User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Endocrine societies push back on discriminatory transgender health policies
Science should be the cornerstone for health policy, and decisions on medical care of transgender and gender-diverse (TGD) individuals should be between a patient and their doctor.

That’s according to a joint policy statement from the Endocrine Society and Pediatric Endocrine Society published in the Journal of Clinical Endocrinology & Metabolism expressing concern about recent proposed legislation that would limit access to medical care for TGD individuals.
“The main emphasis is that we are simply medical people trying to be conservative and science driven in the care of our patients,” Joshua D. Safer, MD, coauthor and executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System, and professor of medicine at Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Why the health care for a particular group of people should be considered political is a mystery to me.”
TGD individuals have seen a recent uptick in efforts to limit or restrict their access to medical care at the federal and state levels. In June 2020, the Department of Health & Human Services finalized a revision to Section 1557 of the Affordable Care Act, rolling back a 2016 rule that determined the phrase “on the basis of sex” included nondiscrimination based on a person’s sex and gender identity. The Endocrine Society opposed this rule revision, arguing that it would allow “providers to deny care to TGD persons as well as discourage patients from seeking routine and gender-affirming care or reporting discrimination.”
Over a dozen U.S. states have introduced proposed legislation concerning medical care of TGD individuals that contain erroneous and misleading information. Proposed laws in Alabama, Missouri, and Texas, for example, would prohibit any use of medical treatments for minors for the purpose of gender-affirming medical care, including “gonadotropin-releasing hormone agonist therapy for pubertal suppression and gender-affirming hormonal therapy,” the authors of the joint statement wrote. In some cases, medical professionals who provide medical care for TGD patients could face criminal charges.
Outside the United States, three High Court judges in the United Kingdom recently ruled that minors aged under 16 years could not legally consent to pubertal suppression. “The recent U.K. court decision could be very disruptive because it would raise a barrier to transgender children receiving puberty blockers at exactly the ages that puberty blockers would be typically used,” Dr. Safer said.
Misleading characterizations of gender-affirming medical care for TGD individuals have also been spread to the general public. A recent Republican primary ballot proposition in Texas asked whether the state should ban “chemical castration, puberty blockers, cross-sex hormones, and genital mutilation surgery on all minor children for transition purposes,” falsely asserting that “Texas children as young as 3 are being transitioned from their biological sex to the opposite sex,” referencing a high-profile custody battle of a transgender child in Texas.
There are several tiers of misinformation that exist within these statements, Dr. Safer noted. “Some statements have suggested that gender-affirming treatment for young children can include hormone therapy or even surgery. Of course, there are no medical treatments for transgender and gender-diverse children prior to puberty.”
For adolescents aged under 18 years, Endocrine Society guidelines released in 2017 state that pubertal suppression is fully reversible and “offered to adolescents who meet diagnostic and treatment criteria, and are requesting care, for gender dysphoria/gender incongruence after they exhibit physical changes of puberty,” Dr. Safer and coauthors wrote in the joint policy statement. Other, more permanent – but still partially reversible – treatments such as hormone therapy are available as options for adolescents with confirmed and persistent gender dysphoria/gender incongruence, after meeting with a team of medical and mental health professionals and giving informed consent, according to the guidelines.
Dr. Safer expressed surprise at the opposition to puberty blockers in proposed state legislation. “Puberty blockers are the conservative option so that we can avoid permanent changes while thoughtful decisions are being made by our adolescent patients with their families and health care providers,” he said.
The perception that puberty blockers will lead to hormone therapy is another misunderstanding and source of misinformation, Dr. Safer explained.
“The fear is that these data suggest that puberty blockers are a ‘gateway drug’ of some sort. But that is false. The reason that most adolescents who take puberty blockers go on to hormone therapy is because most of the adolescents who are identified in our conservative systems are actually transgender and interested in more gender-affirming care as they age,” he said.
Effects of discrimination on TGD persons
Many of these proposed state bills have not advanced through state legislatures, but a few – such as the proposed laws in Alabama, Missouri, and Texas – are still currently under consideration.
“In the United States, most recent efforts to single out transgender and gender-diverse people for discrimination in health care have failed. However, the demonization of trans people and attempts to disrupt care have been the source of much stress among our patients,” Dr. Safer said.
Restricting access to health care has “multiple implications” for TGD patients. “In the era when we did not provide care to transgender youth, we had a situation where approximately 40% of transgender people had considered suicide in their lives,” Dr. Safer said. In contrast, having access to these treatments has been shown to improve mental health outcomes in these patients, according to an Endocrine Society position statement.
The purpose of earlier interventions such as puberty blockers is to allow an adolescent to “explore options and live in the experienced gender before making a decision to proceed with gender-affirming hormone therapy,” the authors of the Endocrine Society and Pediatric Endocrine Society joint statement said.
Blocking access to puberty blockers, on the other hand, forces transgender youth to experience a puberty that doesn’t match their gender identity, Dr. Safer noted. “The puberty will include permanent changes which will then have to be reversed with surgery. Why would we intentionally allow that to happen?”
Dr. Safer reported that his spouse is an employee of Parexel. Dr. Tangpricha is the current president of the World Association for Transgender Health and a board member of the American Association of Clinical Endocrinology. The other authors reported no relevant conflicts of interests.
SOURCE: Safer JD et al. J Clin Endocrinol Metab. 2020. doi: 10.1210/clinem/dgaa816.
Science should be the cornerstone for health policy, and decisions on medical care of transgender and gender-diverse (TGD) individuals should be between a patient and their doctor.
That’s according to a joint policy statement from the Endocrine Society and Pediatric Endocrine Society published in the Journal of Clinical Endocrinology & Metabolism expressing concern about recent proposed legislation that would limit access to medical care for TGD individuals.
“The main emphasis is that we are simply medical people trying to be conservative and science driven in the care of our patients,” Joshua D. Safer, MD, coauthor and executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System, and professor of medicine at Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Why the health care for a particular group of people should be considered political is a mystery to me.”
TGD individuals have seen a recent uptick in efforts to limit or restrict their access to medical care at the federal and state levels. In June 2020, the Department of Health & Human Services finalized a revision to Section 1557 of the Affordable Care Act, rolling back a 2016 rule that determined the phrase “on the basis of sex” included nondiscrimination based on a person’s sex and gender identity. The Endocrine Society opposed this rule revision, arguing that it would allow “providers to deny care to TGD persons as well as discourage patients from seeking routine and gender-affirming care or reporting discrimination.”
Over a dozen U.S. states have introduced proposed legislation concerning medical care of TGD individuals that contain erroneous and misleading information. Proposed laws in Alabama, Missouri, and Texas, for example, would prohibit any use of medical treatments for minors for the purpose of gender-affirming medical care, including “gonadotropin-releasing hormone agonist therapy for pubertal suppression and gender-affirming hormonal therapy,” the authors of the joint statement wrote. In some cases, medical professionals who provide medical care for TGD patients could face criminal charges.
Outside the United States, three High Court judges in the United Kingdom recently ruled that minors aged under 16 years could not legally consent to pubertal suppression. “The recent U.K. court decision could be very disruptive because it would raise a barrier to transgender children receiving puberty blockers at exactly the ages that puberty blockers would be typically used,” Dr. Safer said.
Misleading characterizations of gender-affirming medical care for TGD individuals have also been spread to the general public. A recent Republican primary ballot proposition in Texas asked whether the state should ban “chemical castration, puberty blockers, cross-sex hormones, and genital mutilation surgery on all minor children for transition purposes,” falsely asserting that “Texas children as young as 3 are being transitioned from their biological sex to the opposite sex,” referencing a high-profile custody battle of a transgender child in Texas.
There are several tiers of misinformation that exist within these statements, Dr. Safer noted. “Some statements have suggested that gender-affirming treatment for young children can include hormone therapy or even surgery. Of course, there are no medical treatments for transgender and gender-diverse children prior to puberty.”
For adolescents aged under 18 years, Endocrine Society guidelines released in 2017 state that pubertal suppression is fully reversible and “offered to adolescents who meet diagnostic and treatment criteria, and are requesting care, for gender dysphoria/gender incongruence after they exhibit physical changes of puberty,” Dr. Safer and coauthors wrote in the joint policy statement. Other, more permanent – but still partially reversible – treatments such as hormone therapy are available as options for adolescents with confirmed and persistent gender dysphoria/gender incongruence, after meeting with a team of medical and mental health professionals and giving informed consent, according to the guidelines.
Dr. Safer expressed surprise at the opposition to puberty blockers in proposed state legislation. “Puberty blockers are the conservative option so that we can avoid permanent changes while thoughtful decisions are being made by our adolescent patients with their families and health care providers,” he said.
The perception that puberty blockers will lead to hormone therapy is another misunderstanding and source of misinformation, Dr. Safer explained.
“The fear is that these data suggest that puberty blockers are a ‘gateway drug’ of some sort. But that is false. The reason that most adolescents who take puberty blockers go on to hormone therapy is because most of the adolescents who are identified in our conservative systems are actually transgender and interested in more gender-affirming care as they age,” he said.
Effects of discrimination on TGD persons
Many of these proposed state bills have not advanced through state legislatures, but a few – such as the proposed laws in Alabama, Missouri, and Texas – are still currently under consideration.
“In the United States, most recent efforts to single out transgender and gender-diverse people for discrimination in health care have failed. However, the demonization of trans people and attempts to disrupt care have been the source of much stress among our patients,” Dr. Safer said.
Restricting access to health care has “multiple implications” for TGD patients. “In the era when we did not provide care to transgender youth, we had a situation where approximately 40% of transgender people had considered suicide in their lives,” Dr. Safer said. In contrast, having access to these treatments has been shown to improve mental health outcomes in these patients, according to an Endocrine Society position statement.
The purpose of earlier interventions such as puberty blockers is to allow an adolescent to “explore options and live in the experienced gender before making a decision to proceed with gender-affirming hormone therapy,” the authors of the Endocrine Society and Pediatric Endocrine Society joint statement said.
Blocking access to puberty blockers, on the other hand, forces transgender youth to experience a puberty that doesn’t match their gender identity, Dr. Safer noted. “The puberty will include permanent changes which will then have to be reversed with surgery. Why would we intentionally allow that to happen?”
Dr. Safer reported that his spouse is an employee of Parexel. Dr. Tangpricha is the current president of the World Association for Transgender Health and a board member of the American Association of Clinical Endocrinology. The other authors reported no relevant conflicts of interests.
SOURCE: Safer JD et al. J Clin Endocrinol Metab. 2020. doi: 10.1210/clinem/dgaa816.
Science should be the cornerstone for health policy, and decisions on medical care of transgender and gender-diverse (TGD) individuals should be between a patient and their doctor.
That’s according to a joint policy statement from the Endocrine Society and Pediatric Endocrine Society published in the Journal of Clinical Endocrinology & Metabolism expressing concern about recent proposed legislation that would limit access to medical care for TGD individuals.
“The main emphasis is that we are simply medical people trying to be conservative and science driven in the care of our patients,” Joshua D. Safer, MD, coauthor and executive director of the Center for Transgender Medicine and Surgery at Mount Sinai Health System, and professor of medicine at Icahn School of Medicine at Mount Sinai, New York, said in an interview. “Why the health care for a particular group of people should be considered political is a mystery to me.”
TGD individuals have seen a recent uptick in efforts to limit or restrict their access to medical care at the federal and state levels. In June 2020, the Department of Health & Human Services finalized a revision to Section 1557 of the Affordable Care Act, rolling back a 2016 rule that determined the phrase “on the basis of sex” included nondiscrimination based on a person’s sex and gender identity. The Endocrine Society opposed this rule revision, arguing that it would allow “providers to deny care to TGD persons as well as discourage patients from seeking routine and gender-affirming care or reporting discrimination.”
Over a dozen U.S. states have introduced proposed legislation concerning medical care of TGD individuals that contain erroneous and misleading information. Proposed laws in Alabama, Missouri, and Texas, for example, would prohibit any use of medical treatments for minors for the purpose of gender-affirming medical care, including “gonadotropin-releasing hormone agonist therapy for pubertal suppression and gender-affirming hormonal therapy,” the authors of the joint statement wrote. In some cases, medical professionals who provide medical care for TGD patients could face criminal charges.
Outside the United States, three High Court judges in the United Kingdom recently ruled that minors aged under 16 years could not legally consent to pubertal suppression. “The recent U.K. court decision could be very disruptive because it would raise a barrier to transgender children receiving puberty blockers at exactly the ages that puberty blockers would be typically used,” Dr. Safer said.
Misleading characterizations of gender-affirming medical care for TGD individuals have also been spread to the general public. A recent Republican primary ballot proposition in Texas asked whether the state should ban “chemical castration, puberty blockers, cross-sex hormones, and genital mutilation surgery on all minor children for transition purposes,” falsely asserting that “Texas children as young as 3 are being transitioned from their biological sex to the opposite sex,” referencing a high-profile custody battle of a transgender child in Texas.
There are several tiers of misinformation that exist within these statements, Dr. Safer noted. “Some statements have suggested that gender-affirming treatment for young children can include hormone therapy or even surgery. Of course, there are no medical treatments for transgender and gender-diverse children prior to puberty.”
For adolescents aged under 18 years, Endocrine Society guidelines released in 2017 state that pubertal suppression is fully reversible and “offered to adolescents who meet diagnostic and treatment criteria, and are requesting care, for gender dysphoria/gender incongruence after they exhibit physical changes of puberty,” Dr. Safer and coauthors wrote in the joint policy statement. Other, more permanent – but still partially reversible – treatments such as hormone therapy are available as options for adolescents with confirmed and persistent gender dysphoria/gender incongruence, after meeting with a team of medical and mental health professionals and giving informed consent, according to the guidelines.
Dr. Safer expressed surprise at the opposition to puberty blockers in proposed state legislation. “Puberty blockers are the conservative option so that we can avoid permanent changes while thoughtful decisions are being made by our adolescent patients with their families and health care providers,” he said.
The perception that puberty blockers will lead to hormone therapy is another misunderstanding and source of misinformation, Dr. Safer explained.
“The fear is that these data suggest that puberty blockers are a ‘gateway drug’ of some sort. But that is false. The reason that most adolescents who take puberty blockers go on to hormone therapy is because most of the adolescents who are identified in our conservative systems are actually transgender and interested in more gender-affirming care as they age,” he said.
Effects of discrimination on TGD persons
Many of these proposed state bills have not advanced through state legislatures, but a few – such as the proposed laws in Alabama, Missouri, and Texas – are still currently under consideration.
“In the United States, most recent efforts to single out transgender and gender-diverse people for discrimination in health care have failed. However, the demonization of trans people and attempts to disrupt care have been the source of much stress among our patients,” Dr. Safer said.
Restricting access to health care has “multiple implications” for TGD patients. “In the era when we did not provide care to transgender youth, we had a situation where approximately 40% of transgender people had considered suicide in their lives,” Dr. Safer said. In contrast, having access to these treatments has been shown to improve mental health outcomes in these patients, according to an Endocrine Society position statement.
The purpose of earlier interventions such as puberty blockers is to allow an adolescent to “explore options and live in the experienced gender before making a decision to proceed with gender-affirming hormone therapy,” the authors of the Endocrine Society and Pediatric Endocrine Society joint statement said.
Blocking access to puberty blockers, on the other hand, forces transgender youth to experience a puberty that doesn’t match their gender identity, Dr. Safer noted. “The puberty will include permanent changes which will then have to be reversed with surgery. Why would we intentionally allow that to happen?”
Dr. Safer reported that his spouse is an employee of Parexel. Dr. Tangpricha is the current president of the World Association for Transgender Health and a board member of the American Association of Clinical Endocrinology. The other authors reported no relevant conflicts of interests.
SOURCE: Safer JD et al. J Clin Endocrinol Metab. 2020. doi: 10.1210/clinem/dgaa816.
FROM THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM

Six big changes coming for office-visit coding
Betsy Nicoletti, MS, a nationally recognized coding expert, will take your coding questions via email and provide guidance on how to code properly to maximize reimbursement. Have a question about coding? Send it to gtwachtman@mdedge.com.
thanks to the American Medical Association.
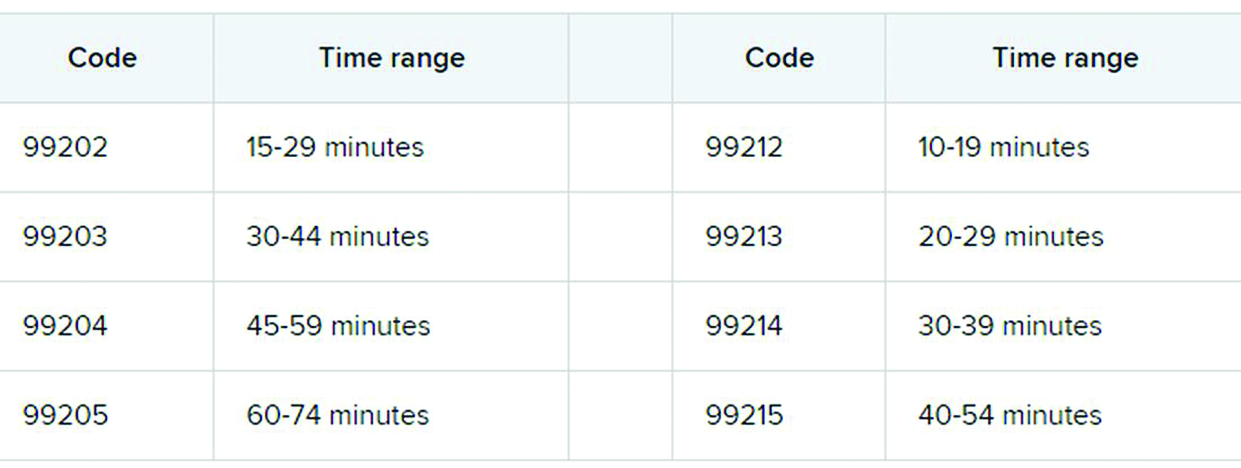
The first major changes to the definitions for E/M services will be in effect as of Jan. 1, 2021, with all payers expected to adopt these new guidelines. In particular, the AMA has revised the definitions for E/M codes 99202-99215 in the Current Procedural Terminology (CPT) 2021 codebook. The existing guidelines were developed in 1995 and 1997 and remain in effect for all other E/M services determined by history, exam, and medical decision-making (MDM).
What do the new changes mean to you? In 2021, for new and established office and other outpatient services reported with codes 99202-99215, a clinician may select the code on the basis of time or MDM.
There are three elements in MDM, and two of three are required. These elements are the number and complexity of problems addressed, amount and/or complexity of data to be reviewed and analyzed, and risk of complications and/or morbidity or mortality of patient management.
Make sure you familiarize yourself with these six big changes. It may take a bit of time to integrate these new processes into your daily routine, but wrapping your head around them as soon as possible can help boost your bottom line:
1. History and exam don’t count toward level of service
Physicians, advanced practice registered nurses, and physician assistants won’t use history or exam to select what level of code to bill for office visits 99202-99215, as they did in the past. They need only document a medically appropriate history and exam. The history may be obtained by staff members and reviewed by the billing practitioner.
While specific history and exam requirements disappear for office visit codes, they remain for all other types of visits, selected on the basis of history, exam, and MDM, such as hospital services, nursing facility services, and home and domiciliary care. So, say goodbye to “all other systems reviewed and negative” in office notes, but keep it handy for those other E/M codes.
2. All time spent caring for the patient on a particular day counts
This includes all time spent on the day of service, including preparing to see the patient, seeing the patient, phone calls or other work done after the visit (if not billed with a care management or other CPT code), and documenting in the medical record. The AMA developed new guidelines for using time for office and other outpatient services. For codes 99202-99215, count all of the face-to-face and non–face-to-face time spent by the billing clinician on the day of the visit. Counseling does not need to be more than 50% of the total time.
Do not include any staff time or time spent on any days before or after the visit. This allows clinicians to capture the work when a significant amount of it takes place before or after the visit with the patient, and to bill for it on the day of the visit.
According to the 2021 CPT codebook, physician or other qualified health care professional time includes the following activities:
- Preparing to see the patient (e.g., review of tests).
- Obtaining and/or reviewing separately obtained history.
- Performing a medically appropriate examination and/or evaluation.
- Counseling and educating the patient/family/caregiver.
- Ordering medications, tests, or procedures.
- Referring and communicating with other health care professionals (when not separately reported).
- Documenting clinical information in the electronic or other health record.
- Independently interpreting results (not separately reported) and communicating results to the patient/family/caregiver.
- Care coordination (not separately reported).
3. Soon to be gone: ‘new to the examiner’ and ‘workup planned’
The current guidelines don’t differentiate between a new problem to the clinician or an established problem to the clinician. So it doesn’t matter whether you’re hearing about a particular problem for the first time or the fifth time. The new office and outpatient services guidelines define problems only as they relate to the patient. For example, when selecting a level of service, a chronic problem with a mild exacerbation is the same level whether it’s the primary care physician seeing the patient for the 10th time to help manage her diabetes or the endocrinologist seeing the patient for the first time.
In the current guidelines (1995 and 1997), additional weight is given in selecting the level of MDM for a problem that’s new to the examiner with a workup planned, yet when the diagnostic test couldn’t be completed at the visit. This concept is gone from element of number and complexity of new problems. Ordering diagnostic tests is part of the second element, the amount and/or complexity of data to be reviewed.
4. Different guidelines if you need a history from a parent or other source
The new guidelines recognize the additional work required by the clinician when the patient is unable to give a history or when the practitioner doesn’t find the history to be reliable.
For example, in the case of a baby or child who is unable to give a history, the parent counts as an “independent historian,” according to the new guidelines. Likewise, for a patient with dementia, the caregiver counts as a historian. Note, however, that the criteria is not met simply because the patient is accompanied by another person. The additional weight in selecting the level of service is based on the patient being unable to give a reliable history.
Bottom line: In cases where patients are unable to communicate clearly, physicians or other providers should document the necessity of getting a complete history and who provided it.
5. A new spin on social determinants of health (SDoH)
In the risk of morbidity and/or mortality element, conditions described as “social determinants of health” are considered moderate complexity. SDoH are social and environmental factors that affect a patient’s health and medical outcomes. These include homelessness, inability to afford medications, food insecurity, and occupational exposure to risk factors. These circumstances are reported with codes in categories Z55-Z65.
In the past, physicians often documented this information in their office notes but rarely added a diagnosis code that described the patient’s situation. The ICD-10-CM code set includes codes that describe these factors. Using them allows the practice to track patients who have increased needs, and it communicates to payers the complexity of caring for these patients.
6. Risks related to surgery are defined
The current guidelines assign different levels of risk to minor and major surgery. They also include differentiation for “minor surgery with no identified risk factors,” “minor surgery with identified risk factors,” “elective major surgery with no identified risk factors,” and “elective major surgery with identified risk factors.” The old guidelines didn’t state whether the risk factors pertained to the patient – such as smoking, heart disease, or high body mass index – or to the procedure itself.
The new guidelines specifically say that it’s both. In the risk column, “decision regarding minor surgery with identified patient or procedure risk factors” and “decision regarding elective major surgery without patient or procedure risk factors” are both considered moderate. “Decision regarding elective major surgery with identified patient or procedure risk factors” and “decision regarding emergency major surgery” are in the high complexity column for risk.
Keep in mind that two of three elements are required: the number and complexity of problems, amount of data, and morbidity/mortality risk. Risk of morbidity/mortality alone doesn’t count as the basis for selecting the code. Of course, when surgeons see this, they ask, “What major procedures don’t have identified risk factors?”
Note, too, that these new CPT guidelines do not define the terms “minor” and “major” surgery. For payment reasons related to the postop period, the Centers for Medicare & Medicaid Services defines minor surgery as a procedure with 0-10 global days and a major surgery as a procedure with 90 global days. However, there are many procedures with 0 global days (endoscopy, cardiac catheterization) that are not minor procedures. Hopefully, the AMA will clarify this in 2021.
What’s the take-away for clinicians?
There are sure to be shifts in coding patterns based on these new guidelines. Some specialties will find that not being able to select a service based on history and exam alone will lower the level of service for which they can bill. Some practices, on the other hand, will be able to code for more high-level visits, without the need for a complete review of systems or a comprehensive exam.
The biggest challenge will be for practices that provide services both in the hospital and in the office, because they’ll have to use both sets of guidelines, depending on which type of service they’re performing.
For more details on what’s coming your way beginning on New Year’s Day, you may want to read the 16-page AMA document .
A version of this article first appeared on Medscape.com.
Betsy Nicoletti, MS, a nationally recognized coding expert, will take your coding questions via email and provide guidance on how to code properly to maximize reimbursement. Have a question about coding? Send it to gtwachtman@mdedge.com.
thanks to the American Medical Association.
The first major changes to the definitions for E/M services will be in effect as of Jan. 1, 2021, with all payers expected to adopt these new guidelines. In particular, the AMA has revised the definitions for E/M codes 99202-99215 in the Current Procedural Terminology (CPT) 2021 codebook. The existing guidelines were developed in 1995 and 1997 and remain in effect for all other E/M services determined by history, exam, and medical decision-making (MDM).
What do the new changes mean to you? In 2021, for new and established office and other outpatient services reported with codes 99202-99215, a clinician may select the code on the basis of time or MDM.
There are three elements in MDM, and two of three are required. These elements are the number and complexity of problems addressed, amount and/or complexity of data to be reviewed and analyzed, and risk of complications and/or morbidity or mortality of patient management.
Make sure you familiarize yourself with these six big changes. It may take a bit of time to integrate these new processes into your daily routine, but wrapping your head around them as soon as possible can help boost your bottom line:
1. History and exam don’t count toward level of service
Physicians, advanced practice registered nurses, and physician assistants won’t use history or exam to select what level of code to bill for office visits 99202-99215, as they did in the past. They need only document a medically appropriate history and exam. The history may be obtained by staff members and reviewed by the billing practitioner.
While specific history and exam requirements disappear for office visit codes, they remain for all other types of visits, selected on the basis of history, exam, and MDM, such as hospital services, nursing facility services, and home and domiciliary care. So, say goodbye to “all other systems reviewed and negative” in office notes, but keep it handy for those other E/M codes.
2. All time spent caring for the patient on a particular day counts
This includes all time spent on the day of service, including preparing to see the patient, seeing the patient, phone calls or other work done after the visit (if not billed with a care management or other CPT code), and documenting in the medical record. The AMA developed new guidelines for using time for office and other outpatient services. For codes 99202-99215, count all of the face-to-face and non–face-to-face time spent by the billing clinician on the day of the visit. Counseling does not need to be more than 50% of the total time.
Do not include any staff time or time spent on any days before or after the visit. This allows clinicians to capture the work when a significant amount of it takes place before or after the visit with the patient, and to bill for it on the day of the visit.
According to the 2021 CPT codebook, physician or other qualified health care professional time includes the following activities:
- Preparing to see the patient (e.g., review of tests).
- Obtaining and/or reviewing separately obtained history.
- Performing a medically appropriate examination and/or evaluation.
- Counseling and educating the patient/family/caregiver.
- Ordering medications, tests, or procedures.
- Referring and communicating with other health care professionals (when not separately reported).
- Documenting clinical information in the electronic or other health record.
- Independently interpreting results (not separately reported) and communicating results to the patient/family/caregiver.
- Care coordination (not separately reported).
3. Soon to be gone: ‘new to the examiner’ and ‘workup planned’
The current guidelines don’t differentiate between a new problem to the clinician or an established problem to the clinician. So it doesn’t matter whether you’re hearing about a particular problem for the first time or the fifth time. The new office and outpatient services guidelines define problems only as they relate to the patient. For example, when selecting a level of service, a chronic problem with a mild exacerbation is the same level whether it’s the primary care physician seeing the patient for the 10th time to help manage her diabetes or the endocrinologist seeing the patient for the first time.
In the current guidelines (1995 and 1997), additional weight is given in selecting the level of MDM for a problem that’s new to the examiner with a workup planned, yet when the diagnostic test couldn’t be completed at the visit. This concept is gone from element of number and complexity of new problems. Ordering diagnostic tests is part of the second element, the amount and/or complexity of data to be reviewed.
4. Different guidelines if you need a history from a parent or other source
The new guidelines recognize the additional work required by the clinician when the patient is unable to give a history or when the practitioner doesn’t find the history to be reliable.
For example, in the case of a baby or child who is unable to give a history, the parent counts as an “independent historian,” according to the new guidelines. Likewise, for a patient with dementia, the caregiver counts as a historian. Note, however, that the criteria is not met simply because the patient is accompanied by another person. The additional weight in selecting the level of service is based on the patient being unable to give a reliable history.
Bottom line: In cases where patients are unable to communicate clearly, physicians or other providers should document the necessity of getting a complete history and who provided it.
5. A new spin on social determinants of health (SDoH)
In the risk of morbidity and/or mortality element, conditions described as “social determinants of health” are considered moderate complexity. SDoH are social and environmental factors that affect a patient’s health and medical outcomes. These include homelessness, inability to afford medications, food insecurity, and occupational exposure to risk factors. These circumstances are reported with codes in categories Z55-Z65.
In the past, physicians often documented this information in their office notes but rarely added a diagnosis code that described the patient’s situation. The ICD-10-CM code set includes codes that describe these factors. Using them allows the practice to track patients who have increased needs, and it communicates to payers the complexity of caring for these patients.
6. Risks related to surgery are defined
The current guidelines assign different levels of risk to minor and major surgery. They also include differentiation for “minor surgery with no identified risk factors,” “minor surgery with identified risk factors,” “elective major surgery with no identified risk factors,” and “elective major surgery with identified risk factors.” The old guidelines didn’t state whether the risk factors pertained to the patient – such as smoking, heart disease, or high body mass index – or to the procedure itself.
The new guidelines specifically say that it’s both. In the risk column, “decision regarding minor surgery with identified patient or procedure risk factors” and “decision regarding elective major surgery without patient or procedure risk factors” are both considered moderate. “Decision regarding elective major surgery with identified patient or procedure risk factors” and “decision regarding emergency major surgery” are in the high complexity column for risk.
Keep in mind that two of three elements are required: the number and complexity of problems, amount of data, and morbidity/mortality risk. Risk of morbidity/mortality alone doesn’t count as the basis for selecting the code. Of course, when surgeons see this, they ask, “What major procedures don’t have identified risk factors?”
Note, too, that these new CPT guidelines do not define the terms “minor” and “major” surgery. For payment reasons related to the postop period, the Centers for Medicare & Medicaid Services defines minor surgery as a procedure with 0-10 global days and a major surgery as a procedure with 90 global days. However, there are many procedures with 0 global days (endoscopy, cardiac catheterization) that are not minor procedures. Hopefully, the AMA will clarify this in 2021.
What’s the take-away for clinicians?
There are sure to be shifts in coding patterns based on these new guidelines. Some specialties will find that not being able to select a service based on history and exam alone will lower the level of service for which they can bill. Some practices, on the other hand, will be able to code for more high-level visits, without the need for a complete review of systems or a comprehensive exam.
The biggest challenge will be for practices that provide services both in the hospital and in the office, because they’ll have to use both sets of guidelines, depending on which type of service they’re performing.
For more details on what’s coming your way beginning on New Year’s Day, you may want to read the 16-page AMA document .
A version of this article first appeared on Medscape.com.
Betsy Nicoletti, MS, a nationally recognized coding expert, will take your coding questions via email and provide guidance on how to code properly to maximize reimbursement. Have a question about coding? Send it to gtwachtman@mdedge.com.
thanks to the American Medical Association.
The first major changes to the definitions for E/M services will be in effect as of Jan. 1, 2021, with all payers expected to adopt these new guidelines. In particular, the AMA has revised the definitions for E/M codes 99202-99215 in the Current Procedural Terminology (CPT) 2021 codebook. The existing guidelines were developed in 1995 and 1997 and remain in effect for all other E/M services determined by history, exam, and medical decision-making (MDM).
What do the new changes mean to you? In 2021, for new and established office and other outpatient services reported with codes 99202-99215, a clinician may select the code on the basis of time or MDM.
There are three elements in MDM, and two of three are required. These elements are the number and complexity of problems addressed, amount and/or complexity of data to be reviewed and analyzed, and risk of complications and/or morbidity or mortality of patient management.
Make sure you familiarize yourself with these six big changes. It may take a bit of time to integrate these new processes into your daily routine, but wrapping your head around them as soon as possible can help boost your bottom line:
1. History and exam don’t count toward level of service
Physicians, advanced practice registered nurses, and physician assistants won’t use history or exam to select what level of code to bill for office visits 99202-99215, as they did in the past. They need only document a medically appropriate history and exam. The history may be obtained by staff members and reviewed by the billing practitioner.
While specific history and exam requirements disappear for office visit codes, they remain for all other types of visits, selected on the basis of history, exam, and MDM, such as hospital services, nursing facility services, and home and domiciliary care. So, say goodbye to “all other systems reviewed and negative” in office notes, but keep it handy for those other E/M codes.
2. All time spent caring for the patient on a particular day counts
This includes all time spent on the day of service, including preparing to see the patient, seeing the patient, phone calls or other work done after the visit (if not billed with a care management or other CPT code), and documenting in the medical record. The AMA developed new guidelines for using time for office and other outpatient services. For codes 99202-99215, count all of the face-to-face and non–face-to-face time spent by the billing clinician on the day of the visit. Counseling does not need to be more than 50% of the total time.
Do not include any staff time or time spent on any days before or after the visit. This allows clinicians to capture the work when a significant amount of it takes place before or after the visit with the patient, and to bill for it on the day of the visit.
According to the 2021 CPT codebook, physician or other qualified health care professional time includes the following activities:
- Preparing to see the patient (e.g., review of tests).
- Obtaining and/or reviewing separately obtained history.
- Performing a medically appropriate examination and/or evaluation.
- Counseling and educating the patient/family/caregiver.
- Ordering medications, tests, or procedures.
- Referring and communicating with other health care professionals (when not separately reported).
- Documenting clinical information in the electronic or other health record.
- Independently interpreting results (not separately reported) and communicating results to the patient/family/caregiver.
- Care coordination (not separately reported).
3. Soon to be gone: ‘new to the examiner’ and ‘workup planned’
The current guidelines don’t differentiate between a new problem to the clinician or an established problem to the clinician. So it doesn’t matter whether you’re hearing about a particular problem for the first time or the fifth time. The new office and outpatient services guidelines define problems only as they relate to the patient. For example, when selecting a level of service, a chronic problem with a mild exacerbation is the same level whether it’s the primary care physician seeing the patient for the 10th time to help manage her diabetes or the endocrinologist seeing the patient for the first time.
In the current guidelines (1995 and 1997), additional weight is given in selecting the level of MDM for a problem that’s new to the examiner with a workup planned, yet when the diagnostic test couldn’t be completed at the visit. This concept is gone from element of number and complexity of new problems. Ordering diagnostic tests is part of the second element, the amount and/or complexity of data to be reviewed.
4. Different guidelines if you need a history from a parent or other source
The new guidelines recognize the additional work required by the clinician when the patient is unable to give a history or when the practitioner doesn’t find the history to be reliable.
For example, in the case of a baby or child who is unable to give a history, the parent counts as an “independent historian,” according to the new guidelines. Likewise, for a patient with dementia, the caregiver counts as a historian. Note, however, that the criteria is not met simply because the patient is accompanied by another person. The additional weight in selecting the level of service is based on the patient being unable to give a reliable history.
Bottom line: In cases where patients are unable to communicate clearly, physicians or other providers should document the necessity of getting a complete history and who provided it.
5. A new spin on social determinants of health (SDoH)
In the risk of morbidity and/or mortality element, conditions described as “social determinants of health” are considered moderate complexity. SDoH are social and environmental factors that affect a patient’s health and medical outcomes. These include homelessness, inability to afford medications, food insecurity, and occupational exposure to risk factors. These circumstances are reported with codes in categories Z55-Z65.
In the past, physicians often documented this information in their office notes but rarely added a diagnosis code that described the patient’s situation. The ICD-10-CM code set includes codes that describe these factors. Using them allows the practice to track patients who have increased needs, and it communicates to payers the complexity of caring for these patients.
6. Risks related to surgery are defined
The current guidelines assign different levels of risk to minor and major surgery. They also include differentiation for “minor surgery with no identified risk factors,” “minor surgery with identified risk factors,” “elective major surgery with no identified risk factors,” and “elective major surgery with identified risk factors.” The old guidelines didn’t state whether the risk factors pertained to the patient – such as smoking, heart disease, or high body mass index – or to the procedure itself.
The new guidelines specifically say that it’s both. In the risk column, “decision regarding minor surgery with identified patient or procedure risk factors” and “decision regarding elective major surgery without patient or procedure risk factors” are both considered moderate. “Decision regarding elective major surgery with identified patient or procedure risk factors” and “decision regarding emergency major surgery” are in the high complexity column for risk.
Keep in mind that two of three elements are required: the number and complexity of problems, amount of data, and morbidity/mortality risk. Risk of morbidity/mortality alone doesn’t count as the basis for selecting the code. Of course, when surgeons see this, they ask, “What major procedures don’t have identified risk factors?”
Note, too, that these new CPT guidelines do not define the terms “minor” and “major” surgery. For payment reasons related to the postop period, the Centers for Medicare & Medicaid Services defines minor surgery as a procedure with 0-10 global days and a major surgery as a procedure with 90 global days. However, there are many procedures with 0 global days (endoscopy, cardiac catheterization) that are not minor procedures. Hopefully, the AMA will clarify this in 2021.
What’s the take-away for clinicians?
There are sure to be shifts in coding patterns based on these new guidelines. Some specialties will find that not being able to select a service based on history and exam alone will lower the level of service for which they can bill. Some practices, on the other hand, will be able to code for more high-level visits, without the need for a complete review of systems or a comprehensive exam.
The biggest challenge will be for practices that provide services both in the hospital and in the office, because they’ll have to use both sets of guidelines, depending on which type of service they’re performing.
For more details on what’s coming your way beginning on New Year’s Day, you may want to read the 16-page AMA document .
A version of this article first appeared on Medscape.com.
Endocrine-disrupting plastics pose growing health threat
Many types of plastics pose an unrecognized threat to human health by leaching endocrine-disrupting chemicals, and a new report from the Endocrine Society and the International Pollutants Elimination Network presents their dangers and risks.

Written in a consumer-friendly form designed to guide public interest groups and policy makers, the report also can be used by clinicians to inform discussions with patients about the potential dangers of plastics and how they can reduce their exposure to endocrine-disrupting chemicals.
The report, Plastics, EDCs, & Health, defines endocrine-disrupting chemicals (EDCs) as “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action.” Hormones in the body must be released at specific times, and therefore interference with their normal activity can have profound effects on health in areas including growth and reproductive development, according to the report.
The available data show “more and more information about the different chemicals and the different effects they are having,” said lead author, Jodi Flaws, PhD, of the University of Illinois at Urbana-Champaign, in a virtual press conference accompanying the release of the report.
Although numerous EDCs have been identified, a recent study suggested that many potentially dangerous chemical additives remain unknown because they are identified as confidential or simply not well described, the report authors said. In addition, creation of more plastic products will likely lead to increased exposure to EDCs and make health problems worse, said report coauthor Pauliina Damdimopoulou, PhD, of the Karolinska Institutet in Stockholm.
Lesser-known EDCs populate consumer products
Most consumers are aware of bisphenol A and phthalates as known EDCs, said Dr. Flaws, but the report identifies other lesser-known EDCs including per- and polyfluoroalkyl substances (PFAS), dioxins, flame retardants, and UV stabilizers.
For example, PFAS have been used for decades in a range of consumer products including stain resistant clothes, fast food wrappers, carpet and furniture treatments, cookware, and firefighting foams, according to the report. Consequently, PFAS have become common in many water sources including surface water, drinking water, and ground water because of how they are disposed. “Consumption of fish and other aquatic creatures caught in waterways contaminated with PFAS also poses heightened risks due to bioaccumulation of persistent chemicals in these animals,” the report authors noted. Human exposures to PFAS have been documented in urine, serum, plasma, placenta, umbilical cord, breast milk, and fetal tissues, they added.
Brominated flame retardants are another lesser-known EDC highlighted in the report. These chemical additives are used in plastics such as electronics cases to reduce the spread of fire, as well as in furniture foam and other building materials, the authors wrote. UV stabilizers, which also have been linked to health problems, often are used in manufacturing cars and other machinery.
Microplastics create large risk
Microplastics, defined as plastic particles less than 5 mm in diameter, are another source of exposure to EDCs that is not well publicized, according to the report. Plastic waste disposal often leads to the release of microplastics, which can infiltrate soil and water. Plastic waste is often dumped or burned; outdoor burning of plastic causes emission of dioxins into the air and ground.
“Not only do microplastics contain endogenous chemical additives, which are not bound to the microplastic and can leach out of the microplastic and expose the population, they can also bind and accumulate toxic chemicals from the surrounding environment such as sea water and sediment,” the report authors said.
Recycling is not an easy answer, either. Often more chemicals are created and released during the process of using plastics to make other plastics, according to the report.
Overall, more awareness of the potential for increased exposure to EDCs and support of strategies to seek out alternatives to hazardous chemicals is needed at the global level, the authors wrote. For example, the European Union has proposed a chemicals strategy that includes improved classification of EDCs and banning identified EDCs in consumer products.
New data support ongoing dangers
“It was important to produce the report at this time because several new studies came out on the effects of EDCs from plastics on human health,” Dr. Flaws said in an interview. “Further, there was not previously a single source that brought together all the information in a manner that was targeted towards the public, policy makers, and others,” she said.
Dr. Flaws said that what has surprised her most in the recent research is the fact that plastics contain such a range of chemicals and EDCs.
“A good take-home message [from the report] is that plastics can contain endocrine-disrupting chemicals that can interfere with normal hormones and lead to adverse health outcomes,” she said. “I suggest limiting the use of plastics as much as possible. I know this is very hard to do, so if someone needs to use plastic, they should not heat food or drink in plastic containers,” she emphasized. Individuals also can limit reuse of plastics over and over,” she said. “Heating and repeated use/washing often causes plastics to leach EDCs into food and drink that we then get into our bodies.”
Additional research is needed to understand the mechanisms by which EDCs from plastics cause damage, Dr. Flaws emphasized. “Given that it is not possible to eliminate plastics at this time, if we understood mechanisms of action, we could develop ways to prevent toxicity or treat EDC-induced adverse health outcomes,” she said. “We also need research designed to develop plastics or ‘green materials’ that do not contain endocrine disruptors and do not cause health problems or damage the environment,” she noted.
The report was produced as a joint effort of the Endocrine Society and International Pollutants Elimination Network. The report authors had no financial conflicts to disclose.
Many types of plastics pose an unrecognized threat to human health by leaching endocrine-disrupting chemicals, and a new report from the Endocrine Society and the International Pollutants Elimination Network presents their dangers and risks.
Written in a consumer-friendly form designed to guide public interest groups and policy makers, the report also can be used by clinicians to inform discussions with patients about the potential dangers of plastics and how they can reduce their exposure to endocrine-disrupting chemicals.
The report, Plastics, EDCs, & Health, defines endocrine-disrupting chemicals (EDCs) as “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action.” Hormones in the body must be released at specific times, and therefore interference with their normal activity can have profound effects on health in areas including growth and reproductive development, according to the report.
The available data show “more and more information about the different chemicals and the different effects they are having,” said lead author, Jodi Flaws, PhD, of the University of Illinois at Urbana-Champaign, in a virtual press conference accompanying the release of the report.
Although numerous EDCs have been identified, a recent study suggested that many potentially dangerous chemical additives remain unknown because they are identified as confidential or simply not well described, the report authors said. In addition, creation of more plastic products will likely lead to increased exposure to EDCs and make health problems worse, said report coauthor Pauliina Damdimopoulou, PhD, of the Karolinska Institutet in Stockholm.
Lesser-known EDCs populate consumer products
Most consumers are aware of bisphenol A and phthalates as known EDCs, said Dr. Flaws, but the report identifies other lesser-known EDCs including per- and polyfluoroalkyl substances (PFAS), dioxins, flame retardants, and UV stabilizers.
For example, PFAS have been used for decades in a range of consumer products including stain resistant clothes, fast food wrappers, carpet and furniture treatments, cookware, and firefighting foams, according to the report. Consequently, PFAS have become common in many water sources including surface water, drinking water, and ground water because of how they are disposed. “Consumption of fish and other aquatic creatures caught in waterways contaminated with PFAS also poses heightened risks due to bioaccumulation of persistent chemicals in these animals,” the report authors noted. Human exposures to PFAS have been documented in urine, serum, plasma, placenta, umbilical cord, breast milk, and fetal tissues, they added.
Brominated flame retardants are another lesser-known EDC highlighted in the report. These chemical additives are used in plastics such as electronics cases to reduce the spread of fire, as well as in furniture foam and other building materials, the authors wrote. UV stabilizers, which also have been linked to health problems, often are used in manufacturing cars and other machinery.
Microplastics create large risk
Microplastics, defined as plastic particles less than 5 mm in diameter, are another source of exposure to EDCs that is not well publicized, according to the report. Plastic waste disposal often leads to the release of microplastics, which can infiltrate soil and water. Plastic waste is often dumped or burned; outdoor burning of plastic causes emission of dioxins into the air and ground.
“Not only do microplastics contain endogenous chemical additives, which are not bound to the microplastic and can leach out of the microplastic and expose the population, they can also bind and accumulate toxic chemicals from the surrounding environment such as sea water and sediment,” the report authors said.
Recycling is not an easy answer, either. Often more chemicals are created and released during the process of using plastics to make other plastics, according to the report.
Overall, more awareness of the potential for increased exposure to EDCs and support of strategies to seek out alternatives to hazardous chemicals is needed at the global level, the authors wrote. For example, the European Union has proposed a chemicals strategy that includes improved classification of EDCs and banning identified EDCs in consumer products.
New data support ongoing dangers
“It was important to produce the report at this time because several new studies came out on the effects of EDCs from plastics on human health,” Dr. Flaws said in an interview. “Further, there was not previously a single source that brought together all the information in a manner that was targeted towards the public, policy makers, and others,” she said.
Dr. Flaws said that what has surprised her most in the recent research is the fact that plastics contain such a range of chemicals and EDCs.
“A good take-home message [from the report] is that plastics can contain endocrine-disrupting chemicals that can interfere with normal hormones and lead to adverse health outcomes,” she said. “I suggest limiting the use of plastics as much as possible. I know this is very hard to do, so if someone needs to use plastic, they should not heat food or drink in plastic containers,” she emphasized. Individuals also can limit reuse of plastics over and over,” she said. “Heating and repeated use/washing often causes plastics to leach EDCs into food and drink that we then get into our bodies.”
Additional research is needed to understand the mechanisms by which EDCs from plastics cause damage, Dr. Flaws emphasized. “Given that it is not possible to eliminate plastics at this time, if we understood mechanisms of action, we could develop ways to prevent toxicity or treat EDC-induced adverse health outcomes,” she said. “We also need research designed to develop plastics or ‘green materials’ that do not contain endocrine disruptors and do not cause health problems or damage the environment,” she noted.
The report was produced as a joint effort of the Endocrine Society and International Pollutants Elimination Network. The report authors had no financial conflicts to disclose.
Many types of plastics pose an unrecognized threat to human health by leaching endocrine-disrupting chemicals, and a new report from the Endocrine Society and the International Pollutants Elimination Network presents their dangers and risks.
Written in a consumer-friendly form designed to guide public interest groups and policy makers, the report also can be used by clinicians to inform discussions with patients about the potential dangers of plastics and how they can reduce their exposure to endocrine-disrupting chemicals.
The report, Plastics, EDCs, & Health, defines endocrine-disrupting chemicals (EDCs) as “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action.” Hormones in the body must be released at specific times, and therefore interference with their normal activity can have profound effects on health in areas including growth and reproductive development, according to the report.
The available data show “more and more information about the different chemicals and the different effects they are having,” said lead author, Jodi Flaws, PhD, of the University of Illinois at Urbana-Champaign, in a virtual press conference accompanying the release of the report.
Although numerous EDCs have been identified, a recent study suggested that many potentially dangerous chemical additives remain unknown because they are identified as confidential or simply not well described, the report authors said. In addition, creation of more plastic products will likely lead to increased exposure to EDCs and make health problems worse, said report coauthor Pauliina Damdimopoulou, PhD, of the Karolinska Institutet in Stockholm.
Lesser-known EDCs populate consumer products
Most consumers are aware of bisphenol A and phthalates as known EDCs, said Dr. Flaws, but the report identifies other lesser-known EDCs including per- and polyfluoroalkyl substances (PFAS), dioxins, flame retardants, and UV stabilizers.
For example, PFAS have been used for decades in a range of consumer products including stain resistant clothes, fast food wrappers, carpet and furniture treatments, cookware, and firefighting foams, according to the report. Consequently, PFAS have become common in many water sources including surface water, drinking water, and ground water because of how they are disposed. “Consumption of fish and other aquatic creatures caught in waterways contaminated with PFAS also poses heightened risks due to bioaccumulation of persistent chemicals in these animals,” the report authors noted. Human exposures to PFAS have been documented in urine, serum, plasma, placenta, umbilical cord, breast milk, and fetal tissues, they added.
Brominated flame retardants are another lesser-known EDC highlighted in the report. These chemical additives are used in plastics such as electronics cases to reduce the spread of fire, as well as in furniture foam and other building materials, the authors wrote. UV stabilizers, which also have been linked to health problems, often are used in manufacturing cars and other machinery.
Microplastics create large risk
Microplastics, defined as plastic particles less than 5 mm in diameter, are another source of exposure to EDCs that is not well publicized, according to the report. Plastic waste disposal often leads to the release of microplastics, which can infiltrate soil and water. Plastic waste is often dumped or burned; outdoor burning of plastic causes emission of dioxins into the air and ground.
“Not only do microplastics contain endogenous chemical additives, which are not bound to the microplastic and can leach out of the microplastic and expose the population, they can also bind and accumulate toxic chemicals from the surrounding environment such as sea water and sediment,” the report authors said.
Recycling is not an easy answer, either. Often more chemicals are created and released during the process of using plastics to make other plastics, according to the report.
Overall, more awareness of the potential for increased exposure to EDCs and support of strategies to seek out alternatives to hazardous chemicals is needed at the global level, the authors wrote. For example, the European Union has proposed a chemicals strategy that includes improved classification of EDCs and banning identified EDCs in consumer products.
New data support ongoing dangers
“It was important to produce the report at this time because several new studies came out on the effects of EDCs from plastics on human health,” Dr. Flaws said in an interview. “Further, there was not previously a single source that brought together all the information in a manner that was targeted towards the public, policy makers, and others,” she said.
Dr. Flaws said that what has surprised her most in the recent research is the fact that plastics contain such a range of chemicals and EDCs.
“A good take-home message [from the report] is that plastics can contain endocrine-disrupting chemicals that can interfere with normal hormones and lead to adverse health outcomes,” she said. “I suggest limiting the use of plastics as much as possible. I know this is very hard to do, so if someone needs to use plastic, they should not heat food or drink in plastic containers,” she emphasized. Individuals also can limit reuse of plastics over and over,” she said. “Heating and repeated use/washing often causes plastics to leach EDCs into food and drink that we then get into our bodies.”
Additional research is needed to understand the mechanisms by which EDCs from plastics cause damage, Dr. Flaws emphasized. “Given that it is not possible to eliminate plastics at this time, if we understood mechanisms of action, we could develop ways to prevent toxicity or treat EDC-induced adverse health outcomes,” she said. “We also need research designed to develop plastics or ‘green materials’ that do not contain endocrine disruptors and do not cause health problems or damage the environment,” she noted.
The report was produced as a joint effort of the Endocrine Society and International Pollutants Elimination Network. The report authors had no financial conflicts to disclose.
COVID-related harm to HCWs must be tracked more rigorously: NAS panel
A panel of scientific experts is urging the nation to do more to track morbidity and mortality among health care workers (HCWs), given the large and disproportionate number who have been infected with or died from SARS-CoV-2.
The National Academies of Sciences, Engineering, and Medicine’s Standing Committee on Emerging Infectious Diseases and 21st Century Health Threats issued a 10-page “rapid expert consultation” on what is known about deaths and mental health problems among HCWs associated with the COVID-19 pandemic and how to protect workers.
“The absence of a uniform national framework and inconsistent requirements across states for collecting, recording, and reporting HCW mortality and morbidity data associated with COVID-19 impairs anyone’s ability to make comparisons, do combined analyses, or draw conclusions about the scale of the problem,” says the panel in the report.
Mental health, in particular, needs to be examined, it says. Although the data are still limited, the prevalence of burnout and suicide “points to a serious concern,” according to the report.
“As with mortality due to COVID-19, there are currently no national systems nor reporting standards for morbidity measures related to the pandemic, such as mental health status, provider well-being, and other psychological effects on HCWs,” the report says.
A more robust national system that collected data on circumstances and interventions that may raise or lower risk, as well as on where the infection occurred, “would support the adoption of effective mitigation strategies,” says the report. It would also facilitate epidemiologic studies on risk factors, such as face-to-face contact with COVID-19 patients and the availability and use of personal protective equipment (PPE). Studies could also examine the impact of institutional requirements for masking.
Studies have consistently shown that universal mask wearing and access to appropriate PPE support the physical safety and mental health of HCWs, says the report.
Track scale of crisis
The committee cited many gaps in the current system. The Occupational Safety and Health Administration, for instance, doesn’t count deaths from occupationally acquired infection. Many states don’t report COVID-19 deaths by profession. The Centers for Disease Control and Prevention (CDC) relies on case report forms from local health departments for all COVID-19 cases, which typically are lacking in specifics, such as occupation or job setting, says the committee’s report.
As of Nov. 3, the CDC had reported 786 deaths among HCWs that were attributable to COVID-19 – a far lower number than other sources have reported.
The committee notes that much could be done immediately. A National Academy of Medicine panel on clinician well-being and resilience in August recommended that the CDC establish a national epidemiologic tracking program to measure HCWs’ well-being, assess the acute and long-term effects of COVID-19 on those workers, and report on the outcomes of interventions.
Such a program “is needed to comprehensively acknowledge the scale of the COVID-19 crisis and protect the health care workforce that is already stretched to the breaking point in many locations,” the committee says in its report.
A version of this article originally appeared on Medscape.com.
A panel of scientific experts is urging the nation to do more to track morbidity and mortality among health care workers (HCWs), given the large and disproportionate number who have been infected with or died from SARS-CoV-2.
The National Academies of Sciences, Engineering, and Medicine’s Standing Committee on Emerging Infectious Diseases and 21st Century Health Threats issued a 10-page “rapid expert consultation” on what is known about deaths and mental health problems among HCWs associated with the COVID-19 pandemic and how to protect workers.
“The absence of a uniform national framework and inconsistent requirements across states for collecting, recording, and reporting HCW mortality and morbidity data associated with COVID-19 impairs anyone’s ability to make comparisons, do combined analyses, or draw conclusions about the scale of the problem,” says the panel in the report.
Mental health, in particular, needs to be examined, it says. Although the data are still limited, the prevalence of burnout and suicide “points to a serious concern,” according to the report.
“As with mortality due to COVID-19, there are currently no national systems nor reporting standards for morbidity measures related to the pandemic, such as mental health status, provider well-being, and other psychological effects on HCWs,” the report says.
A more robust national system that collected data on circumstances and interventions that may raise or lower risk, as well as on where the infection occurred, “would support the adoption of effective mitigation strategies,” says the report. It would also facilitate epidemiologic studies on risk factors, such as face-to-face contact with COVID-19 patients and the availability and use of personal protective equipment (PPE). Studies could also examine the impact of institutional requirements for masking.
Studies have consistently shown that universal mask wearing and access to appropriate PPE support the physical safety and mental health of HCWs, says the report.
Track scale of crisis
The committee cited many gaps in the current system. The Occupational Safety and Health Administration, for instance, doesn’t count deaths from occupationally acquired infection. Many states don’t report COVID-19 deaths by profession. The Centers for Disease Control and Prevention (CDC) relies on case report forms from local health departments for all COVID-19 cases, which typically are lacking in specifics, such as occupation or job setting, says the committee’s report.
As of Nov. 3, the CDC had reported 786 deaths among HCWs that were attributable to COVID-19 – a far lower number than other sources have reported.
The committee notes that much could be done immediately. A National Academy of Medicine panel on clinician well-being and resilience in August recommended that the CDC establish a national epidemiologic tracking program to measure HCWs’ well-being, assess the acute and long-term effects of COVID-19 on those workers, and report on the outcomes of interventions.
Such a program “is needed to comprehensively acknowledge the scale of the COVID-19 crisis and protect the health care workforce that is already stretched to the breaking point in many locations,” the committee says in its report.
A version of this article originally appeared on Medscape.com.
A panel of scientific experts is urging the nation to do more to track morbidity and mortality among health care workers (HCWs), given the large and disproportionate number who have been infected with or died from SARS-CoV-2.
The National Academies of Sciences, Engineering, and Medicine’s Standing Committee on Emerging Infectious Diseases and 21st Century Health Threats issued a 10-page “rapid expert consultation” on what is known about deaths and mental health problems among HCWs associated with the COVID-19 pandemic and how to protect workers.
“The absence of a uniform national framework and inconsistent requirements across states for collecting, recording, and reporting HCW mortality and morbidity data associated with COVID-19 impairs anyone’s ability to make comparisons, do combined analyses, or draw conclusions about the scale of the problem,” says the panel in the report.
Mental health, in particular, needs to be examined, it says. Although the data are still limited, the prevalence of burnout and suicide “points to a serious concern,” according to the report.
“As with mortality due to COVID-19, there are currently no national systems nor reporting standards for morbidity measures related to the pandemic, such as mental health status, provider well-being, and other psychological effects on HCWs,” the report says.
A more robust national system that collected data on circumstances and interventions that may raise or lower risk, as well as on where the infection occurred, “would support the adoption of effective mitigation strategies,” says the report. It would also facilitate epidemiologic studies on risk factors, such as face-to-face contact with COVID-19 patients and the availability and use of personal protective equipment (PPE). Studies could also examine the impact of institutional requirements for masking.
Studies have consistently shown that universal mask wearing and access to appropriate PPE support the physical safety and mental health of HCWs, says the report.
Track scale of crisis
The committee cited many gaps in the current system. The Occupational Safety and Health Administration, for instance, doesn’t count deaths from occupationally acquired infection. Many states don’t report COVID-19 deaths by profession. The Centers for Disease Control and Prevention (CDC) relies on case report forms from local health departments for all COVID-19 cases, which typically are lacking in specifics, such as occupation or job setting, says the committee’s report.
As of Nov. 3, the CDC had reported 786 deaths among HCWs that were attributable to COVID-19 – a far lower number than other sources have reported.
The committee notes that much could be done immediately. A National Academy of Medicine panel on clinician well-being and resilience in August recommended that the CDC establish a national epidemiologic tracking program to measure HCWs’ well-being, assess the acute and long-term effects of COVID-19 on those workers, and report on the outcomes of interventions.
Such a program “is needed to comprehensively acknowledge the scale of the COVID-19 crisis and protect the health care workforce that is already stretched to the breaking point in many locations,” the committee says in its report.
A version of this article originally appeared on Medscape.com.
Proposed HIPAA overhaul to ease access to patient health info
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
ADA 2021 standards address financial hardship in diabetes
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
Medicare payments could get tougher for docs
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
Sac/val heart failure benefit extends to diabetes patients
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).

Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).
Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).
Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
FROM JACC: HEART FAILURE
Understanding messenger RNA and other SARS-CoV-2 vaccines
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.

Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at pdnews@mdedge.com.
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.
Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at pdnews@mdedge.com.
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.
Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at pdnews@mdedge.com.
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
Twincretin ‘impressive’: Topline data from phase 3 trial in diabetes
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.