User login
CHMP recommends CAR T for ALL, DLBCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
CHMP recommends CAR T for DLBCL, PMBCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
European Medicines Agency recommends CAR T-cell approvals
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
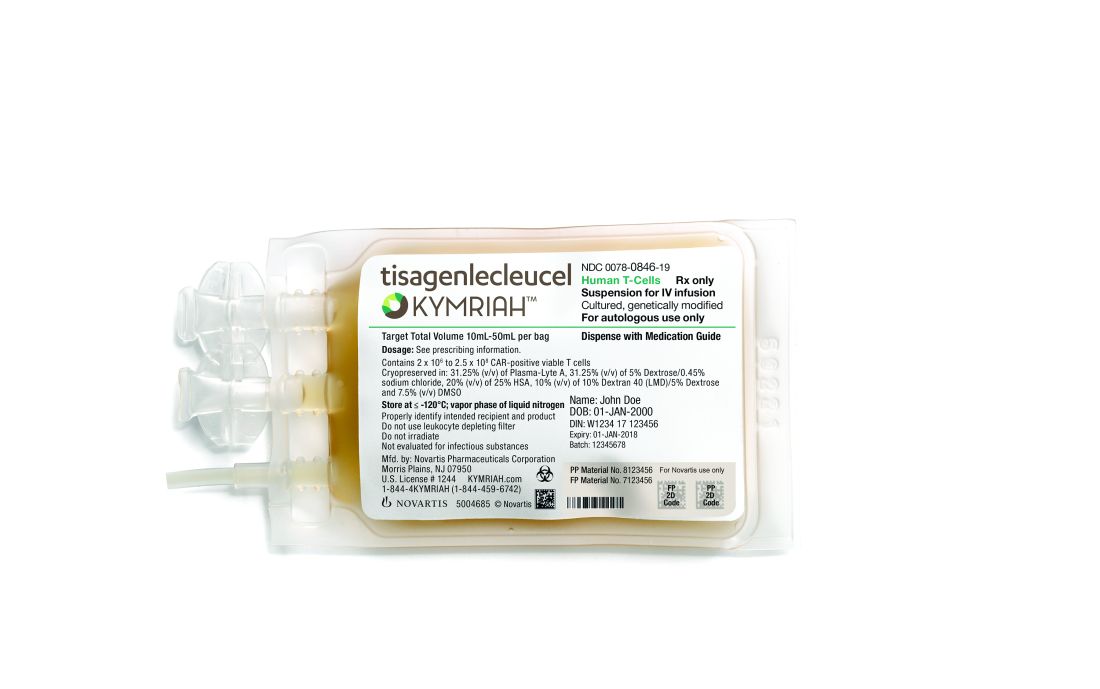
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
Bortezomib plus vorinostat shows modest response in MCL
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
FROM CLINICAL LYMPHOMA, MYELOMA AND LEUKEMIA
Doc reports favorable results from trial on hold

STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
CPI-613 receives orphan designation for BL

The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of Burkitt lymphoma (BL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
The drug is in development as a treatment for hematologic malignancies and solid tumors.
In a phase 1 trial of patients with advanced hematologic malignancies, CPI-613 produced a response in a patient with relapsed BL.
Now, Rafael Pharmaceuticals, Inc., the company developing CPI-613, is launching a phase 2 trial of the drug in patients with relapsed or refractory BL and high-grade B-cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6.
In the phase 1 trial, the patient with relapsed BL achieved a partial response to CPI-613 monotherapy and was ultimately cleared of disease after surgery.
The patient, a 19-year-old female, began taking CPI-613 (2940 mg/m2) after her second relapse. She achieved a radiographic partial response after the third cycle of CPI-613.
The patient completed 17 cycles of CPI-613 over 51 weeks. She decided to stop treatment after the 17th cycle to pursue a surgical resection of residual tumor. The pathology of the surgical specimen revealed BL with extensive necrosis.
Clinical follow-up on the patient showed no evidence of disease more than 36 months later. And CPI-613 was considered well tolerated in this patient.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of Burkitt lymphoma (BL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
The drug is in development as a treatment for hematologic malignancies and solid tumors.
In a phase 1 trial of patients with advanced hematologic malignancies, CPI-613 produced a response in a patient with relapsed BL.
Now, Rafael Pharmaceuticals, Inc., the company developing CPI-613, is launching a phase 2 trial of the drug in patients with relapsed or refractory BL and high-grade B-cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6.
In the phase 1 trial, the patient with relapsed BL achieved a partial response to CPI-613 monotherapy and was ultimately cleared of disease after surgery.
The patient, a 19-year-old female, began taking CPI-613 (2940 mg/m2) after her second relapse. She achieved a radiographic partial response after the third cycle of CPI-613.
The patient completed 17 cycles of CPI-613 over 51 weeks. She decided to stop treatment after the 17th cycle to pursue a surgical resection of residual tumor. The pathology of the surgical specimen revealed BL with extensive necrosis.
Clinical follow-up on the patient showed no evidence of disease more than 36 months later. And CPI-613 was considered well tolerated in this patient.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of Burkitt lymphoma (BL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
The drug is in development as a treatment for hematologic malignancies and solid tumors.
In a phase 1 trial of patients with advanced hematologic malignancies, CPI-613 produced a response in a patient with relapsed BL.
Now, Rafael Pharmaceuticals, Inc., the company developing CPI-613, is launching a phase 2 trial of the drug in patients with relapsed or refractory BL and high-grade B-cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6.
In the phase 1 trial, the patient with relapsed BL achieved a partial response to CPI-613 monotherapy and was ultimately cleared of disease after surgery.
The patient, a 19-year-old female, began taking CPI-613 (2940 mg/m2) after her second relapse. She achieved a radiographic partial response after the third cycle of CPI-613.
The patient completed 17 cycles of CPI-613 over 51 weeks. She decided to stop treatment after the 17th cycle to pursue a surgical resection of residual tumor. The pathology of the surgical specimen revealed BL with extensive necrosis.
Clinical follow-up on the patient showed no evidence of disease more than 36 months later. And CPI-613 was considered well tolerated in this patient.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
CAR T in DLBCL: Liso-cel has ‘remarkable’ efficacy in cohort
CHICAGO – The CD19–directed chimeric antigen receptor (CAR) T-cell product lisocabtagene maraleucel (liso-cel, JCAR017) produced durable responses in poor-prognosis patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), follow-up results of a phase 1 trial show.
Nearly 90% of DLBCL patients who achieved complete response as their best response on liso-cel were alive at 1 year in the study, according to investigator Jeremy S. Abramson, MD, of Massachusetts General Hospital Cancer Center in Boston.
That result is “far superior to what we would have anticipated with conventional therapies in a largely chemorefractory DLBCL population,” Dr. Abramson said at the annual meeting of the American Society of Clinical Oncology.
The new data on liso-cel come on the heels of a second approval of a CAR T-cell therapy for DLBCL, noted Caron Jacobson, MD, of Dana-Farber Cancer Institute, Boston.
Axicabtagene ciloleucel (Yescarta) was approved in October 2017 by the U.S. Food and Drug Administration for relapsed or refractory large B-cell lymphomas, including DLBCL. In May 2018, tisagenlecleucel (Kymriah) received its second FDA approval to treat relapsed or refractory large B-cell lymphomas, including DLBCL.
CAR T-cell therapy “really has transformed outcomes for a group of patients who previously had no other standard of care and who… have a relatively short overall survival,” Dr. Jacobson said.
At the meeting, Dr. Abramson presented findings on DLBCL patients in TRANSCEND NHL 001, a phase 1, multicenter, open-label study of the CD-19 targeted CAR T-cell therapy in relapsed and refractory B-cell non-Hodgkin lymphoma.
About 90% of treated DLBCL patients had one or more poor-risk disease features, such as ECOG performance status 2 and primary refractory disease, which predict poor overall survival, according to Dr. Abramson.
Dr. Abramson’s presentation focused on 102 evaluable DLBCL patients in the dose-finding and dose-expansion cohorts of the TRANSCEND study, including a subset analysis of a core group of 73 patients who met the criteria for pivotal dose cohort of the study (1 x 108 cells given as a single dose).
For the full set of 102 DLBCL patients, the best overall response rate was 75%, including a best complete remission rate of 55%, according to presented data. In the core group of 73 DLBCL patients, best overall response and complete remission rates were 80% and 59%, respectively.
Investigators saw “encouraging” durable response rates at 6 months and beyond in the core DLBCL population, according to Dr. Abramson. Of patients with a complete remission at 3 months, 88% remained in complete remission at the 6-month follow-up, and 93% of those in remission at the 6-month time point were in ongoing response at a median follow-up of 8 months.
Median overall survival had not been reached in either the full or core DLBCL cohorts with a median of 12 months follow-up, he added, noting that 90% of patients who achieved complete remission as their best response remained alive at 1 year.
In terms of adverse effects, liso-cel is showing a low and manageable toxicity profile, with very low rates of severe cytokine release syndrome (CRS) and neurotoxicity at 1% and 13%, respectively, Dr. Abramson reported.
“This anti-CD19 CAR T cell has remarkable efficacy in a group of highly refractory aggressive B-cell non-Hodgkin lymphoma patients,” said Dr. Jacobson, commenting on results of the DLBCL subset.
Based on the data presented, liso-cel is “clearly competitive” with the approved CAR T-cell therapies, though she advised caution in comparing across studies. “I don’t think that there will be a randomized study of all three agents, but I do think that we’ll start to get comparative data from single institution experiences that are using all three products,” she said.
The pivotal DLBCL cohort of TRANSCEND NHL 001 has completed accrual and results will be presented at a future meeting, Dr. Abramson said.
Dr. Abramson reported disclosures related to Celgene, Genentech/Roche, Gilead Sciences, Novartis, Seattle Genetics, and Millennium.
SOURCE: Abramson JS et al. ASCO 2018. Abstract 7505.
CHICAGO – The CD19–directed chimeric antigen receptor (CAR) T-cell product lisocabtagene maraleucel (liso-cel, JCAR017) produced durable responses in poor-prognosis patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), follow-up results of a phase 1 trial show.
Nearly 90% of DLBCL patients who achieved complete response as their best response on liso-cel were alive at 1 year in the study, according to investigator Jeremy S. Abramson, MD, of Massachusetts General Hospital Cancer Center in Boston.
That result is “far superior to what we would have anticipated with conventional therapies in a largely chemorefractory DLBCL population,” Dr. Abramson said at the annual meeting of the American Society of Clinical Oncology.
The new data on liso-cel come on the heels of a second approval of a CAR T-cell therapy for DLBCL, noted Caron Jacobson, MD, of Dana-Farber Cancer Institute, Boston.
Axicabtagene ciloleucel (Yescarta) was approved in October 2017 by the U.S. Food and Drug Administration for relapsed or refractory large B-cell lymphomas, including DLBCL. In May 2018, tisagenlecleucel (Kymriah) received its second FDA approval to treat relapsed or refractory large B-cell lymphomas, including DLBCL.
CAR T-cell therapy “really has transformed outcomes for a group of patients who previously had no other standard of care and who… have a relatively short overall survival,” Dr. Jacobson said.
At the meeting, Dr. Abramson presented findings on DLBCL patients in TRANSCEND NHL 001, a phase 1, multicenter, open-label study of the CD-19 targeted CAR T-cell therapy in relapsed and refractory B-cell non-Hodgkin lymphoma.
About 90% of treated DLBCL patients had one or more poor-risk disease features, such as ECOG performance status 2 and primary refractory disease, which predict poor overall survival, according to Dr. Abramson.
Dr. Abramson’s presentation focused on 102 evaluable DLBCL patients in the dose-finding and dose-expansion cohorts of the TRANSCEND study, including a subset analysis of a core group of 73 patients who met the criteria for pivotal dose cohort of the study (1 x 108 cells given as a single dose).
For the full set of 102 DLBCL patients, the best overall response rate was 75%, including a best complete remission rate of 55%, according to presented data. In the core group of 73 DLBCL patients, best overall response and complete remission rates were 80% and 59%, respectively.
Investigators saw “encouraging” durable response rates at 6 months and beyond in the core DLBCL population, according to Dr. Abramson. Of patients with a complete remission at 3 months, 88% remained in complete remission at the 6-month follow-up, and 93% of those in remission at the 6-month time point were in ongoing response at a median follow-up of 8 months.
Median overall survival had not been reached in either the full or core DLBCL cohorts with a median of 12 months follow-up, he added, noting that 90% of patients who achieved complete remission as their best response remained alive at 1 year.
In terms of adverse effects, liso-cel is showing a low and manageable toxicity profile, with very low rates of severe cytokine release syndrome (CRS) and neurotoxicity at 1% and 13%, respectively, Dr. Abramson reported.
“This anti-CD19 CAR T cell has remarkable efficacy in a group of highly refractory aggressive B-cell non-Hodgkin lymphoma patients,” said Dr. Jacobson, commenting on results of the DLBCL subset.
Based on the data presented, liso-cel is “clearly competitive” with the approved CAR T-cell therapies, though she advised caution in comparing across studies. “I don’t think that there will be a randomized study of all three agents, but I do think that we’ll start to get comparative data from single institution experiences that are using all three products,” she said.
The pivotal DLBCL cohort of TRANSCEND NHL 001 has completed accrual and results will be presented at a future meeting, Dr. Abramson said.
Dr. Abramson reported disclosures related to Celgene, Genentech/Roche, Gilead Sciences, Novartis, Seattle Genetics, and Millennium.
SOURCE: Abramson JS et al. ASCO 2018. Abstract 7505.
CHICAGO – The CD19–directed chimeric antigen receptor (CAR) T-cell product lisocabtagene maraleucel (liso-cel, JCAR017) produced durable responses in poor-prognosis patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), follow-up results of a phase 1 trial show.
Nearly 90% of DLBCL patients who achieved complete response as their best response on liso-cel were alive at 1 year in the study, according to investigator Jeremy S. Abramson, MD, of Massachusetts General Hospital Cancer Center in Boston.
That result is “far superior to what we would have anticipated with conventional therapies in a largely chemorefractory DLBCL population,” Dr. Abramson said at the annual meeting of the American Society of Clinical Oncology.
The new data on liso-cel come on the heels of a second approval of a CAR T-cell therapy for DLBCL, noted Caron Jacobson, MD, of Dana-Farber Cancer Institute, Boston.
Axicabtagene ciloleucel (Yescarta) was approved in October 2017 by the U.S. Food and Drug Administration for relapsed or refractory large B-cell lymphomas, including DLBCL. In May 2018, tisagenlecleucel (Kymriah) received its second FDA approval to treat relapsed or refractory large B-cell lymphomas, including DLBCL.
CAR T-cell therapy “really has transformed outcomes for a group of patients who previously had no other standard of care and who… have a relatively short overall survival,” Dr. Jacobson said.
At the meeting, Dr. Abramson presented findings on DLBCL patients in TRANSCEND NHL 001, a phase 1, multicenter, open-label study of the CD-19 targeted CAR T-cell therapy in relapsed and refractory B-cell non-Hodgkin lymphoma.
About 90% of treated DLBCL patients had one or more poor-risk disease features, such as ECOG performance status 2 and primary refractory disease, which predict poor overall survival, according to Dr. Abramson.
Dr. Abramson’s presentation focused on 102 evaluable DLBCL patients in the dose-finding and dose-expansion cohorts of the TRANSCEND study, including a subset analysis of a core group of 73 patients who met the criteria for pivotal dose cohort of the study (1 x 108 cells given as a single dose).
For the full set of 102 DLBCL patients, the best overall response rate was 75%, including a best complete remission rate of 55%, according to presented data. In the core group of 73 DLBCL patients, best overall response and complete remission rates were 80% and 59%, respectively.
Investigators saw “encouraging” durable response rates at 6 months and beyond in the core DLBCL population, according to Dr. Abramson. Of patients with a complete remission at 3 months, 88% remained in complete remission at the 6-month follow-up, and 93% of those in remission at the 6-month time point were in ongoing response at a median follow-up of 8 months.
Median overall survival had not been reached in either the full or core DLBCL cohorts with a median of 12 months follow-up, he added, noting that 90% of patients who achieved complete remission as their best response remained alive at 1 year.
In terms of adverse effects, liso-cel is showing a low and manageable toxicity profile, with very low rates of severe cytokine release syndrome (CRS) and neurotoxicity at 1% and 13%, respectively, Dr. Abramson reported.
“This anti-CD19 CAR T cell has remarkable efficacy in a group of highly refractory aggressive B-cell non-Hodgkin lymphoma patients,” said Dr. Jacobson, commenting on results of the DLBCL subset.
Based on the data presented, liso-cel is “clearly competitive” with the approved CAR T-cell therapies, though she advised caution in comparing across studies. “I don’t think that there will be a randomized study of all three agents, but I do think that we’ll start to get comparative data from single institution experiences that are using all three products,” she said.
The pivotal DLBCL cohort of TRANSCEND NHL 001 has completed accrual and results will be presented at a future meeting, Dr. Abramson said.
Dr. Abramson reported disclosures related to Celgene, Genentech/Roche, Gilead Sciences, Novartis, Seattle Genetics, and Millennium.
SOURCE: Abramson JS et al. ASCO 2018. Abstract 7505.
REPORTING FROM ASCO 2018
Key clinical point: (DLBCL).
Major finding: Among DLBCL patients treated with the pivotal dose of liso-cel, 88% who were in complete remission at 3 months remained in complete remission at the 6 month follow-up.
Study details: Follow-up report on a cohort of DLBCL patients from TRANSCEND NHL 001, a phase 1 trial of liso-cel in relapsed and refractory B-cell NHL.
Disclosures: Dr. Abramson reported disclosures related to Celgene, Genentech/Roche, Gilead Sciences, Novartis, Seattle Genetics, and Millennium.
Source: Abramson JS et al. ASCO 2018. Abstract 7505.
FDA Approves a Cannabinoid Medicine for Two Forms of Epilepsy
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.”

CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.”
CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.”
CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.
Inhibitor exhibits activity in B- and T-cell NHLs

STOCKHOLM—The dual SYK/JAK inhibitor cerdulatinib has demonstrated efficacy in a phase 2 trial of patients with heavily pretreated B- and T-cell non-Hodgkin lymphomas (NHLs).
There were a few deaths due to sepsis or septic shock that were considered related to cerdulatinib, but investigators have taken steps to prevent additional deaths.
Cerdulatinib produced responses in patients with peripheral T-cell lymphoma (PTCL), cutaneous T-cell lymphoma (CTCL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), follicular lymphoma (FL), and other NHLs.
The 5 deaths due to sepsis or septic shock (3 concomitant with pneumonia) occurred early on in the trial, and dose reductions, monitoring, and antibiotic prophylaxis appeared to be effective in preventing additional deaths.
Results from this trial were presented in a poster (abstract PF437) at the 23rd Congress of the European Hematology Association (EHA).
The research was sponsored by Portola Pharmaceuticals, Inc.
The trial enrolled 114 patients. They had FL (grade 1-3A; n=39), PTCL (n=25), CTCL (n=5), CLL/SLL (n=28), other indolent NHLs (Waldenstrom’s macroglobulinemia and marginal zone lymphoma; n=12), or aggressive NHL (defined as diffuse large B-cell lymphoma [DLBCL], grade 3B FL, mantle cell lymphoma, and transformed NHL with relapsed disease; n=5).
The patients’ median age was 68 (range, 34-93), and 59% were male. The median number of prior treatment regimens was 3 (range, 1-13), and 37% of patients had refractory disease.
Patients received cerdulatinib at 25, 30, or 35 mg twice daily (BID). A total of 101 patients were evaluable as of May 4, 2018.
Response
The objective response rate (ORR) was 47% in the entire population. Thirteen patients achieved a complete response (CR), and 34 had a partial response (PR). Thirty-four patients remained on cerdulatinib at the data cut-off.
The ORR was 46% in the FL patients, with 3 patients achieving a CR and 13 achieving a PR. Thirteen FL patients remained on cerdulatinib.
In the CLL/SLL patients, the ORR was 61%. Two patients had a CR, and 15 had a PR. Four CLL/SLL patients remained on cerdulatinib.
In PTCL, the ORR was 35%. All 7 responders had a CR. Eleven PTCL patients remained on cerdulatinib.
Only 1 CTCL patient was evaluable, but this patient achieved a CR and remained on cerdulatinib.
The ORR was 42% for patients with other indolent NHLs, with 5 PRs and no CRs. Only 1 patient in this group remained on cerdulatinib.
For aggressive NHL, the ORR was 20%, with 1 PR and no CRs. None of the patients in this group stayed on cerdulatinib.
“Cerdulatinib continues to demonstrate promising results across a wide range of B- and T-cell malignancies, including early indications of the potential for durable responses,” said study investigator Paul Hamlin, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The new signals in relapsed/refractory PTCL and CTCL are particularly compelling when you consider the limited treatment options for patients that fail frontline therapy.”
Safety
Grade 3 or higher adverse events included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
The 5 deaths due to sepsis or septic shock (3 of which were concomitant with pneumonia) were considered related to cerdulatinib. Three of the deaths occurred in patients with CLL, 1 in a DLBCL patient, and 1 in an FL patient.
“One of the things we have seen [with CLL patients] is the background infection rate is quite a bit higher,” said John T. Curnutte, MD, PhD, interim co-president and head of research and development at Portola Pharmaceuticals.
“You see this with multiple other agents, so we were not particularly surprised to see [sepsis/septic shock in CLL].”
Dr Curnutte noted that grade 5 sepsis/septic shock tended to occur in patients who were pre-colonized and/or had high plasma levels of cerdulatinib.
So, to prevent these adverse events, the starting dose of cerdulatinib was lowered, investigators began monitoring patients’ plasma levels, and all patients began receiving antibiotic prophylaxis. (Previously, only CLL patients had received this prophylaxis.)
The investigators found that lowering the starting dose from 35 mg BID to 30 mg or even 25 mg BID reduced plasma levels.
“At the 35 mg dose, 1 out of every 4 patients showed accumulation of the drug,” Dr Curnutte said. “In general, the use of the 30 mg BID or step-down 25 mg BID did not result in accumulation of drug.”
Dr Curnutte also noted that enrollment of CLL/SLL patients is complete, and investigators would be “very careful” if the study were to be re-opened to these patients.
For now, Portola is focused on completing enrollment in other patient groups on this phase 2 trial. The company is also hoping to conduct a phase 3 trial of cerdulatinib in PTCL that could begin as early as the end of this year.
STOCKHOLM—The dual SYK/JAK inhibitor cerdulatinib has demonstrated efficacy in a phase 2 trial of patients with heavily pretreated B- and T-cell non-Hodgkin lymphomas (NHLs).
There were a few deaths due to sepsis or septic shock that were considered related to cerdulatinib, but investigators have taken steps to prevent additional deaths.
Cerdulatinib produced responses in patients with peripheral T-cell lymphoma (PTCL), cutaneous T-cell lymphoma (CTCL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), follicular lymphoma (FL), and other NHLs.
The 5 deaths due to sepsis or septic shock (3 concomitant with pneumonia) occurred early on in the trial, and dose reductions, monitoring, and antibiotic prophylaxis appeared to be effective in preventing additional deaths.
Results from this trial were presented in a poster (abstract PF437) at the 23rd Congress of the European Hematology Association (EHA).
The research was sponsored by Portola Pharmaceuticals, Inc.
The trial enrolled 114 patients. They had FL (grade 1-3A; n=39), PTCL (n=25), CTCL (n=5), CLL/SLL (n=28), other indolent NHLs (Waldenstrom’s macroglobulinemia and marginal zone lymphoma; n=12), or aggressive NHL (defined as diffuse large B-cell lymphoma [DLBCL], grade 3B FL, mantle cell lymphoma, and transformed NHL with relapsed disease; n=5).
The patients’ median age was 68 (range, 34-93), and 59% were male. The median number of prior treatment regimens was 3 (range, 1-13), and 37% of patients had refractory disease.
Patients received cerdulatinib at 25, 30, or 35 mg twice daily (BID). A total of 101 patients were evaluable as of May 4, 2018.
Response
The objective response rate (ORR) was 47% in the entire population. Thirteen patients achieved a complete response (CR), and 34 had a partial response (PR). Thirty-four patients remained on cerdulatinib at the data cut-off.
The ORR was 46% in the FL patients, with 3 patients achieving a CR and 13 achieving a PR. Thirteen FL patients remained on cerdulatinib.
In the CLL/SLL patients, the ORR was 61%. Two patients had a CR, and 15 had a PR. Four CLL/SLL patients remained on cerdulatinib.
In PTCL, the ORR was 35%. All 7 responders had a CR. Eleven PTCL patients remained on cerdulatinib.
Only 1 CTCL patient was evaluable, but this patient achieved a CR and remained on cerdulatinib.
The ORR was 42% for patients with other indolent NHLs, with 5 PRs and no CRs. Only 1 patient in this group remained on cerdulatinib.
For aggressive NHL, the ORR was 20%, with 1 PR and no CRs. None of the patients in this group stayed on cerdulatinib.
“Cerdulatinib continues to demonstrate promising results across a wide range of B- and T-cell malignancies, including early indications of the potential for durable responses,” said study investigator Paul Hamlin, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The new signals in relapsed/refractory PTCL and CTCL are particularly compelling when you consider the limited treatment options for patients that fail frontline therapy.”
Safety
Grade 3 or higher adverse events included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
The 5 deaths due to sepsis or septic shock (3 of which were concomitant with pneumonia) were considered related to cerdulatinib. Three of the deaths occurred in patients with CLL, 1 in a DLBCL patient, and 1 in an FL patient.
“One of the things we have seen [with CLL patients] is the background infection rate is quite a bit higher,” said John T. Curnutte, MD, PhD, interim co-president and head of research and development at Portola Pharmaceuticals.
“You see this with multiple other agents, so we were not particularly surprised to see [sepsis/septic shock in CLL].”
Dr Curnutte noted that grade 5 sepsis/septic shock tended to occur in patients who were pre-colonized and/or had high plasma levels of cerdulatinib.
So, to prevent these adverse events, the starting dose of cerdulatinib was lowered, investigators began monitoring patients’ plasma levels, and all patients began receiving antibiotic prophylaxis. (Previously, only CLL patients had received this prophylaxis.)
The investigators found that lowering the starting dose from 35 mg BID to 30 mg or even 25 mg BID reduced plasma levels.
“At the 35 mg dose, 1 out of every 4 patients showed accumulation of the drug,” Dr Curnutte said. “In general, the use of the 30 mg BID or step-down 25 mg BID did not result in accumulation of drug.”
Dr Curnutte also noted that enrollment of CLL/SLL patients is complete, and investigators would be “very careful” if the study were to be re-opened to these patients.
For now, Portola is focused on completing enrollment in other patient groups on this phase 2 trial. The company is also hoping to conduct a phase 3 trial of cerdulatinib in PTCL that could begin as early as the end of this year.
STOCKHOLM—The dual SYK/JAK inhibitor cerdulatinib has demonstrated efficacy in a phase 2 trial of patients with heavily pretreated B- and T-cell non-Hodgkin lymphomas (NHLs).
There were a few deaths due to sepsis or septic shock that were considered related to cerdulatinib, but investigators have taken steps to prevent additional deaths.
Cerdulatinib produced responses in patients with peripheral T-cell lymphoma (PTCL), cutaneous T-cell lymphoma (CTCL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), follicular lymphoma (FL), and other NHLs.
The 5 deaths due to sepsis or septic shock (3 concomitant with pneumonia) occurred early on in the trial, and dose reductions, monitoring, and antibiotic prophylaxis appeared to be effective in preventing additional deaths.
Results from this trial were presented in a poster (abstract PF437) at the 23rd Congress of the European Hematology Association (EHA).
The research was sponsored by Portola Pharmaceuticals, Inc.
The trial enrolled 114 patients. They had FL (grade 1-3A; n=39), PTCL (n=25), CTCL (n=5), CLL/SLL (n=28), other indolent NHLs (Waldenstrom’s macroglobulinemia and marginal zone lymphoma; n=12), or aggressive NHL (defined as diffuse large B-cell lymphoma [DLBCL], grade 3B FL, mantle cell lymphoma, and transformed NHL with relapsed disease; n=5).
The patients’ median age was 68 (range, 34-93), and 59% were male. The median number of prior treatment regimens was 3 (range, 1-13), and 37% of patients had refractory disease.
Patients received cerdulatinib at 25, 30, or 35 mg twice daily (BID). A total of 101 patients were evaluable as of May 4, 2018.
Response
The objective response rate (ORR) was 47% in the entire population. Thirteen patients achieved a complete response (CR), and 34 had a partial response (PR). Thirty-four patients remained on cerdulatinib at the data cut-off.
The ORR was 46% in the FL patients, with 3 patients achieving a CR and 13 achieving a PR. Thirteen FL patients remained on cerdulatinib.
In the CLL/SLL patients, the ORR was 61%. Two patients had a CR, and 15 had a PR. Four CLL/SLL patients remained on cerdulatinib.
In PTCL, the ORR was 35%. All 7 responders had a CR. Eleven PTCL patients remained on cerdulatinib.
Only 1 CTCL patient was evaluable, but this patient achieved a CR and remained on cerdulatinib.
The ORR was 42% for patients with other indolent NHLs, with 5 PRs and no CRs. Only 1 patient in this group remained on cerdulatinib.
For aggressive NHL, the ORR was 20%, with 1 PR and no CRs. None of the patients in this group stayed on cerdulatinib.
“Cerdulatinib continues to demonstrate promising results across a wide range of B- and T-cell malignancies, including early indications of the potential for durable responses,” said study investigator Paul Hamlin, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“The new signals in relapsed/refractory PTCL and CTCL are particularly compelling when you consider the limited treatment options for patients that fail frontline therapy.”
Safety
Grade 3 or higher adverse events included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
The 5 deaths due to sepsis or septic shock (3 of which were concomitant with pneumonia) were considered related to cerdulatinib. Three of the deaths occurred in patients with CLL, 1 in a DLBCL patient, and 1 in an FL patient.
“One of the things we have seen [with CLL patients] is the background infection rate is quite a bit higher,” said John T. Curnutte, MD, PhD, interim co-president and head of research and development at Portola Pharmaceuticals.
“You see this with multiple other agents, so we were not particularly surprised to see [sepsis/septic shock in CLL].”
Dr Curnutte noted that grade 5 sepsis/septic shock tended to occur in patients who were pre-colonized and/or had high plasma levels of cerdulatinib.
So, to prevent these adverse events, the starting dose of cerdulatinib was lowered, investigators began monitoring patients’ plasma levels, and all patients began receiving antibiotic prophylaxis. (Previously, only CLL patients had received this prophylaxis.)
The investigators found that lowering the starting dose from 35 mg BID to 30 mg or even 25 mg BID reduced plasma levels.
“At the 35 mg dose, 1 out of every 4 patients showed accumulation of the drug,” Dr Curnutte said. “In general, the use of the 30 mg BID or step-down 25 mg BID did not result in accumulation of drug.”
Dr Curnutte also noted that enrollment of CLL/SLL patients is complete, and investigators would be “very careful” if the study were to be re-opened to these patients.
For now, Portola is focused on completing enrollment in other patient groups on this phase 2 trial. The company is also hoping to conduct a phase 3 trial of cerdulatinib in PTCL that could begin as early as the end of this year.
Is CLL chemoimmunotherapy dead? Not yet
CHICAGO – Chemoimmunotherapy for chronic lymphocytic leukemia is on the way out, but there’s one scenario where it still plays a key role, according to one leukemia expert.
That scenario is not in relapsed or refractory chronic lymphocytic leukemia (CLL), where the use of fludarabine, cyclophosphamide, and rituximab (FCR) may be hard to justify today. Data supporting use of FCR in relapsed CLL show a median progression-free survival (PFS) of about 21 months, Susan M. O’Brien, MD, of the University of California, Irvine, said at the annual meeting of the American Society of Clinical Oncology. There is also data for bendamustine-rituximab retreatment showing a median event-free survival of about 15 months, she added.
By contrast, the 5-year follow-up data for the Bruton tyrosine kinase inhibitor ibrutinib in the relapsed/refractory setting shows a median PFS of 52 months, which is “extraordinary,” given that the patients had a median of four prior regimens, Dr. O’Brien said.
Similarly, recently published results from the randomized, phase 3 MURANO study of venetoclax plus rituximab in relapsed/refractory CLL showed that median PFS was not reached at a median follow-up of 23.8 months, versus a median of 17 months for the bendamustine-rituximab comparison arm (N Engl J Med. 2018;378[12]:1107-20).
“Thanks to the MURANO study, we likely will have an expanded label for venetoclax that includes the combination of venetoclax and rituximab,” Dr. O’Brien said. “I think it’s quite clear that either of these is dramatically better than what you get with retreatment with chemotherapy, so I personally don’t think there is a role for chemoimmunotherapy in the relapsed patient.”
On June 8, 2018, the Food and Drug Administration granted regular approval for venetoclax for patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who have received at least one prior therapy. The FDA also approved its use in combination with rituximab.*
But frontline CLL treatment is currently a little bit more complicated, Dr. O’Brien said.
Recent studies show favorable long-term outcomes with FCR frontline therapy in the immunoglobulin heavy chain variable gene (IgHV) –mutated subgroup of patients, she noted.
The longest follow-up comes from a study from investigators at the University of Texas MD Anderson Cancer Center, Houston, published in 2016. In that study, the 12.8-year PFS was 53.9% for IgHV-mutated patients, versus just 8.7% for patients with unmutated IgHV. Of the IgHV-mutated group, more than half achieved minimal residual disease (MRD) negativity after treatment (Blood. 2016 Jan 21; 127[3]: 303-9).
“I’m going to go out on a limb and I’m going to suggest that I think there is a cure fraction here,” Dr. O’Brien said. “On the other hand, if there’s not a cure fraction and they’re going to relapse after 17 years, that’s a pretty attractive endpoint, even if it’s not a cure fraction.”
Clinical practice guidelines now recognize IgHV mutation status as an important marker that should be obtained when deciding on treatment, Dr. O’Brien noted.
For unmutated patients, the RESONATE-2 trial showed that ibrutinib was superior to chlorambucil in older patients, many of whom had comorbid conditions. In the 3-year update, median PFS was approximately 15 months for chlorambucil, while for ibrutinib the median PFS was “nowhere near” being reached, Dr. O’Brien said.
Those data may not be so relevant for fit, unmutated patients, and two randomized trials comparing FCR with bendamustine and rituximab have yet to report data. However, one recent cross-trial comparison found fairly overlapping survival curves for the two chemoimmunotherapy approaches.
Dr. O’Brien said she would put older patients with comorbidities on ibrutinib if a clinical trial was not available, and for fit, unmutated patients, while more data are needed, she would also use ibrutinib. However, patient preference sometimes tips the scale in favor of FCR.
“The discussions sometimes are quite long about whether the patient should opt to take ibrutinib or FCR,” Dr. O’Brien said. “The last patient I had that discussion with elected to take FCR. When I asked him why, he said because he liked the idea of being finished in six cycles, off all therapy, and hopefully in remission.”
While Dr. O’Brien said she views chemoimmunotherapy as still relevant in IgHV-mutated patients, eventually it will go away, she concluded. Toward that end, there is considerable interest in venetoclax plus ibrutinib, a combination that, in early reports, has yielded very encouraging MRD results in first-line CLL.
“We have no long-term data, but very, very exciting MRD negativity data,” Dr. O’Brien said.
Dr. O’Brien reported relationships with Abbvie, Amgen, Celgene, Gilead Sciences, Janssen, Pfizer, Pharmacyclics, Sunesis Pharmaceuticals, and others.
*This story was updated 6/25/2018.
CHICAGO – Chemoimmunotherapy for chronic lymphocytic leukemia is on the way out, but there’s one scenario where it still plays a key role, according to one leukemia expert.
That scenario is not in relapsed or refractory chronic lymphocytic leukemia (CLL), where the use of fludarabine, cyclophosphamide, and rituximab (FCR) may be hard to justify today. Data supporting use of FCR in relapsed CLL show a median progression-free survival (PFS) of about 21 months, Susan M. O’Brien, MD, of the University of California, Irvine, said at the annual meeting of the American Society of Clinical Oncology. There is also data for bendamustine-rituximab retreatment showing a median event-free survival of about 15 months, she added.
By contrast, the 5-year follow-up data for the Bruton tyrosine kinase inhibitor ibrutinib in the relapsed/refractory setting shows a median PFS of 52 months, which is “extraordinary,” given that the patients had a median of four prior regimens, Dr. O’Brien said.
Similarly, recently published results from the randomized, phase 3 MURANO study of venetoclax plus rituximab in relapsed/refractory CLL showed that median PFS was not reached at a median follow-up of 23.8 months, versus a median of 17 months for the bendamustine-rituximab comparison arm (N Engl J Med. 2018;378[12]:1107-20).
“Thanks to the MURANO study, we likely will have an expanded label for venetoclax that includes the combination of venetoclax and rituximab,” Dr. O’Brien said. “I think it’s quite clear that either of these is dramatically better than what you get with retreatment with chemotherapy, so I personally don’t think there is a role for chemoimmunotherapy in the relapsed patient.”
On June 8, 2018, the Food and Drug Administration granted regular approval for venetoclax for patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who have received at least one prior therapy. The FDA also approved its use in combination with rituximab.*
But frontline CLL treatment is currently a little bit more complicated, Dr. O’Brien said.
Recent studies show favorable long-term outcomes with FCR frontline therapy in the immunoglobulin heavy chain variable gene (IgHV) –mutated subgroup of patients, she noted.
The longest follow-up comes from a study from investigators at the University of Texas MD Anderson Cancer Center, Houston, published in 2016. In that study, the 12.8-year PFS was 53.9% for IgHV-mutated patients, versus just 8.7% for patients with unmutated IgHV. Of the IgHV-mutated group, more than half achieved minimal residual disease (MRD) negativity after treatment (Blood. 2016 Jan 21; 127[3]: 303-9).
“I’m going to go out on a limb and I’m going to suggest that I think there is a cure fraction here,” Dr. O’Brien said. “On the other hand, if there’s not a cure fraction and they’re going to relapse after 17 years, that’s a pretty attractive endpoint, even if it’s not a cure fraction.”
Clinical practice guidelines now recognize IgHV mutation status as an important marker that should be obtained when deciding on treatment, Dr. O’Brien noted.
For unmutated patients, the RESONATE-2 trial showed that ibrutinib was superior to chlorambucil in older patients, many of whom had comorbid conditions. In the 3-year update, median PFS was approximately 15 months for chlorambucil, while for ibrutinib the median PFS was “nowhere near” being reached, Dr. O’Brien said.
Those data may not be so relevant for fit, unmutated patients, and two randomized trials comparing FCR with bendamustine and rituximab have yet to report data. However, one recent cross-trial comparison found fairly overlapping survival curves for the two chemoimmunotherapy approaches.
Dr. O’Brien said she would put older patients with comorbidities on ibrutinib if a clinical trial was not available, and for fit, unmutated patients, while more data are needed, she would also use ibrutinib. However, patient preference sometimes tips the scale in favor of FCR.
“The discussions sometimes are quite long about whether the patient should opt to take ibrutinib or FCR,” Dr. O’Brien said. “The last patient I had that discussion with elected to take FCR. When I asked him why, he said because he liked the idea of being finished in six cycles, off all therapy, and hopefully in remission.”
While Dr. O’Brien said she views chemoimmunotherapy as still relevant in IgHV-mutated patients, eventually it will go away, she concluded. Toward that end, there is considerable interest in venetoclax plus ibrutinib, a combination that, in early reports, has yielded very encouraging MRD results in first-line CLL.
“We have no long-term data, but very, very exciting MRD negativity data,” Dr. O’Brien said.
Dr. O’Brien reported relationships with Abbvie, Amgen, Celgene, Gilead Sciences, Janssen, Pfizer, Pharmacyclics, Sunesis Pharmaceuticals, and others.
*This story was updated 6/25/2018.
CHICAGO – Chemoimmunotherapy for chronic lymphocytic leukemia is on the way out, but there’s one scenario where it still plays a key role, according to one leukemia expert.
That scenario is not in relapsed or refractory chronic lymphocytic leukemia (CLL), where the use of fludarabine, cyclophosphamide, and rituximab (FCR) may be hard to justify today. Data supporting use of FCR in relapsed CLL show a median progression-free survival (PFS) of about 21 months, Susan M. O’Brien, MD, of the University of California, Irvine, said at the annual meeting of the American Society of Clinical Oncology. There is also data for bendamustine-rituximab retreatment showing a median event-free survival of about 15 months, she added.
By contrast, the 5-year follow-up data for the Bruton tyrosine kinase inhibitor ibrutinib in the relapsed/refractory setting shows a median PFS of 52 months, which is “extraordinary,” given that the patients had a median of four prior regimens, Dr. O’Brien said.
Similarly, recently published results from the randomized, phase 3 MURANO study of venetoclax plus rituximab in relapsed/refractory CLL showed that median PFS was not reached at a median follow-up of 23.8 months, versus a median of 17 months for the bendamustine-rituximab comparison arm (N Engl J Med. 2018;378[12]:1107-20).
“Thanks to the MURANO study, we likely will have an expanded label for venetoclax that includes the combination of venetoclax and rituximab,” Dr. O’Brien said. “I think it’s quite clear that either of these is dramatically better than what you get with retreatment with chemotherapy, so I personally don’t think there is a role for chemoimmunotherapy in the relapsed patient.”
On June 8, 2018, the Food and Drug Administration granted regular approval for venetoclax for patients with CLL or small lymphocytic lymphoma, with or without 17p deletion, who have received at least one prior therapy. The FDA also approved its use in combination with rituximab.*
But frontline CLL treatment is currently a little bit more complicated, Dr. O’Brien said.
Recent studies show favorable long-term outcomes with FCR frontline therapy in the immunoglobulin heavy chain variable gene (IgHV) –mutated subgroup of patients, she noted.
The longest follow-up comes from a study from investigators at the University of Texas MD Anderson Cancer Center, Houston, published in 2016. In that study, the 12.8-year PFS was 53.9% for IgHV-mutated patients, versus just 8.7% for patients with unmutated IgHV. Of the IgHV-mutated group, more than half achieved minimal residual disease (MRD) negativity after treatment (Blood. 2016 Jan 21; 127[3]: 303-9).
“I’m going to go out on a limb and I’m going to suggest that I think there is a cure fraction here,” Dr. O’Brien said. “On the other hand, if there’s not a cure fraction and they’re going to relapse after 17 years, that’s a pretty attractive endpoint, even if it’s not a cure fraction.”
Clinical practice guidelines now recognize IgHV mutation status as an important marker that should be obtained when deciding on treatment, Dr. O’Brien noted.
For unmutated patients, the RESONATE-2 trial showed that ibrutinib was superior to chlorambucil in older patients, many of whom had comorbid conditions. In the 3-year update, median PFS was approximately 15 months for chlorambucil, while for ibrutinib the median PFS was “nowhere near” being reached, Dr. O’Brien said.
Those data may not be so relevant for fit, unmutated patients, and two randomized trials comparing FCR with bendamustine and rituximab have yet to report data. However, one recent cross-trial comparison found fairly overlapping survival curves for the two chemoimmunotherapy approaches.
Dr. O’Brien said she would put older patients with comorbidities on ibrutinib if a clinical trial was not available, and for fit, unmutated patients, while more data are needed, she would also use ibrutinib. However, patient preference sometimes tips the scale in favor of FCR.
“The discussions sometimes are quite long about whether the patient should opt to take ibrutinib or FCR,” Dr. O’Brien said. “The last patient I had that discussion with elected to take FCR. When I asked him why, he said because he liked the idea of being finished in six cycles, off all therapy, and hopefully in remission.”
While Dr. O’Brien said she views chemoimmunotherapy as still relevant in IgHV-mutated patients, eventually it will go away, she concluded. Toward that end, there is considerable interest in venetoclax plus ibrutinib, a combination that, in early reports, has yielded very encouraging MRD results in first-line CLL.
“We have no long-term data, but very, very exciting MRD negativity data,” Dr. O’Brien said.
Dr. O’Brien reported relationships with Abbvie, Amgen, Celgene, Gilead Sciences, Janssen, Pfizer, Pharmacyclics, Sunesis Pharmaceuticals, and others.
*This story was updated 6/25/2018.
EXPERT ANALYSIS FROM ASCO 2018