User login
Richard Bogan, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Lifetime Achievement Award Presented
The Society for Vascular Surgery’s highest honor, the Lifetime Achievement Award, was presented during the Vascular Annual Meeting to Dr. Jack Cronenwett.
This honor recognizes an individual’s outstanding and sustained contributions to the profession and SVS, as well as his or her exemplary professional practice and leadership.
Dr. Cronenwett’s achievements include building the vascular surgery division at Dartmouth-Hitchcock Center into a “powerhouse, a division of vascular surgery that has national and international recognition,” as one fellow SVS member wrote in his nomination. In addition, he was largely responsible for the development at Dartmouth of a new vascular surgery training paradigm – the 0 + 5 residency. He spearheaded the Vascular Study Group of New England, which became the model for the future Vascular Quality Initiative; with Dr. Tom Riles, then president of the American Association for Vascular Surgery, led the merger of AAVS with SVS, creating today’s more inclusive SVS; was senior co-editor of the Journal of Vascular Surgery, was co-editor of the 7th and 8th editions of Rutherford’s Vascular Surgery textbook and helped launch the SVS-PSO as an official new entity and served as its medical director.
View the video of him receiving this award and a story of Dr. Cronenwett’s accomplishments.
The Society for Vascular Surgery’s highest honor, the Lifetime Achievement Award, was presented during the Vascular Annual Meeting to Dr. Jack Cronenwett.
This honor recognizes an individual’s outstanding and sustained contributions to the profession and SVS, as well as his or her exemplary professional practice and leadership.
Dr. Cronenwett’s achievements include building the vascular surgery division at Dartmouth-Hitchcock Center into a “powerhouse, a division of vascular surgery that has national and international recognition,” as one fellow SVS member wrote in his nomination. In addition, he was largely responsible for the development at Dartmouth of a new vascular surgery training paradigm – the 0 + 5 residency. He spearheaded the Vascular Study Group of New England, which became the model for the future Vascular Quality Initiative; with Dr. Tom Riles, then president of the American Association for Vascular Surgery, led the merger of AAVS with SVS, creating today’s more inclusive SVS; was senior co-editor of the Journal of Vascular Surgery, was co-editor of the 7th and 8th editions of Rutherford’s Vascular Surgery textbook and helped launch the SVS-PSO as an official new entity and served as its medical director.
View the video of him receiving this award and a story of Dr. Cronenwett’s accomplishments.
The Society for Vascular Surgery’s highest honor, the Lifetime Achievement Award, was presented during the Vascular Annual Meeting to Dr. Jack Cronenwett.
This honor recognizes an individual’s outstanding and sustained contributions to the profession and SVS, as well as his or her exemplary professional practice and leadership.
Dr. Cronenwett’s achievements include building the vascular surgery division at Dartmouth-Hitchcock Center into a “powerhouse, a division of vascular surgery that has national and international recognition,” as one fellow SVS member wrote in his nomination. In addition, he was largely responsible for the development at Dartmouth of a new vascular surgery training paradigm – the 0 + 5 residency. He spearheaded the Vascular Study Group of New England, which became the model for the future Vascular Quality Initiative; with Dr. Tom Riles, then president of the American Association for Vascular Surgery, led the merger of AAVS with SVS, creating today’s more inclusive SVS; was senior co-editor of the Journal of Vascular Surgery, was co-editor of the 7th and 8th editions of Rutherford’s Vascular Surgery textbook and helped launch the SVS-PSO as an official new entity and served as its medical director.
View the video of him receiving this award and a story of Dr. Cronenwett’s accomplishments.

Bone Ache and Stiffness
1. This patient presented with rapid onset of severe pain in the MTP joint of the great toe accompanied swelling, redness, and fever. The pain was most severe in the first two to 24 hours and peaked within 24 hours. The history was remarkable for a trauma to the toe and a first-degree relative who had experienced the same symptoms. The patient admitted to drinking alcohol regularly.
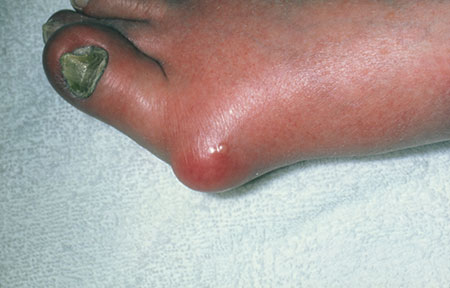
Diagnosis: A presumptive diagnosis of gout may be acceptable in a patient with the classic presentation: rapid onset of severe pain in a swollen, erythematous joint (classically described in Latin as calor, rubor, dolor, et tumor) and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout. In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.
For more information, see “Gout: A Clinical Overview.” Clin Rev. 2011;21(7):29-35.
For the next photograph, proceed to the next page >>
2. A 75-year-old woman presented to her family physician (FP) with pain in both hands. She said that she’d had the pain for a year and that it got worse with cold weather and better with ibuprofen.

Diagnosis: Osteoarthritis (OA) was diagnosed after noting Heberden’s nodes and Bouchard’s nodes at the distal and proximal interphalangeal joints, respectively. Arthritis is the most common cause of disability in the United States. Twenty-one million adults have functional limitations because of arthritis. Fifty percent of adults ages 65 years or older have been given an arthritis diagnosis. The most commonly affected joints are the knees, hips, hands (distal and proximal interphalangeal joints), and spine. If the diagnosis is uncertain, plain x-rays can often differentiate between OA and other forms of arthritis.
For more information, read “Hand pain.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
3. A 45-year-old man with longstanding psoriasis began experiencing finger pain and swelling. The patient, who had a history of hepatitis C, noted that his fingernails had recently begun crumbling and his fingers were stiff in the morning.
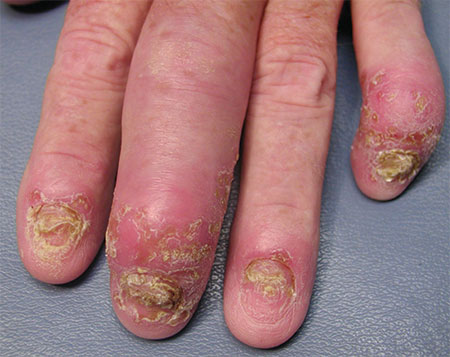
Diagnosis: Psoriatic arthritis (with dactylitis and significant distal interphalangeal joint [DIP] involvement) was diagnosed in both hands, but the left hand was worse. The condition of the patient’s nails was not surprising, given that almost all patients with psoriatic arthritis have nail involvement. Radiographs of the patient’s hands showed periarticular erosions and new bone formation. There was also telescoping of the third DIP joint.
For more information, read “Finger pain and swelling.” J Fam Pract. 2015.
Photo courtesy of Ricardo Zuniga-Montes, MD; text courtesy of Richard P. Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
4. A 62-year-old woman was concerned about her fingers, which weren’t straight. She’d had increasing pain in both hands (specifically the metacarpophalangeal [MCP] joints) over the past 2 years. Her fingers were stiff in the morning, but she was able to gain some relief with ibuprofen and naproxen. On physical exam, her fingers had an ulnar deviation and there was swelling of the MCP joints on both hands.
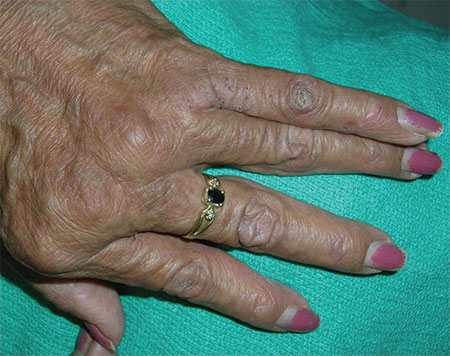
Diagnosis: Rheumatoid arthritis (RA) was diagnosed, based on the patient’s bilateral MCP joint swelling, ulnar deviation, and morning stiffness. Patients with RA initially experience swelling and stiffness in their wrists, as well as their MCP and metatarsophalangeal joints. Later, the larger joints are affected. When RA is advanced, severe destruction and subluxation occur.
“Crooked fingers.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
1. This patient presented with rapid onset of severe pain in the MTP joint of the great toe accompanied swelling, redness, and fever. The pain was most severe in the first two to 24 hours and peaked within 24 hours. The history was remarkable for a trauma to the toe and a first-degree relative who had experienced the same symptoms. The patient admitted to drinking alcohol regularly.
Diagnosis: A presumptive diagnosis of gout may be acceptable in a patient with the classic presentation: rapid onset of severe pain in a swollen, erythematous joint (classically described in Latin as calor, rubor, dolor, et tumor) and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout. In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.
For more information, see “Gout: A Clinical Overview.” Clin Rev. 2011;21(7):29-35.
For the next photograph, proceed to the next page >>
2. A 75-year-old woman presented to her family physician (FP) with pain in both hands. She said that she’d had the pain for a year and that it got worse with cold weather and better with ibuprofen.
Diagnosis: Osteoarthritis (OA) was diagnosed after noting Heberden’s nodes and Bouchard’s nodes at the distal and proximal interphalangeal joints, respectively. Arthritis is the most common cause of disability in the United States. Twenty-one million adults have functional limitations because of arthritis. Fifty percent of adults ages 65 years or older have been given an arthritis diagnosis. The most commonly affected joints are the knees, hips, hands (distal and proximal interphalangeal joints), and spine. If the diagnosis is uncertain, plain x-rays can often differentiate between OA and other forms of arthritis.
For more information, read “Hand pain.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
3. A 45-year-old man with longstanding psoriasis began experiencing finger pain and swelling. The patient, who had a history of hepatitis C, noted that his fingernails had recently begun crumbling and his fingers were stiff in the morning.
Diagnosis: Psoriatic arthritis (with dactylitis and significant distal interphalangeal joint [DIP] involvement) was diagnosed in both hands, but the left hand was worse. The condition of the patient’s nails was not surprising, given that almost all patients with psoriatic arthritis have nail involvement. Radiographs of the patient’s hands showed periarticular erosions and new bone formation. There was also telescoping of the third DIP joint.
For more information, read “Finger pain and swelling.” J Fam Pract. 2015.
Photo courtesy of Ricardo Zuniga-Montes, MD; text courtesy of Richard P. Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
4. A 62-year-old woman was concerned about her fingers, which weren’t straight. She’d had increasing pain in both hands (specifically the metacarpophalangeal [MCP] joints) over the past 2 years. Her fingers were stiff in the morning, but she was able to gain some relief with ibuprofen and naproxen. On physical exam, her fingers had an ulnar deviation and there was swelling of the MCP joints on both hands.
Diagnosis: Rheumatoid arthritis (RA) was diagnosed, based on the patient’s bilateral MCP joint swelling, ulnar deviation, and morning stiffness. Patients with RA initially experience swelling and stiffness in their wrists, as well as their MCP and metatarsophalangeal joints. Later, the larger joints are affected. When RA is advanced, severe destruction and subluxation occur.
“Crooked fingers.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
1. This patient presented with rapid onset of severe pain in the MTP joint of the great toe accompanied swelling, redness, and fever. The pain was most severe in the first two to 24 hours and peaked within 24 hours. The history was remarkable for a trauma to the toe and a first-degree relative who had experienced the same symptoms. The patient admitted to drinking alcohol regularly.
Diagnosis: A presumptive diagnosis of gout may be acceptable in a patient with the classic presentation: rapid onset of severe pain in a swollen, erythematous joint (classically described in Latin as calor, rubor, dolor, et tumor) and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout. In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.
For more information, see “Gout: A Clinical Overview.” Clin Rev. 2011;21(7):29-35.
For the next photograph, proceed to the next page >>
2. A 75-year-old woman presented to her family physician (FP) with pain in both hands. She said that she’d had the pain for a year and that it got worse with cold weather and better with ibuprofen.
Diagnosis: Osteoarthritis (OA) was diagnosed after noting Heberden’s nodes and Bouchard’s nodes at the distal and proximal interphalangeal joints, respectively. Arthritis is the most common cause of disability in the United States. Twenty-one million adults have functional limitations because of arthritis. Fifty percent of adults ages 65 years or older have been given an arthritis diagnosis. The most commonly affected joints are the knees, hips, hands (distal and proximal interphalangeal joints), and spine. If the diagnosis is uncertain, plain x-rays can often differentiate between OA and other forms of arthritis.
For more information, read “Hand pain.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
3. A 45-year-old man with longstanding psoriasis began experiencing finger pain and swelling. The patient, who had a history of hepatitis C, noted that his fingernails had recently begun crumbling and his fingers were stiff in the morning.
Diagnosis: Psoriatic arthritis (with dactylitis and significant distal interphalangeal joint [DIP] involvement) was diagnosed in both hands, but the left hand was worse. The condition of the patient’s nails was not surprising, given that almost all patients with psoriatic arthritis have nail involvement. Radiographs of the patient’s hands showed periarticular erosions and new bone formation. There was also telescoping of the third DIP joint.
For more information, read “Finger pain and swelling.” J Fam Pract. 2015.
Photo courtesy of Ricardo Zuniga-Montes, MD; text courtesy of Richard P. Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
For the next photograph, proceed to the next page >>
4. A 62-year-old woman was concerned about her fingers, which weren’t straight. She’d had increasing pain in both hands (specifically the metacarpophalangeal [MCP] joints) over the past 2 years. Her fingers were stiff in the morning, but she was able to gain some relief with ibuprofen and naproxen. On physical exam, her fingers had an ulnar deviation and there was swelling of the MCP joints on both hands.
Diagnosis: Rheumatoid arthritis (RA) was diagnosed, based on the patient’s bilateral MCP joint swelling, ulnar deviation, and morning stiffness. Patients with RA initially experience swelling and stiffness in their wrists, as well as their MCP and metatarsophalangeal joints. Later, the larger joints are affected. When RA is advanced, severe destruction and subluxation occur.
“Crooked fingers.” J Fam Pract. 2015.
Photo courtesy of Dr. Richard Usatine, MD. The case was adapted with permission from Chumley H, Usatine R. Arthritis overview. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:562-568.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Lisinopril tied to 19% drop in conduction system disease
Compared with chlorthalidone, the angiotensin-converting enzyme inhibitor lisinopril was associated with a 19% drop in new-onset conduction system disease among high-risk patients with hypertension, based on a secondary analysis of the randomized, double-blind Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT).
Amlodipine besylate and pravastatin did not affect rates of conduction defects, added Dr. Thomas A. Dewland of Oregon Health and Science University, Portland, and his associates, who reported the findings online June 27 in JAMA Internal Medicine.
ALLHAT was a randomized, double-blind trial comparing chlorthalidone, amlodipine, lisinopril, or doxazosin mesylate in patients with hypertension and at least one other cardiac risk factor. Patients with elevated fasting low-density lipoprotein levels also were randomized to pravastatin sodium or usual care. Most (56%) patients were men, and the average age was about 67 years (standard deviation, 7.3 years). Twelve-lead electrocardiograms were obtained every 2 years, enabling the current analysis, the researchers said. In all, 21,004 patients without a baseline pacemaker or conduction system disease were followed for an average of 5 years (JAMA Intern Med. 2016 Jun 27. doi: 10.1001/jamainternmed.2016.250).
A total of 1,114 patients developed new-onset conduction system disease (3 events per 1,000 person-years), including 389 patients with left bundle branch block (4.5 events per 1,000 person-years), 570 patients with right bundle branch block (6.6 events per 1,000 person-years), and 155 patients with intraventricular conduction delay. The risk of conduction system disease was 19% lower with lisinopril than chlorthalidone (hazard ratio, 0.81; 95% confidence interval, 0.69-0.95; P = .01). Neither amlodipine nor pravastatin significantly affected the chances of conduction system disease, but patients who were older, white, or had diabetes or left ventricular hypertrophy were at significantly greater risk than the overall cohort.
“The antifibrotic effects of angiotensin-converting enzyme inhibitors may prevent or slow the development of conduction system disease, and future research is warranted to understand whether this treatment affects other conduction-related clinical outcomes,” including the need for pacemaker implantation, the researchers concluded.
The study was supported by the American Heart Association, the Joseph Drown Foundation, the National Heart, Lung, and Blood Institute of the National Institutes of Health, and by Pfizer. The investigators had no disclosures.
To our knowledge, this is the first report that incident conduction system disease may be prevented by drug therapy, in this case lisinopril. This important observation is consistent with established knowledge that angiotensin-converting enzyme inhibition has salutary effects on inflammation, hypertrophy, and fibrosis. We look forward to future studies to confirm that ECG conduction disease can indeed be prevented or progression slowed by relatively simple interventions, to define those individuals most likely to benefit, and to assess whether such preventive strategies can indeed ward off the need for invasive procedures such as pacemakers in selected patient subsets. This is indeed an exciting prospect!
Dr. Sanjiv M. Narayan, Dr. Tina Baykaner, and Dr. David J. Maron are at Falk Cardiovascular Research Center, Stanford University, Palo Alto, California. Dr. Narayan reported receiving research support from the National Institutes of Health; being coinventor of intellectual property owned by the University of California and licensed to Topera, in which he held equity; and receiving consulting fees from the American College of Cardiology and Uptodate.com and speaking fees from St. Jude Medical and Medtronic. The other authors had no disclosures. These comments are from their editorial (JAMA Intern Med. 2016 Jun 27. doi: 10.1001 /jamainternmed.2016.2502).
To our knowledge, this is the first report that incident conduction system disease may be prevented by drug therapy, in this case lisinopril. This important observation is consistent with established knowledge that angiotensin-converting enzyme inhibition has salutary effects on inflammation, hypertrophy, and fibrosis. We look forward to future studies to confirm that ECG conduction disease can indeed be prevented or progression slowed by relatively simple interventions, to define those individuals most likely to benefit, and to assess whether such preventive strategies can indeed ward off the need for invasive procedures such as pacemakers in selected patient subsets. This is indeed an exciting prospect!
Dr. Sanjiv M. Narayan, Dr. Tina Baykaner, and Dr. David J. Maron are at Falk Cardiovascular Research Center, Stanford University, Palo Alto, California. Dr. Narayan reported receiving research support from the National Institutes of Health; being coinventor of intellectual property owned by the University of California and licensed to Topera, in which he held equity; and receiving consulting fees from the American College of Cardiology and Uptodate.com and speaking fees from St. Jude Medical and Medtronic. The other authors had no disclosures. These comments are from their editorial (JAMA Intern Med. 2016 Jun 27. doi: 10.1001 /jamainternmed.2016.2502).
To our knowledge, this is the first report that incident conduction system disease may be prevented by drug therapy, in this case lisinopril. This important observation is consistent with established knowledge that angiotensin-converting enzyme inhibition has salutary effects on inflammation, hypertrophy, and fibrosis. We look forward to future studies to confirm that ECG conduction disease can indeed be prevented or progression slowed by relatively simple interventions, to define those individuals most likely to benefit, and to assess whether such preventive strategies can indeed ward off the need for invasive procedures such as pacemakers in selected patient subsets. This is indeed an exciting prospect!
Dr. Sanjiv M. Narayan, Dr. Tina Baykaner, and Dr. David J. Maron are at Falk Cardiovascular Research Center, Stanford University, Palo Alto, California. Dr. Narayan reported receiving research support from the National Institutes of Health; being coinventor of intellectual property owned by the University of California and licensed to Topera, in which he held equity; and receiving consulting fees from the American College of Cardiology and Uptodate.com and speaking fees from St. Jude Medical and Medtronic. The other authors had no disclosures. These comments are from their editorial (JAMA Intern Med. 2016 Jun 27. doi: 10.1001 /jamainternmed.2016.2502).
Compared with chlorthalidone, the angiotensin-converting enzyme inhibitor lisinopril was associated with a 19% drop in new-onset conduction system disease among high-risk patients with hypertension, based on a secondary analysis of the randomized, double-blind Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT).
Amlodipine besylate and pravastatin did not affect rates of conduction defects, added Dr. Thomas A. Dewland of Oregon Health and Science University, Portland, and his associates, who reported the findings online June 27 in JAMA Internal Medicine.
ALLHAT was a randomized, double-blind trial comparing chlorthalidone, amlodipine, lisinopril, or doxazosin mesylate in patients with hypertension and at least one other cardiac risk factor. Patients with elevated fasting low-density lipoprotein levels also were randomized to pravastatin sodium or usual care. Most (56%) patients were men, and the average age was about 67 years (standard deviation, 7.3 years). Twelve-lead electrocardiograms were obtained every 2 years, enabling the current analysis, the researchers said. In all, 21,004 patients without a baseline pacemaker or conduction system disease were followed for an average of 5 years (JAMA Intern Med. 2016 Jun 27. doi: 10.1001/jamainternmed.2016.250).
A total of 1,114 patients developed new-onset conduction system disease (3 events per 1,000 person-years), including 389 patients with left bundle branch block (4.5 events per 1,000 person-years), 570 patients with right bundle branch block (6.6 events per 1,000 person-years), and 155 patients with intraventricular conduction delay. The risk of conduction system disease was 19% lower with lisinopril than chlorthalidone (hazard ratio, 0.81; 95% confidence interval, 0.69-0.95; P = .01). Neither amlodipine nor pravastatin significantly affected the chances of conduction system disease, but patients who were older, white, or had diabetes or left ventricular hypertrophy were at significantly greater risk than the overall cohort.
“The antifibrotic effects of angiotensin-converting enzyme inhibitors may prevent or slow the development of conduction system disease, and future research is warranted to understand whether this treatment affects other conduction-related clinical outcomes,” including the need for pacemaker implantation, the researchers concluded.
The study was supported by the American Heart Association, the Joseph Drown Foundation, the National Heart, Lung, and Blood Institute of the National Institutes of Health, and by Pfizer. The investigators had no disclosures.
Compared with chlorthalidone, the angiotensin-converting enzyme inhibitor lisinopril was associated with a 19% drop in new-onset conduction system disease among high-risk patients with hypertension, based on a secondary analysis of the randomized, double-blind Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT).
Amlodipine besylate and pravastatin did not affect rates of conduction defects, added Dr. Thomas A. Dewland of Oregon Health and Science University, Portland, and his associates, who reported the findings online June 27 in JAMA Internal Medicine.
ALLHAT was a randomized, double-blind trial comparing chlorthalidone, amlodipine, lisinopril, or doxazosin mesylate in patients with hypertension and at least one other cardiac risk factor. Patients with elevated fasting low-density lipoprotein levels also were randomized to pravastatin sodium or usual care. Most (56%) patients were men, and the average age was about 67 years (standard deviation, 7.3 years). Twelve-lead electrocardiograms were obtained every 2 years, enabling the current analysis, the researchers said. In all, 21,004 patients without a baseline pacemaker or conduction system disease were followed for an average of 5 years (JAMA Intern Med. 2016 Jun 27. doi: 10.1001/jamainternmed.2016.250).
A total of 1,114 patients developed new-onset conduction system disease (3 events per 1,000 person-years), including 389 patients with left bundle branch block (4.5 events per 1,000 person-years), 570 patients with right bundle branch block (6.6 events per 1,000 person-years), and 155 patients with intraventricular conduction delay. The risk of conduction system disease was 19% lower with lisinopril than chlorthalidone (hazard ratio, 0.81; 95% confidence interval, 0.69-0.95; P = .01). Neither amlodipine nor pravastatin significantly affected the chances of conduction system disease, but patients who were older, white, or had diabetes or left ventricular hypertrophy were at significantly greater risk than the overall cohort.
“The antifibrotic effects of angiotensin-converting enzyme inhibitors may prevent or slow the development of conduction system disease, and future research is warranted to understand whether this treatment affects other conduction-related clinical outcomes,” including the need for pacemaker implantation, the researchers concluded.
The study was supported by the American Heart Association, the Joseph Drown Foundation, the National Heart, Lung, and Blood Institute of the National Institutes of Health, and by Pfizer. The investigators had no disclosures.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Compared with chlorthalidone, the angiotensin-converting enzyme inhibitor lisinopril was associated with a significant drop in new-onset conduction system disease among high-risk patients with hypertension.
Major finding: The risk of conduction system disease was 19% lower with lisinopril than chlorthalidone (hazard ratio, 0.81; 95% CI, 0.69 to 0.95; P = .01).
Data source: Serial 12-lead electrocardiograms of 21,004 patients with hypertension and at least one other cardiac risk factor, with an average of 5 years of follow-up.
Disclosures: The study was supported by the American Heart Association, the Joseph Drown Foundation, the National Heart, Lung, and Blood Institute of the National Institutes of Health, and by Pfizer, Inc. The investigators had no disclosures.
RT + ADT linked with improved survival in mPC
The addition of radiotherapy (RT) to androgen deprivation therapy (ADT) appears to boost overall survival in men with metastatic prostate cancer. However, it is more common for men to receive ADT alone than ADT plus RT.
“In this large contemporary analysis, men receiving prostate RT plus ADT lived substantially longer than men treated with ADT alone,” noted Dr. Chad Rusthoven of the University of Colorado, Aurora, and his associates (J Clin Oncol. 2016 June doi: 10.1200/JCO.2016.67.4788).
Investigators reviewed the National Cancer Data Base and identified 6,382 men with metastatic prostate cancer who received ADT as first-line therapy. Of those men, 5,844 (91.6%) received ADT alone, and the remaining 538 (8.4%) men received ADT plus prostate RT. The median age of the study population was 69, 75% were white, the most common T stage was T2, N0 was the most common N stage, and 6% of the entire cohort received chemotherapy. The median follow-up time was 5.1 years, and the median time from diagnosis to initiation of RT was 101 days. Patients were excluded from the study if they died within a month of diagnosis or if they were receiving prostatectomy, cryotherapy, or brachytherapy.
Among men receiving ADT plus RT, 48% were on Medicare and 41% were privately insured. Among men receiving ADT alone, 56% were on Medicare and 27% were privately insured.
Univariate analysis revealed that, compared with ADT alone, RT plus ADT was associated with longer median overall survival (29 vs. 53 months) as well as improved 3-year (43% vs. 62%), 5-year (25% vs. 49%), and 8-year (13% vs. 33%) overall survival estimates.
Multivariate analysis also found an independent association between the addition of radiotherapy with improved overall survival (HR, 0.624; 95% confidence interval, 0.551-0.706; P less than .001). In addition, RT to the prostate only and RT to the prostate and pelvis were both associated with longer overall survival times compared with ADT alone.
The funding source for this study was not listed. Six investigators reported serving in advisory roles for, receiving honoraria or financial compensation from, or holding patents in accordance with multiple companies.
On Twitter @jessnicolecraig
The addition of radiotherapy (RT) to androgen deprivation therapy (ADT) appears to boost overall survival in men with metastatic prostate cancer. However, it is more common for men to receive ADT alone than ADT plus RT.
“In this large contemporary analysis, men receiving prostate RT plus ADT lived substantially longer than men treated with ADT alone,” noted Dr. Chad Rusthoven of the University of Colorado, Aurora, and his associates (J Clin Oncol. 2016 June doi: 10.1200/JCO.2016.67.4788).
Investigators reviewed the National Cancer Data Base and identified 6,382 men with metastatic prostate cancer who received ADT as first-line therapy. Of those men, 5,844 (91.6%) received ADT alone, and the remaining 538 (8.4%) men received ADT plus prostate RT. The median age of the study population was 69, 75% were white, the most common T stage was T2, N0 was the most common N stage, and 6% of the entire cohort received chemotherapy. The median follow-up time was 5.1 years, and the median time from diagnosis to initiation of RT was 101 days. Patients were excluded from the study if they died within a month of diagnosis or if they were receiving prostatectomy, cryotherapy, or brachytherapy.
Among men receiving ADT plus RT, 48% were on Medicare and 41% were privately insured. Among men receiving ADT alone, 56% were on Medicare and 27% were privately insured.
Univariate analysis revealed that, compared with ADT alone, RT plus ADT was associated with longer median overall survival (29 vs. 53 months) as well as improved 3-year (43% vs. 62%), 5-year (25% vs. 49%), and 8-year (13% vs. 33%) overall survival estimates.
Multivariate analysis also found an independent association between the addition of radiotherapy with improved overall survival (HR, 0.624; 95% confidence interval, 0.551-0.706; P less than .001). In addition, RT to the prostate only and RT to the prostate and pelvis were both associated with longer overall survival times compared with ADT alone.
The funding source for this study was not listed. Six investigators reported serving in advisory roles for, receiving honoraria or financial compensation from, or holding patents in accordance with multiple companies.
On Twitter @jessnicolecraig
The addition of radiotherapy (RT) to androgen deprivation therapy (ADT) appears to boost overall survival in men with metastatic prostate cancer. However, it is more common for men to receive ADT alone than ADT plus RT.
“In this large contemporary analysis, men receiving prostate RT plus ADT lived substantially longer than men treated with ADT alone,” noted Dr. Chad Rusthoven of the University of Colorado, Aurora, and his associates (J Clin Oncol. 2016 June doi: 10.1200/JCO.2016.67.4788).
Investigators reviewed the National Cancer Data Base and identified 6,382 men with metastatic prostate cancer who received ADT as first-line therapy. Of those men, 5,844 (91.6%) received ADT alone, and the remaining 538 (8.4%) men received ADT plus prostate RT. The median age of the study population was 69, 75% were white, the most common T stage was T2, N0 was the most common N stage, and 6% of the entire cohort received chemotherapy. The median follow-up time was 5.1 years, and the median time from diagnosis to initiation of RT was 101 days. Patients were excluded from the study if they died within a month of diagnosis or if they were receiving prostatectomy, cryotherapy, or brachytherapy.
Among men receiving ADT plus RT, 48% were on Medicare and 41% were privately insured. Among men receiving ADT alone, 56% were on Medicare and 27% were privately insured.
Univariate analysis revealed that, compared with ADT alone, RT plus ADT was associated with longer median overall survival (29 vs. 53 months) as well as improved 3-year (43% vs. 62%), 5-year (25% vs. 49%), and 8-year (13% vs. 33%) overall survival estimates.
Multivariate analysis also found an independent association between the addition of radiotherapy with improved overall survival (HR, 0.624; 95% confidence interval, 0.551-0.706; P less than .001). In addition, RT to the prostate only and RT to the prostate and pelvis were both associated with longer overall survival times compared with ADT alone.
The funding source for this study was not listed. Six investigators reported serving in advisory roles for, receiving honoraria or financial compensation from, or holding patents in accordance with multiple companies.
On Twitter @jessnicolecraig
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The addition of radiotherapy (RT) to androgen deprivation therapy (ADT) was associated with improved survival in men with prostate cancer.
Major finding: The addition of radiotherapy to androgen deprivation therapy was independently associated with improved overall survival (HR, 0.624; 95% confidence interval, 0.551-0.706; P less than .001).
Data source: A review of the National Cancer Data Base on 6,382 men with metastatic prostate cancer.
Disclosures: The funding source for this study was not listed. Six investigators reported serving in advisory roles for, receiving honoraria or financial compensation from, or holding patents in accordance with multiple companies.
Single Dose of Dexamethasone Not an Alternative to ‘Steroid Burst’ for Acute Asthma Treatment
Clinical question: Is one dose of dexamethasone comparable to five days of prednisone for treating mild-to-moderate asthma exacerbations?

Background: Corticosteroids are the mainstay of initial treatment for asthma exacerbations. The National Heart, Lung, and Blood Institute recommends a minimum of five days of prednisone, though studies have shown incomplete adherence to prolonged therapies. Dexamethasone has a longer duration of action than prednisone.
Study design: Randomized, controlled, double-blinded trial.
Setting: Urban, safety-net, teaching hospital.
Synopsis: The study included 376 adults ages 18–55 presenting to the emergency department for a mild-to-moderate asthma exacerbation who were randomized to two treatment courses of corticosteroids: one 12 mg dose of oral dexamethasone followed by four days of placebo versus five days of 60 mg of oral prednisone. Two weeks later, a telephone survey asked if they had relapsed and had to seek medical attention. This study did not show noninferiority of the dexamethasone option compared to the standard of care. Specifically, it showed a 12.1% relapse rate in the dexamethasone group versus a 9.8% relapse rate for prednisone (95% CI, -4.1% to 8.6%).
This was a small study looking at adults without other chronic lung diseases or diabetes. The authors did not include those patients who were either lost to follow-up (20% of those initially randomized) or ultimately admitted after their emergency department course.
Hospitalists who care for patients with asthma should look to the current standards of corticosteroid selection and duration to minimize clinical relapses and possibly readmissions.
Bottom line: One large dose of dexamethasone is inferior to the standard five days of prednisone for treating acute asthma exacerbations in adults.
Citation: Rehrer MW, Liu B, Rodriguez M, Lam J, Alter HJ. A randomized controlled noninferiority trial of single dose of oral dexamethasone versus 5 days of oral prednisone in acute adult asthma [published online ahead of print April 14, 2016]. Ann Emerg Med. doi:10.1016/j.annemergmed.2016.03.017.
Short Take
Guideline Recommends ED Asthma Management Associated with Shorter Inpatient Stay
Observational study found ED treatment concordance with four guideline-based processes for acute asthma treatment (inhaled beta-agonists, inhaled anticholinergics, systemic corticosteroids, and avoidance of methylxanthines) is associated with a 17% shorter hospital length of stay.
Citation: Hasegawa K, Brenner BE, Nowak RM, et al. Association of guideline-concordant acute asthma care in the emergency department with shorter hospital length of stay: a multicenter observational study. Acad Emerg Med. 2016;23(5):616-622.
Clinical question: Is one dose of dexamethasone comparable to five days of prednisone for treating mild-to-moderate asthma exacerbations?
Background: Corticosteroids are the mainstay of initial treatment for asthma exacerbations. The National Heart, Lung, and Blood Institute recommends a minimum of five days of prednisone, though studies have shown incomplete adherence to prolonged therapies. Dexamethasone has a longer duration of action than prednisone.
Study design: Randomized, controlled, double-blinded trial.
Setting: Urban, safety-net, teaching hospital.
Synopsis: The study included 376 adults ages 18–55 presenting to the emergency department for a mild-to-moderate asthma exacerbation who were randomized to two treatment courses of corticosteroids: one 12 mg dose of oral dexamethasone followed by four days of placebo versus five days of 60 mg of oral prednisone. Two weeks later, a telephone survey asked if they had relapsed and had to seek medical attention. This study did not show noninferiority of the dexamethasone option compared to the standard of care. Specifically, it showed a 12.1% relapse rate in the dexamethasone group versus a 9.8% relapse rate for prednisone (95% CI, -4.1% to 8.6%).
This was a small study looking at adults without other chronic lung diseases or diabetes. The authors did not include those patients who were either lost to follow-up (20% of those initially randomized) or ultimately admitted after their emergency department course.
Hospitalists who care for patients with asthma should look to the current standards of corticosteroid selection and duration to minimize clinical relapses and possibly readmissions.
Bottom line: One large dose of dexamethasone is inferior to the standard five days of prednisone for treating acute asthma exacerbations in adults.
Citation: Rehrer MW, Liu B, Rodriguez M, Lam J, Alter HJ. A randomized controlled noninferiority trial of single dose of oral dexamethasone versus 5 days of oral prednisone in acute adult asthma [published online ahead of print April 14, 2016]. Ann Emerg Med. doi:10.1016/j.annemergmed.2016.03.017.
Short Take
Guideline Recommends ED Asthma Management Associated with Shorter Inpatient Stay
Observational study found ED treatment concordance with four guideline-based processes for acute asthma treatment (inhaled beta-agonists, inhaled anticholinergics, systemic corticosteroids, and avoidance of methylxanthines) is associated with a 17% shorter hospital length of stay.
Citation: Hasegawa K, Brenner BE, Nowak RM, et al. Association of guideline-concordant acute asthma care in the emergency department with shorter hospital length of stay: a multicenter observational study. Acad Emerg Med. 2016;23(5):616-622.
Clinical question: Is one dose of dexamethasone comparable to five days of prednisone for treating mild-to-moderate asthma exacerbations?
Background: Corticosteroids are the mainstay of initial treatment for asthma exacerbations. The National Heart, Lung, and Blood Institute recommends a minimum of five days of prednisone, though studies have shown incomplete adherence to prolonged therapies. Dexamethasone has a longer duration of action than prednisone.
Study design: Randomized, controlled, double-blinded trial.
Setting: Urban, safety-net, teaching hospital.
Synopsis: The study included 376 adults ages 18–55 presenting to the emergency department for a mild-to-moderate asthma exacerbation who were randomized to two treatment courses of corticosteroids: one 12 mg dose of oral dexamethasone followed by four days of placebo versus five days of 60 mg of oral prednisone. Two weeks later, a telephone survey asked if they had relapsed and had to seek medical attention. This study did not show noninferiority of the dexamethasone option compared to the standard of care. Specifically, it showed a 12.1% relapse rate in the dexamethasone group versus a 9.8% relapse rate for prednisone (95% CI, -4.1% to 8.6%).
This was a small study looking at adults without other chronic lung diseases or diabetes. The authors did not include those patients who were either lost to follow-up (20% of those initially randomized) or ultimately admitted after their emergency department course.
Hospitalists who care for patients with asthma should look to the current standards of corticosteroid selection and duration to minimize clinical relapses and possibly readmissions.
Bottom line: One large dose of dexamethasone is inferior to the standard five days of prednisone for treating acute asthma exacerbations in adults.
Citation: Rehrer MW, Liu B, Rodriguez M, Lam J, Alter HJ. A randomized controlled noninferiority trial of single dose of oral dexamethasone versus 5 days of oral prednisone in acute adult asthma [published online ahead of print April 14, 2016]. Ann Emerg Med. doi:10.1016/j.annemergmed.2016.03.017.
Short Take
Guideline Recommends ED Asthma Management Associated with Shorter Inpatient Stay
Observational study found ED treatment concordance with four guideline-based processes for acute asthma treatment (inhaled beta-agonists, inhaled anticholinergics, systemic corticosteroids, and avoidance of methylxanthines) is associated with a 17% shorter hospital length of stay.
Citation: Hasegawa K, Brenner BE, Nowak RM, et al. Association of guideline-concordant acute asthma care in the emergency department with shorter hospital length of stay: a multicenter observational study. Acad Emerg Med. 2016;23(5):616-622.

High-Flow Oxygen after Extubation Reduces Reintubation
Clinical question: Does nasal high-flow (NHF) oxygen after extubation reduce reintubation rates in low-risk patients?
Background: NHF oxygen devices deliver warmed and humidified oxygen up to 60 liters per minutes. NHF provides positive end-expiratory pressure and dead-space washout. NHF in higher-risk post-extubation patients has been shown to have clinical benefits. Whether NHF post-extubation benefits patients at low risk of reintubation is unknown.
Study design: Randomized control trial (RCT).
Setting: Seven ICUs in Spain.
Synopsis: In this RCT, post-extubation NHF oxygen for 24 hours reduced the risk of reintubation among 527 ICU adults at low risk of reintubation when compared to conventional oxygen therapy (by nasal cannula or face mask). Patients with hypercapnia during weaning trials were excluded. The risk of reintubation was 4.9% versus 12.2% in NHF versus standard oxygen therapy, with an absolute difference of 7.2% (95% CI, 2.5–12.2%; P=0.004). ICU length of stay and mortality were not significantly different between the groups. The strengths of the study were adequate sample size, prespecified criteria for reintubation, and low number of crossover patients.
Limitations of the trial were the high percentage of surgical and neurologic cases, exclusion of patients with a variety of common comorbidities, and the inability to blind the physicians to the treatment arm of the subjects. Select patients may benefit from noninvasive ventilation to prevent reintubation, which was not studied. These results are highly relevant to post-extubation patients, with the optimum therapy for low-risk patients now appearing to be NHF.
Bottom line: NHF oxygen reduced reintubation compared to conventional oxygen therapy (nasal cannula or face mask) in extubated patients at low risk of reintubation.
Citation: Hernández G, Vaquero C, González P, et al. Effect of postextubation high-flow nasal cannula vs conventional oxygen therapy on reintubation in low-risk patients: a randomized clinical trial. JAMA. 2016;315(13):1354-1361. doi:10.1001/jama.2016.2711.
Clinical question: Does nasal high-flow (NHF) oxygen after extubation reduce reintubation rates in low-risk patients?
Background: NHF oxygen devices deliver warmed and humidified oxygen up to 60 liters per minutes. NHF provides positive end-expiratory pressure and dead-space washout. NHF in higher-risk post-extubation patients has been shown to have clinical benefits. Whether NHF post-extubation benefits patients at low risk of reintubation is unknown.
Study design: Randomized control trial (RCT).
Setting: Seven ICUs in Spain.
Synopsis: In this RCT, post-extubation NHF oxygen for 24 hours reduced the risk of reintubation among 527 ICU adults at low risk of reintubation when compared to conventional oxygen therapy (by nasal cannula or face mask). Patients with hypercapnia during weaning trials were excluded. The risk of reintubation was 4.9% versus 12.2% in NHF versus standard oxygen therapy, with an absolute difference of 7.2% (95% CI, 2.5–12.2%; P=0.004). ICU length of stay and mortality were not significantly different between the groups. The strengths of the study were adequate sample size, prespecified criteria for reintubation, and low number of crossover patients.
Limitations of the trial were the high percentage of surgical and neurologic cases, exclusion of patients with a variety of common comorbidities, and the inability to blind the physicians to the treatment arm of the subjects. Select patients may benefit from noninvasive ventilation to prevent reintubation, which was not studied. These results are highly relevant to post-extubation patients, with the optimum therapy for low-risk patients now appearing to be NHF.
Bottom line: NHF oxygen reduced reintubation compared to conventional oxygen therapy (nasal cannula or face mask) in extubated patients at low risk of reintubation.
Citation: Hernández G, Vaquero C, González P, et al. Effect of postextubation high-flow nasal cannula vs conventional oxygen therapy on reintubation in low-risk patients: a randomized clinical trial. JAMA. 2016;315(13):1354-1361. doi:10.1001/jama.2016.2711.
Clinical question: Does nasal high-flow (NHF) oxygen after extubation reduce reintubation rates in low-risk patients?
Background: NHF oxygen devices deliver warmed and humidified oxygen up to 60 liters per minutes. NHF provides positive end-expiratory pressure and dead-space washout. NHF in higher-risk post-extubation patients has been shown to have clinical benefits. Whether NHF post-extubation benefits patients at low risk of reintubation is unknown.
Study design: Randomized control trial (RCT).
Setting: Seven ICUs in Spain.
Synopsis: In this RCT, post-extubation NHF oxygen for 24 hours reduced the risk of reintubation among 527 ICU adults at low risk of reintubation when compared to conventional oxygen therapy (by nasal cannula or face mask). Patients with hypercapnia during weaning trials were excluded. The risk of reintubation was 4.9% versus 12.2% in NHF versus standard oxygen therapy, with an absolute difference of 7.2% (95% CI, 2.5–12.2%; P=0.004). ICU length of stay and mortality were not significantly different between the groups. The strengths of the study were adequate sample size, prespecified criteria for reintubation, and low number of crossover patients.
Limitations of the trial were the high percentage of surgical and neurologic cases, exclusion of patients with a variety of common comorbidities, and the inability to blind the physicians to the treatment arm of the subjects. Select patients may benefit from noninvasive ventilation to prevent reintubation, which was not studied. These results are highly relevant to post-extubation patients, with the optimum therapy for low-risk patients now appearing to be NHF.
Bottom line: NHF oxygen reduced reintubation compared to conventional oxygen therapy (nasal cannula or face mask) in extubated patients at low risk of reintubation.
Citation: Hernández G, Vaquero C, González P, et al. Effect of postextubation high-flow nasal cannula vs conventional oxygen therapy on reintubation in low-risk patients: a randomized clinical trial. JAMA. 2016;315(13):1354-1361. doi:10.1001/jama.2016.2711.
Study explains how a mutation spurs AML development
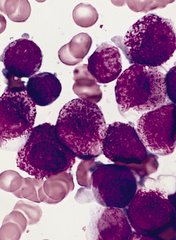
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
EMA recommends authorization of T-cell product

Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
Febrile, Immunocompromised Man With Rash
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
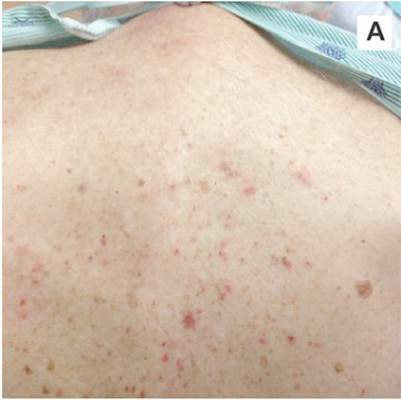
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
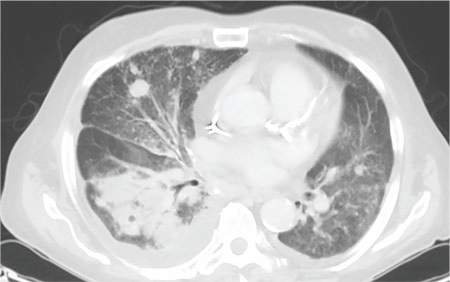
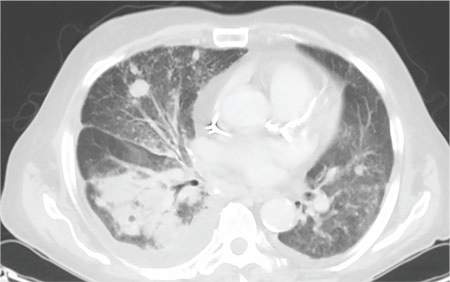
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
| 
|
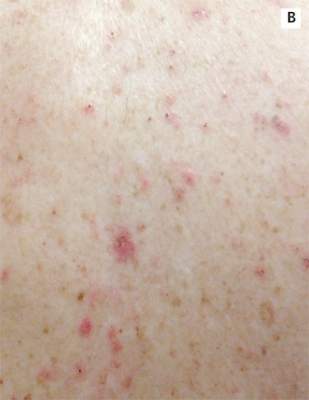
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
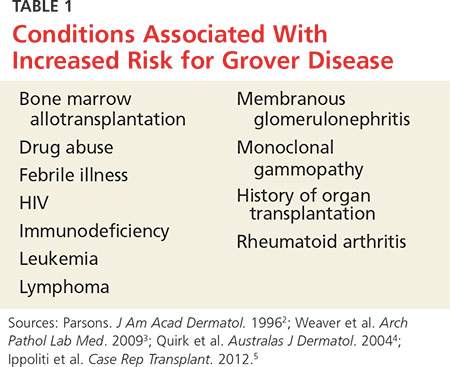
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.

Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
|
|
|
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.
Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
|
|
|
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.
Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.