User login
Stella Fitzgibbons, MD, FHM, Relishes the Variety, Interactions of Hospitalist Practice
Stella Fitzgibbons, MD, FHM, was an engineer for several years after college. But there wasn’t enough working with people for her taste. So she moved into internal medicine. But then there was, how to put this, something lacking in office work.

“I realized how bored I was with office practice and how much more interesting were the problems at the hospital than outpatient ones,” Dr. Fitzgibbons says.
So she went to work in hospitals. She hasn’t left.
Dr. Fitzgibbons is a hospitalist and ED practitioner with Mint Physician Staffing, primarily in the Apollo Hospital System in The Woodlands, Texas. And the best part of the job for Dr. Fitzgibbons, one of eight new members of Team Hospitalist, the volunteer editorial advisory board of The Hospitalist newsmagazine, is easy to pick.
“Seeing sick people get better,” she says.
Question: Switching careers from engineering to medicine is a big step. What motivated that?
Answer: I wanted to see my efforts helping people I could actually see, and I thought—and still do—that medicine uses my talents better and is far more interesting.
Q: You say office practice became a bit boring. How so? What appealed about the inpatient setting?
A: An internist in the office only sees a tiny fraction of the interesting problems that our field covers. Rheumatoid arthritis is diagnosed by a rheumatologist, who then makes all the decisions. Abdominal pain gets sent to the hospital, where all the diagnostic tests are done.
Fortunately, my multispecialty group arranged for about a quarter of its internists [the youngest quarter in most cases] to manage hospital patients; I figured out pretty quickly that it was only there that I got to see pulmonary hypertension, congestive heart failure, and acute abdomens. Even night call was better at the hospital since office doctors only answered phone calls and never had a chance to do any real evaluation and treatment no matter how sick the caller was.
And a problem at the office was something that made me run behind that odious and impractical appointment schedule; at the hospital I was seeing real illnesses, not people who wanted a prescription med for their sore throat so they wouldn’t be bothered with it on their vacation.
Q: What is your biggest professional challenge?
A: EHRs.
Q: What is your biggest professional reward?
A: When a patient says, “Thanks for taking care of me, doctor.”
Q: What does teaching mean to you, and how has it been gratifying in your career?
A: Teaching means paying it forward, in gratitude to those who taught me, with the reward of seeing light bulbs go off behind the eyes of students and younger doctors who are eager to learn.
Q: When you aren’t working, what is important to you?
A: Family and music and church.
Q: Faith is obviously important to you. How does that help your work as a care provider?
A: I don’t think anybody goes around being religious all the time. But it sometimes makes all the difference knowing that a higher power is looking out for me and the patients.
Q: You’ve described mentoring as fun for you. What exactly do you mean by fun?
A: Mentoring is what we do. Patients, nurses—anybody we work with—need explanations and clarifications. About the third day of med school, docs in training realize that anybody who can help us understand and retain the huge stream of information directed at us is performing a necessary service. Throughout the training period, residents teach students, fellows teach residents, and attending faculty teach everybody. Doctors in training are bright people who want to learn both the facts and how to deal with patients’ side of things, and feeding their desires is very enjoyable.
Q: You’d like to see more physicians than MBAs in decision-making positions. Why? What real changes do you think that would effectuate?
A: Physicians and nurses were administrators for decades before insurance company penny-pinching and government regulations led hospitals to hire “bean counters” to replace them. It is a tremendous change for the worse, to have people making decisions for patients whose primary consideration is the bottom line.
Q: What’s next professionally?
A: Small-volume ERs, where I don’t have to do discharge planning while being harassed by insurance company reps.
Q: Where do you see yourself in 10 years?
A: Retired.
Q: If you weren’t a doctor, what would you be doing right now?
A: Law enforcement.
Q: Devices like iPhones and tablets can take away from patient face time. But they can also be valuable. How do you balance that? How do you encourage younger docs to do so, particularly when they’re much more used to having smartphones glued to their hands?
A: I use my iPhone when I’m with patients … but only when they can see the reason I need it to help them, such as looking up the side effects of a medication. Electronic health records can work on an iPad, but I hesitate to use them unless the patient knows just what I am doing, such as looking up results of a lab test that concerns them. Taking a computer on wheels into a patient’s room means that I spend part of the visit looking at a screen instead of at the patient, and I prefer to avoid it if at all possible.
Q: What’s the best book you’ve read recently? Why?
A: The House of Silk by Anthony Horowitz. Great continuation of the Holmes stories, with a seamless link to [Sir Arthur] Conan Doyle’s style.
Q: How many Apple products (phones, iPods, tablets, iTunes, etc.) do you interface with in a given week?
A: Three.
Q: What’s your favorite social network? Do you use it all for work or professional development?
A: Facebook. Heck no, it’s just fun.
Q: What’s next in your Netflix queue?
A: Last two episodes of Game of Thrones season 5.
Richard Quinn is a freelance writer in New Jersey.
Stella Fitzgibbons, MD, FHM, was an engineer for several years after college. But there wasn’t enough working with people for her taste. So she moved into internal medicine. But then there was, how to put this, something lacking in office work.
“I realized how bored I was with office practice and how much more interesting were the problems at the hospital than outpatient ones,” Dr. Fitzgibbons says.
So she went to work in hospitals. She hasn’t left.
Dr. Fitzgibbons is a hospitalist and ED practitioner with Mint Physician Staffing, primarily in the Apollo Hospital System in The Woodlands, Texas. And the best part of the job for Dr. Fitzgibbons, one of eight new members of Team Hospitalist, the volunteer editorial advisory board of The Hospitalist newsmagazine, is easy to pick.
“Seeing sick people get better,” she says.
Question: Switching careers from engineering to medicine is a big step. What motivated that?
Answer: I wanted to see my efforts helping people I could actually see, and I thought—and still do—that medicine uses my talents better and is far more interesting.
Q: You say office practice became a bit boring. How so? What appealed about the inpatient setting?
A: An internist in the office only sees a tiny fraction of the interesting problems that our field covers. Rheumatoid arthritis is diagnosed by a rheumatologist, who then makes all the decisions. Abdominal pain gets sent to the hospital, where all the diagnostic tests are done.
Fortunately, my multispecialty group arranged for about a quarter of its internists [the youngest quarter in most cases] to manage hospital patients; I figured out pretty quickly that it was only there that I got to see pulmonary hypertension, congestive heart failure, and acute abdomens. Even night call was better at the hospital since office doctors only answered phone calls and never had a chance to do any real evaluation and treatment no matter how sick the caller was.
And a problem at the office was something that made me run behind that odious and impractical appointment schedule; at the hospital I was seeing real illnesses, not people who wanted a prescription med for their sore throat so they wouldn’t be bothered with it on their vacation.
Q: What is your biggest professional challenge?
A: EHRs.
Q: What is your biggest professional reward?
A: When a patient says, “Thanks for taking care of me, doctor.”
Q: What does teaching mean to you, and how has it been gratifying in your career?
A: Teaching means paying it forward, in gratitude to those who taught me, with the reward of seeing light bulbs go off behind the eyes of students and younger doctors who are eager to learn.
Q: When you aren’t working, what is important to you?
A: Family and music and church.
Q: Faith is obviously important to you. How does that help your work as a care provider?
A: I don’t think anybody goes around being religious all the time. But it sometimes makes all the difference knowing that a higher power is looking out for me and the patients.
Q: You’ve described mentoring as fun for you. What exactly do you mean by fun?
A: Mentoring is what we do. Patients, nurses—anybody we work with—need explanations and clarifications. About the third day of med school, docs in training realize that anybody who can help us understand and retain the huge stream of information directed at us is performing a necessary service. Throughout the training period, residents teach students, fellows teach residents, and attending faculty teach everybody. Doctors in training are bright people who want to learn both the facts and how to deal with patients’ side of things, and feeding their desires is very enjoyable.
Q: You’d like to see more physicians than MBAs in decision-making positions. Why? What real changes do you think that would effectuate?
A: Physicians and nurses were administrators for decades before insurance company penny-pinching and government regulations led hospitals to hire “bean counters” to replace them. It is a tremendous change for the worse, to have people making decisions for patients whose primary consideration is the bottom line.
Q: What’s next professionally?
A: Small-volume ERs, where I don’t have to do discharge planning while being harassed by insurance company reps.
Q: Where do you see yourself in 10 years?
A: Retired.
Q: If you weren’t a doctor, what would you be doing right now?
A: Law enforcement.
Q: Devices like iPhones and tablets can take away from patient face time. But they can also be valuable. How do you balance that? How do you encourage younger docs to do so, particularly when they’re much more used to having smartphones glued to their hands?
A: I use my iPhone when I’m with patients … but only when they can see the reason I need it to help them, such as looking up the side effects of a medication. Electronic health records can work on an iPad, but I hesitate to use them unless the patient knows just what I am doing, such as looking up results of a lab test that concerns them. Taking a computer on wheels into a patient’s room means that I spend part of the visit looking at a screen instead of at the patient, and I prefer to avoid it if at all possible.
Q: What’s the best book you’ve read recently? Why?
A: The House of Silk by Anthony Horowitz. Great continuation of the Holmes stories, with a seamless link to [Sir Arthur] Conan Doyle’s style.
Q: How many Apple products (phones, iPods, tablets, iTunes, etc.) do you interface with in a given week?
A: Three.
Q: What’s your favorite social network? Do you use it all for work or professional development?
A: Facebook. Heck no, it’s just fun.
Q: What’s next in your Netflix queue?
A: Last two episodes of Game of Thrones season 5.
Richard Quinn is a freelance writer in New Jersey.
Stella Fitzgibbons, MD, FHM, was an engineer for several years after college. But there wasn’t enough working with people for her taste. So she moved into internal medicine. But then there was, how to put this, something lacking in office work.
“I realized how bored I was with office practice and how much more interesting were the problems at the hospital than outpatient ones,” Dr. Fitzgibbons says.
So she went to work in hospitals. She hasn’t left.
Dr. Fitzgibbons is a hospitalist and ED practitioner with Mint Physician Staffing, primarily in the Apollo Hospital System in The Woodlands, Texas. And the best part of the job for Dr. Fitzgibbons, one of eight new members of Team Hospitalist, the volunteer editorial advisory board of The Hospitalist newsmagazine, is easy to pick.
“Seeing sick people get better,” she says.
Question: Switching careers from engineering to medicine is a big step. What motivated that?
Answer: I wanted to see my efforts helping people I could actually see, and I thought—and still do—that medicine uses my talents better and is far more interesting.
Q: You say office practice became a bit boring. How so? What appealed about the inpatient setting?
A: An internist in the office only sees a tiny fraction of the interesting problems that our field covers. Rheumatoid arthritis is diagnosed by a rheumatologist, who then makes all the decisions. Abdominal pain gets sent to the hospital, where all the diagnostic tests are done.
Fortunately, my multispecialty group arranged for about a quarter of its internists [the youngest quarter in most cases] to manage hospital patients; I figured out pretty quickly that it was only there that I got to see pulmonary hypertension, congestive heart failure, and acute abdomens. Even night call was better at the hospital since office doctors only answered phone calls and never had a chance to do any real evaluation and treatment no matter how sick the caller was.
And a problem at the office was something that made me run behind that odious and impractical appointment schedule; at the hospital I was seeing real illnesses, not people who wanted a prescription med for their sore throat so they wouldn’t be bothered with it on their vacation.
Q: What is your biggest professional challenge?
A: EHRs.
Q: What is your biggest professional reward?
A: When a patient says, “Thanks for taking care of me, doctor.”
Q: What does teaching mean to you, and how has it been gratifying in your career?
A: Teaching means paying it forward, in gratitude to those who taught me, with the reward of seeing light bulbs go off behind the eyes of students and younger doctors who are eager to learn.
Q: When you aren’t working, what is important to you?
A: Family and music and church.
Q: Faith is obviously important to you. How does that help your work as a care provider?
A: I don’t think anybody goes around being religious all the time. But it sometimes makes all the difference knowing that a higher power is looking out for me and the patients.
Q: You’ve described mentoring as fun for you. What exactly do you mean by fun?
A: Mentoring is what we do. Patients, nurses—anybody we work with—need explanations and clarifications. About the third day of med school, docs in training realize that anybody who can help us understand and retain the huge stream of information directed at us is performing a necessary service. Throughout the training period, residents teach students, fellows teach residents, and attending faculty teach everybody. Doctors in training are bright people who want to learn both the facts and how to deal with patients’ side of things, and feeding their desires is very enjoyable.
Q: You’d like to see more physicians than MBAs in decision-making positions. Why? What real changes do you think that would effectuate?
A: Physicians and nurses were administrators for decades before insurance company penny-pinching and government regulations led hospitals to hire “bean counters” to replace them. It is a tremendous change for the worse, to have people making decisions for patients whose primary consideration is the bottom line.
Q: What’s next professionally?
A: Small-volume ERs, where I don’t have to do discharge planning while being harassed by insurance company reps.
Q: Where do you see yourself in 10 years?
A: Retired.
Q: If you weren’t a doctor, what would you be doing right now?
A: Law enforcement.
Q: Devices like iPhones and tablets can take away from patient face time. But they can also be valuable. How do you balance that? How do you encourage younger docs to do so, particularly when they’re much more used to having smartphones glued to their hands?
A: I use my iPhone when I’m with patients … but only when they can see the reason I need it to help them, such as looking up the side effects of a medication. Electronic health records can work on an iPad, but I hesitate to use them unless the patient knows just what I am doing, such as looking up results of a lab test that concerns them. Taking a computer on wheels into a patient’s room means that I spend part of the visit looking at a screen instead of at the patient, and I prefer to avoid it if at all possible.
Q: What’s the best book you’ve read recently? Why?
A: The House of Silk by Anthony Horowitz. Great continuation of the Holmes stories, with a seamless link to [Sir Arthur] Conan Doyle’s style.
Q: How many Apple products (phones, iPods, tablets, iTunes, etc.) do you interface with in a given week?
A: Three.
Q: What’s your favorite social network? Do you use it all for work or professional development?
A: Facebook. Heck no, it’s just fun.
Q: What’s next in your Netflix queue?
A: Last two episodes of Game of Thrones season 5.
Richard Quinn is a freelance writer in New Jersey.
IMF denounces report on newer MM drugs

Photo by Bill Branson
A report assessing the value of newer multiple myeloma (MM) treatments “dangerously oversimplifies” a complex issue and could limit patients’ access to treatment, according to the International Myeloma Foundation (IMF).
The report, which was drafted by the Institute for Clinical and Economic Review (ICER), is scheduled to be discussed at the inaugural meeting of the Midwest Comparative Effectiveness Public Advisory Council (Midwest CEPAC) in St. Louis, Missouri, on May 26.
The main conclusion of ICER’s report was that newer second- and third-line treatment regimens for MM appear to confer clinical benefits, but the estimated cost-effectiveness of these regimens exceeds commonly cited thresholds.
For example, ICER said that, based on the available data, there was “moderate certainty” that carfilzomib (CFZ), ixazomib (IX), or elotuzumab (ELO) given in combination with lenalidomide and dexamethasone (LEN+DEX) can provide an incremental or better net health benefit for second-line, third-line, or subsequent therapy in adults with relapsed/refractory MM, relative to LEN+DEX alone.
However, the estimated cost-effectiveness, compared to LEN+DEX, was $200,000 per quality-adjusted life year (QALY) gained for CFZ+LEN+DEX, $428,000 for ELO+LEN+DEX, and $434,000 for IX+LEN+DEX. All of these exceed commonly cited thresholds of $50,000 to $150,000 per QALY.
ICER said achieving levels of value more closely aligned with patient benefit would require substantial discounts from the list price in many cases. In other cases, there is no realistic price for the newest agents that would achieve these thresholds.
IMF’s response
IMF said ICER’s report has a few flaws—namely, the absence of many newer MM drugs and combinations, the use of inaccurate data, and an underestimation of QALYs.
Furthermore, IMF said it is concerned that, if ICER's recommendations were to be adopted by the Centers for Medicare & Medicaid Services, patients might be required to “fail first” before other, possibly more effective drugs would be an option.
“We believe that the IMF’s research body, the International Myeloma Working Group (IMWG), will produce superior patient-centered and research-supported guidelines to effectively impact drug costs at our annual summit in June,” said IMF Chairman Brian Durie.
IMWG plans to focus on healthcare cost containment in a special session at the 2016 IMWG Summit, which is scheduled to take place June 7-9 in Copenhagen, Denmark.
IMF said the guidelines resulting from this session should be available in about 2 months. And they will spell out primary and secondary recommendations that allow for individualized therapy choices based on unique features of the disease, patient and/or physician preference, and local and/or regional access issues. ![]()
Photo by Bill Branson
A report assessing the value of newer multiple myeloma (MM) treatments “dangerously oversimplifies” a complex issue and could limit patients’ access to treatment, according to the International Myeloma Foundation (IMF).
The report, which was drafted by the Institute for Clinical and Economic Review (ICER), is scheduled to be discussed at the inaugural meeting of the Midwest Comparative Effectiveness Public Advisory Council (Midwest CEPAC) in St. Louis, Missouri, on May 26.
The main conclusion of ICER’s report was that newer second- and third-line treatment regimens for MM appear to confer clinical benefits, but the estimated cost-effectiveness of these regimens exceeds commonly cited thresholds.
For example, ICER said that, based on the available data, there was “moderate certainty” that carfilzomib (CFZ), ixazomib (IX), or elotuzumab (ELO) given in combination with lenalidomide and dexamethasone (LEN+DEX) can provide an incremental or better net health benefit for second-line, third-line, or subsequent therapy in adults with relapsed/refractory MM, relative to LEN+DEX alone.
However, the estimated cost-effectiveness, compared to LEN+DEX, was $200,000 per quality-adjusted life year (QALY) gained for CFZ+LEN+DEX, $428,000 for ELO+LEN+DEX, and $434,000 for IX+LEN+DEX. All of these exceed commonly cited thresholds of $50,000 to $150,000 per QALY.
ICER said achieving levels of value more closely aligned with patient benefit would require substantial discounts from the list price in many cases. In other cases, there is no realistic price for the newest agents that would achieve these thresholds.
IMF’s response
IMF said ICER’s report has a few flaws—namely, the absence of many newer MM drugs and combinations, the use of inaccurate data, and an underestimation of QALYs.
Furthermore, IMF said it is concerned that, if ICER's recommendations were to be adopted by the Centers for Medicare & Medicaid Services, patients might be required to “fail first” before other, possibly more effective drugs would be an option.
“We believe that the IMF’s research body, the International Myeloma Working Group (IMWG), will produce superior patient-centered and research-supported guidelines to effectively impact drug costs at our annual summit in June,” said IMF Chairman Brian Durie.
IMWG plans to focus on healthcare cost containment in a special session at the 2016 IMWG Summit, which is scheduled to take place June 7-9 in Copenhagen, Denmark.
IMF said the guidelines resulting from this session should be available in about 2 months. And they will spell out primary and secondary recommendations that allow for individualized therapy choices based on unique features of the disease, patient and/or physician preference, and local and/or regional access issues. ![]()
Photo by Bill Branson
A report assessing the value of newer multiple myeloma (MM) treatments “dangerously oversimplifies” a complex issue and could limit patients’ access to treatment, according to the International Myeloma Foundation (IMF).
The report, which was drafted by the Institute for Clinical and Economic Review (ICER), is scheduled to be discussed at the inaugural meeting of the Midwest Comparative Effectiveness Public Advisory Council (Midwest CEPAC) in St. Louis, Missouri, on May 26.
The main conclusion of ICER’s report was that newer second- and third-line treatment regimens for MM appear to confer clinical benefits, but the estimated cost-effectiveness of these regimens exceeds commonly cited thresholds.
For example, ICER said that, based on the available data, there was “moderate certainty” that carfilzomib (CFZ), ixazomib (IX), or elotuzumab (ELO) given in combination with lenalidomide and dexamethasone (LEN+DEX) can provide an incremental or better net health benefit for second-line, third-line, or subsequent therapy in adults with relapsed/refractory MM, relative to LEN+DEX alone.
However, the estimated cost-effectiveness, compared to LEN+DEX, was $200,000 per quality-adjusted life year (QALY) gained for CFZ+LEN+DEX, $428,000 for ELO+LEN+DEX, and $434,000 for IX+LEN+DEX. All of these exceed commonly cited thresholds of $50,000 to $150,000 per QALY.
ICER said achieving levels of value more closely aligned with patient benefit would require substantial discounts from the list price in many cases. In other cases, there is no realistic price for the newest agents that would achieve these thresholds.
IMF’s response
IMF said ICER’s report has a few flaws—namely, the absence of many newer MM drugs and combinations, the use of inaccurate data, and an underestimation of QALYs.
Furthermore, IMF said it is concerned that, if ICER's recommendations were to be adopted by the Centers for Medicare & Medicaid Services, patients might be required to “fail first” before other, possibly more effective drugs would be an option.
“We believe that the IMF’s research body, the International Myeloma Working Group (IMWG), will produce superior patient-centered and research-supported guidelines to effectively impact drug costs at our annual summit in June,” said IMF Chairman Brian Durie.
IMWG plans to focus on healthcare cost containment in a special session at the 2016 IMWG Summit, which is scheduled to take place June 7-9 in Copenhagen, Denmark.
IMF said the guidelines resulting from this session should be available in about 2 months. And they will spell out primary and secondary recommendations that allow for individualized therapy choices based on unique features of the disease, patient and/or physician preference, and local and/or regional access issues. ![]()
Team simplifies synthesis of anticancer agent

Photo courtesy of Jeff Fitlow
and Rice University
Researchers say they have streamlined synthesis of delta12-prostaglandin J3, a molecule that has been shown to kill leukemia cells.
Total synthesis of the molecule now requires only 6 steps from commercially available starting materials.
The researchers say this work sets the stage for large-scale synthesis of the molecule—a lipid found in nearly all animal tissues—and related compounds that can be produced as potential anticancer agents.
K.C. Nicolaou, PhD, of Rice University in Houston, Texas, and his colleagues described the work in Chemistry - A European Journal.
The prostaglandin the researchers synthesized had been isolated in 2011 as a secondary metabolite formed from eicosapentaenoic acid, which is found primarily in fish oil.
The team reported the first total synthesis of the molecule in 2014. That allowed them to confirm its structure.
Now, the researchers have established techniques to develop related disease-fighting compounds and ramp up bulk production if necessary.
Several such prostaglandin derivatives under consideration as preclinical drug candidates were detailed in a second paper published in the Journal of the American Chemical Society.
That publication described the synthesis of dozens of prostaglandin derivatives that were tested against a range of cancer cells by the National Cancer Institute.
One such derivative, macrolactone 11, is currently under evaluation as a preclinical drug candidate. Related compounds macrolactone 33 and 44 showed evidence of even higher potency against leukemia, lung cancer, colon cancer, melanoma, renal, and prostate cancer.
“The macrolactones are very good—better than the natural product—and now we’re following this lead to optimize the potency while minimizing toxicity,” Dr Nicolaou said. “It’s a balancing act.”
In addition, he and his colleagues are developing other drug candidates based on prostaglandin.
“In the process, we’ve developed a lot of nice chemistry, and we know a lot more about the biology of this compound,” Dr Nicolaou said. “We’ve advanced organic synthesis in general and also enriched the knowledge about how these kinds of molecules behave. We hope the papers provide some ideas and leads and inspiration for others to follow.” ![]()
Photo courtesy of Jeff Fitlow
and Rice University
Researchers say they have streamlined synthesis of delta12-prostaglandin J3, a molecule that has been shown to kill leukemia cells.
Total synthesis of the molecule now requires only 6 steps from commercially available starting materials.
The researchers say this work sets the stage for large-scale synthesis of the molecule—a lipid found in nearly all animal tissues—and related compounds that can be produced as potential anticancer agents.
K.C. Nicolaou, PhD, of Rice University in Houston, Texas, and his colleagues described the work in Chemistry - A European Journal.
The prostaglandin the researchers synthesized had been isolated in 2011 as a secondary metabolite formed from eicosapentaenoic acid, which is found primarily in fish oil.
The team reported the first total synthesis of the molecule in 2014. That allowed them to confirm its structure.
Now, the researchers have established techniques to develop related disease-fighting compounds and ramp up bulk production if necessary.
Several such prostaglandin derivatives under consideration as preclinical drug candidates were detailed in a second paper published in the Journal of the American Chemical Society.
That publication described the synthesis of dozens of prostaglandin derivatives that were tested against a range of cancer cells by the National Cancer Institute.
One such derivative, macrolactone 11, is currently under evaluation as a preclinical drug candidate. Related compounds macrolactone 33 and 44 showed evidence of even higher potency against leukemia, lung cancer, colon cancer, melanoma, renal, and prostate cancer.
“The macrolactones are very good—better than the natural product—and now we’re following this lead to optimize the potency while minimizing toxicity,” Dr Nicolaou said. “It’s a balancing act.”
In addition, he and his colleagues are developing other drug candidates based on prostaglandin.
“In the process, we’ve developed a lot of nice chemistry, and we know a lot more about the biology of this compound,” Dr Nicolaou said. “We’ve advanced organic synthesis in general and also enriched the knowledge about how these kinds of molecules behave. We hope the papers provide some ideas and leads and inspiration for others to follow.” ![]()
Photo courtesy of Jeff Fitlow
and Rice University
Researchers say they have streamlined synthesis of delta12-prostaglandin J3, a molecule that has been shown to kill leukemia cells.
Total synthesis of the molecule now requires only 6 steps from commercially available starting materials.
The researchers say this work sets the stage for large-scale synthesis of the molecule—a lipid found in nearly all animal tissues—and related compounds that can be produced as potential anticancer agents.
K.C. Nicolaou, PhD, of Rice University in Houston, Texas, and his colleagues described the work in Chemistry - A European Journal.
The prostaglandin the researchers synthesized had been isolated in 2011 as a secondary metabolite formed from eicosapentaenoic acid, which is found primarily in fish oil.
The team reported the first total synthesis of the molecule in 2014. That allowed them to confirm its structure.
Now, the researchers have established techniques to develop related disease-fighting compounds and ramp up bulk production if necessary.
Several such prostaglandin derivatives under consideration as preclinical drug candidates were detailed in a second paper published in the Journal of the American Chemical Society.
That publication described the synthesis of dozens of prostaglandin derivatives that were tested against a range of cancer cells by the National Cancer Institute.
One such derivative, macrolactone 11, is currently under evaluation as a preclinical drug candidate. Related compounds macrolactone 33 and 44 showed evidence of even higher potency against leukemia, lung cancer, colon cancer, melanoma, renal, and prostate cancer.
“The macrolactones are very good—better than the natural product—and now we’re following this lead to optimize the potency while minimizing toxicity,” Dr Nicolaou said. “It’s a balancing act.”
In addition, he and his colleagues are developing other drug candidates based on prostaglandin.
“In the process, we’ve developed a lot of nice chemistry, and we know a lot more about the biology of this compound,” Dr Nicolaou said. “We’ve advanced organic synthesis in general and also enriched the knowledge about how these kinds of molecules behave. We hope the papers provide some ideas and leads and inspiration for others to follow.” ![]()
Proteins may be targets for malaria vaccines
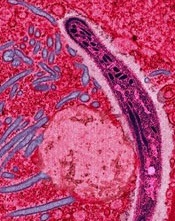
Image courtesy of Ute Frevert
and Margaret Shear
A study published in PLOS Pathogens has revealed proteins that may be viable targets for malaria vaccines.
Investigators identified 42 proteins that can be found on the surface of Plasmodium falciparum sporozoites and could be targeted by vaccines.
However, the team also found evidence to suggest that 2 other surface proteins should not be targeted, as they may be able to evade the immune system.
“We used a method that we developed in a previous paper to identify which proteins of the malaria parasite, Plasmodium falciparum, might be visible to the human immune system on the outside of the parasite and therefore are good potential targets for the development of new malaria vaccines,” said study author Scott E. Lindner, PhD, of Pennsylvania State University in University Park.
“Current experimental malaria vaccines target single proteins and do not provide the level of protection necessary to prevent the spread of the disease. Our new list of potential vaccine targets will allow the development of more effective vaccines that target several proteins on the surface of the parasite.”
To identify these targets, Dr Lindner and his colleagues collected malaria sporozoites from the salivary glands of thousands of infected mosquitoes.
The investigators then marked proteins on the surface of the sporozoites with a chemical label that could not cross through the outer membrane of the parasite. The team identified and characterized the labeled proteins using mass spectrometry.
“We focused on the transmission stage of the parasite because, at this point in an infection, the number of parasites is low, and if we can design effective vaccines for this stage, we can stop the progress of the disease before it causes symptoms,” Dr Lindner said. “Once the parasites are in the liver, they can hide from our immune system by residing inside of liver cells.”
Based on multiple replications of their experiments, the investigators identified 42 proteins that are highly likely to be exposed on the surface of the parasite and are therefore potential targets for vaccines.
The team noted that many of the proteins they identified had been thought to be located exclusively on the inside of the parasite. They suggest that these proteins may become exposed as the parasite moves from the site of a mosquito bite toward the liver.
“Malaria is still one of the great global health issues today, with hundreds of millions of new infections and half a million deaths each year, most of which occur in children under the age of 5,” Dr Lindner said.
“The parasite quickly and efficiently develops resistance to the drugs that we use to treat the disease, so what’s really needed to make eradication of malaria possible is a better vaccine. Our research provides an experimentally validated list of protein targets that could be used to develop new, more effective malaria vaccines.”
The investigators also discovered that 2 surface proteins—CSP and TRAP—are glycosylated in sporozoites, which changes the way the proteins are recognized by the immune system.
The team believes this discovery will affect the way future vaccines are designed as well.
“Our goal was to identify proteins that are present on the surface of sporozoites in hopes of finding targets for new vaccines,” said study author Kristian E. Swearingen, PhD, of the Institute for Systems Biology in Seattle, Washington.
“In addition to the potential new targets we’ve found, we’re also excited about the discovery that 2 of the major sporozoite surface proteins are glycosylated. The presence of sugars on these proteins almost certainly affects the way they are recognized by antibodies, something that will need to be factored in for future vaccine efforts based on these proteins.” ![]()
Image courtesy of Ute Frevert
and Margaret Shear
A study published in PLOS Pathogens has revealed proteins that may be viable targets for malaria vaccines.
Investigators identified 42 proteins that can be found on the surface of Plasmodium falciparum sporozoites and could be targeted by vaccines.
However, the team also found evidence to suggest that 2 other surface proteins should not be targeted, as they may be able to evade the immune system.
“We used a method that we developed in a previous paper to identify which proteins of the malaria parasite, Plasmodium falciparum, might be visible to the human immune system on the outside of the parasite and therefore are good potential targets for the development of new malaria vaccines,” said study author Scott E. Lindner, PhD, of Pennsylvania State University in University Park.
“Current experimental malaria vaccines target single proteins and do not provide the level of protection necessary to prevent the spread of the disease. Our new list of potential vaccine targets will allow the development of more effective vaccines that target several proteins on the surface of the parasite.”
To identify these targets, Dr Lindner and his colleagues collected malaria sporozoites from the salivary glands of thousands of infected mosquitoes.
The investigators then marked proteins on the surface of the sporozoites with a chemical label that could not cross through the outer membrane of the parasite. The team identified and characterized the labeled proteins using mass spectrometry.
“We focused on the transmission stage of the parasite because, at this point in an infection, the number of parasites is low, and if we can design effective vaccines for this stage, we can stop the progress of the disease before it causes symptoms,” Dr Lindner said. “Once the parasites are in the liver, they can hide from our immune system by residing inside of liver cells.”
Based on multiple replications of their experiments, the investigators identified 42 proteins that are highly likely to be exposed on the surface of the parasite and are therefore potential targets for vaccines.
The team noted that many of the proteins they identified had been thought to be located exclusively on the inside of the parasite. They suggest that these proteins may become exposed as the parasite moves from the site of a mosquito bite toward the liver.
“Malaria is still one of the great global health issues today, with hundreds of millions of new infections and half a million deaths each year, most of which occur in children under the age of 5,” Dr Lindner said.
“The parasite quickly and efficiently develops resistance to the drugs that we use to treat the disease, so what’s really needed to make eradication of malaria possible is a better vaccine. Our research provides an experimentally validated list of protein targets that could be used to develop new, more effective malaria vaccines.”
The investigators also discovered that 2 surface proteins—CSP and TRAP—are glycosylated in sporozoites, which changes the way the proteins are recognized by the immune system.
The team believes this discovery will affect the way future vaccines are designed as well.
“Our goal was to identify proteins that are present on the surface of sporozoites in hopes of finding targets for new vaccines,” said study author Kristian E. Swearingen, PhD, of the Institute for Systems Biology in Seattle, Washington.
“In addition to the potential new targets we’ve found, we’re also excited about the discovery that 2 of the major sporozoite surface proteins are glycosylated. The presence of sugars on these proteins almost certainly affects the way they are recognized by antibodies, something that will need to be factored in for future vaccine efforts based on these proteins.” ![]()
Image courtesy of Ute Frevert
and Margaret Shear
A study published in PLOS Pathogens has revealed proteins that may be viable targets for malaria vaccines.
Investigators identified 42 proteins that can be found on the surface of Plasmodium falciparum sporozoites and could be targeted by vaccines.
However, the team also found evidence to suggest that 2 other surface proteins should not be targeted, as they may be able to evade the immune system.
“We used a method that we developed in a previous paper to identify which proteins of the malaria parasite, Plasmodium falciparum, might be visible to the human immune system on the outside of the parasite and therefore are good potential targets for the development of new malaria vaccines,” said study author Scott E. Lindner, PhD, of Pennsylvania State University in University Park.
“Current experimental malaria vaccines target single proteins and do not provide the level of protection necessary to prevent the spread of the disease. Our new list of potential vaccine targets will allow the development of more effective vaccines that target several proteins on the surface of the parasite.”
To identify these targets, Dr Lindner and his colleagues collected malaria sporozoites from the salivary glands of thousands of infected mosquitoes.
The investigators then marked proteins on the surface of the sporozoites with a chemical label that could not cross through the outer membrane of the parasite. The team identified and characterized the labeled proteins using mass spectrometry.
“We focused on the transmission stage of the parasite because, at this point in an infection, the number of parasites is low, and if we can design effective vaccines for this stage, we can stop the progress of the disease before it causes symptoms,” Dr Lindner said. “Once the parasites are in the liver, they can hide from our immune system by residing inside of liver cells.”
Based on multiple replications of their experiments, the investigators identified 42 proteins that are highly likely to be exposed on the surface of the parasite and are therefore potential targets for vaccines.
The team noted that many of the proteins they identified had been thought to be located exclusively on the inside of the parasite. They suggest that these proteins may become exposed as the parasite moves from the site of a mosquito bite toward the liver.
“Malaria is still one of the great global health issues today, with hundreds of millions of new infections and half a million deaths each year, most of which occur in children under the age of 5,” Dr Lindner said.
“The parasite quickly and efficiently develops resistance to the drugs that we use to treat the disease, so what’s really needed to make eradication of malaria possible is a better vaccine. Our research provides an experimentally validated list of protein targets that could be used to develop new, more effective malaria vaccines.”
The investigators also discovered that 2 surface proteins—CSP and TRAP—are glycosylated in sporozoites, which changes the way the proteins are recognized by the immune system.
The team believes this discovery will affect the way future vaccines are designed as well.
“Our goal was to identify proteins that are present on the surface of sporozoites in hopes of finding targets for new vaccines,” said study author Kristian E. Swearingen, PhD, of the Institute for Systems Biology in Seattle, Washington.
“In addition to the potential new targets we’ve found, we’re also excited about the discovery that 2 of the major sporozoite surface proteins are glycosylated. The presence of sugars on these proteins almost certainly affects the way they are recognized by antibodies, something that will need to be factored in for future vaccine efforts based on these proteins.” ![]()
One-size-fits-all approach no good for low-risk ET
Photo by Sage Ross
Results of a retrospective study suggest that patients with low-risk essential thrombocythemia (ET) may benefit from a genotype-based approach to
antiplatelet therapy.
The study showed that, overall, neither CALR-mutated nor JAK2V617F-positive patients derived a significant benefit from treatment with low-dose aspirin.
JAK2V617F-positive patients had a somewhat lower risk of thrombosis while on the therapy than during observation.
But CALR-mutated patients had a significantly increased risk of major bleeding and no decrease in the risk of thrombosis while on antiplatelet therapy.
Carlos Besses, MD, PhD, of Hospital del Mar in Barcelona, Spain, and his colleagues reported these results in haematologica.
The researchers evaluated 433 patients with low-risk ET—271 with CALR mutations and 162 with JAK2V617F. In all, 353 patients received low-dose aspirin (81-100 mg/day), but the treatment was withdrawn in 50 patients (permanently in 46 of them).
Two hundred and thirty-one patients received cytoreductive therapy, including hydroxyurea (n=143), anagrelide (n=66), interferon (n=18), and busulfan (n=4).
The projected time from diagnosis to the start of cytoreductive therapy was significantly shorter in patients with CALR-mutated ET than in JAK2V617F-positive patients—a median of 5.0 years and 9.8 years, respectively (P=0.002). The most common reason for cytoreduction in CALR-mutated patients was extreme thrombocytosis.
Thrombosis
After 2215 person-years of follow-up free from cytoreduction, there were 25 arterial or venous thrombotic events.
Fourteen thrombotic events occurred while patients were receiving low-dose aspirin, and 11 occurred while patients were under observation only. The incidence rates were 10.7 and 12.1 events x 1000 person-years, respectively (P=0.7).
Among CALR-mutated patients, there were more thrombotic events during antiplatelet therapy than during observation—9.7 and 6.9 events x 1000 person-years, respectively (P=0.6).
Among JAK2V617F-positive patients, there were fewer thrombotic events during antiplatelet therapy than during observation, but the difference was not significant—11.6 and 21.1 events x 1000 person-years, respectively (P=0.3).
Coexistence of the JAK2V617F mutation and cardiovascular risk factors increased the risk of thrombosis, even after the researchers adjusted for treatment with low-dose aspirin. The incidence rate ratio (IRR) was 9.8 (P=0.02).
Bleeding
After 2215 person-years of follow-up free from cytoreduction, there were 17 major bleeding episodes.
Thirteen occurred while patients were on antiplatelet therapy, and 4 occurred while patients were on observation. The incidence rates were 9.9 and 4.6 events x 1000 person-years, respectively (P=0.2).
There was no significant difference in major bleeding episodes between the treatment groups for JAK2V617F-positive patients.
But CALR-mutated patients had a significantly higher rate of major bleeding while on antiplatelet therapy than on observation—12.9 and 1.8 events per 1000 person-years, respectively (P=0.03).
In CALR-mutated patients, antiplatelet therapy was associated with a tendency toward an increased risk of bleeding (IRR: 6.9, P=0.06), but extreme thrombocytosis was not (IRR: 2.7, P=0.1).
In JAK2V617F-positive patients, extreme thrombocytosis was associated with an increased risk of bleeding (IRR: 9.8, P=0.002), but antiplatelet therapy was not (IRR: 0.9, P=0.9).
Potential treatment recommendations
The researchers said this retrospective study suggests a genotype-based approach to antiplatelet therapy may be effective for patients with low-risk ET. However, this needs to be confirmed in prospective trials.
The failure of antiplatelet therapy to prevent thrombosis in CALR-mutated patients and their increased need for cytoreductive therapy suggest these patients require a different approach from that used in JAK2V617F-positive patients.
The researchers said the data suggest that patients with CALR-mutated ET who have a low risk of thrombosis and no symptoms should simply be observed. And CALR-mutated patients with symptoms or marked thrombocytosis should receive cytoreductive therapy, as it poses a lower risk of bleeding than antiplatelet therapy.
Patients with JAK2V617F-positive ET should receive antiplatelet therapy rather than undergoing observation, as antiplatelet therapy may reduce the risk of thrombosis in these patients and does not pose an increased risk of bleeding.
However, in JAK2V617F-positive patients with concomitant cardiovascular risk factors and/or leukocytosis, antiplatelet therapy may not be sufficient. These patients might be candidates for cytoreductive therapy, especially if they have marked thrombocytosis. ![]()
Photo by Sage Ross
Results of a retrospective study suggest that patients with low-risk essential thrombocythemia (ET) may benefit from a genotype-based approach to
antiplatelet therapy.
The study showed that, overall, neither CALR-mutated nor JAK2V617F-positive patients derived a significant benefit from treatment with low-dose aspirin.
JAK2V617F-positive patients had a somewhat lower risk of thrombosis while on the therapy than during observation.
But CALR-mutated patients had a significantly increased risk of major bleeding and no decrease in the risk of thrombosis while on antiplatelet therapy.
Carlos Besses, MD, PhD, of Hospital del Mar in Barcelona, Spain, and his colleagues reported these results in haematologica.
The researchers evaluated 433 patients with low-risk ET—271 with CALR mutations and 162 with JAK2V617F. In all, 353 patients received low-dose aspirin (81-100 mg/day), but the treatment was withdrawn in 50 patients (permanently in 46 of them).
Two hundred and thirty-one patients received cytoreductive therapy, including hydroxyurea (n=143), anagrelide (n=66), interferon (n=18), and busulfan (n=4).
The projected time from diagnosis to the start of cytoreductive therapy was significantly shorter in patients with CALR-mutated ET than in JAK2V617F-positive patients—a median of 5.0 years and 9.8 years, respectively (P=0.002). The most common reason for cytoreduction in CALR-mutated patients was extreme thrombocytosis.
Thrombosis
After 2215 person-years of follow-up free from cytoreduction, there were 25 arterial or venous thrombotic events.
Fourteen thrombotic events occurred while patients were receiving low-dose aspirin, and 11 occurred while patients were under observation only. The incidence rates were 10.7 and 12.1 events x 1000 person-years, respectively (P=0.7).
Among CALR-mutated patients, there were more thrombotic events during antiplatelet therapy than during observation—9.7 and 6.9 events x 1000 person-years, respectively (P=0.6).
Among JAK2V617F-positive patients, there were fewer thrombotic events during antiplatelet therapy than during observation, but the difference was not significant—11.6 and 21.1 events x 1000 person-years, respectively (P=0.3).
Coexistence of the JAK2V617F mutation and cardiovascular risk factors increased the risk of thrombosis, even after the researchers adjusted for treatment with low-dose aspirin. The incidence rate ratio (IRR) was 9.8 (P=0.02).
Bleeding
After 2215 person-years of follow-up free from cytoreduction, there were 17 major bleeding episodes.
Thirteen occurred while patients were on antiplatelet therapy, and 4 occurred while patients were on observation. The incidence rates were 9.9 and 4.6 events x 1000 person-years, respectively (P=0.2).
There was no significant difference in major bleeding episodes between the treatment groups for JAK2V617F-positive patients.
But CALR-mutated patients had a significantly higher rate of major bleeding while on antiplatelet therapy than on observation—12.9 and 1.8 events per 1000 person-years, respectively (P=0.03).
In CALR-mutated patients, antiplatelet therapy was associated with a tendency toward an increased risk of bleeding (IRR: 6.9, P=0.06), but extreme thrombocytosis was not (IRR: 2.7, P=0.1).
In JAK2V617F-positive patients, extreme thrombocytosis was associated with an increased risk of bleeding (IRR: 9.8, P=0.002), but antiplatelet therapy was not (IRR: 0.9, P=0.9).
Potential treatment recommendations
The researchers said this retrospective study suggests a genotype-based approach to antiplatelet therapy may be effective for patients with low-risk ET. However, this needs to be confirmed in prospective trials.
The failure of antiplatelet therapy to prevent thrombosis in CALR-mutated patients and their increased need for cytoreductive therapy suggest these patients require a different approach from that used in JAK2V617F-positive patients.
The researchers said the data suggest that patients with CALR-mutated ET who have a low risk of thrombosis and no symptoms should simply be observed. And CALR-mutated patients with symptoms or marked thrombocytosis should receive cytoreductive therapy, as it poses a lower risk of bleeding than antiplatelet therapy.
Patients with JAK2V617F-positive ET should receive antiplatelet therapy rather than undergoing observation, as antiplatelet therapy may reduce the risk of thrombosis in these patients and does not pose an increased risk of bleeding.
However, in JAK2V617F-positive patients with concomitant cardiovascular risk factors and/or leukocytosis, antiplatelet therapy may not be sufficient. These patients might be candidates for cytoreductive therapy, especially if they have marked thrombocytosis. ![]()
Photo by Sage Ross
Results of a retrospective study suggest that patients with low-risk essential thrombocythemia (ET) may benefit from a genotype-based approach to
antiplatelet therapy.
The study showed that, overall, neither CALR-mutated nor JAK2V617F-positive patients derived a significant benefit from treatment with low-dose aspirin.
JAK2V617F-positive patients had a somewhat lower risk of thrombosis while on the therapy than during observation.
But CALR-mutated patients had a significantly increased risk of major bleeding and no decrease in the risk of thrombosis while on antiplatelet therapy.
Carlos Besses, MD, PhD, of Hospital del Mar in Barcelona, Spain, and his colleagues reported these results in haematologica.
The researchers evaluated 433 patients with low-risk ET—271 with CALR mutations and 162 with JAK2V617F. In all, 353 patients received low-dose aspirin (81-100 mg/day), but the treatment was withdrawn in 50 patients (permanently in 46 of them).
Two hundred and thirty-one patients received cytoreductive therapy, including hydroxyurea (n=143), anagrelide (n=66), interferon (n=18), and busulfan (n=4).
The projected time from diagnosis to the start of cytoreductive therapy was significantly shorter in patients with CALR-mutated ET than in JAK2V617F-positive patients—a median of 5.0 years and 9.8 years, respectively (P=0.002). The most common reason for cytoreduction in CALR-mutated patients was extreme thrombocytosis.
Thrombosis
After 2215 person-years of follow-up free from cytoreduction, there were 25 arterial or venous thrombotic events.
Fourteen thrombotic events occurred while patients were receiving low-dose aspirin, and 11 occurred while patients were under observation only. The incidence rates were 10.7 and 12.1 events x 1000 person-years, respectively (P=0.7).
Among CALR-mutated patients, there were more thrombotic events during antiplatelet therapy than during observation—9.7 and 6.9 events x 1000 person-years, respectively (P=0.6).
Among JAK2V617F-positive patients, there were fewer thrombotic events during antiplatelet therapy than during observation, but the difference was not significant—11.6 and 21.1 events x 1000 person-years, respectively (P=0.3).
Coexistence of the JAK2V617F mutation and cardiovascular risk factors increased the risk of thrombosis, even after the researchers adjusted for treatment with low-dose aspirin. The incidence rate ratio (IRR) was 9.8 (P=0.02).
Bleeding
After 2215 person-years of follow-up free from cytoreduction, there were 17 major bleeding episodes.
Thirteen occurred while patients were on antiplatelet therapy, and 4 occurred while patients were on observation. The incidence rates were 9.9 and 4.6 events x 1000 person-years, respectively (P=0.2).
There was no significant difference in major bleeding episodes between the treatment groups for JAK2V617F-positive patients.
But CALR-mutated patients had a significantly higher rate of major bleeding while on antiplatelet therapy than on observation—12.9 and 1.8 events per 1000 person-years, respectively (P=0.03).
In CALR-mutated patients, antiplatelet therapy was associated with a tendency toward an increased risk of bleeding (IRR: 6.9, P=0.06), but extreme thrombocytosis was not (IRR: 2.7, P=0.1).
In JAK2V617F-positive patients, extreme thrombocytosis was associated with an increased risk of bleeding (IRR: 9.8, P=0.002), but antiplatelet therapy was not (IRR: 0.9, P=0.9).
Potential treatment recommendations
The researchers said this retrospective study suggests a genotype-based approach to antiplatelet therapy may be effective for patients with low-risk ET. However, this needs to be confirmed in prospective trials.
The failure of antiplatelet therapy to prevent thrombosis in CALR-mutated patients and their increased need for cytoreductive therapy suggest these patients require a different approach from that used in JAK2V617F-positive patients.
The researchers said the data suggest that patients with CALR-mutated ET who have a low risk of thrombosis and no symptoms should simply be observed. And CALR-mutated patients with symptoms or marked thrombocytosis should receive cytoreductive therapy, as it poses a lower risk of bleeding than antiplatelet therapy.
Patients with JAK2V617F-positive ET should receive antiplatelet therapy rather than undergoing observation, as antiplatelet therapy may reduce the risk of thrombosis in these patients and does not pose an increased risk of bleeding.
However, in JAK2V617F-positive patients with concomitant cardiovascular risk factors and/or leukocytosis, antiplatelet therapy may not be sufficient. These patients might be candidates for cytoreductive therapy, especially if they have marked thrombocytosis. ![]()
Advances in the management of multiple myeloma
Multiple myeloma (MM) is a bone marrow- based malignancy of plasma cells that is diagnosed in over 30,000 patients annually in the United States. Despite the many recent advances in the treatment of MM, it remains an incurable disease. Thus, the need for the development of new effective therapies remains critical for these patients.
Smoldering MM
In general, it has not been shown that patients with smoldering MM (SMM) benefit from early treatment, but recent studies have identified a subset of patients who are at high-risk and may require therapy more quickly. Recent guidelines from the International Myeloma Working Group recommend immediate treatment of this subgroup of SMM.1 However, although findings in a Spanish study suggested that early treatment of high-risk SMM patients with the immunomodulatory agent (IMiD) lenalidomide and dexamethasone improves overall survival (OS),2 the design of that study limits its clinical applicability, and no other randomized trials have been completed to show the advantage of early therapy for these patients.
Specific drugs
The development of novel agents such as proteasome inhibitors (PIs), IMiDs, histone deacetylase inhibitors (HDACIs), and monoclonal antibodies (mAbs) in recent years has vastly changed the approach to the treatment of MM patients.
PIs that are cytotoxic to MM cells, such as bortezomib, have become a foundation for MM treatment over the past decade. However, patients develop drug resistance to bortezomib by acquiring gene mutations and through other mechanisms. In recent years, newer forms of PIs such as carfilzomib and the oral formulations ixazomib and oprozomib have been and are currently being developed.3 Preclinical studies have shown that resistance to one PI can be overcome with treatment with another PI.4
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma (MM) is a bone marrow- based malignancy of plasma cells that is diagnosed in over 30,000 patients annually in the United States. Despite the many recent advances in the treatment of MM, it remains an incurable disease. Thus, the need for the development of new effective therapies remains critical for these patients.
Smoldering MM
In general, it has not been shown that patients with smoldering MM (SMM) benefit from early treatment, but recent studies have identified a subset of patients who are at high-risk and may require therapy more quickly. Recent guidelines from the International Myeloma Working Group recommend immediate treatment of this subgroup of SMM.1 However, although findings in a Spanish study suggested that early treatment of high-risk SMM patients with the immunomodulatory agent (IMiD) lenalidomide and dexamethasone improves overall survival (OS),2 the design of that study limits its clinical applicability, and no other randomized trials have been completed to show the advantage of early therapy for these patients.
Specific drugs
The development of novel agents such as proteasome inhibitors (PIs), IMiDs, histone deacetylase inhibitors (HDACIs), and monoclonal antibodies (mAbs) in recent years has vastly changed the approach to the treatment of MM patients.
PIs that are cytotoxic to MM cells, such as bortezomib, have become a foundation for MM treatment over the past decade. However, patients develop drug resistance to bortezomib by acquiring gene mutations and through other mechanisms. In recent years, newer forms of PIs such as carfilzomib and the oral formulations ixazomib and oprozomib have been and are currently being developed.3 Preclinical studies have shown that resistance to one PI can be overcome with treatment with another PI.4
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma (MM) is a bone marrow- based malignancy of plasma cells that is diagnosed in over 30,000 patients annually in the United States. Despite the many recent advances in the treatment of MM, it remains an incurable disease. Thus, the need for the development of new effective therapies remains critical for these patients.
Smoldering MM
In general, it has not been shown that patients with smoldering MM (SMM) benefit from early treatment, but recent studies have identified a subset of patients who are at high-risk and may require therapy more quickly. Recent guidelines from the International Myeloma Working Group recommend immediate treatment of this subgroup of SMM.1 However, although findings in a Spanish study suggested that early treatment of high-risk SMM patients with the immunomodulatory agent (IMiD) lenalidomide and dexamethasone improves overall survival (OS),2 the design of that study limits its clinical applicability, and no other randomized trials have been completed to show the advantage of early therapy for these patients.
Specific drugs
The development of novel agents such as proteasome inhibitors (PIs), IMiDs, histone deacetylase inhibitors (HDACIs), and monoclonal antibodies (mAbs) in recent years has vastly changed the approach to the treatment of MM patients.
PIs that are cytotoxic to MM cells, such as bortezomib, have become a foundation for MM treatment over the past decade. However, patients develop drug resistance to bortezomib by acquiring gene mutations and through other mechanisms. In recent years, newer forms of PIs such as carfilzomib and the oral formulations ixazomib and oprozomib have been and are currently being developed.3 Preclinical studies have shown that resistance to one PI can be overcome with treatment with another PI.4
Click on the PDF icon at the top of this introduction to read the full article.
Paraneoplastic syndrome and underlying breast cancer: a worsening rash despite initiation of chemotherapy
Culture-broker and medical decoder: contributions of caregivers in American Indian cancer trajectories
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Prognostic significance of HPV status in postoperative squamous-cell carcinoma of the head and neck
Background There are limited data on the prognostic significance of human papillomavirus (HPV) status in relation to traditional risk factors for head and neck squamous-cell carcinoma (HNSCC) in the postoperative setting.
Objective To clarify the impact of HPV status on the risk for HNSCC in the postoperative setting.
Methods We retrospectively evaluated an institutional cohort of 128 patients with HNSCC patients who had been treated with definitive surgery with or without adjuvant radiotherapy or chemoradiotherapy. Patient, disease, and treatment factors were analyzed as potential prognostic indicators.
Results Lymph node extracapsular extension (ECE), perineural invasion (PNI), and lymphovascular space invasion (LVSI) positivity predicted poorer locoregional control (LRC), disease-free survival (DFS), and overall survival (OS). Positive margins related to poorer DFS and OS. HPV status alone did not predict LRC, DFS, or OS. Compared with patients who were HPV-positive and ECE-negative, both HPV-positive and HPV-negative patients with ECE experienced significantly poorer OS (78.6%, 60%, and 43.7%, respectively; P = .010 and P = .018, respectively).
Limitations Retrospective, single-institution study; small patient cohort; short follow-up time
Conclusion The influence of HPV in postoperative HNSCC seems limited compared with traditional risk factors such as ECE, LVSI, and PNI. De-escalation of postoperative treatment based on HPV status alone should be approached with caution.
Click on the PDF icon at the top of this introduction to read the full article.
Background There are limited data on the prognostic significance of human papillomavirus (HPV) status in relation to traditional risk factors for head and neck squamous-cell carcinoma (HNSCC) in the postoperative setting.
Objective To clarify the impact of HPV status on the risk for HNSCC in the postoperative setting.
Methods We retrospectively evaluated an institutional cohort of 128 patients with HNSCC patients who had been treated with definitive surgery with or without adjuvant radiotherapy or chemoradiotherapy. Patient, disease, and treatment factors were analyzed as potential prognostic indicators.
Results Lymph node extracapsular extension (ECE), perineural invasion (PNI), and lymphovascular space invasion (LVSI) positivity predicted poorer locoregional control (LRC), disease-free survival (DFS), and overall survival (OS). Positive margins related to poorer DFS and OS. HPV status alone did not predict LRC, DFS, or OS. Compared with patients who were HPV-positive and ECE-negative, both HPV-positive and HPV-negative patients with ECE experienced significantly poorer OS (78.6%, 60%, and 43.7%, respectively; P = .010 and P = .018, respectively).
Limitations Retrospective, single-institution study; small patient cohort; short follow-up time
Conclusion The influence of HPV in postoperative HNSCC seems limited compared with traditional risk factors such as ECE, LVSI, and PNI. De-escalation of postoperative treatment based on HPV status alone should be approached with caution.
Click on the PDF icon at the top of this introduction to read the full article.
Background There are limited data on the prognostic significance of human papillomavirus (HPV) status in relation to traditional risk factors for head and neck squamous-cell carcinoma (HNSCC) in the postoperative setting.
Objective To clarify the impact of HPV status on the risk for HNSCC in the postoperative setting.
Methods We retrospectively evaluated an institutional cohort of 128 patients with HNSCC patients who had been treated with definitive surgery with or without adjuvant radiotherapy or chemoradiotherapy. Patient, disease, and treatment factors were analyzed as potential prognostic indicators.
Results Lymph node extracapsular extension (ECE), perineural invasion (PNI), and lymphovascular space invasion (LVSI) positivity predicted poorer locoregional control (LRC), disease-free survival (DFS), and overall survival (OS). Positive margins related to poorer DFS and OS. HPV status alone did not predict LRC, DFS, or OS. Compared with patients who were HPV-positive and ECE-negative, both HPV-positive and HPV-negative patients with ECE experienced significantly poorer OS (78.6%, 60%, and 43.7%, respectively; P = .010 and P = .018, respectively).
Limitations Retrospective, single-institution study; small patient cohort; short follow-up time
Conclusion The influence of HPV in postoperative HNSCC seems limited compared with traditional risk factors such as ECE, LVSI, and PNI. De-escalation of postoperative treatment based on HPV status alone should be approached with caution.
Click on the PDF icon at the top of this introduction to read the full article.
Painful Losses
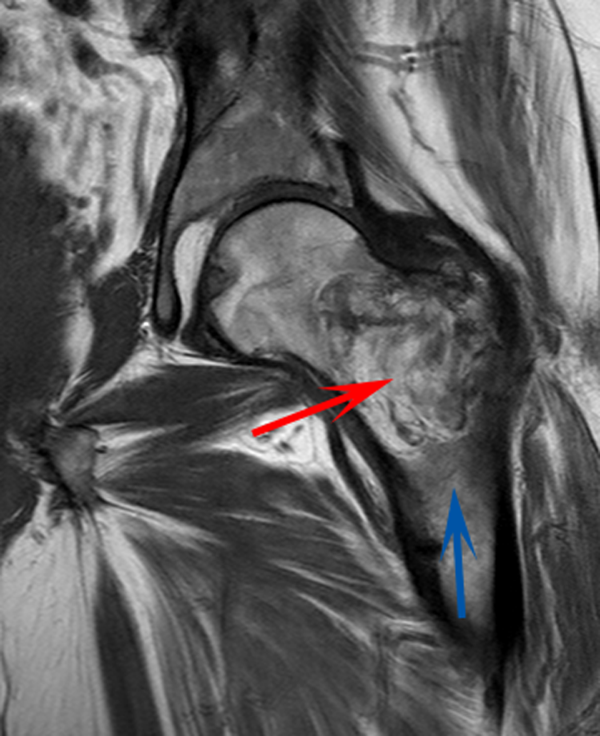
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
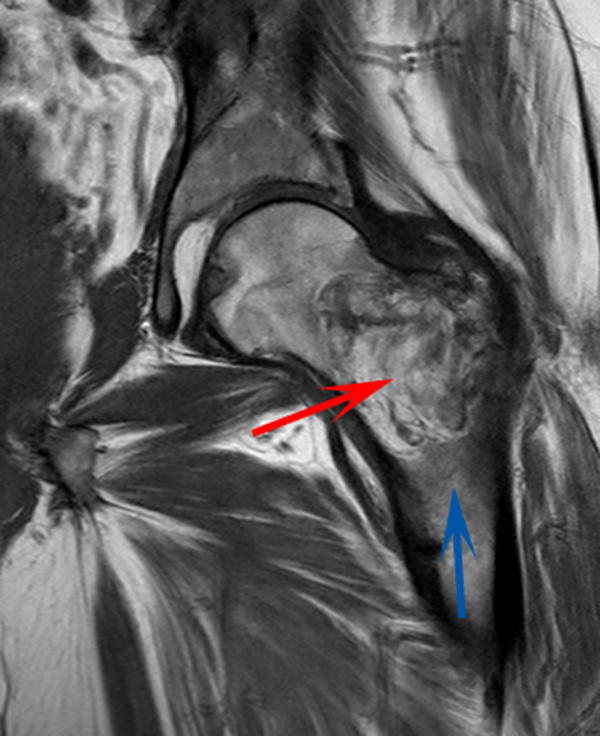
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
A 58‐year‐old man presented to the emergency department with a 1‐month history of progressive, severe left hip pain that had become unbearable. The pain was constant and significantly worse with weight‐bearing, and the patient was now confined to bed. He denied back pain, falls, or trauma.
Although hip pain is a common complaint and a frequent manifestation of chronic degenerative joint disease, the debilitating and subacute nature of the pain suggests a potentially more serious underlying cause. Patients and even clinicians may refer to hip pain when the actual symptoms are periarticular, often presenting over the trochanter laterally, or muscular, presenting as posterior pain. The true hip joint is located in the anterior hip and groin area and often causes symptoms that radiate to the buttock. Pain can also be referred to the hip area from the spine, pelvis, or retroperitoneum, so it is crucial not to restrict the differential diagnosis to hip pathology.
Key diagnostic considerations include (1) inflammatory conditions such as trochanteric bursitis or gout; (2) bacterial infection of the hip joint, adjacent bone, or a nearby structure; (3) benign nerve compression (such as meralgia paresthetica); and (4) tumor (particularly myeloma or metastatic disease to the bone, but also potentially a pelvic or spinal mass with nerve compression). Polymyalgia rheumatica and other systemic rheumatologic complaints are a consideration, but because a single joint is involved, these conditions are less likely. The hip would be an unusual location for a first gout flare, and the duration of symptoms would be unusually long for gout. Avascular necrosis should be considered if the patient has received glucocorticoids for his previously diagnosed rheumatologic disease. If the patient is anticoagulated, consideration of spontaneous hematoma is reasonable, but usually this would present over a course of days, not weeks. The absence of trauma makes a fracture of the hip or pelvis less likely, and the insidious progression of symptoms makes a pathologic fracture less likely.
The patient reported 6 months of worsening proximal upper and lower extremity myalgia and weakness, with arthralgia of the hips and shoulders. The weakness was most notable in his proximal lower extremities, although he had remained ambulatory until the hip pain became limiting. He maintained normal use of his arms. The patient denied current rash but noted photosensitivity and a mild facial rash several months earlier. He described having transient mouth sores intermittently for several years. He denied fever, chills, night sweats, weight loss, dyspnea, recent travel, and outdoor exposures. Several months previously, he had been evaluated for these symptoms at another institution and given the diagnoses of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). At that time, he had initiated treatment with weekly dosing of methotrexate and etanercept.
The patient's medical history was also notable for hypertension, Graves' disease treated previously with radioiodine ablation, quiescent ulcerative colitis, and depression. Current medications included methotrexate, etanercept, levothyroxine, enalapril, hydrochlorothiazide, fluoxetine, ibuprofen, and oxycodone‐acetaminophen. He denied tobacco, alcohol, and recreational drug use.
Weakness occurring in the proximal lower extremities is the classic distribution for polymyositis and dermatomyositis. In contrast to polymyalgia rheumatica, dermatomyositis and polymyositis do not generally feature severe muscle pain, but they can be associated with a painful polyarthritis. Oral ulcers, photosensitivity, and facial rash are consistent with SLE, but dermatomyositis can also lead to a symmetrical erythema of the eyelids (commonly referred to as a heliotrope rash, named after the flower bearing that name) and sometimes can be associated with photosensitivity. Oral ulcers, particularly the painful ones known as canker sores, are extraordinarily common in the general population, and patients and providers may miss the mucosal lesions of SLE because they are usually painless. As methotrexate and etanercept are immunosuppressive, opportunistic pathogens such as typical or atypical mycobacteria and disseminated fungal infections should be considered, with special attention to the possibility of infection in or near the left hip. Given that SLE and RA rarely coexist, it would be helpful to seek outside medical records to know what the prior serologic evaluation entailed, but it is unlikely that this presentation is a manifestation of a diffuse connective tissue process.
Physical examination should focus on the features of dermatomyositis including heliotrope rash, truncal erythema, and papules over the knuckles (Gottron's papules); objective proximal muscle weakness in the shoulder and hip girdle; and findings that might suggest antisynthetase syndrome such as hyperkeratotic mechanic hand palmar and digital changes, and interstitial crackles on lung exam. If necrotic skin lesions are found, this would raise concern for a disseminated infection. The joints should be examined for inflammation and effusions.
His temperature was 36.6C, heart rate 74 beats per minute, blood pressure 134/76 mm Hg, respiratory rate 16 breaths per minute, and O2 saturation 97% on room air. He was obese but did not have moon facies or a buffalo hump. There were no rashes or mucosal lesions. Active and passive motion of his left hip joint elicited pain with both flexion/extension and internal/external rotation. Muscle strength was limited by pain in the left hip flexors and extenders, but was 5/5 in all other muscle groups. Palpation of the proximal muscles of his arms and legs did not elicit pain. His extremities were without edema, and examination of his shoulders, elbows, wrists, hands, knees, ankles, and feet did not reveal any erythema, synovial thickening, effusion, or deformity. Examination of the heart, chest, and abdomen was normal.
Given the reassuring strength examination, the absence of rashes or skin lesions, and the reassuring joint exam aside from the left hip, a focal infectious, inflammatory, or malignant process seems most likely. The pain with range of motion of the hip does not definitively localize the pathology to the hip joint, because pathology of the nearby structures can lead to pain when the hip is moved. Laboratory evaluation should include a complete blood count to screen for evidence of infection or marrow suppression, complete metabolic panel, and creatine kinase. The history of ulcerative colitis raises the possibility of an enthesitis (inflammation of tendons or ligaments) occurring near the hip. Enthesitis is sometimes a feature of the seronegative spondyloarthropathy‐associated conditions and can occur in the absence of sacroiliitis or spondyloarthropathy.
The patient's myalgias and arthralgias had recently been evaluated in the rheumatology clinic. Laboratory evaluation from that visit was remarkable only for an antinuclear antibody (ANA) test that was positive at a titer of 1:320 in a homogeneous pattern, creatine phosphokinase 366 IU/L (normal range [NR] 38240), and alkaline phosphatase 203 IU/L (NR 30130). All of the following labs from that visit were within normal ranges: cyclic citrullinated peptide, rheumatoid factor, antidouble stranded DNA, aldolase, complement levels, serum and urine protein electrophoresis, thyroglobulin antibody, thyroid microsomal antibody, thyroid‐stimulating hormone, erythrocyte sedimentation rate (10 mm/h), and C‐reactive protein (0.3 mg/dL).
The patient was admitted to the hospital. Initial blood test results on admission included sodium 139 mEq/L, potassium 3.9 mEq/L, chloride 105 mEq/L, bicarbonate 27 mEq/L, urea nitrogen 16 mg/dL, creatinine 0.6 mg/dL, glucose 85 mg/dL, calcium 9.2 mg/dL (NR 8.810.3), phosphate 1.3 mg/dL (NR 2.74.6), albumin 4.7 g/dL (NR 3.54.9), and alkaline phosphatase 195 IU/L (NR 30130). The remainder of a comprehensive metabolic profile, complete blood count with differential, and coagulation studies were all normal.
The homogeneous ANA titer of 1:320 is high enough to raise eyebrows, but is nonspecific for lupus and other ANA‐associated rheumatologic conditions, and may be a red herring, particularly given the low likelihood of a systemic inflammatory process explaining this new focal hip pain. The alkaline phosphatase is only mildly elevated and could be of bone or liver origin. The reassuringly low inflammatory markers are potentially helpful, because if checked now and substantially increased from the prior outpatient visit, they would be suggestive of a new inflammatory process. However, this would not point to a specific cause of inflammation.
Given the focality of the symptoms, imaging is warranted. As opposed to plain films, contrast‐enhanced computed tomography (CT) of the pelvis or magnetic resonance imaging (MRI) may be an efficient first step, because there is low suspicion for fracture and high suspicion for an inflammatory, neoplastic, or infectious process. MRI is more expensive and usually cannot be obtained as rapidly as CT. There is a chance that CT imaging alone will provide enough information to guide the next diagnostic steps, such as aspiration of an abscess or joint, or biopsy of a suspicious lesion. However, for soft tissue lesions and many bone lesions, including osteomyelitis, MRI offers better delineation of pathology.
CT scan of the left femur demonstrated a large lytic lesion in the femoral neck that contained macroscopic fat and had an aggressive appearance with significant thinning of the cortex. MRI confirmed these findings and demonstrated a nondisplaced subtrochanteric femur fracture in the proximity of the lesion (Figure 1). Contrast‐enhanced CT scans of the thorax, abdomen, and pelvis revealed no other neoplastic lesions. Prostate‐specific antigen level was normal. The patient's significant hypophosphatemia persisted, with levels dropping to as low as 0.9 mg/dL despite aggressive oral phosphate replacement.
Although hypophosphatemia is often a nonspecific finding in hospitalized patients and is usually of little clinical importance, profound hypophosphatemia that is refractory to supplementation suggests an underlying metabolic disorder. Phosphate levels less than 1.0 mg/dL, particularly if prolonged, can lead to decreased adenosine triphosphate production and subsequent weakness of respiratory and cardiac muscles. Parathyroid hormone (PTH) excess and production of parathyroid hormone‐related protein (PTHrP) by a malignancy can cause profound hypophosphatemia, but are generally associated with hypercalcemia, a finding not seen in this case. Occasionally, tumors can lead to renal phosphate wasting via nonPTH‐related mechanisms. The best characterized example of this is the paraneoplastic syndrome oncogenic osteomalacia caused by tumor production of a fibroblast growth factor. Tumors that lead to this syndrome are usually benign mesenchymal tumors. This patient's tumor may be of this type, causing local destruction and metabolic disturbance. The next step would be consultation with orthopedic surgery for resection of the tumor and total hip arthroplasty with aggressive perioperative repletion of phosphate. Assessment of intact PTH, ionized calcium, 24‐hour urinary phosphate excretion, and even PTHrP levels may help to rule out other etiologies of hypophosphatemia, but given that surgery is needed regardless, it might be reasonable to proceed to the operating room without these diagnostics. If the phosphate levels return to normal postoperatively, then the diagnosis is clear and no further metabolic testing is needed.
PTH level was 47 pg/mL (NR 1065), 25‐hydroxyvitamin D level was 25 ng/mL (NR 2580), and 1,25‐dihydroxyvitamin D level was 18 pg/mL (NR 1872). Urinalysis was normal without proteinuria or glucosuria. A 24‐hour urine collection contained 1936 mg of phosphate (NR 4001200). The ratio of maximum rate of renal tubular reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.3 mg/dL (NR 2.44.2). Tissue obtained by CT‐guided needle biopsy of the femoral mass was consistent with a benign spindle cell neoplasm.
With normal calcium levels, the PTH level is appropriate, and hyperparathyroidism is excluded. The levels of 25‐hydroxyvitamin D and 1‐25‐dihydroxyvitamin D are not low enough to suggest that vitamin D deficiency is driving the impressive hypophosphatemia. What is impressive is the phosphate wasting demonstrated by the 24‐hour urine collection, consistent with paraneoplastic overproduction of fibroblast growth factor 23 (FGF23) by the benign bone tumor. Overproduction of this protein can be detected by blood tests or staining of the tumor specimen, but surgery should be performed as soon as possible independent of any further test results. Once the tumor is resected, phosphate metabolism should normalize.
FGF23 level was 266 RU/mL (NR 180). The patient was diagnosed with tumor‐induced osteomalacia (TIO). He underwent complete resection of the femoral tumor as well as open reduction and internal fixation of the fracture. After surgery, his symptoms of pain and subjective muscle weakness improved, his serum phosphate level normalized, his need for phosphate supplementation resolved, and his blood levels of FGF23 decreased into the normal range (111 RU/mL). The rapid improvement of his symptoms after surgery suggested that they were related to TIO, and not manifestations of SLE or RA. His immunosuppressant medications were discontinued. Surgical pathology demonstrated a heterogeneous tumor consisting of sheets of uniform spindle cells interspersed with mature adipose tissue. This was diagnosed descriptively as a benign spindle cell and lipomatous neoplasm without further classification. Two months later, the patient was ambulating without pain, and muscle strength was subjectively normal.
DISCUSSION
TIO is a rare paraneoplastic syndrome affecting phosphate and vitamin D metabolism, leading to hypophosphatemia and osteomalacia.[1] TIO is caused by the inappropriate tumor secretion of the phosphatonin hormone, FGF23.
The normal physiology of FGF23 is illustrated in Figure 2. Osteocytes appear to be the primary source of FGF23, but the regulation of FGF23 production is not completely understood. FGF23 production may be influenced by several factors, including 1,25 dihydroxyvitamin D levels, and serum phosphate and PTH concentrations. This hormone binds to the FGF receptor and its coreceptor, Klotho,[2] causing 2 major physiological effects. First, it decreases the expression of the sodium‐phosphate cotransporters in the renal proximal tubular cells,[3, 4] resulting in increased tubular phosphate wasting. This effect appears to be partly PTH dependent.[5] Second, it has effects on vitamin D metabolism, decreasing renal production of activated vitamin D.[3, 4, 6]
In overproduction states, the elevated FGF23 leads to chronically low serum phosphate levels (with renal phosphate wasting) and the clinical syndrome of osteomalacia, manifested by bone pain, fractures, and deformities. Hypophosphatemia can also lead to painful proximal myopathy, cardiorespiratory dysfunction, and a spectrum of neuropsychiatric findings. The clinical findings in TIO are similar to those seen in genetic diseases in which hypophosphatemia results from the same mechanism.[3, 4]
In this case, measurement of the serum phosphate level was important in reaching the diagnosis. Although hypophosphatemia in the hospitalized patient is often easily explained, severe or persistent hypophosphatemia requires a focused evaluation. Causes of hypophosphatemia are categorized in Table 1.[7, 8, 9] In patients with hypophosphatemia that is not explained by the clinical situation (eg, osmotic diuresis, insulin treatment, refeeding syndrome, postparathyroidectomy, chronic diarrhea), measurement of serum calcium, PTH, and 25‐hydroxyvitamin D are used to investigate possible primary or secondary hyperparathyroidism. In addition, low‐normal or low serum 1,25‐dihydroxyvitamin D with normal PTH, normal 25‐hydroxyvitamin D stores, and normal renal function are clues to the presence of TIO. Urine phosphate wasting can be measured by collecting a 24‐hour urine sample. Calculation of the TmP/GFR (a measure of the maximum tubular resorption of phosphate relative to the glomerular filtration rate), as described by the nomogram of Walton and Bijvoet, may improve the accuracy of this assessment and confirm a renal source of the hypophosphatemia.[10]
|
| Internal redistribution |
| Insulin or catecholamine effect (including that related to refeeding syndrome, and infusion of glucose or TPN) |
| Acute respiratory alkalosis |
| Accelerated bone formation or rapid cell proliferation (eg, hungry bone syndrome, leukemic blast crisis, erythropoietin, or granulocyte colony stimulating factor administration) |
| Decreased absorption |
| Poor intake (including that seen in alcoholism*) |
| Vitamin D deficiency |
| Gastrointestinal losses (eg, chronic diarrhea) |
| Malabsorption (eg, phosphate‐binding antacids) |
| Urinary losses |
| Osmotic diuresis (eg, poorly controlled diabetes, acetazolamide) or volume expansion |
| Other diuretics: thiazides, indapamide |
| Hyperparathyroidism |
| Primary |
| Secondary (including vitamin D or calcium deficiency) |
| Parathyroid hormone‐related peptide |
| Renal tubular disease |
| Medications (eg, ethanol,* high‐dose glucocorticoids, cisplatin, bisphosphonates, estrogens, imatinib, acyclovir) |
| Fanconi syndrome |
| Medications inducing Fanconi syndrome: tenofovir, cidofovir, adefovir, aminoglycosides, ifosfamide, tetracyclines, valproic acid |
| Other (eg, postrenal transplant) |
| Excessive phosphatonin hormone activity (eg, hereditary syndromes [rickets], tumor‐induced osteomalacia) |
| Multifactorial causes |
| Alcoholism* |
| Acetaminophen toxicity |
| Parenteral iron administration |
The patient presented here had inappropriate urinary phosphate losses, and laboratory testing ruled out primary and secondary hyperparathyroidism and Fanconi syndrome. The patient was not taking medications known to cause tubular phosphate wasting. The patient's age and clinical history made hereditary syndromes unlikely. Therefore, the urinary phosphate wasting had to be related to an acquired defect in phosphate metabolism. The diagnostic characteristics of TIO are summarized in Table 2.
|
| Patients may present with symptoms of osteomalacia (eg, bone pain, fractures), hypophosphatemia (eg, proximal myopathy), and/or neoplasm. |
| Hypophosphatemia with urinary phosphate wasting. |
| Serum calcium level is usually normal. |
| Serum 1,25‐dihydroxyvitamin D level is usually low or low‐normal. |
| Parathyroid hormone is usually normal. |
| Plasma FGF23 level is elevated. |
| A neoplasm with the appropriate histology is identified, although the osteomalacia syndrome may precede identification of the tumor, which may be occult. |
| The syndrome resolves after complete resection of the tumor. |
The presence of a known neoplasm makes the diagnosis of TIO considerably easier. However, osteomalacia often precedes the tumor diagnosis. In these cases, the discovery of this clinical syndrome necessitates a search for the tumor. The tumors can be small, occult, and often located in the extremities. In addition to standard cross‐sectional imaging, specialized diagnostic modalities can be helpful in localizing culprit tumors. These include F‐18 flourodeoxyglucose positron emission tomography with computed tomography, 111‐Indium octreotide single photon emission CT/CT, 68‐Gallium‐DOTA‐octreotide positron emission tomography with computed tomography, and even selective venous sampling for FGF23 levels.[1, 11] The octreotide tests capitalize on the fact that culprit tumors often express somatostatin receptors.
TIO is most often associated with mesenchymal tumors of the bone or soft tissue. It has also been reported in association with several malignancies (small cell carcinoma, hematologic malignancies, prostate cancer), and with polyostotic fibrous dysplasia, neurofibromatosis, and the epidermal nevus syndrome. The mesenchymal tumors are heterogeneous in appearance and can be variably classified as hemangiopericytomas, hemangiomas, sarcomas, ossifying fibromas, granulomas, giant cell tumors, or osteoblastomas.[1] However, 1 review suggests that most of these tumors actually represent a distinct but heterogeneous, under‐recognized entity that is best classified as a phosphaturic mesenchymal tumor.[11]
TIO is only cured by complete resection of the tumor.[1] Local recurrences have been described, as have rare occurrences of metastatic disease.[1, 12] Medical treatment can be used to normalize serum phosphate levels in patients who are unable to be cured by surgery. The goal is to bring serum phosphate into the low‐normal range via phosphate supplementation (typically 13 g/day of elemental phosphorus is required) and treatment with either calcitriol or alfacalcidol. Due to the inhibition of 1,25‐dihydroxyvitamin D activation in TIO, relatively large doses of calcitriol are needed. A reasonable starting dose of calcitriol is 1.5 g/day, and most patients require 15 to 60 ng/kg per day. Because PTH action is involved in FGF23‐mediated hypophosphatemia, suppression of PTH may also be useful in these patients.[13]
This patient presented with a painful femoral tumor in the setting of muscle and joint pain that had been erroneously attributed to connective tissue disease. However, recognition and thorough evaluation of the patient's hypophosphatemia led to a unifying diagnosis of TIO. This diagnosis altered the surgical approach (emphasizing complete resection to eradicate the FGF23 production) and helped alleviate the patient's painful losses of phosphate.
TEACHING POINTS
- Hypophosphatemia, especially if severe or persistent, should not be dismissed as an unimportant laboratory finding. A focused evaluation should be performed to determine the etiology.
- In patients with unexplained hypophosphatemia, the measurement of serum calcium, parathyroid hormone, and vitamin D levels can identify primary or secondary hyperparathyroidism.
- The differential diagnosis of hypophosphatemia is narrowed if there is clinical evidence of inappropriate urinary phosphate wasting (ie, urinary phosphate levels remain high, despite low serum levels).
- TIO is a rare paraneoplastic syndrome caused by FGF23, a phosphatonin hormone that causes renal phosphate wasting, hypophosphatemia, and osteomalacia.
Disclosure: Nothing to report.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.
- , , , . Tumor‐induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–R77.
- . The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619.
- , . Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399–2409.
- . The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9.
- , , , , . FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492.
- , , , et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
- , . Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
- , , . In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: Elsevier; 2011:1237–1304.
- , , . Medication‐induced hypophosphatemia: a review. QJM. 2010;103:449–459.
- , . Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
- , , , et al. Improving diagnosis of tumor‐induced osteomalacia with gallium‐68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013; 98:687–694.
- , , , et al. Most osteomalacia‐associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
- , , , , , . Cinacalcet in the management of tumor‐induced osteomalacia. J Bone Miner Res. 2007;22:931–937.