User login
Drug-eluting stent recipients can safely have surgery sooner
CHICAGO – Current U.S. and European guidelines recommending postponement of noncardiac surgery for 6-12 months after drug-eluting stent implantation appear to be excessive, Dr. Gro Egholm reported at the annual meeting of the American College of Cardiology.
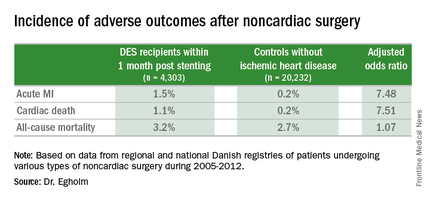
She presented a large retrospective observational study of outcomes in patients undergoing various types of noncardiac surgery in western Denmark during 2005-2012. Among 4,303 patients who had noncardiac surgery within 12 months after receiving a drug-eluting stent (DES), only those whose operations took place during the first month post stenting had increased risks of acute MI and cardiac death within 30 days post surgery.

Risks of major adverse cardiac events among the DES recipients who had noncardiac surgery within that first month post–percutaneous coronary intervention were increased roughly 7.5-fold compared with controls, but for surgery performed after that the risks of MI and cardiac death dropped off abruptly and were no different from rates in 20,232 controls without ischemic heart disease or stents who were matched for age, gender, surgical procedure, and Charlson Comorbidity Index, according to Dr. Egholm of Aarhus (Denmark) University.
Moreover, even in DES recipients undergoing noncardiac surgery during the first month post stenting, all-cause mortality was no greater than in controls.
“Surgery could be performed much earlier than recommended,” she concluded.
Her study was carried out by linking data from comprehensive regional and national Danish health care registries. Most patients with DES remained on dual antiplatelet therapy periprocedurally. The exceptions were neurosurgical operations and others where it’s standard that dual antiplatelet therapy must be stopped.
“If you can continue only one antiplatelet agent, aspirin would be the most appealing,” she said.
Of the DES participants, 56% received their device as treatment for an acute coronary syndrome. The average time from stent placement to noncardiac surgery in this large series was 147 days.
Session co-chair Dr. Sunil V. Rao of Duke University in Durham, N.C., called this work “a very important study that’s relevant to daily practice.” However, he found the 23% incidence of noncardiac surgery within 12 months following DES implantation reported in Dr. Egholm’s study to be “shockingly high.” She agreed, noting that rates in some non-Danish registries she’s looked at are more in the 8%-15% range. But Denmark’s health care registries are known for rigorous accuracy and completeness.
Dr. Egholm reported having no financial conflicts regarding her study.
CHICAGO – Current U.S. and European guidelines recommending postponement of noncardiac surgery for 6-12 months after drug-eluting stent implantation appear to be excessive, Dr. Gro Egholm reported at the annual meeting of the American College of Cardiology.
She presented a large retrospective observational study of outcomes in patients undergoing various types of noncardiac surgery in western Denmark during 2005-2012. Among 4,303 patients who had noncardiac surgery within 12 months after receiving a drug-eluting stent (DES), only those whose operations took place during the first month post stenting had increased risks of acute MI and cardiac death within 30 days post surgery.
Risks of major adverse cardiac events among the DES recipients who had noncardiac surgery within that first month post–percutaneous coronary intervention were increased roughly 7.5-fold compared with controls, but for surgery performed after that the risks of MI and cardiac death dropped off abruptly and were no different from rates in 20,232 controls without ischemic heart disease or stents who were matched for age, gender, surgical procedure, and Charlson Comorbidity Index, according to Dr. Egholm of Aarhus (Denmark) University.
Moreover, even in DES recipients undergoing noncardiac surgery during the first month post stenting, all-cause mortality was no greater than in controls.
“Surgery could be performed much earlier than recommended,” she concluded.
Her study was carried out by linking data from comprehensive regional and national Danish health care registries. Most patients with DES remained on dual antiplatelet therapy periprocedurally. The exceptions were neurosurgical operations and others where it’s standard that dual antiplatelet therapy must be stopped.
“If you can continue only one antiplatelet agent, aspirin would be the most appealing,” she said.
Of the DES participants, 56% received their device as treatment for an acute coronary syndrome. The average time from stent placement to noncardiac surgery in this large series was 147 days.
Session co-chair Dr. Sunil V. Rao of Duke University in Durham, N.C., called this work “a very important study that’s relevant to daily practice.” However, he found the 23% incidence of noncardiac surgery within 12 months following DES implantation reported in Dr. Egholm’s study to be “shockingly high.” She agreed, noting that rates in some non-Danish registries she’s looked at are more in the 8%-15% range. But Denmark’s health care registries are known for rigorous accuracy and completeness.
Dr. Egholm reported having no financial conflicts regarding her study.
CHICAGO – Current U.S. and European guidelines recommending postponement of noncardiac surgery for 6-12 months after drug-eluting stent implantation appear to be excessive, Dr. Gro Egholm reported at the annual meeting of the American College of Cardiology.
She presented a large retrospective observational study of outcomes in patients undergoing various types of noncardiac surgery in western Denmark during 2005-2012. Among 4,303 patients who had noncardiac surgery within 12 months after receiving a drug-eluting stent (DES), only those whose operations took place during the first month post stenting had increased risks of acute MI and cardiac death within 30 days post surgery.
Risks of major adverse cardiac events among the DES recipients who had noncardiac surgery within that first month post–percutaneous coronary intervention were increased roughly 7.5-fold compared with controls, but for surgery performed after that the risks of MI and cardiac death dropped off abruptly and were no different from rates in 20,232 controls without ischemic heart disease or stents who were matched for age, gender, surgical procedure, and Charlson Comorbidity Index, according to Dr. Egholm of Aarhus (Denmark) University.
Moreover, even in DES recipients undergoing noncardiac surgery during the first month post stenting, all-cause mortality was no greater than in controls.
“Surgery could be performed much earlier than recommended,” she concluded.
Her study was carried out by linking data from comprehensive regional and national Danish health care registries. Most patients with DES remained on dual antiplatelet therapy periprocedurally. The exceptions were neurosurgical operations and others where it’s standard that dual antiplatelet therapy must be stopped.
“If you can continue only one antiplatelet agent, aspirin would be the most appealing,” she said.
Of the DES participants, 56% received their device as treatment for an acute coronary syndrome. The average time from stent placement to noncardiac surgery in this large series was 147 days.
Session co-chair Dr. Sunil V. Rao of Duke University in Durham, N.C., called this work “a very important study that’s relevant to daily practice.” However, he found the 23% incidence of noncardiac surgery within 12 months following DES implantation reported in Dr. Egholm’s study to be “shockingly high.” She agreed, noting that rates in some non-Danish registries she’s looked at are more in the 8%-15% range. But Denmark’s health care registries are known for rigorous accuracy and completeness.
Dr. Egholm reported having no financial conflicts regarding her study.
AT ACC 16
Key clinical point: The risk of noncardiac surgery is elevated only when the operation occurs during the first month after stenting.
Major finding: Danish drug-eluting stent recipients who underwent noncardiac surgery within 1 month after stent placement were at 7.5-fold increased risks of acute MI and cardiac death, but surgery performed 2-12 months post stenting carried no increased risks.
Data source: This retrospective observational study based upon large Danish patient registries compared outcomes of noncardiac surgery performed within 12 months after drug-eluting stent placement in 4,303 patients with 20,232 matched controls without ischemic heart disease who underwent the same operations.
Disclosures: The study was supported by Danish research funds. The presenter reported having no financial conflicts of interest.

FDG-PET guides need for eBEACOPP in advanced Hodgkin’s
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).

In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).
In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).
In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Using FDG-PET imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to the eBEACOPP regimen.
Major finding: Progression-free survival at 2 years for those with early interim positive PET scans was 64%; the historical progression-free survival for this group is 15%-30%.
Data source: Evaluations of 331 patients in the Southwest Oncology Group S0816 study.
Disclosures: The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.

CUDC-907 enters phase II for relapsed or refractory lymphoma and multiple myeloma
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
FROM THE LANCET ONCOLOGY
Key clinical point: The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule.
Major finding: Two complete responses and three partial responses were seen in the subgroup of nine patients with diffuse large B-cell lymphoma; three occurred in the five patients with transformed follicular disease.
Data source: A dose-escalation study involving 44 patients at four cancer centers.
Disclosures: The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
Modifying our behavior
“Just say no to overprescribing!” It has such a straightforward Nancy Reagan-ish sound to it. But when it comes to drugs, whether it is crack cocaine or a prescription antibiotic, simple slogans don’t alter behavior.
While most physicians aren’t drug addicts, we do share something in common with other substance abusers. We are all human, and we are all influenced by the social contexts that we inhabit. The global health problems rippling out from the overuse of antibiotics are significant, unmistakable, and well documented. Certainly, we physicians must share some of the blame with the food industry for this unfortunate situation. There is some glimmer of hope that pressure from consumers has begun to convince a few food producers to be more judicious in their use of antibiotics.

However, there seems to be little or no pressure from patients on physicians to curtail our antibiotic prescribing habits. If physicians feel any pressure from patients, it is in the form of stated or more often unstated requests for antibiotics to treat conditions for which we know they are inappropriate. There is some question as to how often this perception of patient pressure actually occurs. It may be that the pressure physicians are feeling could be better described as fear – fear that the patient will die because of an undiscovered and untreated infection. Regardless of what motivates physicians to overprescribe antibiotics, the fact is that this kind of clinical misbehavior is difficult to change.
I recently read an article in which three medical school professors describe several behavior modification strategies that they have found to be effective in discouraging overprescribing (“How to Stop Overprescribing Antibiotics,” by Craig R. Fox, Jeffrey A. Linder, and Jason N. Doctor, New York Times, March 25, 2016). In one study, the researchers found that physicians who posted a pledge to follow antibiotic guidelines reduced inappropriate prescribing by 20%. In another study the investigators found that when physicians were presented with a list of medications in a format that presented the “more aggressive” drugs in a group, as opposed to singly in a vertical column, the physicians were 12% less likely to prescribe those medications.
Better results were achieved when physicians were provided with monthly reports of their prescribing habits in comparison with those of their peers. The physicians whose prescribing patterns followed accepted guidelines most closely were complimented as being “top performers.” Those physicians who did less well were told, “You are not a top performer.” This strategy nearly eliminated inappropriate prescribing. Similar improvement occurred when physicians who clicked their mouse on an antibiotic in a clinical scenario where it was not appropriate were given a screen prompt asking them to type in a short “antibiotic justification note.”
What all of these strategies have in common is that none of them uses financial gain as a motivator. Previous studies have shown that if financial rewards work, it is only for short periods of time. Instead, these strategies leverage our inherent competitive nature and take advantage of the fact that most of us want to do the right thing. We just need a little nudge every now and then. It is also encouraging to learn that none of these strategies incorporates a punishment.
I suspect that further studies will show that a screen prompt in the medical record requiring the overprescribing physician to justify his or her prescription will be the most effective in the long run. In my experience, physicians will do anything to shorten the amount of time they spend at their office computers.
At least two of these strategies hold the promise of being very powerful behavior modifiers. Those wielding these powerful tools must exercise that power carefully and be sure that evidence supporting their target behaviors is solid and continually updated. More importantly, those of us whose behavior is being modified should have a voice in the choice of which behaviors are to be modified.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
“Just say no to overprescribing!” It has such a straightforward Nancy Reagan-ish sound to it. But when it comes to drugs, whether it is crack cocaine or a prescription antibiotic, simple slogans don’t alter behavior.
While most physicians aren’t drug addicts, we do share something in common with other substance abusers. We are all human, and we are all influenced by the social contexts that we inhabit. The global health problems rippling out from the overuse of antibiotics are significant, unmistakable, and well documented. Certainly, we physicians must share some of the blame with the food industry for this unfortunate situation. There is some glimmer of hope that pressure from consumers has begun to convince a few food producers to be more judicious in their use of antibiotics.
However, there seems to be little or no pressure from patients on physicians to curtail our antibiotic prescribing habits. If physicians feel any pressure from patients, it is in the form of stated or more often unstated requests for antibiotics to treat conditions for which we know they are inappropriate. There is some question as to how often this perception of patient pressure actually occurs. It may be that the pressure physicians are feeling could be better described as fear – fear that the patient will die because of an undiscovered and untreated infection. Regardless of what motivates physicians to overprescribe antibiotics, the fact is that this kind of clinical misbehavior is difficult to change.
I recently read an article in which three medical school professors describe several behavior modification strategies that they have found to be effective in discouraging overprescribing (“How to Stop Overprescribing Antibiotics,” by Craig R. Fox, Jeffrey A. Linder, and Jason N. Doctor, New York Times, March 25, 2016). In one study, the researchers found that physicians who posted a pledge to follow antibiotic guidelines reduced inappropriate prescribing by 20%. In another study the investigators found that when physicians were presented with a list of medications in a format that presented the “more aggressive” drugs in a group, as opposed to singly in a vertical column, the physicians were 12% less likely to prescribe those medications.
Better results were achieved when physicians were provided with monthly reports of their prescribing habits in comparison with those of their peers. The physicians whose prescribing patterns followed accepted guidelines most closely were complimented as being “top performers.” Those physicians who did less well were told, “You are not a top performer.” This strategy nearly eliminated inappropriate prescribing. Similar improvement occurred when physicians who clicked their mouse on an antibiotic in a clinical scenario where it was not appropriate were given a screen prompt asking them to type in a short “antibiotic justification note.”
What all of these strategies have in common is that none of them uses financial gain as a motivator. Previous studies have shown that if financial rewards work, it is only for short periods of time. Instead, these strategies leverage our inherent competitive nature and take advantage of the fact that most of us want to do the right thing. We just need a little nudge every now and then. It is also encouraging to learn that none of these strategies incorporates a punishment.
I suspect that further studies will show that a screen prompt in the medical record requiring the overprescribing physician to justify his or her prescription will be the most effective in the long run. In my experience, physicians will do anything to shorten the amount of time they spend at their office computers.
At least two of these strategies hold the promise of being very powerful behavior modifiers. Those wielding these powerful tools must exercise that power carefully and be sure that evidence supporting their target behaviors is solid and continually updated. More importantly, those of us whose behavior is being modified should have a voice in the choice of which behaviors are to be modified.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
“Just say no to overprescribing!” It has such a straightforward Nancy Reagan-ish sound to it. But when it comes to drugs, whether it is crack cocaine or a prescription antibiotic, simple slogans don’t alter behavior.
While most physicians aren’t drug addicts, we do share something in common with other substance abusers. We are all human, and we are all influenced by the social contexts that we inhabit. The global health problems rippling out from the overuse of antibiotics are significant, unmistakable, and well documented. Certainly, we physicians must share some of the blame with the food industry for this unfortunate situation. There is some glimmer of hope that pressure from consumers has begun to convince a few food producers to be more judicious in their use of antibiotics.
However, there seems to be little or no pressure from patients on physicians to curtail our antibiotic prescribing habits. If physicians feel any pressure from patients, it is in the form of stated or more often unstated requests for antibiotics to treat conditions for which we know they are inappropriate. There is some question as to how often this perception of patient pressure actually occurs. It may be that the pressure physicians are feeling could be better described as fear – fear that the patient will die because of an undiscovered and untreated infection. Regardless of what motivates physicians to overprescribe antibiotics, the fact is that this kind of clinical misbehavior is difficult to change.
I recently read an article in which three medical school professors describe several behavior modification strategies that they have found to be effective in discouraging overprescribing (“How to Stop Overprescribing Antibiotics,” by Craig R. Fox, Jeffrey A. Linder, and Jason N. Doctor, New York Times, March 25, 2016). In one study, the researchers found that physicians who posted a pledge to follow antibiotic guidelines reduced inappropriate prescribing by 20%. In another study the investigators found that when physicians were presented with a list of medications in a format that presented the “more aggressive” drugs in a group, as opposed to singly in a vertical column, the physicians were 12% less likely to prescribe those medications.
Better results were achieved when physicians were provided with monthly reports of their prescribing habits in comparison with those of their peers. The physicians whose prescribing patterns followed accepted guidelines most closely were complimented as being “top performers.” Those physicians who did less well were told, “You are not a top performer.” This strategy nearly eliminated inappropriate prescribing. Similar improvement occurred when physicians who clicked their mouse on an antibiotic in a clinical scenario where it was not appropriate were given a screen prompt asking them to type in a short “antibiotic justification note.”
What all of these strategies have in common is that none of them uses financial gain as a motivator. Previous studies have shown that if financial rewards work, it is only for short periods of time. Instead, these strategies leverage our inherent competitive nature and take advantage of the fact that most of us want to do the right thing. We just need a little nudge every now and then. It is also encouraging to learn that none of these strategies incorporates a punishment.
I suspect that further studies will show that a screen prompt in the medical record requiring the overprescribing physician to justify his or her prescription will be the most effective in the long run. In my experience, physicians will do anything to shorten the amount of time they spend at their office computers.
At least two of these strategies hold the promise of being very powerful behavior modifiers. Those wielding these powerful tools must exercise that power carefully and be sure that evidence supporting their target behaviors is solid and continually updated. More importantly, those of us whose behavior is being modified should have a voice in the choice of which behaviors are to be modified.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”

Failure of Promising Treatments for Controlling Cholesterol Leads to More Studies
(Reuters) - New ways of controlling cholesterol, including possibly directly injecting "good" HDL cholesterol into patients, need to be studied following the failure of promising treatments from Eli Lilly, Pfizer Inc and Roche Holding AG, according to top heart researchers.
Lilly in October halted a 12,000-patient study of its experimental drug evacetrapib, an oral medication that in smaller earlier studies slashed "bad" LDL cholesterol and doubled levels of HDL.
But improved cholesterol levels did not prevent heart attacks and strokes, diminishing hopes for the approach to treating heart disease - by raising HDL through blockage of a protein called CETP.
Roche in 2012 scrapped its own CETP inhibitor after it also failed to help patients. Pfizer's similar drug was discontinued in 2006 after being linked to deaths in trials.
Although Merck & Co continues to develop its own CETP inhibitor in a 30,000-patient study expected to be completed next year, researchers on Sunday said the failures of the Lilly, Roche and Pfizer drugs bode poorly for it.
"Merck's drug is the fourth shot on goal for CETP inhibitors, but with disappointment or lack of success for the other agents you have to be increasingly pessimistic" about the class of drugs, said Dr. Stephen Nicholls, deputy director of the South Australian Health and Medical Research Institute in Adelaide, Australia. He was a lead investigator for the failed trial of Lilly's drug.
Nicholls and Dr. Steve Nissen, the head of cardiology for the Cleveland Clinic, who co-lead the evacetrapib study, on Sunday reviewed the baffling evacetrapib data in a presentation at the annual scientific sessions of the American College of Cardiology in Chicago.
"This drug lowered LDL by 37 percent and raised HDL by 130 percent and had absolutely no effect" on preventing deaths and heart attacks, Nissen said in an interview.
Although other ways of raising HDL cholesterol might eventually prove protective, Nissen said all attempts so far have been fruitless.
Nicholls said he remains hopeful of future HDL therapies and is testing whether artificial HDL can be made in the laboratory and injected directly into high-risk heart patients. "There is enthusiasm it may be able to shrink plaque" in heart arteries, he said.
He said he is studying variations of that approach with French drugmaker Cerenis Therapeutics and the Medicines Company. Nicholls said another possible approach would be to instruct the liver to make more HDL.
(Reuters) - New ways of controlling cholesterol, including possibly directly injecting "good" HDL cholesterol into patients, need to be studied following the failure of promising treatments from Eli Lilly, Pfizer Inc and Roche Holding AG, according to top heart researchers.
Lilly in October halted a 12,000-patient study of its experimental drug evacetrapib, an oral medication that in smaller earlier studies slashed "bad" LDL cholesterol and doubled levels of HDL.
But improved cholesterol levels did not prevent heart attacks and strokes, diminishing hopes for the approach to treating heart disease - by raising HDL through blockage of a protein called CETP.
Roche in 2012 scrapped its own CETP inhibitor after it also failed to help patients. Pfizer's similar drug was discontinued in 2006 after being linked to deaths in trials.
Although Merck & Co continues to develop its own CETP inhibitor in a 30,000-patient study expected to be completed next year, researchers on Sunday said the failures of the Lilly, Roche and Pfizer drugs bode poorly for it.
"Merck's drug is the fourth shot on goal for CETP inhibitors, but with disappointment or lack of success for the other agents you have to be increasingly pessimistic" about the class of drugs, said Dr. Stephen Nicholls, deputy director of the South Australian Health and Medical Research Institute in Adelaide, Australia. He was a lead investigator for the failed trial of Lilly's drug.
Nicholls and Dr. Steve Nissen, the head of cardiology for the Cleveland Clinic, who co-lead the evacetrapib study, on Sunday reviewed the baffling evacetrapib data in a presentation at the annual scientific sessions of the American College of Cardiology in Chicago.
"This drug lowered LDL by 37 percent and raised HDL by 130 percent and had absolutely no effect" on preventing deaths and heart attacks, Nissen said in an interview.
Although other ways of raising HDL cholesterol might eventually prove protective, Nissen said all attempts so far have been fruitless.
Nicholls said he remains hopeful of future HDL therapies and is testing whether artificial HDL can be made in the laboratory and injected directly into high-risk heart patients. "There is enthusiasm it may be able to shrink plaque" in heart arteries, he said.
He said he is studying variations of that approach with French drugmaker Cerenis Therapeutics and the Medicines Company. Nicholls said another possible approach would be to instruct the liver to make more HDL.
(Reuters) - New ways of controlling cholesterol, including possibly directly injecting "good" HDL cholesterol into patients, need to be studied following the failure of promising treatments from Eli Lilly, Pfizer Inc and Roche Holding AG, according to top heart researchers.
Lilly in October halted a 12,000-patient study of its experimental drug evacetrapib, an oral medication that in smaller earlier studies slashed "bad" LDL cholesterol and doubled levels of HDL.
But improved cholesterol levels did not prevent heart attacks and strokes, diminishing hopes for the approach to treating heart disease - by raising HDL through blockage of a protein called CETP.
Roche in 2012 scrapped its own CETP inhibitor after it also failed to help patients. Pfizer's similar drug was discontinued in 2006 after being linked to deaths in trials.
Although Merck & Co continues to develop its own CETP inhibitor in a 30,000-patient study expected to be completed next year, researchers on Sunday said the failures of the Lilly, Roche and Pfizer drugs bode poorly for it.
"Merck's drug is the fourth shot on goal for CETP inhibitors, but with disappointment or lack of success for the other agents you have to be increasingly pessimistic" about the class of drugs, said Dr. Stephen Nicholls, deputy director of the South Australian Health and Medical Research Institute in Adelaide, Australia. He was a lead investigator for the failed trial of Lilly's drug.
Nicholls and Dr. Steve Nissen, the head of cardiology for the Cleveland Clinic, who co-lead the evacetrapib study, on Sunday reviewed the baffling evacetrapib data in a presentation at the annual scientific sessions of the American College of Cardiology in Chicago.
"This drug lowered LDL by 37 percent and raised HDL by 130 percent and had absolutely no effect" on preventing deaths and heart attacks, Nissen said in an interview.
Although other ways of raising HDL cholesterol might eventually prove protective, Nissen said all attempts so far have been fruitless.
Nicholls said he remains hopeful of future HDL therapies and is testing whether artificial HDL can be made in the laboratory and injected directly into high-risk heart patients. "There is enthusiasm it may be able to shrink plaque" in heart arteries, he said.
He said he is studying variations of that approach with French drugmaker Cerenis Therapeutics and the Medicines Company. Nicholls said another possible approach would be to instruct the liver to make more HDL.
HM16 Q&A: How Do You try to Make a Positive Impact on Public Health in Your Daily Work?
U.S. Surgeon General Vivek Murthy, MD, MBA, formerly a hospitalist in Boston, said in a keynote speech that hospitalists have more power than they think to improve public health. The Hospitalist asked HM16 attendees: As a hospitalist, how empowered do you feel in having a positive impact on public health, and how do you try to make such an impact in your daily work?

L. Scott Sussman, MD, associate director of the hospitalist service, Yale-New Haven Hospital, Conn.
“We see patients in a time of crisis, so they’re coming in, they’re scared, they’re sick, and it’s a really teachable moment. Because once we start treating them, they start feeling better … [and] what happens is we start to gain their confidence. We use that as a point where we can say, ‘Let’s talk about your other health issues.’”

Nazima Allaudeen, MD, hospitalist, director of quality improvement, Veterans Affairs Palo Alto Healthcare System, Calif.
“I don’t feel like I do [have an impact] very much, but I think his talk kind of made me feel like we probably can and should do a lot more. … You know how sometimes you’ll get an email that has a standard thing that you can send to your congressperson about something like that? I feel like we were all like, ‘Yes, we should,’ and it would have been a great opportunity. … I feel like we kind of need it spelled out for us, you know? But it’s a good reminder.”
Gilbert Wergowske, MD, TeamHealth hospitalist, Vallejo, Calif.
“We try to arrange the best follow-up you can. That’s a difficult problem. The area that I work in is not an area that really has a lot of options … as far as giving care is concerned. So it’s really important to try to get in touch with the primary care provider directly. If that doesn’t happen, most of our patients will end up using the ED as their primary care provider. It’s important mainly for two reasons: Disjointed care like that is suboptimal; the patients do not get the best benefit from the care. And also it’s very wasteful because tests and procedures tend to be repeated over and over again.”
Cory Ritter, MD, hospitalist, soon to be working at the Veterans Affairs Medical Center, Houston
“After Dr. Murthy’s talk, I feel much more empowered to call up my local government representatives and express what I feel should be done for public health in our area. Before today, I didn’t think there was much we could do except for supporting the Society of Hospital Medicine because I know they’ve been very engaged in Obamacare and CMS. But otherwise, before today, it wasn’t much on my radar. … On a one-on-one level, you have some impact, but I’ve never really thought much about a larger public health–scale type of impact.”
U.S. Surgeon General Vivek Murthy, MD, MBA, formerly a hospitalist in Boston, said in a keynote speech that hospitalists have more power than they think to improve public health. The Hospitalist asked HM16 attendees: As a hospitalist, how empowered do you feel in having a positive impact on public health, and how do you try to make such an impact in your daily work?
L. Scott Sussman, MD, associate director of the hospitalist service, Yale-New Haven Hospital, Conn.
“We see patients in a time of crisis, so they’re coming in, they’re scared, they’re sick, and it’s a really teachable moment. Because once we start treating them, they start feeling better … [and] what happens is we start to gain their confidence. We use that as a point where we can say, ‘Let’s talk about your other health issues.’”
Nazima Allaudeen, MD, hospitalist, director of quality improvement, Veterans Affairs Palo Alto Healthcare System, Calif.
“I don’t feel like I do [have an impact] very much, but I think his talk kind of made me feel like we probably can and should do a lot more. … You know how sometimes you’ll get an email that has a standard thing that you can send to your congressperson about something like that? I feel like we were all like, ‘Yes, we should,’ and it would have been a great opportunity. … I feel like we kind of need it spelled out for us, you know? But it’s a good reminder.”
Gilbert Wergowske, MD, TeamHealth hospitalist, Vallejo, Calif.
“We try to arrange the best follow-up you can. That’s a difficult problem. The area that I work in is not an area that really has a lot of options … as far as giving care is concerned. So it’s really important to try to get in touch with the primary care provider directly. If that doesn’t happen, most of our patients will end up using the ED as their primary care provider. It’s important mainly for two reasons: Disjointed care like that is suboptimal; the patients do not get the best benefit from the care. And also it’s very wasteful because tests and procedures tend to be repeated over and over again.”
Cory Ritter, MD, hospitalist, soon to be working at the Veterans Affairs Medical Center, Houston
“After Dr. Murthy’s talk, I feel much more empowered to call up my local government representatives and express what I feel should be done for public health in our area. Before today, I didn’t think there was much we could do except for supporting the Society of Hospital Medicine because I know they’ve been very engaged in Obamacare and CMS. But otherwise, before today, it wasn’t much on my radar. … On a one-on-one level, you have some impact, but I’ve never really thought much about a larger public health–scale type of impact.”
U.S. Surgeon General Vivek Murthy, MD, MBA, formerly a hospitalist in Boston, said in a keynote speech that hospitalists have more power than they think to improve public health. The Hospitalist asked HM16 attendees: As a hospitalist, how empowered do you feel in having a positive impact on public health, and how do you try to make such an impact in your daily work?
L. Scott Sussman, MD, associate director of the hospitalist service, Yale-New Haven Hospital, Conn.
“We see patients in a time of crisis, so they’re coming in, they’re scared, they’re sick, and it’s a really teachable moment. Because once we start treating them, they start feeling better … [and] what happens is we start to gain their confidence. We use that as a point where we can say, ‘Let’s talk about your other health issues.’”
Nazima Allaudeen, MD, hospitalist, director of quality improvement, Veterans Affairs Palo Alto Healthcare System, Calif.
“I don’t feel like I do [have an impact] very much, but I think his talk kind of made me feel like we probably can and should do a lot more. … You know how sometimes you’ll get an email that has a standard thing that you can send to your congressperson about something like that? I feel like we were all like, ‘Yes, we should,’ and it would have been a great opportunity. … I feel like we kind of need it spelled out for us, you know? But it’s a good reminder.”
Gilbert Wergowske, MD, TeamHealth hospitalist, Vallejo, Calif.
“We try to arrange the best follow-up you can. That’s a difficult problem. The area that I work in is not an area that really has a lot of options … as far as giving care is concerned. So it’s really important to try to get in touch with the primary care provider directly. If that doesn’t happen, most of our patients will end up using the ED as their primary care provider. It’s important mainly for two reasons: Disjointed care like that is suboptimal; the patients do not get the best benefit from the care. And also it’s very wasteful because tests and procedures tend to be repeated over and over again.”
Cory Ritter, MD, hospitalist, soon to be working at the Veterans Affairs Medical Center, Houston
“After Dr. Murthy’s talk, I feel much more empowered to call up my local government representatives and express what I feel should be done for public health in our area. Before today, I didn’t think there was much we could do except for supporting the Society of Hospital Medicine because I know they’ve been very engaged in Obamacare and CMS. But otherwise, before today, it wasn’t much on my radar. … On a one-on-one level, you have some impact, but I’ve never really thought much about a larger public health–scale type of impact.”
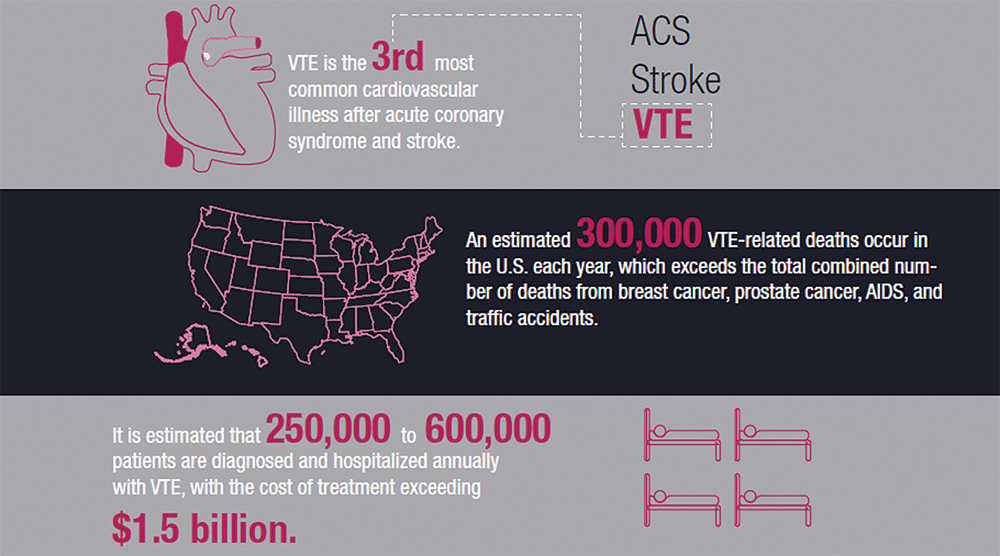
Improve Your Treatment of VTE
It is estimated that 250,000 to 600,000 patients are diagnosed and hospitalized annually with venous thromboembolism (VTE), the third most common cardiovascular illness after acute coronary syndrome and stroke. An estimated 300,000 VTE-related deaths occur in the U.S. each year, which exceeds the total number of deaths from breast cancer, prostate cancer, AIDS, and traffic accidents combined.
Communication among care providers is an essential part of medical care; it influences patients’ quality of life and effective disease treatment. Hospitalists educate both patients and providers regarding appropriate steps to take to improve care transitions and reduce any associated risks.
To ensure hospitalists have the latest information about diagnosis, treatment, and transition of inpatients with VTE, SHM is developing:
- An online tool kit, including a literature review; implementation guide; and other references, materials, and tools (e.g., discharge instructions and checklists)
- A webinar series with free CME
To be notified when SHM’s VTE resources become available, visit www.hospitalmedicine.org/VTEtreatment.

It is estimated that 250,000 to 600,000 patients are diagnosed and hospitalized annually with venous thromboembolism (VTE), the third most common cardiovascular illness after acute coronary syndrome and stroke. An estimated 300,000 VTE-related deaths occur in the U.S. each year, which exceeds the total number of deaths from breast cancer, prostate cancer, AIDS, and traffic accidents combined.
Communication among care providers is an essential part of medical care; it influences patients’ quality of life and effective disease treatment. Hospitalists educate both patients and providers regarding appropriate steps to take to improve care transitions and reduce any associated risks.
To ensure hospitalists have the latest information about diagnosis, treatment, and transition of inpatients with VTE, SHM is developing:
- An online tool kit, including a literature review; implementation guide; and other references, materials, and tools (e.g., discharge instructions and checklists)
- A webinar series with free CME
To be notified when SHM’s VTE resources become available, visit www.hospitalmedicine.org/VTEtreatment.
It is estimated that 250,000 to 600,000 patients are diagnosed and hospitalized annually with venous thromboembolism (VTE), the third most common cardiovascular illness after acute coronary syndrome and stroke. An estimated 300,000 VTE-related deaths occur in the U.S. each year, which exceeds the total number of deaths from breast cancer, prostate cancer, AIDS, and traffic accidents combined.
Communication among care providers is an essential part of medical care; it influences patients’ quality of life and effective disease treatment. Hospitalists educate both patients and providers regarding appropriate steps to take to improve care transitions and reduce any associated risks.
To ensure hospitalists have the latest information about diagnosis, treatment, and transition of inpatients with VTE, SHM is developing:
- An online tool kit, including a literature review; implementation guide; and other references, materials, and tools (e.g., discharge instructions and checklists)
- A webinar series with free CME
To be notified when SHM’s VTE resources become available, visit www.hospitalmedicine.org/VTEtreatment.
FDA grants drug accelerated approval for CLL

Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Protein distribution impacts T cells’ fate
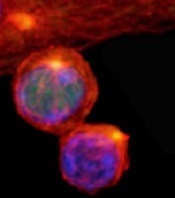
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said. ![]()
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said. ![]()
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said. ![]()
Team creates public repository of xenografts

Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()