User login
Problems with myocardial infarction definitions
To the Editor: In the December 2013 Cleveland Clinic Journal of Medicine, Tehrani and Seto provide a review of the updated definitions of myocardial infarction (MI).1 A key concept incorporated into the structured definitions is that cardiac biomarkers must be interpreted in a clinical context.2 This in turn helps better align the laboratory and clinical findings with the pathophysiologic processes.
However, there is another dimension to the definitions that is sometimes overlooked and requires careful attention: translation of the definitions into codes and comparable databases. Accurate and consistent coding according to the International Statistical Classification of Diseases, ninth edition (ICD-9), and the ICD-10 is critically vital to the appropriate analysis of data, research, quality measurement, and reimbursement of services related to MI. Unfortunately, there is no straightforward translation of the definitions into ICD-9 codes, and the challenge is further confounded when it comes to ICD-10, which will be implemented in October 2014.
The ICD-10-CM Index to Diseases does not yet recognize this nomenclature. ST-elevation MI is the default for the unspecified term “acute MI.” Non-ST-elevation MI requires more explicit documentation and is classified based on whether it occurs during or after a variety of procedures. Type 2 MI is particularly challenging because of the several possible ways to code the condition—for example, as acute subendocardial MI (I21.4), demand ischemia (I24.8), or acute MI, unspecified (I21.9). Coding guidelines are assumed to standardize the approach to coding these conditions, but there is no guarantee that comparability of the data will endure biases of code assignment. Although extreme precision in disease capture by coding may not exist, other clinical conditions have better correlations with coding classifications, such as stages of chronic kidney disease ranging from stage 1 through end-stage renal disease (N18.1 through N18.6). Furthermore, ICD-10 codes are insufficient to clearly distinguish the type of acute MI.3
While the concept of acute MI applies when the stated date of onset is less than 8 weeks in ICD-9,4 it changes to 4 weeks in ICD-10. “Acute” can reference an initial or a subsequent MI in ICD-10, but it does not define the time frame of the MI.5 This is different than in ICD-9, where the concept of “subsequent” refers to a “subsequent episode of care.”
On the surface, these variations may not seem significant. However, the discriminatory efforts to better define a patient’s clinical condition using the new definitions may get diluted by the challenges of the coding process. The implications on comparability of quality metrics and reporting are not to be underestimated and need to be assessed on a national level.
- Tehrani DM, Seto AH. Third universal definition of myocardial infarction: update, caveats, differential diagnoses. Cleve Clin J Med 2013; 80:777–786.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol 2012; 60:1581–1598.
- Alexandrescu R, Bottle A, Jarman B, Aylin P. Current ICD10 codes are insufficient to clearly distinguish acute myocardial infarction type: a descriptive study. BMC Health Serv Res 2013; 13:468.
- ICD-9-CM Addenda, Conversion Table, and Guidelines. www.cdc.gov
- WEDI Strategic National Implementation Process (SNIP). Acute Myocardial Infarction Issue Brief. www.wedi.org. Accessed February 3, 2014.
To the Editor: In the December 2013 Cleveland Clinic Journal of Medicine, Tehrani and Seto provide a review of the updated definitions of myocardial infarction (MI).1 A key concept incorporated into the structured definitions is that cardiac biomarkers must be interpreted in a clinical context.2 This in turn helps better align the laboratory and clinical findings with the pathophysiologic processes.
However, there is another dimension to the definitions that is sometimes overlooked and requires careful attention: translation of the definitions into codes and comparable databases. Accurate and consistent coding according to the International Statistical Classification of Diseases, ninth edition (ICD-9), and the ICD-10 is critically vital to the appropriate analysis of data, research, quality measurement, and reimbursement of services related to MI. Unfortunately, there is no straightforward translation of the definitions into ICD-9 codes, and the challenge is further confounded when it comes to ICD-10, which will be implemented in October 2014.
The ICD-10-CM Index to Diseases does not yet recognize this nomenclature. ST-elevation MI is the default for the unspecified term “acute MI.” Non-ST-elevation MI requires more explicit documentation and is classified based on whether it occurs during or after a variety of procedures. Type 2 MI is particularly challenging because of the several possible ways to code the condition—for example, as acute subendocardial MI (I21.4), demand ischemia (I24.8), or acute MI, unspecified (I21.9). Coding guidelines are assumed to standardize the approach to coding these conditions, but there is no guarantee that comparability of the data will endure biases of code assignment. Although extreme precision in disease capture by coding may not exist, other clinical conditions have better correlations with coding classifications, such as stages of chronic kidney disease ranging from stage 1 through end-stage renal disease (N18.1 through N18.6). Furthermore, ICD-10 codes are insufficient to clearly distinguish the type of acute MI.3
While the concept of acute MI applies when the stated date of onset is less than 8 weeks in ICD-9,4 it changes to 4 weeks in ICD-10. “Acute” can reference an initial or a subsequent MI in ICD-10, but it does not define the time frame of the MI.5 This is different than in ICD-9, where the concept of “subsequent” refers to a “subsequent episode of care.”
On the surface, these variations may not seem significant. However, the discriminatory efforts to better define a patient’s clinical condition using the new definitions may get diluted by the challenges of the coding process. The implications on comparability of quality metrics and reporting are not to be underestimated and need to be assessed on a national level.
To the Editor: In the December 2013 Cleveland Clinic Journal of Medicine, Tehrani and Seto provide a review of the updated definitions of myocardial infarction (MI).1 A key concept incorporated into the structured definitions is that cardiac biomarkers must be interpreted in a clinical context.2 This in turn helps better align the laboratory and clinical findings with the pathophysiologic processes.
However, there is another dimension to the definitions that is sometimes overlooked and requires careful attention: translation of the definitions into codes and comparable databases. Accurate and consistent coding according to the International Statistical Classification of Diseases, ninth edition (ICD-9), and the ICD-10 is critically vital to the appropriate analysis of data, research, quality measurement, and reimbursement of services related to MI. Unfortunately, there is no straightforward translation of the definitions into ICD-9 codes, and the challenge is further confounded when it comes to ICD-10, which will be implemented in October 2014.
The ICD-10-CM Index to Diseases does not yet recognize this nomenclature. ST-elevation MI is the default for the unspecified term “acute MI.” Non-ST-elevation MI requires more explicit documentation and is classified based on whether it occurs during or after a variety of procedures. Type 2 MI is particularly challenging because of the several possible ways to code the condition—for example, as acute subendocardial MI (I21.4), demand ischemia (I24.8), or acute MI, unspecified (I21.9). Coding guidelines are assumed to standardize the approach to coding these conditions, but there is no guarantee that comparability of the data will endure biases of code assignment. Although extreme precision in disease capture by coding may not exist, other clinical conditions have better correlations with coding classifications, such as stages of chronic kidney disease ranging from stage 1 through end-stage renal disease (N18.1 through N18.6). Furthermore, ICD-10 codes are insufficient to clearly distinguish the type of acute MI.3
While the concept of acute MI applies when the stated date of onset is less than 8 weeks in ICD-9,4 it changes to 4 weeks in ICD-10. “Acute” can reference an initial or a subsequent MI in ICD-10, but it does not define the time frame of the MI.5 This is different than in ICD-9, where the concept of “subsequent” refers to a “subsequent episode of care.”
On the surface, these variations may not seem significant. However, the discriminatory efforts to better define a patient’s clinical condition using the new definitions may get diluted by the challenges of the coding process. The implications on comparability of quality metrics and reporting are not to be underestimated and need to be assessed on a national level.
- Tehrani DM, Seto AH. Third universal definition of myocardial infarction: update, caveats, differential diagnoses. Cleve Clin J Med 2013; 80:777–786.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol 2012; 60:1581–1598.
- Alexandrescu R, Bottle A, Jarman B, Aylin P. Current ICD10 codes are insufficient to clearly distinguish acute myocardial infarction type: a descriptive study. BMC Health Serv Res 2013; 13:468.
- ICD-9-CM Addenda, Conversion Table, and Guidelines. www.cdc.gov
- WEDI Strategic National Implementation Process (SNIP). Acute Myocardial Infarction Issue Brief. www.wedi.org. Accessed February 3, 2014.
- Tehrani DM, Seto AH. Third universal definition of myocardial infarction: update, caveats, differential diagnoses. Cleve Clin J Med 2013; 80:777–786.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol 2012; 60:1581–1598.
- Alexandrescu R, Bottle A, Jarman B, Aylin P. Current ICD10 codes are insufficient to clearly distinguish acute myocardial infarction type: a descriptive study. BMC Health Serv Res 2013; 13:468.
- ICD-9-CM Addenda, Conversion Table, and Guidelines. www.cdc.gov
- WEDI Strategic National Implementation Process (SNIP). Acute Myocardial Infarction Issue Brief. www.wedi.org. Accessed February 3, 2014.
In reply: Problems with myocardial infarction definitions
In Reply: We thank Dr. Antonios for his comments regarding the current shortcomings of the ICD-9 and ICD-10 coding systems in describing the acute MI types as defined in the universal definition. We share his concern that accurate and consistent coding of MIs may be difficult when the definition of MI changes over a short period of time. Such changes create a disconnect not only between our clinical terminology and coding systems, but also potentially between our conventional sense of a “heart attack” as an acute coronary syndrome or a clinically significant infarction rather than a small troponin elevation from demand ischemia. This has consequences not only for quality measures and reporting, but also for clinical research trials and clinical care. This is exemplified by reports of recent trials that were possibly prematurely discontinued, as the use of troponin thresholds may conflate large MIs with clinically insignificant ones.1
Recently, the Society for Cardiovascular Angiography and Interventions published a new definition of “clinically relevant” MI after revascularization.2 Rather than relying on troponins, which are elevated in as many as 24.3% of uncomplicated percutaneous coronary interventions and in 42% to 82% of uncomplicated coronary artery bypass grafting procedures (based on the 2007 universal definition), they point to extensive literature documenting that only patients with elevated creatine kinase MB more than 10 times the upper limit of normal after revascularization have a worsened prognosis. We favor this clinically relevant MI definition for post-revascularization MI. We also favor the use of creatine kinase MB as a less sensitive but more specific confirmatory marker for acute coronary syndromes (type 1) or clinically significant supply-demand (type 2) MI, when the symptoms or electrocardiographic signs are nondiagnostic, as they often are.3 However, until there is a consensus around a single definition, clinicians are effectively walking around a Tower of Babel and must take care to be specific when documenting an MI.
- Dangas GD, Kini AS, Sharma SK, et al. Impact of hemodynamic support with Impella 2.5 versus intra-aortic balloon pump on prognostically important clinical outcomes in patients undergoing high-risk percutaneous coronary intervention (from the PROTECT II Randomized Trial). Am J Cardiol 2014; 113:222–228.
- Moussa ID, Klein LW, Shah B, et al. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the society for cardiovascular angiography and interventions (SCAI). Catheter Cardiovasc Interv 2014; 83:27–36.
- Seto A, Tehrani D. Troponins should be confirmed with CK-MB in atypical presentations. J Am Coll Cardiol 2013; 61:1467–1468.
In Reply: We thank Dr. Antonios for his comments regarding the current shortcomings of the ICD-9 and ICD-10 coding systems in describing the acute MI types as defined in the universal definition. We share his concern that accurate and consistent coding of MIs may be difficult when the definition of MI changes over a short period of time. Such changes create a disconnect not only between our clinical terminology and coding systems, but also potentially between our conventional sense of a “heart attack” as an acute coronary syndrome or a clinically significant infarction rather than a small troponin elevation from demand ischemia. This has consequences not only for quality measures and reporting, but also for clinical research trials and clinical care. This is exemplified by reports of recent trials that were possibly prematurely discontinued, as the use of troponin thresholds may conflate large MIs with clinically insignificant ones.1
Recently, the Society for Cardiovascular Angiography and Interventions published a new definition of “clinically relevant” MI after revascularization.2 Rather than relying on troponins, which are elevated in as many as 24.3% of uncomplicated percutaneous coronary interventions and in 42% to 82% of uncomplicated coronary artery bypass grafting procedures (based on the 2007 universal definition), they point to extensive literature documenting that only patients with elevated creatine kinase MB more than 10 times the upper limit of normal after revascularization have a worsened prognosis. We favor this clinically relevant MI definition for post-revascularization MI. We also favor the use of creatine kinase MB as a less sensitive but more specific confirmatory marker for acute coronary syndromes (type 1) or clinically significant supply-demand (type 2) MI, when the symptoms or electrocardiographic signs are nondiagnostic, as they often are.3 However, until there is a consensus around a single definition, clinicians are effectively walking around a Tower of Babel and must take care to be specific when documenting an MI.
In Reply: We thank Dr. Antonios for his comments regarding the current shortcomings of the ICD-9 and ICD-10 coding systems in describing the acute MI types as defined in the universal definition. We share his concern that accurate and consistent coding of MIs may be difficult when the definition of MI changes over a short period of time. Such changes create a disconnect not only between our clinical terminology and coding systems, but also potentially between our conventional sense of a “heart attack” as an acute coronary syndrome or a clinically significant infarction rather than a small troponin elevation from demand ischemia. This has consequences not only for quality measures and reporting, but also for clinical research trials and clinical care. This is exemplified by reports of recent trials that were possibly prematurely discontinued, as the use of troponin thresholds may conflate large MIs with clinically insignificant ones.1
Recently, the Society for Cardiovascular Angiography and Interventions published a new definition of “clinically relevant” MI after revascularization.2 Rather than relying on troponins, which are elevated in as many as 24.3% of uncomplicated percutaneous coronary interventions and in 42% to 82% of uncomplicated coronary artery bypass grafting procedures (based on the 2007 universal definition), they point to extensive literature documenting that only patients with elevated creatine kinase MB more than 10 times the upper limit of normal after revascularization have a worsened prognosis. We favor this clinically relevant MI definition for post-revascularization MI. We also favor the use of creatine kinase MB as a less sensitive but more specific confirmatory marker for acute coronary syndromes (type 1) or clinically significant supply-demand (type 2) MI, when the symptoms or electrocardiographic signs are nondiagnostic, as they often are.3 However, until there is a consensus around a single definition, clinicians are effectively walking around a Tower of Babel and must take care to be specific when documenting an MI.
- Dangas GD, Kini AS, Sharma SK, et al. Impact of hemodynamic support with Impella 2.5 versus intra-aortic balloon pump on prognostically important clinical outcomes in patients undergoing high-risk percutaneous coronary intervention (from the PROTECT II Randomized Trial). Am J Cardiol 2014; 113:222–228.
- Moussa ID, Klein LW, Shah B, et al. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the society for cardiovascular angiography and interventions (SCAI). Catheter Cardiovasc Interv 2014; 83:27–36.
- Seto A, Tehrani D. Troponins should be confirmed with CK-MB in atypical presentations. J Am Coll Cardiol 2013; 61:1467–1468.
- Dangas GD, Kini AS, Sharma SK, et al. Impact of hemodynamic support with Impella 2.5 versus intra-aortic balloon pump on prognostically important clinical outcomes in patients undergoing high-risk percutaneous coronary intervention (from the PROTECT II Randomized Trial). Am J Cardiol 2014; 113:222–228.
- Moussa ID, Klein LW, Shah B, et al. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the society for cardiovascular angiography and interventions (SCAI). Catheter Cardiovasc Interv 2014; 83:27–36.
- Seto A, Tehrani D. Troponins should be confirmed with CK-MB in atypical presentations. J Am Coll Cardiol 2013; 61:1467–1468.
Myasthenia gravis
To the Editor: Dr. Li and colleagues provide a well-written article about what is generally believed regarding myasthenia gravis (MG). However, like most reviews, it perpetuates the myths surrounding current medical practice, resulting in delays in diagnosis, treatment initiation, and insurance approval and reimbursement, and therefore increased morbidity and mortality. Stricter statistical and editorial review is needed and what is known and unknown clearly stated. Patients, in particular those of us who are physicians ourselves, recognize that this is no academic quibble.
Myasthenia gravis was a clinical diagnosis until blood tests began to pick up antibodies. If the blood tests have to be positive to diagnose MG, then everyone diagnosed with MG will have positive blood tests. If the muscle studies have to show particular abnormalities to diagnose MG, then everyone diagnosed with MG will have those abnormalities. It makes doctors more comfortable to have these evidences of their understanding verified, but it does not help any of the patients who do not meet the testing criteria but have the clinical findings.
We know, in contrast to what was thought a number of years ago, that there are “seronegative” patients with clinical evidence of myasthenia who are antibody-positive. For those who are MuSK-positive, their problem is now well described, and although it affects a different part of the neuromuscular junction, it remains under the MG umbrella. We also know there are other antibodies, for which we have no commercial tests, in patients with symptoms of MG who respond to treatment for autoimmune problems. This article is relatively dismissive of the clinical validity of those antibodies, and certainly a degree of skepticism is a good thing as long as the patients remain diagnosed and treated.
It is of more than academic interest that these misconceptions and prejudices be recognized. At the very least, editorial boards should insist that statistics in papers reflect the diagnostic skills of the authors. If over 95% of an author’s diagnosed patients are seropositive, then one can suspect there is heavy reliance on blood studies for diagnosis, and rejection of those who do not meet those criteria. The statistics should read “over 95% of patients we diagnose with MG have positive blood studies” rather than “over 95% of patients with MG have positive blood studies.” Dismissing a significant portion of a patient population will also affect treatment statistics, which then should read that “for those who meet this criteria, —% will respond to…”
If patients meet clinical criteria for the diagnosis of MG and a large percentage do not have positive serology, then more research needs to be done into their particular autoimmune problems, and better testing may become commercially viable. Recognizing the problem will lead to better clinical diagnosis and treatment, and strict diagnostic criteria would permit their inclusion in studies. For many of us this would create a more open and questioning atmosphere as to our understanding of the spectrum of autoimmune myasthenia and the ability and willingness to diagnose and treat “seronegative” autoimmune myasthenia when we see it.
To the Editor: Dr. Li and colleagues provide a well-written article about what is generally believed regarding myasthenia gravis (MG). However, like most reviews, it perpetuates the myths surrounding current medical practice, resulting in delays in diagnosis, treatment initiation, and insurance approval and reimbursement, and therefore increased morbidity and mortality. Stricter statistical and editorial review is needed and what is known and unknown clearly stated. Patients, in particular those of us who are physicians ourselves, recognize that this is no academic quibble.
Myasthenia gravis was a clinical diagnosis until blood tests began to pick up antibodies. If the blood tests have to be positive to diagnose MG, then everyone diagnosed with MG will have positive blood tests. If the muscle studies have to show particular abnormalities to diagnose MG, then everyone diagnosed with MG will have those abnormalities. It makes doctors more comfortable to have these evidences of their understanding verified, but it does not help any of the patients who do not meet the testing criteria but have the clinical findings.
We know, in contrast to what was thought a number of years ago, that there are “seronegative” patients with clinical evidence of myasthenia who are antibody-positive. For those who are MuSK-positive, their problem is now well described, and although it affects a different part of the neuromuscular junction, it remains under the MG umbrella. We also know there are other antibodies, for which we have no commercial tests, in patients with symptoms of MG who respond to treatment for autoimmune problems. This article is relatively dismissive of the clinical validity of those antibodies, and certainly a degree of skepticism is a good thing as long as the patients remain diagnosed and treated.
It is of more than academic interest that these misconceptions and prejudices be recognized. At the very least, editorial boards should insist that statistics in papers reflect the diagnostic skills of the authors. If over 95% of an author’s diagnosed patients are seropositive, then one can suspect there is heavy reliance on blood studies for diagnosis, and rejection of those who do not meet those criteria. The statistics should read “over 95% of patients we diagnose with MG have positive blood studies” rather than “over 95% of patients with MG have positive blood studies.” Dismissing a significant portion of a patient population will also affect treatment statistics, which then should read that “for those who meet this criteria, —% will respond to…”
If patients meet clinical criteria for the diagnosis of MG and a large percentage do not have positive serology, then more research needs to be done into their particular autoimmune problems, and better testing may become commercially viable. Recognizing the problem will lead to better clinical diagnosis and treatment, and strict diagnostic criteria would permit their inclusion in studies. For many of us this would create a more open and questioning atmosphere as to our understanding of the spectrum of autoimmune myasthenia and the ability and willingness to diagnose and treat “seronegative” autoimmune myasthenia when we see it.
To the Editor: Dr. Li and colleagues provide a well-written article about what is generally believed regarding myasthenia gravis (MG). However, like most reviews, it perpetuates the myths surrounding current medical practice, resulting in delays in diagnosis, treatment initiation, and insurance approval and reimbursement, and therefore increased morbidity and mortality. Stricter statistical and editorial review is needed and what is known and unknown clearly stated. Patients, in particular those of us who are physicians ourselves, recognize that this is no academic quibble.
Myasthenia gravis was a clinical diagnosis until blood tests began to pick up antibodies. If the blood tests have to be positive to diagnose MG, then everyone diagnosed with MG will have positive blood tests. If the muscle studies have to show particular abnormalities to diagnose MG, then everyone diagnosed with MG will have those abnormalities. It makes doctors more comfortable to have these evidences of their understanding verified, but it does not help any of the patients who do not meet the testing criteria but have the clinical findings.
We know, in contrast to what was thought a number of years ago, that there are “seronegative” patients with clinical evidence of myasthenia who are antibody-positive. For those who are MuSK-positive, their problem is now well described, and although it affects a different part of the neuromuscular junction, it remains under the MG umbrella. We also know there are other antibodies, for which we have no commercial tests, in patients with symptoms of MG who respond to treatment for autoimmune problems. This article is relatively dismissive of the clinical validity of those antibodies, and certainly a degree of skepticism is a good thing as long as the patients remain diagnosed and treated.
It is of more than academic interest that these misconceptions and prejudices be recognized. At the very least, editorial boards should insist that statistics in papers reflect the diagnostic skills of the authors. If over 95% of an author’s diagnosed patients are seropositive, then one can suspect there is heavy reliance on blood studies for diagnosis, and rejection of those who do not meet those criteria. The statistics should read “over 95% of patients we diagnose with MG have positive blood studies” rather than “over 95% of patients with MG have positive blood studies.” Dismissing a significant portion of a patient population will also affect treatment statistics, which then should read that “for those who meet this criteria, —% will respond to…”
If patients meet clinical criteria for the diagnosis of MG and a large percentage do not have positive serology, then more research needs to be done into their particular autoimmune problems, and better testing may become commercially viable. Recognizing the problem will lead to better clinical diagnosis and treatment, and strict diagnostic criteria would permit their inclusion in studies. For many of us this would create a more open and questioning atmosphere as to our understanding of the spectrum of autoimmune myasthenia and the ability and willingness to diagnose and treat “seronegative” autoimmune myasthenia when we see it.
In reply: Myasthenia gravis
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
Researchers create reversible LMWH
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.” ![]()
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.” ![]()
Scientists say they’ve created a synthetic form of low-molecular-weight heparin (LMWH) that is both reversible and safe for patients with poor kidney function.
In the event of uncontrolled bleeding, this synthetic heparin can be reversed by an existing drug.
And the LMWH is cleared by the liver rather than the kidneys.
The team described their creation of the drug in Nature Chemical Biology.
“When doctors talk to me about the kind of heparin they want to use during and after surgery, they want it reversible, and they want it to not go through the kidneys,” said study author Jian Liu, PhD, of the University of North Carolina, Chapel Hill.
Dr Liu noted that up to 5% of patients receiving heparin experience some form of uncontrolled bleeding. Patients receiving unfractionated heparin are in less danger because there is an existing FDA-approved antidote available, protamine.
But protamine is not as effective in reversing LMWH. So Dr Liu and his colleagues tweaked the drug’s molecular structure so that protamine is able to deactivate LMWH.
The team used a chemo-enzymatic process to synthesize the LMWH, an approach they developed in research on a simpler anticoagulant published in Science in 2011. Synthesizing the LMWH allowed them to make improvements on the animal-derived form of the drug.
That form of LMWH is cleared from the body by the kidneys, which can make it unsuitable for patients with a weakened renal system. So the researchers made changes that allowed their LMWH to bind to receptors that clear it through the liver.
“If a person’s kidneys aren’t effectively clearing heparin from the blood, the drug stays active in the body for longer than expected,” said study author Nigel Key, MB ChB, also of the University of North Carolina.
“That can represent a potentially dangerous situation for the physician, pharmacist, and patient.”
LMWH did prove dangerous in 2008, when more than 80 people died and hundreds of others suffered adverse reactions to the drug. Authorities linked the problems to a contaminant in raw natural heparin from China.
“Whenever you mix the food chain and the drug chain together, you end up with potential for disaster,” said study author Robert Linhardt, PhD, of the Rensselaer Polytechnic Institute in Troy, New York.
“Whether it comes from contamination, adulteration, impurities like viruses or prions—any of those possibilities are much more likely when you make something in an uncontrolled environment. This is a drug that millions of people rely upon, and it’s important to develop a safe, synthetic alternative to the current supply chain.”
LMWH makes up more than half the US market for heparin. The researchers said the new version they created is a safe, economically viable alternative to the existing animal-derived supply.
“The pig stuff has served us well for 50 years and is very inexpensive, but if we cannot control the supply chain, we cannot ensure the safety of the drug,” Dr Liu said. “I am working for the day when synthetic heparin can be brewed in large laboratories at a low cost.” ![]()
Hepatitis C virus: Here comes all-oral treatment
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2

The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES

HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
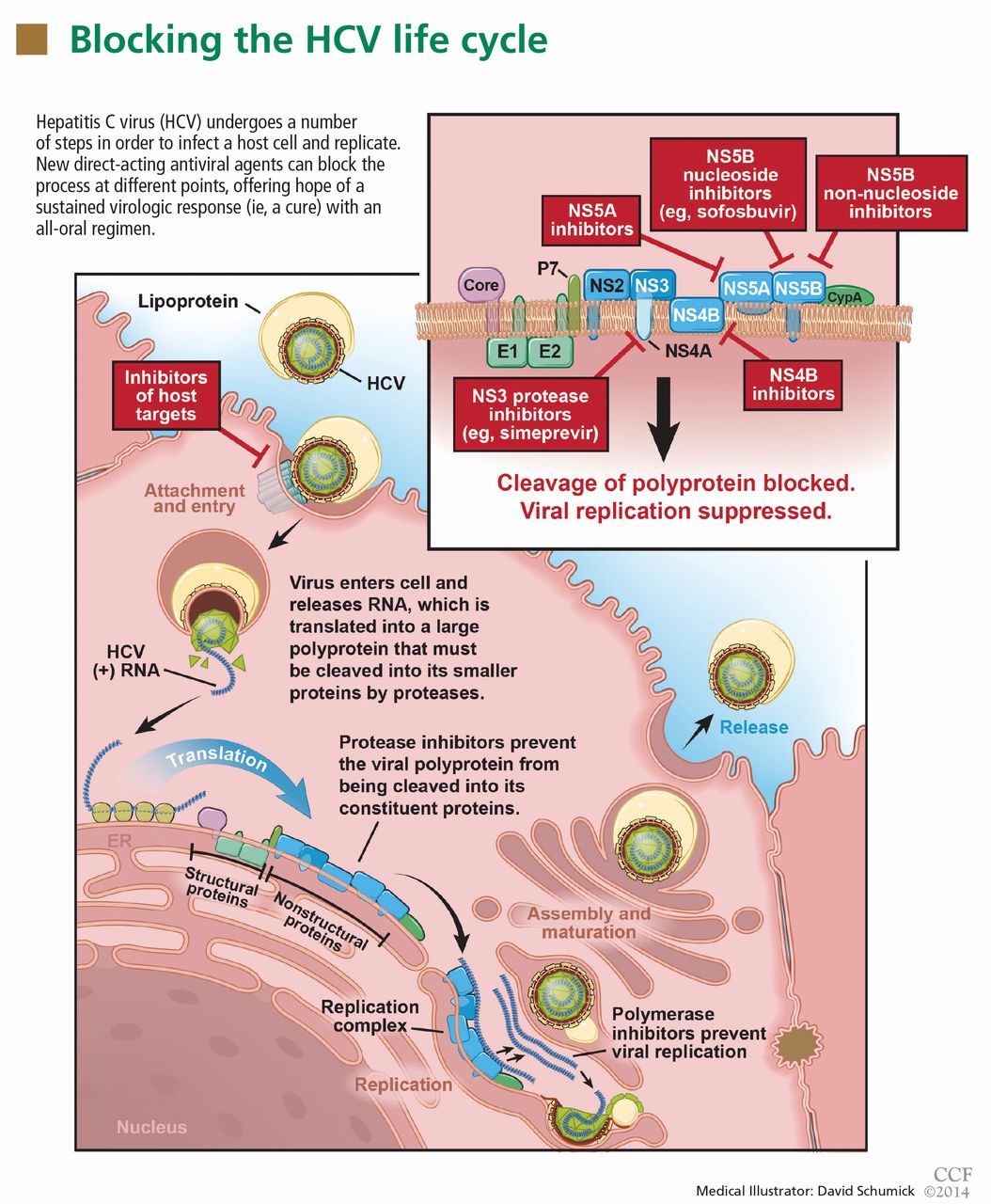
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.

Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir

Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications

Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
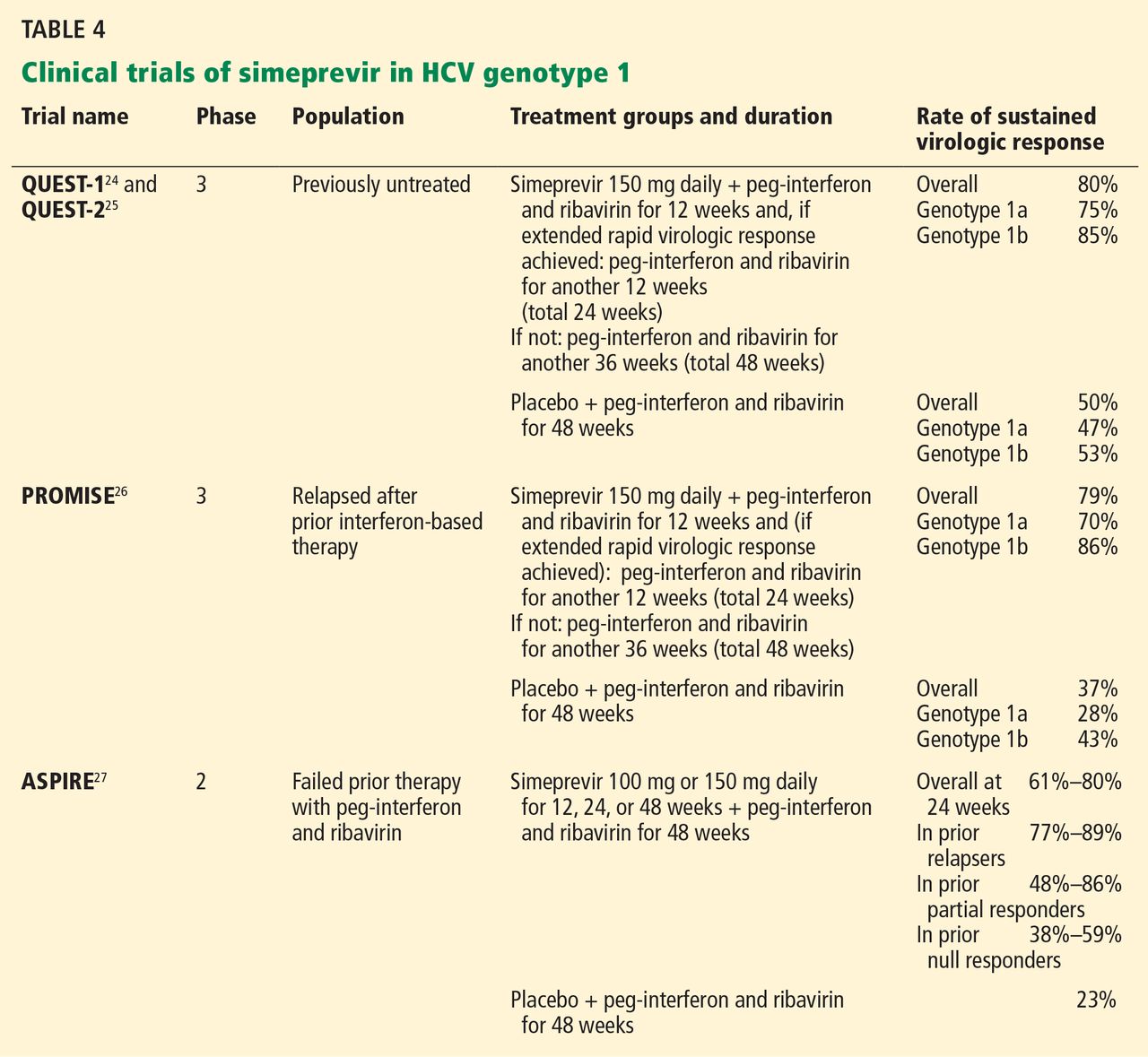
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
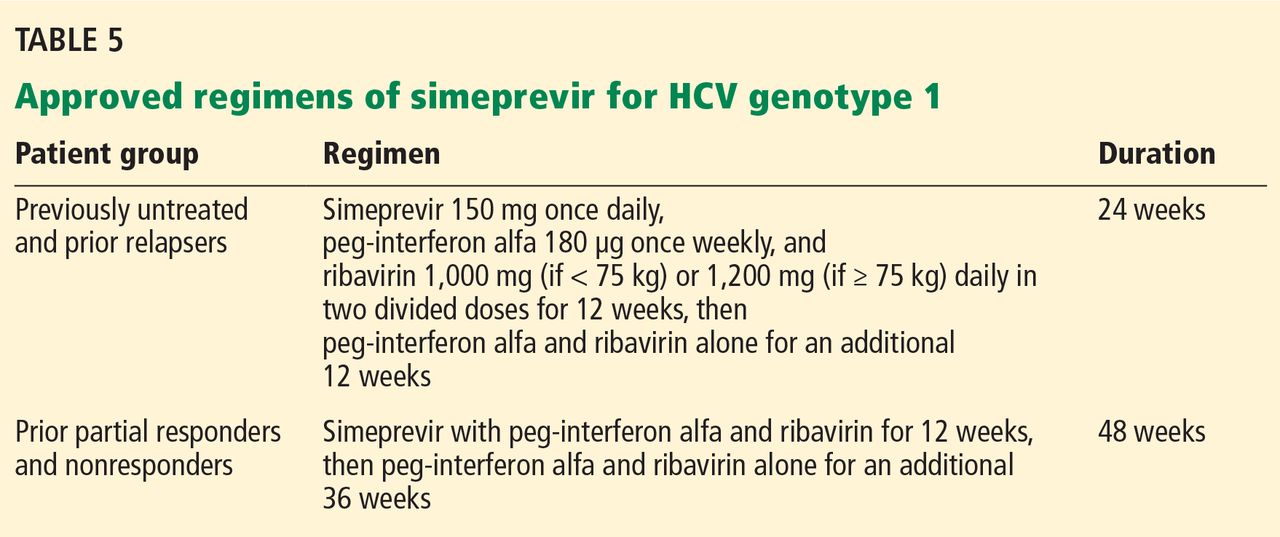
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.

Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining

The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
KEY POINTS
- In clinical trials of treatment for chronic HCV infection, regimens that included a direct-acting antiviral agent were more effective than ones that did not.
- Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3, and in combination with ribavirin and interferon in patients infected with HCV genotype 1 or 4. It is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation.
- Simeprevir is approved in an oral dose of 150 mg once daily in combination with ribavirin and interferon for patients with HCV genotype 1.
- The new drugs are expensive, a potential barrier for many patients. As more direct-acting antiviral agents become available, their cost will likely decrease.
- Combinations of direct-acting antiviral agents of different classes may prove even more effective and could eliminate the need for interferon entirely.
When are effective medications just too expensive?
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
Is hemoglobin A1c an accurate measure of glycemic control in all diabetic patients?
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
New practice guidelines: Constrained or enhanced by the evidence?

Recent guidelines have revisited the management of two major modifiable risk factors for cardiovascular morbidity: hypercholesterolemia and hypertension. The authors of each purposefully emphasized high-grade evidence in generating their recommendations. But, as pointed out by Thomas et al in this issue of the Journal,1 the authors of the hypertension guidelines still resorted to “expert opinion” in five of their 10 recommendations.
The authors of the new hypertension guidelines from the Eighth Joint National Committee (JNC 8),2 as well as the new cholesterol guidelines3 discussed by Raymond et al in the January 2014 issue of the Journal,4 relied on interventional clinical trial evidence for their recommendations. The good news in the context of pay-for-performance metrics is that both of these new guidelines are easier to adhere to than the previous ones. But will the new guidelines really help us achieve better patient outcomes?
Concerns about these guidelines spring directly from their assumed major strength—ie, that they are based on interventional trial data. Well-run, randomized, controlled trials are the brass ring of evidence-based medical practice, presumably providing the cleanest demonstration of therapeutic efficacy. But with “clean” data potentially come sterile, non-real-world conclusions that may advise but should not limit our practice decisions. Most of our patients do not fit neatly into trial inclusion and exclusion criteria, nor do they exactly match the demographics of trial volunteers. Patients who participate in clinical trials are not the same as the patients who populate our clinics. Nor, unfortunately, is the blood pressure measurement technique likely the same in the clinical trial setting as in many of our offices.
In the clinic, it seems obvious not to be overly zealous about blood pressure control in an elderly, frail, hypertensive patient. But at the same time, aiming for a systolic pressure lower than 150 mm Hg (which is looser than in the last set of guidelines) as a target for those over age 60 is incredibly arbitrary, given that physiology and biologic risk rarely act in a step-function manner. Biologic metrics tend to behave as a continuum. If we recognize that the blood pressure can be readily and safely reduced further in a given patient, and if there are observational data to support the concept that risk for cardiovascular events roughly parallels the systolic blood pressure in a continuous manner to lower than 150 mm Hg, why aim to lower it only slightly? Trial-based guidelines are valuable, but they should not replace sound physiologic reasoning and common sense (also known as “expert opinion”). Yet we must temper this logical reasoning with lessons learned from trials such as ACCORD,5 which showed that overly vigorous efforts at reaching theoretical therapeutic targets may be fraught with unexpected adverse outcomes.
Our challenge is to appropriately individualize therapy, usually in the absence of relevant comparative efficacy studies. Trying to apply homogenized clinical trial data to the individual patient in the examination room is not always reasonable. Treating a 59-year-old who has a slowly escalating systolic pressure of 142 mm Hg is not the same as treating a 32-year-old who has a chronic pressure of 138 and an audible S4.
The new hypertension guidelines should be easier to implement than the previous ones in JNC 7. I like some of the specificity of the new recommendations and the disappearance of beta-blockers from the list of recommended early therapies. Yet I think that in the presence of comorbidities and end-organ damage, they may be too lax. And certain groups are left relatively undiscussed, such as patients with cerebrovascular disease, known hypertensive vascular injury, and obstructive sleep apnea, as there were limited trial data to provide guidance (although for some clinical subsets we do have very suggestive observational and experiential data). We can’t always wait for the perfect trial to be done in order to make clinical decisions.
To paraphrase Thomas et al,1 for these guidelines, one size fits many, but we still must do significant custom tailoring in the office. In the months ahead, we will try to provide some guidance on how to effectively deal with those situations where robust trial evidence is insufficient to direct clinical decision-making.
- Thomas G, Shishehbor M, Brill D, Nally JV Jr. New hypertension guidelines: one size fits most? Cleve Clin J Med 2014; 81:178–188.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; doi: 10.1016/jacc.2013.11.002.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
Recent guidelines have revisited the management of two major modifiable risk factors for cardiovascular morbidity: hypercholesterolemia and hypertension. The authors of each purposefully emphasized high-grade evidence in generating their recommendations. But, as pointed out by Thomas et al in this issue of the Journal,1 the authors of the hypertension guidelines still resorted to “expert opinion” in five of their 10 recommendations.
The authors of the new hypertension guidelines from the Eighth Joint National Committee (JNC 8),2 as well as the new cholesterol guidelines3 discussed by Raymond et al in the January 2014 issue of the Journal,4 relied on interventional clinical trial evidence for their recommendations. The good news in the context of pay-for-performance metrics is that both of these new guidelines are easier to adhere to than the previous ones. But will the new guidelines really help us achieve better patient outcomes?
Concerns about these guidelines spring directly from their assumed major strength—ie, that they are based on interventional trial data. Well-run, randomized, controlled trials are the brass ring of evidence-based medical practice, presumably providing the cleanest demonstration of therapeutic efficacy. But with “clean” data potentially come sterile, non-real-world conclusions that may advise but should not limit our practice decisions. Most of our patients do not fit neatly into trial inclusion and exclusion criteria, nor do they exactly match the demographics of trial volunteers. Patients who participate in clinical trials are not the same as the patients who populate our clinics. Nor, unfortunately, is the blood pressure measurement technique likely the same in the clinical trial setting as in many of our offices.
In the clinic, it seems obvious not to be overly zealous about blood pressure control in an elderly, frail, hypertensive patient. But at the same time, aiming for a systolic pressure lower than 150 mm Hg (which is looser than in the last set of guidelines) as a target for those over age 60 is incredibly arbitrary, given that physiology and biologic risk rarely act in a step-function manner. Biologic metrics tend to behave as a continuum. If we recognize that the blood pressure can be readily and safely reduced further in a given patient, and if there are observational data to support the concept that risk for cardiovascular events roughly parallels the systolic blood pressure in a continuous manner to lower than 150 mm Hg, why aim to lower it only slightly? Trial-based guidelines are valuable, but they should not replace sound physiologic reasoning and common sense (also known as “expert opinion”). Yet we must temper this logical reasoning with lessons learned from trials such as ACCORD,5 which showed that overly vigorous efforts at reaching theoretical therapeutic targets may be fraught with unexpected adverse outcomes.
Our challenge is to appropriately individualize therapy, usually in the absence of relevant comparative efficacy studies. Trying to apply homogenized clinical trial data to the individual patient in the examination room is not always reasonable. Treating a 59-year-old who has a slowly escalating systolic pressure of 142 mm Hg is not the same as treating a 32-year-old who has a chronic pressure of 138 and an audible S4.
The new hypertension guidelines should be easier to implement than the previous ones in JNC 7. I like some of the specificity of the new recommendations and the disappearance of beta-blockers from the list of recommended early therapies. Yet I think that in the presence of comorbidities and end-organ damage, they may be too lax. And certain groups are left relatively undiscussed, such as patients with cerebrovascular disease, known hypertensive vascular injury, and obstructive sleep apnea, as there were limited trial data to provide guidance (although for some clinical subsets we do have very suggestive observational and experiential data). We can’t always wait for the perfect trial to be done in order to make clinical decisions.
To paraphrase Thomas et al,1 for these guidelines, one size fits many, but we still must do significant custom tailoring in the office. In the months ahead, we will try to provide some guidance on how to effectively deal with those situations where robust trial evidence is insufficient to direct clinical decision-making.
Recent guidelines have revisited the management of two major modifiable risk factors for cardiovascular morbidity: hypercholesterolemia and hypertension. The authors of each purposefully emphasized high-grade evidence in generating their recommendations. But, as pointed out by Thomas et al in this issue of the Journal,1 the authors of the hypertension guidelines still resorted to “expert opinion” in five of their 10 recommendations.
The authors of the new hypertension guidelines from the Eighth Joint National Committee (JNC 8),2 as well as the new cholesterol guidelines3 discussed by Raymond et al in the January 2014 issue of the Journal,4 relied on interventional clinical trial evidence for their recommendations. The good news in the context of pay-for-performance metrics is that both of these new guidelines are easier to adhere to than the previous ones. But will the new guidelines really help us achieve better patient outcomes?
Concerns about these guidelines spring directly from their assumed major strength—ie, that they are based on interventional trial data. Well-run, randomized, controlled trials are the brass ring of evidence-based medical practice, presumably providing the cleanest demonstration of therapeutic efficacy. But with “clean” data potentially come sterile, non-real-world conclusions that may advise but should not limit our practice decisions. Most of our patients do not fit neatly into trial inclusion and exclusion criteria, nor do they exactly match the demographics of trial volunteers. Patients who participate in clinical trials are not the same as the patients who populate our clinics. Nor, unfortunately, is the blood pressure measurement technique likely the same in the clinical trial setting as in many of our offices.
In the clinic, it seems obvious not to be overly zealous about blood pressure control in an elderly, frail, hypertensive patient. But at the same time, aiming for a systolic pressure lower than 150 mm Hg (which is looser than in the last set of guidelines) as a target for those over age 60 is incredibly arbitrary, given that physiology and biologic risk rarely act in a step-function manner. Biologic metrics tend to behave as a continuum. If we recognize that the blood pressure can be readily and safely reduced further in a given patient, and if there are observational data to support the concept that risk for cardiovascular events roughly parallels the systolic blood pressure in a continuous manner to lower than 150 mm Hg, why aim to lower it only slightly? Trial-based guidelines are valuable, but they should not replace sound physiologic reasoning and common sense (also known as “expert opinion”). Yet we must temper this logical reasoning with lessons learned from trials such as ACCORD,5 which showed that overly vigorous efforts at reaching theoretical therapeutic targets may be fraught with unexpected adverse outcomes.
Our challenge is to appropriately individualize therapy, usually in the absence of relevant comparative efficacy studies. Trying to apply homogenized clinical trial data to the individual patient in the examination room is not always reasonable. Treating a 59-year-old who has a slowly escalating systolic pressure of 142 mm Hg is not the same as treating a 32-year-old who has a chronic pressure of 138 and an audible S4.
The new hypertension guidelines should be easier to implement than the previous ones in JNC 7. I like some of the specificity of the new recommendations and the disappearance of beta-blockers from the list of recommended early therapies. Yet I think that in the presence of comorbidities and end-organ damage, they may be too lax. And certain groups are left relatively undiscussed, such as patients with cerebrovascular disease, known hypertensive vascular injury, and obstructive sleep apnea, as there were limited trial data to provide guidance (although for some clinical subsets we do have very suggestive observational and experiential data). We can’t always wait for the perfect trial to be done in order to make clinical decisions.
To paraphrase Thomas et al,1 for these guidelines, one size fits many, but we still must do significant custom tailoring in the office. In the months ahead, we will try to provide some guidance on how to effectively deal with those situations where robust trial evidence is insufficient to direct clinical decision-making.
- Thomas G, Shishehbor M, Brill D, Nally JV Jr. New hypertension guidelines: one size fits most? Cleve Clin J Med 2014; 81:178–188.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; doi: 10.1016/jacc.2013.11.002.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Thomas G, Shishehbor M, Brill D, Nally JV Jr. New hypertension guidelines: one size fits most? Cleve Clin J Med 2014; 81:178–188.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; doi: 10.1016/jacc.2013.11.002.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
New hypertension guidelines: One size fits most?
The report of the panel appointed to the eighth Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8),1 published in December 2013 after considerable delay, contains some important changes from earlier guidelines from this group.2 For example:
- The blood pressure goal has been changed to less than 150/90 mm Hg in people age 60 and older. Formerly, the goal was less than 140/90 mm Hg.
- The goal has been changed to less than 140/90 mm Hg in all others, including people with diabetes mellitus and chronic kidney disease. Formerly, those two groups had a goal of less than 130/80 mm Hg.
- The initial choice of therapy can be from any of four classes of drugs: thiazide-type diuretics, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers (ARBs). Formerly, the list also contained beta-blockers. Also, thiazide-type diuretics have lost their “preferred” status.
The new guidelines are evidence-based and are intended to simplify the way that hypertension is managed. Below, we summarize them—how they were developed, their strengths and limitations, and the main changes from earlier JNC reports.
WHOSE GUIDELINES ARE THESE?
The JNC has issued guidelines for managing hypertension since 1976, traditionally sanctioned by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health. The guidelines have generally been updated every 4 to 5 years, with the last update, JNC 7,2 published in 2003.
The JNC 8 panel, consisting of 17 members, was commissioned by the NHLBI in 2008. However, in June 2013, the NHLBI announced it was withdrawing from guideline development and was delegating it to selected specialty organizations.3,4 In the interest of bringing the already delayed guidelines to the public in a timely manner, the JNC 8 panel decided to pursue publication independently and submitted the report to a medical journal. It is therefore not an official NHLBI-sanctioned report.
Here, we will refer to the new guidelines as “JNC 8,” but they are officially from “panel members appointed to the Eighth Joint National Committee (JNC 8).”
THREE QUESTIONS THAT GUIDED THE GUIDELINES
Epidemiologic studies clearly show a close relationship between blood pressure and the risk of heart disease, stroke, and kidney disease, these risks being lowest at a blood pressure of around 115/75 mm Hg.5 However, clinical trials have failed to show any evidence to justify treatment with antihypertensive medications to such a low level once hypertension has been diagnosed.
Patients and health care providers thus face questions about when to begin treatment, how low to aim for, and which antihypertensive medications to use. The JNC 8 panel focused on these three questions, believing them to be of greatest relevance to primary care providers.
A RIGOROUS PROCESS OF EVIDENCE REVIEW AND GUIDELINE DEVELOPMENT
The JNC 8 panel followed the guideline-development pathway outlined by the Institute of Medicine report, Clinical Practice Guidelines We Can Trust.6
Studies published from January 1966 through December 2009 that met specified criteria were selected for evidence review. Specifically, the studies had to be randomized controlled trials—no observational studies, systematic reviews, or meta-analyses, which were allowed in the JNC 7 report—with sample sizes of more than 100. Follow-up had to be for more than 1 year. Participants had to be age 18 or older and have hypertension—studies with patients with normal blood pressure or prehypertension were excluded. Health outcomes had to be reported, ie, “hard” end points such as rates of death, myocardial infarction, heart failure, hospitalization for heart failure, stroke, revascularization, and end-stage renal disease. Post hoc analyses were not allowed. The studies had to be rated by the NHLBI’s standardized quality rating tool as “good” (which has the least risk of bias, with valid results) or “fair (which is susceptible to some bias, but not enough to invalidate the results).
Subsequently, another search was conducted for relevant studies published from December 2009 through August 2013. In addition to meeting all the other criteria, this bridging search further restricted selection to major multicenter studies with sample sizes of more than 2,000.
An external methodology team performed the initial literature review and summarized the data. The JNC panel then crafted evidence statements and clinical recommendations using the evidence quality rating and grading systems developed by the NHLBI. In January 2013, the NHLBI submitted the guidelines for external review by individual reviewers with expertise in hypertension and to federal agencies, and a revised document was framed based on their comments and suggestions.
The evidence statements are detailed in an online 300-page supplemental review, and the panel members have indicated that reviewer comments and responses from the presubmission review process will be made available on request.
NINE RECOMMENDATIONS AND ONE COROLLARY
The panel made nine recommendations and one corollary recommendation based on a review of the evidence. Of the 10 total recommendations, five are based on expert opinion. Another two were rated as “moderate” in strength, one was “weak,” and only two were rated as “strong” (ie, based on high-quality evidence).
Recommendation 1: < 150/90 for those 60 and older
In the general population age 60 and older, the JNC 8 recommends starting drug treatment if the systolic pressure is 150 mm Hg or higher or if the diastolic pressure is 90 mm Hg or higher, and aiming for a systolic goal of less than 150 mm Hg and a diastolic goal of less than 90 mm Hg.
Strength of recommendation—strong (grade A).
Comments. Of all the recommendations, this one will probably have the greatest impact on clinical practice. Consider a frail 70-year-old patient at risk of falls who is taking two antihypertensive medications and whose blood pressure is 148/85 mm Hg. This level would have been considered too high under JNC 7 but is now acceptable, and the patient’s therapy does not have to be escalated.
The age cutoff of 60 years for this recommendation is debatable. The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)7 included patients ages 60 to 85 (mean age 74) and found no difference in outcomes comparing a goal systolic pressure of less than 140 mm Hg (this group achieved a mean systolic pressure of 135.9 mm Hg) and a goal systolic pressure of 140 to 160 mm Hg (achieved systolic pressure 145.6 mm Hg).
Similarly, the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) trial8 included patients ages 70 to 84 (mean age 76.1) and found no difference in outcomes between a goal systolic pressure of less than 140 mm Hg (achieved systolic pressure 136.6 mm Hg) and a goal of 140 to 150 mm Hg (achieved systolic pressure 142 mm Hg).
The Hypertension in the Very Elderly Trial (HYVET)9 found lower rates of stroke, death, and heart failure in patients age 80 and older when their systolic pressure was less than 150 mm Hg.
While these trials support a goal pressure of less than 150 mm Hg in the elderly, it is unclear whether this goal should be applied beginning at age 60. Other guidelines, including those recently released jointly by the American Society of Hypertension and the International Society of Hypertension, recommend a systolic goal of less than 150 mm Hg in people age 80 and older—not age 60.10
The recommendation for a goal systolic pressure of less than 150 mm Hg in people age 60 and older was not unanimous; some panel members recommended continuing the JNC 7 goal of less than 140 mm Hg based on expert opinion, as they believed that the evidence was insufficient, especially in high-risk subgroups such as black people and those with cerebrovascular disease and other risk factors.
A subsequent minority report from five panel members discusses in more detail why they believe the systolic target should be kept lower than 140 mm Hg in patients age 60 or older until the risks and benefits of a higher target become clearer.11
Corollary recommendation: No need to down-titrate if lower than 140
In the general population age 60 and older, dosages do not have to be adjusted downward if the patient’s systolic pressure is already lower than 140 mm Hg and treatment is well tolerated without adverse effects on health or quality of life.
Strength of recommendation—expert opinion (grade E).
Comments. In the studies that supported a systolic goal lower than 150 mm Hg, many participants actually achieved a systolic pressure lower than 140 mm Hg without any adverse events. Trials that showed no benefit from a systolic goal lower than 140 mm Hg were graded as lower in quality. Thus, the possibility remains that a systolic goal lower than 140 mm Hg could have a clinically important benefit. Therefore, medications do not have to be adjusted so that blood pressure can “ride up.”
For example, therapy does not need to be down-titrated in a 65-year-old patient whose blood pressure is 138/85 mm Hg on two medications that he or she is tolerating well. On the other hand, based on Recommendation 1, therapy can be down-titrated in a 65-year-old whose pressure is 138/85 mm Hg on four medications that are causing side effects.
Recommendation 2: Diastolic < 90 for those younger than 60
In the general population younger than 60 years, JNC 8 recommends starting pharmacologic treatment if the diastolic pressure is 90 mm Hg or higher and aiming for a goal diastolic pressure of less than 90 mm Hg.
Strength of recommendation—strong (grade A) for ages 30 to 59, expert opinion (grade E) for ages 18 to 29.
Comments. The panel found no evidence to support a goal diastolic pressure of 80 mm Hg or less (or 85 mm Hg or less) compared with 90 mm Hg or less in this population.
It is reasonable to aim for the same diastolic goal in younger persons (under age 30), given the higher prevalence of diastolic hypertension in younger people.
Recommendation 3: Systolic < 140 for those younger than 60
In the general population younger than 60 years, we should start drug treatment at a systolic pressure of 140 mm Hg or higher and treat to a systolic goal of less than 140 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Although evidence was insufficient to support this recommendation, the panel decided to keep the same systolic goal for people younger than 60 as in the JNC 7 recommendations, for the following two reasons.
First, there is strong evidence supporting a diastolic goal of less than 90 mm Hg in this population (Recommendation 2), and many study participants who achieved a diastolic pressure lower than 90 mm Hg also achieved a systolic pressure lower than 140. Therefore, it is not possible to tease out whether the outcome benefits were due to lower systolic pressure or to lower diastolic pressure, or to both.
Second, the panel believed the guidelines would be simpler to implement if the systolic goals were the same in the general population as in those with chronic kidney disease or diabetes (see below).
Recommendation 4: < 140/90 in chronic kidney disease
In patients age 18 and older with chronic kidney disease, JNC 8 recommends starting drug treatment at a systolic pressure of 140 mm Hg or higher or a diastolic pressure of 90 mm Hg or higher and treating to a goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Chronic kidney disease is defined as either a glomerular filtration rate (estimated or measured) less than 60 mL/min/1.73 m2 in people up to age 70, or albuminuria, defined as more than 30 mg/g of creatinine at any glomerular filtration rate at any age.
Strength of recommendation—expert opinion (grade E).
Comments. There was insufficient evidence that aiming for a lower goal of 130/80 mm Hg (as in the JNC 7 recommendations) had any beneficial effect on cardiovascular, cerebrovascular, or mortality outcomes compared with 140/90 mm Hg, and there was moderate-quality evidence showing that treatment to lower goal (< 130/80 mm Hg) did not slow the progression of chronic kidney disease any better than a goal of less than 140/90 mm Hg. (One study that did find better renal outcomes with a lower blood pressure goal was a post hoc analysis of the Modification of Diet in Renal Disease study data in patients with proteinuria of more than 3 g per day.12)
We believe that decisions should be individualized regarding goal blood pressures and pharmacologic therapy in patients with chronic kidney disease and proteinuria, who may benefit from lower blood pressure goals (<130/80 mm Hg), based on low-level evidence.13,14 Risks and benefits should also be weighed in considering the blood pressure goal in the elderly with chronic kidney disease, taking into account functional status, comorbidities, and level of proteinuria.
Recommendation 5: < 140/90 for people with diabetes
In patients with diabetes who are age 18 and older, JNC 8 says to start drug treatment at a systolic pressure of 140 mm Hg or higher or diastolic pressure of 90 mm Hg or higher, and treat to goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Moderate-quality evidence showed cardiovascular, cerebrovascular, and mortality outcome benefits with treatment to a systolic goal of less than 150 mm Hg in patients with diabetes and hypertension.
The panel found no randomized controlled trials that compared a treatment goal of less than 140 mm Hg with one of less than 150 mm Hg for outcome benefits, but decided to base its recommendations on the results of the Action to Control Cardiovascular Risk in Diabetes—Blood-pressure-lowering Arm (ACCORD-BP) trial.15 The control group in this trial had a goal systolic pressure of less than 140 mm Hg and had similar outcomes compared with a lower goal.
The panel found no evidence to support a lower blood pressure goal (< 130/80) as in JNC 7. ACCORD-BP showed no differences in outcomes with a systolic goal lower than 140 mm Hg vs lower than 120 mm Hg except for a small reduction in stroke, and the risks of trying to achieve intensive lowering of blood pressure may outweigh the benefit of a small reduction in stroke.12 There was no evidence for a goal diastolic pressure below 80 mm Hg.
Recommendation 6: In nonblack patients, start with a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB
In the general nonblack population, including those with diabetes, initial drug treatment should include a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB.
Strength of recommendation—moderate (grade B).
Comments. All these drug classes had comparable outcome benefits in terms of rates of death, cardiovascular disease, cerebrovascular disease, and kidney disease, but not heart failure. For improving heart failure outcomes, thiazide-type diuretics are better than ACE inhibitors, which in turn are better than calcium channel blockers.
Thiazide-type diuretics (eg, hydrochlorothiazide, chlorthalidone, and indapamide) were recommended as first-line therapy for most patients in JNC 7, but they no longer carry this preferred status in JNC 8. In addition, the panel did not address preferential use of chlorthalidone as opposed to hydrochlorothiazide, or the use of spironolactone in resistant hypertension.
The panel did not recommend beta-blockers as first-line therapy because there were no differences in outcomes (or insufficient evidence) compared with the above medication classes; additionally, the Losartan Intervention for Endpoint Reduction in Hypertension study16 reported a higher incidence of stroke with a beta-blocker than with an ARB. However, JNC 8 did not consider randomized controlled trials in specific nonhypertensive populations such as patients with coronary artery disease or heart failure. We believe decisions should be individualized as to the use of beta-blockers in these two conditions.
The panel recommended the same approach in patients with diabetes, as there were no differences in major cardiovascular or cerebrovascular outcomes compared with the general population.
Recommendation 7: In black patients, start with a thiazide-type diuretic or calcium channel blocker
In the general black population, including those with diabetes, JNC 8 recommends starting drug treatment with a thiazide-type diuretic or a calcium channel blocker.
Strength of recommendation—moderate (grade B) for the general black population; weak (grade C) for blacks with diabetes.
Comments. In the black subgroup in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack trial (ALLHAT),17 a thiazide-type diuretic (chlorthalidone) was better than an ACE inhibitor (lisinopril) in terms of cerebrovascular, heart failure, and composite outcomes, but similar for mortality rates and cardiovascular, and kidney outcomes. Also in this subgroup, a calcium channel blocker (amlodipine) was better than the ACE inhibitor for cerebrovascular outcomes (there was a 51% higher rate of stroke with the ACE inhibitor as initial therapy than with the calcium channel blocker); the ACE inhibitor was also less effective in reducing blood pressure in blacks than the calcium channel blocker.
For improving heart failure outcomes, the thiazide-type diuretic was better than the ACE inhibitor, which in turn was better than the calcium channel blocker.
Evidence for black patients with diabetes (graded as weak) was extrapolated from ALLHAT, in which 46% had diabetes.17 We would consider using an ACE inhibitor or ARB in this population on an individual basis, especially if the patient had proteinuria.
Recommendation 8: ACEs and ARBs for chronic kidney disease
In patients age 18 and older with chronic kidney disease, irrespective of race, diabetes, or proteinuria, initial or add-on drug treatment should include an ACE inhibitor or ARB to improve kidney outcomes.
Strength of recommendation—moderate (grade B).
Comments. Treatment with an ACE inhibitor or ARB improves kidney outcomes in patients with chronic kidney disease. But in this population, these drugs are no more beneficial than calcium channel blockers or beta-blockers in terms of cardiovascular outcomes.
No randomized controlled trial has compared ACE inhibitors and ARBs for cardiovascular outcomes in chronic kidney disease, and these drugs have similar effects on kidney outcomes.
The panel did not make any recommendations about direct renin inhibitors, as there were no eligible studies demonstrating benefits on cardiovascular or kidney outcomes.
In black patients with chronic kidney disease and proteinuria, the panel recommended initial therapy with an ACE inhibitor or ARB to slow progression to end-stage renal disease (contrast with Recommendation 7).
In black patients with chronic kidney disease and no proteinuria, the panel recommended choosing from a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB. If an ACE inhibitor or ARB is not used as initial therapy, then one can be added on as a second-line medication (contrast with Recommendation 7).
The panel found no evidence to support this recommendation in people over age 75 and noted that although an ACE inhibitor or ARB may be beneficial in this group, a thiazide-type diuretic or calcium channel blocker can be considered.
Recommendation 9: If not at goal, step up
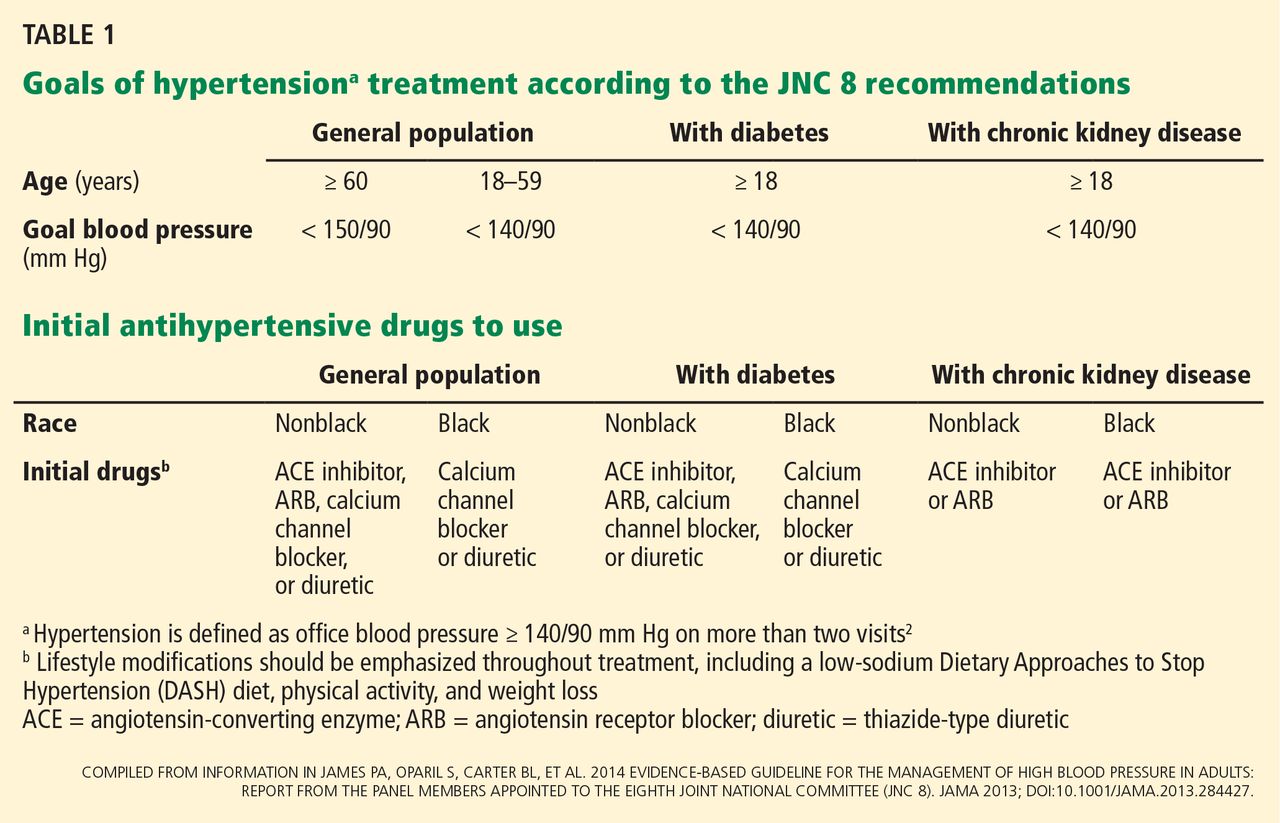
The main objective of pharmacologic treatment of hypertension is to attain and maintain the goal blood pressure. Lifestyle interventions should be maintained throughout treatment (Table 1). Medications can be initiated and titrated according to any of three strategies used in the randomized controlled trials selected by the panel (detailed below). Do not use an ACE inhibitor and ARB together in same patient.
If blood pressure is not at goal using all medication classes as in Recommendation 6 (ie, the triple combination of a thiazide-type diuretic, calcium channel blocker, and either an ACE inhibitor or an ARB), if there is a contraindication to any of these medication classes, or if there is need to use more than three medications to reach the goal, drugs from other classes can be used.
Referral to a hypertension specialist may be indicated for patients who are not at goal using the above strategy or for whom additional clinical consultation is needed.
Strength of recommendation—expert opinion (grade E).
Comments. Blood pressure should be monitored and assessed regularly, treatment adjusted as needed, and lifestyle modifications encouraged.
The panel did not recommend any monitoring schedule before or after goal blood pressure is achieved, and this should be individualized.
ADDITIONAL TOPICS IN JNC 8
A supplemental report covered some additional topics for which formal evidence review was not conducted but which the panel considered important.
Measuring and monitoring blood pressure
The panel recommended measuring the blood pressure with an automated oscillometric device that is properly calibrated and validated, or carefully measuring it manually.
Blood pressure should be measured in a quiet and relaxed environment with the patient seated comfortably for at least 5 minutes in a chair (rather than on an examination table) with feet flat on the floor, back supported, and arm supported at heart level. Blood pressure should be taken on the bare upper arm with an appropriate-sized cuff whose bladder encircles at least 80% of the mid-upper arm circumference, and patients should avoid caffeine, smoking, and physical activity for at least 30 minutes before measurement. In addition, patients should be asked about the need to empty the bladder (and encouraged to do so if they have to).
To establish the diagnosis of hypertension and to assess whether blood pressure goals are being met, two or three measurements should be taken at each visit as outlined above, and the average recorded.
At the first visit, blood pressure should be measured in both arms, and the arm with the higher pressure should be used for subsequent measurements.
Appropriate dosing of antihypertensive medications
Dosing should be individualized for each patient, but in general, target doses can be achieved within 2 to 4 weeks, and generally should not take longer than 2 months.
In general, to minimize potential adverse effects, treatment is started at a lower dose than the target dose and is then titrated up. This is especially important in older patients and patients on multiple medications with other comorbidities, and if two antihypertensive medications are being started simultaneously.
The panel reviewed evidence-based dosing of antihypertensive medications that were shown to improve cardiovascular outcomes from the studies that were selected for review. Hydrochlorothiazide gets a special mention: although doses up to 100 mg were used in some studies, the panel recommended an evidence-based dose of 25 or 50 mg daily to balance efficacy and safety.
Three strategies for dosing antihypertensive medications that were used in the selected randomized controlled trials were provided. These strategies were not compared with each other, nor is it known if one is better than the others in terms of health outcomes. In all cases, avoid combining an ACE inhibitor and an ARB.
- Start one drug from the four classes in Recommendation 6, titrate to the maximum dose, then add a second drug and titrate, then add a third drug and titrate to achieve the goal blood pressure.
- Start one drug from the four classes in Recommendation 6 and add a second drug before increasing the initial drug to its maximal dose. Titrate both to maximal doses, and add a third drug if needed and titrate to achieve the goal blood pressure.
- Start with two drugs at the same time from the four classes in Recommendation 6, either as separate pills or in a fixed-dose combination. Add a third drug if needed to achieve the goal blood pressure.
Lifestyle modification
The panel did not extensively review the evidence for lifestyle modification but endorsed the recommendations of the Lifestyle Work Group, which was convened by the NHLBI to focus on the effects of diet and physical activity on cardiovascular disease risk factors.18
Diet. The Lifestyle Work Group recommends combining the Dietary Approaches to Stop Hypertension (DASH) diet with reduced sodium intake, as there is evidence of a greater blood-pressure-lowering effect when the two are combined. The effect on blood pressure is independent of changes in weight.
The Lifestyle Work Group recommends consuming no more than 2,400 mg of sodium per day, noting that limiting intake to 1,500 mg can result in even greater reduction in blood pressure, and that even without achieving these goals, reducing sodium intake by at least 1,000 mg per day lowers blood pressure.
Physical activity. The Lifestyle Work Group recommends moderate to vigorous physical activity for approximately 160 minutes per week (three to four sessions a week, lasting an average of 40 minutes per session).
Weight loss. The Lifestyle Work Group did not review the blood-pressure-lowering effect of weight loss in those who are overweight or obese. The JNC 8 panel endorsed maintaining a healthy weight in controlling blood pressure.
Alcohol intake received no specific recommendations in JNC 8.
JNC 8 IN PERSPECTIVE
JNC 8 takes a rigorous, evidence-based approach and focuses on a few key questions. Thus, it is very different from the earlier reports: it has a narrower focus and does not address the full range of issues related to hypertension.
Strengths of JNC 8
The panel followed a rigorous process of review and evaluation of evidence from randomized controlled trials, adhering closely to standards set by the Institute of Medicine for guideline development. In contrast, JNC 7 relied on consensus and expert opinion.
The JNC 8 guidelines aim to simplify recommendations, with only two goals to remember: treat to lower than 150/90 mm Hg in patients age 60 and older, and lower than 140/90 mm Hg for everybody else. The initial drug regimen was simplified as well, with any of four choices for initial therapy in nonblacks and two in blacks.
Relaxing the blood pressure goals in elderly patients (although a cutoff of age 60 vs age 80 is likely to be debated) will also allay concerns about overtreating hypertension and causing adverse events in this population that is particularly susceptible to orthostatic changes and is at increased risk of falls.
Limitations and concerns
While the evidence-based nature of the recommendations is a strength, information from observational studies, systematic reviews, and meta-analyses was not incorporated into the formulation of these guidelines. This limits the available evidence, reflected in the fact that despite an extensive attempt to provide recommendations based on good evidence, five of the 10 recommendations (including the corollary recommendation) are still based on expert consensus opinion. Comparing and combining studies from different time periods is also problematic because of different methods of conducting clinical trials and analysis, and also because clinical care in a different period may differ from current standard practices.
Blood pressure targets in some subgroups are not clearly addressed, including those with proteinuria and with a history of stroke. Peterson et al,19 in an editorial accompanying the JNC 8 publication, commented on the need for larger randomized controlled trials to compare different blood pressure thresholds in various patient populations.
Some health care providers will likely be concerned that relaxing blood pressure goals could lead to higher real-world blood pressures, eventually leading to adverse cardiovascular outcomes, particularly on a population level. This is akin to the “speed limit rule”—people are more likely to hover above target, no matter what the target is.
In another editorial, Sox20 raised concerns about the external review process, ie, that the guidelines were not published in draft form to solicit public comment. Additionally, although the recommendations underwent extensive review, they were not endorsed by the specialty societies that the NHLBI designated to develop guidelines. In its defense, however, the JNC 8 panel has offered to share records of the review process on request, and this should serve to increase confidence in the review process.
The original literature search was limited to studies published through December 2009, which is more than 4 years before the publication of the recommendations. Although a bridge search was conducted until August 2013 to identify additional studies, this search used different inclusion criteria than the original criteria.
With its narrow focus, JNC 8 does not address many relevant issues. The American Society of Hypertension/International Society of Hypertension guidelines, published around the same time that the JNC 8 report was released, provide a more comprehensive review that will be of practical use for health care providers in the community.10
Ambulatory blood pressure monitoring is increasingly being used in clinical practice to detect white coat hypertension and, in many cases, to assess hypertension that is resistant to medications. It has also been shown to have better prognostic value in predicting cardiovascular risk and progression of kidney disease than office blood pressures.21,22 The UK National Institute of Health and Care Excellence guideline recommends ambulatory monitoring for the diagnosis of hypertension.23 However, JNC 8 did not provide specific recommendations for the use of this technology. Additionally, the JNC 8 evidence review is based on studies that used office blood pressure readings, and the recommendations are not necessarily applicable to measurements obtained by ambulatory monitoring.
Other topics covered in JNC 7 but not in JNC 8 include:
- Definitions and stages of hypertension (which remain the same)
- Initial treatment of stage 2 hypertension with two medications
- The J-curve phenomenon
- Preferred medications for patients with coronary artery disease or congestive heart failure
- A detailed list of oral antihypertensive agents—JNC 8 confines itself to the drugs and doses used in randomized controlled trials
- Patient evaluation
- Secondary hypertension
- Resistant hypertension
- Adherence issues.
Contrast with other guidelines
While the goal of these recommendations is to make treatment standards more understandable and uniform, contrasting recommendations on blood pressure goals and medications from various groups muddy the waters. Other groups that have issued hypertension guidelines in recent years include:
- The American Diabetes Association24
- The American Society of Hypertension and the International Society of Hypertension10
- The European Society of Hypertension and the European Society of Cardiology25
- The Canadian Hypertension Education Program26
- The Kidney Disease: Improving Global Outcomes initiative14
- The National Institute for Health and Clinical Excellence (UK)23
- The International Society on Hypertension in Blacks27
- The American Heart Association, the American College of Cardiology, and the US Centers for Disease Control and Prevention.28
Future directions
Despite the emphasis on making treatment decisions on an individual basis and using guidelines only as a framework for a safe direction in managing difficult clinical scenarios, guideline recommendations are increasingly being used to assess provider performance and quality of care, and so they assume even more importance in the current health care environment. As specialty organizations review and decide whether to endorse the JNC 8 recommendations, reconciling seemingly disparate recommendations from various groups is needed to send a clear and concise message to practitioners taking care of patients with high blood pressure.
Although a daunting task, integrating guidelines on hypertension management with other cardiovascular risk guidelines (eg, cholesterol, obesity) with assessment of overall cardiovascular risk profile would likely help in developing a more effective cardiovascular prevention strategy.
Despite the panel’s best efforts at providing evidence-based recommendations, many of the recommendations are based on expert opinion, reflecting the need for larger well-conducted studies. It is hoped that ongoing studies such as the Systolic Blood Pressure Intervention Trial29 will provide more clarity about blood pressure goals, especially in the elderly.
Final thoughts
Guidelines are not rules, and while they provide a framework by synthesizing the best available evidence, any treatment plan should be formulated on the basis of individual patient characteristics, including comorbidities, lifestyle factors, medication side effects, patient preferences, cost issues, and adherence.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289: 2560–2572. Erratum in JAMA 2003; 290:197.
- Gibbons GH, Harold JG, Jessup M, Robertson RM, Oetgen WJ. The next steps in developing clinical practice guidelines for prevention. J Am Coll Cardiol 2013; 62:1399–1400.
- Gibbons GH, Shurin SB, Mensah GA, Lauer MS. Refocusing the agenda on cardiovascular guidelines: an announcement from the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2013; 62:1396–1398.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913. Erratum in: Lancet 2003; 361:1060.
- Institute of Medicine. Clinical Practice Guidelines We Can Trust. Washington, DC: National Academies Press; 2011. http://www.iom.edu/Reports/2011/Clinical-Practice-Guide-lines-We-Can-Trust.aspx. Accessed February 4, 2014.
- JATOS Study Group. Principal results of the Japanese Trial To Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56:196–202.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Wright JT, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014 Jan 14. [Epub ahead of print]
- Klahr S, Levey AS, Beck GJ, et al. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N Engl J Med 1994; 330:877–884.
- Upadhyay A, Earley A, Haynes SM, Uhlig K. Systematic review: blood pressure target in chronic kidney disease and proteinuria as an effect modifier. Ann Intern Med 2011; 154:541–548.
- Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int Suppl 2012; 2:337–414.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Hypertension 2003; 42:239–246.
- Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2013 Nov 12. [Epub ahead of print]
- Peterson ED, Gaziano JM, Greenland P. Recommendations for treating hypertension: what are the right goals and purposes? JAMA Editorial. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Sox HC. Assessing the trustworthiness of the guideline for management of high blood pressure in adults (editorial). JAMA. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Dolan E, Stanton A, Thijs L, et al. Superiority of ambulatory over clinic blood pressure measurement in predicting mortality: the Dublin outcome study. Hypertension 2005; 46:156–161.
- Agarwal R, Andersen MJ. Prognostic importance of ambulatory blood pressure recordings in patients with chronic kidney disease. Kidney Int 2006; 69:1175–1180.
- National Institute for Health and Clinical Excellence. Hypertension (CG127). http://publications.nice.org.uk/hypertension-cg127. Accessed February 4, 2014.
- American Diabetes Association. Standards of medical care in diabetes – 2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC practice guidelines for the management of arterial hypertension. Blood Press 2013 Dec 20. [Epub ahead of print]
- Hypertension without compelling indications: 2013 CHEP recommendations. Hypertension Canada website. http://www.hypertension.ca/hypertension-without-compelling-indications. Accessed February 4, 2014.
- Flack JM, Sica DA, Bakris G, et al; International Society on Hypertension in Blacks. Management of high blood pressure in blacks: an update of the International Society on Hypertension in Blacks consensus statement. Hypertension 2010; 56:780–800.
- Go AS, Bauman M, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2013 Nov 15.
- Systolic Blood Pressure Intervention Trial (SPRINT). http://clinicaltrials.gov/ct2/show/NCT01206062. Accessed February 4, 2014.
The report of the panel appointed to the eighth Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8),1 published in December 2013 after considerable delay, contains some important changes from earlier guidelines from this group.2 For example:
- The blood pressure goal has been changed to less than 150/90 mm Hg in people age 60 and older. Formerly, the goal was less than 140/90 mm Hg.
- The goal has been changed to less than 140/90 mm Hg in all others, including people with diabetes mellitus and chronic kidney disease. Formerly, those two groups had a goal of less than 130/80 mm Hg.
- The initial choice of therapy can be from any of four classes of drugs: thiazide-type diuretics, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers (ARBs). Formerly, the list also contained beta-blockers. Also, thiazide-type diuretics have lost their “preferred” status.
The new guidelines are evidence-based and are intended to simplify the way that hypertension is managed. Below, we summarize them—how they were developed, their strengths and limitations, and the main changes from earlier JNC reports.
WHOSE GUIDELINES ARE THESE?
The JNC has issued guidelines for managing hypertension since 1976, traditionally sanctioned by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health. The guidelines have generally been updated every 4 to 5 years, with the last update, JNC 7,2 published in 2003.
The JNC 8 panel, consisting of 17 members, was commissioned by the NHLBI in 2008. However, in June 2013, the NHLBI announced it was withdrawing from guideline development and was delegating it to selected specialty organizations.3,4 In the interest of bringing the already delayed guidelines to the public in a timely manner, the JNC 8 panel decided to pursue publication independently and submitted the report to a medical journal. It is therefore not an official NHLBI-sanctioned report.
Here, we will refer to the new guidelines as “JNC 8,” but they are officially from “panel members appointed to the Eighth Joint National Committee (JNC 8).”
THREE QUESTIONS THAT GUIDED THE GUIDELINES
Epidemiologic studies clearly show a close relationship between blood pressure and the risk of heart disease, stroke, and kidney disease, these risks being lowest at a blood pressure of around 115/75 mm Hg.5 However, clinical trials have failed to show any evidence to justify treatment with antihypertensive medications to such a low level once hypertension has been diagnosed.
Patients and health care providers thus face questions about when to begin treatment, how low to aim for, and which antihypertensive medications to use. The JNC 8 panel focused on these three questions, believing them to be of greatest relevance to primary care providers.
A RIGOROUS PROCESS OF EVIDENCE REVIEW AND GUIDELINE DEVELOPMENT
The JNC 8 panel followed the guideline-development pathway outlined by the Institute of Medicine report, Clinical Practice Guidelines We Can Trust.6
Studies published from January 1966 through December 2009 that met specified criteria were selected for evidence review. Specifically, the studies had to be randomized controlled trials—no observational studies, systematic reviews, or meta-analyses, which were allowed in the JNC 7 report—with sample sizes of more than 100. Follow-up had to be for more than 1 year. Participants had to be age 18 or older and have hypertension—studies with patients with normal blood pressure or prehypertension were excluded. Health outcomes had to be reported, ie, “hard” end points such as rates of death, myocardial infarction, heart failure, hospitalization for heart failure, stroke, revascularization, and end-stage renal disease. Post hoc analyses were not allowed. The studies had to be rated by the NHLBI’s standardized quality rating tool as “good” (which has the least risk of bias, with valid results) or “fair (which is susceptible to some bias, but not enough to invalidate the results).
Subsequently, another search was conducted for relevant studies published from December 2009 through August 2013. In addition to meeting all the other criteria, this bridging search further restricted selection to major multicenter studies with sample sizes of more than 2,000.
An external methodology team performed the initial literature review and summarized the data. The JNC panel then crafted evidence statements and clinical recommendations using the evidence quality rating and grading systems developed by the NHLBI. In January 2013, the NHLBI submitted the guidelines for external review by individual reviewers with expertise in hypertension and to federal agencies, and a revised document was framed based on their comments and suggestions.
The evidence statements are detailed in an online 300-page supplemental review, and the panel members have indicated that reviewer comments and responses from the presubmission review process will be made available on request.
NINE RECOMMENDATIONS AND ONE COROLLARY
The panel made nine recommendations and one corollary recommendation based on a review of the evidence. Of the 10 total recommendations, five are based on expert opinion. Another two were rated as “moderate” in strength, one was “weak,” and only two were rated as “strong” (ie, based on high-quality evidence).
Recommendation 1: < 150/90 for those 60 and older
In the general population age 60 and older, the JNC 8 recommends starting drug treatment if the systolic pressure is 150 mm Hg or higher or if the diastolic pressure is 90 mm Hg or higher, and aiming for a systolic goal of less than 150 mm Hg and a diastolic goal of less than 90 mm Hg.
Strength of recommendation—strong (grade A).
Comments. Of all the recommendations, this one will probably have the greatest impact on clinical practice. Consider a frail 70-year-old patient at risk of falls who is taking two antihypertensive medications and whose blood pressure is 148/85 mm Hg. This level would have been considered too high under JNC 7 but is now acceptable, and the patient’s therapy does not have to be escalated.
The age cutoff of 60 years for this recommendation is debatable. The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)7 included patients ages 60 to 85 (mean age 74) and found no difference in outcomes comparing a goal systolic pressure of less than 140 mm Hg (this group achieved a mean systolic pressure of 135.9 mm Hg) and a goal systolic pressure of 140 to 160 mm Hg (achieved systolic pressure 145.6 mm Hg).
Similarly, the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) trial8 included patients ages 70 to 84 (mean age 76.1) and found no difference in outcomes between a goal systolic pressure of less than 140 mm Hg (achieved systolic pressure 136.6 mm Hg) and a goal of 140 to 150 mm Hg (achieved systolic pressure 142 mm Hg).
The Hypertension in the Very Elderly Trial (HYVET)9 found lower rates of stroke, death, and heart failure in patients age 80 and older when their systolic pressure was less than 150 mm Hg.
While these trials support a goal pressure of less than 150 mm Hg in the elderly, it is unclear whether this goal should be applied beginning at age 60. Other guidelines, including those recently released jointly by the American Society of Hypertension and the International Society of Hypertension, recommend a systolic goal of less than 150 mm Hg in people age 80 and older—not age 60.10
The recommendation for a goal systolic pressure of less than 150 mm Hg in people age 60 and older was not unanimous; some panel members recommended continuing the JNC 7 goal of less than 140 mm Hg based on expert opinion, as they believed that the evidence was insufficient, especially in high-risk subgroups such as black people and those with cerebrovascular disease and other risk factors.
A subsequent minority report from five panel members discusses in more detail why they believe the systolic target should be kept lower than 140 mm Hg in patients age 60 or older until the risks and benefits of a higher target become clearer.11
Corollary recommendation: No need to down-titrate if lower than 140
In the general population age 60 and older, dosages do not have to be adjusted downward if the patient’s systolic pressure is already lower than 140 mm Hg and treatment is well tolerated without adverse effects on health or quality of life.
Strength of recommendation—expert opinion (grade E).
Comments. In the studies that supported a systolic goal lower than 150 mm Hg, many participants actually achieved a systolic pressure lower than 140 mm Hg without any adverse events. Trials that showed no benefit from a systolic goal lower than 140 mm Hg were graded as lower in quality. Thus, the possibility remains that a systolic goal lower than 140 mm Hg could have a clinically important benefit. Therefore, medications do not have to be adjusted so that blood pressure can “ride up.”
For example, therapy does not need to be down-titrated in a 65-year-old patient whose blood pressure is 138/85 mm Hg on two medications that he or she is tolerating well. On the other hand, based on Recommendation 1, therapy can be down-titrated in a 65-year-old whose pressure is 138/85 mm Hg on four medications that are causing side effects.
Recommendation 2: Diastolic < 90 for those younger than 60
In the general population younger than 60 years, JNC 8 recommends starting pharmacologic treatment if the diastolic pressure is 90 mm Hg or higher and aiming for a goal diastolic pressure of less than 90 mm Hg.
Strength of recommendation—strong (grade A) for ages 30 to 59, expert opinion (grade E) for ages 18 to 29.
Comments. The panel found no evidence to support a goal diastolic pressure of 80 mm Hg or less (or 85 mm Hg or less) compared with 90 mm Hg or less in this population.
It is reasonable to aim for the same diastolic goal in younger persons (under age 30), given the higher prevalence of diastolic hypertension in younger people.
Recommendation 3: Systolic < 140 for those younger than 60
In the general population younger than 60 years, we should start drug treatment at a systolic pressure of 140 mm Hg or higher and treat to a systolic goal of less than 140 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Although evidence was insufficient to support this recommendation, the panel decided to keep the same systolic goal for people younger than 60 as in the JNC 7 recommendations, for the following two reasons.
First, there is strong evidence supporting a diastolic goal of less than 90 mm Hg in this population (Recommendation 2), and many study participants who achieved a diastolic pressure lower than 90 mm Hg also achieved a systolic pressure lower than 140. Therefore, it is not possible to tease out whether the outcome benefits were due to lower systolic pressure or to lower diastolic pressure, or to both.
Second, the panel believed the guidelines would be simpler to implement if the systolic goals were the same in the general population as in those with chronic kidney disease or diabetes (see below).
Recommendation 4: < 140/90 in chronic kidney disease
In patients age 18 and older with chronic kidney disease, JNC 8 recommends starting drug treatment at a systolic pressure of 140 mm Hg or higher or a diastolic pressure of 90 mm Hg or higher and treating to a goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Chronic kidney disease is defined as either a glomerular filtration rate (estimated or measured) less than 60 mL/min/1.73 m2 in people up to age 70, or albuminuria, defined as more than 30 mg/g of creatinine at any glomerular filtration rate at any age.
Strength of recommendation—expert opinion (grade E).
Comments. There was insufficient evidence that aiming for a lower goal of 130/80 mm Hg (as in the JNC 7 recommendations) had any beneficial effect on cardiovascular, cerebrovascular, or mortality outcomes compared with 140/90 mm Hg, and there was moderate-quality evidence showing that treatment to lower goal (< 130/80 mm Hg) did not slow the progression of chronic kidney disease any better than a goal of less than 140/90 mm Hg. (One study that did find better renal outcomes with a lower blood pressure goal was a post hoc analysis of the Modification of Diet in Renal Disease study data in patients with proteinuria of more than 3 g per day.12)
We believe that decisions should be individualized regarding goal blood pressures and pharmacologic therapy in patients with chronic kidney disease and proteinuria, who may benefit from lower blood pressure goals (<130/80 mm Hg), based on low-level evidence.13,14 Risks and benefits should also be weighed in considering the blood pressure goal in the elderly with chronic kidney disease, taking into account functional status, comorbidities, and level of proteinuria.
Recommendation 5: < 140/90 for people with diabetes
In patients with diabetes who are age 18 and older, JNC 8 says to start drug treatment at a systolic pressure of 140 mm Hg or higher or diastolic pressure of 90 mm Hg or higher, and treat to goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Moderate-quality evidence showed cardiovascular, cerebrovascular, and mortality outcome benefits with treatment to a systolic goal of less than 150 mm Hg in patients with diabetes and hypertension.
The panel found no randomized controlled trials that compared a treatment goal of less than 140 mm Hg with one of less than 150 mm Hg for outcome benefits, but decided to base its recommendations on the results of the Action to Control Cardiovascular Risk in Diabetes—Blood-pressure-lowering Arm (ACCORD-BP) trial.15 The control group in this trial had a goal systolic pressure of less than 140 mm Hg and had similar outcomes compared with a lower goal.
The panel found no evidence to support a lower blood pressure goal (< 130/80) as in JNC 7. ACCORD-BP showed no differences in outcomes with a systolic goal lower than 140 mm Hg vs lower than 120 mm Hg except for a small reduction in stroke, and the risks of trying to achieve intensive lowering of blood pressure may outweigh the benefit of a small reduction in stroke.12 There was no evidence for a goal diastolic pressure below 80 mm Hg.
Recommendation 6: In nonblack patients, start with a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB
In the general nonblack population, including those with diabetes, initial drug treatment should include a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB.
Strength of recommendation—moderate (grade B).
Comments. All these drug classes had comparable outcome benefits in terms of rates of death, cardiovascular disease, cerebrovascular disease, and kidney disease, but not heart failure. For improving heart failure outcomes, thiazide-type diuretics are better than ACE inhibitors, which in turn are better than calcium channel blockers.
Thiazide-type diuretics (eg, hydrochlorothiazide, chlorthalidone, and indapamide) were recommended as first-line therapy for most patients in JNC 7, but they no longer carry this preferred status in JNC 8. In addition, the panel did not address preferential use of chlorthalidone as opposed to hydrochlorothiazide, or the use of spironolactone in resistant hypertension.
The panel did not recommend beta-blockers as first-line therapy because there were no differences in outcomes (or insufficient evidence) compared with the above medication classes; additionally, the Losartan Intervention for Endpoint Reduction in Hypertension study16 reported a higher incidence of stroke with a beta-blocker than with an ARB. However, JNC 8 did not consider randomized controlled trials in specific nonhypertensive populations such as patients with coronary artery disease or heart failure. We believe decisions should be individualized as to the use of beta-blockers in these two conditions.
The panel recommended the same approach in patients with diabetes, as there were no differences in major cardiovascular or cerebrovascular outcomes compared with the general population.
Recommendation 7: In black patients, start with a thiazide-type diuretic or calcium channel blocker
In the general black population, including those with diabetes, JNC 8 recommends starting drug treatment with a thiazide-type diuretic or a calcium channel blocker.
Strength of recommendation—moderate (grade B) for the general black population; weak (grade C) for blacks with diabetes.
Comments. In the black subgroup in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack trial (ALLHAT),17 a thiazide-type diuretic (chlorthalidone) was better than an ACE inhibitor (lisinopril) in terms of cerebrovascular, heart failure, and composite outcomes, but similar for mortality rates and cardiovascular, and kidney outcomes. Also in this subgroup, a calcium channel blocker (amlodipine) was better than the ACE inhibitor for cerebrovascular outcomes (there was a 51% higher rate of stroke with the ACE inhibitor as initial therapy than with the calcium channel blocker); the ACE inhibitor was also less effective in reducing blood pressure in blacks than the calcium channel blocker.
For improving heart failure outcomes, the thiazide-type diuretic was better than the ACE inhibitor, which in turn was better than the calcium channel blocker.
Evidence for black patients with diabetes (graded as weak) was extrapolated from ALLHAT, in which 46% had diabetes.17 We would consider using an ACE inhibitor or ARB in this population on an individual basis, especially if the patient had proteinuria.
Recommendation 8: ACEs and ARBs for chronic kidney disease
In patients age 18 and older with chronic kidney disease, irrespective of race, diabetes, or proteinuria, initial or add-on drug treatment should include an ACE inhibitor or ARB to improve kidney outcomes.
Strength of recommendation—moderate (grade B).
Comments. Treatment with an ACE inhibitor or ARB improves kidney outcomes in patients with chronic kidney disease. But in this population, these drugs are no more beneficial than calcium channel blockers or beta-blockers in terms of cardiovascular outcomes.
No randomized controlled trial has compared ACE inhibitors and ARBs for cardiovascular outcomes in chronic kidney disease, and these drugs have similar effects on kidney outcomes.
The panel did not make any recommendations about direct renin inhibitors, as there were no eligible studies demonstrating benefits on cardiovascular or kidney outcomes.
In black patients with chronic kidney disease and proteinuria, the panel recommended initial therapy with an ACE inhibitor or ARB to slow progression to end-stage renal disease (contrast with Recommendation 7).
In black patients with chronic kidney disease and no proteinuria, the panel recommended choosing from a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB. If an ACE inhibitor or ARB is not used as initial therapy, then one can be added on as a second-line medication (contrast with Recommendation 7).
The panel found no evidence to support this recommendation in people over age 75 and noted that although an ACE inhibitor or ARB may be beneficial in this group, a thiazide-type diuretic or calcium channel blocker can be considered.
Recommendation 9: If not at goal, step up
The main objective of pharmacologic treatment of hypertension is to attain and maintain the goal blood pressure. Lifestyle interventions should be maintained throughout treatment (Table 1). Medications can be initiated and titrated according to any of three strategies used in the randomized controlled trials selected by the panel (detailed below). Do not use an ACE inhibitor and ARB together in same patient.
If blood pressure is not at goal using all medication classes as in Recommendation 6 (ie, the triple combination of a thiazide-type diuretic, calcium channel blocker, and either an ACE inhibitor or an ARB), if there is a contraindication to any of these medication classes, or if there is need to use more than three medications to reach the goal, drugs from other classes can be used.
Referral to a hypertension specialist may be indicated for patients who are not at goal using the above strategy or for whom additional clinical consultation is needed.
Strength of recommendation—expert opinion (grade E).
Comments. Blood pressure should be monitored and assessed regularly, treatment adjusted as needed, and lifestyle modifications encouraged.
The panel did not recommend any monitoring schedule before or after goal blood pressure is achieved, and this should be individualized.
ADDITIONAL TOPICS IN JNC 8
A supplemental report covered some additional topics for which formal evidence review was not conducted but which the panel considered important.
Measuring and monitoring blood pressure
The panel recommended measuring the blood pressure with an automated oscillometric device that is properly calibrated and validated, or carefully measuring it manually.
Blood pressure should be measured in a quiet and relaxed environment with the patient seated comfortably for at least 5 minutes in a chair (rather than on an examination table) with feet flat on the floor, back supported, and arm supported at heart level. Blood pressure should be taken on the bare upper arm with an appropriate-sized cuff whose bladder encircles at least 80% of the mid-upper arm circumference, and patients should avoid caffeine, smoking, and physical activity for at least 30 minutes before measurement. In addition, patients should be asked about the need to empty the bladder (and encouraged to do so if they have to).
To establish the diagnosis of hypertension and to assess whether blood pressure goals are being met, two or three measurements should be taken at each visit as outlined above, and the average recorded.
At the first visit, blood pressure should be measured in both arms, and the arm with the higher pressure should be used for subsequent measurements.
Appropriate dosing of antihypertensive medications
Dosing should be individualized for each patient, but in general, target doses can be achieved within 2 to 4 weeks, and generally should not take longer than 2 months.
In general, to minimize potential adverse effects, treatment is started at a lower dose than the target dose and is then titrated up. This is especially important in older patients and patients on multiple medications with other comorbidities, and if two antihypertensive medications are being started simultaneously.
The panel reviewed evidence-based dosing of antihypertensive medications that were shown to improve cardiovascular outcomes from the studies that were selected for review. Hydrochlorothiazide gets a special mention: although doses up to 100 mg were used in some studies, the panel recommended an evidence-based dose of 25 or 50 mg daily to balance efficacy and safety.
Three strategies for dosing antihypertensive medications that were used in the selected randomized controlled trials were provided. These strategies were not compared with each other, nor is it known if one is better than the others in terms of health outcomes. In all cases, avoid combining an ACE inhibitor and an ARB.
- Start one drug from the four classes in Recommendation 6, titrate to the maximum dose, then add a second drug and titrate, then add a third drug and titrate to achieve the goal blood pressure.
- Start one drug from the four classes in Recommendation 6 and add a second drug before increasing the initial drug to its maximal dose. Titrate both to maximal doses, and add a third drug if needed and titrate to achieve the goal blood pressure.
- Start with two drugs at the same time from the four classes in Recommendation 6, either as separate pills or in a fixed-dose combination. Add a third drug if needed to achieve the goal blood pressure.
Lifestyle modification
The panel did not extensively review the evidence for lifestyle modification but endorsed the recommendations of the Lifestyle Work Group, which was convened by the NHLBI to focus on the effects of diet and physical activity on cardiovascular disease risk factors.18
Diet. The Lifestyle Work Group recommends combining the Dietary Approaches to Stop Hypertension (DASH) diet with reduced sodium intake, as there is evidence of a greater blood-pressure-lowering effect when the two are combined. The effect on blood pressure is independent of changes in weight.
The Lifestyle Work Group recommends consuming no more than 2,400 mg of sodium per day, noting that limiting intake to 1,500 mg can result in even greater reduction in blood pressure, and that even without achieving these goals, reducing sodium intake by at least 1,000 mg per day lowers blood pressure.
Physical activity. The Lifestyle Work Group recommends moderate to vigorous physical activity for approximately 160 minutes per week (three to four sessions a week, lasting an average of 40 minutes per session).
Weight loss. The Lifestyle Work Group did not review the blood-pressure-lowering effect of weight loss in those who are overweight or obese. The JNC 8 panel endorsed maintaining a healthy weight in controlling blood pressure.
Alcohol intake received no specific recommendations in JNC 8.
JNC 8 IN PERSPECTIVE
JNC 8 takes a rigorous, evidence-based approach and focuses on a few key questions. Thus, it is very different from the earlier reports: it has a narrower focus and does not address the full range of issues related to hypertension.
Strengths of JNC 8
The panel followed a rigorous process of review and evaluation of evidence from randomized controlled trials, adhering closely to standards set by the Institute of Medicine for guideline development. In contrast, JNC 7 relied on consensus and expert opinion.
The JNC 8 guidelines aim to simplify recommendations, with only two goals to remember: treat to lower than 150/90 mm Hg in patients age 60 and older, and lower than 140/90 mm Hg for everybody else. The initial drug regimen was simplified as well, with any of four choices for initial therapy in nonblacks and two in blacks.
Relaxing the blood pressure goals in elderly patients (although a cutoff of age 60 vs age 80 is likely to be debated) will also allay concerns about overtreating hypertension and causing adverse events in this population that is particularly susceptible to orthostatic changes and is at increased risk of falls.
Limitations and concerns
While the evidence-based nature of the recommendations is a strength, information from observational studies, systematic reviews, and meta-analyses was not incorporated into the formulation of these guidelines. This limits the available evidence, reflected in the fact that despite an extensive attempt to provide recommendations based on good evidence, five of the 10 recommendations (including the corollary recommendation) are still based on expert consensus opinion. Comparing and combining studies from different time periods is also problematic because of different methods of conducting clinical trials and analysis, and also because clinical care in a different period may differ from current standard practices.
Blood pressure targets in some subgroups are not clearly addressed, including those with proteinuria and with a history of stroke. Peterson et al,19 in an editorial accompanying the JNC 8 publication, commented on the need for larger randomized controlled trials to compare different blood pressure thresholds in various patient populations.
Some health care providers will likely be concerned that relaxing blood pressure goals could lead to higher real-world blood pressures, eventually leading to adverse cardiovascular outcomes, particularly on a population level. This is akin to the “speed limit rule”—people are more likely to hover above target, no matter what the target is.
In another editorial, Sox20 raised concerns about the external review process, ie, that the guidelines were not published in draft form to solicit public comment. Additionally, although the recommendations underwent extensive review, they were not endorsed by the specialty societies that the NHLBI designated to develop guidelines. In its defense, however, the JNC 8 panel has offered to share records of the review process on request, and this should serve to increase confidence in the review process.
The original literature search was limited to studies published through December 2009, which is more than 4 years before the publication of the recommendations. Although a bridge search was conducted until August 2013 to identify additional studies, this search used different inclusion criteria than the original criteria.
With its narrow focus, JNC 8 does not address many relevant issues. The American Society of Hypertension/International Society of Hypertension guidelines, published around the same time that the JNC 8 report was released, provide a more comprehensive review that will be of practical use for health care providers in the community.10
Ambulatory blood pressure monitoring is increasingly being used in clinical practice to detect white coat hypertension and, in many cases, to assess hypertension that is resistant to medications. It has also been shown to have better prognostic value in predicting cardiovascular risk and progression of kidney disease than office blood pressures.21,22 The UK National Institute of Health and Care Excellence guideline recommends ambulatory monitoring for the diagnosis of hypertension.23 However, JNC 8 did not provide specific recommendations for the use of this technology. Additionally, the JNC 8 evidence review is based on studies that used office blood pressure readings, and the recommendations are not necessarily applicable to measurements obtained by ambulatory monitoring.
Other topics covered in JNC 7 but not in JNC 8 include:
- Definitions and stages of hypertension (which remain the same)
- Initial treatment of stage 2 hypertension with two medications
- The J-curve phenomenon
- Preferred medications for patients with coronary artery disease or congestive heart failure
- A detailed list of oral antihypertensive agents—JNC 8 confines itself to the drugs and doses used in randomized controlled trials
- Patient evaluation
- Secondary hypertension
- Resistant hypertension
- Adherence issues.
Contrast with other guidelines
While the goal of these recommendations is to make treatment standards more understandable and uniform, contrasting recommendations on blood pressure goals and medications from various groups muddy the waters. Other groups that have issued hypertension guidelines in recent years include:
- The American Diabetes Association24
- The American Society of Hypertension and the International Society of Hypertension10
- The European Society of Hypertension and the European Society of Cardiology25
- The Canadian Hypertension Education Program26
- The Kidney Disease: Improving Global Outcomes initiative14
- The National Institute for Health and Clinical Excellence (UK)23
- The International Society on Hypertension in Blacks27
- The American Heart Association, the American College of Cardiology, and the US Centers for Disease Control and Prevention.28
Future directions
Despite the emphasis on making treatment decisions on an individual basis and using guidelines only as a framework for a safe direction in managing difficult clinical scenarios, guideline recommendations are increasingly being used to assess provider performance and quality of care, and so they assume even more importance in the current health care environment. As specialty organizations review and decide whether to endorse the JNC 8 recommendations, reconciling seemingly disparate recommendations from various groups is needed to send a clear and concise message to practitioners taking care of patients with high blood pressure.
Although a daunting task, integrating guidelines on hypertension management with other cardiovascular risk guidelines (eg, cholesterol, obesity) with assessment of overall cardiovascular risk profile would likely help in developing a more effective cardiovascular prevention strategy.
Despite the panel’s best efforts at providing evidence-based recommendations, many of the recommendations are based on expert opinion, reflecting the need for larger well-conducted studies. It is hoped that ongoing studies such as the Systolic Blood Pressure Intervention Trial29 will provide more clarity about blood pressure goals, especially in the elderly.
Final thoughts
Guidelines are not rules, and while they provide a framework by synthesizing the best available evidence, any treatment plan should be formulated on the basis of individual patient characteristics, including comorbidities, lifestyle factors, medication side effects, patient preferences, cost issues, and adherence.
The report of the panel appointed to the eighth Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8),1 published in December 2013 after considerable delay, contains some important changes from earlier guidelines from this group.2 For example:
- The blood pressure goal has been changed to less than 150/90 mm Hg in people age 60 and older. Formerly, the goal was less than 140/90 mm Hg.
- The goal has been changed to less than 140/90 mm Hg in all others, including people with diabetes mellitus and chronic kidney disease. Formerly, those two groups had a goal of less than 130/80 mm Hg.
- The initial choice of therapy can be from any of four classes of drugs: thiazide-type diuretics, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers (ARBs). Formerly, the list also contained beta-blockers. Also, thiazide-type diuretics have lost their “preferred” status.
The new guidelines are evidence-based and are intended to simplify the way that hypertension is managed. Below, we summarize them—how they were developed, their strengths and limitations, and the main changes from earlier JNC reports.
WHOSE GUIDELINES ARE THESE?
The JNC has issued guidelines for managing hypertension since 1976, traditionally sanctioned by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health. The guidelines have generally been updated every 4 to 5 years, with the last update, JNC 7,2 published in 2003.
The JNC 8 panel, consisting of 17 members, was commissioned by the NHLBI in 2008. However, in June 2013, the NHLBI announced it was withdrawing from guideline development and was delegating it to selected specialty organizations.3,4 In the interest of bringing the already delayed guidelines to the public in a timely manner, the JNC 8 panel decided to pursue publication independently and submitted the report to a medical journal. It is therefore not an official NHLBI-sanctioned report.
Here, we will refer to the new guidelines as “JNC 8,” but they are officially from “panel members appointed to the Eighth Joint National Committee (JNC 8).”
THREE QUESTIONS THAT GUIDED THE GUIDELINES
Epidemiologic studies clearly show a close relationship between blood pressure and the risk of heart disease, stroke, and kidney disease, these risks being lowest at a blood pressure of around 115/75 mm Hg.5 However, clinical trials have failed to show any evidence to justify treatment with antihypertensive medications to such a low level once hypertension has been diagnosed.
Patients and health care providers thus face questions about when to begin treatment, how low to aim for, and which antihypertensive medications to use. The JNC 8 panel focused on these three questions, believing them to be of greatest relevance to primary care providers.
A RIGOROUS PROCESS OF EVIDENCE REVIEW AND GUIDELINE DEVELOPMENT
The JNC 8 panel followed the guideline-development pathway outlined by the Institute of Medicine report, Clinical Practice Guidelines We Can Trust.6
Studies published from January 1966 through December 2009 that met specified criteria were selected for evidence review. Specifically, the studies had to be randomized controlled trials—no observational studies, systematic reviews, or meta-analyses, which were allowed in the JNC 7 report—with sample sizes of more than 100. Follow-up had to be for more than 1 year. Participants had to be age 18 or older and have hypertension—studies with patients with normal blood pressure or prehypertension were excluded. Health outcomes had to be reported, ie, “hard” end points such as rates of death, myocardial infarction, heart failure, hospitalization for heart failure, stroke, revascularization, and end-stage renal disease. Post hoc analyses were not allowed. The studies had to be rated by the NHLBI’s standardized quality rating tool as “good” (which has the least risk of bias, with valid results) or “fair (which is susceptible to some bias, but not enough to invalidate the results).
Subsequently, another search was conducted for relevant studies published from December 2009 through August 2013. In addition to meeting all the other criteria, this bridging search further restricted selection to major multicenter studies with sample sizes of more than 2,000.
An external methodology team performed the initial literature review and summarized the data. The JNC panel then crafted evidence statements and clinical recommendations using the evidence quality rating and grading systems developed by the NHLBI. In January 2013, the NHLBI submitted the guidelines for external review by individual reviewers with expertise in hypertension and to federal agencies, and a revised document was framed based on their comments and suggestions.
The evidence statements are detailed in an online 300-page supplemental review, and the panel members have indicated that reviewer comments and responses from the presubmission review process will be made available on request.
NINE RECOMMENDATIONS AND ONE COROLLARY
The panel made nine recommendations and one corollary recommendation based on a review of the evidence. Of the 10 total recommendations, five are based on expert opinion. Another two were rated as “moderate” in strength, one was “weak,” and only two were rated as “strong” (ie, based on high-quality evidence).
Recommendation 1: < 150/90 for those 60 and older
In the general population age 60 and older, the JNC 8 recommends starting drug treatment if the systolic pressure is 150 mm Hg or higher or if the diastolic pressure is 90 mm Hg or higher, and aiming for a systolic goal of less than 150 mm Hg and a diastolic goal of less than 90 mm Hg.
Strength of recommendation—strong (grade A).
Comments. Of all the recommendations, this one will probably have the greatest impact on clinical practice. Consider a frail 70-year-old patient at risk of falls who is taking two antihypertensive medications and whose blood pressure is 148/85 mm Hg. This level would have been considered too high under JNC 7 but is now acceptable, and the patient’s therapy does not have to be escalated.
The age cutoff of 60 years for this recommendation is debatable. The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)7 included patients ages 60 to 85 (mean age 74) and found no difference in outcomes comparing a goal systolic pressure of less than 140 mm Hg (this group achieved a mean systolic pressure of 135.9 mm Hg) and a goal systolic pressure of 140 to 160 mm Hg (achieved systolic pressure 145.6 mm Hg).
Similarly, the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) trial8 included patients ages 70 to 84 (mean age 76.1) and found no difference in outcomes between a goal systolic pressure of less than 140 mm Hg (achieved systolic pressure 136.6 mm Hg) and a goal of 140 to 150 mm Hg (achieved systolic pressure 142 mm Hg).
The Hypertension in the Very Elderly Trial (HYVET)9 found lower rates of stroke, death, and heart failure in patients age 80 and older when their systolic pressure was less than 150 mm Hg.
While these trials support a goal pressure of less than 150 mm Hg in the elderly, it is unclear whether this goal should be applied beginning at age 60. Other guidelines, including those recently released jointly by the American Society of Hypertension and the International Society of Hypertension, recommend a systolic goal of less than 150 mm Hg in people age 80 and older—not age 60.10
The recommendation for a goal systolic pressure of less than 150 mm Hg in people age 60 and older was not unanimous; some panel members recommended continuing the JNC 7 goal of less than 140 mm Hg based on expert opinion, as they believed that the evidence was insufficient, especially in high-risk subgroups such as black people and those with cerebrovascular disease and other risk factors.
A subsequent minority report from five panel members discusses in more detail why they believe the systolic target should be kept lower than 140 mm Hg in patients age 60 or older until the risks and benefits of a higher target become clearer.11
Corollary recommendation: No need to down-titrate if lower than 140
In the general population age 60 and older, dosages do not have to be adjusted downward if the patient’s systolic pressure is already lower than 140 mm Hg and treatment is well tolerated without adverse effects on health or quality of life.
Strength of recommendation—expert opinion (grade E).
Comments. In the studies that supported a systolic goal lower than 150 mm Hg, many participants actually achieved a systolic pressure lower than 140 mm Hg without any adverse events. Trials that showed no benefit from a systolic goal lower than 140 mm Hg were graded as lower in quality. Thus, the possibility remains that a systolic goal lower than 140 mm Hg could have a clinically important benefit. Therefore, medications do not have to be adjusted so that blood pressure can “ride up.”
For example, therapy does not need to be down-titrated in a 65-year-old patient whose blood pressure is 138/85 mm Hg on two medications that he or she is tolerating well. On the other hand, based on Recommendation 1, therapy can be down-titrated in a 65-year-old whose pressure is 138/85 mm Hg on four medications that are causing side effects.
Recommendation 2: Diastolic < 90 for those younger than 60
In the general population younger than 60 years, JNC 8 recommends starting pharmacologic treatment if the diastolic pressure is 90 mm Hg or higher and aiming for a goal diastolic pressure of less than 90 mm Hg.
Strength of recommendation—strong (grade A) for ages 30 to 59, expert opinion (grade E) for ages 18 to 29.
Comments. The panel found no evidence to support a goal diastolic pressure of 80 mm Hg or less (or 85 mm Hg or less) compared with 90 mm Hg or less in this population.
It is reasonable to aim for the same diastolic goal in younger persons (under age 30), given the higher prevalence of diastolic hypertension in younger people.
Recommendation 3: Systolic < 140 for those younger than 60
In the general population younger than 60 years, we should start drug treatment at a systolic pressure of 140 mm Hg or higher and treat to a systolic goal of less than 140 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Although evidence was insufficient to support this recommendation, the panel decided to keep the same systolic goal for people younger than 60 as in the JNC 7 recommendations, for the following two reasons.
First, there is strong evidence supporting a diastolic goal of less than 90 mm Hg in this population (Recommendation 2), and many study participants who achieved a diastolic pressure lower than 90 mm Hg also achieved a systolic pressure lower than 140. Therefore, it is not possible to tease out whether the outcome benefits were due to lower systolic pressure or to lower diastolic pressure, or to both.
Second, the panel believed the guidelines would be simpler to implement if the systolic goals were the same in the general population as in those with chronic kidney disease or diabetes (see below).
Recommendation 4: < 140/90 in chronic kidney disease
In patients age 18 and older with chronic kidney disease, JNC 8 recommends starting drug treatment at a systolic pressure of 140 mm Hg or higher or a diastolic pressure of 90 mm Hg or higher and treating to a goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Chronic kidney disease is defined as either a glomerular filtration rate (estimated or measured) less than 60 mL/min/1.73 m2 in people up to age 70, or albuminuria, defined as more than 30 mg/g of creatinine at any glomerular filtration rate at any age.
Strength of recommendation—expert opinion (grade E).
Comments. There was insufficient evidence that aiming for a lower goal of 130/80 mm Hg (as in the JNC 7 recommendations) had any beneficial effect on cardiovascular, cerebrovascular, or mortality outcomes compared with 140/90 mm Hg, and there was moderate-quality evidence showing that treatment to lower goal (< 130/80 mm Hg) did not slow the progression of chronic kidney disease any better than a goal of less than 140/90 mm Hg. (One study that did find better renal outcomes with a lower blood pressure goal was a post hoc analysis of the Modification of Diet in Renal Disease study data in patients with proteinuria of more than 3 g per day.12)
We believe that decisions should be individualized regarding goal blood pressures and pharmacologic therapy in patients with chronic kidney disease and proteinuria, who may benefit from lower blood pressure goals (<130/80 mm Hg), based on low-level evidence.13,14 Risks and benefits should also be weighed in considering the blood pressure goal in the elderly with chronic kidney disease, taking into account functional status, comorbidities, and level of proteinuria.
Recommendation 5: < 140/90 for people with diabetes
In patients with diabetes who are age 18 and older, JNC 8 says to start drug treatment at a systolic pressure of 140 mm Hg or higher or diastolic pressure of 90 mm Hg or higher, and treat to goal systolic pressure of less than 140 mm Hg and a diastolic pressure of less than 90 mm Hg.
Strength of recommendation—expert opinion (grade E).
Comments. Moderate-quality evidence showed cardiovascular, cerebrovascular, and mortality outcome benefits with treatment to a systolic goal of less than 150 mm Hg in patients with diabetes and hypertension.
The panel found no randomized controlled trials that compared a treatment goal of less than 140 mm Hg with one of less than 150 mm Hg for outcome benefits, but decided to base its recommendations on the results of the Action to Control Cardiovascular Risk in Diabetes—Blood-pressure-lowering Arm (ACCORD-BP) trial.15 The control group in this trial had a goal systolic pressure of less than 140 mm Hg and had similar outcomes compared with a lower goal.
The panel found no evidence to support a lower blood pressure goal (< 130/80) as in JNC 7. ACCORD-BP showed no differences in outcomes with a systolic goal lower than 140 mm Hg vs lower than 120 mm Hg except for a small reduction in stroke, and the risks of trying to achieve intensive lowering of blood pressure may outweigh the benefit of a small reduction in stroke.12 There was no evidence for a goal diastolic pressure below 80 mm Hg.
Recommendation 6: In nonblack patients, start with a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB
In the general nonblack population, including those with diabetes, initial drug treatment should include a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB.
Strength of recommendation—moderate (grade B).
Comments. All these drug classes had comparable outcome benefits in terms of rates of death, cardiovascular disease, cerebrovascular disease, and kidney disease, but not heart failure. For improving heart failure outcomes, thiazide-type diuretics are better than ACE inhibitors, which in turn are better than calcium channel blockers.
Thiazide-type diuretics (eg, hydrochlorothiazide, chlorthalidone, and indapamide) were recommended as first-line therapy for most patients in JNC 7, but they no longer carry this preferred status in JNC 8. In addition, the panel did not address preferential use of chlorthalidone as opposed to hydrochlorothiazide, or the use of spironolactone in resistant hypertension.
The panel did not recommend beta-blockers as first-line therapy because there were no differences in outcomes (or insufficient evidence) compared with the above medication classes; additionally, the Losartan Intervention for Endpoint Reduction in Hypertension study16 reported a higher incidence of stroke with a beta-blocker than with an ARB. However, JNC 8 did not consider randomized controlled trials in specific nonhypertensive populations such as patients with coronary artery disease or heart failure. We believe decisions should be individualized as to the use of beta-blockers in these two conditions.
The panel recommended the same approach in patients with diabetes, as there were no differences in major cardiovascular or cerebrovascular outcomes compared with the general population.
Recommendation 7: In black patients, start with a thiazide-type diuretic or calcium channel blocker
In the general black population, including those with diabetes, JNC 8 recommends starting drug treatment with a thiazide-type diuretic or a calcium channel blocker.
Strength of recommendation—moderate (grade B) for the general black population; weak (grade C) for blacks with diabetes.
Comments. In the black subgroup in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack trial (ALLHAT),17 a thiazide-type diuretic (chlorthalidone) was better than an ACE inhibitor (lisinopril) in terms of cerebrovascular, heart failure, and composite outcomes, but similar for mortality rates and cardiovascular, and kidney outcomes. Also in this subgroup, a calcium channel blocker (amlodipine) was better than the ACE inhibitor for cerebrovascular outcomes (there was a 51% higher rate of stroke with the ACE inhibitor as initial therapy than with the calcium channel blocker); the ACE inhibitor was also less effective in reducing blood pressure in blacks than the calcium channel blocker.
For improving heart failure outcomes, the thiazide-type diuretic was better than the ACE inhibitor, which in turn was better than the calcium channel blocker.
Evidence for black patients with diabetes (graded as weak) was extrapolated from ALLHAT, in which 46% had diabetes.17 We would consider using an ACE inhibitor or ARB in this population on an individual basis, especially if the patient had proteinuria.
Recommendation 8: ACEs and ARBs for chronic kidney disease
In patients age 18 and older with chronic kidney disease, irrespective of race, diabetes, or proteinuria, initial or add-on drug treatment should include an ACE inhibitor or ARB to improve kidney outcomes.
Strength of recommendation—moderate (grade B).
Comments. Treatment with an ACE inhibitor or ARB improves kidney outcomes in patients with chronic kidney disease. But in this population, these drugs are no more beneficial than calcium channel blockers or beta-blockers in terms of cardiovascular outcomes.
No randomized controlled trial has compared ACE inhibitors and ARBs for cardiovascular outcomes in chronic kidney disease, and these drugs have similar effects on kidney outcomes.
The panel did not make any recommendations about direct renin inhibitors, as there were no eligible studies demonstrating benefits on cardiovascular or kidney outcomes.
In black patients with chronic kidney disease and proteinuria, the panel recommended initial therapy with an ACE inhibitor or ARB to slow progression to end-stage renal disease (contrast with Recommendation 7).
In black patients with chronic kidney disease and no proteinuria, the panel recommended choosing from a thiazide-type diuretic, calcium channel blocker, ACE inhibitor, or ARB. If an ACE inhibitor or ARB is not used as initial therapy, then one can be added on as a second-line medication (contrast with Recommendation 7).
The panel found no evidence to support this recommendation in people over age 75 and noted that although an ACE inhibitor or ARB may be beneficial in this group, a thiazide-type diuretic or calcium channel blocker can be considered.
Recommendation 9: If not at goal, step up
The main objective of pharmacologic treatment of hypertension is to attain and maintain the goal blood pressure. Lifestyle interventions should be maintained throughout treatment (Table 1). Medications can be initiated and titrated according to any of three strategies used in the randomized controlled trials selected by the panel (detailed below). Do not use an ACE inhibitor and ARB together in same patient.
If blood pressure is not at goal using all medication classes as in Recommendation 6 (ie, the triple combination of a thiazide-type diuretic, calcium channel blocker, and either an ACE inhibitor or an ARB), if there is a contraindication to any of these medication classes, or if there is need to use more than three medications to reach the goal, drugs from other classes can be used.
Referral to a hypertension specialist may be indicated for patients who are not at goal using the above strategy or for whom additional clinical consultation is needed.
Strength of recommendation—expert opinion (grade E).
Comments. Blood pressure should be monitored and assessed regularly, treatment adjusted as needed, and lifestyle modifications encouraged.
The panel did not recommend any monitoring schedule before or after goal blood pressure is achieved, and this should be individualized.
ADDITIONAL TOPICS IN JNC 8
A supplemental report covered some additional topics for which formal evidence review was not conducted but which the panel considered important.
Measuring and monitoring blood pressure
The panel recommended measuring the blood pressure with an automated oscillometric device that is properly calibrated and validated, or carefully measuring it manually.
Blood pressure should be measured in a quiet and relaxed environment with the patient seated comfortably for at least 5 minutes in a chair (rather than on an examination table) with feet flat on the floor, back supported, and arm supported at heart level. Blood pressure should be taken on the bare upper arm with an appropriate-sized cuff whose bladder encircles at least 80% of the mid-upper arm circumference, and patients should avoid caffeine, smoking, and physical activity for at least 30 minutes before measurement. In addition, patients should be asked about the need to empty the bladder (and encouraged to do so if they have to).
To establish the diagnosis of hypertension and to assess whether blood pressure goals are being met, two or three measurements should be taken at each visit as outlined above, and the average recorded.
At the first visit, blood pressure should be measured in both arms, and the arm with the higher pressure should be used for subsequent measurements.
Appropriate dosing of antihypertensive medications
Dosing should be individualized for each patient, but in general, target doses can be achieved within 2 to 4 weeks, and generally should not take longer than 2 months.
In general, to minimize potential adverse effects, treatment is started at a lower dose than the target dose and is then titrated up. This is especially important in older patients and patients on multiple medications with other comorbidities, and if two antihypertensive medications are being started simultaneously.
The panel reviewed evidence-based dosing of antihypertensive medications that were shown to improve cardiovascular outcomes from the studies that were selected for review. Hydrochlorothiazide gets a special mention: although doses up to 100 mg were used in some studies, the panel recommended an evidence-based dose of 25 or 50 mg daily to balance efficacy and safety.
Three strategies for dosing antihypertensive medications that were used in the selected randomized controlled trials were provided. These strategies were not compared with each other, nor is it known if one is better than the others in terms of health outcomes. In all cases, avoid combining an ACE inhibitor and an ARB.
- Start one drug from the four classes in Recommendation 6, titrate to the maximum dose, then add a second drug and titrate, then add a third drug and titrate to achieve the goal blood pressure.
- Start one drug from the four classes in Recommendation 6 and add a second drug before increasing the initial drug to its maximal dose. Titrate both to maximal doses, and add a third drug if needed and titrate to achieve the goal blood pressure.
- Start with two drugs at the same time from the four classes in Recommendation 6, either as separate pills or in a fixed-dose combination. Add a third drug if needed to achieve the goal blood pressure.
Lifestyle modification
The panel did not extensively review the evidence for lifestyle modification but endorsed the recommendations of the Lifestyle Work Group, which was convened by the NHLBI to focus on the effects of diet and physical activity on cardiovascular disease risk factors.18
Diet. The Lifestyle Work Group recommends combining the Dietary Approaches to Stop Hypertension (DASH) diet with reduced sodium intake, as there is evidence of a greater blood-pressure-lowering effect when the two are combined. The effect on blood pressure is independent of changes in weight.
The Lifestyle Work Group recommends consuming no more than 2,400 mg of sodium per day, noting that limiting intake to 1,500 mg can result in even greater reduction in blood pressure, and that even without achieving these goals, reducing sodium intake by at least 1,000 mg per day lowers blood pressure.
Physical activity. The Lifestyle Work Group recommends moderate to vigorous physical activity for approximately 160 minutes per week (three to four sessions a week, lasting an average of 40 minutes per session).
Weight loss. The Lifestyle Work Group did not review the blood-pressure-lowering effect of weight loss in those who are overweight or obese. The JNC 8 panel endorsed maintaining a healthy weight in controlling blood pressure.
Alcohol intake received no specific recommendations in JNC 8.
JNC 8 IN PERSPECTIVE
JNC 8 takes a rigorous, evidence-based approach and focuses on a few key questions. Thus, it is very different from the earlier reports: it has a narrower focus and does not address the full range of issues related to hypertension.
Strengths of JNC 8
The panel followed a rigorous process of review and evaluation of evidence from randomized controlled trials, adhering closely to standards set by the Institute of Medicine for guideline development. In contrast, JNC 7 relied on consensus and expert opinion.
The JNC 8 guidelines aim to simplify recommendations, with only two goals to remember: treat to lower than 150/90 mm Hg in patients age 60 and older, and lower than 140/90 mm Hg for everybody else. The initial drug regimen was simplified as well, with any of four choices for initial therapy in nonblacks and two in blacks.
Relaxing the blood pressure goals in elderly patients (although a cutoff of age 60 vs age 80 is likely to be debated) will also allay concerns about overtreating hypertension and causing adverse events in this population that is particularly susceptible to orthostatic changes and is at increased risk of falls.
Limitations and concerns
While the evidence-based nature of the recommendations is a strength, information from observational studies, systematic reviews, and meta-analyses was not incorporated into the formulation of these guidelines. This limits the available evidence, reflected in the fact that despite an extensive attempt to provide recommendations based on good evidence, five of the 10 recommendations (including the corollary recommendation) are still based on expert consensus opinion. Comparing and combining studies from different time periods is also problematic because of different methods of conducting clinical trials and analysis, and also because clinical care in a different period may differ from current standard practices.
Blood pressure targets in some subgroups are not clearly addressed, including those with proteinuria and with a history of stroke. Peterson et al,19 in an editorial accompanying the JNC 8 publication, commented on the need for larger randomized controlled trials to compare different blood pressure thresholds in various patient populations.
Some health care providers will likely be concerned that relaxing blood pressure goals could lead to higher real-world blood pressures, eventually leading to adverse cardiovascular outcomes, particularly on a population level. This is akin to the “speed limit rule”—people are more likely to hover above target, no matter what the target is.
In another editorial, Sox20 raised concerns about the external review process, ie, that the guidelines were not published in draft form to solicit public comment. Additionally, although the recommendations underwent extensive review, they were not endorsed by the specialty societies that the NHLBI designated to develop guidelines. In its defense, however, the JNC 8 panel has offered to share records of the review process on request, and this should serve to increase confidence in the review process.
The original literature search was limited to studies published through December 2009, which is more than 4 years before the publication of the recommendations. Although a bridge search was conducted until August 2013 to identify additional studies, this search used different inclusion criteria than the original criteria.
With its narrow focus, JNC 8 does not address many relevant issues. The American Society of Hypertension/International Society of Hypertension guidelines, published around the same time that the JNC 8 report was released, provide a more comprehensive review that will be of practical use for health care providers in the community.10
Ambulatory blood pressure monitoring is increasingly being used in clinical practice to detect white coat hypertension and, in many cases, to assess hypertension that is resistant to medications. It has also been shown to have better prognostic value in predicting cardiovascular risk and progression of kidney disease than office blood pressures.21,22 The UK National Institute of Health and Care Excellence guideline recommends ambulatory monitoring for the diagnosis of hypertension.23 However, JNC 8 did not provide specific recommendations for the use of this technology. Additionally, the JNC 8 evidence review is based on studies that used office blood pressure readings, and the recommendations are not necessarily applicable to measurements obtained by ambulatory monitoring.
Other topics covered in JNC 7 but not in JNC 8 include:
- Definitions and stages of hypertension (which remain the same)
- Initial treatment of stage 2 hypertension with two medications
- The J-curve phenomenon
- Preferred medications for patients with coronary artery disease or congestive heart failure
- A detailed list of oral antihypertensive agents—JNC 8 confines itself to the drugs and doses used in randomized controlled trials
- Patient evaluation
- Secondary hypertension
- Resistant hypertension
- Adherence issues.
Contrast with other guidelines
While the goal of these recommendations is to make treatment standards more understandable and uniform, contrasting recommendations on blood pressure goals and medications from various groups muddy the waters. Other groups that have issued hypertension guidelines in recent years include:
- The American Diabetes Association24
- The American Society of Hypertension and the International Society of Hypertension10
- The European Society of Hypertension and the European Society of Cardiology25
- The Canadian Hypertension Education Program26
- The Kidney Disease: Improving Global Outcomes initiative14
- The National Institute for Health and Clinical Excellence (UK)23
- The International Society on Hypertension in Blacks27
- The American Heart Association, the American College of Cardiology, and the US Centers for Disease Control and Prevention.28
Future directions
Despite the emphasis on making treatment decisions on an individual basis and using guidelines only as a framework for a safe direction in managing difficult clinical scenarios, guideline recommendations are increasingly being used to assess provider performance and quality of care, and so they assume even more importance in the current health care environment. As specialty organizations review and decide whether to endorse the JNC 8 recommendations, reconciling seemingly disparate recommendations from various groups is needed to send a clear and concise message to practitioners taking care of patients with high blood pressure.
Although a daunting task, integrating guidelines on hypertension management with other cardiovascular risk guidelines (eg, cholesterol, obesity) with assessment of overall cardiovascular risk profile would likely help in developing a more effective cardiovascular prevention strategy.
Despite the panel’s best efforts at providing evidence-based recommendations, many of the recommendations are based on expert opinion, reflecting the need for larger well-conducted studies. It is hoped that ongoing studies such as the Systolic Blood Pressure Intervention Trial29 will provide more clarity about blood pressure goals, especially in the elderly.
Final thoughts
Guidelines are not rules, and while they provide a framework by synthesizing the best available evidence, any treatment plan should be formulated on the basis of individual patient characteristics, including comorbidities, lifestyle factors, medication side effects, patient preferences, cost issues, and adherence.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289: 2560–2572. Erratum in JAMA 2003; 290:197.
- Gibbons GH, Harold JG, Jessup M, Robertson RM, Oetgen WJ. The next steps in developing clinical practice guidelines for prevention. J Am Coll Cardiol 2013; 62:1399–1400.
- Gibbons GH, Shurin SB, Mensah GA, Lauer MS. Refocusing the agenda on cardiovascular guidelines: an announcement from the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2013; 62:1396–1398.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913. Erratum in: Lancet 2003; 361:1060.
- Institute of Medicine. Clinical Practice Guidelines We Can Trust. Washington, DC: National Academies Press; 2011. http://www.iom.edu/Reports/2011/Clinical-Practice-Guide-lines-We-Can-Trust.aspx. Accessed February 4, 2014.
- JATOS Study Group. Principal results of the Japanese Trial To Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56:196–202.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Wright JT, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014 Jan 14. [Epub ahead of print]
- Klahr S, Levey AS, Beck GJ, et al. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N Engl J Med 1994; 330:877–884.
- Upadhyay A, Earley A, Haynes SM, Uhlig K. Systematic review: blood pressure target in chronic kidney disease and proteinuria as an effect modifier. Ann Intern Med 2011; 154:541–548.
- Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int Suppl 2012; 2:337–414.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Hypertension 2003; 42:239–246.
- Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2013 Nov 12. [Epub ahead of print]
- Peterson ED, Gaziano JM, Greenland P. Recommendations for treating hypertension: what are the right goals and purposes? JAMA Editorial. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Sox HC. Assessing the trustworthiness of the guideline for management of high blood pressure in adults (editorial). JAMA. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Dolan E, Stanton A, Thijs L, et al. Superiority of ambulatory over clinic blood pressure measurement in predicting mortality: the Dublin outcome study. Hypertension 2005; 46:156–161.
- Agarwal R, Andersen MJ. Prognostic importance of ambulatory blood pressure recordings in patients with chronic kidney disease. Kidney Int 2006; 69:1175–1180.
- National Institute for Health and Clinical Excellence. Hypertension (CG127). http://publications.nice.org.uk/hypertension-cg127. Accessed February 4, 2014.
- American Diabetes Association. Standards of medical care in diabetes – 2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC practice guidelines for the management of arterial hypertension. Blood Press 2013 Dec 20. [Epub ahead of print]
- Hypertension without compelling indications: 2013 CHEP recommendations. Hypertension Canada website. http://www.hypertension.ca/hypertension-without-compelling-indications. Accessed February 4, 2014.
- Flack JM, Sica DA, Bakris G, et al; International Society on Hypertension in Blacks. Management of high blood pressure in blacks: an update of the International Society on Hypertension in Blacks consensus statement. Hypertension 2010; 56:780–800.
- Go AS, Bauman M, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2013 Nov 15.
- Systolic Blood Pressure Intervention Trial (SPRINT). http://clinicaltrials.gov/ct2/show/NCT01206062. Accessed February 4, 2014.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama.2013.284427.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289: 2560–2572. Erratum in JAMA 2003; 290:197.
- Gibbons GH, Harold JG, Jessup M, Robertson RM, Oetgen WJ. The next steps in developing clinical practice guidelines for prevention. J Am Coll Cardiol 2013; 62:1399–1400.
- Gibbons GH, Shurin SB, Mensah GA, Lauer MS. Refocusing the agenda on cardiovascular guidelines: an announcement from the National Heart, Lung, and Blood Institute. J Am Coll Cardiol 2013; 62:1396–1398.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913. Erratum in: Lancet 2003; 361:1060.
- Institute of Medicine. Clinical Practice Guidelines We Can Trust. Washington, DC: National Academies Press; 2011. http://www.iom.edu/Reports/2011/Clinical-Practice-Guide-lines-We-Can-Trust.aspx. Accessed February 4, 2014.
- JATOS Study Group. Principal results of the Japanese Trial To Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56:196–202.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Wright JT, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014 Jan 14. [Epub ahead of print]
- Klahr S, Levey AS, Beck GJ, et al. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N Engl J Med 1994; 330:877–884.
- Upadhyay A, Earley A, Haynes SM, Uhlig K. Systematic review: blood pressure target in chronic kidney disease and proteinuria as an effect modifier. Ann Intern Med 2011; 154:541–548.
- Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group. KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int Suppl 2012; 2:337–414.
- Cushman WC, Evans GW, Byington RP, et al; ACCORD Study Group. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Diuretic versus alpha-blocker as first-step antihypertensive therapy: final results from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Hypertension 2003; 42:239–246.
- Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2013 Nov 12. [Epub ahead of print]
- Peterson ED, Gaziano JM, Greenland P. Recommendations for treating hypertension: what are the right goals and purposes? JAMA Editorial. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Sox HC. Assessing the trustworthiness of the guideline for management of high blood pressure in adults (editorial). JAMA. Published online December 18, 2013. doi: 10.1001/jama.2013.284430.
- Dolan E, Stanton A, Thijs L, et al. Superiority of ambulatory over clinic blood pressure measurement in predicting mortality: the Dublin outcome study. Hypertension 2005; 46:156–161.
- Agarwal R, Andersen MJ. Prognostic importance of ambulatory blood pressure recordings in patients with chronic kidney disease. Kidney Int 2006; 69:1175–1180.
- National Institute for Health and Clinical Excellence. Hypertension (CG127). http://publications.nice.org.uk/hypertension-cg127. Accessed February 4, 2014.
- American Diabetes Association. Standards of medical care in diabetes – 2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC practice guidelines for the management of arterial hypertension. Blood Press 2013 Dec 20. [Epub ahead of print]
- Hypertension without compelling indications: 2013 CHEP recommendations. Hypertension Canada website. http://www.hypertension.ca/hypertension-without-compelling-indications. Accessed February 4, 2014.
- Flack JM, Sica DA, Bakris G, et al; International Society on Hypertension in Blacks. Management of high blood pressure in blacks: an update of the International Society on Hypertension in Blacks consensus statement. Hypertension 2010; 56:780–800.
- Go AS, Bauman M, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2013 Nov 15.
- Systolic Blood Pressure Intervention Trial (SPRINT). http://clinicaltrials.gov/ct2/show/NCT01206062. Accessed February 4, 2014.
KEY POINTS
- JNC 8 focuses on three main questions: when to begin treatment, how low to aim for, and which antihypertensive medications to use. It does not cover many topics that were included in JNC 7.
- In patients age 60 or older, JNC 8 recommends starting antihypertensive treatment if the blood pressure is 150/90 mm Hg or higher, with a goal of less than 150/90.
- For everyone else, including people with diabetes or chronic kidney disease, the threshold is 140/90 mm Hg, and the goal is less than 140/90.
- The recommended classes of drugs for initial therapy in nonblack patients without chronic kidney disease are thiazide-type diuretics, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin receptor blockers (ARBs), although the last two classes should not be used in combination.
- For black patients, the initial classes of drugs are diuretics and calcium channel blockers; patients with chronic kidney disease should receive an ACE inhibitor or ARB.