User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
Link found between sleep disorders and osteoporosis risk
Patients with sleep disorders are much more likely to develop osteoporosis than are those without sleep disorders, according to Dr. Chia-Ming Yen and her associates.
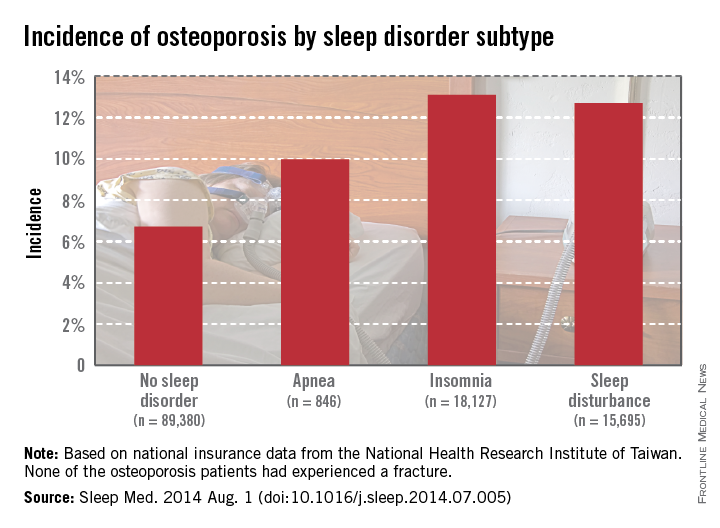
Patients diagnosed with sleep apnea between 1998 and 2001 had an osteoporosis incidence of nearly 10% at the end of 2010, while those without sleep disorders had incidence of 6.7%. Patients with insomnia developed osteoporosis at a rate of 13.1%, and patients with other sleep disturbances had an incidence of 12.7%, Dr. Yen of the National Formosa University in Taiwan, and her associates reported (Sleep Med. 2014 Aug. 1 [doi:10.1016/j.sleep.2014.07.005]).

Women and the elderly were particularly likely to develop osteoporosis if a sleep disorder was present. Of patients aged 64 years and older who were diagnosed with osteoporosis, 36.2% also had sleep apnea, and 31.9% had another sleep disorder. Incidences of osteoporosis in women in all cases were three to five times higher than those in men, and patients with multiple comorbidities also had an increased risk of osteoporosis, the investigators reported.
The study used data collected from 1996-2010 by the National Health Research Institute of Taiwan.
Patients with sleep disorders are much more likely to develop osteoporosis than are those without sleep disorders, according to Dr. Chia-Ming Yen and her associates.
Patients diagnosed with sleep apnea between 1998 and 2001 had an osteoporosis incidence of nearly 10% at the end of 2010, while those without sleep disorders had incidence of 6.7%. Patients with insomnia developed osteoporosis at a rate of 13.1%, and patients with other sleep disturbances had an incidence of 12.7%, Dr. Yen of the National Formosa University in Taiwan, and her associates reported (Sleep Med. 2014 Aug. 1 [doi:10.1016/j.sleep.2014.07.005]).
Women and the elderly were particularly likely to develop osteoporosis if a sleep disorder was present. Of patients aged 64 years and older who were diagnosed with osteoporosis, 36.2% also had sleep apnea, and 31.9% had another sleep disorder. Incidences of osteoporosis in women in all cases were three to five times higher than those in men, and patients with multiple comorbidities also had an increased risk of osteoporosis, the investigators reported.
The study used data collected from 1996-2010 by the National Health Research Institute of Taiwan.
Patients with sleep disorders are much more likely to develop osteoporosis than are those without sleep disorders, according to Dr. Chia-Ming Yen and her associates.
Patients diagnosed with sleep apnea between 1998 and 2001 had an osteoporosis incidence of nearly 10% at the end of 2010, while those without sleep disorders had incidence of 6.7%. Patients with insomnia developed osteoporosis at a rate of 13.1%, and patients with other sleep disturbances had an incidence of 12.7%, Dr. Yen of the National Formosa University in Taiwan, and her associates reported (Sleep Med. 2014 Aug. 1 [doi:10.1016/j.sleep.2014.07.005]).
Women and the elderly were particularly likely to develop osteoporosis if a sleep disorder was present. Of patients aged 64 years and older who were diagnosed with osteoporosis, 36.2% also had sleep apnea, and 31.9% had another sleep disorder. Incidences of osteoporosis in women in all cases were three to five times higher than those in men, and patients with multiple comorbidities also had an increased risk of osteoporosis, the investigators reported.
The study used data collected from 1996-2010 by the National Health Research Institute of Taiwan.
Clinical Guidelines: Obstructive sleep apnea
The Greek word apnea literally translates to “without breath.” More than 18 million American adults experience several moments “without breath” every night, according to the National Sleep Foundation. These pauses in breathing can last from a few seconds to minutes and can occur up to 30 times per hour or more. There are three types of apnea: obstructive, central, and mixed. Of the three, obstructive sleep apnea (OSA) is the most common. If left untreated, sleep apnea can have serious or life-threatening consequences, such as high blood pressure, heart disease, and day-time sleepiness that can lead to car accidents, depression, and headaches.

The significance of this condition has led the American College of Physicians (ACP) to publish guidelines regarding the management of OSA in adults. These guidelines are intended to provide clinicians with evidence-based recommendations that will have positive, long-term effects on patients’ cardiovascular risk, as well overall health and quality of life.
Recommendation 1: Encourage weight loss in all overweight and obese patients diagnosed with OSA.
There is strong evidence that shows how weight loss interventions can reduce the apnea/hypopnea index (AHI) and improve symptoms. The apnea/hypopnea index is a measure of the number of apnea and hypopnea episodes per hour of monitored sleep. According to the American Academy of Sleep Medicine, an OSA diagnosis is defined by ≥ 15 events/hr (with or without OSA symptoms) or ≥ 5 events/hr with OSA symptoms. Severity of OSA is classified as:
• Mild: 5-14 events/hr
• Moderate: 15-30 events/hr
• Severe: >30 events/hr
In patients with mild OSA, weight loss alone can be sufficient to normalize the apnea/hypopnea index, thereby reducing the need for continuous positive airway pressure (CPAP). In those with persistent OSA, weight loss can reduce the amount of PAP pressure required, which can increase tolerance of and adherence to CPAP. However, weight loss alone may not be sufficient to reduce OSA in all patients. In obese patients, weight loss must be encouraged with another primary treatment. In a meta-analysis of 342 patients in 12 published trials, weight loss produced substantial reductions in apnea/hypopnea index; however, the majority of patients continued to have persistent OSA after significant weight reduction. So, while weight loss should always be encouraged in patients with OSA, it should not be assumed that this intervention alone will be sufficient; patients need to be reassessed to determine whether OSA persists and if so, CPAP should be continued.
Recommendation 2: Prescribe CPAP as the initial therapy for patients diagnosed with OSA.
CPAP is as critical as it is effective not only in reducing the AHI but also in improving sleep continuity and architecture and decreasing the sleep hypoxia that is associated with OSA. Because of the lack of adherence to CPAP, many new features have been added including heated humidification, broad pressure adjustments, and expiratory pressure relief; while many of these features mildly improve patients’ preferences, there is no evidence to suggest that these changes improve efficacy or adherence. Again, it is critical that each patient’s needs be taken into consideration. Some patients, particularly those with mild forms of OSA, may not need CPAP and instead, may benefit from positional therapy or weight loss. Other therapy such as surgery or mandibular advancement devices (MADs) may be better suited for some patients despite their lessor effectiveness when compared with CPAP.
Recommendation 3: Consider MAD as an alternate therapy to CPAP for patients diagnosed with OSA who prefer MAD or for those with adverse effects associated with CPAP.
MADs have been recommended for patients with moderate to severe OSA, those with apnea/hypopnea index values between 18-40 events/hour, and for individuals who experienced adverse events on CPAP or who can’t tolerate using it. While CPAP still is considered to be primary therapy, for those who must use a mandibular advancement device, it can often be very effective. Patients may find it easier to be compliant with MAD than with CPAP. One group of researchers observed that MADs were able to provide appropriate decrease in obstructive events in 70% of patients with mild OSA, 48% of those with moderate OSA, and 42% of those with severe OSA (Chest 2011:139:1331-9). The ACP concluded that, without sufficient evidence, it is unclear which patients will benefit from MADs. Data from several studies show that patients who are younger and thinner, with less severe OSA, may benefit more from the use of MADs. Although the ACP does not recommend surgery or pharmaceutical therapy for OSA, the ACP does acknowledge that it may work for certain patients. However, the documented efficacy rate ranges from 20%-100%, thereby making it challenging to determine its true effect.
Bottom Line
The ACP recommends weight loss for all overweight patients with OSA. CPAP is first-line therapy, particularly if weight loss cannot be sustained or does not sufficiently lessen symptoms. For selected patients who are younger and thinner, with less severe OSA, or for those patients who are unable to tolerate CPAP, MADs can be used as an alternative first-line therapy with moderate success.
Reference
Qaseem A., Holty J.C., Owens D. et al. Diagnosis of obstructive sleep apnea in adults: a Clinical Practice Guideline from the American College of Physicians. Ann. Intern. Med. 2014;161:210-20.
Dr. Grover is a second-year resident in the family medicine residency program at Abington Memorial Hospital. Dr. Skolnik is associate director of the family medicine residency program at Abington Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.
The Greek word apnea literally translates to “without breath.” More than 18 million American adults experience several moments “without breath” every night, according to the National Sleep Foundation. These pauses in breathing can last from a few seconds to minutes and can occur up to 30 times per hour or more. There are three types of apnea: obstructive, central, and mixed. Of the three, obstructive sleep apnea (OSA) is the most common. If left untreated, sleep apnea can have serious or life-threatening consequences, such as high blood pressure, heart disease, and day-time sleepiness that can lead to car accidents, depression, and headaches.
The significance of this condition has led the American College of Physicians (ACP) to publish guidelines regarding the management of OSA in adults. These guidelines are intended to provide clinicians with evidence-based recommendations that will have positive, long-term effects on patients’ cardiovascular risk, as well overall health and quality of life.
Recommendation 1: Encourage weight loss in all overweight and obese patients diagnosed with OSA.
There is strong evidence that shows how weight loss interventions can reduce the apnea/hypopnea index (AHI) and improve symptoms. The apnea/hypopnea index is a measure of the number of apnea and hypopnea episodes per hour of monitored sleep. According to the American Academy of Sleep Medicine, an OSA diagnosis is defined by ≥ 15 events/hr (with or without OSA symptoms) or ≥ 5 events/hr with OSA symptoms. Severity of OSA is classified as:
• Mild: 5-14 events/hr
• Moderate: 15-30 events/hr
• Severe: >30 events/hr
In patients with mild OSA, weight loss alone can be sufficient to normalize the apnea/hypopnea index, thereby reducing the need for continuous positive airway pressure (CPAP). In those with persistent OSA, weight loss can reduce the amount of PAP pressure required, which can increase tolerance of and adherence to CPAP. However, weight loss alone may not be sufficient to reduce OSA in all patients. In obese patients, weight loss must be encouraged with another primary treatment. In a meta-analysis of 342 patients in 12 published trials, weight loss produced substantial reductions in apnea/hypopnea index; however, the majority of patients continued to have persistent OSA after significant weight reduction. So, while weight loss should always be encouraged in patients with OSA, it should not be assumed that this intervention alone will be sufficient; patients need to be reassessed to determine whether OSA persists and if so, CPAP should be continued.
Recommendation 2: Prescribe CPAP as the initial therapy for patients diagnosed with OSA.
CPAP is as critical as it is effective not only in reducing the AHI but also in improving sleep continuity and architecture and decreasing the sleep hypoxia that is associated with OSA. Because of the lack of adherence to CPAP, many new features have been added including heated humidification, broad pressure adjustments, and expiratory pressure relief; while many of these features mildly improve patients’ preferences, there is no evidence to suggest that these changes improve efficacy or adherence. Again, it is critical that each patient’s needs be taken into consideration. Some patients, particularly those with mild forms of OSA, may not need CPAP and instead, may benefit from positional therapy or weight loss. Other therapy such as surgery or mandibular advancement devices (MADs) may be better suited for some patients despite their lessor effectiveness when compared with CPAP.
Recommendation 3: Consider MAD as an alternate therapy to CPAP for patients diagnosed with OSA who prefer MAD or for those with adverse effects associated with CPAP.
MADs have been recommended for patients with moderate to severe OSA, those with apnea/hypopnea index values between 18-40 events/hour, and for individuals who experienced adverse events on CPAP or who can’t tolerate using it. While CPAP still is considered to be primary therapy, for those who must use a mandibular advancement device, it can often be very effective. Patients may find it easier to be compliant with MAD than with CPAP. One group of researchers observed that MADs were able to provide appropriate decrease in obstructive events in 70% of patients with mild OSA, 48% of those with moderate OSA, and 42% of those with severe OSA (Chest 2011:139:1331-9). The ACP concluded that, without sufficient evidence, it is unclear which patients will benefit from MADs. Data from several studies show that patients who are younger and thinner, with less severe OSA, may benefit more from the use of MADs. Although the ACP does not recommend surgery or pharmaceutical therapy for OSA, the ACP does acknowledge that it may work for certain patients. However, the documented efficacy rate ranges from 20%-100%, thereby making it challenging to determine its true effect.
Bottom Line
The ACP recommends weight loss for all overweight patients with OSA. CPAP is first-line therapy, particularly if weight loss cannot be sustained or does not sufficiently lessen symptoms. For selected patients who are younger and thinner, with less severe OSA, or for those patients who are unable to tolerate CPAP, MADs can be used as an alternative first-line therapy with moderate success.
Reference
Qaseem A., Holty J.C., Owens D. et al. Diagnosis of obstructive sleep apnea in adults: a Clinical Practice Guideline from the American College of Physicians. Ann. Intern. Med. 2014;161:210-20.
Dr. Grover is a second-year resident in the family medicine residency program at Abington Memorial Hospital. Dr. Skolnik is associate director of the family medicine residency program at Abington Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.
The Greek word apnea literally translates to “without breath.” More than 18 million American adults experience several moments “without breath” every night, according to the National Sleep Foundation. These pauses in breathing can last from a few seconds to minutes and can occur up to 30 times per hour or more. There are three types of apnea: obstructive, central, and mixed. Of the three, obstructive sleep apnea (OSA) is the most common. If left untreated, sleep apnea can have serious or life-threatening consequences, such as high blood pressure, heart disease, and day-time sleepiness that can lead to car accidents, depression, and headaches.
The significance of this condition has led the American College of Physicians (ACP) to publish guidelines regarding the management of OSA in adults. These guidelines are intended to provide clinicians with evidence-based recommendations that will have positive, long-term effects on patients’ cardiovascular risk, as well overall health and quality of life.
Recommendation 1: Encourage weight loss in all overweight and obese patients diagnosed with OSA.
There is strong evidence that shows how weight loss interventions can reduce the apnea/hypopnea index (AHI) and improve symptoms. The apnea/hypopnea index is a measure of the number of apnea and hypopnea episodes per hour of monitored sleep. According to the American Academy of Sleep Medicine, an OSA diagnosis is defined by ≥ 15 events/hr (with or without OSA symptoms) or ≥ 5 events/hr with OSA symptoms. Severity of OSA is classified as:
• Mild: 5-14 events/hr
• Moderate: 15-30 events/hr
• Severe: >30 events/hr
In patients with mild OSA, weight loss alone can be sufficient to normalize the apnea/hypopnea index, thereby reducing the need for continuous positive airway pressure (CPAP). In those with persistent OSA, weight loss can reduce the amount of PAP pressure required, which can increase tolerance of and adherence to CPAP. However, weight loss alone may not be sufficient to reduce OSA in all patients. In obese patients, weight loss must be encouraged with another primary treatment. In a meta-analysis of 342 patients in 12 published trials, weight loss produced substantial reductions in apnea/hypopnea index; however, the majority of patients continued to have persistent OSA after significant weight reduction. So, while weight loss should always be encouraged in patients with OSA, it should not be assumed that this intervention alone will be sufficient; patients need to be reassessed to determine whether OSA persists and if so, CPAP should be continued.
Recommendation 2: Prescribe CPAP as the initial therapy for patients diagnosed with OSA.
CPAP is as critical as it is effective not only in reducing the AHI but also in improving sleep continuity and architecture and decreasing the sleep hypoxia that is associated with OSA. Because of the lack of adherence to CPAP, many new features have been added including heated humidification, broad pressure adjustments, and expiratory pressure relief; while many of these features mildly improve patients’ preferences, there is no evidence to suggest that these changes improve efficacy or adherence. Again, it is critical that each patient’s needs be taken into consideration. Some patients, particularly those with mild forms of OSA, may not need CPAP and instead, may benefit from positional therapy or weight loss. Other therapy such as surgery or mandibular advancement devices (MADs) may be better suited for some patients despite their lessor effectiveness when compared with CPAP.
Recommendation 3: Consider MAD as an alternate therapy to CPAP for patients diagnosed with OSA who prefer MAD or for those with adverse effects associated with CPAP.
MADs have been recommended for patients with moderate to severe OSA, those with apnea/hypopnea index values between 18-40 events/hour, and for individuals who experienced adverse events on CPAP or who can’t tolerate using it. While CPAP still is considered to be primary therapy, for those who must use a mandibular advancement device, it can often be very effective. Patients may find it easier to be compliant with MAD than with CPAP. One group of researchers observed that MADs were able to provide appropriate decrease in obstructive events in 70% of patients with mild OSA, 48% of those with moderate OSA, and 42% of those with severe OSA (Chest 2011:139:1331-9). The ACP concluded that, without sufficient evidence, it is unclear which patients will benefit from MADs. Data from several studies show that patients who are younger and thinner, with less severe OSA, may benefit more from the use of MADs. Although the ACP does not recommend surgery or pharmaceutical therapy for OSA, the ACP does acknowledge that it may work for certain patients. However, the documented efficacy rate ranges from 20%-100%, thereby making it challenging to determine its true effect.
Bottom Line
The ACP recommends weight loss for all overweight patients with OSA. CPAP is first-line therapy, particularly if weight loss cannot be sustained or does not sufficiently lessen symptoms. For selected patients who are younger and thinner, with less severe OSA, or for those patients who are unable to tolerate CPAP, MADs can be used as an alternative first-line therapy with moderate success.
Reference
Qaseem A., Holty J.C., Owens D. et al. Diagnosis of obstructive sleep apnea in adults: a Clinical Practice Guideline from the American College of Physicians. Ann. Intern. Med. 2014;161:210-20.
Dr. Grover is a second-year resident in the family medicine residency program at Abington Memorial Hospital. Dr. Skolnik is associate director of the family medicine residency program at Abington Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.

Recurrent strep throat? Ask two questions before prescribing
SAN DIEGO– If a child presents with what looks like recurrent strep throat, physicians should ask two questions before prescribing antibiotics: Is this a true recurrence of strep infection? And if so, was the child treated adequately the first time?
Viral pharyngitis is in fact the most common cause of what looks like recurrent strep throat, Dr. John Bradley said at the annual meeting of the American Association of Pediatrics. Rapid strep tests can detect very low levels of streptococcus organisms in children who are carriers, so “it can look like a recurrent strep infection when it’s really a virus” that is causing a sore throat, he added.

Physicians should ask whether the patient has the same amount of exudate present at “recurrence” as during the initial infection, said Dr. Bradley, who is a pediatric infectious diseases specialist at the University of California, San Diego. If not, they should order an antistreptolysin O (ASO) titer to prove that there is current streptococcal infection, while keeping in mind that the titer takes 2-3 weeks to increase, he said.
If the infection truly is recurrent strep, amoxicillin may be a better choice than penicillin V because of its superior absorption and tolerability, Dr. Bradley said. Amoxicillin has a longer half-life, better tissue exposure, and also tastes better than penicillin V, which leads to better compliance, he said.
Doctors should base the dose of amoxicillin on the severity of the child’s illness, Dr. Bradley added. Based on his experience, he recommends amoxicillin at a dose of 75 mg/kg three times a day if the affected child has abscessing tonsillitis, even though 40 mg/kg per day may work, he noted.
“Some kids have abscess-riddled tonsils that are so painful that they can’t swallow,” Dr. Bradley emphasized. “The idea that one dose fits everyone breaks down when you have bad strep throat.”
Dr. Bradley reported no conflicts of interest.
SAN DIEGO– If a child presents with what looks like recurrent strep throat, physicians should ask two questions before prescribing antibiotics: Is this a true recurrence of strep infection? And if so, was the child treated adequately the first time?
Viral pharyngitis is in fact the most common cause of what looks like recurrent strep throat, Dr. John Bradley said at the annual meeting of the American Association of Pediatrics. Rapid strep tests can detect very low levels of streptococcus organisms in children who are carriers, so “it can look like a recurrent strep infection when it’s really a virus” that is causing a sore throat, he added.
Physicians should ask whether the patient has the same amount of exudate present at “recurrence” as during the initial infection, said Dr. Bradley, who is a pediatric infectious diseases specialist at the University of California, San Diego. If not, they should order an antistreptolysin O (ASO) titer to prove that there is current streptococcal infection, while keeping in mind that the titer takes 2-3 weeks to increase, he said.
If the infection truly is recurrent strep, amoxicillin may be a better choice than penicillin V because of its superior absorption and tolerability, Dr. Bradley said. Amoxicillin has a longer half-life, better tissue exposure, and also tastes better than penicillin V, which leads to better compliance, he said.
Doctors should base the dose of amoxicillin on the severity of the child’s illness, Dr. Bradley added. Based on his experience, he recommends amoxicillin at a dose of 75 mg/kg three times a day if the affected child has abscessing tonsillitis, even though 40 mg/kg per day may work, he noted.
“Some kids have abscess-riddled tonsils that are so painful that they can’t swallow,” Dr. Bradley emphasized. “The idea that one dose fits everyone breaks down when you have bad strep throat.”
Dr. Bradley reported no conflicts of interest.
SAN DIEGO– If a child presents with what looks like recurrent strep throat, physicians should ask two questions before prescribing antibiotics: Is this a true recurrence of strep infection? And if so, was the child treated adequately the first time?
Viral pharyngitis is in fact the most common cause of what looks like recurrent strep throat, Dr. John Bradley said at the annual meeting of the American Association of Pediatrics. Rapid strep tests can detect very low levels of streptococcus organisms in children who are carriers, so “it can look like a recurrent strep infection when it’s really a virus” that is causing a sore throat, he added.
Physicians should ask whether the patient has the same amount of exudate present at “recurrence” as during the initial infection, said Dr. Bradley, who is a pediatric infectious diseases specialist at the University of California, San Diego. If not, they should order an antistreptolysin O (ASO) titer to prove that there is current streptococcal infection, while keeping in mind that the titer takes 2-3 weeks to increase, he said.
If the infection truly is recurrent strep, amoxicillin may be a better choice than penicillin V because of its superior absorption and tolerability, Dr. Bradley said. Amoxicillin has a longer half-life, better tissue exposure, and also tastes better than penicillin V, which leads to better compliance, he said.
Doctors should base the dose of amoxicillin on the severity of the child’s illness, Dr. Bradley added. Based on his experience, he recommends amoxicillin at a dose of 75 mg/kg three times a day if the affected child has abscessing tonsillitis, even though 40 mg/kg per day may work, he noted.
“Some kids have abscess-riddled tonsils that are so painful that they can’t swallow,” Dr. Bradley emphasized. “The idea that one dose fits everyone breaks down when you have bad strep throat.”
Dr. Bradley reported no conflicts of interest.
AT THE AAP NATIONAL CONFERENCE

Biomarkers identify RA patients at risk of lung disease
An international team of researchers has identified biomarkers that may help pinpoint rheumatoid arthritis patients at risk of interstitial lung disease.
Dr. Juan Chen of the First Hospital of Xiamen (China) University and Dr. Tracy Doyle of Brigham and Women’s Hospital, Boston, classified Chinese rheumatoid arthritis (RA) patients into different clinical and radiographic stages of interstitial lung disease (ILD) – RA with no ILD, RA with mild ILD, and RA with advanced ILD (Arthritis Rheumatol. 2014 [doi:10.1002/art.38904]).
They used multiplex ELISA (enzyme-linked immunosorbent assay) to identify strong correlations between average serum levels of MMP-7 and IP10 and the grade of interstitial lung abnormalities. The average serum concentration of MMP-7 increased from 3.06 ng/mL in the RA patients with no ILD to 5.35 ng/mL in RA patients with ILD (P = .005). Levels of IP10 increased from 173.8 pg/mL in RA patients with no lung disease to 308.6 pg/mL in patients with ILD (P = .0004).
The researchers also found statistically significant elevations of both biomarkers in the patients with mild disease, “strengthening the apparent dose response relationship between these biomarkers and severity of radiographically defined interstitial lung abnormalities.”
Using a replication cohort from two academic centers in the United States, the researchers confirmed their findings by identifying statistically significant correlations between the two biomarkers and the presence of lung disease. The findings held strong even after adjustment for age, sex, smoking history, and 28-joint Disease Activity Score.
“[The results] demonstrate that elevated levels of IP10 and MMP-7 strongly correlate with the presence of RA-ILD, effectively supporting the hypothesis that RA-ILD represents a spectrum of pathology involving parenchymal lung inflammation and dysregulated tissue remodeling,” the study authors concluded.
Longitudinal studies are needed to determine the prognostic value of MMP-7 and IP10 in RA patients with clinically/radiographically established disease, “potentially enabling clinicians to distinguish those individuals most likely to develop progressive fibrosis and an IPF-like clinical course,” they added.
The research was supported in part by the Harvard KL2/Catalyst Medical Research Investigator Training Program as well as grants from the National Institutes of Health, Veterans Affairs Merit Review Program, and the *Rheumatology Research Foundation’s Within Our Reach program. No conflicts of interest were declared.
*Correction 10/30/2014: The article previously listed the Rheumatology Research Foundation under its old name, the American College of Rheumatology Research and Education Foundation.
An international team of researchers has identified biomarkers that may help pinpoint rheumatoid arthritis patients at risk of interstitial lung disease.
Dr. Juan Chen of the First Hospital of Xiamen (China) University and Dr. Tracy Doyle of Brigham and Women’s Hospital, Boston, classified Chinese rheumatoid arthritis (RA) patients into different clinical and radiographic stages of interstitial lung disease (ILD) – RA with no ILD, RA with mild ILD, and RA with advanced ILD (Arthritis Rheumatol. 2014 [doi:10.1002/art.38904]).
They used multiplex ELISA (enzyme-linked immunosorbent assay) to identify strong correlations between average serum levels of MMP-7 and IP10 and the grade of interstitial lung abnormalities. The average serum concentration of MMP-7 increased from 3.06 ng/mL in the RA patients with no ILD to 5.35 ng/mL in RA patients with ILD (P = .005). Levels of IP10 increased from 173.8 pg/mL in RA patients with no lung disease to 308.6 pg/mL in patients with ILD (P = .0004).
The researchers also found statistically significant elevations of both biomarkers in the patients with mild disease, “strengthening the apparent dose response relationship between these biomarkers and severity of radiographically defined interstitial lung abnormalities.”
Using a replication cohort from two academic centers in the United States, the researchers confirmed their findings by identifying statistically significant correlations between the two biomarkers and the presence of lung disease. The findings held strong even after adjustment for age, sex, smoking history, and 28-joint Disease Activity Score.
“[The results] demonstrate that elevated levels of IP10 and MMP-7 strongly correlate with the presence of RA-ILD, effectively supporting the hypothesis that RA-ILD represents a spectrum of pathology involving parenchymal lung inflammation and dysregulated tissue remodeling,” the study authors concluded.
Longitudinal studies are needed to determine the prognostic value of MMP-7 and IP10 in RA patients with clinically/radiographically established disease, “potentially enabling clinicians to distinguish those individuals most likely to develop progressive fibrosis and an IPF-like clinical course,” they added.
The research was supported in part by the Harvard KL2/Catalyst Medical Research Investigator Training Program as well as grants from the National Institutes of Health, Veterans Affairs Merit Review Program, and the *Rheumatology Research Foundation’s Within Our Reach program. No conflicts of interest were declared.
*Correction 10/30/2014: The article previously listed the Rheumatology Research Foundation under its old name, the American College of Rheumatology Research and Education Foundation.
An international team of researchers has identified biomarkers that may help pinpoint rheumatoid arthritis patients at risk of interstitial lung disease.
Dr. Juan Chen of the First Hospital of Xiamen (China) University and Dr. Tracy Doyle of Brigham and Women’s Hospital, Boston, classified Chinese rheumatoid arthritis (RA) patients into different clinical and radiographic stages of interstitial lung disease (ILD) – RA with no ILD, RA with mild ILD, and RA with advanced ILD (Arthritis Rheumatol. 2014 [doi:10.1002/art.38904]).
They used multiplex ELISA (enzyme-linked immunosorbent assay) to identify strong correlations between average serum levels of MMP-7 and IP10 and the grade of interstitial lung abnormalities. The average serum concentration of MMP-7 increased from 3.06 ng/mL in the RA patients with no ILD to 5.35 ng/mL in RA patients with ILD (P = .005). Levels of IP10 increased from 173.8 pg/mL in RA patients with no lung disease to 308.6 pg/mL in patients with ILD (P = .0004).
The researchers also found statistically significant elevations of both biomarkers in the patients with mild disease, “strengthening the apparent dose response relationship between these biomarkers and severity of radiographically defined interstitial lung abnormalities.”
Using a replication cohort from two academic centers in the United States, the researchers confirmed their findings by identifying statistically significant correlations between the two biomarkers and the presence of lung disease. The findings held strong even after adjustment for age, sex, smoking history, and 28-joint Disease Activity Score.
“[The results] demonstrate that elevated levels of IP10 and MMP-7 strongly correlate with the presence of RA-ILD, effectively supporting the hypothesis that RA-ILD represents a spectrum of pathology involving parenchymal lung inflammation and dysregulated tissue remodeling,” the study authors concluded.
Longitudinal studies are needed to determine the prognostic value of MMP-7 and IP10 in RA patients with clinically/radiographically established disease, “potentially enabling clinicians to distinguish those individuals most likely to develop progressive fibrosis and an IPF-like clinical course,” they added.
The research was supported in part by the Harvard KL2/Catalyst Medical Research Investigator Training Program as well as grants from the National Institutes of Health, Veterans Affairs Merit Review Program, and the *Rheumatology Research Foundation’s Within Our Reach program. No conflicts of interest were declared.
*Correction 10/30/2014: The article previously listed the Rheumatology Research Foundation under its old name, the American College of Rheumatology Research and Education Foundation.
FROM ARTHRITIS AND RHEUMATOLOGY
Key clinical point: The discovery of blood biomarkers may lead to early identification of lung complications in RA patients.
Major finding: MMP-7 and IP10 were elevated in the blood of patients with different stages of RA associated interstitial lung disease.
Data source: A Chinese identification cohort (n = 133) and a U.S. replication cohort (n = 86) of RA patients with or without ILD.
Disclosures: The research was supported in part by the Harvard KL2/Catalyst Medical Research Investigator Training Program as well as grants from the National Institutes of Health, Veterans Affairs Merit Review Program, and the *Rheumatology Research Foundation’s Within Our Reach program. No conflicts of interest were declared.
FDA panel supports retaining Chantix boxed warning for now
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
![]()
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.

Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
![]()
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.
Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
![]()
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.
Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA panel supports retaining Chantix boxed warning for now
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.

Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.
Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
SILVER SPRING, MD. – The boxed warning about the risk of serious neuropsychiatric effects associated with the smoking cessation drug varenicline should remain on the drug’s label, and the need for this warning can be reevaluated when the results of a postmarketing safety study become available next year, according to the majority of a Food and Drug Administration advisory panel.
On Oct. 16, at a joint meeting of the FDA’s Psychopharmacologic Drugs and the Drug Safety and Risk Management Advisory committees, 11 of the 18 panelists voted to retain the boxed warning about neuropsychiatric symptoms and suicidality, agreeing that a decision about whether to retain the warning should not be made until the results of the study are available.
Pfizer, which manufactures varenicline, a nicotinic receptor partial agonist in a tablet formulation, as Chantix, is conducting the prospective, randomized, double-blind study comparing neuropsychiatric events in 8,000 smokers, with and without a psychiatric history, treated with varenicline, nicotine replacement therapy, bupropion, or placebo. Although the results are expected in 2015, the company maintained that the boxed warning is no longer justified and can be put in the warnings and precautions section, based on analyses of observational studies and randomized clinical trials. The company submitted those analyses to the FDA earlier this year.
The boxed warning in the drug’s prescribing information states that the serious events reported in patients taking the drug include but “are not limited to” depression, suicidal ideation, suicide attempts, and completed suicides; and that some cases “may have been complicated by the symptoms of nicotine withdrawal in patients who stopped smoking.” The reason the boxed warning is used for this drug is that there is a “serious adverse reaction that can be prevented or reduced in frequency or severity,” the FDA said.
Because the precedent for removing a boxed warning from a drug label is limited and the FDA’s view that the decision should consider the final results of the postmarketing trial, the agency convened the joint meeting to review the analyses submitted by the company. Overall, the panelists were not convinced by the analyses. They cited various weaknesses in the observational studies and the meta-analysis, including questions about whether the correct outcomes were being captured.
Six panelists voted to modify (but not weaken) the language in the boxed warning, and most recommended that the last few lines about the health benefits of quitting smoking be removed and argued that the wording had a promotional tone and did not belong in a boxed warning. Several panelists recommended adding sleep disruption and disorders, and recognized adverse events of the drugs to the boxed warning.
Among those who voted to retain the boxed warning, Dr. Jeanmarie Perrone, director of the medical toxicology division in the emergency medicine department at the University of Pennsylvania, Philadelphia, said there was biologic plausibility for these events. Her biggest concern, she said, was that removing the boxed warning might be interpreted as “an endorsement of safety and that has not been demonstrated.” In the emergency department, she said, she sees many patients who have been prescribed varenicline “by a myriad of clinicians,” and the presence of a boxed warning helps keep this safety issue in mind.
Dr. Rajiv N. Rimal, professor and chair of the department of prevention and community health, George Washington University, Washington, who voted to retain the warning with modified wording, said: “I heard enough compelling evidence to suggest that even some of the more rare events were severe enough” that revisiting the labeling change would be necessary once the other data became available.
Varenicline, approved by the FDA in 2006 as an aid to smoking-cessation treatment, provides a “low level” of dopamine release as people try to quit smoking, according to Pfizer. The boxed warning was added to the label in July 2009, based on the FDA’s review of spontaneous adverse event reports, conducted after an alert about suicidality reports associated with the drug from European regulators about 1 year after FDA approval. About the same time, there was a widely publicized case of a Texas musician who was taking varenicline and was shot and killed by a neighbor while exhibiting uncharacteristic aggressive behavior in the fall of 2007.
The reports provided by the company at that time included 102 suicide-related cases, and 525 reports of aggressive/irrational behavior. A review of the reports in the FDA’s adverse events database included many cases with “hallmarks of drug relatedness,” patients reporting unusual symptoms with “similar distinctive language,” and positive dechallenges and rechallenges, said Dr. Celia Winchell, the medical team leader of addiction products in the FDA’s division of anesthesia, analgesia, and addiction products. Not all cases involved suicidality, depression, or aggression. Postmarketing reports have provided a “rich narrative with detail” about patient experiences, including descriptions that are relatively easy to describe or classify, such as suicide and aggression, while others “defy coding,” such as one person who reported feeling “like a zombie,” she told the panel.
“Based on a review of the cases, there appears to be a syndrome of debilitating symptoms that interfere with people’s ability to function in their daily lives that is associated with the use of Chantix,” Dr. Winchell said.
The company agreed that the spontaneous reports in late 2007 raised concerns about whether the drug was associated with an increase in serious neuropsychiatric events. But since 2009, this issue has been evaluated in controlled studies, which “consistently show no evidence of an increased risk,” Dr. Christopher Wohlberg, the head of Pfizer’s safety surveillance and risk management group, told the panel.
These include a meta-analysis of adverse events in 18 controlled studies of about 5,000 patients treated with varenicline and about 3,500 on placebo, which found no significant difference in the rate of neuropsychiatric events (with the exception of sleep disorders and disturbance) among those on the drug vs. those on placebo; and a meta-analysis of 5 placebo-controlled studies that used the Columbia-Suicide Severity Rating Scale to evaluate suicidal ideation and behavior, which also found no significant differences between the two groups.
In addition, four observational studies that included patients with psychiatric illnesses showed that the rates of serious neuropsychiatric events in patients treated with varenicline were not significantly different from those treated with nicotine replacement therapy or bupropion for smoking cessation. In individual studies of patients with psychiatric illnesses (schizophrenia, schizoaffective disorder, or major depressive disorder), no evidence was found of an increased risk among those treated with varenicline, compared with those on placebo, according to the company.
But the FDA reviewers raised issues with the meta-analysis of the 18 studies and the observational data. The observational data have limitations and “preclude” a conclusion that there is no association of varenicline and an increased risk of neuropsychiatric events, said Natasha Chen, Ph.D., an epidemiologist in the FDA’s office of surveillance and epidemiology. The ongoing postmarketing safety trial “is likely to offer better insights into varenicline’s neuropsychiatric risks,” she added.
The one panelist who voted to remove the warning, Dr. Andrew J. Saxon, said while there “may be some serious adverse or neuropsychiatric effects, the data, while not perfect,” do not show a signal in his view. He added that in his experience working daily with patients who are trying to quit smoking, “patients are afraid to take this medication because of the boxed warning, and it does deter use.” Moreover, in the Veterans Affairs system, there are restrictions on prescribing the drug because of the boxed warning, including a limitation on prescriptions to a 28-day supply, with no refills. Such restrictions increase hassles for patients and prescribers, and often result in patients stopping treatment after 4 weeks.
“If I’m doing a good job as a physician, I’m going to monitor the patient,” said Dr. Saxon, director of the addiction psychiatry residency program, University of Washington, Seattle.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Members of these two panels had no conflicts to disclose. Occasionally, panelists with a conflict are given a waiver, but not at this meeting.
In a statementissued by Pfizer after the meeting, Dr. Steven Romano, senior vice president and head of the medicines development group for Pfizer, said the “completion of our currently ongoing safety study will represent one more step forward in the process of accurately characterizing the neuropsychiatric safety of this important medication.”
The varenicline label is available at http://labeling.pfizer.com/ShowLabeling.aspx?id=557. Serious adverse events associated with this drug should be reported to the FDA’s MedWatch program at http://www.fda.gov/Safety/MedWatch/default.htm.
AT AN FDA ADVISORY COMMITTEE MEETING
Nintedanib, pirfenidone approved for pulmonary fibrosis
The Food and Drug Administration on Oct. 15 approved two new oral medications for idiopathic pulmonary fibrosis, Boehringer Ingelheim’s nintedanib (Ofev) and Roche’s pirfenidone (Esbriet).
The drugs significantly slowed the decline of forced vital capacity (FVC), compared with placebo, in phase III testing. They did not significantly reduce mortality. The FDA granted both agents fast track, priority review, orphan product, and breakthrough designations. Both were also approved ahead of schedule, according to the agency. Each of the drugs should be available for patients within the next 2 weeks, according to their manufacturers.
For pirfenidone, already on the market in Europe and Canada, Roche said it plans “a comprehensive patient support program designed to help with access, financial support, and ongoing education.”
For nintedanib, Boehringer Ingelheim plans “a comprehensive patient support program that will provide a broad range of financial and nursing support services,” called “Open Doors.” In testing, the most frequent serious adverse reactions with nintedanib, versus placebo, were bronchitis (1.2% versus 0.8%) and myocardial infarction (1.5% versus 0.4%). Pneumonia (0.7% versus 0.6%), lung cancer (0.3% versus 0%), and MI (0.3% versus 0.2%) were the most common fatal adverse events.
The most common side effects of nintedanib were diarrhea, nausea, abdominal pain, vomiting, liver enzyme elevation, decreased appetite, headache, weight loss, and high blood pressure. Nintedanib is not recommended for patients who have moderate to severe liver problems or are pregnancy class D. Women of childbearing potential should use contraception for at least 3 months after stopping the drug, according to the FDA.
For pirfenidone, the most serious adverse events versus placebo were liver enzyme elevations (3.7% versus 0.8%), sensitivity to light or rash (9.0% versus 1.0%), and gastrointestinal side effects that caused 2.2 % of patients to discontinue treatment compared with 1.0% of those who received placebo.
The most common side effects of pirfenidone were nausea, rash, abdominal pain, upper respiratory tract infection, diarrhea, fatigue, headache, dyspepsia, dizziness, vomiting, loss of appetite, gastroesophageal reflux, sinusitis, insomnia, decreased weight, and arthralgia.
The FDA warned about use of pirfenidone in patients with severe liver problems or end-stage kidney disease, or in those who require dialysis. Patients taking pirfenidone also must monitor and guard against sun exposure.
Clinical trials of pirfenidone included 1,247 idiopathic pulmonary fibrosis patients. In one with 555 patients, 17% of pirfenidone patients had an FVC decline of at least 10% after a year, compared with 32% who received placebo, Roche said.
Trials of nintedanib included 1,231 patients. One with 513 showed a relative reduction in annual FVC decline of 52% (–115 versus –240 mL for placebo) at 1 year, Boehringer Ingelheim noted.
The Food and Drug Administration on Oct. 15 approved two new oral medications for idiopathic pulmonary fibrosis, Boehringer Ingelheim’s nintedanib (Ofev) and Roche’s pirfenidone (Esbriet).
The drugs significantly slowed the decline of forced vital capacity (FVC), compared with placebo, in phase III testing. They did not significantly reduce mortality. The FDA granted both agents fast track, priority review, orphan product, and breakthrough designations. Both were also approved ahead of schedule, according to the agency. Each of the drugs should be available for patients within the next 2 weeks, according to their manufacturers.
For pirfenidone, already on the market in Europe and Canada, Roche said it plans “a comprehensive patient support program designed to help with access, financial support, and ongoing education.”
For nintedanib, Boehringer Ingelheim plans “a comprehensive patient support program that will provide a broad range of financial and nursing support services,” called “Open Doors.” In testing, the most frequent serious adverse reactions with nintedanib, versus placebo, were bronchitis (1.2% versus 0.8%) and myocardial infarction (1.5% versus 0.4%). Pneumonia (0.7% versus 0.6%), lung cancer (0.3% versus 0%), and MI (0.3% versus 0.2%) were the most common fatal adverse events.
The most common side effects of nintedanib were diarrhea, nausea, abdominal pain, vomiting, liver enzyme elevation, decreased appetite, headache, weight loss, and high blood pressure. Nintedanib is not recommended for patients who have moderate to severe liver problems or are pregnancy class D. Women of childbearing potential should use contraception for at least 3 months after stopping the drug, according to the FDA.
For pirfenidone, the most serious adverse events versus placebo were liver enzyme elevations (3.7% versus 0.8%), sensitivity to light or rash (9.0% versus 1.0%), and gastrointestinal side effects that caused 2.2 % of patients to discontinue treatment compared with 1.0% of those who received placebo.
The most common side effects of pirfenidone were nausea, rash, abdominal pain, upper respiratory tract infection, diarrhea, fatigue, headache, dyspepsia, dizziness, vomiting, loss of appetite, gastroesophageal reflux, sinusitis, insomnia, decreased weight, and arthralgia.
The FDA warned about use of pirfenidone in patients with severe liver problems or end-stage kidney disease, or in those who require dialysis. Patients taking pirfenidone also must monitor and guard against sun exposure.
Clinical trials of pirfenidone included 1,247 idiopathic pulmonary fibrosis patients. In one with 555 patients, 17% of pirfenidone patients had an FVC decline of at least 10% after a year, compared with 32% who received placebo, Roche said.
Trials of nintedanib included 1,231 patients. One with 513 showed a relative reduction in annual FVC decline of 52% (–115 versus –240 mL for placebo) at 1 year, Boehringer Ingelheim noted.
The Food and Drug Administration on Oct. 15 approved two new oral medications for idiopathic pulmonary fibrosis, Boehringer Ingelheim’s nintedanib (Ofev) and Roche’s pirfenidone (Esbriet).
The drugs significantly slowed the decline of forced vital capacity (FVC), compared with placebo, in phase III testing. They did not significantly reduce mortality. The FDA granted both agents fast track, priority review, orphan product, and breakthrough designations. Both were also approved ahead of schedule, according to the agency. Each of the drugs should be available for patients within the next 2 weeks, according to their manufacturers.
For pirfenidone, already on the market in Europe and Canada, Roche said it plans “a comprehensive patient support program designed to help with access, financial support, and ongoing education.”
For nintedanib, Boehringer Ingelheim plans “a comprehensive patient support program that will provide a broad range of financial and nursing support services,” called “Open Doors.” In testing, the most frequent serious adverse reactions with nintedanib, versus placebo, were bronchitis (1.2% versus 0.8%) and myocardial infarction (1.5% versus 0.4%). Pneumonia (0.7% versus 0.6%), lung cancer (0.3% versus 0%), and MI (0.3% versus 0.2%) were the most common fatal adverse events.
The most common side effects of nintedanib were diarrhea, nausea, abdominal pain, vomiting, liver enzyme elevation, decreased appetite, headache, weight loss, and high blood pressure. Nintedanib is not recommended for patients who have moderate to severe liver problems or are pregnancy class D. Women of childbearing potential should use contraception for at least 3 months after stopping the drug, according to the FDA.
For pirfenidone, the most serious adverse events versus placebo were liver enzyme elevations (3.7% versus 0.8%), sensitivity to light or rash (9.0% versus 1.0%), and gastrointestinal side effects that caused 2.2 % of patients to discontinue treatment compared with 1.0% of those who received placebo.
The most common side effects of pirfenidone were nausea, rash, abdominal pain, upper respiratory tract infection, diarrhea, fatigue, headache, dyspepsia, dizziness, vomiting, loss of appetite, gastroesophageal reflux, sinusitis, insomnia, decreased weight, and arthralgia.
The FDA warned about use of pirfenidone in patients with severe liver problems or end-stage kidney disease, or in those who require dialysis. Patients taking pirfenidone also must monitor and guard against sun exposure.
Clinical trials of pirfenidone included 1,247 idiopathic pulmonary fibrosis patients. In one with 555 patients, 17% of pirfenidone patients had an FVC decline of at least 10% after a year, compared with 32% who received placebo, Roche said.
Trials of nintedanib included 1,231 patients. One with 513 showed a relative reduction in annual FVC decline of 52% (–115 versus –240 mL for placebo) at 1 year, Boehringer Ingelheim noted.
CDC program supplies guidance on Ebola patient management
Drawing upon the experience of Emory Healthcare in treating Ebola patients, the Centers for Disease Control and Prevention has issued a Clinician Outreach and Communication Activity program.
As hospitals in the United States begin to treat Ebola patients, a wide variety of processes including biological waste management, shipping processes, and lab testing are coming under scrutiny. At Emory, patients’ liquid wastes were disinfected with bleach or quaternary disinfecting detergents for more than 5 minutes prior to flushing or disposal. Linens and disposable material exposed to patients were handled as regulated medical waste and placed in leakproof containers. Additionally, Emory’s contractor called for a certification that materials were Ebola-free before transporting the waste to its incinerator.
Because sending a possible Ebola-contaminated specimen to the main hospital laboratory could cause that lab to be shut down for hours to address decontamination, Emory put in place a small point-of-care testing lab dedicated solely to specimens related to patients suspected of having Ebola. The dedicated lab included a chemistry analyzer, hematology analyzer, blood-gas analyzer, automated urinalysis analyzer, coagulation analyzer, and a malaria point-of-care device.
The Centers for Disease Control and Prevention has posted several online resources related to Ebola treatment and is expected to have a new web-based on-demand training video posted on Oct. 14.
Drawing upon the experience of Emory Healthcare in treating Ebola patients, the Centers for Disease Control and Prevention has issued a Clinician Outreach and Communication Activity program.
As hospitals in the United States begin to treat Ebola patients, a wide variety of processes including biological waste management, shipping processes, and lab testing are coming under scrutiny. At Emory, patients’ liquid wastes were disinfected with bleach or quaternary disinfecting detergents for more than 5 minutes prior to flushing or disposal. Linens and disposable material exposed to patients were handled as regulated medical waste and placed in leakproof containers. Additionally, Emory’s contractor called for a certification that materials were Ebola-free before transporting the waste to its incinerator.
Because sending a possible Ebola-contaminated specimen to the main hospital laboratory could cause that lab to be shut down for hours to address decontamination, Emory put in place a small point-of-care testing lab dedicated solely to specimens related to patients suspected of having Ebola. The dedicated lab included a chemistry analyzer, hematology analyzer, blood-gas analyzer, automated urinalysis analyzer, coagulation analyzer, and a malaria point-of-care device.
The Centers for Disease Control and Prevention has posted several online resources related to Ebola treatment and is expected to have a new web-based on-demand training video posted on Oct. 14.
Drawing upon the experience of Emory Healthcare in treating Ebola patients, the Centers for Disease Control and Prevention has issued a Clinician Outreach and Communication Activity program.
As hospitals in the United States begin to treat Ebola patients, a wide variety of processes including biological waste management, shipping processes, and lab testing are coming under scrutiny. At Emory, patients’ liquid wastes were disinfected with bleach or quaternary disinfecting detergents for more than 5 minutes prior to flushing or disposal. Linens and disposable material exposed to patients were handled as regulated medical waste and placed in leakproof containers. Additionally, Emory’s contractor called for a certification that materials were Ebola-free before transporting the waste to its incinerator.
Because sending a possible Ebola-contaminated specimen to the main hospital laboratory could cause that lab to be shut down for hours to address decontamination, Emory put in place a small point-of-care testing lab dedicated solely to specimens related to patients suspected of having Ebola. The dedicated lab included a chemistry analyzer, hematology analyzer, blood-gas analyzer, automated urinalysis analyzer, coagulation analyzer, and a malaria point-of-care device.
The Centers for Disease Control and Prevention has posted several online resources related to Ebola treatment and is expected to have a new web-based on-demand training video posted on Oct. 14.
New test will speed enterovirus D68 case confirmation from weeks to days
A new lab test for enterovirus D68 is expected to speed up testing and confirmation of cases, according to a press release from the Centers for Disease Control and Prevention.
The new test is a “real-time” reverse transcription polymerase chain reaction and can identify all strains of EV-D68. The previous test could be used to detect almost any enterovirus, but was labor intensive to perform and not conducive to large-scale testing.

Of the 1,200 samples from 45 states sent to the CDC for EV-D68 testing between Aug. 1 to Oct. 10, less than 200 have been tested and about half have tested positive. The CDC now expects to be able to test around 180 samples a day and complete in 7-10 days the testing on samples received since mid-September, The new method will “reduce what would normally take several weeks to get results to a few days,” Dr. Anne Schuchat, assistant surgeon general and director of the CDC’s National Center for Immunization and Respiratory Diseases, said in the press release.
As with other enteroviruses, the CDC expects new cases of EV-D68 will decrease in the fall, but faster testing will more accurately show trends of the disease and will help to monitor changes in the outbreak as it winds down, according to the CDC press release.
A new lab test for enterovirus D68 is expected to speed up testing and confirmation of cases, according to a press release from the Centers for Disease Control and Prevention.
The new test is a “real-time” reverse transcription polymerase chain reaction and can identify all strains of EV-D68. The previous test could be used to detect almost any enterovirus, but was labor intensive to perform and not conducive to large-scale testing.
Of the 1,200 samples from 45 states sent to the CDC for EV-D68 testing between Aug. 1 to Oct. 10, less than 200 have been tested and about half have tested positive. The CDC now expects to be able to test around 180 samples a day and complete in 7-10 days the testing on samples received since mid-September, The new method will “reduce what would normally take several weeks to get results to a few days,” Dr. Anne Schuchat, assistant surgeon general and director of the CDC’s National Center for Immunization and Respiratory Diseases, said in the press release.
As with other enteroviruses, the CDC expects new cases of EV-D68 will decrease in the fall, but faster testing will more accurately show trends of the disease and will help to monitor changes in the outbreak as it winds down, according to the CDC press release.
A new lab test for enterovirus D68 is expected to speed up testing and confirmation of cases, according to a press release from the Centers for Disease Control and Prevention.
The new test is a “real-time” reverse transcription polymerase chain reaction and can identify all strains of EV-D68. The previous test could be used to detect almost any enterovirus, but was labor intensive to perform and not conducive to large-scale testing.
Of the 1,200 samples from 45 states sent to the CDC for EV-D68 testing between Aug. 1 to Oct. 10, less than 200 have been tested and about half have tested positive. The CDC now expects to be able to test around 180 samples a day and complete in 7-10 days the testing on samples received since mid-September, The new method will “reduce what would normally take several weeks to get results to a few days,” Dr. Anne Schuchat, assistant surgeon general and director of the CDC’s National Center for Immunization and Respiratory Diseases, said in the press release.
As with other enteroviruses, the CDC expects new cases of EV-D68 will decrease in the fall, but faster testing will more accurately show trends of the disease and will help to monitor changes in the outbreak as it winds down, according to the CDC press release.

Inflammatory rheumatic diseases raise venous thromboembolism risk
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).

Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).
Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).
Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point: There is strong evidence for an elevated baseline risk of VTE in patients with inflammatory rheumatic diseases.
Major finding: Patients with rheumatoid arthritis are more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched patients.
Data source: Meta-analysis of 25 studies.
Disclosures: No conflicts of interest were declared.
