User login
Lesions on the hands, high aminotransferase levels
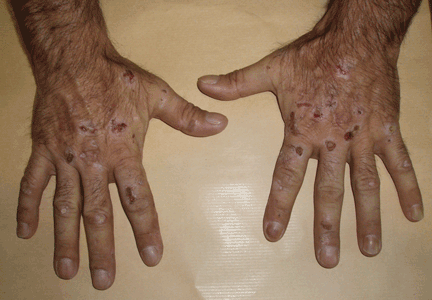
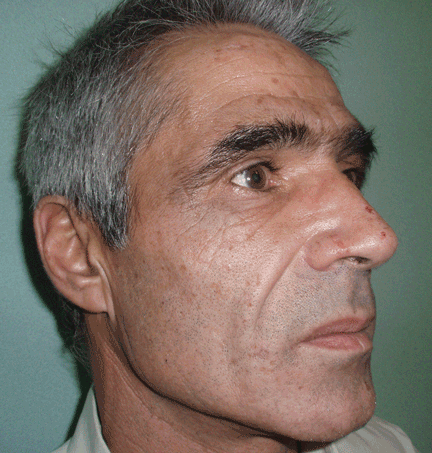
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
Acetaminophen: Old drug, new warnings
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
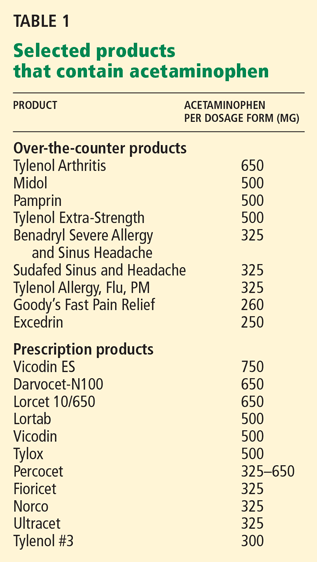
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite

Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
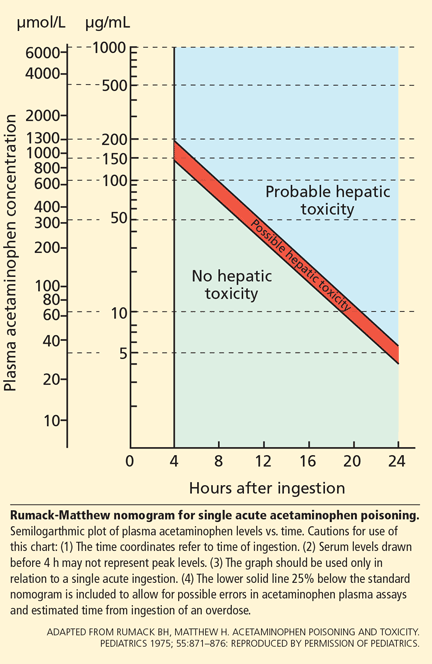
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
FINDINGS FROM LARGE REGISTRIES
Findings in adults
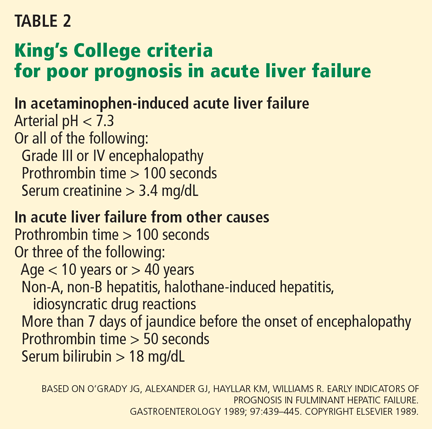
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling

Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite
Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
FINDINGS FROM LARGE REGISTRIES
Findings in adults
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling
Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
Editor’s note: Portions of this article are based on an article previously published in an internal Cleveland Clinic publication, Pharmacotherapy Update. The version here has been revised, updated, and peer-reviewed.
This drug is generally considered safe, but high doses can be toxic. The number of overdoses is worrisome. In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.2 It is responsible for more emergency room visits than any other drug on the market.
According to a position statement from the American Association for the Study of Liver Diseases (AASLD),3 the incidence of acetaminophen-related liver toxicity has been steadily increasing over the past decade, and this drug is now the most common cause of acute liver failure.
MANY OVERDOSES ARE UNINTENTIONAL
Cases of acetaminophen-related liver toxicity can be categorized as either intentional (ie, due to a suicide attempt) or unintentional (ie, due to multiple therapeutic but excessive doses over a period of time, usually more than 3 days).
Up to 50% of cases are unintentional. Bower et al4 reviewed cases of acute liver failure that occurred in the Atlanta, GA, area between November 2000 and October 2004. Acetaminophen was the most common cause in adult patients. Of greater concern is that 61% of the acetaminophen-related cases were due to unintentional overdose. According to the Institute for Safe Medication Practices,5 one hospital (not named) reported that an average of one patient per day was given more than the recommended maximum daily acetaminophen dose of 4 g while in the hospital.
Many patients take more than one acetaminophen product
Unintentional overdoses or “therapeutic misadventures” are most often due to taking multiple products that contain acetaminophen, taking acetaminophen-narcotic combinations, and impulsive behavior involving a lack of understanding of possible injury in consuming multiple acetaminophen-containing products.3
In the US Food and Drug Administration (FDA) Medwatch Database, in 307 cases of unintentional acetaminophen overdose between 1998 and 2001, 25% of patients had been taking more than one acetaminophencontaining product.5
Larson et al6 found that one-third of patients who had had an unintentional acetaminophen overdose were taking an acetaminophen-narcotic combination in addition to another acetaminophen-containing product.
Many consumers don’t know they are taking acetaminophen
Many consumers don’t know that some of the drugs they take contain acetaminophen. This may be because many drug labels contain abbreviations for acetaminophen such as “APAP” or have inconsistent formatting that makes it difficult to determine if the product contains acetaminophen.
Others may not be aware of the total maximum recommended daily dose or may not be able to calculate the total daily intake from the information on the label. The problem is not only with over-the-counter products. For example, if a physician prescribes one or two tablets of hydrocodone/acetaminophen (Vicodin) 5 mg/500 mg every 4 to 6 hours, a patient could easily exceed the recommended maximum daily dose of 4 g of acetaminophen.
Toxicity can occur even at therapeutic doses
Acetaminophen hepatotoxicity can also occur even with therapeutic doses in certain conditions. Risk factors:
- Chronic alcohol use (ie, more than three drinks per day)
- Malnutrition
- Concurrent use of drugs that induce cytochrome P450 (CYP450) enzymes (more on this below).6
FIRST USED IN 1893
Acetaminophen was first used in medicine in 1893, and it became widely used after 1949, when it was found to be a less-toxic metabolite of two parent compounds, acetanilide and phenacetin.1
Acetaminophen is an effective antipyretic and analgesic, but its anti-inflammatory properties are minimal, especially compared with nonsteroidal anti-inflammatory drugs (NSAIDs). Nevertheless, acetaminophen is preferred over NSAIDs in some patients because it carries a lower risk of gastrointestinal toxicity (eg, ulceration, bleeding) and so may be better tolerated.1
INDICATIONS AND DOSAGE
Acetaminophen is indicated for mild to moderate pain or fever, including the pain of osteoarthritis. It is not recommended for chronic inflammatory conditions such as rheumatoid arthritis, since it lacks anti-inflammatory properties.
In adults and in children over age 12, the usual dosage is 325 to 650 mg orally or rectally every 4 to 6 hours, or 1,000 mg three to four times daily.
The current package label recommends that the total daily dose not exceed 4 g in most adults. Lower maximum daily doses (eg, 2 g) are recommended in patients who may be at higher risk of hepatotoxicity, such as those who drink heavily, are malnourished, or take enzyme-inducing drugs. Tylenol products currently include an alcohol warning, advising those who consume three or more alcoholic drinks a day to ask their doctor if they should take acetaminophen.
In children up to 12 years of age, the recommended dosage is 10 to 15 mg/kg orally or rectally every 4 to 6 hours. The maximum dosing for children in this age group should not exceed five doses (or 50 to 75 mg/kg) in 24 hours.7 In children under age 2 or weighing less than 11 kg, acetaminophen should only be used under the direction of a physician.
MOST ACETAMINOPHEN IS CONJUGATED AND THEN EXCRETED IN THE URINE
Acetaminophen usually has excellent bioavailability (up to 98%), but the exact amount absorbed varies, depending on the dosage form and concomitant use of other drugs.8
At therapeutic doses, the elimination halflife is about 2 hours. Peak plasma concentrations are reached 30 to 60 minutes after the dose is taken,1 but taking acetaminophen with opioids, anticholinergic drugs, or even food may delay the time to peak concentration by delaying gastric emptying.8
NAPQI is a toxic metabolite
Under normal circumstances, a small amount of acetaminophen undergoes hepatic metabolism by a different pathway, ie, by CYP450 enzymes, primarily CYP2E1 and to a lesser extent CYP1A2, CYP2A6, and CYP3A4, forming a toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Then, the sulfhydryl groups of glutathione convert NAPQI into harmless metabolites that are excreted in the urine.1,7
Well tolerated and relatively safe
At recommended doses, acetaminophen is well tolerated, and it is considered relatively safe when used according to labeling instructions.
Rarely, patients experience an erythematous or urticarial rash or other allergic complications.1
However, acetaminophen is a dose-dependent hepatotoxin, and excessive doses (intentional or unintentional) may lead to acute liver failure. In addition, even in therapeutic doses, acetaminophen may still cause transient liver enzyme elevations and possibly hepatotoxicity, particularly in people who are malnourished or alcoholic or are taking certain CYP450-inducing drugs.3,6,10
HOW ACETAMINOPHEN CAN INJURE THE LIVER
Glucuronidation and sulfation, the major metabolic pathways, become saturated after an acetaminophen overdose.7 When this happens, more of the toxic metabolite NAPQI is formed by CYP450-mediated N-hydroxylation. When glutathione is depleted after large doses of acetaminophen or in malnourished people, the toxic metabolite accumulates, resulting in liver damage (Figure 1).6
The liver is damaged by two mechanisms. In one, NAPQI binds to hepatic cell macromolecules, causing dysfunction of the enzymatic systems, structural and metabolic disarray, and eventually necrotic cell death. The other mechanism is oxidative stress due to depletion of glutathione.
In children, single acetaminophen doses of 120 to 150 mg/kg of body weight have been associated with hepatotoxicity,11 as have single doses of more than 150 mg/kg or a total dose of greater than 7.5 g in adults. However, the minimal dose associated with liver injury has ranged from 4 to 10 g, and in healthy volunteers even therapeutic doses of 1 g orally every 6 hours resulted in mild liver injury.12
Patients who are malnourished or fasting are thought to be at greater risk of acetaminophen hepatotoxicity because they may be deficient in glutathione at baseline. In addition, even at lower-than-therapeutic doses, induction of CYP450 enzymes by drugs or chronic alcohol consumption may lead to an increase in the formation of NAPQI, increasing the risk of hepatotoxicity. Examples of drugs that induce CYP450 enzymes to produce more NAPQI include the anticonvuslants phenytoin (Dilantin) and phenobarbital.
CLINICAL PRESENTATION OF ACETAMINOPHEN OVERDOSE
The diagnosis of acetaminophen overdose is often established by a thorough history. The pertinent information may be difficult to obtain, however, because the patient may be confused or stuporous at presentation, may not know that the overthe-counter products he or she has been taking contain acetaminophen, or may be embarrassed about taking too much acetaminophen.13
In acute intentional overdose, signs may not be apparent immediately
The symptoms of toxicity may not be apparent immediately after ingestion of an acute overdose of acetaminophen, but early recognition and treatment can prevent more severe liver damage, decreasing morbidity and the risk of death.9
Phase 1 of an acetaminophen overdose begins shortly after ingestion and can last for 12 to 24 hours. Patients may have signs of gastrointestinal upset, nausea, vomiting, anorexia, diaphoresis, and pallor. Although the signs and symptoms show a consistent pattern and are more pronounced after larger acute overdoses, they are not diagnostic or specific.
Phase 2 (up to 48 hours after ingestion). Patients may begin to feel better during this phase. However, the hepatic enzyme levels, the prothrombin time (PT), and the international normalized ratio (INR) may continue to rise, and right upper quadrant pain may develop. Additionally, other laboratory results may be abnormal, and renal insufficiency can occur due to acetaminophen-induced renal tubular necrosis.6 Most patients receive the antidote, acetylcysteine (Mycomyst, Acetadote) before or during this phase, and consequently, liver function gradually returns to normal.
Phase 3, if reached, may be marked by severe hepatic necrosis, typically 3 to 5 days after ingestion. Symptoms during this phase range from less severe (eg, nausea and general malaise) to more severe (eg, confusion and stupor). Also, at this time, liver enzyme levels can be as high as 10,000 IU/L or even higher, and lactic acidosis and coagulopathy may worsen. If death should occur, it is most likely from complications associated with fulminant hepatic failure, including cerebral edema, multiorgan-system failure, or sepsis.6
Phase 4. Patients who recover generally have complete recovery of liver function with no long-term sequelae..
In unintentional overdoses, patients may have low drug levels
Many patients who present with unintentional acetaminophen toxicity have been taking the drug or products that contain the drug over several days to treat an acute or chronic medical condition. They often have low or undetectable serum acetaminophen levels after 2 to 3 days of nonspecific symptoms.12
MANAGING ACETAMINOPHENHEN OVERDOSE
Unintentional overdoses occur over a more prolonged period, and therefore the nomogram is not useful in this situation.
Acetylcysteine is the antidote
Acetylcysteine is the antidote for acetaminophen toxicity and should be given within 8 hours of ingestion for maximal protection against hepatic injury in patients whose serum acetaminophen levels are above the “possible” toxicity line on the nomogram.15 If acetaminophen overdose is suspected but the time elapsed since ingestion cannot be determined, acetylcysteine should be given immediately regardless of the quantity of acetaminophen ingested. 16 In cases of unintentional overdose, it is often given at the discretion of the physician.
Acetylcysteine limits the toxicity of acetaminophen by increasing glutathione stores, binding with NAPQI as a substitute for glutathione, and enhancing sulfate conjugation.6 It may further limit acetaminophen toxicity by nonspecific mechanisms including anti-inflammatory, antioxidant, inotropic, and vasodilating effects.6 In addition, it may prevent further hepatic damage in any patient thought to have acetaminophen-related liver toxicity even beyond the first 12 hours of an overdose.1
Acetylcysteine is available in an oral (Mucomyst) and an intravenous (Acetadote) formulation, which are similar in efficacy. Because many patients find the taste of the oral solution unpleasant and difficult to tolerate, it should be diluted in a 1:3 ratio with cola, orange juice, or other drink to mask its flavor. This mixture should be used within 1 hour of preparation.
Anaphylactoid reactions have occurred in patients receiving intravenous acetylcysteine for acetaminophen overdose soon after the infusion was started, most commonly during the loading dose. The frequency of infusion-related reactions has been reported to be 0.2% to 20.8%.15
The recommended dosage for intravenous acetylcysteine is a loading dose of 150 mg/kg given over 60 minutes, followed by a second dose of 50 mg/kg given over 4 hours, and finally a third dose of 100 mg/kg given over 16 hours.15 For oral acetylcysteine, the loading dose is 140 mg/kg followed by 70 mg/kg every 4 hours for 17 additional doses.17
Other measures
Activated charcoal can be used if the patient presents within 1 to 2 hours after taking acetaminophen. However, the rapid gastrointestinal absorption of acetaminophen makes this treatment ineffective in most cases.9
FINDINGS FROM LARGE REGISTRIES
Findings in adults
Ostapowicz et al,19 as part of the Acute Liver Failure Study Group, in 2002 prospectively characterized the short-term outcomes of acute liver failure in a large number of patients at 17 tertiary care centers in the United States over approximately 41 months. All centers except one performed liver transplants. Eligible patients had to meet criteria for acute liver failure, including an INR higher than 1.5, evidence of hepatic encephalopathy, and presentation within 26 weeks of illness onset without apparent chronic liver disease.
Of the 308 cases of acute liver failure, 120 (39%) were from acetaminophen overdose, making this drug the most common cause of acute liver failure. Forty-four (37%) of the patients with acetaminophen-related acute liver failure were trying to commit suicide, 57% of cases were accidental, and the remaining reasons for acetaminophen overdose were unknown. The median amount ingested was 13.2 g/day (range 2.6–75 g), and 99 (83%) of the 120 patients took more than 4 g/day.
The patients with acetaminophen-related acute liver toxicity differed from those with other causes of acute liver failure such as idiosyncratic drug reactions or indeterminate causes. The acetaminophen group had a shorter duration of disease and higher serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum creatinine than those with acute liver failure from other causes. They also had lower bilirubin levels and a lower arterial pH.
Rates of liver transplantation were 6% in the acetaminophen group, 53% in the group with other drug-induced liver toxicity, 51% in the indeterminate group, and 36% in the remaining groups. Of the acetaminophen group, 47% met the criteria for transplantation, but only 57% of those eligible were listed for transplantation. Those excluded from the transplant list had medical contraindications or were excluded for psychosocial reasons. The short-term transplant-free survival rate was 68% in the acetaminophen group.
Overall, 11% of the patients died, including 28% of the acetaminophen group. The authors concluded that most cases of liver injury in the United States are due to medications and may be preventable.
Larson et al,20 as part of the Acute Liver Failure Study Group, examined the incidence, risk factors, and outcomes of acetaminopheninduced acute liver failure at 22 tertiary care centers in the United States over a 6-year period. Of the 662 patients in the study, 302 had acetaminophen-related toxicity and 275 were included in the final analysis; some of them had been included in the study by Ostapowicz et al.19 During the study period, the number of cases of acute liver toxicity related to acetaminophen increased from 28% to 51%.
Of the patients enrolled in the study, 56% met the criteria for potentially toxic acetaminophen ingestion. Of those who met the criteria, 77% had detectable acetaminophen levels in their serum, and 91% had ALT levels higher than 1,000 IU/L. The time between ingestion and symptom onset ranged from 1 to 32 days, and the median dose was 24 g (range 1.2–180 g).
Of the overdose cases, 48% were unintentional, 44% were intentional, and the remaining 8% had no definable reason. Of the patients in the unintentional-overdose group, 38% were using more than one acetaminophen-containing product and 63% were taking combination products containing narcotics. Overeall, 44% of patients reported using narcotic-acetaminophen combination products. The unintentional-overdose group had lower serum acetaminophen levels than the intentional-overdose group, but they were more likely to present with severe hepatic encephalopathy.
The number of patients with an unintentional overdose was worrisome. Furthermore, one-third of the patients who were receiving an acetaminophen-narcotic combination product were taking an additional acetaminophencontaining product. The authors concluded that unintentional overdose is the leading cause of acetaminophen-related hepatotoxicity, and efforts to limit the over-the-counter package size and to restrict prescriptions of acetaminophen-narcotic combinations may be necessary to decrease the incidence of this preventable cause of acute liver failure.
Findings in children
Squires et al21 and the Pediatric Acute Liver Failure Study Group examined the pathogenesis, treatment, and outcome of acute liver failure in children (any age from birth to 18 years) with no previous evidence of chronic liver disease, evidence of acute liver injury, or hepatic-based coagulopathy. From December 1999 to December 2004, 348 patients were enrolled.
Fourteen percent of the cases were due to acetaminophen. The median dose of acetaminophen ingested was 183 mg/kg. Most of these patients were white and female, and 96% were over age 3. Hepatic encephalopathy was more common in the non-acetaminophen groups than in the acetaminophen group, although this is often difficult to assess in infants and children. Children with acetaminophen toxicity had the highest rate of spontaneous recovery: 45 (94%) of 48 recovered.
Although there are fewer acetaminophenrelated cases of acute liver failure in children than in adults, the use of acetaminophen in children is still worrisome. The authors concluded that if they do not have hepatic encephalopathy, children with acetaminopheninduced liver toxicity have an excellent prognosis.
THE FDA LOOKS AT THE PROBLEM
Why overdoses occur
A recent FDA report22 cited the following reasons for acetaminophen overdose:
- Some patients may experience liver injury at doses only slightly above the recommended 4-g daily limit.
- Some patients may be more prone to liver injury from acetaminophen.
- The symptoms associated with liver injury due to acetaminophen can be nonspecific and tend to evolve over several days.
- Many acetaminophen-containing products are available, including over-the-counter and prescription drugs with many different strengths and indications.
- Consumers are unaware of the risk of liver toxicity with acetaminophen.
- Prescription products are not always clearly labeled with acetaminophen as an ingredient.
- Pediatric dosage forms are available in many different concentrations.
New package labeling
Recommendations from an advisory panel
In addition, an FDA advisory panel recommended decreasing the maximum recommended daily dose (possibly to 3,250 mg/day in adults, although this is not final) to help prevent overdoses, and reducing the maximum amount in a single nonprescription dose of the drug to 650 mg.23 The panel also voted (by a narrow margin) to ban all acetaminophen-narcotic combination products.
As of this writing, the FDA has not adopted these recommendations. (Although the FDA is not obliged to follow the advice of its advisory panels, in most cases it does.) The recommendations about acetaminophen could take years to implement fully.
THE UNITED KINGDOM ACTED IN 1998
In response to a rising number of analgesicrelated deaths, the United Kingdom enacted legislation in 1998 to limit the package size available to consumers.24 Packages sold in general stores can contain no more than 16 capsules, while those sold in pharmacies can contain 24. Blister packs were also introduced to make it harder for people to impulsively take handfuls of tablets.25
Two studies since then both found that the number of deaths related to paracetamol (as acetaminophen is known in the United Kingdom) had fallen since the legislation was implemented.24,26 Greene et al27 found that patients who ingested potentially toxic doses of paracetamol had obtained the drug “in a manner contravening the 1998 legislation.”27 In other words, shops in London were not obeying the law.
SUMMARY
The new labeling and the proposed changes are sensible and draw needed attention to the problem of acetaminophen toxicity. To prevent unintentional acetaminophen overdoses, education of patients and health care professionals is urgently needed so that the dangers of consuming excess acetaminophen daily are understood.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
- Burke A, Smyth EM, Fitzgerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In:Brunton LL, Lazo JS, Parker K, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 11th ed. New York: McGraw-Hill, 2006:671–716.
- Bronstein AC, Spyker DA, Cantilena LR, Green J, Rumack BH, Heard SE. 2006 Annual Report of the American Association of Poison Control Centers National Poison Data System (NPDS). Clin Toxicol (Phila) 2007; 45:815–917.
- Schwartz J, Stravitz T, Lee WM; American Association for the Study of Liver Disease Study Group. AASLD position on acetaminophen. www.aasld.org/about/publicpolicy/Documents/Public%2520Policy%2520Documents/AcetaminophenPosition.pdf.
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Populationbased surveillance for acute liver failure. Am J Gastroenterol 2007; 102:2459–2463.
- Institute for Safe Medication Practices. How are you preventing acetaminophen overdoses? www.ismp.org/newsletters/acutecare/articles/20030808.asp. Accessed 11/17/2009.
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007; 11:525–548.
- Tylenol package insert. Fort Washington, PA: McNeil-PPC Inc.; 1999.
- Bizovi KE, Hendrickson RG. Chapter 34. Acetaminophen. In:Hoffman RS, Nelson LS, Howland MA, Lewin NA, Flomenbaum NE, Goldfrank LR, editors. Goldfrank’s Manual of Toxicologic Emergencies. 3rd ed. McGraw-Hill: New York, 2007. www.accessemergencymedicine.com/content.aspx?aID=88781. Accessed 11/7/2009.
- Hung OL, Nelson LS. Chapter 171. Acetaminophen. In:Tintinalli JE, Kelen GD, Stapcynski S, editors. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 6th ed. McGraw-Hill: New York, 2004. www.accessmedicine.com/content.aspx?aID=602606. Accessed 11/17/2009.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- American Academy of Pediatrics Committee on Drugs. Acetaminophen toxicity in children. Pediatrics 2001; 108:1020–1024.
- Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am 2008; 92:761–794.
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008; 359:285–292.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Acetadote package insert. Nashville, TN: Cumberland Pharmaceuticals; 2008Dec.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- Product Information: acetylcysteine inhalation solution, acetylcysteine inhalation solution. Hospira,Inc, Lake Forest, IL, 2004.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first 348 patients in the Pediatric Acute Liver Failure Study Group. J Pediatr 2006; 148:652–658.
- US Food and Drug Administration. Questions and answers on final rule for labeling changes to over-the-counter pain relievers. www.fda.gov/Drugs/NewsEvents/ucm144068.htm. Accessed 10/30/2009.
- Perrone M. FDA panel recommends smaller doses of painkillers. Associated Press. Adelphi, MD. June 30, 2009.
- Hawton K, Simkin S, Deeks J, et al. UK legislation on analgesic packs: before and after study of long term effect on poisonings. BMJ 2004; 329:1076.
- Hughes B, Durran A, Langford NJ, Mutimer D. Paracetamol poisoning—impact of pack size restrictions. J Clin Pharmacol Ther 2003; 28:307–310.
- Wilkinson S, Taylor G, Templeton L, Mistral W, Salter E, Bennett P. Admissions to hospital for deliberate self-harm in England 1995–2000: an analysis of hospital episode statistics. J Public Health Med 2002; 24:179–183.
- Greene SL, Dargan PI, Leman P, Jones AL. Paracetamol availability and recent changes in paracetamol poisoning: is the 1998 legislation limiting availability of paracetamol being followed? Postgrad Med J 2006; 82:520–523.
KEY POINTS
- Acetaminophen is the leading cause of acute liver failure in the United States, and nearly half of acetaminophenassociated cases are due to unintentional overdose.
- In many cases of unintentional overdose, patients took more than one acetaminophen-containing product and did not know that both products contained this drug.
- Prescribers need to inform all patients, especially vulnerable ones (eg, those taking enzyme-inducing drugs, those who chronically use alcohol, and those who are malnourished) of the risks associated with acetaminophen.
- Although no consensus has been reached on what is a safe dose in patients with liver disease, 4 g/day is too much: a total daily dose of no more than 2 g is recommended to decrease the risk of toxicity in these patients.
Evidence, limes, and cement

In 1753, Lind2 described how he gave fresh fruit vs cider, vinegar, sulfuric acid, seawater, or barley water to 12 sailors aboard the HMS Salisbury in an effort to find a cure for scurvy. This landmark clinical trial has been hailed as an example of how clinical research can dramatically alter clinical practice. Yet practice did not change aboard British naval ships until almost 50 years after Lind’s treatise was published.3
For many reasons, randomized clinical trials may not immediately affect what physicians do. Sometimes, physicians believe that the trials were not well designed or well conducted, or that the results do not apply to their patients. I briefly discussed some limitations of evidence-based clinical decision-making in our September 2009 issue.4
Another reason is that the conclusions from some trials do not jibe with the experience of seasoned clinicians. That is why, this month, I have asked two physicians, a rheumatologist5 and a spine surgeon,6 to comment on how two studies7,8 have influenced their clinical practice. Both studies concluded that vertebroplasty (injecting cement to shore up osteoporotic vertebrae) was no more beneficial than a sham procedure in patients with vertebral compression fractures. Neither physician is ready to completely abandon vertebroplasty on the basis of these two studies. Thus, it seems that published evidence may provide us guidance and fruit for discussion, but does not give us certainty.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t [editorial]. BMJ 1996; 312:71–72.
- Lind J. Treatise of the Scurvy in Three Parts. Containing an inquiry into the Nature, Causes and Cure of that Disease. Together with a Critical and Chronological View of what has been published on the subject. Edinburgh: Sands, Murray, and Cochran, 1753.
- Carpenter K. The History of Scurvy and Vitamin C. Cambridge, UK: Cambridge University Press, 1988.
- Mandell BF. Vertebroplasty, evidence, and health care reform: What is quality care? Cleve Clin J Med 2009; 76:497–502.
- Bolster MA. Consternation and questions about two vertebroplasty trials. Cleve Clin J Med 2010; 77:12–16.
- Orr RD. Vertebroplasty, cognitive dissonance, and evidence-based medicine: what do we do when the ‘evidence’ says we are wrong? Cleve Clin J Med 2010; 77:8–11.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
In 1753, Lind2 described how he gave fresh fruit vs cider, vinegar, sulfuric acid, seawater, or barley water to 12 sailors aboard the HMS Salisbury in an effort to find a cure for scurvy. This landmark clinical trial has been hailed as an example of how clinical research can dramatically alter clinical practice. Yet practice did not change aboard British naval ships until almost 50 years after Lind’s treatise was published.3
For many reasons, randomized clinical trials may not immediately affect what physicians do. Sometimes, physicians believe that the trials were not well designed or well conducted, or that the results do not apply to their patients. I briefly discussed some limitations of evidence-based clinical decision-making in our September 2009 issue.4
Another reason is that the conclusions from some trials do not jibe with the experience of seasoned clinicians. That is why, this month, I have asked two physicians, a rheumatologist5 and a spine surgeon,6 to comment on how two studies7,8 have influenced their clinical practice. Both studies concluded that vertebroplasty (injecting cement to shore up osteoporotic vertebrae) was no more beneficial than a sham procedure in patients with vertebral compression fractures. Neither physician is ready to completely abandon vertebroplasty on the basis of these two studies. Thus, it seems that published evidence may provide us guidance and fruit for discussion, but does not give us certainty.
In 1753, Lind2 described how he gave fresh fruit vs cider, vinegar, sulfuric acid, seawater, or barley water to 12 sailors aboard the HMS Salisbury in an effort to find a cure for scurvy. This landmark clinical trial has been hailed as an example of how clinical research can dramatically alter clinical practice. Yet practice did not change aboard British naval ships until almost 50 years after Lind’s treatise was published.3
For many reasons, randomized clinical trials may not immediately affect what physicians do. Sometimes, physicians believe that the trials were not well designed or well conducted, or that the results do not apply to their patients. I briefly discussed some limitations of evidence-based clinical decision-making in our September 2009 issue.4
Another reason is that the conclusions from some trials do not jibe with the experience of seasoned clinicians. That is why, this month, I have asked two physicians, a rheumatologist5 and a spine surgeon,6 to comment on how two studies7,8 have influenced their clinical practice. Both studies concluded that vertebroplasty (injecting cement to shore up osteoporotic vertebrae) was no more beneficial than a sham procedure in patients with vertebral compression fractures. Neither physician is ready to completely abandon vertebroplasty on the basis of these two studies. Thus, it seems that published evidence may provide us guidance and fruit for discussion, but does not give us certainty.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t [editorial]. BMJ 1996; 312:71–72.
- Lind J. Treatise of the Scurvy in Three Parts. Containing an inquiry into the Nature, Causes and Cure of that Disease. Together with a Critical and Chronological View of what has been published on the subject. Edinburgh: Sands, Murray, and Cochran, 1753.
- Carpenter K. The History of Scurvy and Vitamin C. Cambridge, UK: Cambridge University Press, 1988.
- Mandell BF. Vertebroplasty, evidence, and health care reform: What is quality care? Cleve Clin J Med 2009; 76:497–502.
- Bolster MA. Consternation and questions about two vertebroplasty trials. Cleve Clin J Med 2010; 77:12–16.
- Orr RD. Vertebroplasty, cognitive dissonance, and evidence-based medicine: what do we do when the ‘evidence’ says we are wrong? Cleve Clin J Med 2010; 77:8–11.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t [editorial]. BMJ 1996; 312:71–72.
- Lind J. Treatise of the Scurvy in Three Parts. Containing an inquiry into the Nature, Causes and Cure of that Disease. Together with a Critical and Chronological View of what has been published on the subject. Edinburgh: Sands, Murray, and Cochran, 1753.
- Carpenter K. The History of Scurvy and Vitamin C. Cambridge, UK: Cambridge University Press, 1988.
- Mandell BF. Vertebroplasty, evidence, and health care reform: What is quality care? Cleve Clin J Med 2009; 76:497–502.
- Bolster MA. Consternation and questions about two vertebroplasty trials. Cleve Clin J Med 2010; 77:12–16.
- Orr RD. Vertebroplasty, cognitive dissonance, and evidence-based medicine: what do we do when the ‘evidence’ says we are wrong? Cleve Clin J Med 2010; 77:8–11.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
Consternation and questions about two vertebroplasty trials
Confronted with the unexpected results of two trials of vertebroplasty,1,2 physicians are feeling some consternation, We had thought that percutaneous vertebroplasty helps patients with osteoporosis who sustain a painful vertebral insufficiency fracture. However, the trials found it to be no better than a sham procedure in terms of relieving pain.
How will these findings affect our practice? Should we abandon this popular procedure? Or are there other considerations that may mitigate these negative findings? And what should we tell our patients?
700,000 FRACTURES PER YEAR
Vertebral insufficiency fractures are the most common type of fracture in patients with osteoporosis. Every year in the United States, about 700,000 of them occur.
Nearly two-thirds are asymptomatic. The other one-third typically present with the acute onset of localized pain.
Vertebral insufficiency fractures often lead to chronic pain, impair the ability to walk and to perform daily activities, and accentuate thoracic kyphosis, which in turn can lead to pulmonary restrictive disease, and they raise the risk of death. Also, a patient who has a vertebral insufficiency fracture has a 20% risk of sustaining a new one within 1 year.3
Whether symptomatic or asymptomatic, finding a vertebral insufficiency fracture should prompt one to consider drug therapy for osteoporosis. In addition, until now, a patient who presented with the acute onset of back pain and whose evaluation revealed a vertebral insufficiency fracture would also be considered for a vertebral augmentation procedure, either vertebroplasty or kyphoplasty, to relieve the pain.
Vertebroplasty involves injecting polymethylmethacrylate cement percutaneously into the affected vertebral body. Kyphoplasty, a similar procedure, uses a balloon to create a cavity in the fractured vertebral body. After the balloon is withdrawn, the cavity is filled with cement.
TWO RANDOMIZED TRIALS OF SHAM VS REAL VERTEBROPLASTY
Two teams, Kallmes et al2 and Buchbinder et al,1 independently performed randomized controlled trials to see if vertebroplasty really relieves pain as well as has been reported in open studies, case series, and nonrandomized trials.4–7
In both trials, patients were randomized to undergo either sham vertebroplasty or real vertebroplasty. The sham procedure closely approximated the real procedure, including inserting a needle, infiltrating a local anesthetic, bupivacaine (Marcaine), into the periosteum of the posterior lamina1 or the pedicle of the target vertebrae,2 and opening a vial of polymethylmethacrylate so that the patient would smell the product.
Inclusion criteria
Patients in both trials had to have evidence of a recent (acute) or nonhealed vertebral insufficiency fracture.
Pain was the primary outcome measured
In both trials, the investigators assessed the patients’ pain at baseline and again at several specified intervals, using validated tools.
Kallmes et al assessed pain intensity and functional measures at 1 month (the primary outcome measured), and also at 3, 14, and 90 days and at 1 year.
Buchbinder et al assessed pain at 1 week and at 1, 3, and 6 months. The primary outcome measured was pain at 3 months. Secondary outcomes included quality-of-life measures, pain at rest, and pain at night.
Surprising results
In both trials, the mean pain scores were better than at baseline at all time points after the procedure in both the real-procedure and the sham-procedure groups. Moreover, the effect did not differ between the two treatment groups in either study.
QUESTIONS COMPLICATE THE ISSUE
These two trials should make us consider whether this intervention is warranted. We should, however, also consider some limitations of these studies that raise questions about how the conclusions should or should not alter practice.
Does local anesthetic continue to relieve pain?
In both the sham and the real procedure, the bupivacaine injection may have helped relieve pain to some extent afterward, as its anesthetic effect may last longer than we would expect from its 3-hour half-life. The effect could certainly have contributed to improvements in pain levels at the earlier time points after the procedure.
Was there selection bias?
Both studies were highly rigorous and were done at hospitals that had extensive experience with vertebroplasty. However, they may have harbored selection bias, as many more patients were screened than were randomized.
Buchbinder et al1 screened 468 patients. Of these, 30% declined to participate, and another 53% did not meet the eligibility criteria. In the end, only 78 patients were randomized.
Kallmes et al2 screened 1,813 patients, 300 of whom declined and 1,382 of whom were excluded, leaving 131 patients to be randomized. The reasons for exclusion were not specifically reported in many cases.
In both studies, it would be interesting to know how many of those who declined proceeded to undergo a vertebral augmentation procedure.
Did the trials have enough power?
In the study by Kallmes et al,2 recruitment got off to a slow start. Thus, after three patients were recruited, the inclusion requirements were liberalized. The study was originally designed to include 250 patients, which would have given it a power of greater than 80% to detect differences in primary and secondary outcomes. The design was revised to include 130 patients. The statistical power was still 80%, but this was to detect a greater difference in the outcomes than originally projected.
Had the window of opportunity already closed?
Vertebroplasty may have a window of opportunity within which it is most effective. Sooner is probably better than later, but it would be good to identify this time frame.
Kaufmann et al9 reported that patients with older fractures needed slightly more analgesic drugs after the procedure. It has been shown previously that patients who are the most likely to respond to a vertebral augmentation procedure are those with fractures that occurred between 1 and 12 months prior to the procedure and who have evidence that the fracture was recent, ie, edema on magnetic resonance imaging (MRI) or increased uptake on a bone scan.10
Other studies suggested that intervention works best in patients who have had uncontrolled pain lasting less than 6 weeks.8,11 (In the study by Buchbinder et al,1 only 32% of the patients in either group reported pain lasting less than 6 weeks.)
The study by Kallmes et al included patients whose pain had begun within 1 year previously. However, if the duration of pain (ie, the age of the fracture) was uncertain, MRI was done to look for edema, which would indicate the fracture was fresh. It is thus unclear whether all patients in this study truly had an acute or subacute fracture, since all did not undergo confirmatory MRI.
Why did so many patients cross over from sham to real treatment?
Patients in the Kallmes trial2 could cross over from one treatment group to the other as early as 1 month after the procedure. And, in fact, 43% of patients in the sham-treatment group did choose to cross over by 3 months. In contrast, after real vertebroplasty, significantly fewer—only 12% (P < .001)—crossed over to receive the sham procedure. The patients who crossed over from the sham-procedure group to receive vertebroplasty experienced an early improvement in pain, but this was not sustained at 1 or 3 months of follow-up.
The higher crossover rate in the shamprocedure group suggests they were dissatisfied with this intervention, although their outcomes were not significantly better after they got the real procedure. The patients who first received the sham treatment and elected to cross over to vertebroplasty had higher pain and disability scores at baseline. Thus, they may have had other, more chronic causes of pain or other factors affecting the likelihood of a response, particularly of a durable or sustained response.
How do the interventions compare with medical therapy?
Earlier studies showed that vertebroplasty relieves pain almost immediately.4–6 But the benefit does not last: at 6 weeks and up to 12 months later there is no difference in either pain or functional capacity reported in patients receiving vertebroplasty vs conservative treatment.4,6,7 It would thus appear that pain gradually diminishes over time after a vertebral insufficiency fracture, as the fracture heals.
The recent studies1,2 raise the possibility that the pain relief is due to the local anesthetic, not the vertebroplasty itself. We do not know, however, if either vertebroplasty or the sham procedure is superior to conservative medical management. Prospective multicenter trials are under way to address this question.11
Further complicating the issue, the two trials did not keep track of medical treatments patients were receiving concomitantly during the trial period. It is thus more difficult to compare the pain assessment outcomes following invasive procedures—real or sham.
Would kyphoplasty be better?
These studies addressed one procedure, vertebroplasty, and the results and conclusions should not be generalized to kyphoplasty. A prospective randomized trial of kyphoplasty is clearly warranted.
If kyphoplasty is found to be better than a sham procedure, then vertebroplasty should be re-examined in comparison with kyphoplasty. In any future studies, it will be important to select patients rigorously (eg, to include only patients with recent fractures), to match patients according to concomitant therapies, and to consider other potential superimposed causes of back pain in this elderly population, which has a high prevalence of back pain.
HOW SHOULD MY PRACTICE CHANGE? WHAT SHOULD I TELL PATIENTS?
Having considered the results, conclusions, and limitations of these two randomized trials, particularly in terms of recruitment, I cannot say that my practice has changed in terms of referring patients who have a vertebral compression fracture to an interventionalist. However, the education that I provide to patients has changed.
In my mind, the highest priority for a patient with a vertebral insufficiency fracture is to treat (or to reassess the current treatment of) the underlying systemic disease, ie, osteoporosis. This is especially true since most vertebral insufficiency fractures are asymptomatic.
On the other hand, a patient with a painful vertebral compression fracture needs prompt attention and consideration for interventional pain relief. Rapid pain relief is desirable. And in uncontrolled trials,4–7 vertebroplasty and kyphoplasty rapidly relieved vertebral pain. However, it may be that an anesthetic injection is equivalent to vertebroplasty and could accomplish the goal of immediate pain relief just as well.
The pain relief from sham or real vertebroplasty may not be durable, and 3 to 12 months later the pain benefit may be no greater than if more conservative therapy had been pursued.
It is essential to determine the most appropriate window for treatment as well as the most appropriate candidates on whom to perform a procedure. The recently published studies1,2 may have had significant patient selection bias and may not have optimized the window of opportunity for vertebral augmentation performance. There were many patients who declined the study, and some were excluded because of acute pain requiring hospitalization.
As a rheumatologist treating patients with osteoporosis, it is my responsibility to discuss with the patient and family the potential treatments available, to discuss the associated possible risks and benefits, to report on available evidence, and to refer patients to an appropriate interventional specialist if they desire. In light of the lack of superior pain reduction with vertebroplasty than with a sham procedure, many patients may opt for conservative therapy.
It is thus appropriate to determine the acuity of the fracture and to have a frank discussion with the patient about the options for pain management. Opiate drugs pose risks in elderly patients, particularly altered mentation, somnolence, interference with balance, and risk of falls. Vertebroplasty or anesthetic injection may rapidly relieve the pain and reduce the need for opiate therapy. Not yet subjected to the rigors of a randomized placebocontrolled trial, kyphoplasty may yet prove to be better than a sham intervention.
It is essential to determine if there is a role for vertebral augmentation in a select patient population—perhaps selected on the basis of the time that has elapsed since the fracture occurred (determined objectively), the severity of the fracture, and other factors. Perhaps a subset of patients would gain greater benefit from the procedure, whether it amounts solely to acute pain reduction or perhaps to a more durable response.
The recent studies by Kallmes et al2 and Buchbinder et al1 found vertebroplasty and sham vertebroplasty to be equally effective in reducing pain and improving function. However, given the limitations of each of these studies, particularly the low numbers of patients, it is difficult to establish that vertebral augmentation procedures should no longer be done. And vertebroplasty may still benefit correctly selected patients.
- Buchbinder R, Osborne RH, Ebeling PR, et a.l A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Francis RM, Aspray TJ, Hide G, Sutcliffe AM, Wilkinson P. Back pain in osteoporotic vertebral fractures. Osteoporos Int 2008; 19:895–903.
- Diamond TH, Champion B, Clark WA. Management of acute osteoporotic vertebral fractures: a nonrandomized trial comparing percutaneous vertebroplasty with conservative therapy. Am J Med 2003; 114:257–265.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term clinical outcome of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007; 28:555–560.
- Alvarez L, Alcaraz M, Pérez-Higueras A, et al. Percutaneous vertebroplasty: functional improvement in patients with osteoporotic compression fractures. Spine (Phila PA 1976) 2006; 31:1113–1118.
- Rousing R, Andersen MO, Jespersen SM, Thomsen K, Lauritsen J. Percutaneous vertebroplasty compared to conservative treatment in patients with painful acute or subacute osteoporotic vertebral fractures: three-months follow-up in a clinical randomized study. Spine (Phila PA 1976) 2009; 34:1349–1354.
- Clark W, Lyon S, Burnes J. Trials of vertebroplasty for vertebral fractures. N Engl J Med 2009; 361:2097–2098.
- Kaufmann TJ, Jensen ME, Schweickert PA, Marx WF, Kallmes DF. Age of fracture and clinical outcomes of percutaneous vertebroplasty. AJNR Am J Neuroradiol 2001; 22:1860–1863.
- Maynard AS, Jensen ME, Schweickert PA, Marx WF, Short JG, Kallmes DF. Value of bone scan imaging in predicting pain relief from percutaneous vertebroplasty in osteoporotic vertebral fractures. AJNR Am J Neuroradiol 2000; 21:1807–1812.
- Klazen C, Verhaar H, Lampmann L, et al. VERTOS II: Percutaneous vertebroplasty versus conservative therapy in patients with painful osteoporotic vertebral compression fractures; rationale, objectives and design of a multicenter randomized controlled trial. Trials 2007; 8:33.
Confronted with the unexpected results of two trials of vertebroplasty,1,2 physicians are feeling some consternation, We had thought that percutaneous vertebroplasty helps patients with osteoporosis who sustain a painful vertebral insufficiency fracture. However, the trials found it to be no better than a sham procedure in terms of relieving pain.
How will these findings affect our practice? Should we abandon this popular procedure? Or are there other considerations that may mitigate these negative findings? And what should we tell our patients?
700,000 FRACTURES PER YEAR
Vertebral insufficiency fractures are the most common type of fracture in patients with osteoporosis. Every year in the United States, about 700,000 of them occur.
Nearly two-thirds are asymptomatic. The other one-third typically present with the acute onset of localized pain.
Vertebral insufficiency fractures often lead to chronic pain, impair the ability to walk and to perform daily activities, and accentuate thoracic kyphosis, which in turn can lead to pulmonary restrictive disease, and they raise the risk of death. Also, a patient who has a vertebral insufficiency fracture has a 20% risk of sustaining a new one within 1 year.3
Whether symptomatic or asymptomatic, finding a vertebral insufficiency fracture should prompt one to consider drug therapy for osteoporosis. In addition, until now, a patient who presented with the acute onset of back pain and whose evaluation revealed a vertebral insufficiency fracture would also be considered for a vertebral augmentation procedure, either vertebroplasty or kyphoplasty, to relieve the pain.
Vertebroplasty involves injecting polymethylmethacrylate cement percutaneously into the affected vertebral body. Kyphoplasty, a similar procedure, uses a balloon to create a cavity in the fractured vertebral body. After the balloon is withdrawn, the cavity is filled with cement.
TWO RANDOMIZED TRIALS OF SHAM VS REAL VERTEBROPLASTY
Two teams, Kallmes et al2 and Buchbinder et al,1 independently performed randomized controlled trials to see if vertebroplasty really relieves pain as well as has been reported in open studies, case series, and nonrandomized trials.4–7
In both trials, patients were randomized to undergo either sham vertebroplasty or real vertebroplasty. The sham procedure closely approximated the real procedure, including inserting a needle, infiltrating a local anesthetic, bupivacaine (Marcaine), into the periosteum of the posterior lamina1 or the pedicle of the target vertebrae,2 and opening a vial of polymethylmethacrylate so that the patient would smell the product.
Inclusion criteria
Patients in both trials had to have evidence of a recent (acute) or nonhealed vertebral insufficiency fracture.
Pain was the primary outcome measured
In both trials, the investigators assessed the patients’ pain at baseline and again at several specified intervals, using validated tools.
Kallmes et al assessed pain intensity and functional measures at 1 month (the primary outcome measured), and also at 3, 14, and 90 days and at 1 year.
Buchbinder et al assessed pain at 1 week and at 1, 3, and 6 months. The primary outcome measured was pain at 3 months. Secondary outcomes included quality-of-life measures, pain at rest, and pain at night.
Surprising results
In both trials, the mean pain scores were better than at baseline at all time points after the procedure in both the real-procedure and the sham-procedure groups. Moreover, the effect did not differ between the two treatment groups in either study.
QUESTIONS COMPLICATE THE ISSUE
These two trials should make us consider whether this intervention is warranted. We should, however, also consider some limitations of these studies that raise questions about how the conclusions should or should not alter practice.
Does local anesthetic continue to relieve pain?
In both the sham and the real procedure, the bupivacaine injection may have helped relieve pain to some extent afterward, as its anesthetic effect may last longer than we would expect from its 3-hour half-life. The effect could certainly have contributed to improvements in pain levels at the earlier time points after the procedure.
Was there selection bias?
Both studies were highly rigorous and were done at hospitals that had extensive experience with vertebroplasty. However, they may have harbored selection bias, as many more patients were screened than were randomized.
Buchbinder et al1 screened 468 patients. Of these, 30% declined to participate, and another 53% did not meet the eligibility criteria. In the end, only 78 patients were randomized.
Kallmes et al2 screened 1,813 patients, 300 of whom declined and 1,382 of whom were excluded, leaving 131 patients to be randomized. The reasons for exclusion were not specifically reported in many cases.
In both studies, it would be interesting to know how many of those who declined proceeded to undergo a vertebral augmentation procedure.
Did the trials have enough power?
In the study by Kallmes et al,2 recruitment got off to a slow start. Thus, after three patients were recruited, the inclusion requirements were liberalized. The study was originally designed to include 250 patients, which would have given it a power of greater than 80% to detect differences in primary and secondary outcomes. The design was revised to include 130 patients. The statistical power was still 80%, but this was to detect a greater difference in the outcomes than originally projected.
Had the window of opportunity already closed?
Vertebroplasty may have a window of opportunity within which it is most effective. Sooner is probably better than later, but it would be good to identify this time frame.
Kaufmann et al9 reported that patients with older fractures needed slightly more analgesic drugs after the procedure. It has been shown previously that patients who are the most likely to respond to a vertebral augmentation procedure are those with fractures that occurred between 1 and 12 months prior to the procedure and who have evidence that the fracture was recent, ie, edema on magnetic resonance imaging (MRI) or increased uptake on a bone scan.10
Other studies suggested that intervention works best in patients who have had uncontrolled pain lasting less than 6 weeks.8,11 (In the study by Buchbinder et al,1 only 32% of the patients in either group reported pain lasting less than 6 weeks.)
The study by Kallmes et al included patients whose pain had begun within 1 year previously. However, if the duration of pain (ie, the age of the fracture) was uncertain, MRI was done to look for edema, which would indicate the fracture was fresh. It is thus unclear whether all patients in this study truly had an acute or subacute fracture, since all did not undergo confirmatory MRI.
Why did so many patients cross over from sham to real treatment?
Patients in the Kallmes trial2 could cross over from one treatment group to the other as early as 1 month after the procedure. And, in fact, 43% of patients in the sham-treatment group did choose to cross over by 3 months. In contrast, after real vertebroplasty, significantly fewer—only 12% (P < .001)—crossed over to receive the sham procedure. The patients who crossed over from the sham-procedure group to receive vertebroplasty experienced an early improvement in pain, but this was not sustained at 1 or 3 months of follow-up.
The higher crossover rate in the shamprocedure group suggests they were dissatisfied with this intervention, although their outcomes were not significantly better after they got the real procedure. The patients who first received the sham treatment and elected to cross over to vertebroplasty had higher pain and disability scores at baseline. Thus, they may have had other, more chronic causes of pain or other factors affecting the likelihood of a response, particularly of a durable or sustained response.
How do the interventions compare with medical therapy?
Earlier studies showed that vertebroplasty relieves pain almost immediately.4–6 But the benefit does not last: at 6 weeks and up to 12 months later there is no difference in either pain or functional capacity reported in patients receiving vertebroplasty vs conservative treatment.4,6,7 It would thus appear that pain gradually diminishes over time after a vertebral insufficiency fracture, as the fracture heals.
The recent studies1,2 raise the possibility that the pain relief is due to the local anesthetic, not the vertebroplasty itself. We do not know, however, if either vertebroplasty or the sham procedure is superior to conservative medical management. Prospective multicenter trials are under way to address this question.11
Further complicating the issue, the two trials did not keep track of medical treatments patients were receiving concomitantly during the trial period. It is thus more difficult to compare the pain assessment outcomes following invasive procedures—real or sham.
Would kyphoplasty be better?
These studies addressed one procedure, vertebroplasty, and the results and conclusions should not be generalized to kyphoplasty. A prospective randomized trial of kyphoplasty is clearly warranted.
If kyphoplasty is found to be better than a sham procedure, then vertebroplasty should be re-examined in comparison with kyphoplasty. In any future studies, it will be important to select patients rigorously (eg, to include only patients with recent fractures), to match patients according to concomitant therapies, and to consider other potential superimposed causes of back pain in this elderly population, which has a high prevalence of back pain.
HOW SHOULD MY PRACTICE CHANGE? WHAT SHOULD I TELL PATIENTS?
Having considered the results, conclusions, and limitations of these two randomized trials, particularly in terms of recruitment, I cannot say that my practice has changed in terms of referring patients who have a vertebral compression fracture to an interventionalist. However, the education that I provide to patients has changed.
In my mind, the highest priority for a patient with a vertebral insufficiency fracture is to treat (or to reassess the current treatment of) the underlying systemic disease, ie, osteoporosis. This is especially true since most vertebral insufficiency fractures are asymptomatic.
On the other hand, a patient with a painful vertebral compression fracture needs prompt attention and consideration for interventional pain relief. Rapid pain relief is desirable. And in uncontrolled trials,4–7 vertebroplasty and kyphoplasty rapidly relieved vertebral pain. However, it may be that an anesthetic injection is equivalent to vertebroplasty and could accomplish the goal of immediate pain relief just as well.
The pain relief from sham or real vertebroplasty may not be durable, and 3 to 12 months later the pain benefit may be no greater than if more conservative therapy had been pursued.
It is essential to determine the most appropriate window for treatment as well as the most appropriate candidates on whom to perform a procedure. The recently published studies1,2 may have had significant patient selection bias and may not have optimized the window of opportunity for vertebral augmentation performance. There were many patients who declined the study, and some were excluded because of acute pain requiring hospitalization.
As a rheumatologist treating patients with osteoporosis, it is my responsibility to discuss with the patient and family the potential treatments available, to discuss the associated possible risks and benefits, to report on available evidence, and to refer patients to an appropriate interventional specialist if they desire. In light of the lack of superior pain reduction with vertebroplasty than with a sham procedure, many patients may opt for conservative therapy.
It is thus appropriate to determine the acuity of the fracture and to have a frank discussion with the patient about the options for pain management. Opiate drugs pose risks in elderly patients, particularly altered mentation, somnolence, interference with balance, and risk of falls. Vertebroplasty or anesthetic injection may rapidly relieve the pain and reduce the need for opiate therapy. Not yet subjected to the rigors of a randomized placebocontrolled trial, kyphoplasty may yet prove to be better than a sham intervention.
It is essential to determine if there is a role for vertebral augmentation in a select patient population—perhaps selected on the basis of the time that has elapsed since the fracture occurred (determined objectively), the severity of the fracture, and other factors. Perhaps a subset of patients would gain greater benefit from the procedure, whether it amounts solely to acute pain reduction or perhaps to a more durable response.
The recent studies by Kallmes et al2 and Buchbinder et al1 found vertebroplasty and sham vertebroplasty to be equally effective in reducing pain and improving function. However, given the limitations of each of these studies, particularly the low numbers of patients, it is difficult to establish that vertebral augmentation procedures should no longer be done. And vertebroplasty may still benefit correctly selected patients.
Confronted with the unexpected results of two trials of vertebroplasty,1,2 physicians are feeling some consternation, We had thought that percutaneous vertebroplasty helps patients with osteoporosis who sustain a painful vertebral insufficiency fracture. However, the trials found it to be no better than a sham procedure in terms of relieving pain.
How will these findings affect our practice? Should we abandon this popular procedure? Or are there other considerations that may mitigate these negative findings? And what should we tell our patients?
700,000 FRACTURES PER YEAR
Vertebral insufficiency fractures are the most common type of fracture in patients with osteoporosis. Every year in the United States, about 700,000 of them occur.
Nearly two-thirds are asymptomatic. The other one-third typically present with the acute onset of localized pain.
Vertebral insufficiency fractures often lead to chronic pain, impair the ability to walk and to perform daily activities, and accentuate thoracic kyphosis, which in turn can lead to pulmonary restrictive disease, and they raise the risk of death. Also, a patient who has a vertebral insufficiency fracture has a 20% risk of sustaining a new one within 1 year.3
Whether symptomatic or asymptomatic, finding a vertebral insufficiency fracture should prompt one to consider drug therapy for osteoporosis. In addition, until now, a patient who presented with the acute onset of back pain and whose evaluation revealed a vertebral insufficiency fracture would also be considered for a vertebral augmentation procedure, either vertebroplasty or kyphoplasty, to relieve the pain.
Vertebroplasty involves injecting polymethylmethacrylate cement percutaneously into the affected vertebral body. Kyphoplasty, a similar procedure, uses a balloon to create a cavity in the fractured vertebral body. After the balloon is withdrawn, the cavity is filled with cement.
TWO RANDOMIZED TRIALS OF SHAM VS REAL VERTEBROPLASTY
Two teams, Kallmes et al2 and Buchbinder et al,1 independently performed randomized controlled trials to see if vertebroplasty really relieves pain as well as has been reported in open studies, case series, and nonrandomized trials.4–7
In both trials, patients were randomized to undergo either sham vertebroplasty or real vertebroplasty. The sham procedure closely approximated the real procedure, including inserting a needle, infiltrating a local anesthetic, bupivacaine (Marcaine), into the periosteum of the posterior lamina1 or the pedicle of the target vertebrae,2 and opening a vial of polymethylmethacrylate so that the patient would smell the product.
Inclusion criteria
Patients in both trials had to have evidence of a recent (acute) or nonhealed vertebral insufficiency fracture.
Pain was the primary outcome measured
In both trials, the investigators assessed the patients’ pain at baseline and again at several specified intervals, using validated tools.
Kallmes et al assessed pain intensity and functional measures at 1 month (the primary outcome measured), and also at 3, 14, and 90 days and at 1 year.
Buchbinder et al assessed pain at 1 week and at 1, 3, and 6 months. The primary outcome measured was pain at 3 months. Secondary outcomes included quality-of-life measures, pain at rest, and pain at night.
Surprising results
In both trials, the mean pain scores were better than at baseline at all time points after the procedure in both the real-procedure and the sham-procedure groups. Moreover, the effect did not differ between the two treatment groups in either study.
QUESTIONS COMPLICATE THE ISSUE
These two trials should make us consider whether this intervention is warranted. We should, however, also consider some limitations of these studies that raise questions about how the conclusions should or should not alter practice.
Does local anesthetic continue to relieve pain?
In both the sham and the real procedure, the bupivacaine injection may have helped relieve pain to some extent afterward, as its anesthetic effect may last longer than we would expect from its 3-hour half-life. The effect could certainly have contributed to improvements in pain levels at the earlier time points after the procedure.
Was there selection bias?
Both studies were highly rigorous and were done at hospitals that had extensive experience with vertebroplasty. However, they may have harbored selection bias, as many more patients were screened than were randomized.
Buchbinder et al1 screened 468 patients. Of these, 30% declined to participate, and another 53% did not meet the eligibility criteria. In the end, only 78 patients were randomized.
Kallmes et al2 screened 1,813 patients, 300 of whom declined and 1,382 of whom were excluded, leaving 131 patients to be randomized. The reasons for exclusion were not specifically reported in many cases.
In both studies, it would be interesting to know how many of those who declined proceeded to undergo a vertebral augmentation procedure.
Did the trials have enough power?
In the study by Kallmes et al,2 recruitment got off to a slow start. Thus, after three patients were recruited, the inclusion requirements were liberalized. The study was originally designed to include 250 patients, which would have given it a power of greater than 80% to detect differences in primary and secondary outcomes. The design was revised to include 130 patients. The statistical power was still 80%, but this was to detect a greater difference in the outcomes than originally projected.
Had the window of opportunity already closed?
Vertebroplasty may have a window of opportunity within which it is most effective. Sooner is probably better than later, but it would be good to identify this time frame.
Kaufmann et al9 reported that patients with older fractures needed slightly more analgesic drugs after the procedure. It has been shown previously that patients who are the most likely to respond to a vertebral augmentation procedure are those with fractures that occurred between 1 and 12 months prior to the procedure and who have evidence that the fracture was recent, ie, edema on magnetic resonance imaging (MRI) or increased uptake on a bone scan.10
Other studies suggested that intervention works best in patients who have had uncontrolled pain lasting less than 6 weeks.8,11 (In the study by Buchbinder et al,1 only 32% of the patients in either group reported pain lasting less than 6 weeks.)
The study by Kallmes et al included patients whose pain had begun within 1 year previously. However, if the duration of pain (ie, the age of the fracture) was uncertain, MRI was done to look for edema, which would indicate the fracture was fresh. It is thus unclear whether all patients in this study truly had an acute or subacute fracture, since all did not undergo confirmatory MRI.
Why did so many patients cross over from sham to real treatment?
Patients in the Kallmes trial2 could cross over from one treatment group to the other as early as 1 month after the procedure. And, in fact, 43% of patients in the sham-treatment group did choose to cross over by 3 months. In contrast, after real vertebroplasty, significantly fewer—only 12% (P < .001)—crossed over to receive the sham procedure. The patients who crossed over from the sham-procedure group to receive vertebroplasty experienced an early improvement in pain, but this was not sustained at 1 or 3 months of follow-up.
The higher crossover rate in the shamprocedure group suggests they were dissatisfied with this intervention, although their outcomes were not significantly better after they got the real procedure. The patients who first received the sham treatment and elected to cross over to vertebroplasty had higher pain and disability scores at baseline. Thus, they may have had other, more chronic causes of pain or other factors affecting the likelihood of a response, particularly of a durable or sustained response.
How do the interventions compare with medical therapy?
Earlier studies showed that vertebroplasty relieves pain almost immediately.4–6 But the benefit does not last: at 6 weeks and up to 12 months later there is no difference in either pain or functional capacity reported in patients receiving vertebroplasty vs conservative treatment.4,6,7 It would thus appear that pain gradually diminishes over time after a vertebral insufficiency fracture, as the fracture heals.
The recent studies1,2 raise the possibility that the pain relief is due to the local anesthetic, not the vertebroplasty itself. We do not know, however, if either vertebroplasty or the sham procedure is superior to conservative medical management. Prospective multicenter trials are under way to address this question.11
Further complicating the issue, the two trials did not keep track of medical treatments patients were receiving concomitantly during the trial period. It is thus more difficult to compare the pain assessment outcomes following invasive procedures—real or sham.
Would kyphoplasty be better?
These studies addressed one procedure, vertebroplasty, and the results and conclusions should not be generalized to kyphoplasty. A prospective randomized trial of kyphoplasty is clearly warranted.
If kyphoplasty is found to be better than a sham procedure, then vertebroplasty should be re-examined in comparison with kyphoplasty. In any future studies, it will be important to select patients rigorously (eg, to include only patients with recent fractures), to match patients according to concomitant therapies, and to consider other potential superimposed causes of back pain in this elderly population, which has a high prevalence of back pain.
HOW SHOULD MY PRACTICE CHANGE? WHAT SHOULD I TELL PATIENTS?
Having considered the results, conclusions, and limitations of these two randomized trials, particularly in terms of recruitment, I cannot say that my practice has changed in terms of referring patients who have a vertebral compression fracture to an interventionalist. However, the education that I provide to patients has changed.
In my mind, the highest priority for a patient with a vertebral insufficiency fracture is to treat (or to reassess the current treatment of) the underlying systemic disease, ie, osteoporosis. This is especially true since most vertebral insufficiency fractures are asymptomatic.
On the other hand, a patient with a painful vertebral compression fracture needs prompt attention and consideration for interventional pain relief. Rapid pain relief is desirable. And in uncontrolled trials,4–7 vertebroplasty and kyphoplasty rapidly relieved vertebral pain. However, it may be that an anesthetic injection is equivalent to vertebroplasty and could accomplish the goal of immediate pain relief just as well.
The pain relief from sham or real vertebroplasty may not be durable, and 3 to 12 months later the pain benefit may be no greater than if more conservative therapy had been pursued.
It is essential to determine the most appropriate window for treatment as well as the most appropriate candidates on whom to perform a procedure. The recently published studies1,2 may have had significant patient selection bias and may not have optimized the window of opportunity for vertebral augmentation performance. There were many patients who declined the study, and some were excluded because of acute pain requiring hospitalization.
As a rheumatologist treating patients with osteoporosis, it is my responsibility to discuss with the patient and family the potential treatments available, to discuss the associated possible risks and benefits, to report on available evidence, and to refer patients to an appropriate interventional specialist if they desire. In light of the lack of superior pain reduction with vertebroplasty than with a sham procedure, many patients may opt for conservative therapy.
It is thus appropriate to determine the acuity of the fracture and to have a frank discussion with the patient about the options for pain management. Opiate drugs pose risks in elderly patients, particularly altered mentation, somnolence, interference with balance, and risk of falls. Vertebroplasty or anesthetic injection may rapidly relieve the pain and reduce the need for opiate therapy. Not yet subjected to the rigors of a randomized placebocontrolled trial, kyphoplasty may yet prove to be better than a sham intervention.
It is essential to determine if there is a role for vertebral augmentation in a select patient population—perhaps selected on the basis of the time that has elapsed since the fracture occurred (determined objectively), the severity of the fracture, and other factors. Perhaps a subset of patients would gain greater benefit from the procedure, whether it amounts solely to acute pain reduction or perhaps to a more durable response.
The recent studies by Kallmes et al2 and Buchbinder et al1 found vertebroplasty and sham vertebroplasty to be equally effective in reducing pain and improving function. However, given the limitations of each of these studies, particularly the low numbers of patients, it is difficult to establish that vertebral augmentation procedures should no longer be done. And vertebroplasty may still benefit correctly selected patients.
- Buchbinder R, Osborne RH, Ebeling PR, et a.l A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Francis RM, Aspray TJ, Hide G, Sutcliffe AM, Wilkinson P. Back pain in osteoporotic vertebral fractures. Osteoporos Int 2008; 19:895–903.
- Diamond TH, Champion B, Clark WA. Management of acute osteoporotic vertebral fractures: a nonrandomized trial comparing percutaneous vertebroplasty with conservative therapy. Am J Med 2003; 114:257–265.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term clinical outcome of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007; 28:555–560.
- Alvarez L, Alcaraz M, Pérez-Higueras A, et al. Percutaneous vertebroplasty: functional improvement in patients with osteoporotic compression fractures. Spine (Phila PA 1976) 2006; 31:1113–1118.
- Rousing R, Andersen MO, Jespersen SM, Thomsen K, Lauritsen J. Percutaneous vertebroplasty compared to conservative treatment in patients with painful acute or subacute osteoporotic vertebral fractures: three-months follow-up in a clinical randomized study. Spine (Phila PA 1976) 2009; 34:1349–1354.
- Clark W, Lyon S, Burnes J. Trials of vertebroplasty for vertebral fractures. N Engl J Med 2009; 361:2097–2098.
- Kaufmann TJ, Jensen ME, Schweickert PA, Marx WF, Kallmes DF. Age of fracture and clinical outcomes of percutaneous vertebroplasty. AJNR Am J Neuroradiol 2001; 22:1860–1863.
- Maynard AS, Jensen ME, Schweickert PA, Marx WF, Short JG, Kallmes DF. Value of bone scan imaging in predicting pain relief from percutaneous vertebroplasty in osteoporotic vertebral fractures. AJNR Am J Neuroradiol 2000; 21:1807–1812.
- Klazen C, Verhaar H, Lampmann L, et al. VERTOS II: Percutaneous vertebroplasty versus conservative therapy in patients with painful osteoporotic vertebral compression fractures; rationale, objectives and design of a multicenter randomized controlled trial. Trials 2007; 8:33.
- Buchbinder R, Osborne RH, Ebeling PR, et a.l A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Francis RM, Aspray TJ, Hide G, Sutcliffe AM, Wilkinson P. Back pain in osteoporotic vertebral fractures. Osteoporos Int 2008; 19:895–903.
- Diamond TH, Champion B, Clark WA. Management of acute osteoporotic vertebral fractures: a nonrandomized trial comparing percutaneous vertebroplasty with conservative therapy. Am J Med 2003; 114:257–265.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term clinical outcome of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007; 28:555–560.
- Alvarez L, Alcaraz M, Pérez-Higueras A, et al. Percutaneous vertebroplasty: functional improvement in patients with osteoporotic compression fractures. Spine (Phila PA 1976) 2006; 31:1113–1118.
- Rousing R, Andersen MO, Jespersen SM, Thomsen K, Lauritsen J. Percutaneous vertebroplasty compared to conservative treatment in patients with painful acute or subacute osteoporotic vertebral fractures: three-months follow-up in a clinical randomized study. Spine (Phila PA 1976) 2009; 34:1349–1354.
- Clark W, Lyon S, Burnes J. Trials of vertebroplasty for vertebral fractures. N Engl J Med 2009; 361:2097–2098.
- Kaufmann TJ, Jensen ME, Schweickert PA, Marx WF, Kallmes DF. Age of fracture and clinical outcomes of percutaneous vertebroplasty. AJNR Am J Neuroradiol 2001; 22:1860–1863.
- Maynard AS, Jensen ME, Schweickert PA, Marx WF, Short JG, Kallmes DF. Value of bone scan imaging in predicting pain relief from percutaneous vertebroplasty in osteoporotic vertebral fractures. AJNR Am J Neuroradiol 2000; 21:1807–1812.
- Klazen C, Verhaar H, Lampmann L, et al. VERTOS II: Percutaneous vertebroplasty versus conservative therapy in patients with painful osteoporotic vertebral compression fractures; rationale, objectives and design of a multicenter randomized controlled trial. Trials 2007; 8:33.
Vertebroplasty, cognitive dissonance, and evidence-based medicine: What do we do when the ‘evidence’ says we are wrong?
Cognitive dissonance describes how we respond to conflicting information that challenges our existing belief, the uncomfortable feeling we get when new evidence calls into question things that we “know” are true.
To the point: two recent clinical trials1,2 have called into question the efficacy of vertebroplasty for treating osteoporotic vertebral compression fractures and have led many of us to question many of our assumptions, not only about vertebroplasty but also about evidencebased medicine.
Osteoporotic vertebral compression fractures are very common: more than 700,000 are estimated to occur in the United States annually. 3 They are costly and are associated with a risk of death.4 Fortunately, most heal without problems over 4 to 6 weeks with conventional treatment, ie, activity modification, analgesics, and bracing.
However, some patients do not seem to do so well and are debilitated by the pain of the fracture. Conventional fracture surgery carries very high risk and poor outcomes,5 and so has been reserved mostly for patients with neurologic deficits.
VERTEBROPLASTY GOES MAINSTREAM
Given these facts, investigators began looking for alternative treatments. One that rose to the fore was polymethylmethacrylate cement to stabilize the fracture. This technique, called vertebroplasty, involves injecting liquid cement through a needle into the vertebral body, where it hardens and is thought to restore stability.
Since the first description of vertebroplasty for treating symptomatic hemangiomas,6 many papers have been published about the procedure and about similar ones, now grouped under the general heading of vertebral augmentation. This includes kyphoplasty and other newer proprietary techniques. These procedures have been widely accepted, and their use is growing. They have shown good results in several prospective case series, and nonrandomized and randomized controlled studies have shown them to be more effective than conventional medical treatment.7–25 For example, VERTOS, a small prospective randomized trial, showed that vertebroplasty was superior to conventional medical treatment.25 When Wardlaw et al24 showed that shortterm outcomes were better with kyphoplasty than with conventional medical therapy in a prospective randomized trial, many of us had moved past questioning whether vertebral augmentation is effective and were debating the relative merits of different methods and materials.
On a personal level, most of us became proponents of these procedures because we saw dramatic results—usually unequivocal. Most patients report significant improvement in pain immediately after the procedure, and many bedridden patients are able to leave the hospital within hours. In spine surgery, few procedures give such dramatic results with so few complications.
TWO NEW STUDIES UPSET ESTABLISHED BELIEF
This is why I am having such a hard time digesting the results of the trials by Buchbinder et al2 and Kallmes et al,1 published in the August 9, 2009, issue of the New England Journal of Medicine. Both were randomized controlled trials that used sham surgery rather than conventional medical treatment as the control. The sham procedure in each trial was the same as the intervention, with local anesthetic infiltration of the periosteum and mixing of the cement (so that the patients smelled its distinctive odor), but without placing the needle into the vertebra and injecting the cement.
In the study by Buchbinder et al,2 the real treatment had no benefit in any primary or secondary end point. This study did not allow crossovers.
In the study by Kallmes et al,1 more patients who received the real treatment reported clinically meaningful improvement in pain (a secondary end point), but the difference was not quite statistically significant (64% vs 48%, P = .06). In this trial, patients were allowed to cross over to the other study group after 1 month, and significantly more patients crossed over from the sham surgery group to the active treatment group than the other way around (43% vs 12%, P < .001).
My first instinct was to pick through the papers for flaws that would invalidate the results— and there were some problems. Both studies were initially planned to include more patients and therefore to have greater statistical power, but they were reassessed because of slow enrollment. In the study by Kallmes et al,1 the difference in clinically meaningful improvement might have reached statistical significance if the trial had been larger. The study by Buchbinder et al2 was a multicenter trial, but one center accounted for 53 (69%) of the 78 patients. Could this have biased the results?
The surgeon in me also seized for a while on the idea that since all of the interventions in both studies were done by interventional radiologists, the problem may have been in patient selection and that radiologists are not as astute as we are. However, even a surgeon’s ego cannot support this interpretation.
As I looked in more detail at the response I had written to these trials, I realized these criticisms were hardly fatal flaws, and the fact that two separate well-designed studies reached the same conclusion enhances their validity.
One concern that does bear some scrutiny is that the trials were too small to identify subgroups that may benefit from the procedure. In my experience, vertebral augmentation seems to have better results with certain types of fractures. Patients with a mobile pseudarthrotic cleft pattern of fracture seem to do much better than those with the more common nonmobile fracture.
THE POWERFUL PLACEBO EFFECT
Many commentaries on these two trials have discussed a famous study of a different procedure for a different condition. In this study, Moseley et al26 evaluated the use of arthroscopy to treat osteoarthritis of the knee and found that sham arthroscopy was as effective as real arthroscopy and that both were better than conventional treatment.
I was not long out of my orthopedic residency when this trial was published and was very aware of the debate that preceded it, as I once had to prepare a talk about it for resident rounds. I remember that there was a lively debate in the orthopedic community over the efficacy of the procedure before the results of this trial were released.
In contrast, the vertebral augmentation controversy had become a debate about the relative efficacy and the economics of specific techniques, not about the effectiveness of the entire concept. The mainstream had accepted the validity of the procedure, which was not the case in the knee arthroscopy trial.
In both vertebroplasty studies, the activetreatment groups and the sham-treatment groups all showed significant and rapid improvement in pain and disability, and these results were maintained over the study period. Though most vertebral compression fractures do heal, the clinical improvement is usually gradual over a period of weeks. This raises the possibility that the sham treatment was actualy an active placebo.
There is some evidence to support this possibility. In a randomized trial of the efficacy of selective nerve root blocks for lumbar radiculopathy, Riew et al27 showed that injection with a local anesthetic alone, although not as efficacious as a local anesthetic plus a corticosteroid at allowing patients to avoid surgery, showed an effect long after the expected duration of the anesthetic. The effect persisted even at 5 years of follow-up.28
Is it possible that the local anesthetic in this trial and the vertebroplasty trials acted as some sort of “reset button” for pain sensation? This is an area that may bear further investigation.
WHERE DOES THIS LEAVE US?
So where does this leave us? On one hand, randomized controlled trials comparing vertebral augmentation with conventional medical therapy24,25 showed augmentation to be beneficial. On the other hand, the studies by Kallmes et al1 and Buchbinder et al2 indicate vertebroplasty is no more effective than sham surgery.
It is very difficult for me to look at my own experience with vertebral augmentation and say that, on the basis of these trials, I am no longer going to offer it to my patients. I understand on an intellectual level that these trials call the efficacy of the procedure into question, but on a visceral level I cannot rationalize it. When faced with a patient who is barely ambulatory or in fact bed-bound due to pain, my experience tells me that vertebral augmentation has a very high chance of getting them ambulatory within hours. The trials of vertebroplasty would indicate this is a placebo effect or that local anesthetic alone is as effective, but I am not yet ready to make that leap.
Cognitive dissonance seems to rule.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Carmona RH. Department of Health and Human Services. Office of the Surgeon General. Bone health and osteoporosis: A report of the Surgeon General (2004). www.surgeongeneral.gov/library/bonehealth/content.html. Accessed November 16, 2009.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Hu SS. Internal fixation in the osteoporotic spine. Spine (Phila PA 1976) 1997; 22( suppl 24):43S–48S.
- Galibert P, Deramond H, Rosat P, Le Gars D. Preliminary note on the treatment of vertebral angioma by percutaneous acrylic vertebroplasty. Neurochirurgie 1987; 33:166–168.
- Kasperk C, Hillmeier J, Noldge G, et al. Treatment of painful vertebral fractures by kyphoplasty in patients with primary osteoporosis: a prospective nonrandomized controlled study. J Bone Miner Res 2005; 20:604–612.
- Komp M, Ruetten S, Godolias G. Minimally invasive therapy for functionally unstable osteoporotic vertebral fracture by means of kyphoplasty: prospective comparative study of 19 surgically and 17 conservatively treated patients. J Miner Stoffwechs 2004; 11( suppl 1):13–15.
- Coumans JV, Reinhardt MK, Lieberman IH. Kyphoplasty for vertebral compression fractures: 1-year clinical outcomes from a prospective study. J Neurosurg 2003; 99 (suppl 1):44–50.
- Lieberman IH, Dudeney S, Reinhardt MK, Bell G. Initial outcome and efficacy of ‘kyphoplasty’ in the treatment of painful osteoporotic vertebral compression fractures. Spine (Phila PA 1976) 2001; 26:1631–1638.
- Garfin SR, Buckley RA, Ledlie J; Balloon Kyphoplasty Outcomes Group. Balloon kyphoplasty for symptomatic vertebral body compression fractures results in rapid, significant, and sustained improvements in back pain, function, and quality of life for elderly patients. Spine (Phila PA 1976) 2006; 31:2213–2220.
- Ledlie JT, Renfro MB. Kyphoplasty treatment of vertebral fractures: 2-year outcomes show sustained benefits. Spine (Phila PA 1976) 2006; 31:57–64.
- Majd ME, Farley S, Holt RT. Preliminary outcomes and efficacy of the first 360 consecutive kyphoplasties for the treatment of painful osteoporotic vertebral compression fractures. Spine J 2005; 5:244–255.
- Rhyne A, Banit D, Laxer E, Odum S, Nussman D. Kyphoplasty: report of eighty-two thoracolumbar osteoporotic vertebral fractures. J Orthop Trauma 2004; 18:294–299.
- Theodorou DJ, Theodorou SJ, Duncan TD, Garfin SR, Wong WH. Percutaneous balloon kyphoplasty for the correction of spinal deformity in painful vertebral body compression fractures. Clin Imaging 2002; 26:1–5.
- Berlemann U, Franz T, Orler R, Heini PF. Kyphoplasty for treatment of osteoporotic vertebral fractures: a prospective nonrandomized study. Eur Spine J 2004; 13:496–501.
- McGraw JK, Lippert JA, Minkus KD, Rami PM, Davis TM, Budzik RF. Prospective evaluation of pain relief in 100 patients undergoing percutaneous vertebroplasty: results and followup. J Vasc Interv Radiol 2002; 13:883–886.
- Zoarski GH, Snow P, Olan WJ, et al. Percutaneous vertebroplasty for osteoporotic compression fractures: quantitative prospective evaluation of long-term outcomes. J Vasc Interv Radiol 2002; 13:139–148.
- Evans AJ, Jensen ME, Kip KE, et al. Vertebral compression fractures: pain reduction and improvement in functional mobility after percutaneous polymethylmethacrylate vertebroplasty retrospective report of 245 cases. Radiology 2003; 226:366–372.
- Grohs JG, Matzner M, Trieb K, Krepler P. Minimal invasive stabilization of osteoporotic vertebral fractures: a prospective nonrandomized comparison of vertebroplasty and balloon kyphoplasty. J Spinal Disord Tech 2005; 18:238–242.
- Kallmes DF, Schweickert PA, Marx WF, Jensen ME. Vertebroplasty in the mid-and upper thoracic spine. AJNR Am J Neuroradiol 2002; 23:1117–1120.
- Grados F, Depriester C, Cayrolle G, Hardy N, Deramond H, Fardellone P. Long-term observations of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Rheumatology (Oxford) 2000; 39:1410–1414.
- Legroux-Gérot I, Lormeau C, Boutry N, Cotten A, Duquesnoy B, Cortet B. Long-term follow-up of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Clin Rheumatol 2004; 23:310–317.
- Wardlaw D, Cummings SR, Van Meirhaeghe J, et al. Efficacy and safety of balloon kyphoplasty compared with nonsurgical care for vertebral compression fracture (FREE): a randomised controlled trial. Lancet 2009; 373:1016–1024.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term outcomes of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007: 28:555–560.
- Moseley JB, O'Malley K, Petersen NJ. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med 2002; 347:81–88.
- Riew KD, Yin Y, Gilula L, et al. The effect of nerve-root injections on the need for operative treatment of lumbar radicular pain. A prospective, randomized, controlled, double-blind study. J Bone Joint Surg Am 2000; 82–A:1589–1593.
- Riew KD, Park JB, Cho YS, et al. Nerve root blocks in the treatment of lumbar radicular pain. A minimum five-year follow-up. J Bone Joint Surg Am 2006; 88:1722–1725.
Cognitive dissonance describes how we respond to conflicting information that challenges our existing belief, the uncomfortable feeling we get when new evidence calls into question things that we “know” are true.
To the point: two recent clinical trials1,2 have called into question the efficacy of vertebroplasty for treating osteoporotic vertebral compression fractures and have led many of us to question many of our assumptions, not only about vertebroplasty but also about evidencebased medicine.
Osteoporotic vertebral compression fractures are very common: more than 700,000 are estimated to occur in the United States annually. 3 They are costly and are associated with a risk of death.4 Fortunately, most heal without problems over 4 to 6 weeks with conventional treatment, ie, activity modification, analgesics, and bracing.
However, some patients do not seem to do so well and are debilitated by the pain of the fracture. Conventional fracture surgery carries very high risk and poor outcomes,5 and so has been reserved mostly for patients with neurologic deficits.
VERTEBROPLASTY GOES MAINSTREAM
Given these facts, investigators began looking for alternative treatments. One that rose to the fore was polymethylmethacrylate cement to stabilize the fracture. This technique, called vertebroplasty, involves injecting liquid cement through a needle into the vertebral body, where it hardens and is thought to restore stability.
Since the first description of vertebroplasty for treating symptomatic hemangiomas,6 many papers have been published about the procedure and about similar ones, now grouped under the general heading of vertebral augmentation. This includes kyphoplasty and other newer proprietary techniques. These procedures have been widely accepted, and their use is growing. They have shown good results in several prospective case series, and nonrandomized and randomized controlled studies have shown them to be more effective than conventional medical treatment.7–25 For example, VERTOS, a small prospective randomized trial, showed that vertebroplasty was superior to conventional medical treatment.25 When Wardlaw et al24 showed that shortterm outcomes were better with kyphoplasty than with conventional medical therapy in a prospective randomized trial, many of us had moved past questioning whether vertebral augmentation is effective and were debating the relative merits of different methods and materials.
On a personal level, most of us became proponents of these procedures because we saw dramatic results—usually unequivocal. Most patients report significant improvement in pain immediately after the procedure, and many bedridden patients are able to leave the hospital within hours. In spine surgery, few procedures give such dramatic results with so few complications.
TWO NEW STUDIES UPSET ESTABLISHED BELIEF
This is why I am having such a hard time digesting the results of the trials by Buchbinder et al2 and Kallmes et al,1 published in the August 9, 2009, issue of the New England Journal of Medicine. Both were randomized controlled trials that used sham surgery rather than conventional medical treatment as the control. The sham procedure in each trial was the same as the intervention, with local anesthetic infiltration of the periosteum and mixing of the cement (so that the patients smelled its distinctive odor), but without placing the needle into the vertebra and injecting the cement.
In the study by Buchbinder et al,2 the real treatment had no benefit in any primary or secondary end point. This study did not allow crossovers.
In the study by Kallmes et al,1 more patients who received the real treatment reported clinically meaningful improvement in pain (a secondary end point), but the difference was not quite statistically significant (64% vs 48%, P = .06). In this trial, patients were allowed to cross over to the other study group after 1 month, and significantly more patients crossed over from the sham surgery group to the active treatment group than the other way around (43% vs 12%, P < .001).
My first instinct was to pick through the papers for flaws that would invalidate the results— and there were some problems. Both studies were initially planned to include more patients and therefore to have greater statistical power, but they were reassessed because of slow enrollment. In the study by Kallmes et al,1 the difference in clinically meaningful improvement might have reached statistical significance if the trial had been larger. The study by Buchbinder et al2 was a multicenter trial, but one center accounted for 53 (69%) of the 78 patients. Could this have biased the results?
The surgeon in me also seized for a while on the idea that since all of the interventions in both studies were done by interventional radiologists, the problem may have been in patient selection and that radiologists are not as astute as we are. However, even a surgeon’s ego cannot support this interpretation.
As I looked in more detail at the response I had written to these trials, I realized these criticisms were hardly fatal flaws, and the fact that two separate well-designed studies reached the same conclusion enhances their validity.
One concern that does bear some scrutiny is that the trials were too small to identify subgroups that may benefit from the procedure. In my experience, vertebral augmentation seems to have better results with certain types of fractures. Patients with a mobile pseudarthrotic cleft pattern of fracture seem to do much better than those with the more common nonmobile fracture.
THE POWERFUL PLACEBO EFFECT
Many commentaries on these two trials have discussed a famous study of a different procedure for a different condition. In this study, Moseley et al26 evaluated the use of arthroscopy to treat osteoarthritis of the knee and found that sham arthroscopy was as effective as real arthroscopy and that both were better than conventional treatment.
I was not long out of my orthopedic residency when this trial was published and was very aware of the debate that preceded it, as I once had to prepare a talk about it for resident rounds. I remember that there was a lively debate in the orthopedic community over the efficacy of the procedure before the results of this trial were released.
In contrast, the vertebral augmentation controversy had become a debate about the relative efficacy and the economics of specific techniques, not about the effectiveness of the entire concept. The mainstream had accepted the validity of the procedure, which was not the case in the knee arthroscopy trial.
In both vertebroplasty studies, the activetreatment groups and the sham-treatment groups all showed significant and rapid improvement in pain and disability, and these results were maintained over the study period. Though most vertebral compression fractures do heal, the clinical improvement is usually gradual over a period of weeks. This raises the possibility that the sham treatment was actualy an active placebo.
There is some evidence to support this possibility. In a randomized trial of the efficacy of selective nerve root blocks for lumbar radiculopathy, Riew et al27 showed that injection with a local anesthetic alone, although not as efficacious as a local anesthetic plus a corticosteroid at allowing patients to avoid surgery, showed an effect long after the expected duration of the anesthetic. The effect persisted even at 5 years of follow-up.28
Is it possible that the local anesthetic in this trial and the vertebroplasty trials acted as some sort of “reset button” for pain sensation? This is an area that may bear further investigation.
WHERE DOES THIS LEAVE US?
So where does this leave us? On one hand, randomized controlled trials comparing vertebral augmentation with conventional medical therapy24,25 showed augmentation to be beneficial. On the other hand, the studies by Kallmes et al1 and Buchbinder et al2 indicate vertebroplasty is no more effective than sham surgery.
It is very difficult for me to look at my own experience with vertebral augmentation and say that, on the basis of these trials, I am no longer going to offer it to my patients. I understand on an intellectual level that these trials call the efficacy of the procedure into question, but on a visceral level I cannot rationalize it. When faced with a patient who is barely ambulatory or in fact bed-bound due to pain, my experience tells me that vertebral augmentation has a very high chance of getting them ambulatory within hours. The trials of vertebroplasty would indicate this is a placebo effect or that local anesthetic alone is as effective, but I am not yet ready to make that leap.
Cognitive dissonance seems to rule.
Cognitive dissonance describes how we respond to conflicting information that challenges our existing belief, the uncomfortable feeling we get when new evidence calls into question things that we “know” are true.
To the point: two recent clinical trials1,2 have called into question the efficacy of vertebroplasty for treating osteoporotic vertebral compression fractures and have led many of us to question many of our assumptions, not only about vertebroplasty but also about evidencebased medicine.
Osteoporotic vertebral compression fractures are very common: more than 700,000 are estimated to occur in the United States annually. 3 They are costly and are associated with a risk of death.4 Fortunately, most heal without problems over 4 to 6 weeks with conventional treatment, ie, activity modification, analgesics, and bracing.
However, some patients do not seem to do so well and are debilitated by the pain of the fracture. Conventional fracture surgery carries very high risk and poor outcomes,5 and so has been reserved mostly for patients with neurologic deficits.
VERTEBROPLASTY GOES MAINSTREAM
Given these facts, investigators began looking for alternative treatments. One that rose to the fore was polymethylmethacrylate cement to stabilize the fracture. This technique, called vertebroplasty, involves injecting liquid cement through a needle into the vertebral body, where it hardens and is thought to restore stability.
Since the first description of vertebroplasty for treating symptomatic hemangiomas,6 many papers have been published about the procedure and about similar ones, now grouped under the general heading of vertebral augmentation. This includes kyphoplasty and other newer proprietary techniques. These procedures have been widely accepted, and their use is growing. They have shown good results in several prospective case series, and nonrandomized and randomized controlled studies have shown them to be more effective than conventional medical treatment.7–25 For example, VERTOS, a small prospective randomized trial, showed that vertebroplasty was superior to conventional medical treatment.25 When Wardlaw et al24 showed that shortterm outcomes were better with kyphoplasty than with conventional medical therapy in a prospective randomized trial, many of us had moved past questioning whether vertebral augmentation is effective and were debating the relative merits of different methods and materials.
On a personal level, most of us became proponents of these procedures because we saw dramatic results—usually unequivocal. Most patients report significant improvement in pain immediately after the procedure, and many bedridden patients are able to leave the hospital within hours. In spine surgery, few procedures give such dramatic results with so few complications.
TWO NEW STUDIES UPSET ESTABLISHED BELIEF
This is why I am having such a hard time digesting the results of the trials by Buchbinder et al2 and Kallmes et al,1 published in the August 9, 2009, issue of the New England Journal of Medicine. Both were randomized controlled trials that used sham surgery rather than conventional medical treatment as the control. The sham procedure in each trial was the same as the intervention, with local anesthetic infiltration of the periosteum and mixing of the cement (so that the patients smelled its distinctive odor), but without placing the needle into the vertebra and injecting the cement.
In the study by Buchbinder et al,2 the real treatment had no benefit in any primary or secondary end point. This study did not allow crossovers.
In the study by Kallmes et al,1 more patients who received the real treatment reported clinically meaningful improvement in pain (a secondary end point), but the difference was not quite statistically significant (64% vs 48%, P = .06). In this trial, patients were allowed to cross over to the other study group after 1 month, and significantly more patients crossed over from the sham surgery group to the active treatment group than the other way around (43% vs 12%, P < .001).
My first instinct was to pick through the papers for flaws that would invalidate the results— and there were some problems. Both studies were initially planned to include more patients and therefore to have greater statistical power, but they were reassessed because of slow enrollment. In the study by Kallmes et al,1 the difference in clinically meaningful improvement might have reached statistical significance if the trial had been larger. The study by Buchbinder et al2 was a multicenter trial, but one center accounted for 53 (69%) of the 78 patients. Could this have biased the results?
The surgeon in me also seized for a while on the idea that since all of the interventions in both studies were done by interventional radiologists, the problem may have been in patient selection and that radiologists are not as astute as we are. However, even a surgeon’s ego cannot support this interpretation.
As I looked in more detail at the response I had written to these trials, I realized these criticisms were hardly fatal flaws, and the fact that two separate well-designed studies reached the same conclusion enhances their validity.
One concern that does bear some scrutiny is that the trials were too small to identify subgroups that may benefit from the procedure. In my experience, vertebral augmentation seems to have better results with certain types of fractures. Patients with a mobile pseudarthrotic cleft pattern of fracture seem to do much better than those with the more common nonmobile fracture.
THE POWERFUL PLACEBO EFFECT
Many commentaries on these two trials have discussed a famous study of a different procedure for a different condition. In this study, Moseley et al26 evaluated the use of arthroscopy to treat osteoarthritis of the knee and found that sham arthroscopy was as effective as real arthroscopy and that both were better than conventional treatment.
I was not long out of my orthopedic residency when this trial was published and was very aware of the debate that preceded it, as I once had to prepare a talk about it for resident rounds. I remember that there was a lively debate in the orthopedic community over the efficacy of the procedure before the results of this trial were released.
In contrast, the vertebral augmentation controversy had become a debate about the relative efficacy and the economics of specific techniques, not about the effectiveness of the entire concept. The mainstream had accepted the validity of the procedure, which was not the case in the knee arthroscopy trial.
In both vertebroplasty studies, the activetreatment groups and the sham-treatment groups all showed significant and rapid improvement in pain and disability, and these results were maintained over the study period. Though most vertebral compression fractures do heal, the clinical improvement is usually gradual over a period of weeks. This raises the possibility that the sham treatment was actualy an active placebo.
There is some evidence to support this possibility. In a randomized trial of the efficacy of selective nerve root blocks for lumbar radiculopathy, Riew et al27 showed that injection with a local anesthetic alone, although not as efficacious as a local anesthetic plus a corticosteroid at allowing patients to avoid surgery, showed an effect long after the expected duration of the anesthetic. The effect persisted even at 5 years of follow-up.28
Is it possible that the local anesthetic in this trial and the vertebroplasty trials acted as some sort of “reset button” for pain sensation? This is an area that may bear further investigation.
WHERE DOES THIS LEAVE US?
So where does this leave us? On one hand, randomized controlled trials comparing vertebral augmentation with conventional medical therapy24,25 showed augmentation to be beneficial. On the other hand, the studies by Kallmes et al1 and Buchbinder et al2 indicate vertebroplasty is no more effective than sham surgery.
It is very difficult for me to look at my own experience with vertebral augmentation and say that, on the basis of these trials, I am no longer going to offer it to my patients. I understand on an intellectual level that these trials call the efficacy of the procedure into question, but on a visceral level I cannot rationalize it. When faced with a patient who is barely ambulatory or in fact bed-bound due to pain, my experience tells me that vertebral augmentation has a very high chance of getting them ambulatory within hours. The trials of vertebroplasty would indicate this is a placebo effect or that local anesthetic alone is as effective, but I am not yet ready to make that leap.
Cognitive dissonance seems to rule.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Carmona RH. Department of Health and Human Services. Office of the Surgeon General. Bone health and osteoporosis: A report of the Surgeon General (2004). www.surgeongeneral.gov/library/bonehealth/content.html. Accessed November 16, 2009.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Hu SS. Internal fixation in the osteoporotic spine. Spine (Phila PA 1976) 1997; 22( suppl 24):43S–48S.
- Galibert P, Deramond H, Rosat P, Le Gars D. Preliminary note on the treatment of vertebral angioma by percutaneous acrylic vertebroplasty. Neurochirurgie 1987; 33:166–168.
- Kasperk C, Hillmeier J, Noldge G, et al. Treatment of painful vertebral fractures by kyphoplasty in patients with primary osteoporosis: a prospective nonrandomized controlled study. J Bone Miner Res 2005; 20:604–612.
- Komp M, Ruetten S, Godolias G. Minimally invasive therapy for functionally unstable osteoporotic vertebral fracture by means of kyphoplasty: prospective comparative study of 19 surgically and 17 conservatively treated patients. J Miner Stoffwechs 2004; 11( suppl 1):13–15.
- Coumans JV, Reinhardt MK, Lieberman IH. Kyphoplasty for vertebral compression fractures: 1-year clinical outcomes from a prospective study. J Neurosurg 2003; 99 (suppl 1):44–50.
- Lieberman IH, Dudeney S, Reinhardt MK, Bell G. Initial outcome and efficacy of ‘kyphoplasty’ in the treatment of painful osteoporotic vertebral compression fractures. Spine (Phila PA 1976) 2001; 26:1631–1638.
- Garfin SR, Buckley RA, Ledlie J; Balloon Kyphoplasty Outcomes Group. Balloon kyphoplasty for symptomatic vertebral body compression fractures results in rapid, significant, and sustained improvements in back pain, function, and quality of life for elderly patients. Spine (Phila PA 1976) 2006; 31:2213–2220.
- Ledlie JT, Renfro MB. Kyphoplasty treatment of vertebral fractures: 2-year outcomes show sustained benefits. Spine (Phila PA 1976) 2006; 31:57–64.
- Majd ME, Farley S, Holt RT. Preliminary outcomes and efficacy of the first 360 consecutive kyphoplasties for the treatment of painful osteoporotic vertebral compression fractures. Spine J 2005; 5:244–255.
- Rhyne A, Banit D, Laxer E, Odum S, Nussman D. Kyphoplasty: report of eighty-two thoracolumbar osteoporotic vertebral fractures. J Orthop Trauma 2004; 18:294–299.
- Theodorou DJ, Theodorou SJ, Duncan TD, Garfin SR, Wong WH. Percutaneous balloon kyphoplasty for the correction of spinal deformity in painful vertebral body compression fractures. Clin Imaging 2002; 26:1–5.
- Berlemann U, Franz T, Orler R, Heini PF. Kyphoplasty for treatment of osteoporotic vertebral fractures: a prospective nonrandomized study. Eur Spine J 2004; 13:496–501.
- McGraw JK, Lippert JA, Minkus KD, Rami PM, Davis TM, Budzik RF. Prospective evaluation of pain relief in 100 patients undergoing percutaneous vertebroplasty: results and followup. J Vasc Interv Radiol 2002; 13:883–886.
- Zoarski GH, Snow P, Olan WJ, et al. Percutaneous vertebroplasty for osteoporotic compression fractures: quantitative prospective evaluation of long-term outcomes. J Vasc Interv Radiol 2002; 13:139–148.
- Evans AJ, Jensen ME, Kip KE, et al. Vertebral compression fractures: pain reduction and improvement in functional mobility after percutaneous polymethylmethacrylate vertebroplasty retrospective report of 245 cases. Radiology 2003; 226:366–372.
- Grohs JG, Matzner M, Trieb K, Krepler P. Minimal invasive stabilization of osteoporotic vertebral fractures: a prospective nonrandomized comparison of vertebroplasty and balloon kyphoplasty. J Spinal Disord Tech 2005; 18:238–242.
- Kallmes DF, Schweickert PA, Marx WF, Jensen ME. Vertebroplasty in the mid-and upper thoracic spine. AJNR Am J Neuroradiol 2002; 23:1117–1120.
- Grados F, Depriester C, Cayrolle G, Hardy N, Deramond H, Fardellone P. Long-term observations of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Rheumatology (Oxford) 2000; 39:1410–1414.
- Legroux-Gérot I, Lormeau C, Boutry N, Cotten A, Duquesnoy B, Cortet B. Long-term follow-up of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Clin Rheumatol 2004; 23:310–317.
- Wardlaw D, Cummings SR, Van Meirhaeghe J, et al. Efficacy and safety of balloon kyphoplasty compared with nonsurgical care for vertebral compression fracture (FREE): a randomised controlled trial. Lancet 2009; 373:1016–1024.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term outcomes of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007: 28:555–560.
- Moseley JB, O'Malley K, Petersen NJ. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med 2002; 347:81–88.
- Riew KD, Yin Y, Gilula L, et al. The effect of nerve-root injections on the need for operative treatment of lumbar radicular pain. A prospective, randomized, controlled, double-blind study. J Bone Joint Surg Am 2000; 82–A:1589–1593.
- Riew KD, Park JB, Cho YS, et al. Nerve root blocks in the treatment of lumbar radicular pain. A minimum five-year follow-up. J Bone Joint Surg Am 2006; 88:1722–1725.
- Kallmes DF, Comstock BA, Heagerty PJ, et al. A randomized trial of vertebroplasty for osteoporotic spinal fractures. N Engl J Med 2009; 361:569–579.
- Buchbinder R, Osborne RH, Ebeling PR, et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med 2009; 361:557–568.
- Carmona RH. Department of Health and Human Services. Office of the Surgeon General. Bone health and osteoporosis: A report of the Surgeon General (2004). www.surgeongeneral.gov/library/bonehealth/content.html. Accessed November 16, 2009.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Hu SS. Internal fixation in the osteoporotic spine. Spine (Phila PA 1976) 1997; 22( suppl 24):43S–48S.
- Galibert P, Deramond H, Rosat P, Le Gars D. Preliminary note on the treatment of vertebral angioma by percutaneous acrylic vertebroplasty. Neurochirurgie 1987; 33:166–168.
- Kasperk C, Hillmeier J, Noldge G, et al. Treatment of painful vertebral fractures by kyphoplasty in patients with primary osteoporosis: a prospective nonrandomized controlled study. J Bone Miner Res 2005; 20:604–612.
- Komp M, Ruetten S, Godolias G. Minimally invasive therapy for functionally unstable osteoporotic vertebral fracture by means of kyphoplasty: prospective comparative study of 19 surgically and 17 conservatively treated patients. J Miner Stoffwechs 2004; 11( suppl 1):13–15.
- Coumans JV, Reinhardt MK, Lieberman IH. Kyphoplasty for vertebral compression fractures: 1-year clinical outcomes from a prospective study. J Neurosurg 2003; 99 (suppl 1):44–50.
- Lieberman IH, Dudeney S, Reinhardt MK, Bell G. Initial outcome and efficacy of ‘kyphoplasty’ in the treatment of painful osteoporotic vertebral compression fractures. Spine (Phila PA 1976) 2001; 26:1631–1638.
- Garfin SR, Buckley RA, Ledlie J; Balloon Kyphoplasty Outcomes Group. Balloon kyphoplasty for symptomatic vertebral body compression fractures results in rapid, significant, and sustained improvements in back pain, function, and quality of life for elderly patients. Spine (Phila PA 1976) 2006; 31:2213–2220.
- Ledlie JT, Renfro MB. Kyphoplasty treatment of vertebral fractures: 2-year outcomes show sustained benefits. Spine (Phila PA 1976) 2006; 31:57–64.
- Majd ME, Farley S, Holt RT. Preliminary outcomes and efficacy of the first 360 consecutive kyphoplasties for the treatment of painful osteoporotic vertebral compression fractures. Spine J 2005; 5:244–255.
- Rhyne A, Banit D, Laxer E, Odum S, Nussman D. Kyphoplasty: report of eighty-two thoracolumbar osteoporotic vertebral fractures. J Orthop Trauma 2004; 18:294–299.
- Theodorou DJ, Theodorou SJ, Duncan TD, Garfin SR, Wong WH. Percutaneous balloon kyphoplasty for the correction of spinal deformity in painful vertebral body compression fractures. Clin Imaging 2002; 26:1–5.
- Berlemann U, Franz T, Orler R, Heini PF. Kyphoplasty for treatment of osteoporotic vertebral fractures: a prospective nonrandomized study. Eur Spine J 2004; 13:496–501.
- McGraw JK, Lippert JA, Minkus KD, Rami PM, Davis TM, Budzik RF. Prospective evaluation of pain relief in 100 patients undergoing percutaneous vertebroplasty: results and followup. J Vasc Interv Radiol 2002; 13:883–886.
- Zoarski GH, Snow P, Olan WJ, et al. Percutaneous vertebroplasty for osteoporotic compression fractures: quantitative prospective evaluation of long-term outcomes. J Vasc Interv Radiol 2002; 13:139–148.
- Evans AJ, Jensen ME, Kip KE, et al. Vertebral compression fractures: pain reduction and improvement in functional mobility after percutaneous polymethylmethacrylate vertebroplasty retrospective report of 245 cases. Radiology 2003; 226:366–372.
- Grohs JG, Matzner M, Trieb K, Krepler P. Minimal invasive stabilization of osteoporotic vertebral fractures: a prospective nonrandomized comparison of vertebroplasty and balloon kyphoplasty. J Spinal Disord Tech 2005; 18:238–242.
- Kallmes DF, Schweickert PA, Marx WF, Jensen ME. Vertebroplasty in the mid-and upper thoracic spine. AJNR Am J Neuroradiol 2002; 23:1117–1120.
- Grados F, Depriester C, Cayrolle G, Hardy N, Deramond H, Fardellone P. Long-term observations of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Rheumatology (Oxford) 2000; 39:1410–1414.
- Legroux-Gérot I, Lormeau C, Boutry N, Cotten A, Duquesnoy B, Cortet B. Long-term follow-up of vertebral osteoporotic fractures treated by percutaneous vertebroplasty. Clin Rheumatol 2004; 23:310–317.
- Wardlaw D, Cummings SR, Van Meirhaeghe J, et al. Efficacy and safety of balloon kyphoplasty compared with nonsurgical care for vertebral compression fracture (FREE): a randomised controlled trial. Lancet 2009; 373:1016–1024.
- Voormolen MH, Mali WP, Lohle PN, et al. Percutaneous vertebroplasty compared with optimal pain medication treatment: short-term outcomes of patients with subacute or chronic painful osteoporotic vertebral compression fractures. The VERTOS study. AJNR Am J Neuroradiol 2007: 28:555–560.
- Moseley JB, O'Malley K, Petersen NJ. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med 2002; 347:81–88.
- Riew KD, Yin Y, Gilula L, et al. The effect of nerve-root injections on the need for operative treatment of lumbar radicular pain. A prospective, randomized, controlled, double-blind study. J Bone Joint Surg Am 2000; 82–A:1589–1593.
- Riew KD, Park JB, Cho YS, et al. Nerve root blocks in the treatment of lumbar radicular pain. A minimum five-year follow-up. J Bone Joint Surg Am 2006; 88:1722–1725.
Chondromyxoid Fibroma of the Radial Shaft Treated With Nonvascularized Fibular Autograft
A Review of the Anti-inflammatory Properties of Clindamycin in the Treatment of Acne Vulgaris
What's Eating You? Cat Flea (Ctenocephalides felis)
CPT changes for ObGyns are minor in 2010; the big news is Medicare’s toss of consult codes
Current Procedural Terminology (CPT) 2010, which took effect January 1, doesn’t bring many changes for ObGyn practice, but there’s been a major backpedaling in Medicare coverage of consultations that you must be aware of. In conjunction with this move by the Centers for Medicare & Medicaid Services (CMS), CPT has added a definition of “transfer of care” and established two possible reasons for providing a consultation. I’ll have more to report about these important developments later in this article.
Among the changes to billing codes for the work performed in ObGyn: rebundling of commonly performed urodynamics procedures and new codes for revision of a vaginal graft. There is also a new (and unpublished) code for administering the H1N1 influenza vaccine.
Last, CPT has revised the explanation of non–face-to-face prolonged services. Read on!
New codes bundle urodynamic studies—a product of joint CMS and CPT input
The biggest changes in coding for ObGyn procedures are urodynamics study codes. The American Medical Association (AMA) has 1) created three new codes that represent test bundles and, in the process, 2) deleted the stand-alone urodynamics codes 51772 (urethral pressure profile studies [UPP] [urethral closure pressure profile], any technique) and 51795 (voiding pressure studies; bladder voiding pressure, any technique).
These changes were made because the most commonly reported codes for a female patient were billed together 90% of the time (51726, 51772, 51795, and 51797); the AMA reasoned that the most frequent combinations were considered overvalued when billed separately—that is, there was no repeat of pre-test and post-test work when these combinations were performed and there was no duplication in the cost of supplies and staff time.
The new bundles were therefore considered to better reflect current medical practice, and the Relative Value Update Committee (RUC) recommended, and CMS accepted, the relative value units (RVU) for the combination codes to reflect the true physician work value and practice expense of the combined procedures.
New and revised codes are:
51726 Complex cystometrogram (i.e., calibrated electronic equipment)
51727 …with urethral pressure profile studies (i.e., urethral closure pressure profile), any technique
51728 …with voiding pressure studies (i.e., bladder voiding pressure), any technique
51729 …with voiding pressure studies (i.e., bladder voiding pressure) and urethral pressure profile studies (i.e., urethral closure pressure profile), any technique.
According to the clinical vignette submitted to the AMA for code 51727, this procedure will include a sustained Valsalva maneuver as part of the urethral closure pressure profile. CPT did, however, retain the add-on code +51797 (voiding pressure studies, intra-abdominal [i.e., rectal, gastric, intraperitoneal]) and has clarified that 51797 may be billed in addition to 51728 and 51729 if a rectal catheter is placed to determine if the patient is straining during the voiding event.
In other words, the add-on code may be reported only when the primary procedure includes a voiding pressure study.
RVU for these new procedures have also been revised (see the TABLE ). Notable is the seeming discrepancy in RVU between code 51726 (cystometrogram alone) and the bundled tests. This is the case because the practice expense for 51726 has not reached its final level (the practice expense RVU are being increased or decreased in increments over several years); for 2010 only, therefore, this code will have a higher total RVU value than the new codes (51727, 51728, 51729), despite having a lower physician work relative value.
The discrepancy will be corrected in 2011, when 51726 will have lower RVU than the other urodynamics combination test codes.
TABLE
Changes in 2010 to RVU for urodynamic studies
| 2009 | 2010 | |||
|---|---|---|---|---|
| CPT code | Work RVU | Total RVU | Work RVU | Total RVU |
| 51726 | 1.71 | 9.02 | 1.71 | 8.71 |
| 51727 | Not applicable (NA) | NA | 2.11 | 8.07 |
| 51728 | NA | NA | 2.11 | 8.06 |
| 51729 | NA | NA | 2.11 | 8.14 |
Laparoscopic revision of a vaginal graft
In 2006, the AMA added the code for a vaginal approach to revising a graft (57295, revision [including removal] of prosthetic vaginal graft; vaginal approach). Then, in 2007, it added a code for an abdominal approach (57296, revision [including removal] of prosthetic vaginal graft; open abdominal approach).
Now, you have a code for a laparoscopic approach, completing the code set for this procedure. As with 57295 and 57296, report the new code when the graft is either revised or removed entirely.
57426 Revision (including removal) of prosthetic vaginal graft, laparoscopic approach
Other, miscellaneous changes take effect
OBSTETRIC PANEL
Although code 80055 comprises a battery of tests that are performed routinely on obstetric patients, a new code, 86780, was created to report syphilis screening using a treponemal antibody method, in which IgM and IgG antibodies are measured. This test is not the same syphilis test that is now part of the 80055 panel. CPT has therefore cautioned that, when you use code 86780 instead of the standard syphilis test code 86592, you should not report the obstetrics panel but, instead, separately report each test performed.
REPRODUCTIVE MEDICINE
New code 89398 (unlisted reproductive medicine laboratory procedure) has been added, but CPT still directs billers to use the unlisted miscellaneous pathology test code 89240 to report cryopreservation of reproductive ovarian tissues.
BILLING FOR THE H1N1 INFLUENZA VACCINE
Because of the urgency of collecting data on the H1N1 influenza epidemic, CPT has revised code 90663 to include the H1N1 formulation of the flu vaccine product. In addition, CPT has created a new code, 90470, for administering the H1N1 flu vaccine, which became valid in September (but which isn’t included in the hard-copy version of CPT 2010). The new code is to be used for intramuscular injection or intranasal administration, and includes any time spent counseling.
In addition:
- Do not report established code 90471 (immunization administration [includes percutaneous, intradermal, subcutaneous, or intramuscular injections]; one vaccine [single or combination vaccine/toxoid]) when you administer the H1N1 flu vaccine
- Report the vaccine product code only when your practice has purchased the vaccine, or when the payer requires the code with a 0 charge to match the administration code.
- Medicare coding for administering the H1N1 flu vaccine is different than what I’ve just described. Do not use CPT codes for Medicare patients; instead, code H1N1 flu immunization as:
G9141 Influenza A (H1N1) immunization administration (includes the physician counseling the patient/family)
G9142 Influenza A (H1N1) vaccine, any route of administration
Medicare will not reimburse for the vaccine product because it is being given to its providers without cost. Some carriers may require that the new vaccine product code be listed with a 0 charge.
Prolonged inpatient E/M services
CPT has revised guidelines for prolonged services that do not involve direct face-to-face contact with a patient. Keep in mind, however, that, although these changes are welcome, many payers don’t reimburse separately for work that isn’t performed face to face.
These codes are no longer considered add-on codes; they can be reported on a different date than the related E/M service.
According to CPT, codes 99358 and 99359 are reported when the prolonged time:
- is greater than would be expected for normal pre-service and post-service work associated with the E/M service
- exceeds 30 minutes
- is related to an E/M service that has already occurred, or to one that will occur and represents ongoing patient management (for example, your review of extensive patient records that weren’t available at the time of the visit)
- is in addition to any telephone services codes (99441–99443)—but not with more specific codes, such as medical team conferences, online medical evaluation, or care plan oversight services, which have no upper limit to the time required to accomplish the service.
Consultation codes and clarifications
Two changes of note, from a CPT perspective, have been made in the area of consultations. CPT has:
- added a definition for a transfer of care
- defined two circumstances under which a consultation can be coded. These revisions come at the same time Medicare has made the decision to no longer pay for consultations other than tele-health consults (see following section).
For 2010, CPT defines transfer of care as
…the process whereby a physician who is providing management for some or all of a patient’s problems relinquishes this responsibility to another physician who agrees to accept this responsibility and who, from the initial encounter, is not providing consultative services.
The guidelines also explain that 1) a transferring physician is no longer responsible for caring for the problem for which the patient was referred and 2) the consultation codes should not be reported by the physician who accepts care.
Two alternative conditions must now apply for a consultation to be considered provided:
- A physician requested an opinion or advice for a specific condition or problem, or
- The consulting physician saw the patient first to determine whether to accept ongoing management of her entire care or of a specific condition or problem (i.e., transfer of care).
The second condition is new; it remains to be seen if payers will accept it as a valid reason to bill for consultation.
As with all billable services, you should ensure that the criteria required by the payer you are billing have been met. CPT also directs that the written request for consultation can be documented by either the requesting or the receiving physician—something that was unacceptable under Medicare guidelines.
Last, CPT has added instructions to clarify the type of consultation code to bill under certain circumstances:
- When the patient is admitted after an outpatient consultation but the physician does not see the patient on the unit on the date of admission, bill only for outpatient consultation
- When the patient is seen for an office visit, emergency room visit, or outpatient consult on the date of admission and the physician then sees the patient on the unit that day, bill only the inpatient consultation or initial hospital care code, whichever applies. All services that day are used to determine the final level of service.
Medicare tilts the playing field on consultations
Although CPT has retained all consultation codes, and although the hope is that commercial payers will continue to reimburse for such services in the near future, the big news is that Medicare has announced that it will no longer recognize (or reimburse for) codes for outpatient or inpatient consultations. (Note: This story is still unfolding, however. The changes announced by Medicare that I discuss below are still before Congress as this article goes to press. Although Medicare has, in fact, released the transmittal letter to all carriers instructing them about the changes, Senator Arlen Specter [D-Pa] has introduced an amendment to the Patient Protection and Affordable Care Act [H.R. 3590] to postpone the policy change for 1 year. If Congress has not passed this bill before the end of 2009, the changes go through as planned. Stay tuned for developments!)
Assuming the changes go through, here is what is expected of you in the circumstances of providing consultations and billing Medicare (Medicaid payers aren’t required to follow this policy change but may opt to do so).
Outpatients. Document, and report, the appropriate level of visit for a new or established Medicare patient using outpatient codes 99201–99215
Inpatients. If you are a non-admitting physician asked to see a patient for the first time, report the appropriate level of initial hospital care (codes 99221–99223). Note the following three points:
- Initial hospital care includes only three levels of service—not the five levels from which you choose for consultation codes
- The lowest level of history and exam for these initial visit codes is a detailed history and examination—no matter the level of medical decision-making. If the level of history or exam is documented lower than “detailed”—say, as “expanded problem-focused”—you are required to report the unlisted E/M code 99499.
- The admitting physician adds the new Healthcare Common Procedure Coding System (HCPCS) modifier –AI (that is, “‘A’ upper-case ‘i’”) to the initial visit code, so that Medicare can distinguish the admitting physician from others providing care for the patient.
- All subsequent visits with the inpatient continue to be billed with the subsequent care inpatient codes (99231–99233).
Fallout from this change? Medicare is studying the implications of its new policy on secondary payments—that is, when Medicare is the primary payer and there is a supplemental carrier, or when Medicare is the secondary payer. Note: Medicare strongly advises all providers to check with their primary payers, because 1) Medicare will not accept a consultation code when a primary insurer has paid on that code and 2) it’s doubtful that a commercial payer will accept a consultation code when Medicare has paid for a new or established patient service.
To add to the turmoil…
The CMS has announced that, as a result of the changes in Medicare policy on consultations, it is increasing the relative values for all new and established patient services and initial hospital care. CMS is doing this, however, by reducing the relative values of some consultation codes.
In addition, all surgical procedure codes that carry a 10- or 90-day global period will see an increase in work RVU because of the increase in E/M services that are a part of all global care. Keep in mind that payers who use the Resource-Based Relative Value Scale (RBRVS) to reimburse services will probably adopt the new values when contracts are up for renewal, although many will be unable to do so in the short term.
It also remains to be seen if any commercial payers adopt Medicare policy or continue to pay for consultations. This area might be a contract issue with payers.
Current Procedural Terminology (CPT) 2010, which took effect January 1, doesn’t bring many changes for ObGyn practice, but there’s been a major backpedaling in Medicare coverage of consultations that you must be aware of. In conjunction with this move by the Centers for Medicare & Medicaid Services (CMS), CPT has added a definition of “transfer of care” and established two possible reasons for providing a consultation. I’ll have more to report about these important developments later in this article.
Among the changes to billing codes for the work performed in ObGyn: rebundling of commonly performed urodynamics procedures and new codes for revision of a vaginal graft. There is also a new (and unpublished) code for administering the H1N1 influenza vaccine.
Last, CPT has revised the explanation of non–face-to-face prolonged services. Read on!
New codes bundle urodynamic studies—a product of joint CMS and CPT input
The biggest changes in coding for ObGyn procedures are urodynamics study codes. The American Medical Association (AMA) has 1) created three new codes that represent test bundles and, in the process, 2) deleted the stand-alone urodynamics codes 51772 (urethral pressure profile studies [UPP] [urethral closure pressure profile], any technique) and 51795 (voiding pressure studies; bladder voiding pressure, any technique).
These changes were made because the most commonly reported codes for a female patient were billed together 90% of the time (51726, 51772, 51795, and 51797); the AMA reasoned that the most frequent combinations were considered overvalued when billed separately—that is, there was no repeat of pre-test and post-test work when these combinations were performed and there was no duplication in the cost of supplies and staff time.
The new bundles were therefore considered to better reflect current medical practice, and the Relative Value Update Committee (RUC) recommended, and CMS accepted, the relative value units (RVU) for the combination codes to reflect the true physician work value and practice expense of the combined procedures.
New and revised codes are:
51726 Complex cystometrogram (i.e., calibrated electronic equipment)
51727 …with urethral pressure profile studies (i.e., urethral closure pressure profile), any technique
51728 …with voiding pressure studies (i.e., bladder voiding pressure), any technique
51729 …with voiding pressure studies (i.e., bladder voiding pressure) and urethral pressure profile studies (i.e., urethral closure pressure profile), any technique.
According to the clinical vignette submitted to the AMA for code 51727, this procedure will include a sustained Valsalva maneuver as part of the urethral closure pressure profile. CPT did, however, retain the add-on code +51797 (voiding pressure studies, intra-abdominal [i.e., rectal, gastric, intraperitoneal]) and has clarified that 51797 may be billed in addition to 51728 and 51729 if a rectal catheter is placed to determine if the patient is straining during the voiding event.
In other words, the add-on code may be reported only when the primary procedure includes a voiding pressure study.
RVU for these new procedures have also been revised (see the TABLE ). Notable is the seeming discrepancy in RVU between code 51726 (cystometrogram alone) and the bundled tests. This is the case because the practice expense for 51726 has not reached its final level (the practice expense RVU are being increased or decreased in increments over several years); for 2010 only, therefore, this code will have a higher total RVU value than the new codes (51727, 51728, 51729), despite having a lower physician work relative value.
The discrepancy will be corrected in 2011, when 51726 will have lower RVU than the other urodynamics combination test codes.
TABLE
Changes in 2010 to RVU for urodynamic studies
| 2009 | 2010 | |||
|---|---|---|---|---|
| CPT code | Work RVU | Total RVU | Work RVU | Total RVU |
| 51726 | 1.71 | 9.02 | 1.71 | 8.71 |
| 51727 | Not applicable (NA) | NA | 2.11 | 8.07 |
| 51728 | NA | NA | 2.11 | 8.06 |
| 51729 | NA | NA | 2.11 | 8.14 |
Laparoscopic revision of a vaginal graft
In 2006, the AMA added the code for a vaginal approach to revising a graft (57295, revision [including removal] of prosthetic vaginal graft; vaginal approach). Then, in 2007, it added a code for an abdominal approach (57296, revision [including removal] of prosthetic vaginal graft; open abdominal approach).
Now, you have a code for a laparoscopic approach, completing the code set for this procedure. As with 57295 and 57296, report the new code when the graft is either revised or removed entirely.
57426 Revision (including removal) of prosthetic vaginal graft, laparoscopic approach
Other, miscellaneous changes take effect
OBSTETRIC PANEL
Although code 80055 comprises a battery of tests that are performed routinely on obstetric patients, a new code, 86780, was created to report syphilis screening using a treponemal antibody method, in which IgM and IgG antibodies are measured. This test is not the same syphilis test that is now part of the 80055 panel. CPT has therefore cautioned that, when you use code 86780 instead of the standard syphilis test code 86592, you should not report the obstetrics panel but, instead, separately report each test performed.
REPRODUCTIVE MEDICINE
New code 89398 (unlisted reproductive medicine laboratory procedure) has been added, but CPT still directs billers to use the unlisted miscellaneous pathology test code 89240 to report cryopreservation of reproductive ovarian tissues.
BILLING FOR THE H1N1 INFLUENZA VACCINE
Because of the urgency of collecting data on the H1N1 influenza epidemic, CPT has revised code 90663 to include the H1N1 formulation of the flu vaccine product. In addition, CPT has created a new code, 90470, for administering the H1N1 flu vaccine, which became valid in September (but which isn’t included in the hard-copy version of CPT 2010). The new code is to be used for intramuscular injection or intranasal administration, and includes any time spent counseling.
In addition:
- Do not report established code 90471 (immunization administration [includes percutaneous, intradermal, subcutaneous, or intramuscular injections]; one vaccine [single or combination vaccine/toxoid]) when you administer the H1N1 flu vaccine
- Report the vaccine product code only when your practice has purchased the vaccine, or when the payer requires the code with a 0 charge to match the administration code.
- Medicare coding for administering the H1N1 flu vaccine is different than what I’ve just described. Do not use CPT codes for Medicare patients; instead, code H1N1 flu immunization as:
G9141 Influenza A (H1N1) immunization administration (includes the physician counseling the patient/family)
G9142 Influenza A (H1N1) vaccine, any route of administration
Medicare will not reimburse for the vaccine product because it is being given to its providers without cost. Some carriers may require that the new vaccine product code be listed with a 0 charge.
Prolonged inpatient E/M services
CPT has revised guidelines for prolonged services that do not involve direct face-to-face contact with a patient. Keep in mind, however, that, although these changes are welcome, many payers don’t reimburse separately for work that isn’t performed face to face.
These codes are no longer considered add-on codes; they can be reported on a different date than the related E/M service.
According to CPT, codes 99358 and 99359 are reported when the prolonged time:
- is greater than would be expected for normal pre-service and post-service work associated with the E/M service
- exceeds 30 minutes
- is related to an E/M service that has already occurred, or to one that will occur and represents ongoing patient management (for example, your review of extensive patient records that weren’t available at the time of the visit)
- is in addition to any telephone services codes (99441–99443)—but not with more specific codes, such as medical team conferences, online medical evaluation, or care plan oversight services, which have no upper limit to the time required to accomplish the service.
Consultation codes and clarifications
Two changes of note, from a CPT perspective, have been made in the area of consultations. CPT has:
- added a definition for a transfer of care
- defined two circumstances under which a consultation can be coded. These revisions come at the same time Medicare has made the decision to no longer pay for consultations other than tele-health consults (see following section).
For 2010, CPT defines transfer of care as
…the process whereby a physician who is providing management for some or all of a patient’s problems relinquishes this responsibility to another physician who agrees to accept this responsibility and who, from the initial encounter, is not providing consultative services.
The guidelines also explain that 1) a transferring physician is no longer responsible for caring for the problem for which the patient was referred and 2) the consultation codes should not be reported by the physician who accepts care.
Two alternative conditions must now apply for a consultation to be considered provided:
- A physician requested an opinion or advice for a specific condition or problem, or
- The consulting physician saw the patient first to determine whether to accept ongoing management of her entire care or of a specific condition or problem (i.e., transfer of care).
The second condition is new; it remains to be seen if payers will accept it as a valid reason to bill for consultation.
As with all billable services, you should ensure that the criteria required by the payer you are billing have been met. CPT also directs that the written request for consultation can be documented by either the requesting or the receiving physician—something that was unacceptable under Medicare guidelines.
Last, CPT has added instructions to clarify the type of consultation code to bill under certain circumstances:
- When the patient is admitted after an outpatient consultation but the physician does not see the patient on the unit on the date of admission, bill only for outpatient consultation
- When the patient is seen for an office visit, emergency room visit, or outpatient consult on the date of admission and the physician then sees the patient on the unit that day, bill only the inpatient consultation or initial hospital care code, whichever applies. All services that day are used to determine the final level of service.
Medicare tilts the playing field on consultations
Although CPT has retained all consultation codes, and although the hope is that commercial payers will continue to reimburse for such services in the near future, the big news is that Medicare has announced that it will no longer recognize (or reimburse for) codes for outpatient or inpatient consultations. (Note: This story is still unfolding, however. The changes announced by Medicare that I discuss below are still before Congress as this article goes to press. Although Medicare has, in fact, released the transmittal letter to all carriers instructing them about the changes, Senator Arlen Specter [D-Pa] has introduced an amendment to the Patient Protection and Affordable Care Act [H.R. 3590] to postpone the policy change for 1 year. If Congress has not passed this bill before the end of 2009, the changes go through as planned. Stay tuned for developments!)
Assuming the changes go through, here is what is expected of you in the circumstances of providing consultations and billing Medicare (Medicaid payers aren’t required to follow this policy change but may opt to do so).
Outpatients. Document, and report, the appropriate level of visit for a new or established Medicare patient using outpatient codes 99201–99215
Inpatients. If you are a non-admitting physician asked to see a patient for the first time, report the appropriate level of initial hospital care (codes 99221–99223). Note the following three points:
- Initial hospital care includes only three levels of service—not the five levels from which you choose for consultation codes
- The lowest level of history and exam for these initial visit codes is a detailed history and examination—no matter the level of medical decision-making. If the level of history or exam is documented lower than “detailed”—say, as “expanded problem-focused”—you are required to report the unlisted E/M code 99499.
- The admitting physician adds the new Healthcare Common Procedure Coding System (HCPCS) modifier –AI (that is, “‘A’ upper-case ‘i’”) to the initial visit code, so that Medicare can distinguish the admitting physician from others providing care for the patient.
- All subsequent visits with the inpatient continue to be billed with the subsequent care inpatient codes (99231–99233).
Fallout from this change? Medicare is studying the implications of its new policy on secondary payments—that is, when Medicare is the primary payer and there is a supplemental carrier, or when Medicare is the secondary payer. Note: Medicare strongly advises all providers to check with their primary payers, because 1) Medicare will not accept a consultation code when a primary insurer has paid on that code and 2) it’s doubtful that a commercial payer will accept a consultation code when Medicare has paid for a new or established patient service.
To add to the turmoil…
The CMS has announced that, as a result of the changes in Medicare policy on consultations, it is increasing the relative values for all new and established patient services and initial hospital care. CMS is doing this, however, by reducing the relative values of some consultation codes.
In addition, all surgical procedure codes that carry a 10- or 90-day global period will see an increase in work RVU because of the increase in E/M services that are a part of all global care. Keep in mind that payers who use the Resource-Based Relative Value Scale (RBRVS) to reimburse services will probably adopt the new values when contracts are up for renewal, although many will be unable to do so in the short term.
It also remains to be seen if any commercial payers adopt Medicare policy or continue to pay for consultations. This area might be a contract issue with payers.
Current Procedural Terminology (CPT) 2010, which took effect January 1, doesn’t bring many changes for ObGyn practice, but there’s been a major backpedaling in Medicare coverage of consultations that you must be aware of. In conjunction with this move by the Centers for Medicare & Medicaid Services (CMS), CPT has added a definition of “transfer of care” and established two possible reasons for providing a consultation. I’ll have more to report about these important developments later in this article.
Among the changes to billing codes for the work performed in ObGyn: rebundling of commonly performed urodynamics procedures and new codes for revision of a vaginal graft. There is also a new (and unpublished) code for administering the H1N1 influenza vaccine.
Last, CPT has revised the explanation of non–face-to-face prolonged services. Read on!
New codes bundle urodynamic studies—a product of joint CMS and CPT input
The biggest changes in coding for ObGyn procedures are urodynamics study codes. The American Medical Association (AMA) has 1) created three new codes that represent test bundles and, in the process, 2) deleted the stand-alone urodynamics codes 51772 (urethral pressure profile studies [UPP] [urethral closure pressure profile], any technique) and 51795 (voiding pressure studies; bladder voiding pressure, any technique).
These changes were made because the most commonly reported codes for a female patient were billed together 90% of the time (51726, 51772, 51795, and 51797); the AMA reasoned that the most frequent combinations were considered overvalued when billed separately—that is, there was no repeat of pre-test and post-test work when these combinations were performed and there was no duplication in the cost of supplies and staff time.
The new bundles were therefore considered to better reflect current medical practice, and the Relative Value Update Committee (RUC) recommended, and CMS accepted, the relative value units (RVU) for the combination codes to reflect the true physician work value and practice expense of the combined procedures.
New and revised codes are:
51726 Complex cystometrogram (i.e., calibrated electronic equipment)
51727 …with urethral pressure profile studies (i.e., urethral closure pressure profile), any technique
51728 …with voiding pressure studies (i.e., bladder voiding pressure), any technique
51729 …with voiding pressure studies (i.e., bladder voiding pressure) and urethral pressure profile studies (i.e., urethral closure pressure profile), any technique.
According to the clinical vignette submitted to the AMA for code 51727, this procedure will include a sustained Valsalva maneuver as part of the urethral closure pressure profile. CPT did, however, retain the add-on code +51797 (voiding pressure studies, intra-abdominal [i.e., rectal, gastric, intraperitoneal]) and has clarified that 51797 may be billed in addition to 51728 and 51729 if a rectal catheter is placed to determine if the patient is straining during the voiding event.
In other words, the add-on code may be reported only when the primary procedure includes a voiding pressure study.
RVU for these new procedures have also been revised (see the TABLE ). Notable is the seeming discrepancy in RVU between code 51726 (cystometrogram alone) and the bundled tests. This is the case because the practice expense for 51726 has not reached its final level (the practice expense RVU are being increased or decreased in increments over several years); for 2010 only, therefore, this code will have a higher total RVU value than the new codes (51727, 51728, 51729), despite having a lower physician work relative value.
The discrepancy will be corrected in 2011, when 51726 will have lower RVU than the other urodynamics combination test codes.
TABLE
Changes in 2010 to RVU for urodynamic studies
| 2009 | 2010 | |||
|---|---|---|---|---|
| CPT code | Work RVU | Total RVU | Work RVU | Total RVU |
| 51726 | 1.71 | 9.02 | 1.71 | 8.71 |
| 51727 | Not applicable (NA) | NA | 2.11 | 8.07 |
| 51728 | NA | NA | 2.11 | 8.06 |
| 51729 | NA | NA | 2.11 | 8.14 |
Laparoscopic revision of a vaginal graft
In 2006, the AMA added the code for a vaginal approach to revising a graft (57295, revision [including removal] of prosthetic vaginal graft; vaginal approach). Then, in 2007, it added a code for an abdominal approach (57296, revision [including removal] of prosthetic vaginal graft; open abdominal approach).
Now, you have a code for a laparoscopic approach, completing the code set for this procedure. As with 57295 and 57296, report the new code when the graft is either revised or removed entirely.
57426 Revision (including removal) of prosthetic vaginal graft, laparoscopic approach
Other, miscellaneous changes take effect
OBSTETRIC PANEL
Although code 80055 comprises a battery of tests that are performed routinely on obstetric patients, a new code, 86780, was created to report syphilis screening using a treponemal antibody method, in which IgM and IgG antibodies are measured. This test is not the same syphilis test that is now part of the 80055 panel. CPT has therefore cautioned that, when you use code 86780 instead of the standard syphilis test code 86592, you should not report the obstetrics panel but, instead, separately report each test performed.
REPRODUCTIVE MEDICINE
New code 89398 (unlisted reproductive medicine laboratory procedure) has been added, but CPT still directs billers to use the unlisted miscellaneous pathology test code 89240 to report cryopreservation of reproductive ovarian tissues.
BILLING FOR THE H1N1 INFLUENZA VACCINE
Because of the urgency of collecting data on the H1N1 influenza epidemic, CPT has revised code 90663 to include the H1N1 formulation of the flu vaccine product. In addition, CPT has created a new code, 90470, for administering the H1N1 flu vaccine, which became valid in September (but which isn’t included in the hard-copy version of CPT 2010). The new code is to be used for intramuscular injection or intranasal administration, and includes any time spent counseling.
In addition:
- Do not report established code 90471 (immunization administration [includes percutaneous, intradermal, subcutaneous, or intramuscular injections]; one vaccine [single or combination vaccine/toxoid]) when you administer the H1N1 flu vaccine
- Report the vaccine product code only when your practice has purchased the vaccine, or when the payer requires the code with a 0 charge to match the administration code.
- Medicare coding for administering the H1N1 flu vaccine is different than what I’ve just described. Do not use CPT codes for Medicare patients; instead, code H1N1 flu immunization as:
G9141 Influenza A (H1N1) immunization administration (includes the physician counseling the patient/family)
G9142 Influenza A (H1N1) vaccine, any route of administration
Medicare will not reimburse for the vaccine product because it is being given to its providers without cost. Some carriers may require that the new vaccine product code be listed with a 0 charge.
Prolonged inpatient E/M services
CPT has revised guidelines for prolonged services that do not involve direct face-to-face contact with a patient. Keep in mind, however, that, although these changes are welcome, many payers don’t reimburse separately for work that isn’t performed face to face.
These codes are no longer considered add-on codes; they can be reported on a different date than the related E/M service.
According to CPT, codes 99358 and 99359 are reported when the prolonged time:
- is greater than would be expected for normal pre-service and post-service work associated with the E/M service
- exceeds 30 minutes
- is related to an E/M service that has already occurred, or to one that will occur and represents ongoing patient management (for example, your review of extensive patient records that weren’t available at the time of the visit)
- is in addition to any telephone services codes (99441–99443)—but not with more specific codes, such as medical team conferences, online medical evaluation, or care plan oversight services, which have no upper limit to the time required to accomplish the service.
Consultation codes and clarifications
Two changes of note, from a CPT perspective, have been made in the area of consultations. CPT has:
- added a definition for a transfer of care
- defined two circumstances under which a consultation can be coded. These revisions come at the same time Medicare has made the decision to no longer pay for consultations other than tele-health consults (see following section).
For 2010, CPT defines transfer of care as
…the process whereby a physician who is providing management for some or all of a patient’s problems relinquishes this responsibility to another physician who agrees to accept this responsibility and who, from the initial encounter, is not providing consultative services.
The guidelines also explain that 1) a transferring physician is no longer responsible for caring for the problem for which the patient was referred and 2) the consultation codes should not be reported by the physician who accepts care.
Two alternative conditions must now apply for a consultation to be considered provided:
- A physician requested an opinion or advice for a specific condition or problem, or
- The consulting physician saw the patient first to determine whether to accept ongoing management of her entire care or of a specific condition or problem (i.e., transfer of care).
The second condition is new; it remains to be seen if payers will accept it as a valid reason to bill for consultation.
As with all billable services, you should ensure that the criteria required by the payer you are billing have been met. CPT also directs that the written request for consultation can be documented by either the requesting or the receiving physician—something that was unacceptable under Medicare guidelines.
Last, CPT has added instructions to clarify the type of consultation code to bill under certain circumstances:
- When the patient is admitted after an outpatient consultation but the physician does not see the patient on the unit on the date of admission, bill only for outpatient consultation
- When the patient is seen for an office visit, emergency room visit, or outpatient consult on the date of admission and the physician then sees the patient on the unit that day, bill only the inpatient consultation or initial hospital care code, whichever applies. All services that day are used to determine the final level of service.
Medicare tilts the playing field on consultations
Although CPT has retained all consultation codes, and although the hope is that commercial payers will continue to reimburse for such services in the near future, the big news is that Medicare has announced that it will no longer recognize (or reimburse for) codes for outpatient or inpatient consultations. (Note: This story is still unfolding, however. The changes announced by Medicare that I discuss below are still before Congress as this article goes to press. Although Medicare has, in fact, released the transmittal letter to all carriers instructing them about the changes, Senator Arlen Specter [D-Pa] has introduced an amendment to the Patient Protection and Affordable Care Act [H.R. 3590] to postpone the policy change for 1 year. If Congress has not passed this bill before the end of 2009, the changes go through as planned. Stay tuned for developments!)
Assuming the changes go through, here is what is expected of you in the circumstances of providing consultations and billing Medicare (Medicaid payers aren’t required to follow this policy change but may opt to do so).
Outpatients. Document, and report, the appropriate level of visit for a new or established Medicare patient using outpatient codes 99201–99215
Inpatients. If you are a non-admitting physician asked to see a patient for the first time, report the appropriate level of initial hospital care (codes 99221–99223). Note the following three points:
- Initial hospital care includes only three levels of service—not the five levels from which you choose for consultation codes
- The lowest level of history and exam for these initial visit codes is a detailed history and examination—no matter the level of medical decision-making. If the level of history or exam is documented lower than “detailed”—say, as “expanded problem-focused”—you are required to report the unlisted E/M code 99499.
- The admitting physician adds the new Healthcare Common Procedure Coding System (HCPCS) modifier –AI (that is, “‘A’ upper-case ‘i’”) to the initial visit code, so that Medicare can distinguish the admitting physician from others providing care for the patient.
- All subsequent visits with the inpatient continue to be billed with the subsequent care inpatient codes (99231–99233).
Fallout from this change? Medicare is studying the implications of its new policy on secondary payments—that is, when Medicare is the primary payer and there is a supplemental carrier, or when Medicare is the secondary payer. Note: Medicare strongly advises all providers to check with their primary payers, because 1) Medicare will not accept a consultation code when a primary insurer has paid on that code and 2) it’s doubtful that a commercial payer will accept a consultation code when Medicare has paid for a new or established patient service.
To add to the turmoil…
The CMS has announced that, as a result of the changes in Medicare policy on consultations, it is increasing the relative values for all new and established patient services and initial hospital care. CMS is doing this, however, by reducing the relative values of some consultation codes.
In addition, all surgical procedure codes that carry a 10- or 90-day global period will see an increase in work RVU because of the increase in E/M services that are a part of all global care. Keep in mind that payers who use the Resource-Based Relative Value Scale (RBRVS) to reimburse services will probably adopt the new values when contracts are up for renewal, although many will be unable to do so in the short term.
It also remains to be seen if any commercial payers adopt Medicare policy or continue to pay for consultations. This area might be a contract issue with payers.
Guanfacine extended release for ADHD
Guanfacine extended release (GXR)—a selective α-2 adrenergic agonist FDA-approved for the treatment of attention-deficit/hyperactivity disorder (ADHD)—has demonstrated efficacy for inattentive and hyperactive/impulsive symptom domains in 2 large trials lasting 8 and 9 weeks.1,2 GXR’s once-daily formulation may increase adherence and deliver consistent control of symptoms across a full day ( Table 1 ).
Table 1
Guanfacine extended release: Fast facts
| Brand name: Intuniv |
| Indication: Attention-deficit/hyperactivity disorder |
| Approval date: September 3, 2009 |
| Availability date: November 2009 |
| Manufacturer: Shire |
| Dosing forms: 1-mg, 2-mg, 3-mg, and 4-mg extended-release tablets |
| Recommended dosage: 0.05 to 0.12 mg/kg once daily |
Clinical implications
GXR exhibits enhancement of noradrenergic pathways through selective direct receptor action in the prefrontal cortex.3 This mechanism of action is different from that of other FDA-approved ADHD medications. GXR can be used alone or in combination with stimulants or atomoxetine for treating complex ADHD, such as cases accompanied by oppositional features and emotional dysregulation or characterized by partial stimulant response.
How it works
Guanfacine—originally developed as an immediate-release (IR) antihypertensive—reduces sympathetic tone, causing centrally mediated vasodilation and reduced heart rate. Although GXR’s mechanism of action in ADHD is not known, the drug is a selective α-2A receptor agonist thought to directly engage postsynaptic receptors in the prefrontal cortex (PFC), an area of the brain believed to play a major role in attentional and organizational functions that preclinical research has linked to ADHD.3
The postsynaptic α-2A receptor is thought to play a central role in the optimal functioning of the PFC as illustrated by the “inverted U hypothesis of PFC activation.”4 In this model, cyclic adenosine monophosphate (cAMP) levels build within the prefrontal cortical neurons and cause specific ion channels—hyperpolarization-activated cyclic nucleotide gated (HCN) channels—to open on dendritic spines of these neurons.5 Activation of HCN channels effectively reduces membrane resistance, cutting off synaptic inputs and disconnecting PFC network connections. Because α-2A receptors are located in proximity to HCN channels, their stimulation by GXR closes HCN channels, inhibits further production of cAMP, and reestablishes synaptic function and the resulting network connectivity.5 Blockade of α-2A receptors by yohimbine reverses this process, eroding network connectivity, and in monkeys has been demonstrated to impair working memory,6 damage inhibition/impulse control, and produce locomotor hyperactivity.
Direct stimulation by GXR of the postsynaptic α-2A receptors is thought to:
- strengthen working memory
- reduce susceptibility to distraction
- improve attention regulation
- improve behavioral inhibition
- enhance impulse control.7
Pharmacokinetics
GXR offers enhanced pharmaceutics relative to IR guanfacine. IR guanfacine exhibits poor absorption characteristics—peak plasma concentration is achieved too rapidly and then declines precipitously, with considerable inter-individual variation.
GXR’s once-daily formulation is implemented by a proprietary enteric-coated sustained release mechanism8 that is meant to:
- control absorption
- provide a broad but flat plasma concentration profile
- reduce inter-individual variation of guanfacine exposure.
Compared with IR guanfacine, GXR exhibits delayed time of maximum concentration (Tmax) and reduced maximum concentration (Cmax). Therapeutic concentrations can be sustained over longer periods with reduced peak-to-trough fluctuation,8 which tends to improve tolerability and symptom control throughout the day. The convenience of once-daily dosing also may increase adherence.
GXR’s pharmacokinetic characteristics do not change with dose, but high-fat meals will increase absorption of the drug—Cmax increases by 75% and area under the plasma concentration time curve increases by 40%. Because GXR primarily is metabolized through cytochrome P450 (CYP) 3A4, CYP3A4 inhibitors such as ketoconazole will increase guanfacine plasma concentrations and elevate the risk of adverse events such as bradycardia, hypotension, and sedation. Conversely, CYP3A4 inducers such as rifampin will significantly reduce total guanfacine exposure. Coadministration of valproic acid with GXR can result in increased valproic acid levels, producing additive CNS side effects.
Efficacy
GXR reduced both inattentive and hyperactive/impulsive symptoms in 2 phase III, forced-dose, parallel-design, randomized, placebo-controlled trials ( Table 2 ). In the first trial,1 345 children age 6 to 17 received placebo or GXR, 2 mg, 3 mg, or 4 mg once daily for 8 weeks. In the second study,2 324 children age 6 to 17 received placebo or GXR, 1 mg, 2 mg, 3 mg, or 4 mg, once daily for 9 weeks; the 1-mg dose was given only to patients weighing <50 kg (<110 lbs).
In both trials, doses were increased in increments of 1 mg/week, and investigators evaluated participants’ ADHD signs and symptoms once a week using the clinician administered and scored ADHD Rating Scale-IV (ADHD-RS-IV). The primary outcome was change in total ADHD-RS-IV score from baseline to endpoint.
In both trials, patients taking GXR demonstrated statistically signifcant improvements in ADHD-RS-IV score starting 1 to 2 weeks after they began receiving once-daily GXR:
- In the first trial, the mean reduction in ADHD-RS-IV total score at endpoint was –16.7 for GXR compared with –8.9 for placebo (P < .0001).
- In the second, the reduction was –19.6 for GXR and –12.2 for placebo (P=.004).
Placebo-adjusted least squares mean changes from baseline were statistically significant for all GXR doses in the randomized treatment groups in both studies.
Secondary efficacy outcome measures included the Conners’ Parent Rating Scale-Revised: Short Form (CPRS-R) and the Conners’ Teacher Rating Scale-Revised: Short Form (CTRS-R).
Significant improvements were seen on both scales. On the CPRS-R, parents reported significant improvement across a full day (as measured at 6 PM, 8 PM, and 6 AM the next day). On the CTRS-R—which was used only in the first trial—teachers reported significant improvement throughout the school day (as measured at 10 AM and 2 PM).
Treating oppositional symptoms. In a collateral study,9 GXR was evaluated in complex ADHD patients age 6 to 12 who exhibited oppositional symptoms. The primary efficacy measure was change from baseline to endpoint in the oppositional subscale of the Conners’ Parent Rating Scale-Revised: Long Form (CPRS-R:L) score.
All subjects randomized to GXR started on a dose of 1 mg/d—which could be titrated by 1 mg/week during the 5-week, dose-optimization period to a maximum of 4 mg/d—and were maintained at their optimal doses for 3 additional weeks. Among the 217 subjects enrolled, 138 received GXR and 79, placebo.
Least-squares mean reductions from baseline to endpoint in CPRS-R:L oppositional subscale scores were –10.9 in the GXR group compared with –6.8 in the placebo group (P < .001; effect size 0.590). The GXR-treated group showed a significantly greater reduction in ADHD-RS-IV total score from baseline to endpoint compared with the placebo group (–23.8 vs –11.4, respectively, P < .001; effect size 0.916).
Table 2
Randomized, controlled trials supporting GXR’s effectiveness
for treating ADHD symptoms
| Study | Subjects | GXR dosages | Results |
|---|---|---|---|
| Biederman et al, 20087 ; phase III, forced-dose parallel-design | 345 ADHD patients age 6 to 17 | 2, 3, or 4 mg given once daily for 8 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-16.7 vs -8.9) |
| Sallee et al, 20098 ; phase III, forced-dose parallel-design | 324 ADHD patients age 6 to 17 | 1,* 2, 3, or 4 mg given once daily for 9 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-19.6 vs -12.2) |
| Connor et al, 20099 ; collateral study | 217 complex ADHD patients age 6 to 12 with oppositional symptoms | Starting dose 1 mg/d, titrated to a maximum of 4 mg/d for a total of 8 weeks | GXR was associated with significantly lower scores on CPRS-R:L oppositional subscale (-10.9 vs -6.8) and ADHD-RS-IV (-23.8 vs -11.4) compared with placebo |
| *1-mg dose was given only to subjects weighing <50 kg (<110 lbs) | |||
| ADHD: attention-deficit/hyperactivity disorder; ADHD-RS-IV: Attention-Deficit/Hyperactivity Disorder Rating Scale-IV; CPRS-R:L: Conners’ Parent Rating Scale-Revised: Long Form; GXR: guanfacine extended release | |||
Tolerability
In the phase III trials, the most commonly reported drug-related adverse reactions (occurring in ≥2% of patients) were:
- somnolence (38%)
- headache (24%)
- fatigue (14%)
- upper abdominal pain (10%)
- nausea, lethargy, dizziness, hypotension/decreased blood pressure, irritability (6% for each)
- decreased appetite (5%)
- dry mouth (4%)
- constipation (3%).
Many of these adverse reactions appear to be dose-related, particularly somnolence, sedation, abdominal pain, dizziness, and hypotension/decreased blood pressure.
Overall, GXR was well tolerated; clinicians rated most events as mild to moderate. Twelve percent of GXR patients discontinued the clinical studies because of adverse events, compared with 4% in the placebo groups. The most common adverse reactions leading to discontinuation were somnolence/sedation (6%) and fatigue (2%). Less common adverse reactions leading to discontinuation (occurring in 1% of patients) included hypotension/decreased blood pressure, headache, and dizziness.
Open-label safety trial. Sallee et al10 conducted a longer-term, open-label, flexible-dose safety continuation study of 259 GXR-treated patients (mean exposure 10 months), some of whom also received a psychostimulant. Common adverse reactions (occurring in ≥5% of subjects) included somnolence (45%), headache (26%), fatigue (16%), upper abdominal pain (11%), hypotension/decreased blood pressure (10%), vomiting (9%), dizziness (7%), nausea (7%), weight gain (7%), and irritability (6%).10 In a subset of patients, the onset of sedative events typically occurred within the first 3 weeks of GXR treatment and then declined with maintenance to a frequency of approximately 16%. The rates of somnolence, sedation, or fatigue were lowest among patients who also received a psychostimulant ( Figure ).
Distribution of GXR doses before the end of this study was 37% of patients on 4 mg, 33% on 3 mg, 27% on 2 mg, and 3% on 1 mg, suggesting a preference for maintenance doses of 3 to 4 mg/d. The most frequent adverse reactions leading to discontinuation were somnolence (3%), syncopal events (2%), increased weight (2%), depression (2%), and fatigue (2%). Other adverse reactions leading to discontinuation (occurring in approximately 1% of patients) included hypotension/decreased blood pressure, sedation, headache, and lethargy. Serious adverse reactions in the longer-term study in >1 patient included syncope (2%) and convulsion (0.4%).
Figure: Incidence of somnolence, sedation, and fatigue in study patients receiving GXR
with or without psychostimulants
In an open-label continuation study of 259 patients treated with guanfacine extended release (GXR), somnolence, sedation, or fatigue was reported by 49% of subjects overall, 59% of those who received GXR monotherapy, and 11% of those given GXR with a psychostimulant.
GXR: guanfacine extended release
Source: Reprinted with permission from Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226 Safety warnings relating to the likelihood of hypotension, bradycardia, and possible syncope when prescribing GXR should be understood in the context of its pharmacologic action to lower heart rate and blood pressure. In the short-term (8 to 9 weeks) controlled trials, the maximum mean changes from baseline in systolic blood pressure, diastolic blood pressure, and pulse were -5 mm Hg, -3 mm Hg, and -6 bpm, respectively, for all dose groups combined. These changes, which generally occurred 1 week after reaching target doses of 1 to 4 mg/d, were dose-dependent but usually modest and did not cause other symptoms; however, hypotension and bradycardia can occur.
In the longer-term, open-label safety study,10 maximum decreases in systolic and diastolic blood pressure occurred in the first month of treatment; decreases were less pronounced over time. Syncope occurred in 1% of pediatric subjects but was not dose-dependent. Guanfacine IR can increase QT interval but not in a dose-dependent fashion.
Dosing
The approved dose range for GXR is 1 to 4 mg once daily in the morning. Initiate treatment at 1 mg/d, and adjust the dose in increments of no more than 1 mg/week, evaluating the patient weekly. GXR maintenance therapy is frequently in the range of 2 to 4 mg/d.
Because adverse events such as hypotension, bradycardia, and sedation are dose-related, evaluate benefit and risk using mg/kg range approximation. GXR efficacy on a weight-adjusted (mg/kg) basis is consistent across a dosage range of 0.01 to 0.17 mg/kg/d. Clinically relevant improvements are usually observed beginning at doses of 0.05 to 0.08 mg/kg/d. In clinical trials, efficacy increased with increasing weight-adjusted dose (mg/kg), so if GXR is well-tolerated, doses up to 0.12 mg/kg once daily may provide additional benefit up to the maximum of 4 mg/d.
Instruct patients to swallow GXR whole because crushing, chewing, or otherwise breaking the tablet’s enteric coating will markedly enhance guanfacine release.
Abruptly discontinuing GXR is associated with infrequent, transient elevations in blood pressure above the patient’s baseline (ie, rebound). To minimize these effects, GXR should be gradually tapered in decrements of no more than 1 mg every 3 to 7 days. Isolated missed doses of GXR generally are not a problem, but ≥2 consecutive missed doses may warrant reinitiation of the titration schedule.
Related resource
- Guanfacine extended release (Intuniv) prescribing information. www.intuniv.com/documents/INTUNIV_Full_Prescribing_Information.pdf.
Drug brand names
- Atomoxetine • Strattera
- Guanfacine extended release • Intuniv
- Guanfacine immediate release • Tenex
- Ketoconazole • Nizoral
- Rifampin • Rifadin, Rimactane
- Valproic acid • Depakene, Depakote
Disclosure
Dr. Sallee receives grant/research support from the National Institutes of Health. He is a consultant to Otsuka, Nextwave, and Sepracor and a consultant to and speaker for Shire. Dr. Sallee is a consultant to, shareholder of, and member of the board of directors of P2D Inc. and a principal in Satiety Solutions.
1. Biederman J, Melmed RD, Patel A, et al. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e73-e84.
2. Sallee F, McGough J, Wigal T, et al. For the SPD503 Study Group Guanfacine extended release in children and adolescents with attention deficit hyperactivity disorder: a placebo-controlled trial. J Am Acad Child Adolesc Psychiatry. 2009;48(2):155-165.
3. Arnsten AF, Cai JX, Goldman-Rakic PS. The α-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for α-2 receptor subtypes. J Neurosci. 1988;8:4287-4298.
4. Vijayraghavan S, Wang M, Birnbaum SG, et al. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376-384.
5. Wang M, Ramos BP, Paspalas CD, et al. α 2-A adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397-410.
6. Li BM, Mei ZT. Delayed-response deficit induced by local injection of the α 2-adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134-139.
7. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158:1067-1074.
8. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29:617-625.
9. Connor D, Spencer T, Kratochvil C, et al. Effects of guanfacine extended release on secondary measures in children with attention-deficit/hyperactivity disorder and oppositional symptoms. Abstract presented at: Annual Meeting of the American Psychiatric Association; May 18, 2009; San Francisco, CA.
10. Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226.
Guanfacine extended release (GXR)—a selective α-2 adrenergic agonist FDA-approved for the treatment of attention-deficit/hyperactivity disorder (ADHD)—has demonstrated efficacy for inattentive and hyperactive/impulsive symptom domains in 2 large trials lasting 8 and 9 weeks.1,2 GXR’s once-daily formulation may increase adherence and deliver consistent control of symptoms across a full day ( Table 1 ).
Table 1
Guanfacine extended release: Fast facts
| Brand name: Intuniv |
| Indication: Attention-deficit/hyperactivity disorder |
| Approval date: September 3, 2009 |
| Availability date: November 2009 |
| Manufacturer: Shire |
| Dosing forms: 1-mg, 2-mg, 3-mg, and 4-mg extended-release tablets |
| Recommended dosage: 0.05 to 0.12 mg/kg once daily |
Clinical implications
GXR exhibits enhancement of noradrenergic pathways through selective direct receptor action in the prefrontal cortex.3 This mechanism of action is different from that of other FDA-approved ADHD medications. GXR can be used alone or in combination with stimulants or atomoxetine for treating complex ADHD, such as cases accompanied by oppositional features and emotional dysregulation or characterized by partial stimulant response.
How it works
Guanfacine—originally developed as an immediate-release (IR) antihypertensive—reduces sympathetic tone, causing centrally mediated vasodilation and reduced heart rate. Although GXR’s mechanism of action in ADHD is not known, the drug is a selective α-2A receptor agonist thought to directly engage postsynaptic receptors in the prefrontal cortex (PFC), an area of the brain believed to play a major role in attentional and organizational functions that preclinical research has linked to ADHD.3
The postsynaptic α-2A receptor is thought to play a central role in the optimal functioning of the PFC as illustrated by the “inverted U hypothesis of PFC activation.”4 In this model, cyclic adenosine monophosphate (cAMP) levels build within the prefrontal cortical neurons and cause specific ion channels—hyperpolarization-activated cyclic nucleotide gated (HCN) channels—to open on dendritic spines of these neurons.5 Activation of HCN channels effectively reduces membrane resistance, cutting off synaptic inputs and disconnecting PFC network connections. Because α-2A receptors are located in proximity to HCN channels, their stimulation by GXR closes HCN channels, inhibits further production of cAMP, and reestablishes synaptic function and the resulting network connectivity.5 Blockade of α-2A receptors by yohimbine reverses this process, eroding network connectivity, and in monkeys has been demonstrated to impair working memory,6 damage inhibition/impulse control, and produce locomotor hyperactivity.
Direct stimulation by GXR of the postsynaptic α-2A receptors is thought to:
- strengthen working memory
- reduce susceptibility to distraction
- improve attention regulation
- improve behavioral inhibition
- enhance impulse control.7
Pharmacokinetics
GXR offers enhanced pharmaceutics relative to IR guanfacine. IR guanfacine exhibits poor absorption characteristics—peak plasma concentration is achieved too rapidly and then declines precipitously, with considerable inter-individual variation.
GXR’s once-daily formulation is implemented by a proprietary enteric-coated sustained release mechanism8 that is meant to:
- control absorption
- provide a broad but flat plasma concentration profile
- reduce inter-individual variation of guanfacine exposure.
Compared with IR guanfacine, GXR exhibits delayed time of maximum concentration (Tmax) and reduced maximum concentration (Cmax). Therapeutic concentrations can be sustained over longer periods with reduced peak-to-trough fluctuation,8 which tends to improve tolerability and symptom control throughout the day. The convenience of once-daily dosing also may increase adherence.
GXR’s pharmacokinetic characteristics do not change with dose, but high-fat meals will increase absorption of the drug—Cmax increases by 75% and area under the plasma concentration time curve increases by 40%. Because GXR primarily is metabolized through cytochrome P450 (CYP) 3A4, CYP3A4 inhibitors such as ketoconazole will increase guanfacine plasma concentrations and elevate the risk of adverse events such as bradycardia, hypotension, and sedation. Conversely, CYP3A4 inducers such as rifampin will significantly reduce total guanfacine exposure. Coadministration of valproic acid with GXR can result in increased valproic acid levels, producing additive CNS side effects.
Efficacy
GXR reduced both inattentive and hyperactive/impulsive symptoms in 2 phase III, forced-dose, parallel-design, randomized, placebo-controlled trials ( Table 2 ). In the first trial,1 345 children age 6 to 17 received placebo or GXR, 2 mg, 3 mg, or 4 mg once daily for 8 weeks. In the second study,2 324 children age 6 to 17 received placebo or GXR, 1 mg, 2 mg, 3 mg, or 4 mg, once daily for 9 weeks; the 1-mg dose was given only to patients weighing <50 kg (<110 lbs).
In both trials, doses were increased in increments of 1 mg/week, and investigators evaluated participants’ ADHD signs and symptoms once a week using the clinician administered and scored ADHD Rating Scale-IV (ADHD-RS-IV). The primary outcome was change in total ADHD-RS-IV score from baseline to endpoint.
In both trials, patients taking GXR demonstrated statistically signifcant improvements in ADHD-RS-IV score starting 1 to 2 weeks after they began receiving once-daily GXR:
- In the first trial, the mean reduction in ADHD-RS-IV total score at endpoint was –16.7 for GXR compared with –8.9 for placebo (P < .0001).
- In the second, the reduction was –19.6 for GXR and –12.2 for placebo (P=.004).
Placebo-adjusted least squares mean changes from baseline were statistically significant for all GXR doses in the randomized treatment groups in both studies.
Secondary efficacy outcome measures included the Conners’ Parent Rating Scale-Revised: Short Form (CPRS-R) and the Conners’ Teacher Rating Scale-Revised: Short Form (CTRS-R).
Significant improvements were seen on both scales. On the CPRS-R, parents reported significant improvement across a full day (as measured at 6 PM, 8 PM, and 6 AM the next day). On the CTRS-R—which was used only in the first trial—teachers reported significant improvement throughout the school day (as measured at 10 AM and 2 PM).
Treating oppositional symptoms. In a collateral study,9 GXR was evaluated in complex ADHD patients age 6 to 12 who exhibited oppositional symptoms. The primary efficacy measure was change from baseline to endpoint in the oppositional subscale of the Conners’ Parent Rating Scale-Revised: Long Form (CPRS-R:L) score.
All subjects randomized to GXR started on a dose of 1 mg/d—which could be titrated by 1 mg/week during the 5-week, dose-optimization period to a maximum of 4 mg/d—and were maintained at their optimal doses for 3 additional weeks. Among the 217 subjects enrolled, 138 received GXR and 79, placebo.
Least-squares mean reductions from baseline to endpoint in CPRS-R:L oppositional subscale scores were –10.9 in the GXR group compared with –6.8 in the placebo group (P < .001; effect size 0.590). The GXR-treated group showed a significantly greater reduction in ADHD-RS-IV total score from baseline to endpoint compared with the placebo group (–23.8 vs –11.4, respectively, P < .001; effect size 0.916).
Table 2
Randomized, controlled trials supporting GXR’s effectiveness
for treating ADHD symptoms
| Study | Subjects | GXR dosages | Results |
|---|---|---|---|
| Biederman et al, 20087 ; phase III, forced-dose parallel-design | 345 ADHD patients age 6 to 17 | 2, 3, or 4 mg given once daily for 8 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-16.7 vs -8.9) |
| Sallee et al, 20098 ; phase III, forced-dose parallel-design | 324 ADHD patients age 6 to 17 | 1,* 2, 3, or 4 mg given once daily for 9 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-19.6 vs -12.2) |
| Connor et al, 20099 ; collateral study | 217 complex ADHD patients age 6 to 12 with oppositional symptoms | Starting dose 1 mg/d, titrated to a maximum of 4 mg/d for a total of 8 weeks | GXR was associated with significantly lower scores on CPRS-R:L oppositional subscale (-10.9 vs -6.8) and ADHD-RS-IV (-23.8 vs -11.4) compared with placebo |
| *1-mg dose was given only to subjects weighing <50 kg (<110 lbs) | |||
| ADHD: attention-deficit/hyperactivity disorder; ADHD-RS-IV: Attention-Deficit/Hyperactivity Disorder Rating Scale-IV; CPRS-R:L: Conners’ Parent Rating Scale-Revised: Long Form; GXR: guanfacine extended release | |||
Tolerability
In the phase III trials, the most commonly reported drug-related adverse reactions (occurring in ≥2% of patients) were:
- somnolence (38%)
- headache (24%)
- fatigue (14%)
- upper abdominal pain (10%)
- nausea, lethargy, dizziness, hypotension/decreased blood pressure, irritability (6% for each)
- decreased appetite (5%)
- dry mouth (4%)
- constipation (3%).
Many of these adverse reactions appear to be dose-related, particularly somnolence, sedation, abdominal pain, dizziness, and hypotension/decreased blood pressure.
Overall, GXR was well tolerated; clinicians rated most events as mild to moderate. Twelve percent of GXR patients discontinued the clinical studies because of adverse events, compared with 4% in the placebo groups. The most common adverse reactions leading to discontinuation were somnolence/sedation (6%) and fatigue (2%). Less common adverse reactions leading to discontinuation (occurring in 1% of patients) included hypotension/decreased blood pressure, headache, and dizziness.
Open-label safety trial. Sallee et al10 conducted a longer-term, open-label, flexible-dose safety continuation study of 259 GXR-treated patients (mean exposure 10 months), some of whom also received a psychostimulant. Common adverse reactions (occurring in ≥5% of subjects) included somnolence (45%), headache (26%), fatigue (16%), upper abdominal pain (11%), hypotension/decreased blood pressure (10%), vomiting (9%), dizziness (7%), nausea (7%), weight gain (7%), and irritability (6%).10 In a subset of patients, the onset of sedative events typically occurred within the first 3 weeks of GXR treatment and then declined with maintenance to a frequency of approximately 16%. The rates of somnolence, sedation, or fatigue were lowest among patients who also received a psychostimulant ( Figure ).
Distribution of GXR doses before the end of this study was 37% of patients on 4 mg, 33% on 3 mg, 27% on 2 mg, and 3% on 1 mg, suggesting a preference for maintenance doses of 3 to 4 mg/d. The most frequent adverse reactions leading to discontinuation were somnolence (3%), syncopal events (2%), increased weight (2%), depression (2%), and fatigue (2%). Other adverse reactions leading to discontinuation (occurring in approximately 1% of patients) included hypotension/decreased blood pressure, sedation, headache, and lethargy. Serious adverse reactions in the longer-term study in >1 patient included syncope (2%) and convulsion (0.4%).
Figure: Incidence of somnolence, sedation, and fatigue in study patients receiving GXR
with or without psychostimulants
In an open-label continuation study of 259 patients treated with guanfacine extended release (GXR), somnolence, sedation, or fatigue was reported by 49% of subjects overall, 59% of those who received GXR monotherapy, and 11% of those given GXR with a psychostimulant.
GXR: guanfacine extended release
Source: Reprinted with permission from Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226 Safety warnings relating to the likelihood of hypotension, bradycardia, and possible syncope when prescribing GXR should be understood in the context of its pharmacologic action to lower heart rate and blood pressure. In the short-term (8 to 9 weeks) controlled trials, the maximum mean changes from baseline in systolic blood pressure, diastolic blood pressure, and pulse were -5 mm Hg, -3 mm Hg, and -6 bpm, respectively, for all dose groups combined. These changes, which generally occurred 1 week after reaching target doses of 1 to 4 mg/d, were dose-dependent but usually modest and did not cause other symptoms; however, hypotension and bradycardia can occur.
In the longer-term, open-label safety study,10 maximum decreases in systolic and diastolic blood pressure occurred in the first month of treatment; decreases were less pronounced over time. Syncope occurred in 1% of pediatric subjects but was not dose-dependent. Guanfacine IR can increase QT interval but not in a dose-dependent fashion.
Dosing
The approved dose range for GXR is 1 to 4 mg once daily in the morning. Initiate treatment at 1 mg/d, and adjust the dose in increments of no more than 1 mg/week, evaluating the patient weekly. GXR maintenance therapy is frequently in the range of 2 to 4 mg/d.
Because adverse events such as hypotension, bradycardia, and sedation are dose-related, evaluate benefit and risk using mg/kg range approximation. GXR efficacy on a weight-adjusted (mg/kg) basis is consistent across a dosage range of 0.01 to 0.17 mg/kg/d. Clinically relevant improvements are usually observed beginning at doses of 0.05 to 0.08 mg/kg/d. In clinical trials, efficacy increased with increasing weight-adjusted dose (mg/kg), so if GXR is well-tolerated, doses up to 0.12 mg/kg once daily may provide additional benefit up to the maximum of 4 mg/d.
Instruct patients to swallow GXR whole because crushing, chewing, or otherwise breaking the tablet’s enteric coating will markedly enhance guanfacine release.
Abruptly discontinuing GXR is associated with infrequent, transient elevations in blood pressure above the patient’s baseline (ie, rebound). To minimize these effects, GXR should be gradually tapered in decrements of no more than 1 mg every 3 to 7 days. Isolated missed doses of GXR generally are not a problem, but ≥2 consecutive missed doses may warrant reinitiation of the titration schedule.
Related resource
- Guanfacine extended release (Intuniv) prescribing information. www.intuniv.com/documents/INTUNIV_Full_Prescribing_Information.pdf.
Drug brand names
- Atomoxetine • Strattera
- Guanfacine extended release • Intuniv
- Guanfacine immediate release • Tenex
- Ketoconazole • Nizoral
- Rifampin • Rifadin, Rimactane
- Valproic acid • Depakene, Depakote
Disclosure
Dr. Sallee receives grant/research support from the National Institutes of Health. He is a consultant to Otsuka, Nextwave, and Sepracor and a consultant to and speaker for Shire. Dr. Sallee is a consultant to, shareholder of, and member of the board of directors of P2D Inc. and a principal in Satiety Solutions.
Guanfacine extended release (GXR)—a selective α-2 adrenergic agonist FDA-approved for the treatment of attention-deficit/hyperactivity disorder (ADHD)—has demonstrated efficacy for inattentive and hyperactive/impulsive symptom domains in 2 large trials lasting 8 and 9 weeks.1,2 GXR’s once-daily formulation may increase adherence and deliver consistent control of symptoms across a full day ( Table 1 ).
Table 1
Guanfacine extended release: Fast facts
| Brand name: Intuniv |
| Indication: Attention-deficit/hyperactivity disorder |
| Approval date: September 3, 2009 |
| Availability date: November 2009 |
| Manufacturer: Shire |
| Dosing forms: 1-mg, 2-mg, 3-mg, and 4-mg extended-release tablets |
| Recommended dosage: 0.05 to 0.12 mg/kg once daily |
Clinical implications
GXR exhibits enhancement of noradrenergic pathways through selective direct receptor action in the prefrontal cortex.3 This mechanism of action is different from that of other FDA-approved ADHD medications. GXR can be used alone or in combination with stimulants or atomoxetine for treating complex ADHD, such as cases accompanied by oppositional features and emotional dysregulation or characterized by partial stimulant response.
How it works
Guanfacine—originally developed as an immediate-release (IR) antihypertensive—reduces sympathetic tone, causing centrally mediated vasodilation and reduced heart rate. Although GXR’s mechanism of action in ADHD is not known, the drug is a selective α-2A receptor agonist thought to directly engage postsynaptic receptors in the prefrontal cortex (PFC), an area of the brain believed to play a major role in attentional and organizational functions that preclinical research has linked to ADHD.3
The postsynaptic α-2A receptor is thought to play a central role in the optimal functioning of the PFC as illustrated by the “inverted U hypothesis of PFC activation.”4 In this model, cyclic adenosine monophosphate (cAMP) levels build within the prefrontal cortical neurons and cause specific ion channels—hyperpolarization-activated cyclic nucleotide gated (HCN) channels—to open on dendritic spines of these neurons.5 Activation of HCN channels effectively reduces membrane resistance, cutting off synaptic inputs and disconnecting PFC network connections. Because α-2A receptors are located in proximity to HCN channels, their stimulation by GXR closes HCN channels, inhibits further production of cAMP, and reestablishes synaptic function and the resulting network connectivity.5 Blockade of α-2A receptors by yohimbine reverses this process, eroding network connectivity, and in monkeys has been demonstrated to impair working memory,6 damage inhibition/impulse control, and produce locomotor hyperactivity.
Direct stimulation by GXR of the postsynaptic α-2A receptors is thought to:
- strengthen working memory
- reduce susceptibility to distraction
- improve attention regulation
- improve behavioral inhibition
- enhance impulse control.7
Pharmacokinetics
GXR offers enhanced pharmaceutics relative to IR guanfacine. IR guanfacine exhibits poor absorption characteristics—peak plasma concentration is achieved too rapidly and then declines precipitously, with considerable inter-individual variation.
GXR’s once-daily formulation is implemented by a proprietary enteric-coated sustained release mechanism8 that is meant to:
- control absorption
- provide a broad but flat plasma concentration profile
- reduce inter-individual variation of guanfacine exposure.
Compared with IR guanfacine, GXR exhibits delayed time of maximum concentration (Tmax) and reduced maximum concentration (Cmax). Therapeutic concentrations can be sustained over longer periods with reduced peak-to-trough fluctuation,8 which tends to improve tolerability and symptom control throughout the day. The convenience of once-daily dosing also may increase adherence.
GXR’s pharmacokinetic characteristics do not change with dose, but high-fat meals will increase absorption of the drug—Cmax increases by 75% and area under the plasma concentration time curve increases by 40%. Because GXR primarily is metabolized through cytochrome P450 (CYP) 3A4, CYP3A4 inhibitors such as ketoconazole will increase guanfacine plasma concentrations and elevate the risk of adverse events such as bradycardia, hypotension, and sedation. Conversely, CYP3A4 inducers such as rifampin will significantly reduce total guanfacine exposure. Coadministration of valproic acid with GXR can result in increased valproic acid levels, producing additive CNS side effects.
Efficacy
GXR reduced both inattentive and hyperactive/impulsive symptoms in 2 phase III, forced-dose, parallel-design, randomized, placebo-controlled trials ( Table 2 ). In the first trial,1 345 children age 6 to 17 received placebo or GXR, 2 mg, 3 mg, or 4 mg once daily for 8 weeks. In the second study,2 324 children age 6 to 17 received placebo or GXR, 1 mg, 2 mg, 3 mg, or 4 mg, once daily for 9 weeks; the 1-mg dose was given only to patients weighing <50 kg (<110 lbs).
In both trials, doses were increased in increments of 1 mg/week, and investigators evaluated participants’ ADHD signs and symptoms once a week using the clinician administered and scored ADHD Rating Scale-IV (ADHD-RS-IV). The primary outcome was change in total ADHD-RS-IV score from baseline to endpoint.
In both trials, patients taking GXR demonstrated statistically signifcant improvements in ADHD-RS-IV score starting 1 to 2 weeks after they began receiving once-daily GXR:
- In the first trial, the mean reduction in ADHD-RS-IV total score at endpoint was –16.7 for GXR compared with –8.9 for placebo (P < .0001).
- In the second, the reduction was –19.6 for GXR and –12.2 for placebo (P=.004).
Placebo-adjusted least squares mean changes from baseline were statistically significant for all GXR doses in the randomized treatment groups in both studies.
Secondary efficacy outcome measures included the Conners’ Parent Rating Scale-Revised: Short Form (CPRS-R) and the Conners’ Teacher Rating Scale-Revised: Short Form (CTRS-R).
Significant improvements were seen on both scales. On the CPRS-R, parents reported significant improvement across a full day (as measured at 6 PM, 8 PM, and 6 AM the next day). On the CTRS-R—which was used only in the first trial—teachers reported significant improvement throughout the school day (as measured at 10 AM and 2 PM).
Treating oppositional symptoms. In a collateral study,9 GXR was evaluated in complex ADHD patients age 6 to 12 who exhibited oppositional symptoms. The primary efficacy measure was change from baseline to endpoint in the oppositional subscale of the Conners’ Parent Rating Scale-Revised: Long Form (CPRS-R:L) score.
All subjects randomized to GXR started on a dose of 1 mg/d—which could be titrated by 1 mg/week during the 5-week, dose-optimization period to a maximum of 4 mg/d—and were maintained at their optimal doses for 3 additional weeks. Among the 217 subjects enrolled, 138 received GXR and 79, placebo.
Least-squares mean reductions from baseline to endpoint in CPRS-R:L oppositional subscale scores were –10.9 in the GXR group compared with –6.8 in the placebo group (P < .001; effect size 0.590). The GXR-treated group showed a significantly greater reduction in ADHD-RS-IV total score from baseline to endpoint compared with the placebo group (–23.8 vs –11.4, respectively, P < .001; effect size 0.916).
Table 2
Randomized, controlled trials supporting GXR’s effectiveness
for treating ADHD symptoms
| Study | Subjects | GXR dosages | Results |
|---|---|---|---|
| Biederman et al, 20087 ; phase III, forced-dose parallel-design | 345 ADHD patients age 6 to 17 | 2, 3, or 4 mg given once daily for 8 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-16.7 vs -8.9) |
| Sallee et al, 20098 ; phase III, forced-dose parallel-design | 324 ADHD patients age 6 to 17 | 1,* 2, 3, or 4 mg given once daily for 9 weeks | GXR was associated with significantly lower ADHD-RS-IV score compared with placebo (-19.6 vs -12.2) |
| Connor et al, 20099 ; collateral study | 217 complex ADHD patients age 6 to 12 with oppositional symptoms | Starting dose 1 mg/d, titrated to a maximum of 4 mg/d for a total of 8 weeks | GXR was associated with significantly lower scores on CPRS-R:L oppositional subscale (-10.9 vs -6.8) and ADHD-RS-IV (-23.8 vs -11.4) compared with placebo |
| *1-mg dose was given only to subjects weighing <50 kg (<110 lbs) | |||
| ADHD: attention-deficit/hyperactivity disorder; ADHD-RS-IV: Attention-Deficit/Hyperactivity Disorder Rating Scale-IV; CPRS-R:L: Conners’ Parent Rating Scale-Revised: Long Form; GXR: guanfacine extended release | |||
Tolerability
In the phase III trials, the most commonly reported drug-related adverse reactions (occurring in ≥2% of patients) were:
- somnolence (38%)
- headache (24%)
- fatigue (14%)
- upper abdominal pain (10%)
- nausea, lethargy, dizziness, hypotension/decreased blood pressure, irritability (6% for each)
- decreased appetite (5%)
- dry mouth (4%)
- constipation (3%).
Many of these adverse reactions appear to be dose-related, particularly somnolence, sedation, abdominal pain, dizziness, and hypotension/decreased blood pressure.
Overall, GXR was well tolerated; clinicians rated most events as mild to moderate. Twelve percent of GXR patients discontinued the clinical studies because of adverse events, compared with 4% in the placebo groups. The most common adverse reactions leading to discontinuation were somnolence/sedation (6%) and fatigue (2%). Less common adverse reactions leading to discontinuation (occurring in 1% of patients) included hypotension/decreased blood pressure, headache, and dizziness.
Open-label safety trial. Sallee et al10 conducted a longer-term, open-label, flexible-dose safety continuation study of 259 GXR-treated patients (mean exposure 10 months), some of whom also received a psychostimulant. Common adverse reactions (occurring in ≥5% of subjects) included somnolence (45%), headache (26%), fatigue (16%), upper abdominal pain (11%), hypotension/decreased blood pressure (10%), vomiting (9%), dizziness (7%), nausea (7%), weight gain (7%), and irritability (6%).10 In a subset of patients, the onset of sedative events typically occurred within the first 3 weeks of GXR treatment and then declined with maintenance to a frequency of approximately 16%. The rates of somnolence, sedation, or fatigue were lowest among patients who also received a psychostimulant ( Figure ).
Distribution of GXR doses before the end of this study was 37% of patients on 4 mg, 33% on 3 mg, 27% on 2 mg, and 3% on 1 mg, suggesting a preference for maintenance doses of 3 to 4 mg/d. The most frequent adverse reactions leading to discontinuation were somnolence (3%), syncopal events (2%), increased weight (2%), depression (2%), and fatigue (2%). Other adverse reactions leading to discontinuation (occurring in approximately 1% of patients) included hypotension/decreased blood pressure, sedation, headache, and lethargy. Serious adverse reactions in the longer-term study in >1 patient included syncope (2%) and convulsion (0.4%).
Figure: Incidence of somnolence, sedation, and fatigue in study patients receiving GXR
with or without psychostimulants
In an open-label continuation study of 259 patients treated with guanfacine extended release (GXR), somnolence, sedation, or fatigue was reported by 49% of subjects overall, 59% of those who received GXR monotherapy, and 11% of those given GXR with a psychostimulant.
GXR: guanfacine extended release
Source: Reprinted with permission from Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226 Safety warnings relating to the likelihood of hypotension, bradycardia, and possible syncope when prescribing GXR should be understood in the context of its pharmacologic action to lower heart rate and blood pressure. In the short-term (8 to 9 weeks) controlled trials, the maximum mean changes from baseline in systolic blood pressure, diastolic blood pressure, and pulse were -5 mm Hg, -3 mm Hg, and -6 bpm, respectively, for all dose groups combined. These changes, which generally occurred 1 week after reaching target doses of 1 to 4 mg/d, were dose-dependent but usually modest and did not cause other symptoms; however, hypotension and bradycardia can occur.
In the longer-term, open-label safety study,10 maximum decreases in systolic and diastolic blood pressure occurred in the first month of treatment; decreases were less pronounced over time. Syncope occurred in 1% of pediatric subjects but was not dose-dependent. Guanfacine IR can increase QT interval but not in a dose-dependent fashion.
Dosing
The approved dose range for GXR is 1 to 4 mg once daily in the morning. Initiate treatment at 1 mg/d, and adjust the dose in increments of no more than 1 mg/week, evaluating the patient weekly. GXR maintenance therapy is frequently in the range of 2 to 4 mg/d.
Because adverse events such as hypotension, bradycardia, and sedation are dose-related, evaluate benefit and risk using mg/kg range approximation. GXR efficacy on a weight-adjusted (mg/kg) basis is consistent across a dosage range of 0.01 to 0.17 mg/kg/d. Clinically relevant improvements are usually observed beginning at doses of 0.05 to 0.08 mg/kg/d. In clinical trials, efficacy increased with increasing weight-adjusted dose (mg/kg), so if GXR is well-tolerated, doses up to 0.12 mg/kg once daily may provide additional benefit up to the maximum of 4 mg/d.
Instruct patients to swallow GXR whole because crushing, chewing, or otherwise breaking the tablet’s enteric coating will markedly enhance guanfacine release.
Abruptly discontinuing GXR is associated with infrequent, transient elevations in blood pressure above the patient’s baseline (ie, rebound). To minimize these effects, GXR should be gradually tapered in decrements of no more than 1 mg every 3 to 7 days. Isolated missed doses of GXR generally are not a problem, but ≥2 consecutive missed doses may warrant reinitiation of the titration schedule.
Related resource
- Guanfacine extended release (Intuniv) prescribing information. www.intuniv.com/documents/INTUNIV_Full_Prescribing_Information.pdf.
Drug brand names
- Atomoxetine • Strattera
- Guanfacine extended release • Intuniv
- Guanfacine immediate release • Tenex
- Ketoconazole • Nizoral
- Rifampin • Rifadin, Rimactane
- Valproic acid • Depakene, Depakote
Disclosure
Dr. Sallee receives grant/research support from the National Institutes of Health. He is a consultant to Otsuka, Nextwave, and Sepracor and a consultant to and speaker for Shire. Dr. Sallee is a consultant to, shareholder of, and member of the board of directors of P2D Inc. and a principal in Satiety Solutions.
1. Biederman J, Melmed RD, Patel A, et al. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e73-e84.
2. Sallee F, McGough J, Wigal T, et al. For the SPD503 Study Group Guanfacine extended release in children and adolescents with attention deficit hyperactivity disorder: a placebo-controlled trial. J Am Acad Child Adolesc Psychiatry. 2009;48(2):155-165.
3. Arnsten AF, Cai JX, Goldman-Rakic PS. The α-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for α-2 receptor subtypes. J Neurosci. 1988;8:4287-4298.
4. Vijayraghavan S, Wang M, Birnbaum SG, et al. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376-384.
5. Wang M, Ramos BP, Paspalas CD, et al. α 2-A adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397-410.
6. Li BM, Mei ZT. Delayed-response deficit induced by local injection of the α 2-adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134-139.
7. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158:1067-1074.
8. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29:617-625.
9. Connor D, Spencer T, Kratochvil C, et al. Effects of guanfacine extended release on secondary measures in children with attention-deficit/hyperactivity disorder and oppositional symptoms. Abstract presented at: Annual Meeting of the American Psychiatric Association; May 18, 2009; San Francisco, CA.
10. Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226.
1. Biederman J, Melmed RD, Patel A, et al. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e73-e84.
2. Sallee F, McGough J, Wigal T, et al. For the SPD503 Study Group Guanfacine extended release in children and adolescents with attention deficit hyperactivity disorder: a placebo-controlled trial. J Am Acad Child Adolesc Psychiatry. 2009;48(2):155-165.
3. Arnsten AF, Cai JX, Goldman-Rakic PS. The α-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for α-2 receptor subtypes. J Neurosci. 1988;8:4287-4298.
4. Vijayraghavan S, Wang M, Birnbaum SG, et al. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376-384.
5. Wang M, Ramos BP, Paspalas CD, et al. α 2-A adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397-410.
6. Li BM, Mei ZT. Delayed-response deficit induced by local injection of the α 2-adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134-139.
7. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158:1067-1074.
8. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29:617-625.
9. Connor D, Spencer T, Kratochvil C, et al. Effects of guanfacine extended release on secondary measures in children with attention-deficit/hyperactivity disorder and oppositional symptoms. Abstract presented at: Annual Meeting of the American Psychiatric Association; May 18, 2009; San Francisco, CA.
10. Sallee FR, Lyne A, Wigal T, et al. Long-term safety and efficacy of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2009;19(3):215-226.