User login
Patients’ Out-of-Pocket Spending Increasing
Clinical Question: How much are insured nonelderly adult patients paying out of pocket for inpatient care, and does that amount vary over time or by patient characteristics, region, or type of insurance?

Background: Prior estimates have been based on patient-reported survey data. This is the first study to find nationwide out-of-pocket expenditure for inpatient hospitalizations.
Study Design: Retrospective analysis.
Setting: Medical claims data from Aetna, UnitedHealthcare, and Humana including 7.3 million hospitalizations from 2009 to 2013.
Synopsis: Authors used the Health Care Cost Institute (HCCI) database and studied inpatient hospitalization for ages 18–64. The adjusted total cost sharing per inpatient hospitalization increased by 37% (from $738 in 2009 to $1,013 in 2013). Both the mean amount of coinsurance and deductibles increased during this period by 33% (from $518 to $688) and 86% (from $145 to $270), respectively. The mean copayment decreased by 27% (from $75 to $55).
Increase in cost sharing was lowest in individual-market and consumer-directed health plans, although both had highest cost sharing.
Total cost sharing increased in every state. The largest increases were seen in Georgia, Louisiana, and Colorado. In 2013, the states with the highest cost sharing were Utah, Alaska, and Oregon.
Acute myocardial infarction and acute appendicitis saw maximum rise in out-of-pocket spending; both surpassed $1,500 in 2013. Cost sharing associated with procedures was lower.
Bottom Line: Even after adjusting for inflation and case-mix differences, the total cost sharing per inpatient hospitalization increased between 2009 and 2013. Policymakers and patients need to pay attention to these trends.
Citation: Adrion ER, Ryan AM, Seltzer AC, Chen LM, Ayanian JZ, Nallamothu BK. Out-of-pocket spending for hospitalizations among nonelderly adults. JAMA Intern Med. 2016;176(9)1325-1332.
Short Take
Aspirin Is Being Used Instead of Anticoagulation in Afib
Despite recommendations to anticoagulate patients with CHADS2 /CHA2DS2-VASc scores of ≥2, more than one-third of the patients in a large population of cardiology outpatients were treated with aspirin alone.
Citation: Hsu JC, Maddox TM, Kennedy K, et al. Aspirin instead of oral anticoagulant prescription in atrial fibrillation patients at risk for stroke. J Am Coll Cardiol. 2016;67(25):2913-2923.
Clinical Question: How much are insured nonelderly adult patients paying out of pocket for inpatient care, and does that amount vary over time or by patient characteristics, region, or type of insurance?
Background: Prior estimates have been based on patient-reported survey data. This is the first study to find nationwide out-of-pocket expenditure for inpatient hospitalizations.
Study Design: Retrospective analysis.
Setting: Medical claims data from Aetna, UnitedHealthcare, and Humana including 7.3 million hospitalizations from 2009 to 2013.
Synopsis: Authors used the Health Care Cost Institute (HCCI) database and studied inpatient hospitalization for ages 18–64. The adjusted total cost sharing per inpatient hospitalization increased by 37% (from $738 in 2009 to $1,013 in 2013). Both the mean amount of coinsurance and deductibles increased during this period by 33% (from $518 to $688) and 86% (from $145 to $270), respectively. The mean copayment decreased by 27% (from $75 to $55).
Increase in cost sharing was lowest in individual-market and consumer-directed health plans, although both had highest cost sharing.
Total cost sharing increased in every state. The largest increases were seen in Georgia, Louisiana, and Colorado. In 2013, the states with the highest cost sharing were Utah, Alaska, and Oregon.
Acute myocardial infarction and acute appendicitis saw maximum rise in out-of-pocket spending; both surpassed $1,500 in 2013. Cost sharing associated with procedures was lower.
Bottom Line: Even after adjusting for inflation and case-mix differences, the total cost sharing per inpatient hospitalization increased between 2009 and 2013. Policymakers and patients need to pay attention to these trends.
Citation: Adrion ER, Ryan AM, Seltzer AC, Chen LM, Ayanian JZ, Nallamothu BK. Out-of-pocket spending for hospitalizations among nonelderly adults. JAMA Intern Med. 2016;176(9)1325-1332.
Short Take
Aspirin Is Being Used Instead of Anticoagulation in Afib
Despite recommendations to anticoagulate patients with CHADS2 /CHA2DS2-VASc scores of ≥2, more than one-third of the patients in a large population of cardiology outpatients were treated with aspirin alone.
Citation: Hsu JC, Maddox TM, Kennedy K, et al. Aspirin instead of oral anticoagulant prescription in atrial fibrillation patients at risk for stroke. J Am Coll Cardiol. 2016;67(25):2913-2923.
Clinical Question: How much are insured nonelderly adult patients paying out of pocket for inpatient care, and does that amount vary over time or by patient characteristics, region, or type of insurance?
Background: Prior estimates have been based on patient-reported survey data. This is the first study to find nationwide out-of-pocket expenditure for inpatient hospitalizations.
Study Design: Retrospective analysis.
Setting: Medical claims data from Aetna, UnitedHealthcare, and Humana including 7.3 million hospitalizations from 2009 to 2013.
Synopsis: Authors used the Health Care Cost Institute (HCCI) database and studied inpatient hospitalization for ages 18–64. The adjusted total cost sharing per inpatient hospitalization increased by 37% (from $738 in 2009 to $1,013 in 2013). Both the mean amount of coinsurance and deductibles increased during this period by 33% (from $518 to $688) and 86% (from $145 to $270), respectively. The mean copayment decreased by 27% (from $75 to $55).
Increase in cost sharing was lowest in individual-market and consumer-directed health plans, although both had highest cost sharing.
Total cost sharing increased in every state. The largest increases were seen in Georgia, Louisiana, and Colorado. In 2013, the states with the highest cost sharing were Utah, Alaska, and Oregon.
Acute myocardial infarction and acute appendicitis saw maximum rise in out-of-pocket spending; both surpassed $1,500 in 2013. Cost sharing associated with procedures was lower.
Bottom Line: Even after adjusting for inflation and case-mix differences, the total cost sharing per inpatient hospitalization increased between 2009 and 2013. Policymakers and patients need to pay attention to these trends.
Citation: Adrion ER, Ryan AM, Seltzer AC, Chen LM, Ayanian JZ, Nallamothu BK. Out-of-pocket spending for hospitalizations among nonelderly adults. JAMA Intern Med. 2016;176(9)1325-1332.
Short Take
Aspirin Is Being Used Instead of Anticoagulation in Afib
Despite recommendations to anticoagulate patients with CHADS2 /CHA2DS2-VASc scores of ≥2, more than one-third of the patients in a large population of cardiology outpatients were treated with aspirin alone.
Citation: Hsu JC, Maddox TM, Kennedy K, et al. Aspirin instead of oral anticoagulant prescription in atrial fibrillation patients at risk for stroke. J Am Coll Cardiol. 2016;67(25):2913-2923.

Finally, Some Good News!
August 2016 provided 2 impressive news stories. These stories have far more salience and granularity than I can began to entertain in this brief editorial, but they show that the VA with all its systemic problems has unrivaled potential to promote what Aristotle called human flourishing.
A past director of mine greeted any small success or positive accomplishment of the facility and its employees with the folksy aphorism “You have to celebrate when you can in this outfit.” He was wise, for he knew that taking a respite to recognize a job well done is crucial to the emotional wellness of the workforce. And after that moment of satisfaction, everyone gets back to work at least a little bit recharged. So in this editorial, I will praise a few recent, unique VA achievements that underscore the importance of keeping the organization not only upright, but also doing right.
On August 1, President Obama announced that since 2010 veteran homelessness had been reduced by almost half. VA Secretary Robert A. McDonald also applauded a 56% decrease in unsheltered homeless veterans. Yet just as quickly, he refocused the collaborating agencies on the goal of ending veteran homelessness, which seemed a long shot when initially announced but now seems to have a realistic chance of success. “Although this achievement is noteworthy, we will not rest until every veteran in need is permanently housed,” McDonald said.
Three large government agencies and extensive partnerships cooperated to keep 360,000 veterans and their families from being homeless. But each veteran also had the outreach and support of a HUD-VASH (U.S. Department of Housing and Urban Development and VA Supportive Housing) worker and counterparts in the community. It is hard to see how any other health care organization could leverage this large an effort or would choose to dedicate its federal, state, and city resources to meet a need so basic that without it few persons can move up Maslow’s hierarchy of human actualization.
The same week the VA Research and Development program gave all of us in federal service a reason to hold up our collective heads a little higher announcing that the Million Veteran Program (MVP) had enrolled its 500,000th participant, making it the largest genomic database in the world. Once again, it is difficult to imagine any other health care organization, except another federal agency like the National Institutes of Health, mounting such an ambitious research initiative.
The MVP offers a databank—the likes of which has never been assembled—to study some of the most common and debilitating conditions, such as mental illness, substance use, and kidney and heart disease among many others. The combination of environmental genetics and clinical and psychosocial data will open doors of discoveries for thousands of people, veteran and nonveteran alike. Secretary McDonald applauded the most important ethical aspect of the project, the incomparable altruism of veterans, “Many of our veterans have saved lives on the battlefield and because of their participation in MVP, their participation has the potential to save countless lives—now and for generations to come.”
These 2 amazing initiatives have more in common than may seem apparent at first glance. Besides their intrinsic worth in humanist service and scientific creativity, respectively, putting veterans in homes and constructing a repository of scientific knowledge show that the VA—once accused of being a dinosaur ignoring the plummeting temperatures of its own ice age—has demonstrated remarkable instantiation of the I CARE Core Characteristics of agility and innovation (available at http://www.va.gov/icare) in the campaign to end homelessness and the MVP initiative.
These good-news stories celebrate the immense power of the VA to change the world for the better. This is reason enough to keep the faith that VA will emerge from the hearings and the headlines as a workforce proud of their privilege to care for veterans and contribute to the common good.
August 2016 provided 2 impressive news stories. These stories have far more salience and granularity than I can began to entertain in this brief editorial, but they show that the VA with all its systemic problems has unrivaled potential to promote what Aristotle called human flourishing.
A past director of mine greeted any small success or positive accomplishment of the facility and its employees with the folksy aphorism “You have to celebrate when you can in this outfit.” He was wise, for he knew that taking a respite to recognize a job well done is crucial to the emotional wellness of the workforce. And after that moment of satisfaction, everyone gets back to work at least a little bit recharged. So in this editorial, I will praise a few recent, unique VA achievements that underscore the importance of keeping the organization not only upright, but also doing right.
On August 1, President Obama announced that since 2010 veteran homelessness had been reduced by almost half. VA Secretary Robert A. McDonald also applauded a 56% decrease in unsheltered homeless veterans. Yet just as quickly, he refocused the collaborating agencies on the goal of ending veteran homelessness, which seemed a long shot when initially announced but now seems to have a realistic chance of success. “Although this achievement is noteworthy, we will not rest until every veteran in need is permanently housed,” McDonald said.
Three large government agencies and extensive partnerships cooperated to keep 360,000 veterans and their families from being homeless. But each veteran also had the outreach and support of a HUD-VASH (U.S. Department of Housing and Urban Development and VA Supportive Housing) worker and counterparts in the community. It is hard to see how any other health care organization could leverage this large an effort or would choose to dedicate its federal, state, and city resources to meet a need so basic that without it few persons can move up Maslow’s hierarchy of human actualization.
The same week the VA Research and Development program gave all of us in federal service a reason to hold up our collective heads a little higher announcing that the Million Veteran Program (MVP) had enrolled its 500,000th participant, making it the largest genomic database in the world. Once again, it is difficult to imagine any other health care organization, except another federal agency like the National Institutes of Health, mounting such an ambitious research initiative.
The MVP offers a databank—the likes of which has never been assembled—to study some of the most common and debilitating conditions, such as mental illness, substance use, and kidney and heart disease among many others. The combination of environmental genetics and clinical and psychosocial data will open doors of discoveries for thousands of people, veteran and nonveteran alike. Secretary McDonald applauded the most important ethical aspect of the project, the incomparable altruism of veterans, “Many of our veterans have saved lives on the battlefield and because of their participation in MVP, their participation has the potential to save countless lives—now and for generations to come.”
These 2 amazing initiatives have more in common than may seem apparent at first glance. Besides their intrinsic worth in humanist service and scientific creativity, respectively, putting veterans in homes and constructing a repository of scientific knowledge show that the VA—once accused of being a dinosaur ignoring the plummeting temperatures of its own ice age—has demonstrated remarkable instantiation of the I CARE Core Characteristics of agility and innovation (available at http://www.va.gov/icare) in the campaign to end homelessness and the MVP initiative.
These good-news stories celebrate the immense power of the VA to change the world for the better. This is reason enough to keep the faith that VA will emerge from the hearings and the headlines as a workforce proud of their privilege to care for veterans and contribute to the common good.
August 2016 provided 2 impressive news stories. These stories have far more salience and granularity than I can began to entertain in this brief editorial, but they show that the VA with all its systemic problems has unrivaled potential to promote what Aristotle called human flourishing.
A past director of mine greeted any small success or positive accomplishment of the facility and its employees with the folksy aphorism “You have to celebrate when you can in this outfit.” He was wise, for he knew that taking a respite to recognize a job well done is crucial to the emotional wellness of the workforce. And after that moment of satisfaction, everyone gets back to work at least a little bit recharged. So in this editorial, I will praise a few recent, unique VA achievements that underscore the importance of keeping the organization not only upright, but also doing right.
On August 1, President Obama announced that since 2010 veteran homelessness had been reduced by almost half. VA Secretary Robert A. McDonald also applauded a 56% decrease in unsheltered homeless veterans. Yet just as quickly, he refocused the collaborating agencies on the goal of ending veteran homelessness, which seemed a long shot when initially announced but now seems to have a realistic chance of success. “Although this achievement is noteworthy, we will not rest until every veteran in need is permanently housed,” McDonald said.
Three large government agencies and extensive partnerships cooperated to keep 360,000 veterans and their families from being homeless. But each veteran also had the outreach and support of a HUD-VASH (U.S. Department of Housing and Urban Development and VA Supportive Housing) worker and counterparts in the community. It is hard to see how any other health care organization could leverage this large an effort or would choose to dedicate its federal, state, and city resources to meet a need so basic that without it few persons can move up Maslow’s hierarchy of human actualization.
The same week the VA Research and Development program gave all of us in federal service a reason to hold up our collective heads a little higher announcing that the Million Veteran Program (MVP) had enrolled its 500,000th participant, making it the largest genomic database in the world. Once again, it is difficult to imagine any other health care organization, except another federal agency like the National Institutes of Health, mounting such an ambitious research initiative.
The MVP offers a databank—the likes of which has never been assembled—to study some of the most common and debilitating conditions, such as mental illness, substance use, and kidney and heart disease among many others. The combination of environmental genetics and clinical and psychosocial data will open doors of discoveries for thousands of people, veteran and nonveteran alike. Secretary McDonald applauded the most important ethical aspect of the project, the incomparable altruism of veterans, “Many of our veterans have saved lives on the battlefield and because of their participation in MVP, their participation has the potential to save countless lives—now and for generations to come.”
These 2 amazing initiatives have more in common than may seem apparent at first glance. Besides their intrinsic worth in humanist service and scientific creativity, respectively, putting veterans in homes and constructing a repository of scientific knowledge show that the VA—once accused of being a dinosaur ignoring the plummeting temperatures of its own ice age—has demonstrated remarkable instantiation of the I CARE Core Characteristics of agility and innovation (available at http://www.va.gov/icare) in the campaign to end homelessness and the MVP initiative.
These good-news stories celebrate the immense power of the VA to change the world for the better. This is reason enough to keep the faith that VA will emerge from the hearings and the headlines as a workforce proud of their privilege to care for veterans and contribute to the common good.

Immunotherapy produces CRs in kids with rel/ref ALL
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
Speaker outlines importance of cell of origin in DLBCL

NEW YORK—The importance of cell of origin in choosing a treatment for diffuse large B-cell lymphoma (DLBCL) is a topic “that has been kicking around for 16 years,” according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Cell of origin was first described in the year 2000 as a distinguishing factor in large-cell lymphoma, said the speaker, Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York.
The cell of origin in DLBCL—whether it’s germinal center B cell (GCB), activated B cell (ABC), or unclassified—contributes to biological and clinical heterogeneity of the disease.
“And more importantly, activated B-cell diffuse large B-cell lymphoma and germinal center diffuse large B-cell lymphoma are simply different diseases,” Dr Zelenetz said.
He then elaborated on the importance of cell of origin in treating DLBCL.
Biology
Dr Zelenetz noted that ABC and GCB lymphomas have different molecular pathways. ABC lymphomas are very dependent on the NF-kB pathway and have more active signaling through the B-cell receptor.
The GCB lymphomas tend to have more tonic regulation, and the PI3 kinase/mTOR pathway is more critical. GCB lymphomas have more genomic instability.
“So cell of origin determination identifies tumors with distinct biology, may provide prognostic information, and may be predictive for treatment selection,” Dr Zelenetz said. “Unfortunately, cell of origin is not the whole story.”
Gene mutations occur in large-cell lymphoma “just like every other cancer,” Dr Zelenetz said. And the vast majority occur in both lymphoma subtypes, he added, further complicating our understanding of the biology of these tumors.
Some of these mutations predict for sensitivity to treatment, while others predict for resistance. For example, CARD11 predicts for resistance to ibrutinib, while CD79b predicts for sensitivity.
Determining the cell of origin
Gene-expression profiling on fresh tissue is considered the gold standard, but “it is clearly not a clinical tool,” Dr Zelenetz said. It requires the Wright classifier, a statistical method based on Bayes’ rule, to make patient-level assignments to 1 of the 3 subgroups.
Immunohistochemistry is widely available, but reproducibility may be difficult. Many assays exist, such as the Hans, Choi, and Muris assays, but, in many studies, there may be a lack of correlation with gene-expression profiling.
In the last few years, gene-expression profiling of formalin‐fixed paraffin‐embedded (FFPE) tissue has emerged as a reliable method. The assay is reproducible between laboratories, and it’s reproducible between different sets of reagents.
“[T]here is tremendous correlation between the Lymph2Cx assay and the gold standard,” Dr Zelenetz added.
“So here we have a robust assay,” he said, which allows investigators to explore whether the cell of origin is prognostic in large-cell lymphoma.
Prognosis
In a data set of 339 patients with de novo DLBCL treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the Lymph2Cx assay showing cell of origin was predictive of overall and progression-free survival.
However, the same exact assay applied to the RICOVER-60 data from the German High-Grade Non-Hodgkin Lymphoma Study group was not predictive, Dr Zelenetz reported, based on a personal communication from one of the investigators. There was a slight trend in favor of GCB tumors, but it was not statistically significant.
And the REMoDL-B study, using gene-expression profiling of FFPE tissue with the DASL assay, also didn’t show any difference in outcome between ABC or GCB tumors.
So gene-expression profiling of FFPE tissue does not universally show a prognostic difference, Dr Zelenetz said.
Influence of chemotherapy by cell of origin
CALGB 59910 showed that GCB tumors had a superior event-free, progression-free, and overall survival with dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) compared to ABC tumors.
“However, this is a phase 2, hypothesis-generating experiment,” Dr Zelenetz pointed out, and the results of the confirmatory study comparing dose-adjusted EPOCH-R and R-CHOP21 (CALGB 50303) will be presented later this year at the ASH Annual Meeting.
Sequential, non-cross-resistant chemotherapy
Data from the Memorial Sloan Kettering study (MSKCC 01-142/08-146; NCT00712582) of sequential therapy with R-CHOP followed by ICE (ifosfamide, carboplatin, and etoposide) demonstrate “excellent” progression-free and overall survival, Dr Zelenetz said.
“When we analyzed the outcome by cell of origin, there was a suggestion that the patients with the non-germinal center tumors were actually doing better than the germinal center tumors,” he added.
He pointed out one of the limitations of the study is that the cell of origin was determined by the Hans model. Nevertheless, the study raised another testable hypothesis: sequential therapy might overcome the adverse impact of the non-germinal center tumors.
Cell of origin analysis of the prospective, randomized study (LNH 03-2B) comparing R-ACVBP (rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, and prednisone) to R-CHOP showed that whether patients with GCB tumors received CHOP or ACVBP didn’t make “a whit of difference,” Dr Zelenetz said, in terms of progression-free and overall survival.
However, patients with ABC tumors demonstrated an “enormous difference in favor of R-ACVBP,” he said.
“Again, evidence that you can overcome the adverse effect of the ABC tumors with chemotherapy.”
Dr Zelenetz pointed out that R-ACVBP and R-CHOP followed by ICE are “actually remarkably similar regimens.” Both are sequential, both include consolidation, and both incorporate high-dose ifosfamide and etoposide.
“[S]o they actually reinforce each other,” he said, “demonstrating a similar result.”
Lenalidomide
Lenalidomide in the relapsed/refractory setting has modest activity in DLBCL, with most of the benefit accruing to patients with non-germinal center tumors.
Two clinical studies evaluated the impact of adding lenalidomide to standard chemotherapy.
In an Italian series using lenalidomide (L) plus R-CHOP21 in elderly untreated patients, the combination produced outstanding progression-free and event-free survival, but with no significant differences between the subtypes.
A US study of RL-CHOP versus R-CHOP included 87 matched historical controls treated with R-CHOP and 64 patients treated with RL-CHOP. Patients with non-germinal center tumors treated with RL-CHOP fared much better than historical controls treated with R-CHOP.
However, among germinal center tumors, “there was not a hint of any difference,” Dr Zelenetz noted.
Two studies—E1412, using an unselected population, and the international ROBUST study, selecting for patients with ABC tumors—are underway to confirm that the benefit with lenalidomide is in patients with activated B-cell tumors.
Ibrutinib
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, also has modest activity as a single agent in an unselected patient population with relapsed/refractory DLBCL. And most of the patients who demonstrated benefit had activated B-cell tumors.
Upon further analysis, investigators found that response was enhanced by the CD79b mutation, but it was not necessary for a response. And patients with CARD11 had no response.
MYD88 mutations seemed to cause resistance to ibrutinib, unless the mutation was associated with the CD79b mutation, and then patients had a “great” response, Dr Zelenetz explained.
In the upfront setting, a phase 1b study of R-CHOP plus ibrutinib demonstrated the safety of the combination, which had an overall survival rate of 100% and a complete response rate of 91%.
The prospective, randomized, phase 3 PHOENIX trial (NCT01855750) evaluating the combination in newly diagnosed non-germinal center DLBCL has completed accrual, but analysis is still pending.
Conclusion
“The prognostic significance of cell of origin is still controversial,” Dr Zelenetz wrapped up, “although I actually believe there is a prognostic difference in unselected registry patients.”
Sequential chemotherapy with ifosfamide and etoposide consolidation does very well in activated B-cell tumors, both in phase 2 and phase 3 studies.
“Importantly, small molecules seem to have differential effects totally predictable based on the biology of the difference between activated B-cell and germinal center tumors,” Dr Zelenetz said.
“But the big wild card here is somatic mutations further complicate things and will have to be incorporated into our understanding in the selection of patients.” ![]()
NEW YORK—The importance of cell of origin in choosing a treatment for diffuse large B-cell lymphoma (DLBCL) is a topic “that has been kicking around for 16 years,” according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Cell of origin was first described in the year 2000 as a distinguishing factor in large-cell lymphoma, said the speaker, Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York.
The cell of origin in DLBCL—whether it’s germinal center B cell (GCB), activated B cell (ABC), or unclassified—contributes to biological and clinical heterogeneity of the disease.
“And more importantly, activated B-cell diffuse large B-cell lymphoma and germinal center diffuse large B-cell lymphoma are simply different diseases,” Dr Zelenetz said.
He then elaborated on the importance of cell of origin in treating DLBCL.
Biology
Dr Zelenetz noted that ABC and GCB lymphomas have different molecular pathways. ABC lymphomas are very dependent on the NF-kB pathway and have more active signaling through the B-cell receptor.
The GCB lymphomas tend to have more tonic regulation, and the PI3 kinase/mTOR pathway is more critical. GCB lymphomas have more genomic instability.
“So cell of origin determination identifies tumors with distinct biology, may provide prognostic information, and may be predictive for treatment selection,” Dr Zelenetz said. “Unfortunately, cell of origin is not the whole story.”
Gene mutations occur in large-cell lymphoma “just like every other cancer,” Dr Zelenetz said. And the vast majority occur in both lymphoma subtypes, he added, further complicating our understanding of the biology of these tumors.
Some of these mutations predict for sensitivity to treatment, while others predict for resistance. For example, CARD11 predicts for resistance to ibrutinib, while CD79b predicts for sensitivity.
Determining the cell of origin
Gene-expression profiling on fresh tissue is considered the gold standard, but “it is clearly not a clinical tool,” Dr Zelenetz said. It requires the Wright classifier, a statistical method based on Bayes’ rule, to make patient-level assignments to 1 of the 3 subgroups.
Immunohistochemistry is widely available, but reproducibility may be difficult. Many assays exist, such as the Hans, Choi, and Muris assays, but, in many studies, there may be a lack of correlation with gene-expression profiling.
In the last few years, gene-expression profiling of formalin‐fixed paraffin‐embedded (FFPE) tissue has emerged as a reliable method. The assay is reproducible between laboratories, and it’s reproducible between different sets of reagents.
“[T]here is tremendous correlation between the Lymph2Cx assay and the gold standard,” Dr Zelenetz added.
“So here we have a robust assay,” he said, which allows investigators to explore whether the cell of origin is prognostic in large-cell lymphoma.
Prognosis
In a data set of 339 patients with de novo DLBCL treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the Lymph2Cx assay showing cell of origin was predictive of overall and progression-free survival.
However, the same exact assay applied to the RICOVER-60 data from the German High-Grade Non-Hodgkin Lymphoma Study group was not predictive, Dr Zelenetz reported, based on a personal communication from one of the investigators. There was a slight trend in favor of GCB tumors, but it was not statistically significant.
And the REMoDL-B study, using gene-expression profiling of FFPE tissue with the DASL assay, also didn’t show any difference in outcome between ABC or GCB tumors.
So gene-expression profiling of FFPE tissue does not universally show a prognostic difference, Dr Zelenetz said.
Influence of chemotherapy by cell of origin
CALGB 59910 showed that GCB tumors had a superior event-free, progression-free, and overall survival with dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) compared to ABC tumors.
“However, this is a phase 2, hypothesis-generating experiment,” Dr Zelenetz pointed out, and the results of the confirmatory study comparing dose-adjusted EPOCH-R and R-CHOP21 (CALGB 50303) will be presented later this year at the ASH Annual Meeting.
Sequential, non-cross-resistant chemotherapy
Data from the Memorial Sloan Kettering study (MSKCC 01-142/08-146; NCT00712582) of sequential therapy with R-CHOP followed by ICE (ifosfamide, carboplatin, and etoposide) demonstrate “excellent” progression-free and overall survival, Dr Zelenetz said.
“When we analyzed the outcome by cell of origin, there was a suggestion that the patients with the non-germinal center tumors were actually doing better than the germinal center tumors,” he added.
He pointed out one of the limitations of the study is that the cell of origin was determined by the Hans model. Nevertheless, the study raised another testable hypothesis: sequential therapy might overcome the adverse impact of the non-germinal center tumors.
Cell of origin analysis of the prospective, randomized study (LNH 03-2B) comparing R-ACVBP (rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, and prednisone) to R-CHOP showed that whether patients with GCB tumors received CHOP or ACVBP didn’t make “a whit of difference,” Dr Zelenetz said, in terms of progression-free and overall survival.
However, patients with ABC tumors demonstrated an “enormous difference in favor of R-ACVBP,” he said.
“Again, evidence that you can overcome the adverse effect of the ABC tumors with chemotherapy.”
Dr Zelenetz pointed out that R-ACVBP and R-CHOP followed by ICE are “actually remarkably similar regimens.” Both are sequential, both include consolidation, and both incorporate high-dose ifosfamide and etoposide.
“[S]o they actually reinforce each other,” he said, “demonstrating a similar result.”
Lenalidomide
Lenalidomide in the relapsed/refractory setting has modest activity in DLBCL, with most of the benefit accruing to patients with non-germinal center tumors.
Two clinical studies evaluated the impact of adding lenalidomide to standard chemotherapy.
In an Italian series using lenalidomide (L) plus R-CHOP21 in elderly untreated patients, the combination produced outstanding progression-free and event-free survival, but with no significant differences between the subtypes.
A US study of RL-CHOP versus R-CHOP included 87 matched historical controls treated with R-CHOP and 64 patients treated with RL-CHOP. Patients with non-germinal center tumors treated with RL-CHOP fared much better than historical controls treated with R-CHOP.
However, among germinal center tumors, “there was not a hint of any difference,” Dr Zelenetz noted.
Two studies—E1412, using an unselected population, and the international ROBUST study, selecting for patients with ABC tumors—are underway to confirm that the benefit with lenalidomide is in patients with activated B-cell tumors.
Ibrutinib
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, also has modest activity as a single agent in an unselected patient population with relapsed/refractory DLBCL. And most of the patients who demonstrated benefit had activated B-cell tumors.
Upon further analysis, investigators found that response was enhanced by the CD79b mutation, but it was not necessary for a response. And patients with CARD11 had no response.
MYD88 mutations seemed to cause resistance to ibrutinib, unless the mutation was associated with the CD79b mutation, and then patients had a “great” response, Dr Zelenetz explained.
In the upfront setting, a phase 1b study of R-CHOP plus ibrutinib demonstrated the safety of the combination, which had an overall survival rate of 100% and a complete response rate of 91%.
The prospective, randomized, phase 3 PHOENIX trial (NCT01855750) evaluating the combination in newly diagnosed non-germinal center DLBCL has completed accrual, but analysis is still pending.
Conclusion
“The prognostic significance of cell of origin is still controversial,” Dr Zelenetz wrapped up, “although I actually believe there is a prognostic difference in unselected registry patients.”
Sequential chemotherapy with ifosfamide and etoposide consolidation does very well in activated B-cell tumors, both in phase 2 and phase 3 studies.
“Importantly, small molecules seem to have differential effects totally predictable based on the biology of the difference between activated B-cell and germinal center tumors,” Dr Zelenetz said.
“But the big wild card here is somatic mutations further complicate things and will have to be incorporated into our understanding in the selection of patients.” ![]()
NEW YORK—The importance of cell of origin in choosing a treatment for diffuse large B-cell lymphoma (DLBCL) is a topic “that has been kicking around for 16 years,” according to a speaker at the NCCN 11th Annual Congress: Hematologic Malignancies.
Cell of origin was first described in the year 2000 as a distinguishing factor in large-cell lymphoma, said the speaker, Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York.
The cell of origin in DLBCL—whether it’s germinal center B cell (GCB), activated B cell (ABC), or unclassified—contributes to biological and clinical heterogeneity of the disease.
“And more importantly, activated B-cell diffuse large B-cell lymphoma and germinal center diffuse large B-cell lymphoma are simply different diseases,” Dr Zelenetz said.
He then elaborated on the importance of cell of origin in treating DLBCL.
Biology
Dr Zelenetz noted that ABC and GCB lymphomas have different molecular pathways. ABC lymphomas are very dependent on the NF-kB pathway and have more active signaling through the B-cell receptor.
The GCB lymphomas tend to have more tonic regulation, and the PI3 kinase/mTOR pathway is more critical. GCB lymphomas have more genomic instability.
“So cell of origin determination identifies tumors with distinct biology, may provide prognostic information, and may be predictive for treatment selection,” Dr Zelenetz said. “Unfortunately, cell of origin is not the whole story.”
Gene mutations occur in large-cell lymphoma “just like every other cancer,” Dr Zelenetz said. And the vast majority occur in both lymphoma subtypes, he added, further complicating our understanding of the biology of these tumors.
Some of these mutations predict for sensitivity to treatment, while others predict for resistance. For example, CARD11 predicts for resistance to ibrutinib, while CD79b predicts for sensitivity.
Determining the cell of origin
Gene-expression profiling on fresh tissue is considered the gold standard, but “it is clearly not a clinical tool,” Dr Zelenetz said. It requires the Wright classifier, a statistical method based on Bayes’ rule, to make patient-level assignments to 1 of the 3 subgroups.
Immunohistochemistry is widely available, but reproducibility may be difficult. Many assays exist, such as the Hans, Choi, and Muris assays, but, in many studies, there may be a lack of correlation with gene-expression profiling.
In the last few years, gene-expression profiling of formalin‐fixed paraffin‐embedded (FFPE) tissue has emerged as a reliable method. The assay is reproducible between laboratories, and it’s reproducible between different sets of reagents.
“[T]here is tremendous correlation between the Lymph2Cx assay and the gold standard,” Dr Zelenetz added.
“So here we have a robust assay,” he said, which allows investigators to explore whether the cell of origin is prognostic in large-cell lymphoma.
Prognosis
In a data set of 339 patients with de novo DLBCL treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the Lymph2Cx assay showing cell of origin was predictive of overall and progression-free survival.
However, the same exact assay applied to the RICOVER-60 data from the German High-Grade Non-Hodgkin Lymphoma Study group was not predictive, Dr Zelenetz reported, based on a personal communication from one of the investigators. There was a slight trend in favor of GCB tumors, but it was not statistically significant.
And the REMoDL-B study, using gene-expression profiling of FFPE tissue with the DASL assay, also didn’t show any difference in outcome between ABC or GCB tumors.
So gene-expression profiling of FFPE tissue does not universally show a prognostic difference, Dr Zelenetz said.
Influence of chemotherapy by cell of origin
CALGB 59910 showed that GCB tumors had a superior event-free, progression-free, and overall survival with dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) compared to ABC tumors.
“However, this is a phase 2, hypothesis-generating experiment,” Dr Zelenetz pointed out, and the results of the confirmatory study comparing dose-adjusted EPOCH-R and R-CHOP21 (CALGB 50303) will be presented later this year at the ASH Annual Meeting.
Sequential, non-cross-resistant chemotherapy
Data from the Memorial Sloan Kettering study (MSKCC 01-142/08-146; NCT00712582) of sequential therapy with R-CHOP followed by ICE (ifosfamide, carboplatin, and etoposide) demonstrate “excellent” progression-free and overall survival, Dr Zelenetz said.
“When we analyzed the outcome by cell of origin, there was a suggestion that the patients with the non-germinal center tumors were actually doing better than the germinal center tumors,” he added.
He pointed out one of the limitations of the study is that the cell of origin was determined by the Hans model. Nevertheless, the study raised another testable hypothesis: sequential therapy might overcome the adverse impact of the non-germinal center tumors.
Cell of origin analysis of the prospective, randomized study (LNH 03-2B) comparing R-ACVBP (rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, and prednisone) to R-CHOP showed that whether patients with GCB tumors received CHOP or ACVBP didn’t make “a whit of difference,” Dr Zelenetz said, in terms of progression-free and overall survival.
However, patients with ABC tumors demonstrated an “enormous difference in favor of R-ACVBP,” he said.
“Again, evidence that you can overcome the adverse effect of the ABC tumors with chemotherapy.”
Dr Zelenetz pointed out that R-ACVBP and R-CHOP followed by ICE are “actually remarkably similar regimens.” Both are sequential, both include consolidation, and both incorporate high-dose ifosfamide and etoposide.
“[S]o they actually reinforce each other,” he said, “demonstrating a similar result.”
Lenalidomide
Lenalidomide in the relapsed/refractory setting has modest activity in DLBCL, with most of the benefit accruing to patients with non-germinal center tumors.
Two clinical studies evaluated the impact of adding lenalidomide to standard chemotherapy.
In an Italian series using lenalidomide (L) plus R-CHOP21 in elderly untreated patients, the combination produced outstanding progression-free and event-free survival, but with no significant differences between the subtypes.
A US study of RL-CHOP versus R-CHOP included 87 matched historical controls treated with R-CHOP and 64 patients treated with RL-CHOP. Patients with non-germinal center tumors treated with RL-CHOP fared much better than historical controls treated with R-CHOP.
However, among germinal center tumors, “there was not a hint of any difference,” Dr Zelenetz noted.
Two studies—E1412, using an unselected population, and the international ROBUST study, selecting for patients with ABC tumors—are underway to confirm that the benefit with lenalidomide is in patients with activated B-cell tumors.
Ibrutinib
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, also has modest activity as a single agent in an unselected patient population with relapsed/refractory DLBCL. And most of the patients who demonstrated benefit had activated B-cell tumors.
Upon further analysis, investigators found that response was enhanced by the CD79b mutation, but it was not necessary for a response. And patients with CARD11 had no response.
MYD88 mutations seemed to cause resistance to ibrutinib, unless the mutation was associated with the CD79b mutation, and then patients had a “great” response, Dr Zelenetz explained.
In the upfront setting, a phase 1b study of R-CHOP plus ibrutinib demonstrated the safety of the combination, which had an overall survival rate of 100% and a complete response rate of 91%.
The prospective, randomized, phase 3 PHOENIX trial (NCT01855750) evaluating the combination in newly diagnosed non-germinal center DLBCL has completed accrual, but analysis is still pending.
Conclusion
“The prognostic significance of cell of origin is still controversial,” Dr Zelenetz wrapped up, “although I actually believe there is a prognostic difference in unselected registry patients.”
Sequential chemotherapy with ifosfamide and etoposide consolidation does very well in activated B-cell tumors, both in phase 2 and phase 3 studies.
“Importantly, small molecules seem to have differential effects totally predictable based on the biology of the difference between activated B-cell and germinal center tumors,” Dr Zelenetz said.
“But the big wild card here is somatic mutations further complicate things and will have to be incorporated into our understanding in the selection of patients.” ![]()
Study shows more bleeding with rivaroxaban than dabigatran

A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
Triplet improves PFS in relapsed/refractory MM

Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Fast Track to Abdominal Pain
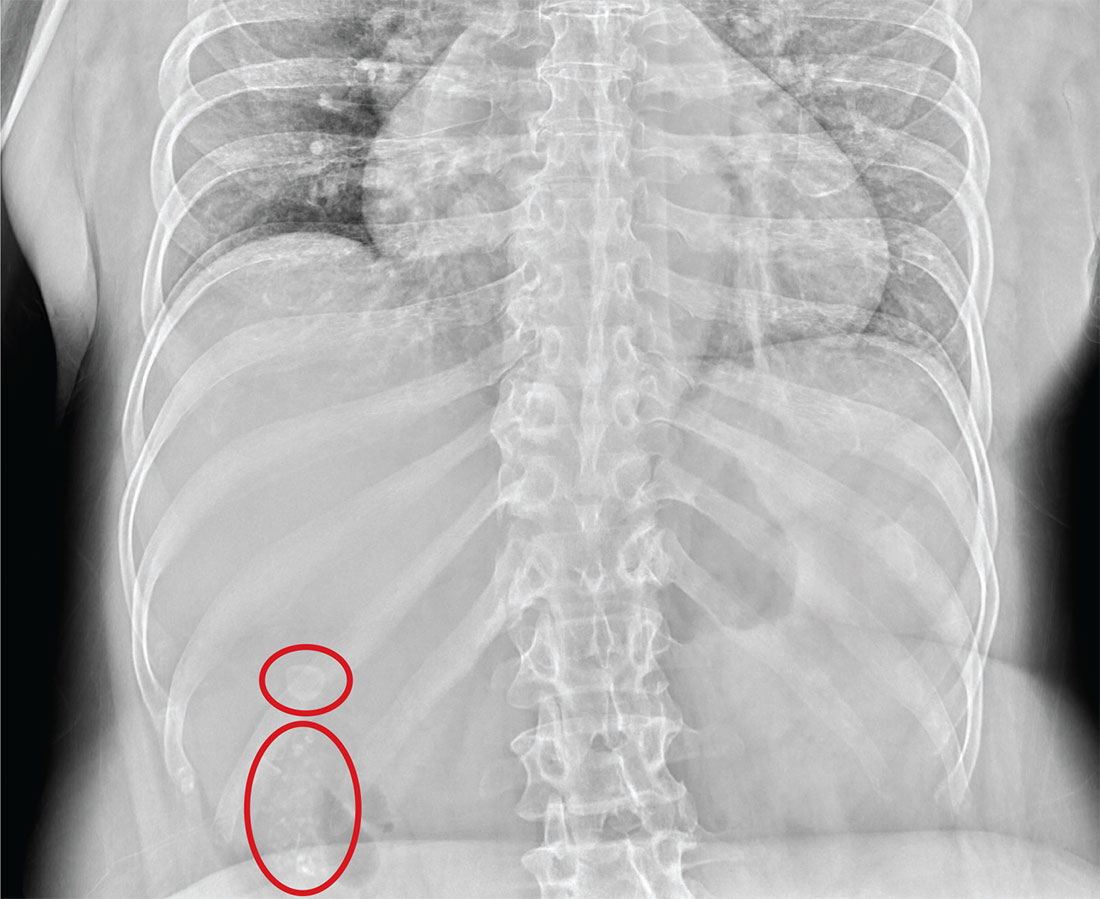
ANSWER
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
ANSWER
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
ANSWER
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
An NP student you are precepting in the emergency department fast track area presents her patient to you: a 60-year-old woman with abdominal pain. The pain is chronic but has worsened slightly, prompting the patient, who does not have a primary care provider, to present today. She experiences occasional nausea but no fever, and she denies any bowel or bladder complaints other than constipation. Her medical history is significant for mild hypertension.
On exam, your student notes an obese female who is in no obvious distress. The patient’s vital signs are all within normal limits. The abdominal exam is unimpressive, revealing a soft abdomen with good bowel sounds. Although she does have mild diffuse tenderness, she has no rebound or guarding.
Although your student suspects the patient is just constipated, she orders blood work and urinalysis. An abdominal survey is obtained as well. What is your impression?
In favorable intermediate-risk prostate cancer, brachytherapy alone suffices
BOSTON – In a finding hailed as “paradigm changing,” men with favorable intermediate-risk prostate cancer may have good disease control with brachytherapy alone, with fewer late-term toxicities than with brachytherapy combined with external-beam radiation, investigators reported.
After 5 years of follow-up, there were no significant differences in rates of freedom from disease progression among patients assigned to receive combined external-beam radiation (EBRT) and transperineal interstitial permanent brachytherapy (PB) or PB alone in selected patients with intermediate-risk prostatic carcinoma.

“I think you’re underselling your results – I’m pretty excited about these results,” Colleen Lawton, MD, professor and vice chair of radiation oncology at the Medical College of Wisconsin in Milwaukee, told Dr. Prestidge at the annual meeting of the American Society of Radiation Oncology.
The results show that, “for the intermediate-risk group, not the worst of intermediate [risk] but the vast majority, they don’t need the toxicity of that external beam, and they don’t need the cost of it,” she said at a briefing following his presentation of the data in a plenary session.
Dr. Lawton said that, when the NRG Oncology/RTOG 0232study was initiated (the first patients were enrolled in 2003), “there were factions that believed you could not treat intermediate-risk prostate cancer – which isn’t the best, isn’t the worse, but is somewhere in the middle – with anything but the combination, and this shows that’s not true.”
The trial was designed to test the hypothesis that patients with intermediate-risk prostate cancer treated with combined EBRT and PB would have a 10% improvement in freedom from progression (FFP), compared with men treated with PB alone.
A total of 579 patients with histologically confirmed prostate cancer, stages T1c-2b and Zubrod performance score 0 or 1 were enrolled. The patients also had Gleason score 2-6 and prostate-specific antigen (PSA) from 10 to less than 20 ng/mL; Gleason score and PSA less than 10 ng/mL; or prostate volume less than 60 cc.
In addition, androgen deprivation therapy (ADT) was not allowed on the study, and patients could not have distant metastases or radiographically suspicious nodes at the time of enrollment.
Patients were stratified by stage, Gleason score, PSA and history of ADT, and then randomized to the combined therapy, consisting of 45 Gy in a partial pelvis dose delivered in 1.8 Gy fractions for 5 weeks, followed 2-4 weeks later with brachytherapy using either a radioactive iodine (I-125) source prescribed as a 110-Gy boost dose, or palladium 103 in a 100 Gy boost dose; or brachytherapy alone (145-Gy dose with I-I25, 125-Gy dose with palladium 103). EBRT was delivered by either intensity-modulated radiation (43%) or 3D conformal radiation.
Five years after randomization, there were a total of 66 cases of treatment failure (measured as either biochemical failure according to ASTRO criteria, local progression, distant metastases, or death from any cause), 34 occurring in men treated with the combination, and 32 in men treated with brachytherapy alone. Biochemical failures accounted for 68% of events among patients on the combined modalities and 53% of those on brachytherapy alone. There were 9 deaths in the combined arm and 14 deaths in the brachytherapy-only arm, but none were prostate cancer related, Dr. Prestidge said.
In multivariate analysis adjusted for treatment type, age, race, prior hormonal therapy, Gleason/PSA, and tumor stage, the only significant predictor of FFP was Gleason 7/PSA less than 10 ng/mL, which was associated with better outcomes, compared with Gleason score less than 7/PSA 10-20 ng/mL (odds ratio, 0.5; P = .046).
There was no difference in 5-year overall survival between the groups.
As noted before, there were no significant differences in acute toxicities, but there were significantly more late grade 2 or greater toxicities among patients who underwent EBRT and PB (53% vs. 37%; P less than .0001), and more late grade 3 or greater toxicities (12% vs. 7%; P = .039).
Michael J Zelefsky, MD, professor of radiation oncology and chief of the brachytherapy service at Memorial Sloan Kettering Cancer Center in New York, the invited discussant, said that the study suggests that “combining EBRT with brachytherapy for favorable intermediate-risk disease may constitute unnecessary overkill.”
He asserted, however, that favorable intermediate-risk disease behaves clinically more like low-risk disease, whereas unfavorable intermediate-risk disease is closer to high-risk disease in its clinical behavior.
It is plausible that, for patients with unfavorable intermediate-risk disease, added dose intensification associated with combined modality therapy could result in improved FFP and local tumor control, he said.
The study was funded by the National Cancer Institute. Dr. Prestidge disclosed consulting for Varian, Elekta, and IsoRay. Dr. Zelefsky disclosed receiving honoraria from Bebig Brachytherapy, Augmenix, and as editor-in-chief of Brachytherapy. Dr. Lawton reported no relevant disclosures.
BOSTON – In a finding hailed as “paradigm changing,” men with favorable intermediate-risk prostate cancer may have good disease control with brachytherapy alone, with fewer late-term toxicities than with brachytherapy combined with external-beam radiation, investigators reported.
After 5 years of follow-up, there were no significant differences in rates of freedom from disease progression among patients assigned to receive combined external-beam radiation (EBRT) and transperineal interstitial permanent brachytherapy (PB) or PB alone in selected patients with intermediate-risk prostatic carcinoma.
“I think you’re underselling your results – I’m pretty excited about these results,” Colleen Lawton, MD, professor and vice chair of radiation oncology at the Medical College of Wisconsin in Milwaukee, told Dr. Prestidge at the annual meeting of the American Society of Radiation Oncology.
The results show that, “for the intermediate-risk group, not the worst of intermediate [risk] but the vast majority, they don’t need the toxicity of that external beam, and they don’t need the cost of it,” she said at a briefing following his presentation of the data in a plenary session.
Dr. Lawton said that, when the NRG Oncology/RTOG 0232study was initiated (the first patients were enrolled in 2003), “there were factions that believed you could not treat intermediate-risk prostate cancer – which isn’t the best, isn’t the worse, but is somewhere in the middle – with anything but the combination, and this shows that’s not true.”
The trial was designed to test the hypothesis that patients with intermediate-risk prostate cancer treated with combined EBRT and PB would have a 10% improvement in freedom from progression (FFP), compared with men treated with PB alone.
A total of 579 patients with histologically confirmed prostate cancer, stages T1c-2b and Zubrod performance score 0 or 1 were enrolled. The patients also had Gleason score 2-6 and prostate-specific antigen (PSA) from 10 to less than 20 ng/mL; Gleason score and PSA less than 10 ng/mL; or prostate volume less than 60 cc.
In addition, androgen deprivation therapy (ADT) was not allowed on the study, and patients could not have distant metastases or radiographically suspicious nodes at the time of enrollment.
Patients were stratified by stage, Gleason score, PSA and history of ADT, and then randomized to the combined therapy, consisting of 45 Gy in a partial pelvis dose delivered in 1.8 Gy fractions for 5 weeks, followed 2-4 weeks later with brachytherapy using either a radioactive iodine (I-125) source prescribed as a 110-Gy boost dose, or palladium 103 in a 100 Gy boost dose; or brachytherapy alone (145-Gy dose with I-I25, 125-Gy dose with palladium 103). EBRT was delivered by either intensity-modulated radiation (43%) or 3D conformal radiation.
Five years after randomization, there were a total of 66 cases of treatment failure (measured as either biochemical failure according to ASTRO criteria, local progression, distant metastases, or death from any cause), 34 occurring in men treated with the combination, and 32 in men treated with brachytherapy alone. Biochemical failures accounted for 68% of events among patients on the combined modalities and 53% of those on brachytherapy alone. There were 9 deaths in the combined arm and 14 deaths in the brachytherapy-only arm, but none were prostate cancer related, Dr. Prestidge said.
In multivariate analysis adjusted for treatment type, age, race, prior hormonal therapy, Gleason/PSA, and tumor stage, the only significant predictor of FFP was Gleason 7/PSA less than 10 ng/mL, which was associated with better outcomes, compared with Gleason score less than 7/PSA 10-20 ng/mL (odds ratio, 0.5; P = .046).
There was no difference in 5-year overall survival between the groups.
As noted before, there were no significant differences in acute toxicities, but there were significantly more late grade 2 or greater toxicities among patients who underwent EBRT and PB (53% vs. 37%; P less than .0001), and more late grade 3 or greater toxicities (12% vs. 7%; P = .039).
Michael J Zelefsky, MD, professor of radiation oncology and chief of the brachytherapy service at Memorial Sloan Kettering Cancer Center in New York, the invited discussant, said that the study suggests that “combining EBRT with brachytherapy for favorable intermediate-risk disease may constitute unnecessary overkill.”
He asserted, however, that favorable intermediate-risk disease behaves clinically more like low-risk disease, whereas unfavorable intermediate-risk disease is closer to high-risk disease in its clinical behavior.
It is plausible that, for patients with unfavorable intermediate-risk disease, added dose intensification associated with combined modality therapy could result in improved FFP and local tumor control, he said.
The study was funded by the National Cancer Institute. Dr. Prestidge disclosed consulting for Varian, Elekta, and IsoRay. Dr. Zelefsky disclosed receiving honoraria from Bebig Brachytherapy, Augmenix, and as editor-in-chief of Brachytherapy. Dr. Lawton reported no relevant disclosures.
BOSTON – In a finding hailed as “paradigm changing,” men with favorable intermediate-risk prostate cancer may have good disease control with brachytherapy alone, with fewer late-term toxicities than with brachytherapy combined with external-beam radiation, investigators reported.
After 5 years of follow-up, there were no significant differences in rates of freedom from disease progression among patients assigned to receive combined external-beam radiation (EBRT) and transperineal interstitial permanent brachytherapy (PB) or PB alone in selected patients with intermediate-risk prostatic carcinoma.
“I think you’re underselling your results – I’m pretty excited about these results,” Colleen Lawton, MD, professor and vice chair of radiation oncology at the Medical College of Wisconsin in Milwaukee, told Dr. Prestidge at the annual meeting of the American Society of Radiation Oncology.
The results show that, “for the intermediate-risk group, not the worst of intermediate [risk] but the vast majority, they don’t need the toxicity of that external beam, and they don’t need the cost of it,” she said at a briefing following his presentation of the data in a plenary session.
Dr. Lawton said that, when the NRG Oncology/RTOG 0232study was initiated (the first patients were enrolled in 2003), “there were factions that believed you could not treat intermediate-risk prostate cancer – which isn’t the best, isn’t the worse, but is somewhere in the middle – with anything but the combination, and this shows that’s not true.”
The trial was designed to test the hypothesis that patients with intermediate-risk prostate cancer treated with combined EBRT and PB would have a 10% improvement in freedom from progression (FFP), compared with men treated with PB alone.
A total of 579 patients with histologically confirmed prostate cancer, stages T1c-2b and Zubrod performance score 0 or 1 were enrolled. The patients also had Gleason score 2-6 and prostate-specific antigen (PSA) from 10 to less than 20 ng/mL; Gleason score and PSA less than 10 ng/mL; or prostate volume less than 60 cc.
In addition, androgen deprivation therapy (ADT) was not allowed on the study, and patients could not have distant metastases or radiographically suspicious nodes at the time of enrollment.
Patients were stratified by stage, Gleason score, PSA and history of ADT, and then randomized to the combined therapy, consisting of 45 Gy in a partial pelvis dose delivered in 1.8 Gy fractions for 5 weeks, followed 2-4 weeks later with brachytherapy using either a radioactive iodine (I-125) source prescribed as a 110-Gy boost dose, or palladium 103 in a 100 Gy boost dose; or brachytherapy alone (145-Gy dose with I-I25, 125-Gy dose with palladium 103). EBRT was delivered by either intensity-modulated radiation (43%) or 3D conformal radiation.
Five years after randomization, there were a total of 66 cases of treatment failure (measured as either biochemical failure according to ASTRO criteria, local progression, distant metastases, or death from any cause), 34 occurring in men treated with the combination, and 32 in men treated with brachytherapy alone. Biochemical failures accounted for 68% of events among patients on the combined modalities and 53% of those on brachytherapy alone. There were 9 deaths in the combined arm and 14 deaths in the brachytherapy-only arm, but none were prostate cancer related, Dr. Prestidge said.
In multivariate analysis adjusted for treatment type, age, race, prior hormonal therapy, Gleason/PSA, and tumor stage, the only significant predictor of FFP was Gleason 7/PSA less than 10 ng/mL, which was associated with better outcomes, compared with Gleason score less than 7/PSA 10-20 ng/mL (odds ratio, 0.5; P = .046).
There was no difference in 5-year overall survival between the groups.
As noted before, there were no significant differences in acute toxicities, but there were significantly more late grade 2 or greater toxicities among patients who underwent EBRT and PB (53% vs. 37%; P less than .0001), and more late grade 3 or greater toxicities (12% vs. 7%; P = .039).
Michael J Zelefsky, MD, professor of radiation oncology and chief of the brachytherapy service at Memorial Sloan Kettering Cancer Center in New York, the invited discussant, said that the study suggests that “combining EBRT with brachytherapy for favorable intermediate-risk disease may constitute unnecessary overkill.”
He asserted, however, that favorable intermediate-risk disease behaves clinically more like low-risk disease, whereas unfavorable intermediate-risk disease is closer to high-risk disease in its clinical behavior.
It is plausible that, for patients with unfavorable intermediate-risk disease, added dose intensification associated with combined modality therapy could result in improved FFP and local tumor control, he said.
The study was funded by the National Cancer Institute. Dr. Prestidge disclosed consulting for Varian, Elekta, and IsoRay. Dr. Zelefsky disclosed receiving honoraria from Bebig Brachytherapy, Augmenix, and as editor-in-chief of Brachytherapy. Dr. Lawton reported no relevant disclosures.
AT ASTRO 2016
Key clinical point: It may be clinically unnecessary to add external-beam radiation to brachytherapy in men with favorable intermediate-risk prostate cancer.
Major finding: Outcomes were comparable for men with favorable intermediate-risk prostate cancer treated with external-beam radiation plus brachytherapy or brachytherapy alone.
Data source: Randomized, controlled trial in 579 men with intermediate-risk prostate cancer.
Disclosures: The study was funded by the National Cancer Institute. Dr. Prestidge disclosed consulting for Varian, Elekta, and IsoRay. Dr. Zelefsky disclosed receiving honoraria from Bebig Brachytherapy, Augmenix, and as editor-in-chief of Brachytherapy. Dr. Lawton reported no relevant disclosures.
Ear-worn device helps Parkinson’s patients to improve their voice
PORTLAND, ORE. – A new device worn on one ear may help individuals with Parkinson’s disease to overcome speech deficits associated with the disease. When triggered by the wearer’s voice, the device, SpeechVive, provides a simulated room crowd noise or “babble” only when they are talking. Because it capitalizes on natural reflexes to this simulated background noise, improvement can occur independent of speech training.
Researchers led by Jessica Huber, PhD, a professor of speech, language, and hearing sciences at Purdue University in West Lafayette, Ind., investigated changes to speech after 3 months of daily use of the SpeechVive device by 16 individuals. The cohort had a mean age of 64.5 years (range: 56-78 years) and a mean disease duration of 8.1 years (range: 2-17 years). Six participants had previously had speech therapy (five with LSVT [Lee Silverman Voice Treatment] LOUD speech training exercises), and two had deep brain stimulation implants.

Similar to an earlier study of 39 participants, “We found a nice increase in loudness, that they just can wear the device, and they’re louder in their everyday communication,” Dr. Huber said in an interview during a poster session at the World Parkinson Congress. New to this study was a finding that the melody of speech was a little better, “so their questions are more like questions, and their statements are more like statements, so are the rising intonation on questions and the falling intonation on a statement.” Participants were also able to say more on one breath and to sound more natural with appropriate pauses because of the longer utterances.
After 3 months of use, participants as a group increased their sound pressure level (loudness) from 76.6 dB to 78.8 dB (a gain of 2.2 dB) from having the device OFF to ON. When it was then turned off, they had an intermediate sound pressure level of 77.7 dB (both P less than .0001 vs. pretest OFF). Intonation variability and intonation range also showed significant improvements for both statements and questions with the use of the device (all P less than or equal to .01). There were also improvements in a correct statement or question being produced, pausing patterns, and utterance length. No adverse events occurred in the study.
Dr. Huber said patients in the study were quite variable in their responses to SpeechVive for the different measures of speech. In general, she has found that about 75% of users get louder with the device, and some have clearer articulation or slower speech. Another 10%-15% have slower speech or better articulation without an increase in loudness.
A total of 50% of the users have a “carry-over” effect after using the device when they are not wearing it. “After about 8 weeks, they don’t need to keep wearing the device every day,” Dr. Huber noted. Others may wear it in the morning and have a lasting effect for the rest of the day, and some lose any benefit as soon as taking the device off. Not all patients reach a normal speech loudness, “but they’re going to get way better” than where they started, she said.
Available speech therapies
People with Parkinson’s disease (PD) often have reduced vocal loudness, increased speech rate, and slurred articulation. Available speech therapies have included adduction exercises, vocal function exercises, and LSVT LOUD speech training exercises. For some individuals with PD, these behavioral treatments may not carry over into daily life.
The SpeechVive consists of a piece placed into the ear canal and a piece that sits behind the ear, similar to a slightly larger version of a modern hearing aid. It is designed large enough that many patients, who have motor deficits, can don it themselves. Because it is on just one ear, users can hear other people talking and what is going on around them.
Dr. Huber noted several strengths of the device. First, it does not impose any cognitive load on the user because its effect is based on an automatic reflex. Second, little training is necessary. It is easy for the user to put on the device, and many of its beneficial effects occur as soon as it is activated. Third, compliance can be tracked using data stored by the device itself.
In a previous study, she saw that after using the device for 8 weeks, participants used more effective patterns with their respiratory and laryngeal mechanisms to produce louder speech. “It’s not like they’re working super hard after 8 weeks.” She thinks they are just not usually using their speech apparatus in the most efficient manner, and they become more efficient after using the device. They may also not sense how loud they are without it, so it helps patients “recalibrate” a sense of their own voices, something the LSVT LOUD training also does.
Jori Fleisher, MD, a movement disorders neurologist at the New York University Langone Medical Center, commented that the study shows an interesting approach to the common and difficult problem of hypophonia and dysphonia in people with Parkinson’s disease. “It can be very difficult to speak loud enough to be heard, and it often leads to people withdrawing socially and not speaking as much in conversation as they normally would,” she said. “LSVT LOUD works in practice when you’re actually doing the exercises, but often when people stop doing the exercises, their voice goes back toward their normal. So this is something that you can keep with you and sort of have constant prompting to overcome that tendency toward quieter voice. It has a lot of promise.”
She said she would like to see the data on compliance or adherence with using the device “because it seems like a great idea, but how much are people actually using it? ... Having more long-term follow-up to see whether this is something that people want to put into practice in their daily life would be very useful.” She also said she would like to see how long the carry-over effect lasts once people take off the device to be able to compare it with sessions of speech therapy or doing speech exercises.
SpeechVive is $2,495, which includes the device, a charger, and earpiece fittings. It is available only in the United States, but the company now has clearance to market it in Canada.
The research was funded by a grant from SpeechVive. Dr. Huber is the inventor of SpeechVive, has a patent on it, has a financial interest in the SpeechVive company, and sits on its board of directors. Dr. Fleisher reported having no financial conflicts.
PORTLAND, ORE. – A new device worn on one ear may help individuals with Parkinson’s disease to overcome speech deficits associated with the disease. When triggered by the wearer’s voice, the device, SpeechVive, provides a simulated room crowd noise or “babble” only when they are talking. Because it capitalizes on natural reflexes to this simulated background noise, improvement can occur independent of speech training.
Researchers led by Jessica Huber, PhD, a professor of speech, language, and hearing sciences at Purdue University in West Lafayette, Ind., investigated changes to speech after 3 months of daily use of the SpeechVive device by 16 individuals. The cohort had a mean age of 64.5 years (range: 56-78 years) and a mean disease duration of 8.1 years (range: 2-17 years). Six participants had previously had speech therapy (five with LSVT [Lee Silverman Voice Treatment] LOUD speech training exercises), and two had deep brain stimulation implants.
Similar to an earlier study of 39 participants, “We found a nice increase in loudness, that they just can wear the device, and they’re louder in their everyday communication,” Dr. Huber said in an interview during a poster session at the World Parkinson Congress. New to this study was a finding that the melody of speech was a little better, “so their questions are more like questions, and their statements are more like statements, so are the rising intonation on questions and the falling intonation on a statement.” Participants were also able to say more on one breath and to sound more natural with appropriate pauses because of the longer utterances.
After 3 months of use, participants as a group increased their sound pressure level (loudness) from 76.6 dB to 78.8 dB (a gain of 2.2 dB) from having the device OFF to ON. When it was then turned off, they had an intermediate sound pressure level of 77.7 dB (both P less than .0001 vs. pretest OFF). Intonation variability and intonation range also showed significant improvements for both statements and questions with the use of the device (all P less than or equal to .01). There were also improvements in a correct statement or question being produced, pausing patterns, and utterance length. No adverse events occurred in the study.
Dr. Huber said patients in the study were quite variable in their responses to SpeechVive for the different measures of speech. In general, she has found that about 75% of users get louder with the device, and some have clearer articulation or slower speech. Another 10%-15% have slower speech or better articulation without an increase in loudness.
A total of 50% of the users have a “carry-over” effect after using the device when they are not wearing it. “After about 8 weeks, they don’t need to keep wearing the device every day,” Dr. Huber noted. Others may wear it in the morning and have a lasting effect for the rest of the day, and some lose any benefit as soon as taking the device off. Not all patients reach a normal speech loudness, “but they’re going to get way better” than where they started, she said.
Available speech therapies
People with Parkinson’s disease (PD) often have reduced vocal loudness, increased speech rate, and slurred articulation. Available speech therapies have included adduction exercises, vocal function exercises, and LSVT LOUD speech training exercises. For some individuals with PD, these behavioral treatments may not carry over into daily life.
The SpeechVive consists of a piece placed into the ear canal and a piece that sits behind the ear, similar to a slightly larger version of a modern hearing aid. It is designed large enough that many patients, who have motor deficits, can don it themselves. Because it is on just one ear, users can hear other people talking and what is going on around them.
Dr. Huber noted several strengths of the device. First, it does not impose any cognitive load on the user because its effect is based on an automatic reflex. Second, little training is necessary. It is easy for the user to put on the device, and many of its beneficial effects occur as soon as it is activated. Third, compliance can be tracked using data stored by the device itself.
In a previous study, she saw that after using the device for 8 weeks, participants used more effective patterns with their respiratory and laryngeal mechanisms to produce louder speech. “It’s not like they’re working super hard after 8 weeks.” She thinks they are just not usually using their speech apparatus in the most efficient manner, and they become more efficient after using the device. They may also not sense how loud they are without it, so it helps patients “recalibrate” a sense of their own voices, something the LSVT LOUD training also does.
Jori Fleisher, MD, a movement disorders neurologist at the New York University Langone Medical Center, commented that the study shows an interesting approach to the common and difficult problem of hypophonia and dysphonia in people with Parkinson’s disease. “It can be very difficult to speak loud enough to be heard, and it often leads to people withdrawing socially and not speaking as much in conversation as they normally would,” she said. “LSVT LOUD works in practice when you’re actually doing the exercises, but often when people stop doing the exercises, their voice goes back toward their normal. So this is something that you can keep with you and sort of have constant prompting to overcome that tendency toward quieter voice. It has a lot of promise.”
She said she would like to see the data on compliance or adherence with using the device “because it seems like a great idea, but how much are people actually using it? ... Having more long-term follow-up to see whether this is something that people want to put into practice in their daily life would be very useful.” She also said she would like to see how long the carry-over effect lasts once people take off the device to be able to compare it with sessions of speech therapy or doing speech exercises.
SpeechVive is $2,495, which includes the device, a charger, and earpiece fittings. It is available only in the United States, but the company now has clearance to market it in Canada.
The research was funded by a grant from SpeechVive. Dr. Huber is the inventor of SpeechVive, has a patent on it, has a financial interest in the SpeechVive company, and sits on its board of directors. Dr. Fleisher reported having no financial conflicts.
PORTLAND, ORE. – A new device worn on one ear may help individuals with Parkinson’s disease to overcome speech deficits associated with the disease. When triggered by the wearer’s voice, the device, SpeechVive, provides a simulated room crowd noise or “babble” only when they are talking. Because it capitalizes on natural reflexes to this simulated background noise, improvement can occur independent of speech training.
Researchers led by Jessica Huber, PhD, a professor of speech, language, and hearing sciences at Purdue University in West Lafayette, Ind., investigated changes to speech after 3 months of daily use of the SpeechVive device by 16 individuals. The cohort had a mean age of 64.5 years (range: 56-78 years) and a mean disease duration of 8.1 years (range: 2-17 years). Six participants had previously had speech therapy (five with LSVT [Lee Silverman Voice Treatment] LOUD speech training exercises), and two had deep brain stimulation implants.
Similar to an earlier study of 39 participants, “We found a nice increase in loudness, that they just can wear the device, and they’re louder in their everyday communication,” Dr. Huber said in an interview during a poster session at the World Parkinson Congress. New to this study was a finding that the melody of speech was a little better, “so their questions are more like questions, and their statements are more like statements, so are the rising intonation on questions and the falling intonation on a statement.” Participants were also able to say more on one breath and to sound more natural with appropriate pauses because of the longer utterances.
After 3 months of use, participants as a group increased their sound pressure level (loudness) from 76.6 dB to 78.8 dB (a gain of 2.2 dB) from having the device OFF to ON. When it was then turned off, they had an intermediate sound pressure level of 77.7 dB (both P less than .0001 vs. pretest OFF). Intonation variability and intonation range also showed significant improvements for both statements and questions with the use of the device (all P less than or equal to .01). There were also improvements in a correct statement or question being produced, pausing patterns, and utterance length. No adverse events occurred in the study.
Dr. Huber said patients in the study were quite variable in their responses to SpeechVive for the different measures of speech. In general, she has found that about 75% of users get louder with the device, and some have clearer articulation or slower speech. Another 10%-15% have slower speech or better articulation without an increase in loudness.
A total of 50% of the users have a “carry-over” effect after using the device when they are not wearing it. “After about 8 weeks, they don’t need to keep wearing the device every day,” Dr. Huber noted. Others may wear it in the morning and have a lasting effect for the rest of the day, and some lose any benefit as soon as taking the device off. Not all patients reach a normal speech loudness, “but they’re going to get way better” than where they started, she said.
Available speech therapies
People with Parkinson’s disease (PD) often have reduced vocal loudness, increased speech rate, and slurred articulation. Available speech therapies have included adduction exercises, vocal function exercises, and LSVT LOUD speech training exercises. For some individuals with PD, these behavioral treatments may not carry over into daily life.
The SpeechVive consists of a piece placed into the ear canal and a piece that sits behind the ear, similar to a slightly larger version of a modern hearing aid. It is designed large enough that many patients, who have motor deficits, can don it themselves. Because it is on just one ear, users can hear other people talking and what is going on around them.
Dr. Huber noted several strengths of the device. First, it does not impose any cognitive load on the user because its effect is based on an automatic reflex. Second, little training is necessary. It is easy for the user to put on the device, and many of its beneficial effects occur as soon as it is activated. Third, compliance can be tracked using data stored by the device itself.
In a previous study, she saw that after using the device for 8 weeks, participants used more effective patterns with their respiratory and laryngeal mechanisms to produce louder speech. “It’s not like they’re working super hard after 8 weeks.” She thinks they are just not usually using their speech apparatus in the most efficient manner, and they become more efficient after using the device. They may also not sense how loud they are without it, so it helps patients “recalibrate” a sense of their own voices, something the LSVT LOUD training also does.
Jori Fleisher, MD, a movement disorders neurologist at the New York University Langone Medical Center, commented that the study shows an interesting approach to the common and difficult problem of hypophonia and dysphonia in people with Parkinson’s disease. “It can be very difficult to speak loud enough to be heard, and it often leads to people withdrawing socially and not speaking as much in conversation as they normally would,” she said. “LSVT LOUD works in practice when you’re actually doing the exercises, but often when people stop doing the exercises, their voice goes back toward their normal. So this is something that you can keep with you and sort of have constant prompting to overcome that tendency toward quieter voice. It has a lot of promise.”
She said she would like to see the data on compliance or adherence with using the device “because it seems like a great idea, but how much are people actually using it? ... Having more long-term follow-up to see whether this is something that people want to put into practice in their daily life would be very useful.” She also said she would like to see how long the carry-over effect lasts once people take off the device to be able to compare it with sessions of speech therapy or doing speech exercises.
SpeechVive is $2,495, which includes the device, a charger, and earpiece fittings. It is available only in the United States, but the company now has clearance to market it in Canada.
The research was funded by a grant from SpeechVive. Dr. Huber is the inventor of SpeechVive, has a patent on it, has a financial interest in the SpeechVive company, and sits on its board of directors. Dr. Fleisher reported having no financial conflicts.
AT WPC 2016
Key clinical point:
Major finding: Loudness increased 2.2 dB from 76.6 dB to 78.8 dB after 3 months of use. Intonation also improved.
Data source: A prospective study of 16 participants with hypophonia comparing speech parameters with and without the device after 3 months of use.
Disclosures: The research was funded by a grant from SpeechVive. Dr. Huber is the inventor of SpeechVive, has a patent on it, has a financial interest in the SpeechVive company, and sits on its board of directors. Dr. Fleisher reported having no financial conflicts.
Calcium channel blocker reduces cardiac iron loading in thalassemia major
The calcium channel blocker amlodipine, added to iron chelation therapy, significantly reduced excess myocardial iron concentration in patients with thalassemia major, compared with chelation alone, according to results from a randomized trial.
The findings (Blood. 2016;128[12]:1555-61) suggest that amlodipine, a cheap, widely available drug with a well-established safety profile, may serve as an adjunct to standard treatment for people with thalassemia major and cardiac siderosis. Cardiovascular disease caused by excess myocardial iron remains a major cause of morbidity and mortality in thalassemia major.
Juliano L. Fernandes, MD, PhD, of the Jose Michel Kalaf Research Institute in Campinas, Brazil, led the study, which randomized 62 patients already receiving chelation treatment for thalassemia major to 1 year of chelation plus placebo (n = 31) or chelation plus 5 mg daily amlodipine (n = 31).
Patients in each arm were subdivided into two subgroups: those whose baseline myocardial iron concentration was within normal thresholds, and those with excess myocardial iron concentration as measured by magnetic resonance imaging (above 0.59 mg/g dry weight or with a cardiac T2* below 35 milliseconds).
In the amlodipine arm, patients with excess cardiac iron at baseline (n = 15) saw significant reductions in myocardial iron concentrations at 1 year, compared with those randomized to placebo (n = 15). The former had a median reduction of –0.26 mg/g (95% confidence interval, –1.02 to –0.01) while the placebo group saw an increase of 0.01 mg/g (95% CI, 20.13 to 20.23; P = .02).
The investigators acknowledged that some of the findings were limited by the study’s short observation period.
Patients without excess myocardial iron concentration at baseline did not see significant changes associated with amlodipine. While Dr. Fernandes and his colleagues could not conclude that the drug prevented excess cardiac iron from accumulating, “our data cannot rule out the possibility that extended use of amlodipine might prevent myocardial iron accumulation with a longer observation period.”
Secondary endpoints of the study included measurements of iron storage in the liver and of serum ferritin, neither of which appeared to be affected by amlodipine treatment, which the investigators said was consistent with the drug’s known mechanism of action. No serious adverse effects were reported related to amlodipine treatment.
Dr. Fernandes and his colleagues also did not find improvements in left ventricular ejection fraction associated with amlodipine use at 12 months. This may be due, they wrote in their analysis, to a “relatively low prevalence of reduced ejection fraction or severe myocardial siderosis upon trial enrollment, limiting the power of the study to assess these outcomes.”
The government of Brazil and the Sultan Bin Khalifa Translational Research Scholarship sponsored the study. Dr. Fernandes reported receiving fees from Novartis and Sanofi. The remaining 12 authors disclosed no conflicts of interest.
Why is this small clinical trial of such pivotal importance in this day and age of massive multicenter prospective randomized studies? The answer is that it tells us that iron entry into the heart through L-type calcium channels, a mechanism that has been clearly demonstrated in vitro, seems to be actually occurring in humans. As an added bonus, we have a possible new adjunctive treatment of iron cardiomyopathy. More clinical studies are needed, and certainly biochemical studies need to continue because all calcium channel blockers do not have the same effect in vitro, but at least the “channels” for more progress on both clinical and biochemical fronts are now open.
Thomas D. Coates, MD, is with Children’s Hospital of Los Angeles and University of Southern California, Los Angeles. He made his remarks in an editorial that accompanied the published study.
Why is this small clinical trial of such pivotal importance in this day and age of massive multicenter prospective randomized studies? The answer is that it tells us that iron entry into the heart through L-type calcium channels, a mechanism that has been clearly demonstrated in vitro, seems to be actually occurring in humans. As an added bonus, we have a possible new adjunctive treatment of iron cardiomyopathy. More clinical studies are needed, and certainly biochemical studies need to continue because all calcium channel blockers do not have the same effect in vitro, but at least the “channels” for more progress on both clinical and biochemical fronts are now open.
Thomas D. Coates, MD, is with Children’s Hospital of Los Angeles and University of Southern California, Los Angeles. He made his remarks in an editorial that accompanied the published study.
Why is this small clinical trial of such pivotal importance in this day and age of massive multicenter prospective randomized studies? The answer is that it tells us that iron entry into the heart through L-type calcium channels, a mechanism that has been clearly demonstrated in vitro, seems to be actually occurring in humans. As an added bonus, we have a possible new adjunctive treatment of iron cardiomyopathy. More clinical studies are needed, and certainly biochemical studies need to continue because all calcium channel blockers do not have the same effect in vitro, but at least the “channels” for more progress on both clinical and biochemical fronts are now open.
Thomas D. Coates, MD, is with Children’s Hospital of Los Angeles and University of Southern California, Los Angeles. He made his remarks in an editorial that accompanied the published study.
The calcium channel blocker amlodipine, added to iron chelation therapy, significantly reduced excess myocardial iron concentration in patients with thalassemia major, compared with chelation alone, according to results from a randomized trial.
The findings (Blood. 2016;128[12]:1555-61) suggest that amlodipine, a cheap, widely available drug with a well-established safety profile, may serve as an adjunct to standard treatment for people with thalassemia major and cardiac siderosis. Cardiovascular disease caused by excess myocardial iron remains a major cause of morbidity and mortality in thalassemia major.
Juliano L. Fernandes, MD, PhD, of the Jose Michel Kalaf Research Institute in Campinas, Brazil, led the study, which randomized 62 patients already receiving chelation treatment for thalassemia major to 1 year of chelation plus placebo (n = 31) or chelation plus 5 mg daily amlodipine (n = 31).
Patients in each arm were subdivided into two subgroups: those whose baseline myocardial iron concentration was within normal thresholds, and those with excess myocardial iron concentration as measured by magnetic resonance imaging (above 0.59 mg/g dry weight or with a cardiac T2* below 35 milliseconds).
In the amlodipine arm, patients with excess cardiac iron at baseline (n = 15) saw significant reductions in myocardial iron concentrations at 1 year, compared with those randomized to placebo (n = 15). The former had a median reduction of –0.26 mg/g (95% confidence interval, –1.02 to –0.01) while the placebo group saw an increase of 0.01 mg/g (95% CI, 20.13 to 20.23; P = .02).
The investigators acknowledged that some of the findings were limited by the study’s short observation period.
Patients without excess myocardial iron concentration at baseline did not see significant changes associated with amlodipine. While Dr. Fernandes and his colleagues could not conclude that the drug prevented excess cardiac iron from accumulating, “our data cannot rule out the possibility that extended use of amlodipine might prevent myocardial iron accumulation with a longer observation period.”
Secondary endpoints of the study included measurements of iron storage in the liver and of serum ferritin, neither of which appeared to be affected by amlodipine treatment, which the investigators said was consistent with the drug’s known mechanism of action. No serious adverse effects were reported related to amlodipine treatment.
Dr. Fernandes and his colleagues also did not find improvements in left ventricular ejection fraction associated with amlodipine use at 12 months. This may be due, they wrote in their analysis, to a “relatively low prevalence of reduced ejection fraction or severe myocardial siderosis upon trial enrollment, limiting the power of the study to assess these outcomes.”
The government of Brazil and the Sultan Bin Khalifa Translational Research Scholarship sponsored the study. Dr. Fernandes reported receiving fees from Novartis and Sanofi. The remaining 12 authors disclosed no conflicts of interest.
The calcium channel blocker amlodipine, added to iron chelation therapy, significantly reduced excess myocardial iron concentration in patients with thalassemia major, compared with chelation alone, according to results from a randomized trial.
The findings (Blood. 2016;128[12]:1555-61) suggest that amlodipine, a cheap, widely available drug with a well-established safety profile, may serve as an adjunct to standard treatment for people with thalassemia major and cardiac siderosis. Cardiovascular disease caused by excess myocardial iron remains a major cause of morbidity and mortality in thalassemia major.
Juliano L. Fernandes, MD, PhD, of the Jose Michel Kalaf Research Institute in Campinas, Brazil, led the study, which randomized 62 patients already receiving chelation treatment for thalassemia major to 1 year of chelation plus placebo (n = 31) or chelation plus 5 mg daily amlodipine (n = 31).
Patients in each arm were subdivided into two subgroups: those whose baseline myocardial iron concentration was within normal thresholds, and those with excess myocardial iron concentration as measured by magnetic resonance imaging (above 0.59 mg/g dry weight or with a cardiac T2* below 35 milliseconds).
In the amlodipine arm, patients with excess cardiac iron at baseline (n = 15) saw significant reductions in myocardial iron concentrations at 1 year, compared with those randomized to placebo (n = 15). The former had a median reduction of –0.26 mg/g (95% confidence interval, –1.02 to –0.01) while the placebo group saw an increase of 0.01 mg/g (95% CI, 20.13 to 20.23; P = .02).
The investigators acknowledged that some of the findings were limited by the study’s short observation period.
Patients without excess myocardial iron concentration at baseline did not see significant changes associated with amlodipine. While Dr. Fernandes and his colleagues could not conclude that the drug prevented excess cardiac iron from accumulating, “our data cannot rule out the possibility that extended use of amlodipine might prevent myocardial iron accumulation with a longer observation period.”
Secondary endpoints of the study included measurements of iron storage in the liver and of serum ferritin, neither of which appeared to be affected by amlodipine treatment, which the investigators said was consistent with the drug’s known mechanism of action. No serious adverse effects were reported related to amlodipine treatment.
Dr. Fernandes and his colleagues also did not find improvements in left ventricular ejection fraction associated with amlodipine use at 12 months. This may be due, they wrote in their analysis, to a “relatively low prevalence of reduced ejection fraction or severe myocardial siderosis upon trial enrollment, limiting the power of the study to assess these outcomes.”
The government of Brazil and the Sultan Bin Khalifa Translational Research Scholarship sponsored the study. Dr. Fernandes reported receiving fees from Novartis and Sanofi. The remaining 12 authors disclosed no conflicts of interest.
FROM BLOOD
Key clinical point: Amlodipine added to standard chelation therapy significantly reduced cardiac iron in thalassemia major patients with cardiac siderosis.
Major finding: At 12 months, cardiac iron was a median 0.26 mg/g lower in subjects with myocardial iron overload treated with 5 mg daily amlodipine plus chelation, while patients treated with chelation alone saw a 0.01 mg/g increase (P = .02).
Data source: A randomized, double-blind, placebo-controlled trial enrolling from 62 patients with TM from six centers in Brazil, about half with cardiac siderosis at baseline.
Disclosures: The Brazil government and the Sultan Bin Khalifa Translational Research Scholarship sponsored the investigation. Its lead author reported receiving fees from Novartis and Sanofi. Other study investigators and the author of a linked editorial declared no conflicts of interest.