensis. The product’s website highlights these ingredients as being a safe and effective treatment for mole removal, with claims that the product will remove the mole or skin tag without irritating the surrounding skin and can be safely used anywhere on the body without scarring.6 A Google search at the time this article was written using the term skin tag remover revealed similar products marketed as all-natural “skin tag remover and mole corrector creams.” These similar products containing zinc chloride and S canadensis were available in the United States at the time of our initial research but have since been removed and only are available outside of the United States.7
Prior reports of melanoma masked by zinc chloride and S canadensis described the use of topical agents marketed as black salve. This new wave of products marketed as all-natural creams makes continued education on the available products and their associated risks necessary for clinicians. The lack of US Food and Drug Administration oversight for these products and their frequent introduction and discontinuation in the market makes keeping updated even more challenging. Because many patients self-treat without prior evaluation by a health care provider, treatment with these products can lead to a delay in diagnosis or inaccurate staging due to scars from the chemical destruction, both of which may have occurred in our patient.5 Until these products become regulated by the US Food and Drug Administration, it is imperative that clinicians continue to educate their patients on the lack of documented benefit and clear risks of their use as well as remain up-to-date on product trends.
- Cohen DK. Mohs micrographic surgery: past, present, and future. Dermatol Surg. 2019;45:329-339. doi:10.1097/DSS.0000000000001701
- Eastman KL. A review of topical corrosive black salve. J Altern Complement Med. 2014;20:284-289. doi:10.1089/acm.2012.0377
- Sivyer GW, Rosendahl C. Application of black salve to a thin melanoma that subsequently progressed to metastatic melanoma: a case study. Dermatol Pract Concept. 2014;4:77-80. doi:10.5826/dpc.0403a16
- McDaniel S. Consequences of using escharotic agents as primary treatment for nonmelanoma skin cancer. Arch Dermatol. 2002;138:1593-1596.
- Clark JJ. Community perceptions about the use of black salve. J Am Acad Dermatol. 2016;74:1021-1023. doi:10.1016/j.jaad.2015.10.016
- Skinprov Cream. Skinprov. Accessed February 22, 2022. https://skinprov.net
- HaloDerm. HaloDerm Inc. Accessed February 22, 2022. https://haloderm.com/
To the Editor:
Zinc chloride originally was used by Dr. Frederic Mohs as an in vivo tissue fixative during the early phases of Mohs micrographic surgery.1 Although this technique has since been replaced with fresh frozen tissue fixation, zinc chloride still is found in topical preparations that are readily available to patients. Specifically, black salve describes variably composed topical preparations that share the common ingredients zinc chloride and Sanguinaria canadensis (bloodroot).2 Patients self-treat with these unregulated compounds, but the majority do not have their lesions evaluated by a clinician prior to use and are unaware of the potential risks.3-5 Products containing zinc chloride and S canadensis that are not marketed as black salve present a new problem for the dermatology community.
A 73-year-old man presented to our dermatology clinic for the focused evaluation of scaly lesions on the face and nose. At this visit, it was recommended he undergo a total-body skin examination for skin cancer screening given his age and substantial photodamage.
Physical examination revealed more than 20 superficial, 3- to 10-mm scars predominantly over the trunk. One scar over the left mid-back had a large, 1.2-cm peripheral rim of dark brown pigment that was clinically concerning for a melanocytic neoplasm. Shave removal of this lesion was performed. Histologic examination showed melanoma in situ with a central scar. The central scar spanned the depth of the dermis, and the melanocytic component was absent in this area, raising the question if prior biopsy or treatment had been performed on this lesion. During a discussion of the results with the patient, he was questioned about prior biopsy or treatment of this lesion. He reported prior use of a topical all-natural cream containing zinc chloride and S canadensis that he purchased online, which he had used to treat this lesion as well as numerous presumed moles.
The trend of at-home mole removal products containing the traditional ingredients in black salve seems to be one of rapidly shifting product availability as well as a departure from marketing items as black salve. Many prior black salve products are no longer available.4 The product that our patient used is a topical cream marketed as a treatment for moles and skin tags.6 Despite not being marketed as black salve, it does contain zinc chloride and S canadensis. The product’s website highlights these ingredients as being a safe and effective treatment for mole removal, with claims that the product will remove the mole or skin tag without irritating the surrounding skin and can be safely used anywhere on the body without scarring.6 A Google search at the time this article was written using the term skin tag remover revealed similar products marketed as all-natural “skin tag remover and mole corrector creams.” These similar products containing zinc chloride and S canadensis were available in the United States at the time of our initial research but have since been removed and only are available outside of the United States.7
Prior reports of melanoma masked by zinc chloride and S canadensis described the use of topical agents marketed as black salve. This new wave of products marketed as all-natural creams makes continued education on the available products and their associated risks necessary for clinicians. The lack of US Food and Drug Administration oversight for these products and their frequent introduction and discontinuation in the market makes keeping updated even more challenging. Because many patients self-treat without prior evaluation by a health care provider, treatment with these products can lead to a delay in diagnosis or inaccurate staging due to scars from the chemical destruction, both of which may have occurred in our patient.5 Until these products become regulated by the US Food and Drug Administration, it is imperative that clinicians continue to educate their patients on the lack of documented benefit and clear risks of their use as well as remain up-to-date on product trends.
To the Editor:
Zinc chloride originally was used by Dr. Frederic Mohs as an in vivo tissue fixative during the early phases of Mohs micrographic surgery.1 Although this technique has since been replaced with fresh frozen tissue fixation, zinc chloride still is found in topical preparations that are readily available to patients. Specifically, black salve describes variably composed topical preparations that share the common ingredients zinc chloride and Sanguinaria canadensis (bloodroot).2 Patients self-treat with these unregulated compounds, but the majority do not have their lesions evaluated by a clinician prior to use and are unaware of the potential risks.3-5 Products containing zinc chloride and S canadensis that are not marketed as black salve present a new problem for the dermatology community.
A 73-year-old man presented to our dermatology clinic for the focused evaluation of scaly lesions on the face and nose. At this visit, it was recommended he undergo a total-body skin examination for skin cancer screening given his age and substantial photodamage.
Physical examination revealed more than 20 superficial, 3- to 10-mm scars predominantly over the trunk. One scar over the left mid-back had a large, 1.2-cm peripheral rim of dark brown pigment that was clinically concerning for a melanocytic neoplasm. Shave removal of this lesion was performed. Histologic examination showed melanoma in situ with a central scar. The central scar spanned the depth of the dermis, and the melanocytic component was absent in this area, raising the question if prior biopsy or treatment had been performed on this lesion. During a discussion of the results with the patient, he was questioned about prior biopsy or treatment of this lesion. He reported prior use of a topical all-natural cream containing zinc chloride and S canadensis that he purchased online, which he had used to treat this lesion as well as numerous presumed moles.
The trend of at-home mole removal products containing the traditional ingredients in black salve seems to be one of rapidly shifting product availability as well as a departure from marketing items as black salve. Many prior black salve products are no longer available.4 The product that our patient used is a topical cream marketed as a treatment for moles and skin tags.6 Despite not being marketed as black salve, it does contain zinc chloride and S canadensis. The product’s website highlights these ingredients as being a safe and effective treatment for mole removal, with claims that the product will remove the mole or skin tag without irritating the surrounding skin and can be safely used anywhere on the body without scarring.6 A Google search at the time this article was written using the term skin tag remover revealed similar products marketed as all-natural “skin tag remover and mole corrector creams.” These similar products containing zinc chloride and S canadensis were available in the United States at the time of our initial research but have since been removed and only are available outside of the United States.7
Prior reports of melanoma masked by zinc chloride and S canadensis described the use of topical agents marketed as black salve. This new wave of products marketed as all-natural creams makes continued education on the available products and their associated risks necessary for clinicians. The lack of US Food and Drug Administration oversight for these products and their frequent introduction and discontinuation in the market makes keeping updated even more challenging. Because many patients self-treat without prior evaluation by a health care provider, treatment with these products can lead to a delay in diagnosis or inaccurate staging due to scars from the chemical destruction, both of which may have occurred in our patient.5 Until these products become regulated by the US Food and Drug Administration, it is imperative that clinicians continue to educate their patients on the lack of documented benefit and clear risks of their use as well as remain up-to-date on product trends.
- Cohen DK. Mohs micrographic surgery: past, present, and future. Dermatol Surg. 2019;45:329-339. doi:10.1097/DSS.0000000000001701
- Eastman KL. A review of topical corrosive black salve. J Altern Complement Med. 2014;20:284-289. doi:10.1089/acm.2012.0377
- Sivyer GW, Rosendahl C. Application of black salve to a thin melanoma that subsequently progressed to metastatic melanoma: a case study. Dermatol Pract Concept. 2014;4:77-80. doi:10.5826/dpc.0403a16
- McDaniel S. Consequences of using escharotic agents as primary treatment for nonmelanoma skin cancer. Arch Dermatol. 2002;138:1593-1596.
- Clark JJ. Community perceptions about the use of black salve. J Am Acad Dermatol. 2016;74:1021-1023. doi:10.1016/j.jaad.2015.10.016
- Skinprov Cream. Skinprov. Accessed February 22, 2022. https://skinprov.net
- HaloDerm. HaloDerm Inc. Accessed February 22, 2022. https://haloderm.com/
- Cohen DK. Mohs micrographic surgery: past, present, and future. Dermatol Surg. 2019;45:329-339. doi:10.1097/DSS.0000000000001701
- Eastman KL. A review of topical corrosive black salve. J Altern Complement Med. 2014;20:284-289. doi:10.1089/acm.2012.0377
- Sivyer GW, Rosendahl C. Application of black salve to a thin melanoma that subsequently progressed to metastatic melanoma: a case study. Dermatol Pract Concept. 2014;4:77-80. doi:10.5826/dpc.0403a16
- McDaniel S. Consequences of using escharotic agents as primary treatment for nonmelanoma skin cancer. Arch Dermatol. 2002;138:1593-1596.
- Clark JJ. Community perceptions about the use of black salve. J Am Acad Dermatol. 2016;74:1021-1023. doi:10.1016/j.jaad.2015.10.016
- Skinprov Cream. Skinprov. Accessed February 22, 2022. https://skinprov.net
- HaloDerm. HaloDerm Inc. Accessed February 22, 2022. https://haloderm.com/
Practice Points
- Zinc chloride preparations are readily available over the counter and unregulated.
- Patients may attempt to self-treat pigmented lesions based on claims they see online.
- When asking patients about prior treatments, it may be prudent to specifically ask about over-the-counter products and their ingredients.
Leukemia Cutis Manifesting as Nonpalpable Purpura
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.

The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).

Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
 and numerous notched nuclei")
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.

Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
Practice Points
- Leukemia cutis complicates 5% to 15% of all cases of acute myeloid leukemia (AML) in adults.
- The appearance of leukemia cutis may be highly variable. Therefore, it should be included in the differential diagnosis for any cutaneous presentation in patients with an existing diagnosis or high likelihood of AML.
- Leukemic infiltrates are associated with sites of inflammation.

Treatment of Elephantiasic Pretibial Myxedema With Rituximab Therapy
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.

The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
. B, Colloidal iron staining highlighted the prominent mucin within the dermis")
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
Practice Points
- Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet.
- Although many therapeutic modalities have been described for the management of the elephantiasis variant of PTM, few treatments have shown notable efficacy.
- Rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.

Sarcoidosis Presenting as Telangiectatic Macules
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.

Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
.")
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.
Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
To the Editor:
Sarcoidosis is a multisystem, noncaseating, granulomatous disorder thought to occur from a combination of immunologic, genetic, and environmental factors.1 Often referred to as the “great imitator,” the cutaneous manifestations of sarcoidosis encompass many morphologies, including papules, plaques, nodules, and scars.1 We report an unusual case of sarcoidosis presenting as telangiectatic macules on the lower extremities.
A woman in her early 30s presented with a burning, pruritic, erythematous, telangiectatic eruption on the lower extremities with concurrent ankle swelling of 4 weeks’ duration. The patient denied any fevers, chills, recent infections, or new medications. Evaluation by her primary care physician during the time of the eruption included unremarkable antinuclear antibodies, thyroid stimulating hormone level, complete blood cell count, comprehensive metabolic panel, urinalysis, chest radiography, and lower-extremity Doppler ultrasonography.
Physical examination at the current presentation revealed numerous scattered, faint, erythematous, blanchable macules on the lower extremities along with mild pitting edema (Figure 1). The patient’s current medications included cetirizine, which she had been taking for years, as well as an intrauterine device. A punch biopsy from the right lower leg revealed small, well-demarcated sarcoidal granulomatous inflammation surrounding vascular structures and skin appendages (Figure 2). No foreign bodies were observed with polarized light microscopy. Microscopic findings suggestive of an infection, including caseation necrosis and suppurative inflammation, also were absent. Angiotensin-converting enzyme levels were normal. Myeloperoxidase and proteinase 3 IgG antibody levels were evaluated due to potential vascular involvement but were negative. An infectious cause of the sarcoidal granulomas was unlikely given histopathologic findings and negative tuberculosis skin testing, which the patient underwent annually for her job, so a tissue culture was not performed. The patient was prescribed triamcinolone acetonide cream 0.1% for the itching and burning at the initial visit and was continued on this treatment after the diagnosis of sarcoidosis was made. At 2-month follow-up, the patient’s eruption had nearly resolved with topical therapy.
Cutaneous manifestation occurs in 20% to 35% of sarcoidosis cases and may develop in the presence or absence of systemic disease. Approximately 60% of individuals with cutaneous sarcoidosis are found to have systemic involvement; therefore, careful monitoring and diagnostic workup are important in the management of these patients.2 While most cases of cutaneous sarcoidosis are papular, it is important for clinicians to maintain a level of suspicion for sarcoidosis in any uncertain dermatologic presentation.1,2 Evidence of telangiectasias has been shown in rarer forms of sarcoidosis (eg, angiolupoid), but the lesions usually are confined to the face, ears, or neck.3 Granulomatous vasculitis has been reported in a small number of individuals with ulcerative sarcoidosis.4 In our case, no ulcerations were present, possibly indicating an early lesion or an entirely novel process. Lastly, although reticular dermal granulomas are found in drug-induced interstitial granulomatous dermatitis, these lesions often are dispersed interstitially amongst collagen bundles and are associated with necrobiosis of collagen and eosinophilic/neutrophilic infiltrates.5 The lack of these characteristic pathologic findings in our patient along with no known reported cases of cetirizine-induced granulomatous dermatitis led us to rule out reticular dermal granulomas as a diagnosis. We present our case as a reminder of the diversity of cutaneous sarcoidosis manifestations and the importance of early diagnosis of these lesions.
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Haimovic A, Sanchez M, Judson MA, et al. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. cutaneous disease. J Am Acad Dermatol. 2012;66:699.E1-E18.
- Yanardag H, Tetikkurt C, Bilir M, et al. Diagnosis of cutaneous sarcoidosis; clinical and the prognostic significance of skin lesions. Multidiscip Respir Med. 2013;8:26.
- Arias-Santiago S, Fernández-Pugnaire MA, Aneiros- Fernández J, et al. Recurrent telangiectasias on the cheek: angiolupoid sarcoidosis. Am J Med. 2010;123:E7-E8.
- Wei C-H, Huang Y-H, Shih Y-C, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
Practice Points
- Cutaneous manifestations of sarcoidosis can encompass numerous morphologies. A high degree of suspicion should be maintained for any uncertain dermatologic presentation.
- Although papular eruptions are the most common cutaneous findings in sarcoidosis, this case report illustrates a less common vascular-appearing presentation.
- A systemic workup is indicated in any presentation of sarcoidosis.

Graft-vs-host Disease and Toxic Epidermal Necrolysis Following Hematopoietic Stem Cell Transplantation
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.

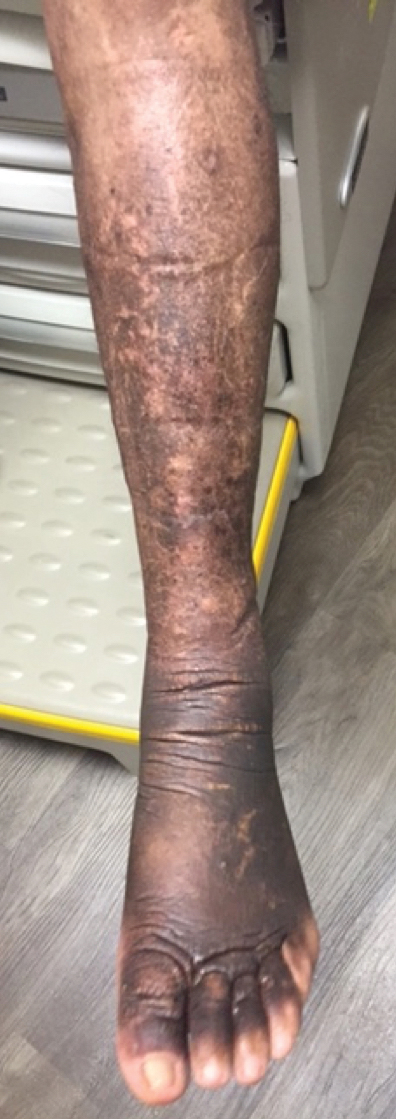
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.

A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.

Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8

Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10

Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
Practice Points
- Graft-vs-host disease (GVHD) and toxic epidermal necrolysis (TEN) are rare life-threatening complications seen in patients with allogeneic hematopoietic stem cell transplantation.
- Although mild acute GVHD easily is distinguished from TEN, severe acute GVHD and TEN share overlapping features and present a diagnostic challenge.
- Therapeutic decisions and associated outcomes hinge on accurate diagnosis, as high-dose systemic corticosteroids have been associated with higher mortality rates in TEN.

Phototoxic Contact Dermatitis From Over-the-counter 8-Methoxypsoralen
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.

The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
Practice Points
- Phototoxic contact dermatitis is an irritant reaction resembling an exaggerated sunburn that occurs with the use of a photosensitizing agent and UV light exposure.
- A range of topical and systemic medications, plants, and natural products can elicit phototoxic reactions.
- With the wide availability of prescription-strength over-the-counter medications, a detailed history often is necessary to identify the causative agents of phototoxic contact dermatitis and ensure future avoidance.

Pencil-core Granuloma Forming 62 Years After Initial Injury
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.

An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
.")
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.
An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.
An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
Practice Points
- Pencil-core granulomas can arise even decades after the lead is embedded in the skin.
- It is important to biopsy to confirm the diagnosis, as pencil-core granulomas can very closely mimic melanomas.

Lower Leg Hyperpigmentation in MYH9-Related Disorder
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.

Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.
Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.
Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
Practice Points
- MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.
- Leg hyperpigmentation can occur secondary to hemosiderin deposition from MYH9-related disorder.
- The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder.
- Lasers and intense pulsed light therapy are modalities to consider for the hyperpigmentation of MYH9- related disorder.

Scleral Plaques in Nephrogenic Systemic Fibrosis
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.

Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2

- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
Practice Points
- It is important to examine the eyes in a patient with sclerotic skin changes on physical examination.
- The presence of yellow scleral plaques strongly is associated with a diagnosis of nephrogenic systemic fibrosis.

Febrile Ulceronecrotic Mucha-Habermann Disease: A Rare Form of Pityriasis Lichenoides et Varioliformis Acuta
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.

A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.

The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.

At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.

Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
Practice Points
- Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare variant of pityriasis lichenoides et varioliformis acuta, characterized by ulceronecrotic lesions, fever, and systemic symptoms.
- A variety of treatments including immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been used in patients with FUMHD.
