User login
News and Views that Matter to Pediatricians
The leading independent newspaper covering news and commentary in pediatrics.
COVID-19 vaccines: Preparing for patient questions
With U.S. approval of one coronavirus vaccine likely imminent and approval of a second one expected soon after, physicians will likely be deluged with questions. Public attitudes about the vaccines vary by demographics, with a recent poll showing that men and older adults are more likely to choose vaccination, and women and people of color evincing more wariness.
Although the reasons for reluctance may vary, questions from patient will likely be similar. Some are related to the “warp speed” language about the vaccines. Other concerns arise from the fact that the platform – mRNA – has not been used in human vaccines before. And as with any vaccine, there are rumors and false claims making the rounds on social media.
In anticipation of the most common questions physicians may encounter, two experts, Krutika Kuppalli, MD, assistant professor of medicine in the division of infectious diseases at the Medical University of South Carolina, Charleston, and Angela Rasmussen, PhD, virologist and nonresident affiliate at Georgetown University’s Center for Global Health Science and Security, Washington, talked in an interview about what clinicians can expect and what evidence-based – as well as compassionate – answers might look like.
Q: Will this vaccine give me COVID-19?
“There is not an intact virus in there,” Dr. Rasmussen said. The mRNA-based vaccines cannot cause COVID-19 because they don’t use any part of the coronavirus itself. Instead, the Moderna and Pfizer vaccines contain manufactured mRNA molecules that carry the instructions for building the virus’ spike protein. After vaccine administration, the recipient’s own cells take up this mRNA, use it to build this bit of protein, and display it on their surfaces. The foreign protein flag triggers the immune system response.
The mRNA does not enter the cell nucleus or interact with the recipient’s DNA. And because it’s so fragile, it degrades quite quickly. To keep that from happening before cell entry, the mRNAs are cushioned in protective fats.
Q: Was this vaccine made too quickly?
“People have been working on this platform for 30 years, so it’s not that this is brand new,” Dr. Kuppalli said.
Researchers began working on mRNA vaccines in the 1990s. Technological developments in the last decade have meant that their use has become feasible, and they have been tested in animals against many viral diseases. The mRNA vaccines are attractive because they’re expected to be safe and easily manufactured from common materials. That’s what we’ve seen in the COVID-19 pandemic, the Centers for Disease Control and Prevention says on its website. Design of the spike protein mRNA component began as soon as the viral genome became available in January.
Usually, rolling out a vaccine takes years, so less than a year under a program called Operation Warp Speed can seem like moving too fast, Dr. Rasmussen acknowledged. “The name has given people the impression that by going at warp speed, we’re cutting all the corners. [But] the reality is that Operation Warp Speed is mostly for manufacturing and distribution.”
What underlies the speed is a restructuring of the normal vaccine development process, Dr. Kuppalli said. The same phases of development – animal testing, a small initial human phase, a second for safety testing, a third large phase for efficacy – were all conducted as for any vaccine. But in this case, some phases were completed in parallel, rather than sequentially. This approach has proved so successful that there is already talk about making it the model for developing future vaccines.
Two other factors contributed to the speed, said Dr. Kuppalli and Dr. Rasmussen. First, gearing up production can slow a rollout, but with these vaccines, companies ramped up production even before anyone knew if the vaccines would work – the “warp speed” part. The second factor has been the large number of cases, making exposures more likely and thus accelerating the results of the efficacy trials. “There is so much COVID being transmitted everywhere in the United States that it did not take long to hit the threshold of events to read out phase 3,” Dr. Rasmussen said.
Q: This vaccine has never been used in humans. How do we know it’s safe?
The Pfizer phase 3 trial included more than 43,000 people, and Moderna’s had more than 30,000. The first humans received mRNA-based COVID-19 vaccines in March. The most common adverse events emerge right after a vaccination, Dr. Kuppalli said.
As with any vaccine that gains approval, monitoring will continue.
UK health officials have reported that two health care workers vaccinated in the initial rollout of the Pfizer vaccine had what seems to have been a severe allergic response. Both recipients had a history of anaphylactic allergic responses and carried EpiPens, and both recovered. During the trial, allergic reaction rates were 0.63% in the vaccine group and 0.51% in the placebo group.
As a result of the two reactions, UK regulators are now recommending that patients with a history of severe allergies not receive the vaccine at the current time.
Q: What are the likely side effects?
So far, the most common side effects are pain at the injection site and an achy, flu-like feeling, Dr. Kuppalli said. More severe reactions have been reported, but were not common in the trials.
Dr. Rasmussen noted that the common side effects are a good sign, and signal that the recipient is generating “a robust immune response.”
“Everybody I’ve talked to who’s had the response has said they would go through it again,” Dr. Kruppalli said. “I definitely plan on lining up and being one of the first people to get the vaccine.”
Q: I already had COVID-19 or had a positive antibody test. Do I still need to get the vaccine?
Dr. Rasmussen said that there are “too many unknowns” to say if a history of COVID-19 would make a difference. “We don’t know how long neutralizing antibodies last” after infection, she said. “What we know is that the vaccine tends to produce antibody titers towards the higher end of the spectrum,” suggesting better immunity with vaccination than after natural infection.
Q: Can patients of color feel safe getting the vaccine?
“People of color might be understandably reluctant to take a vaccine that was developed in a way that appears to be faster [than past development],” said Dr. Rasmussen. She said physicians should acknowledge and understand the history that has led them to feel that way, “everything from Tuskegee to Henrietta Lacks to today.”
Empathy is key, and “providers should meet patients where they are and not condescend to them.”
Dr. Kuppalli agreed. “Clinicians really need to work on trying to strip away their biases.”
Thus far there are no safety signals that differ by race or ethnicity, according to the companies. The Pfizer phase 3 trial enrolled just over 9% Black participants, 0.5% Native American/Alaska Native, 0.2% Native Hawaiian/Pacific Islander, 2.3% multiracial participants, and 28% Hispanic/Latinx. For its part, Moderna says that approximately 37% of participants in its phase 3 trial come from communities of color.
Q: What about children and pregnant women?
Although the trials included participants from many different age groups and backgrounds, children and pregnant or lactating women were not among them. Pfizer gained approval in October to include participants as young as age 12 years, and a Moderna spokesperson said in an interview that the company planned pediatric inclusion at the end of 2020, pending approval.
“Unfortunately, we don’t have data on pregnant and lactating women,” Dr. Kuppalli said. She said she hopes that public health organizations such as the CDC will address that in the coming weeks. Dr. Rasmussen called the lack of data in pregnant women and children “a big oversight.”
Dr. Rasmussen has disclosed no relevant financial relationships. Dr. Kuppalli is a consultant with GlaxoSmithKline.
A version of this article originally appeared on Medscape.com.
With U.S. approval of one coronavirus vaccine likely imminent and approval of a second one expected soon after, physicians will likely be deluged with questions. Public attitudes about the vaccines vary by demographics, with a recent poll showing that men and older adults are more likely to choose vaccination, and women and people of color evincing more wariness.
Although the reasons for reluctance may vary, questions from patient will likely be similar. Some are related to the “warp speed” language about the vaccines. Other concerns arise from the fact that the platform – mRNA – has not been used in human vaccines before. And as with any vaccine, there are rumors and false claims making the rounds on social media.
In anticipation of the most common questions physicians may encounter, two experts, Krutika Kuppalli, MD, assistant professor of medicine in the division of infectious diseases at the Medical University of South Carolina, Charleston, and Angela Rasmussen, PhD, virologist and nonresident affiliate at Georgetown University’s Center for Global Health Science and Security, Washington, talked in an interview about what clinicians can expect and what evidence-based – as well as compassionate – answers might look like.
Q: Will this vaccine give me COVID-19?
“There is not an intact virus in there,” Dr. Rasmussen said. The mRNA-based vaccines cannot cause COVID-19 because they don’t use any part of the coronavirus itself. Instead, the Moderna and Pfizer vaccines contain manufactured mRNA molecules that carry the instructions for building the virus’ spike protein. After vaccine administration, the recipient’s own cells take up this mRNA, use it to build this bit of protein, and display it on their surfaces. The foreign protein flag triggers the immune system response.
The mRNA does not enter the cell nucleus or interact with the recipient’s DNA. And because it’s so fragile, it degrades quite quickly. To keep that from happening before cell entry, the mRNAs are cushioned in protective fats.
Q: Was this vaccine made too quickly?
“People have been working on this platform for 30 years, so it’s not that this is brand new,” Dr. Kuppalli said.
Researchers began working on mRNA vaccines in the 1990s. Technological developments in the last decade have meant that their use has become feasible, and they have been tested in animals against many viral diseases. The mRNA vaccines are attractive because they’re expected to be safe and easily manufactured from common materials. That’s what we’ve seen in the COVID-19 pandemic, the Centers for Disease Control and Prevention says on its website. Design of the spike protein mRNA component began as soon as the viral genome became available in January.
Usually, rolling out a vaccine takes years, so less than a year under a program called Operation Warp Speed can seem like moving too fast, Dr. Rasmussen acknowledged. “The name has given people the impression that by going at warp speed, we’re cutting all the corners. [But] the reality is that Operation Warp Speed is mostly for manufacturing and distribution.”
What underlies the speed is a restructuring of the normal vaccine development process, Dr. Kuppalli said. The same phases of development – animal testing, a small initial human phase, a second for safety testing, a third large phase for efficacy – were all conducted as for any vaccine. But in this case, some phases were completed in parallel, rather than sequentially. This approach has proved so successful that there is already talk about making it the model for developing future vaccines.
Two other factors contributed to the speed, said Dr. Kuppalli and Dr. Rasmussen. First, gearing up production can slow a rollout, but with these vaccines, companies ramped up production even before anyone knew if the vaccines would work – the “warp speed” part. The second factor has been the large number of cases, making exposures more likely and thus accelerating the results of the efficacy trials. “There is so much COVID being transmitted everywhere in the United States that it did not take long to hit the threshold of events to read out phase 3,” Dr. Rasmussen said.
Q: This vaccine has never been used in humans. How do we know it’s safe?
The Pfizer phase 3 trial included more than 43,000 people, and Moderna’s had more than 30,000. The first humans received mRNA-based COVID-19 vaccines in March. The most common adverse events emerge right after a vaccination, Dr. Kuppalli said.
As with any vaccine that gains approval, monitoring will continue.
UK health officials have reported that two health care workers vaccinated in the initial rollout of the Pfizer vaccine had what seems to have been a severe allergic response. Both recipients had a history of anaphylactic allergic responses and carried EpiPens, and both recovered. During the trial, allergic reaction rates were 0.63% in the vaccine group and 0.51% in the placebo group.
As a result of the two reactions, UK regulators are now recommending that patients with a history of severe allergies not receive the vaccine at the current time.
Q: What are the likely side effects?
So far, the most common side effects are pain at the injection site and an achy, flu-like feeling, Dr. Kuppalli said. More severe reactions have been reported, but were not common in the trials.
Dr. Rasmussen noted that the common side effects are a good sign, and signal that the recipient is generating “a robust immune response.”
“Everybody I’ve talked to who’s had the response has said they would go through it again,” Dr. Kruppalli said. “I definitely plan on lining up and being one of the first people to get the vaccine.”
Q: I already had COVID-19 or had a positive antibody test. Do I still need to get the vaccine?
Dr. Rasmussen said that there are “too many unknowns” to say if a history of COVID-19 would make a difference. “We don’t know how long neutralizing antibodies last” after infection, she said. “What we know is that the vaccine tends to produce antibody titers towards the higher end of the spectrum,” suggesting better immunity with vaccination than after natural infection.
Q: Can patients of color feel safe getting the vaccine?
“People of color might be understandably reluctant to take a vaccine that was developed in a way that appears to be faster [than past development],” said Dr. Rasmussen. She said physicians should acknowledge and understand the history that has led them to feel that way, “everything from Tuskegee to Henrietta Lacks to today.”
Empathy is key, and “providers should meet patients where they are and not condescend to them.”
Dr. Kuppalli agreed. “Clinicians really need to work on trying to strip away their biases.”
Thus far there are no safety signals that differ by race or ethnicity, according to the companies. The Pfizer phase 3 trial enrolled just over 9% Black participants, 0.5% Native American/Alaska Native, 0.2% Native Hawaiian/Pacific Islander, 2.3% multiracial participants, and 28% Hispanic/Latinx. For its part, Moderna says that approximately 37% of participants in its phase 3 trial come from communities of color.
Q: What about children and pregnant women?
Although the trials included participants from many different age groups and backgrounds, children and pregnant or lactating women were not among them. Pfizer gained approval in October to include participants as young as age 12 years, and a Moderna spokesperson said in an interview that the company planned pediatric inclusion at the end of 2020, pending approval.
“Unfortunately, we don’t have data on pregnant and lactating women,” Dr. Kuppalli said. She said she hopes that public health organizations such as the CDC will address that in the coming weeks. Dr. Rasmussen called the lack of data in pregnant women and children “a big oversight.”
Dr. Rasmussen has disclosed no relevant financial relationships. Dr. Kuppalli is a consultant with GlaxoSmithKline.
A version of this article originally appeared on Medscape.com.
With U.S. approval of one coronavirus vaccine likely imminent and approval of a second one expected soon after, physicians will likely be deluged with questions. Public attitudes about the vaccines vary by demographics, with a recent poll showing that men and older adults are more likely to choose vaccination, and women and people of color evincing more wariness.
Although the reasons for reluctance may vary, questions from patient will likely be similar. Some are related to the “warp speed” language about the vaccines. Other concerns arise from the fact that the platform – mRNA – has not been used in human vaccines before. And as with any vaccine, there are rumors and false claims making the rounds on social media.
In anticipation of the most common questions physicians may encounter, two experts, Krutika Kuppalli, MD, assistant professor of medicine in the division of infectious diseases at the Medical University of South Carolina, Charleston, and Angela Rasmussen, PhD, virologist and nonresident affiliate at Georgetown University’s Center for Global Health Science and Security, Washington, talked in an interview about what clinicians can expect and what evidence-based – as well as compassionate – answers might look like.
Q: Will this vaccine give me COVID-19?
“There is not an intact virus in there,” Dr. Rasmussen said. The mRNA-based vaccines cannot cause COVID-19 because they don’t use any part of the coronavirus itself. Instead, the Moderna and Pfizer vaccines contain manufactured mRNA molecules that carry the instructions for building the virus’ spike protein. After vaccine administration, the recipient’s own cells take up this mRNA, use it to build this bit of protein, and display it on their surfaces. The foreign protein flag triggers the immune system response.
The mRNA does not enter the cell nucleus or interact with the recipient’s DNA. And because it’s so fragile, it degrades quite quickly. To keep that from happening before cell entry, the mRNAs are cushioned in protective fats.
Q: Was this vaccine made too quickly?
“People have been working on this platform for 30 years, so it’s not that this is brand new,” Dr. Kuppalli said.
Researchers began working on mRNA vaccines in the 1990s. Technological developments in the last decade have meant that their use has become feasible, and they have been tested in animals against many viral diseases. The mRNA vaccines are attractive because they’re expected to be safe and easily manufactured from common materials. That’s what we’ve seen in the COVID-19 pandemic, the Centers for Disease Control and Prevention says on its website. Design of the spike protein mRNA component began as soon as the viral genome became available in January.
Usually, rolling out a vaccine takes years, so less than a year under a program called Operation Warp Speed can seem like moving too fast, Dr. Rasmussen acknowledged. “The name has given people the impression that by going at warp speed, we’re cutting all the corners. [But] the reality is that Operation Warp Speed is mostly for manufacturing and distribution.”
What underlies the speed is a restructuring of the normal vaccine development process, Dr. Kuppalli said. The same phases of development – animal testing, a small initial human phase, a second for safety testing, a third large phase for efficacy – were all conducted as for any vaccine. But in this case, some phases were completed in parallel, rather than sequentially. This approach has proved so successful that there is already talk about making it the model for developing future vaccines.
Two other factors contributed to the speed, said Dr. Kuppalli and Dr. Rasmussen. First, gearing up production can slow a rollout, but with these vaccines, companies ramped up production even before anyone knew if the vaccines would work – the “warp speed” part. The second factor has been the large number of cases, making exposures more likely and thus accelerating the results of the efficacy trials. “There is so much COVID being transmitted everywhere in the United States that it did not take long to hit the threshold of events to read out phase 3,” Dr. Rasmussen said.
Q: This vaccine has never been used in humans. How do we know it’s safe?
The Pfizer phase 3 trial included more than 43,000 people, and Moderna’s had more than 30,000. The first humans received mRNA-based COVID-19 vaccines in March. The most common adverse events emerge right after a vaccination, Dr. Kuppalli said.
As with any vaccine that gains approval, monitoring will continue.
UK health officials have reported that two health care workers vaccinated in the initial rollout of the Pfizer vaccine had what seems to have been a severe allergic response. Both recipients had a history of anaphylactic allergic responses and carried EpiPens, and both recovered. During the trial, allergic reaction rates were 0.63% in the vaccine group and 0.51% in the placebo group.
As a result of the two reactions, UK regulators are now recommending that patients with a history of severe allergies not receive the vaccine at the current time.
Q: What are the likely side effects?
So far, the most common side effects are pain at the injection site and an achy, flu-like feeling, Dr. Kuppalli said. More severe reactions have been reported, but were not common in the trials.
Dr. Rasmussen noted that the common side effects are a good sign, and signal that the recipient is generating “a robust immune response.”
“Everybody I’ve talked to who’s had the response has said they would go through it again,” Dr. Kruppalli said. “I definitely plan on lining up and being one of the first people to get the vaccine.”
Q: I already had COVID-19 or had a positive antibody test. Do I still need to get the vaccine?
Dr. Rasmussen said that there are “too many unknowns” to say if a history of COVID-19 would make a difference. “We don’t know how long neutralizing antibodies last” after infection, she said. “What we know is that the vaccine tends to produce antibody titers towards the higher end of the spectrum,” suggesting better immunity with vaccination than after natural infection.
Q: Can patients of color feel safe getting the vaccine?
“People of color might be understandably reluctant to take a vaccine that was developed in a way that appears to be faster [than past development],” said Dr. Rasmussen. She said physicians should acknowledge and understand the history that has led them to feel that way, “everything from Tuskegee to Henrietta Lacks to today.”
Empathy is key, and “providers should meet patients where they are and not condescend to them.”
Dr. Kuppalli agreed. “Clinicians really need to work on trying to strip away their biases.”
Thus far there are no safety signals that differ by race or ethnicity, according to the companies. The Pfizer phase 3 trial enrolled just over 9% Black participants, 0.5% Native American/Alaska Native, 0.2% Native Hawaiian/Pacific Islander, 2.3% multiracial participants, and 28% Hispanic/Latinx. For its part, Moderna says that approximately 37% of participants in its phase 3 trial come from communities of color.
Q: What about children and pregnant women?
Although the trials included participants from many different age groups and backgrounds, children and pregnant or lactating women were not among them. Pfizer gained approval in October to include participants as young as age 12 years, and a Moderna spokesperson said in an interview that the company planned pediatric inclusion at the end of 2020, pending approval.
“Unfortunately, we don’t have data on pregnant and lactating women,” Dr. Kuppalli said. She said she hopes that public health organizations such as the CDC will address that in the coming weeks. Dr. Rasmussen called the lack of data in pregnant women and children “a big oversight.”
Dr. Rasmussen has disclosed no relevant financial relationships. Dr. Kuppalli is a consultant with GlaxoSmithKline.
A version of this article originally appeared on Medscape.com.
Vitamin D deficiency in COVID-19 quadrupled death rate
Vitamin D deficiency on admission to hospital was associated with a 3.7-fold increase in the odds of dying from COVID-19, according to an observational study looking back at data from the first wave of the pandemic.
Nearly 60% of patients with COVID-19 were vitamin D deficient upon hospitalization, with men in the advanced stages of COVID-19 pneumonia showing the greatest deficit.
Importantly, the results were independent of comorbidities known to be affected by vitamin D deficiency, wrote the authors, led by Dieter De Smet, MD, from AZ Delta General Hospital, Roeselare, Belgium.
“[The findings] highlight the need for randomized, controlled trials specifically targeting vitamin D–deficient patients at intake, and make a call for general avoidance of vitamin D deficiency as a safe and inexpensive possible mitigation of the SARS-CoV-2 pandemic,” Dr. De Smet and colleagues wrote in their article, published online Nov. 25 in the American Journal of Clinical Pathology.
A search of ClinicalTrials.gov reveals there are currently close to 40 ongoing intervention trials with vitamin D in COVID-19 around the world for varying purposes, including prevention, and varying forms of treatment.
Consider vitamin D to prevent COVID-19 infection
With regard to the potential role in prevention, “Numerous observational studies have shown that low vitamin D levels are a major predictor for poor COVID outcomes,” noted Jacob Teitelbaum, MD, an internist who specializes in treating chronic fatigue syndrome and fibromyalgia who also has an interest in COVID-19.
“This study shows how severe a problem this is,” Dr. Teitelbaum said in an interview. “A 3.7-fold increase in death rate if someone’s vitamin D level was below 20 [ng/mL] is staggering. It is arguably one of the most important risk factors to consider.”
“What is not clear is whether vitamin D levels are acting as an acute-phase reactant, dropping because of the infection, with larger drops indicating more severe disease, or whether vitamin D deficiency is causing worse outcomes,” added Dr. Teitelbaum, who is director of the Center for Effective CFIDS/Fibromyalgia Therapies, Kailua-Kona, Hawaii.
Also asked to comment, Andrea Giustina, MD, president of the European Society of Endocrinology, said: “The paper by De Smet et al confirms what we already hypothesized in BMJ last March: that patients with low vitamin D levels are at high risk of hospitalization for COVID-19 and developing severe and lethal disease. This is likely due to the loss in the protective action of vitamin D on the immune system and against the SARS-CoV-2–induced cytokine storm.”
He said it is particularly interesting that the authors of the new study had reported more prevalent vitamin D deficiency among men than women, most likely because women are more often treated with vitamin D for osteoporosis.
The new study should prompt all clinicians and health authorities to seriously consider vitamin D supplementation as an additional tool in the fight against COVID-19, particularly for the prevention of infection in those at high risk of both COVID-19 and hypovitaminosis D, such the elderly, urged Dr. Giustina, of San Raffaele Vita-Salute University, Milan.
Results adjusted for multiple confounders
Dr. De Smet and colleagues looked at serum 25-hydroxyvitamin D (25[OH]D) levels in 186 patients hospitalized for severe COVID-19 infection as a function of radiologic stage of COVID-19 pneumonia as well as the association between vitamin D status on admission and COVID-19 mortality.
Cognizant of the potential for confounding by multiple factors, they adjusted for age, sex, and known vitamin D–affected comorbidities such as diabetes, chronic lung disease, and coronary artery disease.
Patients were hospitalized from March 1 to April 7, 2020 (the peak of the first wave of the pandemic) at their institution, AZ Delta General Hospital, a tertiary network hospital.
The mean age of patients was 69 years, 41% were women, and 59% had coronary artery disease. Upon admission to hospital, median vitamin D level was 18 ng/mL (women, 20.7 ng/mL; men, 17.6 ng/mL).
A remarkably high percentage (59%, 109/186) of patients with COVID-19 were vitamin D deficient (25[OH]D <20 ng/mL) when admitted (47% of women and 67% of men), wrote the authors.
“What surprises me,” said Dr. Teitelbaum, is that almost 60% “of these patients had 25(OH)D under 20 ng/mL but most clinicians consider under 50 to be low.”
All patients had a chest CT scan to determine the radiologic stage of COVID-19 pneumonia and serum vitamin D measurement on admission. Radiologic stage of pneumonia was used as a proxy for immunologic phase of COVID-19.
Vitamin D deficiency correlated with worsening pneumonia
Among men, rates of vitamin D deficiency increased with advancing disease, with rates of 55% in stage 1, 67% in stage 2, and up to 74% in stage 3 pneumonia.
There is therefore “a clear correlation between 25(OH)D level and temporal stages of viral pneumonia, particularly in male patients,” the authors wrote.
“Vitamin D dampens excessive inflammation,” said Dr. Teitelbaum. “In these patients with acute respiratory distress syndrome, the immune system has gone wild.”
“The study was carried out in Belgium, so there’s less sunlight there than some other places, but even here in Hawaii, with plenty of sunshine, we have vitamin D deficiency,” he added.
“More studies are needed, but I think there are enough data to suggest a multivitamin should be used to aid prophylaxis, and this is reflected in [some] infectious disease recommendations,” he noted.
A version of this article originally appeared on Medscape.com.
Vitamin D deficiency on admission to hospital was associated with a 3.7-fold increase in the odds of dying from COVID-19, according to an observational study looking back at data from the first wave of the pandemic.
Nearly 60% of patients with COVID-19 were vitamin D deficient upon hospitalization, with men in the advanced stages of COVID-19 pneumonia showing the greatest deficit.
Importantly, the results were independent of comorbidities known to be affected by vitamin D deficiency, wrote the authors, led by Dieter De Smet, MD, from AZ Delta General Hospital, Roeselare, Belgium.
“[The findings] highlight the need for randomized, controlled trials specifically targeting vitamin D–deficient patients at intake, and make a call for general avoidance of vitamin D deficiency as a safe and inexpensive possible mitigation of the SARS-CoV-2 pandemic,” Dr. De Smet and colleagues wrote in their article, published online Nov. 25 in the American Journal of Clinical Pathology.
A search of ClinicalTrials.gov reveals there are currently close to 40 ongoing intervention trials with vitamin D in COVID-19 around the world for varying purposes, including prevention, and varying forms of treatment.
Consider vitamin D to prevent COVID-19 infection
With regard to the potential role in prevention, “Numerous observational studies have shown that low vitamin D levels are a major predictor for poor COVID outcomes,” noted Jacob Teitelbaum, MD, an internist who specializes in treating chronic fatigue syndrome and fibromyalgia who also has an interest in COVID-19.
“This study shows how severe a problem this is,” Dr. Teitelbaum said in an interview. “A 3.7-fold increase in death rate if someone’s vitamin D level was below 20 [ng/mL] is staggering. It is arguably one of the most important risk factors to consider.”
“What is not clear is whether vitamin D levels are acting as an acute-phase reactant, dropping because of the infection, with larger drops indicating more severe disease, or whether vitamin D deficiency is causing worse outcomes,” added Dr. Teitelbaum, who is director of the Center for Effective CFIDS/Fibromyalgia Therapies, Kailua-Kona, Hawaii.
Also asked to comment, Andrea Giustina, MD, president of the European Society of Endocrinology, said: “The paper by De Smet et al confirms what we already hypothesized in BMJ last March: that patients with low vitamin D levels are at high risk of hospitalization for COVID-19 and developing severe and lethal disease. This is likely due to the loss in the protective action of vitamin D on the immune system and against the SARS-CoV-2–induced cytokine storm.”
He said it is particularly interesting that the authors of the new study had reported more prevalent vitamin D deficiency among men than women, most likely because women are more often treated with vitamin D for osteoporosis.
The new study should prompt all clinicians and health authorities to seriously consider vitamin D supplementation as an additional tool in the fight against COVID-19, particularly for the prevention of infection in those at high risk of both COVID-19 and hypovitaminosis D, such the elderly, urged Dr. Giustina, of San Raffaele Vita-Salute University, Milan.
Results adjusted for multiple confounders
Dr. De Smet and colleagues looked at serum 25-hydroxyvitamin D (25[OH]D) levels in 186 patients hospitalized for severe COVID-19 infection as a function of radiologic stage of COVID-19 pneumonia as well as the association between vitamin D status on admission and COVID-19 mortality.
Cognizant of the potential for confounding by multiple factors, they adjusted for age, sex, and known vitamin D–affected comorbidities such as diabetes, chronic lung disease, and coronary artery disease.
Patients were hospitalized from March 1 to April 7, 2020 (the peak of the first wave of the pandemic) at their institution, AZ Delta General Hospital, a tertiary network hospital.
The mean age of patients was 69 years, 41% were women, and 59% had coronary artery disease. Upon admission to hospital, median vitamin D level was 18 ng/mL (women, 20.7 ng/mL; men, 17.6 ng/mL).
A remarkably high percentage (59%, 109/186) of patients with COVID-19 were vitamin D deficient (25[OH]D <20 ng/mL) when admitted (47% of women and 67% of men), wrote the authors.
“What surprises me,” said Dr. Teitelbaum, is that almost 60% “of these patients had 25(OH)D under 20 ng/mL but most clinicians consider under 50 to be low.”
All patients had a chest CT scan to determine the radiologic stage of COVID-19 pneumonia and serum vitamin D measurement on admission. Radiologic stage of pneumonia was used as a proxy for immunologic phase of COVID-19.
Vitamin D deficiency correlated with worsening pneumonia
Among men, rates of vitamin D deficiency increased with advancing disease, with rates of 55% in stage 1, 67% in stage 2, and up to 74% in stage 3 pneumonia.
There is therefore “a clear correlation between 25(OH)D level and temporal stages of viral pneumonia, particularly in male patients,” the authors wrote.
“Vitamin D dampens excessive inflammation,” said Dr. Teitelbaum. “In these patients with acute respiratory distress syndrome, the immune system has gone wild.”
“The study was carried out in Belgium, so there’s less sunlight there than some other places, but even here in Hawaii, with plenty of sunshine, we have vitamin D deficiency,” he added.
“More studies are needed, but I think there are enough data to suggest a multivitamin should be used to aid prophylaxis, and this is reflected in [some] infectious disease recommendations,” he noted.
A version of this article originally appeared on Medscape.com.
Vitamin D deficiency on admission to hospital was associated with a 3.7-fold increase in the odds of dying from COVID-19, according to an observational study looking back at data from the first wave of the pandemic.
Nearly 60% of patients with COVID-19 were vitamin D deficient upon hospitalization, with men in the advanced stages of COVID-19 pneumonia showing the greatest deficit.
Importantly, the results were independent of comorbidities known to be affected by vitamin D deficiency, wrote the authors, led by Dieter De Smet, MD, from AZ Delta General Hospital, Roeselare, Belgium.
“[The findings] highlight the need for randomized, controlled trials specifically targeting vitamin D–deficient patients at intake, and make a call for general avoidance of vitamin D deficiency as a safe and inexpensive possible mitigation of the SARS-CoV-2 pandemic,” Dr. De Smet and colleagues wrote in their article, published online Nov. 25 in the American Journal of Clinical Pathology.
A search of ClinicalTrials.gov reveals there are currently close to 40 ongoing intervention trials with vitamin D in COVID-19 around the world for varying purposes, including prevention, and varying forms of treatment.
Consider vitamin D to prevent COVID-19 infection
With regard to the potential role in prevention, “Numerous observational studies have shown that low vitamin D levels are a major predictor for poor COVID outcomes,” noted Jacob Teitelbaum, MD, an internist who specializes in treating chronic fatigue syndrome and fibromyalgia who also has an interest in COVID-19.
“This study shows how severe a problem this is,” Dr. Teitelbaum said in an interview. “A 3.7-fold increase in death rate if someone’s vitamin D level was below 20 [ng/mL] is staggering. It is arguably one of the most important risk factors to consider.”
“What is not clear is whether vitamin D levels are acting as an acute-phase reactant, dropping because of the infection, with larger drops indicating more severe disease, or whether vitamin D deficiency is causing worse outcomes,” added Dr. Teitelbaum, who is director of the Center for Effective CFIDS/Fibromyalgia Therapies, Kailua-Kona, Hawaii.
Also asked to comment, Andrea Giustina, MD, president of the European Society of Endocrinology, said: “The paper by De Smet et al confirms what we already hypothesized in BMJ last March: that patients with low vitamin D levels are at high risk of hospitalization for COVID-19 and developing severe and lethal disease. This is likely due to the loss in the protective action of vitamin D on the immune system and against the SARS-CoV-2–induced cytokine storm.”
He said it is particularly interesting that the authors of the new study had reported more prevalent vitamin D deficiency among men than women, most likely because women are more often treated with vitamin D for osteoporosis.
The new study should prompt all clinicians and health authorities to seriously consider vitamin D supplementation as an additional tool in the fight against COVID-19, particularly for the prevention of infection in those at high risk of both COVID-19 and hypovitaminosis D, such the elderly, urged Dr. Giustina, of San Raffaele Vita-Salute University, Milan.
Results adjusted for multiple confounders
Dr. De Smet and colleagues looked at serum 25-hydroxyvitamin D (25[OH]D) levels in 186 patients hospitalized for severe COVID-19 infection as a function of radiologic stage of COVID-19 pneumonia as well as the association between vitamin D status on admission and COVID-19 mortality.
Cognizant of the potential for confounding by multiple factors, they adjusted for age, sex, and known vitamin D–affected comorbidities such as diabetes, chronic lung disease, and coronary artery disease.
Patients were hospitalized from March 1 to April 7, 2020 (the peak of the first wave of the pandemic) at their institution, AZ Delta General Hospital, a tertiary network hospital.
The mean age of patients was 69 years, 41% were women, and 59% had coronary artery disease. Upon admission to hospital, median vitamin D level was 18 ng/mL (women, 20.7 ng/mL; men, 17.6 ng/mL).
A remarkably high percentage (59%, 109/186) of patients with COVID-19 were vitamin D deficient (25[OH]D <20 ng/mL) when admitted (47% of women and 67% of men), wrote the authors.
“What surprises me,” said Dr. Teitelbaum, is that almost 60% “of these patients had 25(OH)D under 20 ng/mL but most clinicians consider under 50 to be low.”
All patients had a chest CT scan to determine the radiologic stage of COVID-19 pneumonia and serum vitamin D measurement on admission. Radiologic stage of pneumonia was used as a proxy for immunologic phase of COVID-19.
Vitamin D deficiency correlated with worsening pneumonia
Among men, rates of vitamin D deficiency increased with advancing disease, with rates of 55% in stage 1, 67% in stage 2, and up to 74% in stage 3 pneumonia.
There is therefore “a clear correlation between 25(OH)D level and temporal stages of viral pneumonia, particularly in male patients,” the authors wrote.
“Vitamin D dampens excessive inflammation,” said Dr. Teitelbaum. “In these patients with acute respiratory distress syndrome, the immune system has gone wild.”
“The study was carried out in Belgium, so there’s less sunlight there than some other places, but even here in Hawaii, with plenty of sunshine, we have vitamin D deficiency,” he added.
“More studies are needed, but I think there are enough data to suggest a multivitamin should be used to aid prophylaxis, and this is reflected in [some] infectious disease recommendations,” he noted.
A version of this article originally appeared on Medscape.com.
Just under three million will get COVID-19 vaccine in first week
The federal government says it will distribute only enough doses of Pfizer’s COVID-19 vaccine to immunize 2.9 million Americans in the first week after the US Food and Drug Administration (FDA) authorizes it, far less than the initially discussed 6.4 million doses.
Theoretically, states have already formulated plans for distribution based on the revised lower amount. But in a briefing with reporters on December 9, officials from Operation Warp Speed and the Department of Health and Human Services (HHS) didn’t make clear exactly what the states were expecting.
Vaccine will be shipped to and allocated by 64 jurisdictions and five federal agencies — the Bureau of Prisons, the Department of Defense, the Department of State, the Indian Health Service, and the Veterans Health Administration — according to the Centers for Disease Control and Prevention’s COVID-19 Vaccination Program Interim Playbook.
It will be up to states — which will receive a supply prorated to population — and these agencies to determine how to prioritize distribution of the 2.9 million doses. Each state and agency has its own plan. Gen. Gustave Perna, the chief operating officer for Operation Warp Speed, said in the briefing that 30 states have told the federal government they will prioritize initial doses for residents and staff of long-term care facilities.
The distribution is contingent on FDA authorization, which could happen soon. The FDA’s Vaccines and Related Biologics Advisory Committee weighed the effectiveness data for the Pfizer vaccine on December 10 and recommended that the agency grant emergency authorization. The FDA could issue a decision at any time.
Fewer doses out of the gate
Perna said the federal government will begin shipping the Pfizer vaccine within 24 hours of an FDA authorization.
He said those shipments will include a total of 2.9 million doses — not the 6.4 million that will be available. The government is holding 500,000 doses in reserve and another 2.9 million to guarantee that the first few million people who are vaccinated will be able to receive a second dose 21 days later, said Perna.
In part, that is because the FDA labeling will require that a first dose be followed by a second exactly 21 days later, said HHS Secretary Alex Azar in the briefing.
Federal officials have calculated how much to hold back on the basis of Pfizer’s production, said Azar. At least initially, “we will not distribute a vaccine knowing that the booster will not be available either from reserve supply by us or ongoing expected predicted production,” he said.
Even with Pfizer having reduced its estimates of how much vaccine it can deliver in December, Azar said, “There will be enough vaccine available for 20 million first vaccinations in the month of December.”
That estimate is predicated, however, on the idea that a vaccine under development by Moderna will receive clearance shortly after the FDA assesses that vaccine’s safety and effectiveness on December 17.
This article first appeared on Medscape.com.
The federal government says it will distribute only enough doses of Pfizer’s COVID-19 vaccine to immunize 2.9 million Americans in the first week after the US Food and Drug Administration (FDA) authorizes it, far less than the initially discussed 6.4 million doses.
Theoretically, states have already formulated plans for distribution based on the revised lower amount. But in a briefing with reporters on December 9, officials from Operation Warp Speed and the Department of Health and Human Services (HHS) didn’t make clear exactly what the states were expecting.
Vaccine will be shipped to and allocated by 64 jurisdictions and five federal agencies — the Bureau of Prisons, the Department of Defense, the Department of State, the Indian Health Service, and the Veterans Health Administration — according to the Centers for Disease Control and Prevention’s COVID-19 Vaccination Program Interim Playbook.
It will be up to states — which will receive a supply prorated to population — and these agencies to determine how to prioritize distribution of the 2.9 million doses. Each state and agency has its own plan. Gen. Gustave Perna, the chief operating officer for Operation Warp Speed, said in the briefing that 30 states have told the federal government they will prioritize initial doses for residents and staff of long-term care facilities.
The distribution is contingent on FDA authorization, which could happen soon. The FDA’s Vaccines and Related Biologics Advisory Committee weighed the effectiveness data for the Pfizer vaccine on December 10 and recommended that the agency grant emergency authorization. The FDA could issue a decision at any time.
Fewer doses out of the gate
Perna said the federal government will begin shipping the Pfizer vaccine within 24 hours of an FDA authorization.
He said those shipments will include a total of 2.9 million doses — not the 6.4 million that will be available. The government is holding 500,000 doses in reserve and another 2.9 million to guarantee that the first few million people who are vaccinated will be able to receive a second dose 21 days later, said Perna.
In part, that is because the FDA labeling will require that a first dose be followed by a second exactly 21 days later, said HHS Secretary Alex Azar in the briefing.
Federal officials have calculated how much to hold back on the basis of Pfizer’s production, said Azar. At least initially, “we will not distribute a vaccine knowing that the booster will not be available either from reserve supply by us or ongoing expected predicted production,” he said.
Even with Pfizer having reduced its estimates of how much vaccine it can deliver in December, Azar said, “There will be enough vaccine available for 20 million first vaccinations in the month of December.”
That estimate is predicated, however, on the idea that a vaccine under development by Moderna will receive clearance shortly after the FDA assesses that vaccine’s safety and effectiveness on December 17.
This article first appeared on Medscape.com.
The federal government says it will distribute only enough doses of Pfizer’s COVID-19 vaccine to immunize 2.9 million Americans in the first week after the US Food and Drug Administration (FDA) authorizes it, far less than the initially discussed 6.4 million doses.
Theoretically, states have already formulated plans for distribution based on the revised lower amount. But in a briefing with reporters on December 9, officials from Operation Warp Speed and the Department of Health and Human Services (HHS) didn’t make clear exactly what the states were expecting.
Vaccine will be shipped to and allocated by 64 jurisdictions and five federal agencies — the Bureau of Prisons, the Department of Defense, the Department of State, the Indian Health Service, and the Veterans Health Administration — according to the Centers for Disease Control and Prevention’s COVID-19 Vaccination Program Interim Playbook.
It will be up to states — which will receive a supply prorated to population — and these agencies to determine how to prioritize distribution of the 2.9 million doses. Each state and agency has its own plan. Gen. Gustave Perna, the chief operating officer for Operation Warp Speed, said in the briefing that 30 states have told the federal government they will prioritize initial doses for residents and staff of long-term care facilities.
The distribution is contingent on FDA authorization, which could happen soon. The FDA’s Vaccines and Related Biologics Advisory Committee weighed the effectiveness data for the Pfizer vaccine on December 10 and recommended that the agency grant emergency authorization. The FDA could issue a decision at any time.
Fewer doses out of the gate
Perna said the federal government will begin shipping the Pfizer vaccine within 24 hours of an FDA authorization.
He said those shipments will include a total of 2.9 million doses — not the 6.4 million that will be available. The government is holding 500,000 doses in reserve and another 2.9 million to guarantee that the first few million people who are vaccinated will be able to receive a second dose 21 days later, said Perna.
In part, that is because the FDA labeling will require that a first dose be followed by a second exactly 21 days later, said HHS Secretary Alex Azar in the briefing.
Federal officials have calculated how much to hold back on the basis of Pfizer’s production, said Azar. At least initially, “we will not distribute a vaccine knowing that the booster will not be available either from reserve supply by us or ongoing expected predicted production,” he said.
Even with Pfizer having reduced its estimates of how much vaccine it can deliver in December, Azar said, “There will be enough vaccine available for 20 million first vaccinations in the month of December.”
That estimate is predicated, however, on the idea that a vaccine under development by Moderna will receive clearance shortly after the FDA assesses that vaccine’s safety and effectiveness on December 17.
This article first appeared on Medscape.com.
Antihistamine prescribing for AD varies by specialty
(AD), according to an analysis of a national database.
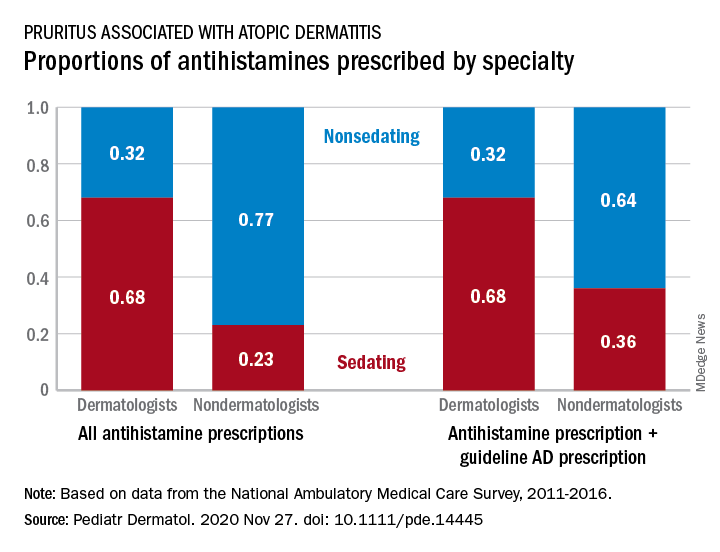
Dermatologists were more likely to prescribe sedating than nonsedating antihistamines (0.68 vs. 0.32) for patients with AD, but the reverse applied to nondermatologists, whose antihistamine distribution was 0.23 sedating and 0.77 nonsedating, based on 2011-2016 data from the National Ambulatory Medical Care Survey.
The numbers were similar for new antihistamine prescriptions, with sedating/nonsedating proportions of 0.60/0.40 for dermatologists and 0.24/0.76 for nondermatologists. Addition of guideline-recommended drugs such as topical corticosteroids and calcineurin inhibitors to the AD equation did not change the result, as dermatologists again showed a preference for sedating antihistamines, compared with nondermatologists, the investigators said.
The data also showed that Black patients with AD were more likely than were White patients to receive prescriptions for first-generation antihistamines and for therapies recommended by the AAD guidelines, and that patients under 21 years received more sedating antihistamines than did patients over age 21, they reported.
The age disparity “may be due to patient preference, as sedation effects may be less desirable to adult patients,” the investigators noted.
SOURCE: Garg S et al. Pediatr Dermatol. 2020 Nov 27. doi: 10.1111/pde.14445.
(AD), according to an analysis of a national database.
Dermatologists were more likely to prescribe sedating than nonsedating antihistamines (0.68 vs. 0.32) for patients with AD, but the reverse applied to nondermatologists, whose antihistamine distribution was 0.23 sedating and 0.77 nonsedating, based on 2011-2016 data from the National Ambulatory Medical Care Survey.
The numbers were similar for new antihistamine prescriptions, with sedating/nonsedating proportions of 0.60/0.40 for dermatologists and 0.24/0.76 for nondermatologists. Addition of guideline-recommended drugs such as topical corticosteroids and calcineurin inhibitors to the AD equation did not change the result, as dermatologists again showed a preference for sedating antihistamines, compared with nondermatologists, the investigators said.
The data also showed that Black patients with AD were more likely than were White patients to receive prescriptions for first-generation antihistamines and for therapies recommended by the AAD guidelines, and that patients under 21 years received more sedating antihistamines than did patients over age 21, they reported.
The age disparity “may be due to patient preference, as sedation effects may be less desirable to adult patients,” the investigators noted.
SOURCE: Garg S et al. Pediatr Dermatol. 2020 Nov 27. doi: 10.1111/pde.14445.
(AD), according to an analysis of a national database.
Dermatologists were more likely to prescribe sedating than nonsedating antihistamines (0.68 vs. 0.32) for patients with AD, but the reverse applied to nondermatologists, whose antihistamine distribution was 0.23 sedating and 0.77 nonsedating, based on 2011-2016 data from the National Ambulatory Medical Care Survey.
The numbers were similar for new antihistamine prescriptions, with sedating/nonsedating proportions of 0.60/0.40 for dermatologists and 0.24/0.76 for nondermatologists. Addition of guideline-recommended drugs such as topical corticosteroids and calcineurin inhibitors to the AD equation did not change the result, as dermatologists again showed a preference for sedating antihistamines, compared with nondermatologists, the investigators said.
The data also showed that Black patients with AD were more likely than were White patients to receive prescriptions for first-generation antihistamines and for therapies recommended by the AAD guidelines, and that patients under 21 years received more sedating antihistamines than did patients over age 21, they reported.
The age disparity “may be due to patient preference, as sedation effects may be less desirable to adult patients,” the investigators noted.
SOURCE: Garg S et al. Pediatr Dermatol. 2020 Nov 27. doi: 10.1111/pde.14445.
FROM PEDIATRIC DERMATOLOGY
Advocate for legislation to improve, protect LGBTQ lives
In January in many states, the start of a new year also means the start of a new legislative session. For LGBTQ youth and their families, these sessions can create a significant amount of anxiety, as legislators in several states introduce legislation to curtail the rights of this population. In some cases, legislators have attempted to criminalize the provision of gender-affirming medical care to the trans and gender-diverse adolescents that many of us provide care to on a daily basis. As pediatricians,
2020 started on a positive note for LGBTQ children and adolescents, with Virginia becoming the 20th state to ban conversion therapy for minors. Legislation was introduced in several other states to prohibit this practice, including Kentucky, Missouri, and Ohio, and but they ultimately died in committee or were never referred. While there is not yet a nationwide ban on conversion therapy, legislation was introduced in the last three U.S. Congress sessions to ban this harmful practice. In June 2020, the Supreme Court decision in Bostock vs. Clayton County stated that employers could not fire an employee solely because of that person’s sexual orientation and/or gender identity.
However, 19 separate bills were introduced in 2020 alone in states across the United States that would prohibit gender-affirming care for adolescents under age 18.1 Many of these bills also would make the provision of gender-affirming medical care codified as felony child abuse, with loss of licensure, fines and/or jail time a possibility for physicians who prescribe hormones or puberty blockers for gender-affirming care to minors. Fortunately, these bills either died in committee or never had a hearing. However, legislation has been prefiled in several states for their 2021 session to again attempt to prohibit minors from obtaining gender-affirming medical care and/or criminalizing the provision of this care by physicians. Other bills were filed or have been prefiled again to allow various medical and mental health providers to refuse to treat LGBTQ patients because of their personal religious beliefs and/or forcing these same providers to tell a parent if a minor reveals to that provider that they are LGBTQ.
Even if this legislation does not pass or get a hearing, the fact that the bills were introduced can have a profound impact on LGBTQ patients and their families. After a bill was introduced in Texas in their 2017 legislative session that would require trans and gender-diverse (TGD) people to use the bathroom based on their sex assigned at birth, the Trevor Project reported that it had an increase of 34% in crisis calls from trans youth who were in distress.2 This was similar, but slightly less, than was reported by the Trevor Project in September 2015 when in the run-up to a vote on Houston’s Equal Rights Ordinance, advertising was run equating trans women as predators who could be lying in wait in bathrooms. On the converse, when LGBTQ youth feel supported in the media, courts, and legislatures, this can have a positive impact on their mental health. A 2017 study found that, in states who enacted same-sex marriage laws prior to the 2015 Supreme Court decision in Obergefell, compared with those who did not, there was a 7% relative reduction in the proportion of high school students who attempted suicide.3
The American Academy of Pediatrics published its policy statement in September 2018 outlining suggestions for pediatricians to provide support to TGD youth.4 In this position statement, recommendation No. 7 states “that pediatricians have a role in advocating for policies and laws that protect youth who identify as TGD from discrimination and violence.” Therefore, it is incumbent upon us to use our voices to support our LGBTQ youth. In 2020, several pediatricians from the South Dakota chapter of the AAP provided testimony – and organized public rallies – against legislation in that state which would have made gender-affirming care to minors under age 16 punishable by a fine and/or up to 10 years in prison.5

So what can you do? First, get to know your local and state legislators. While it was difficult to meet them in person for much of 2020, you can always call their district and/or Capitol offices, email them, or fill out their constituent contact form typically found on their website. Let them know that you oppose bills which introduce discrimination against your LGBTQ patients or threaten to criminalize the care that you provide to these patients.
Second, work with your state medical association or state AAP chapter to encourage them to oppose these harmful laws and support laws that improve the lives of LGBTQ patients. Third, you can write op-eds to your local newspaper, expressing your support for your patients and outlining the detrimental effects that anti-LGBTQ laws have on your patients. Lastly, you can be active on Twitter, Facebook, or other social media platforms sharing stories of how harmful or helpful certain pieces of legislation can be for your patients.
Dr. Cooper is assistant professor of pediatrics at the University of Texas, Dallas, and an adolescent medicine specialist at Children’s Medical Center Dallas. He has no relevant financial disclosures. Email Dr. Cooper at pdnews@mdedge.com.
References
1. “Leglislation affecting LGBT rights across country.” www.aclu.org.
2. “Bathroom Bills Fuel Spike In Calls From Trans Youth To Suicide Hotline.” www.outsmartmagazine.com. 2017 Aug.
3. JAMA Pediatr. 2017 Apr 1. doi: 10.1001/jamapediatrics.2016.4529.
4. Pediatrics. 2018 Oct. doi: 10.1542/peds.2018-2162.
5. Wyckoff AS. “State bills seek to place limits on transgender care, ‘punish’ physicians.” AAP News. 2020 Feb 18.
In January in many states, the start of a new year also means the start of a new legislative session. For LGBTQ youth and their families, these sessions can create a significant amount of anxiety, as legislators in several states introduce legislation to curtail the rights of this population. In some cases, legislators have attempted to criminalize the provision of gender-affirming medical care to the trans and gender-diverse adolescents that many of us provide care to on a daily basis. As pediatricians,
2020 started on a positive note for LGBTQ children and adolescents, with Virginia becoming the 20th state to ban conversion therapy for minors. Legislation was introduced in several other states to prohibit this practice, including Kentucky, Missouri, and Ohio, and but they ultimately died in committee or were never referred. While there is not yet a nationwide ban on conversion therapy, legislation was introduced in the last three U.S. Congress sessions to ban this harmful practice. In June 2020, the Supreme Court decision in Bostock vs. Clayton County stated that employers could not fire an employee solely because of that person’s sexual orientation and/or gender identity.
However, 19 separate bills were introduced in 2020 alone in states across the United States that would prohibit gender-affirming care for adolescents under age 18.1 Many of these bills also would make the provision of gender-affirming medical care codified as felony child abuse, with loss of licensure, fines and/or jail time a possibility for physicians who prescribe hormones or puberty blockers for gender-affirming care to minors. Fortunately, these bills either died in committee or never had a hearing. However, legislation has been prefiled in several states for their 2021 session to again attempt to prohibit minors from obtaining gender-affirming medical care and/or criminalizing the provision of this care by physicians. Other bills were filed or have been prefiled again to allow various medical and mental health providers to refuse to treat LGBTQ patients because of their personal religious beliefs and/or forcing these same providers to tell a parent if a minor reveals to that provider that they are LGBTQ.
Even if this legislation does not pass or get a hearing, the fact that the bills were introduced can have a profound impact on LGBTQ patients and their families. After a bill was introduced in Texas in their 2017 legislative session that would require trans and gender-diverse (TGD) people to use the bathroom based on their sex assigned at birth, the Trevor Project reported that it had an increase of 34% in crisis calls from trans youth who were in distress.2 This was similar, but slightly less, than was reported by the Trevor Project in September 2015 when in the run-up to a vote on Houston’s Equal Rights Ordinance, advertising was run equating trans women as predators who could be lying in wait in bathrooms. On the converse, when LGBTQ youth feel supported in the media, courts, and legislatures, this can have a positive impact on their mental health. A 2017 study found that, in states who enacted same-sex marriage laws prior to the 2015 Supreme Court decision in Obergefell, compared with those who did not, there was a 7% relative reduction in the proportion of high school students who attempted suicide.3
The American Academy of Pediatrics published its policy statement in September 2018 outlining suggestions for pediatricians to provide support to TGD youth.4 In this position statement, recommendation No. 7 states “that pediatricians have a role in advocating for policies and laws that protect youth who identify as TGD from discrimination and violence.” Therefore, it is incumbent upon us to use our voices to support our LGBTQ youth. In 2020, several pediatricians from the South Dakota chapter of the AAP provided testimony – and organized public rallies – against legislation in that state which would have made gender-affirming care to minors under age 16 punishable by a fine and/or up to 10 years in prison.5
So what can you do? First, get to know your local and state legislators. While it was difficult to meet them in person for much of 2020, you can always call their district and/or Capitol offices, email them, or fill out their constituent contact form typically found on their website. Let them know that you oppose bills which introduce discrimination against your LGBTQ patients or threaten to criminalize the care that you provide to these patients.
Second, work with your state medical association or state AAP chapter to encourage them to oppose these harmful laws and support laws that improve the lives of LGBTQ patients. Third, you can write op-eds to your local newspaper, expressing your support for your patients and outlining the detrimental effects that anti-LGBTQ laws have on your patients. Lastly, you can be active on Twitter, Facebook, or other social media platforms sharing stories of how harmful or helpful certain pieces of legislation can be for your patients.
Dr. Cooper is assistant professor of pediatrics at the University of Texas, Dallas, and an adolescent medicine specialist at Children’s Medical Center Dallas. He has no relevant financial disclosures. Email Dr. Cooper at pdnews@mdedge.com.
References
1. “Leglislation affecting LGBT rights across country.” www.aclu.org.
2. “Bathroom Bills Fuel Spike In Calls From Trans Youth To Suicide Hotline.” www.outsmartmagazine.com. 2017 Aug.
3. JAMA Pediatr. 2017 Apr 1. doi: 10.1001/jamapediatrics.2016.4529.
4. Pediatrics. 2018 Oct. doi: 10.1542/peds.2018-2162.
5. Wyckoff AS. “State bills seek to place limits on transgender care, ‘punish’ physicians.” AAP News. 2020 Feb 18.
In January in many states, the start of a new year also means the start of a new legislative session. For LGBTQ youth and their families, these sessions can create a significant amount of anxiety, as legislators in several states introduce legislation to curtail the rights of this population. In some cases, legislators have attempted to criminalize the provision of gender-affirming medical care to the trans and gender-diverse adolescents that many of us provide care to on a daily basis. As pediatricians,
2020 started on a positive note for LGBTQ children and adolescents, with Virginia becoming the 20th state to ban conversion therapy for minors. Legislation was introduced in several other states to prohibit this practice, including Kentucky, Missouri, and Ohio, and but they ultimately died in committee or were never referred. While there is not yet a nationwide ban on conversion therapy, legislation was introduced in the last three U.S. Congress sessions to ban this harmful practice. In June 2020, the Supreme Court decision in Bostock vs. Clayton County stated that employers could not fire an employee solely because of that person’s sexual orientation and/or gender identity.
However, 19 separate bills were introduced in 2020 alone in states across the United States that would prohibit gender-affirming care for adolescents under age 18.1 Many of these bills also would make the provision of gender-affirming medical care codified as felony child abuse, with loss of licensure, fines and/or jail time a possibility for physicians who prescribe hormones or puberty blockers for gender-affirming care to minors. Fortunately, these bills either died in committee or never had a hearing. However, legislation has been prefiled in several states for their 2021 session to again attempt to prohibit minors from obtaining gender-affirming medical care and/or criminalizing the provision of this care by physicians. Other bills were filed or have been prefiled again to allow various medical and mental health providers to refuse to treat LGBTQ patients because of their personal religious beliefs and/or forcing these same providers to tell a parent if a minor reveals to that provider that they are LGBTQ.
Even if this legislation does not pass or get a hearing, the fact that the bills were introduced can have a profound impact on LGBTQ patients and their families. After a bill was introduced in Texas in their 2017 legislative session that would require trans and gender-diverse (TGD) people to use the bathroom based on their sex assigned at birth, the Trevor Project reported that it had an increase of 34% in crisis calls from trans youth who were in distress.2 This was similar, but slightly less, than was reported by the Trevor Project in September 2015 when in the run-up to a vote on Houston’s Equal Rights Ordinance, advertising was run equating trans women as predators who could be lying in wait in bathrooms. On the converse, when LGBTQ youth feel supported in the media, courts, and legislatures, this can have a positive impact on their mental health. A 2017 study found that, in states who enacted same-sex marriage laws prior to the 2015 Supreme Court decision in Obergefell, compared with those who did not, there was a 7% relative reduction in the proportion of high school students who attempted suicide.3
The American Academy of Pediatrics published its policy statement in September 2018 outlining suggestions for pediatricians to provide support to TGD youth.4 In this position statement, recommendation No. 7 states “that pediatricians have a role in advocating for policies and laws that protect youth who identify as TGD from discrimination and violence.” Therefore, it is incumbent upon us to use our voices to support our LGBTQ youth. In 2020, several pediatricians from the South Dakota chapter of the AAP provided testimony – and organized public rallies – against legislation in that state which would have made gender-affirming care to minors under age 16 punishable by a fine and/or up to 10 years in prison.5
So what can you do? First, get to know your local and state legislators. While it was difficult to meet them in person for much of 2020, you can always call their district and/or Capitol offices, email them, or fill out their constituent contact form typically found on their website. Let them know that you oppose bills which introduce discrimination against your LGBTQ patients or threaten to criminalize the care that you provide to these patients.
Second, work with your state medical association or state AAP chapter to encourage them to oppose these harmful laws and support laws that improve the lives of LGBTQ patients. Third, you can write op-eds to your local newspaper, expressing your support for your patients and outlining the detrimental effects that anti-LGBTQ laws have on your patients. Lastly, you can be active on Twitter, Facebook, or other social media platforms sharing stories of how harmful or helpful certain pieces of legislation can be for your patients.
Dr. Cooper is assistant professor of pediatrics at the University of Texas, Dallas, and an adolescent medicine specialist at Children’s Medical Center Dallas. He has no relevant financial disclosures. Email Dr. Cooper at pdnews@mdedge.com.
References
1. “Leglislation affecting LGBT rights across country.” www.aclu.org.
2. “Bathroom Bills Fuel Spike In Calls From Trans Youth To Suicide Hotline.” www.outsmartmagazine.com. 2017 Aug.
3. JAMA Pediatr. 2017 Apr 1. doi: 10.1001/jamapediatrics.2016.4529.
4. Pediatrics. 2018 Oct. doi: 10.1542/peds.2018-2162.
5. Wyckoff AS. “State bills seek to place limits on transgender care, ‘punish’ physicians.” AAP News. 2020 Feb 18.

FDA panel overwhelmingly backs emergency authorization for Pfizer COVID vaccine
Federal advisers on Thursday told US regulators that the benefits of Pfizer's COVID vaccine outweigh its risks for people aged 16 years and older, moving this product closer to a special emergency clearance.
The US Food and Drug Administration (FDA) put Pfizer's application before its Vaccines and Related Biological Products Advisory Committee (VRBPAC), seeking expert feedback on what is likely to be the first COVID-19 vaccine cleared for use in the United States.
New York-based Pfizer is seeking an emergency use authorization (EUA) for its vaccine, known as BNT162b2, which it developed with Germany's BioNTech. The FDA asked its advisers to vote on a single question regarding this product: "Based on the totality of scientific evidence available, do the benefits of the Pfizer-BioNTech COVID-19 Vaccine outweigh its risks for use in individuals 16 years of age and older?"
The members of VRBPAC voted 17-4 in favor of the Pfizer vaccine, with one panelist abstaining. The FDA considers the recommendations of its panels, but is not bound by them. The agency is expected to quickly grant the special clearance to Pfizer's vaccine, with the company then expected to complete work needed for a more complete biologics license application (BLA).
The FDA often allows members of its advisory committees to explain the reasons for their decisions to vote for or against an application after the tallies are publicly counted.
But the FDA did not give VRBPAC members this opportunity on Thursday, leaving the public without detailed insight into their support or objections.
Before the vote, several panelists had asked if the FDA could rephrase the voting question, raising the age for the approved group to perhaps 18 years of age. During the day, panelists also had questioned whether Pfizer's studies give enough information to judge whether the vaccine works against severe cases of COVID. And there was a discussion about how Pfizer could address concerns about the potential for allergic reactions to the vaccine, given the news of two healthcare workers who experienced allergic reactions after having the vaccine but who have since recovered.
In closing the meeting, VRBPAC chairman, Arnold Monto, MD, noted that the panel will on Dec. 17 meet again to offer recommendations on Moderna Inc.'s COVID vaccine.
"I believe most of us are going to be revisiting some of these issues in about a week," he said.
The panelist who abstained was H. Cody Meissner, MD, an expert in pediatric infectious disease from Tufts University. He earlier was among the several panelists who raised questions about the limited data available about the benefit to those ages 16 and 17. Those voting against the application were Michael Kurilla, MD, PhD; Archana Chatterjee, MD, PhD; A. Oveta Fuller, PhD, and David Kim, MD, MA, according to a tally read by the FDA staff after the vote.
Meanwhile, Sheldon Toubman, JD, voted in favor of the application according to the FDA staff's tally. Toubman had been a chief critic among VRBPAC members in reviewing Pfizer's application at the meeting. He'd suggested limiting the EUA to healthcare workers and residents of nursing homes. Members of these two groups are expected to be the first in the US to get Pfizer's vaccine, for which there will be only a limited initial supply. That idea gained no traction.
Toubman also pressed for more evidence that Pfizer's vaccine will work against severe cases of COVID.
The FDA staff on December 8 released a largely positive agency review of Pfizer vaccine. The efficacy of a two-dose administration of the vaccine has been pegged at 95.0%, with eight COVID-19 cases in the vaccine group and 162 COVID-19 cases in the placebo group. The FDA staff said that the 95% credible interval for the vaccine efficacy was 90.3% to 97.6%.
In that review, the FDA staff said there may be a hint from the results observed to date that the Pfizer vaccine may help ward off severe cases of COVID-19. There were 10 study participants that had severe COVID-19 disease after the first dose: one who received the vaccine and nine who received placebo.
"The total number of severe cases is small, which limits the overall conclusions that can be drawn; however, the case split does suggest protection from severe COVID-19 disease," the FDA staff said.
At the meeting today, Doron Fink, MD, PhD, a lead FDA official on the COVID vaccine review, responded directly to Toubman's concerns. There are many examples of vaccines that protect as well if not better against severe disease as they do against mild to moderate disease, Fink said.
"Protecting against disease of any severity is actually a pretty good predictor of protection against severe disease," Fink said, adding that there's already been a "strong result" shown in terms of the efficacy of Pfizer's vaccine.
Rolling out
Canadian health regulators on December 9 announced their nation's conditional approval of Pfizer's vaccine for people ages 16 and older. In the United Kingdom, a widely publicized rollout of Pfizer's vaccine began on Dec. 8. News quickly spread about two workers in the National Health Service having allergic reactions following vaccination. Both of these workers carry adrenaline autoinjectors, suggesting they have suffered reactions in the past, the Guardian reported. These kinds of autoinjectors are well known in the United States under the brand name EpiPen.
A noted vaccine expert serving on VRBPAC, Paul Offit, MD, of Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, urged the FDA and Pfizer to investigate any connection between reaction to the vaccine and known allergies. If not fully addressed, reports of the reactions seen in initial vaccinations in the UK could prove to unnecessarily frighten people who have allergies away from getting the COVID shot, he said.
Offit suggested running tests where people with egg and peanut allergies would get the Pfizer vaccine under close medical observation "to prove that this is not going to be a problem."
"This is a practical solution because this issue is not going to die until we have better data," Offit said.
More than a dozen COVID-19 vaccines have reached advanced stages of testing, including ones developed in Russia and China, according to the World Health Organization (WHO). The two leading candidates for the US market are the Pfizer/BioNTech vaccine and a similar vaccine developed by Moderna and the National Institute of Allergy and Infectious Diseases. Johnson & Johnson and AstraZeneca are among the other companies with COVID-19 vaccines in testing.
The rapid development of COVID vaccines will create challenges in testing these products. A key issue will be how and whether to continue with placebo-controlled trials, even though such research would be helpful, FDA advisers said.
The FDA tasked Steven Goodman, MD, MHS, PhD, of Stanford University with presenting an overview of considerations for continuing a placebo-controlled trial as COVID vaccines become available. Once a COVID-19 vaccine becomes available to the public, people who have received placebo in the Pfizer trial should not be allowed to immediately receive the vaccine, Goodman said.
There isn't a strong medically-based argument against placebo-controlled research in COVID-19, as many people can take steps to reduce their risk for the infection, Goodman said.
"So as long as there are still important things to learn about the vaccine, placebo-controlled trials should not be regarded as unethical," Goodman said. " I think, however, they might be infeasible. And that is a big issue, because people may not be willing to either remain in the study or to enroll."
During the public comment session, a former FDA official spoke of a need for careful consideration of study volunteers' needs in designing trials of COVID-19 vaccines.
"Reasonable people can disagree over whether study subjects should have priority access to a product whose efficacy they helped demonstrate," said Peter Lurie, MD, president of the nonprofit Center for Science in the Public Interest. "But we ought to be able to agree on this: No subject who has put their body on the line in a vaccine study should be at a disadvantage in terms of vaccine access as a result of their participation."
Lurie argued against extended periods of blinded follow-up after authorization of a COVID-19 vaccine. Such a requirement would be "hard to justify ethically, if it is inconsistent with public health recommendations, particularly with rapidly rising case rates and the reported levels of effectiveness" of the Pfizer vaccine, said Lurie, who served as an associate commissioner at FDA from 2014 to 2017.
Lurie also noted the FDA staff's identification of what he called "disproportionate numbers of Bell's Palsy cases (4 in the vaccine groups vs. 0 in the placebo group)" as a matter that should continue to be monitored, including in the postmarketing phase. He raised no objections to the EUA.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen's Health Research Group, also spoke at the public comment session, citing no objection to an EUA for the Pfizer vaccine. Like Lurie, he urged special consideration of people who have or will receive placebo in COVID-19 vaccine trials.
The Thursday advisory committee on the Pfizer vaccine differed from those held for many other products. The discussion focused more on how to monitor and evaluate the vaccine once approved, while advisory committees sometimes include a detailed look at whether a company has proven that its product works. One of the special advisers serving temporarily on VRBPAC, Eric J. Rubin, MD, PhD, also today published an editorial in The New England Journal of Medicine, titled "SARS-CoV-2 Vaccination — An Ounce (Actually, Much Less) of Prevention."
In the editorial, Rubin and coauthor, Dan L. Longo, MD, called the Pfizer vaccine results seen so far "impressive."
"In the primary analysis, only 8 cases of Covid-19 were seen in the vaccine group, as compared with 162 in the placebo group, for an overall efficacy of 95% (with a 95% credible interval of 90.3 to 97.6%)," they write. "Although the trial does not have the statistical power to assess subgroups, efficacy appeared to be similar in low-risk and high-risk persons, including some from communities that have been disproportionately affected by disease, and in participants older than 55 years of age and those younger than 55."
Intense Scrutiny
The FDA has come under intense scrutiny this year in part because of the aggressive — and ultimately unrealistic — timelines for COVID-19 treatments promoted by the Trump administration. President Donald Trump several times suggested a COVID-19 vaccine could be approved before the November election. Many concerned physicians and scientists including Medscape Editor-in-Chief Eric Topol, MD, called on FDA staff to fight back against any bid to inappropriately speed the approval process for political reasons.
"Any shortcuts will not only jeopardize the vaccine programs but betray the public trust, which is already fragile about vaccines, and has been made more so by your lack of autonomy from the Trump administration and its overt politicization of the FDA," Topol wrote in an August open letter to FDA Commissioner Stephen Hahn, MD.
In an October interview with Topol, Hahn noted that there has been some pushback against the idea of an EUA for a COVID-19 vaccine, with some people preferring to wait for a more complete biological license application.
"When you're talking about a pandemic where people are dying, you want to expedite it as much as possible," Hahn told Topol in the interview.
On Thursday, Hahn issued a public statement about the VRBPAC meeting. Hahn said the FDA's "career staff — made up of physicians, biologists, chemists, epidemiologists, statisticians, and other professionals — have been working around the clock to thoroughly evaluate the data and information in the EUA request."
"I can assure you that no vaccine will be authorized for use in the United States until FDA career officials feel confident in allowing their own families to receive it," Hahn said.
Many clinicians offered their views on the FDA meeting during the day on Twitter.
Robert Wachter, MD, chair of the Department of Medicine at the University of California, San Francisco, who has been a vocal opponent of some of Trump's public statements on COVID-19, urged state officials to stick with the FDA's call on the Pfizer vaccine. In a tweet, he noted that officials in California and several other states have called for independent reviews of COVID-19 vaccines.
If such reviews were to delay distribution of vaccines, this would "lead to more harm than good," Wachter tweeted. "Once FDA says 'go', we should go."
This article was updated 12/10/20.
This article originally appeared on Medscape.com.
Federal advisers on Thursday told US regulators that the benefits of Pfizer's COVID vaccine outweigh its risks for people aged 16 years and older, moving this product closer to a special emergency clearance.
The US Food and Drug Administration (FDA) put Pfizer's application before its Vaccines and Related Biological Products Advisory Committee (VRBPAC), seeking expert feedback on what is likely to be the first COVID-19 vaccine cleared for use in the United States.
New York-based Pfizer is seeking an emergency use authorization (EUA) for its vaccine, known as BNT162b2, which it developed with Germany's BioNTech. The FDA asked its advisers to vote on a single question regarding this product: "Based on the totality of scientific evidence available, do the benefits of the Pfizer-BioNTech COVID-19 Vaccine outweigh its risks for use in individuals 16 years of age and older?"
The members of VRBPAC voted 17-4 in favor of the Pfizer vaccine, with one panelist abstaining. The FDA considers the recommendations of its panels, but is not bound by them. The agency is expected to quickly grant the special clearance to Pfizer's vaccine, with the company then expected to complete work needed for a more complete biologics license application (BLA).
The FDA often allows members of its advisory committees to explain the reasons for their decisions to vote for or against an application after the tallies are publicly counted.
But the FDA did not give VRBPAC members this opportunity on Thursday, leaving the public without detailed insight into their support or objections.
Before the vote, several panelists had asked if the FDA could rephrase the voting question, raising the age for the approved group to perhaps 18 years of age. During the day, panelists also had questioned whether Pfizer's studies give enough information to judge whether the vaccine works against severe cases of COVID. And there was a discussion about how Pfizer could address concerns about the potential for allergic reactions to the vaccine, given the news of two healthcare workers who experienced allergic reactions after having the vaccine but who have since recovered.
In closing the meeting, VRBPAC chairman, Arnold Monto, MD, noted that the panel will on Dec. 17 meet again to offer recommendations on Moderna Inc.'s COVID vaccine.
"I believe most of us are going to be revisiting some of these issues in about a week," he said.
The panelist who abstained was H. Cody Meissner, MD, an expert in pediatric infectious disease from Tufts University. He earlier was among the several panelists who raised questions about the limited data available about the benefit to those ages 16 and 17. Those voting against the application were Michael Kurilla, MD, PhD; Archana Chatterjee, MD, PhD; A. Oveta Fuller, PhD, and David Kim, MD, MA, according to a tally read by the FDA staff after the vote.
Meanwhile, Sheldon Toubman, JD, voted in favor of the application according to the FDA staff's tally. Toubman had been a chief critic among VRBPAC members in reviewing Pfizer's application at the meeting. He'd suggested limiting the EUA to healthcare workers and residents of nursing homes. Members of these two groups are expected to be the first in the US to get Pfizer's vaccine, for which there will be only a limited initial supply. That idea gained no traction.
Toubman also pressed for more evidence that Pfizer's vaccine will work against severe cases of COVID.
The FDA staff on December 8 released a largely positive agency review of Pfizer vaccine. The efficacy of a two-dose administration of the vaccine has been pegged at 95.0%, with eight COVID-19 cases in the vaccine group and 162 COVID-19 cases in the placebo group. The FDA staff said that the 95% credible interval for the vaccine efficacy was 90.3% to 97.6%.
In that review, the FDA staff said there may be a hint from the results observed to date that the Pfizer vaccine may help ward off severe cases of COVID-19. There were 10 study participants that had severe COVID-19 disease after the first dose: one who received the vaccine and nine who received placebo.
"The total number of severe cases is small, which limits the overall conclusions that can be drawn; however, the case split does suggest protection from severe COVID-19 disease," the FDA staff said.
At the meeting today, Doron Fink, MD, PhD, a lead FDA official on the COVID vaccine review, responded directly to Toubman's concerns. There are many examples of vaccines that protect as well if not better against severe disease as they do against mild to moderate disease, Fink said.
"Protecting against disease of any severity is actually a pretty good predictor of protection against severe disease," Fink said, adding that there's already been a "strong result" shown in terms of the efficacy of Pfizer's vaccine.
Rolling out
Canadian health regulators on December 9 announced their nation's conditional approval of Pfizer's vaccine for people ages 16 and older. In the United Kingdom, a widely publicized rollout of Pfizer's vaccine began on Dec. 8. News quickly spread about two workers in the National Health Service having allergic reactions following vaccination. Both of these workers carry adrenaline autoinjectors, suggesting they have suffered reactions in the past, the Guardian reported. These kinds of autoinjectors are well known in the United States under the brand name EpiPen.
A noted vaccine expert serving on VRBPAC, Paul Offit, MD, of Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, urged the FDA and Pfizer to investigate any connection between reaction to the vaccine and known allergies. If not fully addressed, reports of the reactions seen in initial vaccinations in the UK could prove to unnecessarily frighten people who have allergies away from getting the COVID shot, he said.
Offit suggested running tests where people with egg and peanut allergies would get the Pfizer vaccine under close medical observation "to prove that this is not going to be a problem."
"This is a practical solution because this issue is not going to die until we have better data," Offit said.
More than a dozen COVID-19 vaccines have reached advanced stages of testing, including ones developed in Russia and China, according to the World Health Organization (WHO). The two leading candidates for the US market are the Pfizer/BioNTech vaccine and a similar vaccine developed by Moderna and the National Institute of Allergy and Infectious Diseases. Johnson & Johnson and AstraZeneca are among the other companies with COVID-19 vaccines in testing.
The rapid development of COVID vaccines will create challenges in testing these products. A key issue will be how and whether to continue with placebo-controlled trials, even though such research would be helpful, FDA advisers said.
The FDA tasked Steven Goodman, MD, MHS, PhD, of Stanford University with presenting an overview of considerations for continuing a placebo-controlled trial as COVID vaccines become available. Once a COVID-19 vaccine becomes available to the public, people who have received placebo in the Pfizer trial should not be allowed to immediately receive the vaccine, Goodman said.
There isn't a strong medically-based argument against placebo-controlled research in COVID-19, as many people can take steps to reduce their risk for the infection, Goodman said.
"So as long as there are still important things to learn about the vaccine, placebo-controlled trials should not be regarded as unethical," Goodman said. " I think, however, they might be infeasible. And that is a big issue, because people may not be willing to either remain in the study or to enroll."
During the public comment session, a former FDA official spoke of a need for careful consideration of study volunteers' needs in designing trials of COVID-19 vaccines.
"Reasonable people can disagree over whether study subjects should have priority access to a product whose efficacy they helped demonstrate," said Peter Lurie, MD, president of the nonprofit Center for Science in the Public Interest. "But we ought to be able to agree on this: No subject who has put their body on the line in a vaccine study should be at a disadvantage in terms of vaccine access as a result of their participation."
Lurie argued against extended periods of blinded follow-up after authorization of a COVID-19 vaccine. Such a requirement would be "hard to justify ethically, if it is inconsistent with public health recommendations, particularly with rapidly rising case rates and the reported levels of effectiveness" of the Pfizer vaccine, said Lurie, who served as an associate commissioner at FDA from 2014 to 2017.
Lurie also noted the FDA staff's identification of what he called "disproportionate numbers of Bell's Palsy cases (4 in the vaccine groups vs. 0 in the placebo group)" as a matter that should continue to be monitored, including in the postmarketing phase. He raised no objections to the EUA.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen's Health Research Group, also spoke at the public comment session, citing no objection to an EUA for the Pfizer vaccine. Like Lurie, he urged special consideration of people who have or will receive placebo in COVID-19 vaccine trials.
The Thursday advisory committee on the Pfizer vaccine differed from those held for many other products. The discussion focused more on how to monitor and evaluate the vaccine once approved, while advisory committees sometimes include a detailed look at whether a company has proven that its product works. One of the special advisers serving temporarily on VRBPAC, Eric J. Rubin, MD, PhD, also today published an editorial in The New England Journal of Medicine, titled "SARS-CoV-2 Vaccination — An Ounce (Actually, Much Less) of Prevention."
In the editorial, Rubin and coauthor, Dan L. Longo, MD, called the Pfizer vaccine results seen so far "impressive."
"In the primary analysis, only 8 cases of Covid-19 were seen in the vaccine group, as compared with 162 in the placebo group, for an overall efficacy of 95% (with a 95% credible interval of 90.3 to 97.6%)," they write. "Although the trial does not have the statistical power to assess subgroups, efficacy appeared to be similar in low-risk and high-risk persons, including some from communities that have been disproportionately affected by disease, and in participants older than 55 years of age and those younger than 55."
Intense Scrutiny
The FDA has come under intense scrutiny this year in part because of the aggressive — and ultimately unrealistic — timelines for COVID-19 treatments promoted by the Trump administration. President Donald Trump several times suggested a COVID-19 vaccine could be approved before the November election. Many concerned physicians and scientists including Medscape Editor-in-Chief Eric Topol, MD, called on FDA staff to fight back against any bid to inappropriately speed the approval process for political reasons.
"Any shortcuts will not only jeopardize the vaccine programs but betray the public trust, which is already fragile about vaccines, and has been made more so by your lack of autonomy from the Trump administration and its overt politicization of the FDA," Topol wrote in an August open letter to FDA Commissioner Stephen Hahn, MD.
In an October interview with Topol, Hahn noted that there has been some pushback against the idea of an EUA for a COVID-19 vaccine, with some people preferring to wait for a more complete biological license application.
"When you're talking about a pandemic where people are dying, you want to expedite it as much as possible," Hahn told Topol in the interview.
On Thursday, Hahn issued a public statement about the VRBPAC meeting. Hahn said the FDA's "career staff — made up of physicians, biologists, chemists, epidemiologists, statisticians, and other professionals — have been working around the clock to thoroughly evaluate the data and information in the EUA request."
"I can assure you that no vaccine will be authorized for use in the United States until FDA career officials feel confident in allowing their own families to receive it," Hahn said.
Many clinicians offered their views on the FDA meeting during the day on Twitter.
Robert Wachter, MD, chair of the Department of Medicine at the University of California, San Francisco, who has been a vocal opponent of some of Trump's public statements on COVID-19, urged state officials to stick with the FDA's call on the Pfizer vaccine. In a tweet, he noted that officials in California and several other states have called for independent reviews of COVID-19 vaccines.
If such reviews were to delay distribution of vaccines, this would "lead to more harm than good," Wachter tweeted. "Once FDA says 'go', we should go."
This article was updated 12/10/20.
This article originally appeared on Medscape.com.
Federal advisers on Thursday told US regulators that the benefits of Pfizer's COVID vaccine outweigh its risks for people aged 16 years and older, moving this product closer to a special emergency clearance.
The US Food and Drug Administration (FDA) put Pfizer's application before its Vaccines and Related Biological Products Advisory Committee (VRBPAC), seeking expert feedback on what is likely to be the first COVID-19 vaccine cleared for use in the United States.
New York-based Pfizer is seeking an emergency use authorization (EUA) for its vaccine, known as BNT162b2, which it developed with Germany's BioNTech. The FDA asked its advisers to vote on a single question regarding this product: "Based on the totality of scientific evidence available, do the benefits of the Pfizer-BioNTech COVID-19 Vaccine outweigh its risks for use in individuals 16 years of age and older?"
The members of VRBPAC voted 17-4 in favor of the Pfizer vaccine, with one panelist abstaining. The FDA considers the recommendations of its panels, but is not bound by them. The agency is expected to quickly grant the special clearance to Pfizer's vaccine, with the company then expected to complete work needed for a more complete biologics license application (BLA).
The FDA often allows members of its advisory committees to explain the reasons for their decisions to vote for or against an application after the tallies are publicly counted.
But the FDA did not give VRBPAC members this opportunity on Thursday, leaving the public without detailed insight into their support or objections.
Before the vote, several panelists had asked if the FDA could rephrase the voting question, raising the age for the approved group to perhaps 18 years of age. During the day, panelists also had questioned whether Pfizer's studies give enough information to judge whether the vaccine works against severe cases of COVID. And there was a discussion about how Pfizer could address concerns about the potential for allergic reactions to the vaccine, given the news of two healthcare workers who experienced allergic reactions after having the vaccine but who have since recovered.
In closing the meeting, VRBPAC chairman, Arnold Monto, MD, noted that the panel will on Dec. 17 meet again to offer recommendations on Moderna Inc.'s COVID vaccine.
"I believe most of us are going to be revisiting some of these issues in about a week," he said.
The panelist who abstained was H. Cody Meissner, MD, an expert in pediatric infectious disease from Tufts University. He earlier was among the several panelists who raised questions about the limited data available about the benefit to those ages 16 and 17. Those voting against the application were Michael Kurilla, MD, PhD; Archana Chatterjee, MD, PhD; A. Oveta Fuller, PhD, and David Kim, MD, MA, according to a tally read by the FDA staff after the vote.
Meanwhile, Sheldon Toubman, JD, voted in favor of the application according to the FDA staff's tally. Toubman had been a chief critic among VRBPAC members in reviewing Pfizer's application at the meeting. He'd suggested limiting the EUA to healthcare workers and residents of nursing homes. Members of these two groups are expected to be the first in the US to get Pfizer's vaccine, for which there will be only a limited initial supply. That idea gained no traction.
Toubman also pressed for more evidence that Pfizer's vaccine will work against severe cases of COVID.
The FDA staff on December 8 released a largely positive agency review of Pfizer vaccine. The efficacy of a two-dose administration of the vaccine has been pegged at 95.0%, with eight COVID-19 cases in the vaccine group and 162 COVID-19 cases in the placebo group. The FDA staff said that the 95% credible interval for the vaccine efficacy was 90.3% to 97.6%.
In that review, the FDA staff said there may be a hint from the results observed to date that the Pfizer vaccine may help ward off severe cases of COVID-19. There were 10 study participants that had severe COVID-19 disease after the first dose: one who received the vaccine and nine who received placebo.
"The total number of severe cases is small, which limits the overall conclusions that can be drawn; however, the case split does suggest protection from severe COVID-19 disease," the FDA staff said.
At the meeting today, Doron Fink, MD, PhD, a lead FDA official on the COVID vaccine review, responded directly to Toubman's concerns. There are many examples of vaccines that protect as well if not better against severe disease as they do against mild to moderate disease, Fink said.
"Protecting against disease of any severity is actually a pretty good predictor of protection against severe disease," Fink said, adding that there's already been a "strong result" shown in terms of the efficacy of Pfizer's vaccine.
Rolling out
Canadian health regulators on December 9 announced their nation's conditional approval of Pfizer's vaccine for people ages 16 and older. In the United Kingdom, a widely publicized rollout of Pfizer's vaccine began on Dec. 8. News quickly spread about two workers in the National Health Service having allergic reactions following vaccination. Both of these workers carry adrenaline autoinjectors, suggesting they have suffered reactions in the past, the Guardian reported. These kinds of autoinjectors are well known in the United States under the brand name EpiPen.
A noted vaccine expert serving on VRBPAC, Paul Offit, MD, of Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, urged the FDA and Pfizer to investigate any connection between reaction to the vaccine and known allergies. If not fully addressed, reports of the reactions seen in initial vaccinations in the UK could prove to unnecessarily frighten people who have allergies away from getting the COVID shot, he said.
Offit suggested running tests where people with egg and peanut allergies would get the Pfizer vaccine under close medical observation "to prove that this is not going to be a problem."
"This is a practical solution because this issue is not going to die until we have better data," Offit said.
More than a dozen COVID-19 vaccines have reached advanced stages of testing, including ones developed in Russia and China, according to the World Health Organization (WHO). The two leading candidates for the US market are the Pfizer/BioNTech vaccine and a similar vaccine developed by Moderna and the National Institute of Allergy and Infectious Diseases. Johnson & Johnson and AstraZeneca are among the other companies with COVID-19 vaccines in testing.
The rapid development of COVID vaccines will create challenges in testing these products. A key issue will be how and whether to continue with placebo-controlled trials, even though such research would be helpful, FDA advisers said.
The FDA tasked Steven Goodman, MD, MHS, PhD, of Stanford University with presenting an overview of considerations for continuing a placebo-controlled trial as COVID vaccines become available. Once a COVID-19 vaccine becomes available to the public, people who have received placebo in the Pfizer trial should not be allowed to immediately receive the vaccine, Goodman said.
There isn't a strong medically-based argument against placebo-controlled research in COVID-19, as many people can take steps to reduce their risk for the infection, Goodman said.
"So as long as there are still important things to learn about the vaccine, placebo-controlled trials should not be regarded as unethical," Goodman said. " I think, however, they might be infeasible. And that is a big issue, because people may not be willing to either remain in the study or to enroll."
During the public comment session, a former FDA official spoke of a need for careful consideration of study volunteers' needs in designing trials of COVID-19 vaccines.
"Reasonable people can disagree over whether study subjects should have priority access to a product whose efficacy they helped demonstrate," said Peter Lurie, MD, president of the nonprofit Center for Science in the Public Interest. "But we ought to be able to agree on this: No subject who has put their body on the line in a vaccine study should be at a disadvantage in terms of vaccine access as a result of their participation."
Lurie argued against extended periods of blinded follow-up after authorization of a COVID-19 vaccine. Such a requirement would be "hard to justify ethically, if it is inconsistent with public health recommendations, particularly with rapidly rising case rates and the reported levels of effectiveness" of the Pfizer vaccine, said Lurie, who served as an associate commissioner at FDA from 2014 to 2017.
Lurie also noted the FDA staff's identification of what he called "disproportionate numbers of Bell's Palsy cases (4 in the vaccine groups vs. 0 in the placebo group)" as a matter that should continue to be monitored, including in the postmarketing phase. He raised no objections to the EUA.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen's Health Research Group, also spoke at the public comment session, citing no objection to an EUA for the Pfizer vaccine. Like Lurie, he urged special consideration of people who have or will receive placebo in COVID-19 vaccine trials.
The Thursday advisory committee on the Pfizer vaccine differed from those held for many other products. The discussion focused more on how to monitor and evaluate the vaccine once approved, while advisory committees sometimes include a detailed look at whether a company has proven that its product works. One of the special advisers serving temporarily on VRBPAC, Eric J. Rubin, MD, PhD, also today published an editorial in The New England Journal of Medicine, titled "SARS-CoV-2 Vaccination — An Ounce (Actually, Much Less) of Prevention."
In the editorial, Rubin and coauthor, Dan L. Longo, MD, called the Pfizer vaccine results seen so far "impressive."
"In the primary analysis, only 8 cases of Covid-19 were seen in the vaccine group, as compared with 162 in the placebo group, for an overall efficacy of 95% (with a 95% credible interval of 90.3 to 97.6%)," they write. "Although the trial does not have the statistical power to assess subgroups, efficacy appeared to be similar in low-risk and high-risk persons, including some from communities that have been disproportionately affected by disease, and in participants older than 55 years of age and those younger than 55."
Intense Scrutiny
The FDA has come under intense scrutiny this year in part because of the aggressive — and ultimately unrealistic — timelines for COVID-19 treatments promoted by the Trump administration. President Donald Trump several times suggested a COVID-19 vaccine could be approved before the November election. Many concerned physicians and scientists including Medscape Editor-in-Chief Eric Topol, MD, called on FDA staff to fight back against any bid to inappropriately speed the approval process for political reasons.
"Any shortcuts will not only jeopardize the vaccine programs but betray the public trust, which is already fragile about vaccines, and has been made more so by your lack of autonomy from the Trump administration and its overt politicization of the FDA," Topol wrote in an August open letter to FDA Commissioner Stephen Hahn, MD.
In an October interview with Topol, Hahn noted that there has been some pushback against the idea of an EUA for a COVID-19 vaccine, with some people preferring to wait for a more complete biological license application.
"When you're talking about a pandemic where people are dying, you want to expedite it as much as possible," Hahn told Topol in the interview.
On Thursday, Hahn issued a public statement about the VRBPAC meeting. Hahn said the FDA's "career staff — made up of physicians, biologists, chemists, epidemiologists, statisticians, and other professionals — have been working around the clock to thoroughly evaluate the data and information in the EUA request."
"I can assure you that no vaccine will be authorized for use in the United States until FDA career officials feel confident in allowing their own families to receive it," Hahn said.
Many clinicians offered their views on the FDA meeting during the day on Twitter.
Robert Wachter, MD, chair of the Department of Medicine at the University of California, San Francisco, who has been a vocal opponent of some of Trump's public statements on COVID-19, urged state officials to stick with the FDA's call on the Pfizer vaccine. In a tweet, he noted that officials in California and several other states have called for independent reviews of COVID-19 vaccines.
If such reviews were to delay distribution of vaccines, this would "lead to more harm than good," Wachter tweeted. "Once FDA says 'go', we should go."
This article was updated 12/10/20.
This article originally appeared on Medscape.com.

Pediatric regimens better for adolescents/young adults with aggressive B-cell NHL
Adolescents and young adults with aggressive mature B-cell non-Hodgkin lymphomas appear to have better outcomes when they’re treated under pediatric protocols rather than adult regimens, Canadian investigators say.
Results of a study of patients from the ages of 15 to 21 years with either diffuse large B-cell lymphoma (DLBCL) or Burkitt’s lymphoma treated at regional or community cancer centers in the province of Ontario indicated that adolescents and young adult (AYA) patients treated at adult centers had a more than fourfold risk for disease relapse or progression, compared with their counterparts who were treated at pediatric centers, reported Sumit Gupta, MD, PhD, from the Hospital for Sick Children in Toronto and colleagues.
“Our data suggest that pediatric approaches are associated with improved event-free survival and overall survival, primarily due to a decrease in the risk of relapse or progression, while still using lower cumulative doses of chemotherapy,” he said in an oral abstract presented at the American Society of Hematology annual meeting, held virtually.
The findings echo those seen in the treatment of patients with acute lymphoblastic leukemia (ALL). As previously reported, a study from Nordic and Baltic countries showed that young adults with ALL who were treated with a pediatric regimen had a 4-year event-free survival rate of 73%, compared with 42% for historical controls.
Similarly, a prospective U.S. study reported in 2014 showed that AYA with ALL treated with a pediatric regimen had better overall and event-free survival rates, compared with historical controls.
As with ALL, pediatric and adult regimens for treatment of patients with aggressive mature B-cell NHL differ substantially, with pediatric patients receiving more intensive short-term therapy with lower cumulative doses.
In addition, while pediatric regimens for DLBCL and Burkitt’s lymphoma are identical, adult regimens differ substantially between the two histologies, Dr. Gupta pointed out.
Adult regimens for DLBCL most often incorporate CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) or CHOP plus rituximab (R-CHOP), whereas Burkitt’s lymphoma in adults is generally treated with more aggressive multidrug regimens, in combination with rituximab.
Rituximab was incorporated into adults’ regimens far earlier than in pediatric regimens, with Food and Drug Administration approval of rituximab in frontline therapy of adults with DLBCL in 2006, “whereas the first pediatric large-scale randomized controlled trial of rituximab in pediatric mature B-cell lymphoma was only published earlier this year,” he noted.
Population-based study
To see how treatment patterns for AYA patients with aggressive mature B-cell non-Hodgkin lymphomas differ between pediatric and adult centers, Dr. Gupta and colleagues conducted a population-based study of all AYA in Ontario diagnosed with Burkitt’s or DLBCL from the ages of 15 to 21 years from 1992 through 2012.
AYA from the ages of 15 to 18 years who were treated at pediatric centers were identified through the Provincial Pediatric Oncology Registry, which includes data on demographics, disease treatment, and outcomes from each of Ontario’s five childhood cancer treatments centers.
Adolescents and young adults from 15 to 21 years who were treated at adult centers with adult regimens were identified through the Ontario Cancer Registry using chart abstraction by trained personnel at all treatment centers, with all data validated by clinician reviewers.
A total of 176 patients were identified, 129 with DLBCL and 47 with Burkitt’s lymphoma. In all, 62 of the 176 patients (35.2%) were treated in pediatric centers. Not surprisingly, multivariable analysis showed that AYA treated in adult centers were older, and more likely to have been treated earlier in the study period.
Comparing treatment patterns by locus of care, the investigators found that patients with DLBCL in pediatric centers received half of the cumulative anthracycline doses as those in adult centers (150 mg/m2 vs. 300 mg/m2; P < .001) and about 75% of cumulative alkylating agent doses (3,300 mg/m2 vs. 4,465 mg/m2; P = .009).
Patients with Burkitt’s lymphoma had identical exposures to anthracyclines in pediatric vs. adult centers (120 mg/m2), but those treated in pediatric centers had half the exposure to alkylators as those treated in adult centers (3,300 mg/m2 vs. 6,600 mg/m2; P = .03).
Among patients with DLBCL, none of those treated at pediatric centers received rituximab, compared with 32.3% of those treated at adult centers (P < .001), whereas only a handful of patients with Burkitt’s lymphoma received rituximab in both pediatric and adult centers (nonsignificant).
Among all patients. 5-year event-free survival was 82.3% for those treated in pediatric centers, compared with 66.7% for those treated in adult centers (P = .02). Respective 5-year overall survival rates were 85.5% and 71.1% (P = .03).
Looking at survival by histology, the investigators saw that 5-year event-free survival for patients with DLBCL was 83.3% when they were treated like children vs. 66.7% when they were treated like adults (P = .04). Respective 5-year overall survival rates were 88.9% and 72% (P = .04).
Both event-free survival (80.8% vs. 66.7%) and overall survival (80.8% vs. 66.7%) were numerically but not statistically higher among patients with Burkitt’s treated at pediatric vs. adult centers.
An analysis adjusting for disease histology, stage, and time period of diagnosis showed that treatment at an adult center was associated with higher risk for death, with a hazard ratio of 2.4 (P = .03).
Additionally, an analysis adjusted for age, disease stage, and histology showed that patients treated in adult centers had a significantly increased risk of relapse or progression, compared with a HR of 4.4 (95% confidence interval; P = .008).
There were no significant differences in the risk of treatment-related mortality between the center types, however.
“It is important to note, however, that pediatric approaches to mature B-cell NHL [non-Hodgkin lymphoma] are associated with increased inpatient needs as compared to adult approaches, and with greater supportive care requirements. Thus the safety of such approaches in adults centers need to be established,” Dr. Gupta said.
Lower doses, better outcomes
In the question and answer session following the presentation, Jennifer Teichman, MD, MSc, a fellow in hematology at the University of Toronto who was not involved in the study asked why patients treated at adult centers would have higher relapse rates despite receiving higher doses of chemotherapy, noting that the poorer outcomes in those patients were not attributable to treatment-related mortality.
“I think one of the distinctions is that higher cumulative doses versus higher intensity of treatment over a shorter period of time are two different things, perhaps, and so giving lower cumulative doses but over a short period of time, and so giving higher intensity within that short period of time, may be what explains the higher success rate in pediatric trials,” Dr. Gupta said.
R. Michael Crump, MD, from the Princess Margaret Cancer Center, also in Toronto, asked whether the study results could have been influenced by differences between the pediatric center and adult center datasets in regard to pathology review, staging information, and International Prognostic Index.
Dr. Gupta acknowledged that, while the pediatric data were captured prospectively at each center by pediatric cancer registry staff and adult data were extracted retrospectively by trained chart reviewers, “the information that we were collecting was relatively basic – basic stage, basic histology, and that is a limitation.”
He also noted that clinicians reviewed the submitted retrospective data for completeness and had the ability to request chart extractors to return to a particular record for additional information or to correct potential errors.
The study was supported by the Canadian Institutes of Health Research, the C17 Council on Children’s Cancer & Blood Disorders, and the Pediatric Oncology Group of Ontario. Dr. Gupta, Dr. Teichman, and Dr. Crump all reported no relevant conflicts of interest.
SOURCE: Gupta S et al. ASH 2020, Abstract 708.
Adolescents and young adults with aggressive mature B-cell non-Hodgkin lymphomas appear to have better outcomes when they’re treated under pediatric protocols rather than adult regimens, Canadian investigators say.
Results of a study of patients from the ages of 15 to 21 years with either diffuse large B-cell lymphoma (DLBCL) or Burkitt’s lymphoma treated at regional or community cancer centers in the province of Ontario indicated that adolescents and young adult (AYA) patients treated at adult centers had a more than fourfold risk for disease relapse or progression, compared with their counterparts who were treated at pediatric centers, reported Sumit Gupta, MD, PhD, from the Hospital for Sick Children in Toronto and colleagues.
“Our data suggest that pediatric approaches are associated with improved event-free survival and overall survival, primarily due to a decrease in the risk of relapse or progression, while still using lower cumulative doses of chemotherapy,” he said in an oral abstract presented at the American Society of Hematology annual meeting, held virtually.
The findings echo those seen in the treatment of patients with acute lymphoblastic leukemia (ALL). As previously reported, a study from Nordic and Baltic countries showed that young adults with ALL who were treated with a pediatric regimen had a 4-year event-free survival rate of 73%, compared with 42% for historical controls.
Similarly, a prospective U.S. study reported in 2014 showed that AYA with ALL treated with a pediatric regimen had better overall and event-free survival rates, compared with historical controls.
As with ALL, pediatric and adult regimens for treatment of patients with aggressive mature B-cell NHL differ substantially, with pediatric patients receiving more intensive short-term therapy with lower cumulative doses.
In addition, while pediatric regimens for DLBCL and Burkitt’s lymphoma are identical, adult regimens differ substantially between the two histologies, Dr. Gupta pointed out.
Adult regimens for DLBCL most often incorporate CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) or CHOP plus rituximab (R-CHOP), whereas Burkitt’s lymphoma in adults is generally treated with more aggressive multidrug regimens, in combination with rituximab.
Rituximab was incorporated into adults’ regimens far earlier than in pediatric regimens, with Food and Drug Administration approval of rituximab in frontline therapy of adults with DLBCL in 2006, “whereas the first pediatric large-scale randomized controlled trial of rituximab in pediatric mature B-cell lymphoma was only published earlier this year,” he noted.
Population-based study
To see how treatment patterns for AYA patients with aggressive mature B-cell non-Hodgkin lymphomas differ between pediatric and adult centers, Dr. Gupta and colleagues conducted a population-based study of all AYA in Ontario diagnosed with Burkitt’s or DLBCL from the ages of 15 to 21 years from 1992 through 2012.
AYA from the ages of 15 to 18 years who were treated at pediatric centers were identified through the Provincial Pediatric Oncology Registry, which includes data on demographics, disease treatment, and outcomes from each of Ontario’s five childhood cancer treatments centers.
Adolescents and young adults from 15 to 21 years who were treated at adult centers with adult regimens were identified through the Ontario Cancer Registry using chart abstraction by trained personnel at all treatment centers, with all data validated by clinician reviewers.
A total of 176 patients were identified, 129 with DLBCL and 47 with Burkitt’s lymphoma. In all, 62 of the 176 patients (35.2%) were treated in pediatric centers. Not surprisingly, multivariable analysis showed that AYA treated in adult centers were older, and more likely to have been treated earlier in the study period.
Comparing treatment patterns by locus of care, the investigators found that patients with DLBCL in pediatric centers received half of the cumulative anthracycline doses as those in adult centers (150 mg/m2 vs. 300 mg/m2; P < .001) and about 75% of cumulative alkylating agent doses (3,300 mg/m2 vs. 4,465 mg/m2; P = .009).
Patients with Burkitt’s lymphoma had identical exposures to anthracyclines in pediatric vs. adult centers (120 mg/m2), but those treated in pediatric centers had half the exposure to alkylators as those treated in adult centers (3,300 mg/m2 vs. 6,600 mg/m2; P = .03).
Among patients with DLBCL, none of those treated at pediatric centers received rituximab, compared with 32.3% of those treated at adult centers (P < .001), whereas only a handful of patients with Burkitt’s lymphoma received rituximab in both pediatric and adult centers (nonsignificant).
Among all patients. 5-year event-free survival was 82.3% for those treated in pediatric centers, compared with 66.7% for those treated in adult centers (P = .02). Respective 5-year overall survival rates were 85.5% and 71.1% (P = .03).
Looking at survival by histology, the investigators saw that 5-year event-free survival for patients with DLBCL was 83.3% when they were treated like children vs. 66.7% when they were treated like adults (P = .04). Respective 5-year overall survival rates were 88.9% and 72% (P = .04).
Both event-free survival (80.8% vs. 66.7%) and overall survival (80.8% vs. 66.7%) were numerically but not statistically higher among patients with Burkitt’s treated at pediatric vs. adult centers.
An analysis adjusting for disease histology, stage, and time period of diagnosis showed that treatment at an adult center was associated with higher risk for death, with a hazard ratio of 2.4 (P = .03).
Additionally, an analysis adjusted for age, disease stage, and histology showed that patients treated in adult centers had a significantly increased risk of relapse or progression, compared with a HR of 4.4 (95% confidence interval; P = .008).
There were no significant differences in the risk of treatment-related mortality between the center types, however.
“It is important to note, however, that pediatric approaches to mature B-cell NHL [non-Hodgkin lymphoma] are associated with increased inpatient needs as compared to adult approaches, and with greater supportive care requirements. Thus the safety of such approaches in adults centers need to be established,” Dr. Gupta said.
Lower doses, better outcomes
In the question and answer session following the presentation, Jennifer Teichman, MD, MSc, a fellow in hematology at the University of Toronto who was not involved in the study asked why patients treated at adult centers would have higher relapse rates despite receiving higher doses of chemotherapy, noting that the poorer outcomes in those patients were not attributable to treatment-related mortality.
“I think one of the distinctions is that higher cumulative doses versus higher intensity of treatment over a shorter period of time are two different things, perhaps, and so giving lower cumulative doses but over a short period of time, and so giving higher intensity within that short period of time, may be what explains the higher success rate in pediatric trials,” Dr. Gupta said.
R. Michael Crump, MD, from the Princess Margaret Cancer Center, also in Toronto, asked whether the study results could have been influenced by differences between the pediatric center and adult center datasets in regard to pathology review, staging information, and International Prognostic Index.
Dr. Gupta acknowledged that, while the pediatric data were captured prospectively at each center by pediatric cancer registry staff and adult data were extracted retrospectively by trained chart reviewers, “the information that we were collecting was relatively basic – basic stage, basic histology, and that is a limitation.”
He also noted that clinicians reviewed the submitted retrospective data for completeness and had the ability to request chart extractors to return to a particular record for additional information or to correct potential errors.
The study was supported by the Canadian Institutes of Health Research, the C17 Council on Children’s Cancer & Blood Disorders, and the Pediatric Oncology Group of Ontario. Dr. Gupta, Dr. Teichman, and Dr. Crump all reported no relevant conflicts of interest.
SOURCE: Gupta S et al. ASH 2020, Abstract 708.
Adolescents and young adults with aggressive mature B-cell non-Hodgkin lymphomas appear to have better outcomes when they’re treated under pediatric protocols rather than adult regimens, Canadian investigators say.
Results of a study of patients from the ages of 15 to 21 years with either diffuse large B-cell lymphoma (DLBCL) or Burkitt’s lymphoma treated at regional or community cancer centers in the province of Ontario indicated that adolescents and young adult (AYA) patients treated at adult centers had a more than fourfold risk for disease relapse or progression, compared with their counterparts who were treated at pediatric centers, reported Sumit Gupta, MD, PhD, from the Hospital for Sick Children in Toronto and colleagues.
“Our data suggest that pediatric approaches are associated with improved event-free survival and overall survival, primarily due to a decrease in the risk of relapse or progression, while still using lower cumulative doses of chemotherapy,” he said in an oral abstract presented at the American Society of Hematology annual meeting, held virtually.
The findings echo those seen in the treatment of patients with acute lymphoblastic leukemia (ALL). As previously reported, a study from Nordic and Baltic countries showed that young adults with ALL who were treated with a pediatric regimen had a 4-year event-free survival rate of 73%, compared with 42% for historical controls.
Similarly, a prospective U.S. study reported in 2014 showed that AYA with ALL treated with a pediatric regimen had better overall and event-free survival rates, compared with historical controls.
As with ALL, pediatric and adult regimens for treatment of patients with aggressive mature B-cell NHL differ substantially, with pediatric patients receiving more intensive short-term therapy with lower cumulative doses.
In addition, while pediatric regimens for DLBCL and Burkitt’s lymphoma are identical, adult regimens differ substantially between the two histologies, Dr. Gupta pointed out.
Adult regimens for DLBCL most often incorporate CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) or CHOP plus rituximab (R-CHOP), whereas Burkitt’s lymphoma in adults is generally treated with more aggressive multidrug regimens, in combination with rituximab.
Rituximab was incorporated into adults’ regimens far earlier than in pediatric regimens, with Food and Drug Administration approval of rituximab in frontline therapy of adults with DLBCL in 2006, “whereas the first pediatric large-scale randomized controlled trial of rituximab in pediatric mature B-cell lymphoma was only published earlier this year,” he noted.
Population-based study
To see how treatment patterns for AYA patients with aggressive mature B-cell non-Hodgkin lymphomas differ between pediatric and adult centers, Dr. Gupta and colleagues conducted a population-based study of all AYA in Ontario diagnosed with Burkitt’s or DLBCL from the ages of 15 to 21 years from 1992 through 2012.
AYA from the ages of 15 to 18 years who were treated at pediatric centers were identified through the Provincial Pediatric Oncology Registry, which includes data on demographics, disease treatment, and outcomes from each of Ontario’s five childhood cancer treatments centers.
Adolescents and young adults from 15 to 21 years who were treated at adult centers with adult regimens were identified through the Ontario Cancer Registry using chart abstraction by trained personnel at all treatment centers, with all data validated by clinician reviewers.
A total of 176 patients were identified, 129 with DLBCL and 47 with Burkitt’s lymphoma. In all, 62 of the 176 patients (35.2%) were treated in pediatric centers. Not surprisingly, multivariable analysis showed that AYA treated in adult centers were older, and more likely to have been treated earlier in the study period.
Comparing treatment patterns by locus of care, the investigators found that patients with DLBCL in pediatric centers received half of the cumulative anthracycline doses as those in adult centers (150 mg/m2 vs. 300 mg/m2; P < .001) and about 75% of cumulative alkylating agent doses (3,300 mg/m2 vs. 4,465 mg/m2; P = .009).
Patients with Burkitt’s lymphoma had identical exposures to anthracyclines in pediatric vs. adult centers (120 mg/m2), but those treated in pediatric centers had half the exposure to alkylators as those treated in adult centers (3,300 mg/m2 vs. 6,600 mg/m2; P = .03).
Among patients with DLBCL, none of those treated at pediatric centers received rituximab, compared with 32.3% of those treated at adult centers (P < .001), whereas only a handful of patients with Burkitt’s lymphoma received rituximab in both pediatric and adult centers (nonsignificant).
Among all patients. 5-year event-free survival was 82.3% for those treated in pediatric centers, compared with 66.7% for those treated in adult centers (P = .02). Respective 5-year overall survival rates were 85.5% and 71.1% (P = .03).
Looking at survival by histology, the investigators saw that 5-year event-free survival for patients with DLBCL was 83.3% when they were treated like children vs. 66.7% when they were treated like adults (P = .04). Respective 5-year overall survival rates were 88.9% and 72% (P = .04).
Both event-free survival (80.8% vs. 66.7%) and overall survival (80.8% vs. 66.7%) were numerically but not statistically higher among patients with Burkitt’s treated at pediatric vs. adult centers.
An analysis adjusting for disease histology, stage, and time period of diagnosis showed that treatment at an adult center was associated with higher risk for death, with a hazard ratio of 2.4 (P = .03).
Additionally, an analysis adjusted for age, disease stage, and histology showed that patients treated in adult centers had a significantly increased risk of relapse or progression, compared with a HR of 4.4 (95% confidence interval; P = .008).
There were no significant differences in the risk of treatment-related mortality between the center types, however.
“It is important to note, however, that pediatric approaches to mature B-cell NHL [non-Hodgkin lymphoma] are associated with increased inpatient needs as compared to adult approaches, and with greater supportive care requirements. Thus the safety of such approaches in adults centers need to be established,” Dr. Gupta said.
Lower doses, better outcomes
In the question and answer session following the presentation, Jennifer Teichman, MD, MSc, a fellow in hematology at the University of Toronto who was not involved in the study asked why patients treated at adult centers would have higher relapse rates despite receiving higher doses of chemotherapy, noting that the poorer outcomes in those patients were not attributable to treatment-related mortality.
“I think one of the distinctions is that higher cumulative doses versus higher intensity of treatment over a shorter period of time are two different things, perhaps, and so giving lower cumulative doses but over a short period of time, and so giving higher intensity within that short period of time, may be what explains the higher success rate in pediatric trials,” Dr. Gupta said.
R. Michael Crump, MD, from the Princess Margaret Cancer Center, also in Toronto, asked whether the study results could have been influenced by differences between the pediatric center and adult center datasets in regard to pathology review, staging information, and International Prognostic Index.
Dr. Gupta acknowledged that, while the pediatric data were captured prospectively at each center by pediatric cancer registry staff and adult data were extracted retrospectively by trained chart reviewers, “the information that we were collecting was relatively basic – basic stage, basic histology, and that is a limitation.”
He also noted that clinicians reviewed the submitted retrospective data for completeness and had the ability to request chart extractors to return to a particular record for additional information or to correct potential errors.
The study was supported by the Canadian Institutes of Health Research, the C17 Council on Children’s Cancer & Blood Disorders, and the Pediatric Oncology Group of Ontario. Dr. Gupta, Dr. Teichman, and Dr. Crump all reported no relevant conflicts of interest.
SOURCE: Gupta S et al. ASH 2020, Abstract 708.
FROM ASH 2020
Key clinical point: Pediatric cancer regimens may offer better outcomes for adolescents/young adults with aggressive mature B-cell lymphomas.
Major finding: The hazard ratio for relapse or progression for patients treated in adults centers was 4.4 (P = .008)
Study details: Retrospective study of 176 adolescents/young adults with diffuse large B-cell lymphoma or Burkitt’s lymphoma.
Disclosures: The study was supported the Canadian Institutes of Health Research, the C17 Council on Children’s Cancer & Blood Disorders, and the Pediatric Oncology Group of Ontario. Dr. Gupta, Dr. Teichman, and Dr. Crump all reported no relevant conflicts of interest.
Source: Gupta S. et al. ASH 2020, Abstract 708.
Pfizer can’t supply additional vaccines to U.S. until June
Pfizer won’t be able to provide more COVID-19 vaccine doses to the United States until late June or July because other countries have bought up the available supply, according to The Washington Post.
The U.S. government signed a deal with the giant pharmaceutical company earlier this year to provide 100 million doses for $1.95 billion – enough for 50 million Americans to receive the two-dose vaccine. At that time, Pfizer officials encouraged Operation Warp Speed officials to purchase an additional 100 million doses, The New York Times first reported Dec. 7, but the federal officials declined.
Since then, other countries have signed vaccine deals with Pfizer, so the U.S. may not be able to receive a second major allotment until the summer of 2021, The Washington Post reported. Without a substantial number of additional doses, the U.S. may not be able to follow its schedule of vaccinating the majority of Americans against COVID-19 by April or May.
However, Trump administration officials told the newspaper that there won’t be issues, citing other vaccine companies such as Moderna.
“I’m not concerned about our ability to buy vaccines to offer to all of the American public,” Gen. Paul Ostrowski, who oversees logistics for Operation Warp Speed, told The Washington Post.
“It’s clear that Pfizer made plans with other countries. Many have been announced. We understand those pieces,” he said.
With Pfizer’s COVID-19 vaccine on the verge of FDA approval, federal officials contacted the company last weekend to buy another 100 million doses, but the company said its current supply is already committed, the newspaper reported.
The vaccine from Pfizer and BioNTech is expected to win emergency approval within days and has been shown to be effective against COVID-19.
Pfizer added that it may be able to provide 50 million doses at the end of the second quarter and another 50 million doses during the third quarter. However, the company can’t offer anything “substantial” until next summer.
Beyond the initial 100 million doses that the U.S. has already secured, Pfizer and federal officials would need to negotiate a new, “separate and mutually acceptable agreement,” Amy Rose, a spokeswoman for Pfizer, told the newspaper.
On Dec. 8, President Donald Trump was expected to sign an executive order prioritizing vaccination for Americans first before providing doses to other countries, according to Fox News.
The order will provide guidelines to the Department of Health and Human Services, the U.S. Agency for International Development and the U.S. International Development Finance Corporation for foreign assistance with vaccines, the news outlet reported.
It’s unclear whether the executive order is related to the Pfizer issue, whether the president can prevent a private company from fulfilling contracts with other countries, and whether President-elect Joe Biden will create his own policy, according to CNBC. The order may prove to be mostly symbolic.
The FDA could issue an emergency use authorization for Pfizer’s coronavirus vaccine this week and will likely approve Moderna’s vaccine next week. The U.S. has signed a contract with Moderna for 100 million doses.
During a call with reporters on Dec. 7, a spokeswoman for the Department of Health and Human Services said, “We are confident that we will have 100 million doses of Pfizer’s vaccine as agreed to in our contract, and beyond that, we have five other vaccine candidates, including 100 million doses on the way from Moderna.”
Federal officials are counting on vaccine candidates from AstraZeneca and Johnson & Johnson to seek FDA approval in January and be ready for shipment in February.
“We could have all of them,” Moncef Slaoui, the chief science adviser for Operation Warp Speed, told The Washington Post on Dec. 7.
“And for this reason, we feel confident we could cover the needs without a specific cliff,” he said. “We have planned things in such a way as we would indeed avoid a cliff.”
This article first appeared on WebMD.com.
Pfizer won’t be able to provide more COVID-19 vaccine doses to the United States until late June or July because other countries have bought up the available supply, according to The Washington Post.
The U.S. government signed a deal with the giant pharmaceutical company earlier this year to provide 100 million doses for $1.95 billion – enough for 50 million Americans to receive the two-dose vaccine. At that time, Pfizer officials encouraged Operation Warp Speed officials to purchase an additional 100 million doses, The New York Times first reported Dec. 7, but the federal officials declined.
Since then, other countries have signed vaccine deals with Pfizer, so the U.S. may not be able to receive a second major allotment until the summer of 2021, The Washington Post reported. Without a substantial number of additional doses, the U.S. may not be able to follow its schedule of vaccinating the majority of Americans against COVID-19 by April or May.
However, Trump administration officials told the newspaper that there won’t be issues, citing other vaccine companies such as Moderna.
“I’m not concerned about our ability to buy vaccines to offer to all of the American public,” Gen. Paul Ostrowski, who oversees logistics for Operation Warp Speed, told The Washington Post.
“It’s clear that Pfizer made plans with other countries. Many have been announced. We understand those pieces,” he said.
With Pfizer’s COVID-19 vaccine on the verge of FDA approval, federal officials contacted the company last weekend to buy another 100 million doses, but the company said its current supply is already committed, the newspaper reported.
The vaccine from Pfizer and BioNTech is expected to win emergency approval within days and has been shown to be effective against COVID-19.
Pfizer added that it may be able to provide 50 million doses at the end of the second quarter and another 50 million doses during the third quarter. However, the company can’t offer anything “substantial” until next summer.
Beyond the initial 100 million doses that the U.S. has already secured, Pfizer and federal officials would need to negotiate a new, “separate and mutually acceptable agreement,” Amy Rose, a spokeswoman for Pfizer, told the newspaper.
On Dec. 8, President Donald Trump was expected to sign an executive order prioritizing vaccination for Americans first before providing doses to other countries, according to Fox News.
The order will provide guidelines to the Department of Health and Human Services, the U.S. Agency for International Development and the U.S. International Development Finance Corporation for foreign assistance with vaccines, the news outlet reported.
It’s unclear whether the executive order is related to the Pfizer issue, whether the president can prevent a private company from fulfilling contracts with other countries, and whether President-elect Joe Biden will create his own policy, according to CNBC. The order may prove to be mostly symbolic.
The FDA could issue an emergency use authorization for Pfizer’s coronavirus vaccine this week and will likely approve Moderna’s vaccine next week. The U.S. has signed a contract with Moderna for 100 million doses.
During a call with reporters on Dec. 7, a spokeswoman for the Department of Health and Human Services said, “We are confident that we will have 100 million doses of Pfizer’s vaccine as agreed to in our contract, and beyond that, we have five other vaccine candidates, including 100 million doses on the way from Moderna.”
Federal officials are counting on vaccine candidates from AstraZeneca and Johnson & Johnson to seek FDA approval in January and be ready for shipment in February.
“We could have all of them,” Moncef Slaoui, the chief science adviser for Operation Warp Speed, told The Washington Post on Dec. 7.
“And for this reason, we feel confident we could cover the needs without a specific cliff,” he said. “We have planned things in such a way as we would indeed avoid a cliff.”
This article first appeared on WebMD.com.
Pfizer won’t be able to provide more COVID-19 vaccine doses to the United States until late June or July because other countries have bought up the available supply, according to The Washington Post.
The U.S. government signed a deal with the giant pharmaceutical company earlier this year to provide 100 million doses for $1.95 billion – enough for 50 million Americans to receive the two-dose vaccine. At that time, Pfizer officials encouraged Operation Warp Speed officials to purchase an additional 100 million doses, The New York Times first reported Dec. 7, but the federal officials declined.
Since then, other countries have signed vaccine deals with Pfizer, so the U.S. may not be able to receive a second major allotment until the summer of 2021, The Washington Post reported. Without a substantial number of additional doses, the U.S. may not be able to follow its schedule of vaccinating the majority of Americans against COVID-19 by April or May.
However, Trump administration officials told the newspaper that there won’t be issues, citing other vaccine companies such as Moderna.
“I’m not concerned about our ability to buy vaccines to offer to all of the American public,” Gen. Paul Ostrowski, who oversees logistics for Operation Warp Speed, told The Washington Post.
“It’s clear that Pfizer made plans with other countries. Many have been announced. We understand those pieces,” he said.
With Pfizer’s COVID-19 vaccine on the verge of FDA approval, federal officials contacted the company last weekend to buy another 100 million doses, but the company said its current supply is already committed, the newspaper reported.
The vaccine from Pfizer and BioNTech is expected to win emergency approval within days and has been shown to be effective against COVID-19.
Pfizer added that it may be able to provide 50 million doses at the end of the second quarter and another 50 million doses during the third quarter. However, the company can’t offer anything “substantial” until next summer.
Beyond the initial 100 million doses that the U.S. has already secured, Pfizer and federal officials would need to negotiate a new, “separate and mutually acceptable agreement,” Amy Rose, a spokeswoman for Pfizer, told the newspaper.
On Dec. 8, President Donald Trump was expected to sign an executive order prioritizing vaccination for Americans first before providing doses to other countries, according to Fox News.
The order will provide guidelines to the Department of Health and Human Services, the U.S. Agency for International Development and the U.S. International Development Finance Corporation for foreign assistance with vaccines, the news outlet reported.
It’s unclear whether the executive order is related to the Pfizer issue, whether the president can prevent a private company from fulfilling contracts with other countries, and whether President-elect Joe Biden will create his own policy, according to CNBC. The order may prove to be mostly symbolic.
The FDA could issue an emergency use authorization for Pfizer’s coronavirus vaccine this week and will likely approve Moderna’s vaccine next week. The U.S. has signed a contract with Moderna for 100 million doses.
During a call with reporters on Dec. 7, a spokeswoman for the Department of Health and Human Services said, “We are confident that we will have 100 million doses of Pfizer’s vaccine as agreed to in our contract, and beyond that, we have five other vaccine candidates, including 100 million doses on the way from Moderna.”
Federal officials are counting on vaccine candidates from AstraZeneca and Johnson & Johnson to seek FDA approval in January and be ready for shipment in February.
“We could have all of them,” Moncef Slaoui, the chief science adviser for Operation Warp Speed, told The Washington Post on Dec. 7.
“And for this reason, we feel confident we could cover the needs without a specific cliff,” he said. “We have planned things in such a way as we would indeed avoid a cliff.”
This article first appeared on WebMD.com.
Can a health care worker refuse the COVID-19 vaccine?
As hospitals across the country develop their plans to vaccinate their health care employees against COVID-19, a key question has come to the fore: What if an employee – whether nurse, physician, or other health care worker – refuses to receive the vaccine? Can hospitals require their employees to be vaccinated against COVID-19? And what consequences could an employee face for refusing the vaccine?
My answer needs to be based, in part, on the law related to previous vaccines – influenza, for example – because at the time of this writing (early December 2020), no vaccine for COVID-19 has been approved, although approval of at least one vaccine is expected within a week. So there have been no offers of vaccine and refusals yet, nor are there any cases to date involving an employee who refused a COVID-19 vaccine. As of December 2020, there are no state or federal laws that either require an employee to be vaccinated against COVID-19 or that protect an employee who refuses vaccination against COVID-19. It will take a while after the vaccine is approved and distributed before refusals, reactions, policies, cases, and laws begin to emerge.
If we look at the law related to health care workers refusing to be vaccinated against the closest relative to COVID-19 – influenza – then the answer would be yes, employers can require employees to be vaccinated.
An employer can fire an employee who refuses influenza vaccination. If an employee who refused and was fired sues the employer for wrongful termination, the employee has more or less chance of success depending on the reason for refusal. Some courts and the Equal Employment Opportunity Commission have held that a refusal on religious grounds is protected by the U.S. Constitution, as in this recent case. The Constitution protects freedom to practice one’s religion. Specific religions may have a range of tenets that support refusal to be vaccinated.
A refusal on medical grounds has been successful if the medical grounds fall under the protections of the Americans with Disabilities Act but may fail when the medical grounds for the claim are not covered by the ADA.
Refusal for secular, nonmedical reasons, such as a health care worker’s policy of treating their body as their temple, has not gone over well with employers or courts. However, in at least one case, a nurse who refused vaccination on secular, nonmedical grounds won her case against her employer, on appeal. The appeals court found that the hospital violated her First Amendment rights.
Employees who refuse vaccination for religious or medical reasons still will need to take measures to protect patients and other employees from infection. An employer such as a hospital can, rather than fire the employee, offer the employee an accommodation, such as requiring that the employee wear a mask or quarantine. There are no cases that have upheld an employee’s right to refuse to wear a mask or quarantine.
The situation with the COVID-19 vaccine is different from the situation surrounding influenza vaccines. There are plenty of data on effectiveness and side effects of influenza vaccines, but there is very little evidence of short- or long-term effects of the COVID-19 vaccines currently being tested and/or considered for approval. One could argue that the process of vaccine development is the same for all virus vaccines. However, public confidence in the vaccine vetting process is not what it once was. It has been widely publicized that the COVID-19 vaccine trials have been rushed. As of December 2020, only 60% of the general population say they would take the vaccine, although researchers say confidence is increasing.
The Centers for Disease Control and Prevention has designated health care workers as first in line to get the vaccine, but some health care workers may not want to be the first to try it. A CDC survey found that 63% of health care workers polled in recent months said they would get a COVID-19 vaccine.
Unions have entered the conversation. A coalition of unions that represent health care workers said, “we need a transparent, evidence-based federal vaccine strategy based on principles of equity, safety, and priority, as well as robust efforts to address a high degree of skepticism about safety of an authorized vaccine.” The organization declined to promote a vaccine until more is known.
As of publication date, the EEOC guidance for employers responding to COVID-19 does not address vaccines.
The CDC’s Interim Guidance for Businesses and Employers Responding to Coronavirus Disease 2019, May 2020, updated Dec. 4, 2020, does not address vaccines. The CDC’s page on COVID-19 vaccination for health care workers does not address a health care worker’s refusal. The site does assure health care workers that the vaccine development process is sound: “The current vaccine safety system is strong and robust, with the capacity to effectively monitor COVID-19 vaccine safety. Existing data systems have validated analytic methods that can rapidly detect statistical signals for possible vaccine safety problems. These systems are being scaled up to fully meet the needs of the nation. Additional systems and data sources are also being developed to further enhance safety monitoring capabilities. CDC is committed to ensuring that COVID-19 vaccines are safe.”
In the coming months, government officials and vaccine manufacturers will be working to reassure the public of the safety of the vaccine and the rigor of the vaccine development process. In November 2020, National Institute of Allergy and Infectious Diseases Director Anthony Fauci, MD, told Kaiser Health News: “The company looks at the data. I look at the data. Then the company puts the data to the FDA. The FDA will make the decision to do an emergency-use authorization or a license application approval. And they have career scientists who are really independent. They’re not beholden to anybody. Then there’s another independent group, the Vaccines and Related Biological Products Advisory Committee. The FDA commissioner has vowed publicly that he will go according to the opinion of the career scientists and the advisory board.” President-elect Joe Biden said he would get a vaccine when Dr. Fauci thinks it is safe.
An employee who, after researching the vaccine and the process, still wants to refuse when offered the vaccine is not likely to be fired for that reason right away, as long as the employee takes other precautions, such as wearing a mask. If the employer does fire the employee and the employee sues the employer, it is impossible to predict how a court would decide the case.
Related legal questions may arise in the coming months. For example:
- Is an employer exempt from paying workers’ compensation to an employee who refuses to be vaccinated and then contracts the virus while on the job?
- Can a prospective employer require COVID-19 vaccination as a precondition of employment?
- Is it within a patient’s rights to receive an answer to the question: Has my health care worker been vaccinated against COVID-19?
- If a hospital allows employees to refuse vaccination and keep working, and an outbreak occurs, and it is suggested through contact tracing that unvaccinated workers infected patients, will a court hold the hospital liable for patients’ damages?
Answers to these questions are yet to be determined.
Carolyn Buppert (www.buppert.com) is an attorney and former nurse practitioner who focuses on the legal issues affecting nurse practitioners.
A version of this article originally appeared on Medscape.com.
As hospitals across the country develop their plans to vaccinate their health care employees against COVID-19, a key question has come to the fore: What if an employee – whether nurse, physician, or other health care worker – refuses to receive the vaccine? Can hospitals require their employees to be vaccinated against COVID-19? And what consequences could an employee face for refusing the vaccine?
My answer needs to be based, in part, on the law related to previous vaccines – influenza, for example – because at the time of this writing (early December 2020), no vaccine for COVID-19 has been approved, although approval of at least one vaccine is expected within a week. So there have been no offers of vaccine and refusals yet, nor are there any cases to date involving an employee who refused a COVID-19 vaccine. As of December 2020, there are no state or federal laws that either require an employee to be vaccinated against COVID-19 or that protect an employee who refuses vaccination against COVID-19. It will take a while after the vaccine is approved and distributed before refusals, reactions, policies, cases, and laws begin to emerge.
If we look at the law related to health care workers refusing to be vaccinated against the closest relative to COVID-19 – influenza – then the answer would be yes, employers can require employees to be vaccinated.
An employer can fire an employee who refuses influenza vaccination. If an employee who refused and was fired sues the employer for wrongful termination, the employee has more or less chance of success depending on the reason for refusal. Some courts and the Equal Employment Opportunity Commission have held that a refusal on religious grounds is protected by the U.S. Constitution, as in this recent case. The Constitution protects freedom to practice one’s religion. Specific religions may have a range of tenets that support refusal to be vaccinated.
A refusal on medical grounds has been successful if the medical grounds fall under the protections of the Americans with Disabilities Act but may fail when the medical grounds for the claim are not covered by the ADA.
Refusal for secular, nonmedical reasons, such as a health care worker’s policy of treating their body as their temple, has not gone over well with employers or courts. However, in at least one case, a nurse who refused vaccination on secular, nonmedical grounds won her case against her employer, on appeal. The appeals court found that the hospital violated her First Amendment rights.
Employees who refuse vaccination for religious or medical reasons still will need to take measures to protect patients and other employees from infection. An employer such as a hospital can, rather than fire the employee, offer the employee an accommodation, such as requiring that the employee wear a mask or quarantine. There are no cases that have upheld an employee’s right to refuse to wear a mask or quarantine.
The situation with the COVID-19 vaccine is different from the situation surrounding influenza vaccines. There are plenty of data on effectiveness and side effects of influenza vaccines, but there is very little evidence of short- or long-term effects of the COVID-19 vaccines currently being tested and/or considered for approval. One could argue that the process of vaccine development is the same for all virus vaccines. However, public confidence in the vaccine vetting process is not what it once was. It has been widely publicized that the COVID-19 vaccine trials have been rushed. As of December 2020, only 60% of the general population say they would take the vaccine, although researchers say confidence is increasing.
The Centers for Disease Control and Prevention has designated health care workers as first in line to get the vaccine, but some health care workers may not want to be the first to try it. A CDC survey found that 63% of health care workers polled in recent months said they would get a COVID-19 vaccine.
Unions have entered the conversation. A coalition of unions that represent health care workers said, “we need a transparent, evidence-based federal vaccine strategy based on principles of equity, safety, and priority, as well as robust efforts to address a high degree of skepticism about safety of an authorized vaccine.” The organization declined to promote a vaccine until more is known.
As of publication date, the EEOC guidance for employers responding to COVID-19 does not address vaccines.
The CDC’s Interim Guidance for Businesses and Employers Responding to Coronavirus Disease 2019, May 2020, updated Dec. 4, 2020, does not address vaccines. The CDC’s page on COVID-19 vaccination for health care workers does not address a health care worker’s refusal. The site does assure health care workers that the vaccine development process is sound: “The current vaccine safety system is strong and robust, with the capacity to effectively monitor COVID-19 vaccine safety. Existing data systems have validated analytic methods that can rapidly detect statistical signals for possible vaccine safety problems. These systems are being scaled up to fully meet the needs of the nation. Additional systems and data sources are also being developed to further enhance safety monitoring capabilities. CDC is committed to ensuring that COVID-19 vaccines are safe.”
In the coming months, government officials and vaccine manufacturers will be working to reassure the public of the safety of the vaccine and the rigor of the vaccine development process. In November 2020, National Institute of Allergy and Infectious Diseases Director Anthony Fauci, MD, told Kaiser Health News: “The company looks at the data. I look at the data. Then the company puts the data to the FDA. The FDA will make the decision to do an emergency-use authorization or a license application approval. And they have career scientists who are really independent. They’re not beholden to anybody. Then there’s another independent group, the Vaccines and Related Biological Products Advisory Committee. The FDA commissioner has vowed publicly that he will go according to the opinion of the career scientists and the advisory board.” President-elect Joe Biden said he would get a vaccine when Dr. Fauci thinks it is safe.
An employee who, after researching the vaccine and the process, still wants to refuse when offered the vaccine is not likely to be fired for that reason right away, as long as the employee takes other precautions, such as wearing a mask. If the employer does fire the employee and the employee sues the employer, it is impossible to predict how a court would decide the case.
Related legal questions may arise in the coming months. For example:
- Is an employer exempt from paying workers’ compensation to an employee who refuses to be vaccinated and then contracts the virus while on the job?
- Can a prospective employer require COVID-19 vaccination as a precondition of employment?
- Is it within a patient’s rights to receive an answer to the question: Has my health care worker been vaccinated against COVID-19?
- If a hospital allows employees to refuse vaccination and keep working, and an outbreak occurs, and it is suggested through contact tracing that unvaccinated workers infected patients, will a court hold the hospital liable for patients’ damages?
Answers to these questions are yet to be determined.
Carolyn Buppert (www.buppert.com) is an attorney and former nurse practitioner who focuses on the legal issues affecting nurse practitioners.
A version of this article originally appeared on Medscape.com.
As hospitals across the country develop their plans to vaccinate their health care employees against COVID-19, a key question has come to the fore: What if an employee – whether nurse, physician, or other health care worker – refuses to receive the vaccine? Can hospitals require their employees to be vaccinated against COVID-19? And what consequences could an employee face for refusing the vaccine?
My answer needs to be based, in part, on the law related to previous vaccines – influenza, for example – because at the time of this writing (early December 2020), no vaccine for COVID-19 has been approved, although approval of at least one vaccine is expected within a week. So there have been no offers of vaccine and refusals yet, nor are there any cases to date involving an employee who refused a COVID-19 vaccine. As of December 2020, there are no state or federal laws that either require an employee to be vaccinated against COVID-19 or that protect an employee who refuses vaccination against COVID-19. It will take a while after the vaccine is approved and distributed before refusals, reactions, policies, cases, and laws begin to emerge.
If we look at the law related to health care workers refusing to be vaccinated against the closest relative to COVID-19 – influenza – then the answer would be yes, employers can require employees to be vaccinated.
An employer can fire an employee who refuses influenza vaccination. If an employee who refused and was fired sues the employer for wrongful termination, the employee has more or less chance of success depending on the reason for refusal. Some courts and the Equal Employment Opportunity Commission have held that a refusal on religious grounds is protected by the U.S. Constitution, as in this recent case. The Constitution protects freedom to practice one’s religion. Specific religions may have a range of tenets that support refusal to be vaccinated.
A refusal on medical grounds has been successful if the medical grounds fall under the protections of the Americans with Disabilities Act but may fail when the medical grounds for the claim are not covered by the ADA.
Refusal for secular, nonmedical reasons, such as a health care worker’s policy of treating their body as their temple, has not gone over well with employers or courts. However, in at least one case, a nurse who refused vaccination on secular, nonmedical grounds won her case against her employer, on appeal. The appeals court found that the hospital violated her First Amendment rights.
Employees who refuse vaccination for religious or medical reasons still will need to take measures to protect patients and other employees from infection. An employer such as a hospital can, rather than fire the employee, offer the employee an accommodation, such as requiring that the employee wear a mask or quarantine. There are no cases that have upheld an employee’s right to refuse to wear a mask or quarantine.
The situation with the COVID-19 vaccine is different from the situation surrounding influenza vaccines. There are plenty of data on effectiveness and side effects of influenza vaccines, but there is very little evidence of short- or long-term effects of the COVID-19 vaccines currently being tested and/or considered for approval. One could argue that the process of vaccine development is the same for all virus vaccines. However, public confidence in the vaccine vetting process is not what it once was. It has been widely publicized that the COVID-19 vaccine trials have been rushed. As of December 2020, only 60% of the general population say they would take the vaccine, although researchers say confidence is increasing.
The Centers for Disease Control and Prevention has designated health care workers as first in line to get the vaccine, but some health care workers may not want to be the first to try it. A CDC survey found that 63% of health care workers polled in recent months said they would get a COVID-19 vaccine.
Unions have entered the conversation. A coalition of unions that represent health care workers said, “we need a transparent, evidence-based federal vaccine strategy based on principles of equity, safety, and priority, as well as robust efforts to address a high degree of skepticism about safety of an authorized vaccine.” The organization declined to promote a vaccine until more is known.
As of publication date, the EEOC guidance for employers responding to COVID-19 does not address vaccines.
The CDC’s Interim Guidance for Businesses and Employers Responding to Coronavirus Disease 2019, May 2020, updated Dec. 4, 2020, does not address vaccines. The CDC’s page on COVID-19 vaccination for health care workers does not address a health care worker’s refusal. The site does assure health care workers that the vaccine development process is sound: “The current vaccine safety system is strong and robust, with the capacity to effectively monitor COVID-19 vaccine safety. Existing data systems have validated analytic methods that can rapidly detect statistical signals for possible vaccine safety problems. These systems are being scaled up to fully meet the needs of the nation. Additional systems and data sources are also being developed to further enhance safety monitoring capabilities. CDC is committed to ensuring that COVID-19 vaccines are safe.”
In the coming months, government officials and vaccine manufacturers will be working to reassure the public of the safety of the vaccine and the rigor of the vaccine development process. In November 2020, National Institute of Allergy and Infectious Diseases Director Anthony Fauci, MD, told Kaiser Health News: “The company looks at the data. I look at the data. Then the company puts the data to the FDA. The FDA will make the decision to do an emergency-use authorization or a license application approval. And they have career scientists who are really independent. They’re not beholden to anybody. Then there’s another independent group, the Vaccines and Related Biological Products Advisory Committee. The FDA commissioner has vowed publicly that he will go according to the opinion of the career scientists and the advisory board.” President-elect Joe Biden said he would get a vaccine when Dr. Fauci thinks it is safe.
An employee who, after researching the vaccine and the process, still wants to refuse when offered the vaccine is not likely to be fired for that reason right away, as long as the employee takes other precautions, such as wearing a mask. If the employer does fire the employee and the employee sues the employer, it is impossible to predict how a court would decide the case.
Related legal questions may arise in the coming months. For example:
- Is an employer exempt from paying workers’ compensation to an employee who refuses to be vaccinated and then contracts the virus while on the job?
- Can a prospective employer require COVID-19 vaccination as a precondition of employment?
- Is it within a patient’s rights to receive an answer to the question: Has my health care worker been vaccinated against COVID-19?
- If a hospital allows employees to refuse vaccination and keep working, and an outbreak occurs, and it is suggested through contact tracing that unvaccinated workers infected patients, will a court hold the hospital liable for patients’ damages?
Answers to these questions are yet to be determined.
Carolyn Buppert (www.buppert.com) is an attorney and former nurse practitioner who focuses on the legal issues affecting nurse practitioners.
A version of this article originally appeared on Medscape.com.
Peripheral neuropathy tied to mortality in adults without diabetes
researchers reported in Annals of Internal Medicine.

The findings do not necessarily mean that doctors should implement broader screening for peripheral neuropathy at this time, however, the investigators said.
“Doctors don’t typically screen for peripheral neuropathy in persons without diabetes,” senior author Elizabeth Selvin, PhD, MPH, professor of epidemiology at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview.
“Our study shows that peripheral neuropathy – as assessed by decreased sensation in the feet – is common, even in people without diabetes,” Dr. Selvin explained. “It is not yet clear whether we should be screening people without diabetes since we don’t have clear treatments, but our study does suggest that this condition is an underrecognized condition that is associated with poor outcomes.”
Patients with diabetes typically undergo annual foot examinations that include screening for peripheral neuropathy, but that’s not the case for most adults in the absence of diabetes.
“I don’t know if we can make the jump that we should be screening people without diabetes,” said first author Caitlin W. Hicks, MD, assistant professor of surgery, division of vascular surgery and endovascular therapy, Johns Hopkins University, Baltimore. “Right now, we do not exactly know what it means in the people without diabetes, and we definitely do not know how to treat it. So, screening for it will tell us that this person has this and is at higher risk of mortality than someone who doesn’t, but we do not know what to do with that information yet.”
Nevertheless, the study raises the question of whether physicians should pay more attention to peripheral neuropathy in people without diabetes, said Dr. Hicks, director of research at the university’s diabetic foot and wound service.
Heightened risk
To examine associations between peripheral neuropathy and all-cause and cardiovascular mortality in U.S. adults, Dr. Hicks and colleagues analyzed data from 7,116 adults aged 40 years or older who participated in the National Health and Nutrition Examination Survey (NHANES) between 1999 and 2004.
The study included participants who underwent monofilament testing for peripheral neuropathy. During testing, technicians used a standard 5.07 Semmes-Weinstein nylon monofilament to apply slight pressure to the bottom of each foot at three sites. If participants could not correctly identify where pressure was applied, the test was repeated. After participants gave two incorrect or undeterminable responses for a site, the site was defined as insensate. The researchers defined peripheral neuropathy as at least one insensate site on either foot.
The researchers determined deaths and causes of death using death certificate records from the National Death Index through 2015.
In all, 13.5% of the participants had peripheral neuropathy, including 27% of adults with diabetes and 11.6% of adults without diabetes. Those with peripheral neuropathy were older, were more likely to be male, and had lower levels of education, compared with participants without peripheral neuropathy. They also had higher body mass index, were more often former or current smokers, and had a higher prevalence of hypertension, hypercholesterolemia, and cardiovascular disease.
During a median follow-up of 13 years, 2,128 participants died, including 488 who died of cardiovascular causes.
The incidence rate of all-cause mortality per 1,000 person-years was 57.6 in adults with diabetes and peripheral neuropathy, 34.3 in adults with peripheral neuropathy but no diabetes, 27.1 in adults with diabetes but no peripheral neuropathy, and 13.0 in adults without diabetes or peripheral neuropathy.
Among participants with diabetes, the leading cause of death was cardiovascular disease (31% of deaths), whereas among participants without diabetes, the leading cause of death was malignant neoplasms (27% of deaths).
After adjustment for age, sex, race, or ethnicity, and risk factors such as cardiovascular disease, peripheral neuropathy was significantly associated with all-cause mortality (hazard ratio [HR], 1.49) and cardiovascular mortality (HR, 1.66) in participants with diabetes. In participants without diabetes, peripheral neuropathy was significantly associated with all-cause mortality (HR, 1.31), but its association with cardiovascular mortality was not statistically significant.
The association between peripheral neuropathy and all-cause mortality persisted in a sensitivity analysis that focused on adults with normoglycemia.
Related conditions
The study confirms findings from prior studies that examined the prevalence of loss of peripheral sensation in populations of older adults with and without diabetes, said Elsa S. Strotmeyer, PhD, MPH, associate professor of epidemiology at the University of Pittsburgh. “The clinical significance of the loss of peripheral sensation in older adults without diabetes is not fully appreciated,” she said.
A limitation of the study is that peripheral neuropathy was not a clinical diagnosis. “Monofilament testing at the foot is a quick clinical screen for decreased lower-extremity sensation that likely is a result of sensory peripheral nerve decline,” Dr. Strotmeyer said.
Another limitation is that death certificates are less accurate than medical records for determining cause of death.
“Past studies have indicated that peripheral nerve decline is related to common conditions in aging such as the metabolic syndrome and cardiovascular disease, cancer treatment, and physical function loss,” Dr. Strotmeyer said. “Therefore it is not surprising that is related to mortality as these conditions in aging are associated with increased mortality. Loss of peripheral sensation at the foot may also be related to fall injuries, and mortality from fall injuries has increased dramatically in older adults over the past several decades.”
Prior research has suggested that monofilament testing may play a role in screening for fall risk in older adults without diabetes, Dr. Strotmeyer added.
“For older adults both with and without diabetes, past studies have recommended monofilament testing be incorporated in geriatric screening for fall risk. Therefore, this article expands implications of clinical importance to understanding the pathology and consequences of loss of sensation at the foot in older patients,” she said.
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute. Dr. Hicks, Dr. Selvin, and a coauthor, Kunihiro Matsushita, MD, PhD, disclosed NIH grants. In addition, Dr. Selvin disclosed personal fees from Novo Nordisk and grants from the Foundation for the National Institutes of Health outside the submitted work, and Dr. Matsushita disclosed grants and personal fees from Fukuda Denshi outside the submitted work. Dr. Strotmeyer receives funding from the National Institute on Aging and the National Institute of Arthritis and Musculoskeletal and Skin Diseases and is chair of the health sciences section of the Gerontological Society of America.
A version of this article originally appeared on Medscape.com.
researchers reported in Annals of Internal Medicine.
The findings do not necessarily mean that doctors should implement broader screening for peripheral neuropathy at this time, however, the investigators said.
“Doctors don’t typically screen for peripheral neuropathy in persons without diabetes,” senior author Elizabeth Selvin, PhD, MPH, professor of epidemiology at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview.
“Our study shows that peripheral neuropathy – as assessed by decreased sensation in the feet – is common, even in people without diabetes,” Dr. Selvin explained. “It is not yet clear whether we should be screening people without diabetes since we don’t have clear treatments, but our study does suggest that this condition is an underrecognized condition that is associated with poor outcomes.”
Patients with diabetes typically undergo annual foot examinations that include screening for peripheral neuropathy, but that’s not the case for most adults in the absence of diabetes.
“I don’t know if we can make the jump that we should be screening people without diabetes,” said first author Caitlin W. Hicks, MD, assistant professor of surgery, division of vascular surgery and endovascular therapy, Johns Hopkins University, Baltimore. “Right now, we do not exactly know what it means in the people without diabetes, and we definitely do not know how to treat it. So, screening for it will tell us that this person has this and is at higher risk of mortality than someone who doesn’t, but we do not know what to do with that information yet.”
Nevertheless, the study raises the question of whether physicians should pay more attention to peripheral neuropathy in people without diabetes, said Dr. Hicks, director of research at the university’s diabetic foot and wound service.
Heightened risk
To examine associations between peripheral neuropathy and all-cause and cardiovascular mortality in U.S. adults, Dr. Hicks and colleagues analyzed data from 7,116 adults aged 40 years or older who participated in the National Health and Nutrition Examination Survey (NHANES) between 1999 and 2004.
The study included participants who underwent monofilament testing for peripheral neuropathy. During testing, technicians used a standard 5.07 Semmes-Weinstein nylon monofilament to apply slight pressure to the bottom of each foot at three sites. If participants could not correctly identify where pressure was applied, the test was repeated. After participants gave two incorrect or undeterminable responses for a site, the site was defined as insensate. The researchers defined peripheral neuropathy as at least one insensate site on either foot.
The researchers determined deaths and causes of death using death certificate records from the National Death Index through 2015.
In all, 13.5% of the participants had peripheral neuropathy, including 27% of adults with diabetes and 11.6% of adults without diabetes. Those with peripheral neuropathy were older, were more likely to be male, and had lower levels of education, compared with participants without peripheral neuropathy. They also had higher body mass index, were more often former or current smokers, and had a higher prevalence of hypertension, hypercholesterolemia, and cardiovascular disease.
During a median follow-up of 13 years, 2,128 participants died, including 488 who died of cardiovascular causes.
The incidence rate of all-cause mortality per 1,000 person-years was 57.6 in adults with diabetes and peripheral neuropathy, 34.3 in adults with peripheral neuropathy but no diabetes, 27.1 in adults with diabetes but no peripheral neuropathy, and 13.0 in adults without diabetes or peripheral neuropathy.
Among participants with diabetes, the leading cause of death was cardiovascular disease (31% of deaths), whereas among participants without diabetes, the leading cause of death was malignant neoplasms (27% of deaths).
After adjustment for age, sex, race, or ethnicity, and risk factors such as cardiovascular disease, peripheral neuropathy was significantly associated with all-cause mortality (hazard ratio [HR], 1.49) and cardiovascular mortality (HR, 1.66) in participants with diabetes. In participants without diabetes, peripheral neuropathy was significantly associated with all-cause mortality (HR, 1.31), but its association with cardiovascular mortality was not statistically significant.
The association between peripheral neuropathy and all-cause mortality persisted in a sensitivity analysis that focused on adults with normoglycemia.
Related conditions
The study confirms findings from prior studies that examined the prevalence of loss of peripheral sensation in populations of older adults with and without diabetes, said Elsa S. Strotmeyer, PhD, MPH, associate professor of epidemiology at the University of Pittsburgh. “The clinical significance of the loss of peripheral sensation in older adults without diabetes is not fully appreciated,” she said.
A limitation of the study is that peripheral neuropathy was not a clinical diagnosis. “Monofilament testing at the foot is a quick clinical screen for decreased lower-extremity sensation that likely is a result of sensory peripheral nerve decline,” Dr. Strotmeyer said.
Another limitation is that death certificates are less accurate than medical records for determining cause of death.
“Past studies have indicated that peripheral nerve decline is related to common conditions in aging such as the metabolic syndrome and cardiovascular disease, cancer treatment, and physical function loss,” Dr. Strotmeyer said. “Therefore it is not surprising that is related to mortality as these conditions in aging are associated with increased mortality. Loss of peripheral sensation at the foot may also be related to fall injuries, and mortality from fall injuries has increased dramatically in older adults over the past several decades.”
Prior research has suggested that monofilament testing may play a role in screening for fall risk in older adults without diabetes, Dr. Strotmeyer added.
“For older adults both with and without diabetes, past studies have recommended monofilament testing be incorporated in geriatric screening for fall risk. Therefore, this article expands implications of clinical importance to understanding the pathology and consequences of loss of sensation at the foot in older patients,” she said.
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute. Dr. Hicks, Dr. Selvin, and a coauthor, Kunihiro Matsushita, MD, PhD, disclosed NIH grants. In addition, Dr. Selvin disclosed personal fees from Novo Nordisk and grants from the Foundation for the National Institutes of Health outside the submitted work, and Dr. Matsushita disclosed grants and personal fees from Fukuda Denshi outside the submitted work. Dr. Strotmeyer receives funding from the National Institute on Aging and the National Institute of Arthritis and Musculoskeletal and Skin Diseases and is chair of the health sciences section of the Gerontological Society of America.
A version of this article originally appeared on Medscape.com.
researchers reported in Annals of Internal Medicine.
The findings do not necessarily mean that doctors should implement broader screening for peripheral neuropathy at this time, however, the investigators said.
“Doctors don’t typically screen for peripheral neuropathy in persons without diabetes,” senior author Elizabeth Selvin, PhD, MPH, professor of epidemiology at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview.
“Our study shows that peripheral neuropathy – as assessed by decreased sensation in the feet – is common, even in people without diabetes,” Dr. Selvin explained. “It is not yet clear whether we should be screening people without diabetes since we don’t have clear treatments, but our study does suggest that this condition is an underrecognized condition that is associated with poor outcomes.”
Patients with diabetes typically undergo annual foot examinations that include screening for peripheral neuropathy, but that’s not the case for most adults in the absence of diabetes.
“I don’t know if we can make the jump that we should be screening people without diabetes,” said first author Caitlin W. Hicks, MD, assistant professor of surgery, division of vascular surgery and endovascular therapy, Johns Hopkins University, Baltimore. “Right now, we do not exactly know what it means in the people without diabetes, and we definitely do not know how to treat it. So, screening for it will tell us that this person has this and is at higher risk of mortality than someone who doesn’t, but we do not know what to do with that information yet.”
Nevertheless, the study raises the question of whether physicians should pay more attention to peripheral neuropathy in people without diabetes, said Dr. Hicks, director of research at the university’s diabetic foot and wound service.
Heightened risk
To examine associations between peripheral neuropathy and all-cause and cardiovascular mortality in U.S. adults, Dr. Hicks and colleagues analyzed data from 7,116 adults aged 40 years or older who participated in the National Health and Nutrition Examination Survey (NHANES) between 1999 and 2004.
The study included participants who underwent monofilament testing for peripheral neuropathy. During testing, technicians used a standard 5.07 Semmes-Weinstein nylon monofilament to apply slight pressure to the bottom of each foot at three sites. If participants could not correctly identify where pressure was applied, the test was repeated. After participants gave two incorrect or undeterminable responses for a site, the site was defined as insensate. The researchers defined peripheral neuropathy as at least one insensate site on either foot.
The researchers determined deaths and causes of death using death certificate records from the National Death Index through 2015.
In all, 13.5% of the participants had peripheral neuropathy, including 27% of adults with diabetes and 11.6% of adults without diabetes. Those with peripheral neuropathy were older, were more likely to be male, and had lower levels of education, compared with participants without peripheral neuropathy. They also had higher body mass index, were more often former or current smokers, and had a higher prevalence of hypertension, hypercholesterolemia, and cardiovascular disease.
During a median follow-up of 13 years, 2,128 participants died, including 488 who died of cardiovascular causes.
The incidence rate of all-cause mortality per 1,000 person-years was 57.6 in adults with diabetes and peripheral neuropathy, 34.3 in adults with peripheral neuropathy but no diabetes, 27.1 in adults with diabetes but no peripheral neuropathy, and 13.0 in adults without diabetes or peripheral neuropathy.
Among participants with diabetes, the leading cause of death was cardiovascular disease (31% of deaths), whereas among participants without diabetes, the leading cause of death was malignant neoplasms (27% of deaths).
After adjustment for age, sex, race, or ethnicity, and risk factors such as cardiovascular disease, peripheral neuropathy was significantly associated with all-cause mortality (hazard ratio [HR], 1.49) and cardiovascular mortality (HR, 1.66) in participants with diabetes. In participants without diabetes, peripheral neuropathy was significantly associated with all-cause mortality (HR, 1.31), but its association with cardiovascular mortality was not statistically significant.
The association between peripheral neuropathy and all-cause mortality persisted in a sensitivity analysis that focused on adults with normoglycemia.
Related conditions
The study confirms findings from prior studies that examined the prevalence of loss of peripheral sensation in populations of older adults with and without diabetes, said Elsa S. Strotmeyer, PhD, MPH, associate professor of epidemiology at the University of Pittsburgh. “The clinical significance of the loss of peripheral sensation in older adults without diabetes is not fully appreciated,” she said.
A limitation of the study is that peripheral neuropathy was not a clinical diagnosis. “Monofilament testing at the foot is a quick clinical screen for decreased lower-extremity sensation that likely is a result of sensory peripheral nerve decline,” Dr. Strotmeyer said.
Another limitation is that death certificates are less accurate than medical records for determining cause of death.
“Past studies have indicated that peripheral nerve decline is related to common conditions in aging such as the metabolic syndrome and cardiovascular disease, cancer treatment, and physical function loss,” Dr. Strotmeyer said. “Therefore it is not surprising that is related to mortality as these conditions in aging are associated with increased mortality. Loss of peripheral sensation at the foot may also be related to fall injuries, and mortality from fall injuries has increased dramatically in older adults over the past several decades.”
Prior research has suggested that monofilament testing may play a role in screening for fall risk in older adults without diabetes, Dr. Strotmeyer added.
“For older adults both with and without diabetes, past studies have recommended monofilament testing be incorporated in geriatric screening for fall risk. Therefore, this article expands implications of clinical importance to understanding the pathology and consequences of loss of sensation at the foot in older patients,” she said.
The study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute. Dr. Hicks, Dr. Selvin, and a coauthor, Kunihiro Matsushita, MD, PhD, disclosed NIH grants. In addition, Dr. Selvin disclosed personal fees from Novo Nordisk and grants from the Foundation for the National Institutes of Health outside the submitted work, and Dr. Matsushita disclosed grants and personal fees from Fukuda Denshi outside the submitted work. Dr. Strotmeyer receives funding from the National Institute on Aging and the National Institute of Arthritis and Musculoskeletal and Skin Diseases and is chair of the health sciences section of the Gerontological Society of America.
A version of this article originally appeared on Medscape.com.