User login
Ipilimumab’s survival benefit extends to 5 years
Among patients with advanced melanoma, twice as many survived to 5 years if they received ipilimumab plus dacarbazine than if they received placebo plus dacarbazine as part of a phase III randomized clinical trial, according to a report published online Feb. 23 in the Journal of Clinical Oncology.
The investigators described their analysis as the first examination of long-term survival after a course of ipilimumab therapy, as well as the first analysis of safety outcomes in a small subset of patients who continued to receive ipilimumab as maintenance therapy. They performed this milestone survival analysis “to confirm prior reports of long-term survival in a proportion of patients from nonrandomized phase II studies,” and their findings do in fact confirm those reports, said Dr. Michele Maio of University Hospital of Siena (Italy) and her associates.
The industry-sponsored study (CA184-024) involved 502 adults with treatment-naive unresectable stage III or IV melanoma who were randomly assigned to receive ipilimumab plus dacarbazine (250 patients) or placebo plus dacarbazine (252 patients) for 10 weeks. Those who showed stable disease, a partial response, or a complete response at week 24 were permitted to continue on ipilimumab or placebo as maintenance therapy every 12 weeks thereafter.
At a minimum follow-up of 5 years, 40 patients (18.2%) in the ipilimumab group and 20 (8.8%) in the placebo group were still alive. Five-year survival with ipilimumab was not only double that with placebo, it also was roughly double the historical survival rate of patients with stage IV melanoma, which is approximately 10%, Dr. Maio and her associates reported (J. Clin. Oncol. 2015 Feb. 23 [doi:10.1200/JCO.2014.56.6018]).
With ipilimumab, 3 long-term survivors (7.5%) achieved a complete response, 17 (42.5%) had a partial response, and 11 (27.5%) achieved stable disease. In contrast, with placebo no long-term survivors achieved a complete response, 7 (35%) had a partial response, and 8 (40%) had stable disease.
There were no grade 5 adverse events. Grade 3-4 adverse events were confined to the skin and comprised pruritus and rash. Low-grade adverse events included rash, vitiligo, pruritus, GI problems, and an increased ALT or AST level.
The subset of study participants who used ipilimumab maintenance therapy was small (seven patients) but “provides some insight into the safety profile of ipilimumab during extended treatment.” The only high-grade adverse events were pruritus and rash, which affected only one patient.
“This analysis demonstrates that a proportion of treatment-naive patients with advanced melanoma can achieve durable, long-term survival with ipilimumab therapy,” Dr. Maio and her associates wrote.
Among patients with advanced melanoma, twice as many survived to 5 years if they received ipilimumab plus dacarbazine than if they received placebo plus dacarbazine as part of a phase III randomized clinical trial, according to a report published online Feb. 23 in the Journal of Clinical Oncology.
The investigators described their analysis as the first examination of long-term survival after a course of ipilimumab therapy, as well as the first analysis of safety outcomes in a small subset of patients who continued to receive ipilimumab as maintenance therapy. They performed this milestone survival analysis “to confirm prior reports of long-term survival in a proportion of patients from nonrandomized phase II studies,” and their findings do in fact confirm those reports, said Dr. Michele Maio of University Hospital of Siena (Italy) and her associates.
The industry-sponsored study (CA184-024) involved 502 adults with treatment-naive unresectable stage III or IV melanoma who were randomly assigned to receive ipilimumab plus dacarbazine (250 patients) or placebo plus dacarbazine (252 patients) for 10 weeks. Those who showed stable disease, a partial response, or a complete response at week 24 were permitted to continue on ipilimumab or placebo as maintenance therapy every 12 weeks thereafter.
At a minimum follow-up of 5 years, 40 patients (18.2%) in the ipilimumab group and 20 (8.8%) in the placebo group were still alive. Five-year survival with ipilimumab was not only double that with placebo, it also was roughly double the historical survival rate of patients with stage IV melanoma, which is approximately 10%, Dr. Maio and her associates reported (J. Clin. Oncol. 2015 Feb. 23 [doi:10.1200/JCO.2014.56.6018]).
With ipilimumab, 3 long-term survivors (7.5%) achieved a complete response, 17 (42.5%) had a partial response, and 11 (27.5%) achieved stable disease. In contrast, with placebo no long-term survivors achieved a complete response, 7 (35%) had a partial response, and 8 (40%) had stable disease.
There were no grade 5 adverse events. Grade 3-4 adverse events were confined to the skin and comprised pruritus and rash. Low-grade adverse events included rash, vitiligo, pruritus, GI problems, and an increased ALT or AST level.
The subset of study participants who used ipilimumab maintenance therapy was small (seven patients) but “provides some insight into the safety profile of ipilimumab during extended treatment.” The only high-grade adverse events were pruritus and rash, which affected only one patient.
“This analysis demonstrates that a proportion of treatment-naive patients with advanced melanoma can achieve durable, long-term survival with ipilimumab therapy,” Dr. Maio and her associates wrote.
Among patients with advanced melanoma, twice as many survived to 5 years if they received ipilimumab plus dacarbazine than if they received placebo plus dacarbazine as part of a phase III randomized clinical trial, according to a report published online Feb. 23 in the Journal of Clinical Oncology.
The investigators described their analysis as the first examination of long-term survival after a course of ipilimumab therapy, as well as the first analysis of safety outcomes in a small subset of patients who continued to receive ipilimumab as maintenance therapy. They performed this milestone survival analysis “to confirm prior reports of long-term survival in a proportion of patients from nonrandomized phase II studies,” and their findings do in fact confirm those reports, said Dr. Michele Maio of University Hospital of Siena (Italy) and her associates.
The industry-sponsored study (CA184-024) involved 502 adults with treatment-naive unresectable stage III or IV melanoma who were randomly assigned to receive ipilimumab plus dacarbazine (250 patients) or placebo plus dacarbazine (252 patients) for 10 weeks. Those who showed stable disease, a partial response, or a complete response at week 24 were permitted to continue on ipilimumab or placebo as maintenance therapy every 12 weeks thereafter.
At a minimum follow-up of 5 years, 40 patients (18.2%) in the ipilimumab group and 20 (8.8%) in the placebo group were still alive. Five-year survival with ipilimumab was not only double that with placebo, it also was roughly double the historical survival rate of patients with stage IV melanoma, which is approximately 10%, Dr. Maio and her associates reported (J. Clin. Oncol. 2015 Feb. 23 [doi:10.1200/JCO.2014.56.6018]).
With ipilimumab, 3 long-term survivors (7.5%) achieved a complete response, 17 (42.5%) had a partial response, and 11 (27.5%) achieved stable disease. In contrast, with placebo no long-term survivors achieved a complete response, 7 (35%) had a partial response, and 8 (40%) had stable disease.
There were no grade 5 adverse events. Grade 3-4 adverse events were confined to the skin and comprised pruritus and rash. Low-grade adverse events included rash, vitiligo, pruritus, GI problems, and an increased ALT or AST level.
The subset of study participants who used ipilimumab maintenance therapy was small (seven patients) but “provides some insight into the safety profile of ipilimumab during extended treatment.” The only high-grade adverse events were pruritus and rash, which affected only one patient.
“This analysis demonstrates that a proportion of treatment-naive patients with advanced melanoma can achieve durable, long-term survival with ipilimumab therapy,” Dr. Maio and her associates wrote.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The short-term survival benefit conferred by ipilimumab therapy in advanced melanoma extends to at least 5 years.
Major finding: Five-year survival with ipilimumab (18.2%) was double that with placebo (8.8%) and roughly double the historical survival rate of patients with stage IV melanoma (approximately 10%).
Data source: An industry-sponsored “milestone survival” analysis to capture 5-year mortality data for 502 adults with advanced melanoma who participated in a phase III clinical trial comparing ipilimumab with placebo.
Disclosures: This study (NCT 00324155) was sponsored by Bristol-Myers Squibb. Dr. Maio reported receiving honoraria, research funding, and other compensation and serving as a consultant/adviser to Bristol-Myers Squibb, GlaxoSmithKline, and Roche. Her associates reported ties to numerous industry sources.
Trastuzumab emtansine investigated for early-stage breast cancer
A 1-year course of trastuzumab emtansine was found to be well tolerated and safe for patients with HER2-positive early breast cancer who had undergone anthracycline-based chemotherapy in an industry-sponsored open-label phase II clinical trial.
The investigators described this as the first study to assess trastuzumab emtansine (T-DM1), a conjugate of the monoclonal antibody trastuzumab and the cytotoxic agent emtansine, in patients with early-stage rather than metastatic disease. They found that the conjugate agent was better tolerated than has been reported for trastuzumab alone in numerous studies, suggesting that it may become a less toxic alternative to the conventional chemotherapy-plus-trastuzumab approach for breast cancer.
“If validated in phase III studies, the favorable tolerability of T-DM1 may allow use of targeted cytotoxic therapy for a longer duration (e.g., 1 year) than is feasible with conventional cytotoxic agents,” wrote Dr. Ian E. Krop of breast oncology, Dana-Farber Cancer Institute, Boston, and his associates (J. Clin. Oncol 2015 Feb. 23 [doi:10.1200/JCO.2014.58.7782]).
Three randomized phase III trials are now underway – NCT01772472 (KATHERINE),NCT01966471, and NCT02131064 – to confirm and extend the results of their study.
T-DM1 delivers the emtansine directly to tumor cells that overexpress HER2, inhibiting microtubule function and leading to cell death. In patients with metastatic breast cancer, the conjugate produces fewer adverse effects typically associated with chemotherapy, such as hair loss and neutropenia, presumably because it spares cells other than tumor cells. However, trastuzumab can be associated with cardiotoxicity, particularly in patients who also receive anthracycline-based regimens.
To assess the safety and tolerability of T-DM1 in early-stage breast cancer, Dr. Krop and his associates examined it in this single-arm trial involving 153 patients with HER2-positive early breast cancer whose left ventricular ejection fraction was 55% or greater at baseline. The patients were treated at 35 sites during a 1-year period and followed for a median of 25 months (range, 0.2-29.0 months).
They received four cycles of (neo)adjuvant doxorubicin plus cyclophosphamide or three to four cycles of fluorouracil plus epirubicin plus cyclophosphamide, then were given four cycles of T-DM1. They could then opt to receive three to four cycles of docetaxel, with or without trastuzumab, at their physician’s discretion. Sequential or concurrent radiotherapy could then be provided, and T-DM1 was resumed for a planned year of treatment.
Of the 148 patients who received at least 1 cycle of T-DM1, 122 (82%) completed the planned duration of therapy – a median of 14 cycles. “Only 17.6% of patients who initiated treatment with T-DM1 did not complete the planned therapy duration,” the investigators wrote, adding that by comparison, 31% of patients in a joint analysis of the NSABP and NCCTG trials discontinued trastuzumab alone before completing 1 year of treatment .
None of the study participants developed a cardiac event or symptomatic heart failure. By comparison, previous trials of adjuvant trastuzumab alone showed 2%-4% rates of CHF after anthracycline-based chemotherapy.
In this study, 2.7% of patients had asymptomatic declines in LVEF. By comparison, rates of LVEF decline ranged from 7% to 34% in previous trials of single-agent trastuzumab. Of the four women in this study who had LVEF decreases, one discontinued T-DM1 when her LVEF dipped to 45%; it subsequently recovered to baseline level.
Other adverse events included nausea (38% of patients) and headache (37%). The most common adverse events of grade 3 and above were thrombocytopenia (8.1%), increased ALT (7.4%), and increased AST (7.4%).
T-DM1 is marketed by Genentech as Kadcyla.Genentech supported the study. Dr. Krop is an employee of Vertex Pharmaceuticals and has received funding from Genentech. His associates reported ties to numerous industry sources
A 1-year course of trastuzumab emtansine was found to be well tolerated and safe for patients with HER2-positive early breast cancer who had undergone anthracycline-based chemotherapy in an industry-sponsored open-label phase II clinical trial.
The investigators described this as the first study to assess trastuzumab emtansine (T-DM1), a conjugate of the monoclonal antibody trastuzumab and the cytotoxic agent emtansine, in patients with early-stage rather than metastatic disease. They found that the conjugate agent was better tolerated than has been reported for trastuzumab alone in numerous studies, suggesting that it may become a less toxic alternative to the conventional chemotherapy-plus-trastuzumab approach for breast cancer.
“If validated in phase III studies, the favorable tolerability of T-DM1 may allow use of targeted cytotoxic therapy for a longer duration (e.g., 1 year) than is feasible with conventional cytotoxic agents,” wrote Dr. Ian E. Krop of breast oncology, Dana-Farber Cancer Institute, Boston, and his associates (J. Clin. Oncol 2015 Feb. 23 [doi:10.1200/JCO.2014.58.7782]).
Three randomized phase III trials are now underway – NCT01772472 (KATHERINE),NCT01966471, and NCT02131064 – to confirm and extend the results of their study.
T-DM1 delivers the emtansine directly to tumor cells that overexpress HER2, inhibiting microtubule function and leading to cell death. In patients with metastatic breast cancer, the conjugate produces fewer adverse effects typically associated with chemotherapy, such as hair loss and neutropenia, presumably because it spares cells other than tumor cells. However, trastuzumab can be associated with cardiotoxicity, particularly in patients who also receive anthracycline-based regimens.
To assess the safety and tolerability of T-DM1 in early-stage breast cancer, Dr. Krop and his associates examined it in this single-arm trial involving 153 patients with HER2-positive early breast cancer whose left ventricular ejection fraction was 55% or greater at baseline. The patients were treated at 35 sites during a 1-year period and followed for a median of 25 months (range, 0.2-29.0 months).
They received four cycles of (neo)adjuvant doxorubicin plus cyclophosphamide or three to four cycles of fluorouracil plus epirubicin plus cyclophosphamide, then were given four cycles of T-DM1. They could then opt to receive three to four cycles of docetaxel, with or without trastuzumab, at their physician’s discretion. Sequential or concurrent radiotherapy could then be provided, and T-DM1 was resumed for a planned year of treatment.
Of the 148 patients who received at least 1 cycle of T-DM1, 122 (82%) completed the planned duration of therapy – a median of 14 cycles. “Only 17.6% of patients who initiated treatment with T-DM1 did not complete the planned therapy duration,” the investigators wrote, adding that by comparison, 31% of patients in a joint analysis of the NSABP and NCCTG trials discontinued trastuzumab alone before completing 1 year of treatment .
None of the study participants developed a cardiac event or symptomatic heart failure. By comparison, previous trials of adjuvant trastuzumab alone showed 2%-4% rates of CHF after anthracycline-based chemotherapy.
In this study, 2.7% of patients had asymptomatic declines in LVEF. By comparison, rates of LVEF decline ranged from 7% to 34% in previous trials of single-agent trastuzumab. Of the four women in this study who had LVEF decreases, one discontinued T-DM1 when her LVEF dipped to 45%; it subsequently recovered to baseline level.
Other adverse events included nausea (38% of patients) and headache (37%). The most common adverse events of grade 3 and above were thrombocytopenia (8.1%), increased ALT (7.4%), and increased AST (7.4%).
T-DM1 is marketed by Genentech as Kadcyla.Genentech supported the study. Dr. Krop is an employee of Vertex Pharmaceuticals and has received funding from Genentech. His associates reported ties to numerous industry sources
A 1-year course of trastuzumab emtansine was found to be well tolerated and safe for patients with HER2-positive early breast cancer who had undergone anthracycline-based chemotherapy in an industry-sponsored open-label phase II clinical trial.
The investigators described this as the first study to assess trastuzumab emtansine (T-DM1), a conjugate of the monoclonal antibody trastuzumab and the cytotoxic agent emtansine, in patients with early-stage rather than metastatic disease. They found that the conjugate agent was better tolerated than has been reported for trastuzumab alone in numerous studies, suggesting that it may become a less toxic alternative to the conventional chemotherapy-plus-trastuzumab approach for breast cancer.
“If validated in phase III studies, the favorable tolerability of T-DM1 may allow use of targeted cytotoxic therapy for a longer duration (e.g., 1 year) than is feasible with conventional cytotoxic agents,” wrote Dr. Ian E. Krop of breast oncology, Dana-Farber Cancer Institute, Boston, and his associates (J. Clin. Oncol 2015 Feb. 23 [doi:10.1200/JCO.2014.58.7782]).
Three randomized phase III trials are now underway – NCT01772472 (KATHERINE),NCT01966471, and NCT02131064 – to confirm and extend the results of their study.
T-DM1 delivers the emtansine directly to tumor cells that overexpress HER2, inhibiting microtubule function and leading to cell death. In patients with metastatic breast cancer, the conjugate produces fewer adverse effects typically associated with chemotherapy, such as hair loss and neutropenia, presumably because it spares cells other than tumor cells. However, trastuzumab can be associated with cardiotoxicity, particularly in patients who also receive anthracycline-based regimens.
To assess the safety and tolerability of T-DM1 in early-stage breast cancer, Dr. Krop and his associates examined it in this single-arm trial involving 153 patients with HER2-positive early breast cancer whose left ventricular ejection fraction was 55% or greater at baseline. The patients were treated at 35 sites during a 1-year period and followed for a median of 25 months (range, 0.2-29.0 months).
They received four cycles of (neo)adjuvant doxorubicin plus cyclophosphamide or three to four cycles of fluorouracil plus epirubicin plus cyclophosphamide, then were given four cycles of T-DM1. They could then opt to receive three to four cycles of docetaxel, with or without trastuzumab, at their physician’s discretion. Sequential or concurrent radiotherapy could then be provided, and T-DM1 was resumed for a planned year of treatment.
Of the 148 patients who received at least 1 cycle of T-DM1, 122 (82%) completed the planned duration of therapy – a median of 14 cycles. “Only 17.6% of patients who initiated treatment with T-DM1 did not complete the planned therapy duration,” the investigators wrote, adding that by comparison, 31% of patients in a joint analysis of the NSABP and NCCTG trials discontinued trastuzumab alone before completing 1 year of treatment .
None of the study participants developed a cardiac event or symptomatic heart failure. By comparison, previous trials of adjuvant trastuzumab alone showed 2%-4% rates of CHF after anthracycline-based chemotherapy.
In this study, 2.7% of patients had asymptomatic declines in LVEF. By comparison, rates of LVEF decline ranged from 7% to 34% in previous trials of single-agent trastuzumab. Of the four women in this study who had LVEF decreases, one discontinued T-DM1 when her LVEF dipped to 45%; it subsequently recovered to baseline level.
Other adverse events included nausea (38% of patients) and headache (37%). The most common adverse events of grade 3 and above were thrombocytopenia (8.1%), increased ALT (7.4%), and increased AST (7.4%).
T-DM1 is marketed by Genentech as Kadcyla.Genentech supported the study. Dr. Krop is an employee of Vertex Pharmaceuticals and has received funding from Genentech. His associates reported ties to numerous industry sources
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Trastuzumab emtansine was well tolerated by patients with HER2-positive early-stage breast cancer.
Major finding: Of the 148 patients who received at least 1 cycle of T-DM1, 122 (82%) completed the planned duration of therapy – a median of 14 cycles.
Data source: An industry-sponsored single-arm open-label phase II clinical trial involving 153 patients with early-stage breast cancer who were followed for a median of 2 years.
Disclosures: Genentech supported the study. Dr. Krop is an employee of Vertex Pharmaceuticals and has received funding from Genentech. His associates reported ties to numerous industry sources.
Propranolol Effective in 60% of Infantile Hemangiomas
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Ventilator driving pressure may predict mortality in ARDS
One variable involved with mechanical ventilation – driving pressure – may predict mortality in adult respiratory distress syndrome, according to a report published online Feb. 19 in the New England Journal of Medicine.
Research suggests that scaling ventilator tidal volumes (VT) to patients’ body weight will minimize ventilator-induced lung injury. But patients with ARDS have a marked decrease in the proportion of lung available for ventilation, as is indicated by their lower respiratory-system compliance (CRS), which is not related to their body weight. “Therefore, we hypothesized that normalizing VT to CRS and using the ratio as an index indicating the ‘functional’ size of the lung would provide a better predictor of outcomes in patients with ARDS than VT alone,” said Dr. Marcelo B. P. Amato of the cardiopulmonary department, University of Sao Paulo (Brazil) Heart Institute, and his associates.
This ratio, also known as the driving pressure, is easily calculated at the bedside. The investigators explored whether driving pressure or other variables related to mechanical ventilation, including variables set by the ventilator operator, could be statistically linked to survival outcomes and therefore serve as a risk predictor.
They first devised a survival-prediction model using data from a cohort of 336 ARDS patients participating in four randomized clinical trials examining different ventilation strategies. They then tested their findings using a validation cohort of 861 patients from a single large trial, then tested them again in a more recent validation cohort of 2,365 patients participating in four more randomized trials comparing different ventilation strategies.
Driving pressure was the only ventilation variable found to be strongly associated with survival. Higher driving pressures strongly predicted higher mortality: Every 1-SD increase in driving pressure was related to increased mortality, with a relative risk of 1.41. Even in patients receiving lung-protective plateau pressures and low tidal volumes, higher driving pressure was associated with increased mortality, with a relative risk of 1.36, Dr. Amato and his associates said (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMsa1410639]).
These findings can only suggest that driving pressure is a critical mediator of various ventilator strategies, since they are derived from a post hoc observational statistical analysis and cannot establish causality. Now, prospective clinical trials are needed to determine whether adjusting ventilator settings to lower driving pressure will improve survival in ARDS; in other words, “whether our observations can be translated into changes that may be implemented at the bedside,” the investigators noted.
We strongly urge caution against accepting the idea that clinicians should now set ventilators to limit driving pressure in patients with ARDS, even though that is an appealing concept.
The findings of Amato et al. derive from a meta-analysis, not from prospective randomized controlled trials. Their results should form the basis for a robust debate regarding the design of future trials so that, before we take action, we first ensure that limiting driving pressure will actually be beneficial.
Dr. Stephen H. Loring is in the department of anesthesia, critical care, and pain medicine at Beth Israel Deaconess Medical Center and at Harvard, both in Boston. Dr. Atul Malhotra is in the division of pulmonary critical care and sleep medicine at the University of California, San Diego, in La Jolla. Both Dr. Loring and Dr. Malhotra reported having no financial disclosures. They made these remarks in an editorial accompanying Dr. Amato’s report (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMe1414218]).
We strongly urge caution against accepting the idea that clinicians should now set ventilators to limit driving pressure in patients with ARDS, even though that is an appealing concept.
The findings of Amato et al. derive from a meta-analysis, not from prospective randomized controlled trials. Their results should form the basis for a robust debate regarding the design of future trials so that, before we take action, we first ensure that limiting driving pressure will actually be beneficial.
Dr. Stephen H. Loring is in the department of anesthesia, critical care, and pain medicine at Beth Israel Deaconess Medical Center and at Harvard, both in Boston. Dr. Atul Malhotra is in the division of pulmonary critical care and sleep medicine at the University of California, San Diego, in La Jolla. Both Dr. Loring and Dr. Malhotra reported having no financial disclosures. They made these remarks in an editorial accompanying Dr. Amato’s report (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMe1414218]).
We strongly urge caution against accepting the idea that clinicians should now set ventilators to limit driving pressure in patients with ARDS, even though that is an appealing concept.
The findings of Amato et al. derive from a meta-analysis, not from prospective randomized controlled trials. Their results should form the basis for a robust debate regarding the design of future trials so that, before we take action, we first ensure that limiting driving pressure will actually be beneficial.
Dr. Stephen H. Loring is in the department of anesthesia, critical care, and pain medicine at Beth Israel Deaconess Medical Center and at Harvard, both in Boston. Dr. Atul Malhotra is in the division of pulmonary critical care and sleep medicine at the University of California, San Diego, in La Jolla. Both Dr. Loring and Dr. Malhotra reported having no financial disclosures. They made these remarks in an editorial accompanying Dr. Amato’s report (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMe1414218]).
One variable involved with mechanical ventilation – driving pressure – may predict mortality in adult respiratory distress syndrome, according to a report published online Feb. 19 in the New England Journal of Medicine.
Research suggests that scaling ventilator tidal volumes (VT) to patients’ body weight will minimize ventilator-induced lung injury. But patients with ARDS have a marked decrease in the proportion of lung available for ventilation, as is indicated by their lower respiratory-system compliance (CRS), which is not related to their body weight. “Therefore, we hypothesized that normalizing VT to CRS and using the ratio as an index indicating the ‘functional’ size of the lung would provide a better predictor of outcomes in patients with ARDS than VT alone,” said Dr. Marcelo B. P. Amato of the cardiopulmonary department, University of Sao Paulo (Brazil) Heart Institute, and his associates.
This ratio, also known as the driving pressure, is easily calculated at the bedside. The investigators explored whether driving pressure or other variables related to mechanical ventilation, including variables set by the ventilator operator, could be statistically linked to survival outcomes and therefore serve as a risk predictor.
They first devised a survival-prediction model using data from a cohort of 336 ARDS patients participating in four randomized clinical trials examining different ventilation strategies. They then tested their findings using a validation cohort of 861 patients from a single large trial, then tested them again in a more recent validation cohort of 2,365 patients participating in four more randomized trials comparing different ventilation strategies.
Driving pressure was the only ventilation variable found to be strongly associated with survival. Higher driving pressures strongly predicted higher mortality: Every 1-SD increase in driving pressure was related to increased mortality, with a relative risk of 1.41. Even in patients receiving lung-protective plateau pressures and low tidal volumes, higher driving pressure was associated with increased mortality, with a relative risk of 1.36, Dr. Amato and his associates said (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMsa1410639]).
These findings can only suggest that driving pressure is a critical mediator of various ventilator strategies, since they are derived from a post hoc observational statistical analysis and cannot establish causality. Now, prospective clinical trials are needed to determine whether adjusting ventilator settings to lower driving pressure will improve survival in ARDS; in other words, “whether our observations can be translated into changes that may be implemented at the bedside,” the investigators noted.
One variable involved with mechanical ventilation – driving pressure – may predict mortality in adult respiratory distress syndrome, according to a report published online Feb. 19 in the New England Journal of Medicine.
Research suggests that scaling ventilator tidal volumes (VT) to patients’ body weight will minimize ventilator-induced lung injury. But patients with ARDS have a marked decrease in the proportion of lung available for ventilation, as is indicated by their lower respiratory-system compliance (CRS), which is not related to their body weight. “Therefore, we hypothesized that normalizing VT to CRS and using the ratio as an index indicating the ‘functional’ size of the lung would provide a better predictor of outcomes in patients with ARDS than VT alone,” said Dr. Marcelo B. P. Amato of the cardiopulmonary department, University of Sao Paulo (Brazil) Heart Institute, and his associates.
This ratio, also known as the driving pressure, is easily calculated at the bedside. The investigators explored whether driving pressure or other variables related to mechanical ventilation, including variables set by the ventilator operator, could be statistically linked to survival outcomes and therefore serve as a risk predictor.
They first devised a survival-prediction model using data from a cohort of 336 ARDS patients participating in four randomized clinical trials examining different ventilation strategies. They then tested their findings using a validation cohort of 861 patients from a single large trial, then tested them again in a more recent validation cohort of 2,365 patients participating in four more randomized trials comparing different ventilation strategies.
Driving pressure was the only ventilation variable found to be strongly associated with survival. Higher driving pressures strongly predicted higher mortality: Every 1-SD increase in driving pressure was related to increased mortality, with a relative risk of 1.41. Even in patients receiving lung-protective plateau pressures and low tidal volumes, higher driving pressure was associated with increased mortality, with a relative risk of 1.36, Dr. Amato and his associates said (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJMsa1410639]).
These findings can only suggest that driving pressure is a critical mediator of various ventilator strategies, since they are derived from a post hoc observational statistical analysis and cannot establish causality. Now, prospective clinical trials are needed to determine whether adjusting ventilator settings to lower driving pressure will improve survival in ARDS; in other words, “whether our observations can be translated into changes that may be implemented at the bedside,” the investigators noted.
Key clinical point: One variable associated with mechanical ventilation – driving pressure – may predict mortality risk in acute respiratory distress syndrome.
Major finding: Higher driving pressures strongly predicted higher mortality, such that every 1-SD increase in driving pressure was related to increased mortality with a relative risk of 1.41.
Data source: A post hoc observational statistical mediation analysis of data from 3,562 ARDS patients enrolled in nine previously reported randomized trials.
Disclosures: This study was supported by Fundacao de Amparo e Pesquisa do Estado de Sao Paulo, Conselho Nacional de Pesquisa e Desenvolvimento, and Financiadora de Estudos e Projectos. Dr. Amato reported having no financial disclosures; his associates reported ties to numerous industry sources.
Propranolol effective in 60% of infantile hemangiomas
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
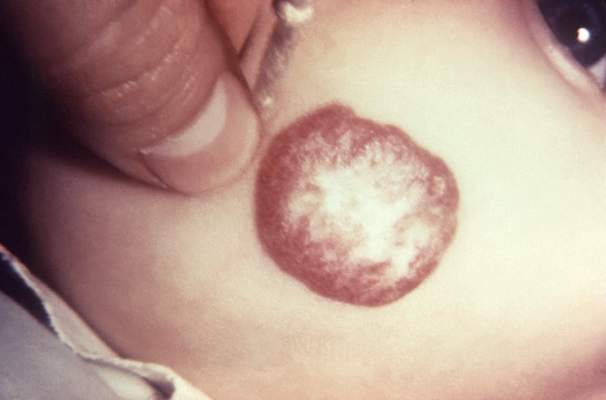
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
A 6-month course of oral propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in 60% of treated babies participating an international clinical trial, according to a study published online Feb. 19 in the New England Journal of Medicine.
A pediatric oral formulation of propranolol, a nonselective beta adrenergic receptor–blocker, was recently introduced and rapidly came to be considered first-line therapy for infantile hemangiomas, even though no large randomized controlled trials have assessed the drug in this patient population. Researchers now report findings from an industry-sponsored randomized, placebo-controlled, double-blind phase II/III trial involving 460 infants aged 35-150 days treated during a 1.5-year period at 56 medical centers in 16 countries.
In the first, dose-finding stage of the trial, patients were randomly assigned in roughly equivalent numbers to receive one of four doses of propranolol or a matching placebo for 3 or 6 months. An interim analysis determined that 6 months of the 3 mg/kg-per-day dose had the best benefit-to-risk ratio, so in the second stage of the study the babies were randomly reassigned to either that dose or a matching placebo for a total of 24 weeks. All were then followed for 72 more weeks to assess the permanence of the treatment effect and adverse events, said Dr. Christine Léauté-Labrèze of the pediatric dermatology unit, Hôspital Pellegrin-Enfants, Bordeaux, France, and her associates.
The primary efficacy endpoint – complete or nearly complete resolution of the target hemangioma at week 24 – occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo. Marked improvement in the lesion was noted by week 5 in 88% of the first group, compared with 5% of the placebo group. Overall, propranolol produced a significantly greater reduction in hemangioma surface area and color intensity than did placebo (N. Engl. J. Med. 2015 Feb. 19 [doi:10.1056/NEJM0a1404710]).
Six patients (10%) who received 3 mg/kg per day of propranolol required a second systemic hemangioma treatment during follow-up. This rate is comparable to the relapse rate reported in the literature, Dr. Léauté-Labrèze and her associates said.
The incidence of adverse events was higher with propranolol than with placebo, but that of serious adverse events was comparable between the two groups. Diarrhea and bronchial hyperreactivity, two adverse events known to be associated with propranolol, were more common with active treatment than with placebo but were mild or moderate in severity. Bronchospasm, bradycardia, hypotension, and hypoglycemia were infrequent but occurred more often in the propranolol groups than in the placebo group, the researchers said. “Only one patient who received propranolol had a serious adverse event (bradycardia in the context of enterocolitis). Heart-rate decreases typically occurred within 1 hour after dose administration.”
Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: A 6-month course of 3 mg/kg per day propranolol induced complete or nearly complete resolution of proliferating infantile hemangioma in more than half of treated babies in an international clinical trial.
Major finding: Complete or nearly complete resolution of the target hemangioma at week 24 occurred in 60% of the 101 patients treated with the selected propranolol regimen, compared with 4% of the 55 patients assigned to placebo.
Data source: An industry-sponsored international randomized double-blind placebo-controlled phase II/III clinical trial of 460 babies with proliferating infantile hemangioma requiring systemic therapy.
Disclosures: Dr. Léauté-Labrèze disclosed ties with Pierre Fabre Dermatologie, which sponsored the study. Some of her associates reported ties to Pierre Fabre, Amgen, Centocor, Shire, Infectopharm, Pfizer, AbbVie, and Galderma.
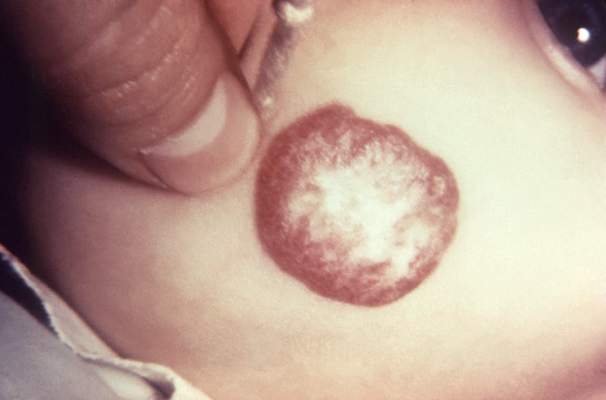
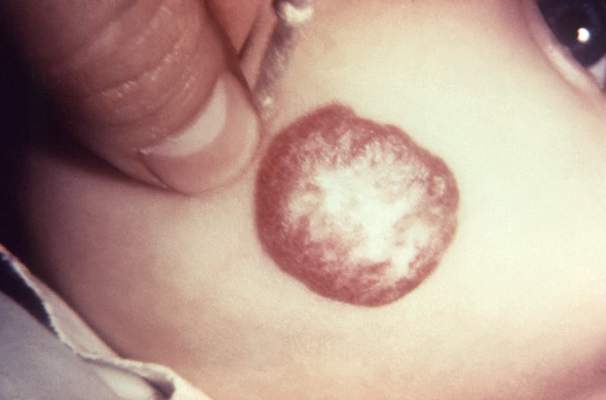
Study confirms value of adding radiotherapy to ADT in advanced prostate cancer
Adding radiotherapy to androgen-deprivation therapy for locally advanced prostate cancer improves overall survival 30% in the long term, according to a report published online Feb. 17 in the Journal of Clinical Oncology.
Radiotherapy also improves disease-specific outcomes when added to androgen-deprivation therapy (ADT), and its benefits “are achieved without major detriment in terms of long-term toxicity,” said Dr. Malcolm D. Mason of Cardiff University and Velindre Hospital, Wales, and his associates.

These long-term results from this international randomized, controlled trial confirm that the previously reported short-term survival benefits are maintained for a median follow-up of 8 years and “firmly establish the role of radiotherapy” for locally advanced prostate cancer, they said.
“Recent data suggest that some men with T3-4 disease are still being managed with ADT alone. Although there are undoubtedly patients for whom [radiotherapy] or indeed any curative therapy would be inappropriate because of age, comorbidity, or other factors, we conclude that patients with clinically node-negative, locally advanced prostate cancer who are suitable for additional radiotherapy should be offered that option – an opinion shared by European and North American guidelines,” Dr. Mason and his associates noted.
The trial, which the investigators described as the largest to compare the two approaches to prostate cancer, enrolled 1,205 patients during 1995-2005 and followed them through the end of 2010. The median patient age at baseline was 70 years. There were 465 deaths.
A total of 260 deaths occurred with ADT alone, compared with 205 with ADT plus radiotherapy. Median overall survival time was 9.7 years for ADT alone, compared with 10.9 years for ADT plus radiotherapy. Ten-year overall survival was 49% for ADT alone, compared with 55% for ADT plus radiotherapy.
Prostate cancer–specific mortality also declined markedly with the addition of radiotherapy to ADT (hazard ratio, 0.46). Time to disease progression was extended with radiotherapy. And the rate of progression-free survival at 10 years was 46% with ADT alone, compared with 74% when radiotherapy was added, Dr. Mason and his associates said (J. Clin. Oncol. 2015 Feb. 17 [doi:10.1200/JCO.2014.57.7510]).
“The radiotherapy technique used in our trial reflects the prevailing treatment philosophies of the time. The study predated outcome data from randomized trials of dose-escalated radiotherapy, and the radiotherapy doses used here are modest by modern standards,” they noted.
The addition of radiotherapy had “a detectable, although modest” impact on toxicity, with small increases in the number of patients who experienced impotence, urinary frequency, ischemia, or hypertension. Bowel-related adverse effects were more frequent with the addition of radiotherapy, but most were grade 1 or 2. Any toxic effects of grade 3 or higher were of short duration, “and we would suggest that the toxicity of radiotherapy should not be regarded as a barrier to its routine use in this patient population,” the researchers said.
The National Cancer Institute, the Canadian Cancer Society Research Institute, the United Kingdom Medical Research Council, and the United Kingdom National Cancer Research Network supported the study. Dr. Mason disclosed ties to Sanofi and Dendreon; his associates reported ties to MIOT Institute Chennai, BiPar/Sanofi-Aventis, Novartis, Pfizer, Janssen Pharmaceuticals, Astellas Pharma, and B&C.
Adding radiotherapy to androgen-deprivation therapy for locally advanced prostate cancer improves overall survival 30% in the long term, according to a report published online Feb. 17 in the Journal of Clinical Oncology.
Radiotherapy also improves disease-specific outcomes when added to androgen-deprivation therapy (ADT), and its benefits “are achieved without major detriment in terms of long-term toxicity,” said Dr. Malcolm D. Mason of Cardiff University and Velindre Hospital, Wales, and his associates.
These long-term results from this international randomized, controlled trial confirm that the previously reported short-term survival benefits are maintained for a median follow-up of 8 years and “firmly establish the role of radiotherapy” for locally advanced prostate cancer, they said.
“Recent data suggest that some men with T3-4 disease are still being managed with ADT alone. Although there are undoubtedly patients for whom [radiotherapy] or indeed any curative therapy would be inappropriate because of age, comorbidity, or other factors, we conclude that patients with clinically node-negative, locally advanced prostate cancer who are suitable for additional radiotherapy should be offered that option – an opinion shared by European and North American guidelines,” Dr. Mason and his associates noted.
The trial, which the investigators described as the largest to compare the two approaches to prostate cancer, enrolled 1,205 patients during 1995-2005 and followed them through the end of 2010. The median patient age at baseline was 70 years. There were 465 deaths.
A total of 260 deaths occurred with ADT alone, compared with 205 with ADT plus radiotherapy. Median overall survival time was 9.7 years for ADT alone, compared with 10.9 years for ADT plus radiotherapy. Ten-year overall survival was 49% for ADT alone, compared with 55% for ADT plus radiotherapy.
Prostate cancer–specific mortality also declined markedly with the addition of radiotherapy to ADT (hazard ratio, 0.46). Time to disease progression was extended with radiotherapy. And the rate of progression-free survival at 10 years was 46% with ADT alone, compared with 74% when radiotherapy was added, Dr. Mason and his associates said (J. Clin. Oncol. 2015 Feb. 17 [doi:10.1200/JCO.2014.57.7510]).
“The radiotherapy technique used in our trial reflects the prevailing treatment philosophies of the time. The study predated outcome data from randomized trials of dose-escalated radiotherapy, and the radiotherapy doses used here are modest by modern standards,” they noted.
The addition of radiotherapy had “a detectable, although modest” impact on toxicity, with small increases in the number of patients who experienced impotence, urinary frequency, ischemia, or hypertension. Bowel-related adverse effects were more frequent with the addition of radiotherapy, but most were grade 1 or 2. Any toxic effects of grade 3 or higher were of short duration, “and we would suggest that the toxicity of radiotherapy should not be regarded as a barrier to its routine use in this patient population,” the researchers said.
The National Cancer Institute, the Canadian Cancer Society Research Institute, the United Kingdom Medical Research Council, and the United Kingdom National Cancer Research Network supported the study. Dr. Mason disclosed ties to Sanofi and Dendreon; his associates reported ties to MIOT Institute Chennai, BiPar/Sanofi-Aventis, Novartis, Pfizer, Janssen Pharmaceuticals, Astellas Pharma, and B&C.
Adding radiotherapy to androgen-deprivation therapy for locally advanced prostate cancer improves overall survival 30% in the long term, according to a report published online Feb. 17 in the Journal of Clinical Oncology.
Radiotherapy also improves disease-specific outcomes when added to androgen-deprivation therapy (ADT), and its benefits “are achieved without major detriment in terms of long-term toxicity,” said Dr. Malcolm D. Mason of Cardiff University and Velindre Hospital, Wales, and his associates.
These long-term results from this international randomized, controlled trial confirm that the previously reported short-term survival benefits are maintained for a median follow-up of 8 years and “firmly establish the role of radiotherapy” for locally advanced prostate cancer, they said.
“Recent data suggest that some men with T3-4 disease are still being managed with ADT alone. Although there are undoubtedly patients for whom [radiotherapy] or indeed any curative therapy would be inappropriate because of age, comorbidity, or other factors, we conclude that patients with clinically node-negative, locally advanced prostate cancer who are suitable for additional radiotherapy should be offered that option – an opinion shared by European and North American guidelines,” Dr. Mason and his associates noted.
The trial, which the investigators described as the largest to compare the two approaches to prostate cancer, enrolled 1,205 patients during 1995-2005 and followed them through the end of 2010. The median patient age at baseline was 70 years. There were 465 deaths.
A total of 260 deaths occurred with ADT alone, compared with 205 with ADT plus radiotherapy. Median overall survival time was 9.7 years for ADT alone, compared with 10.9 years for ADT plus radiotherapy. Ten-year overall survival was 49% for ADT alone, compared with 55% for ADT plus radiotherapy.
Prostate cancer–specific mortality also declined markedly with the addition of radiotherapy to ADT (hazard ratio, 0.46). Time to disease progression was extended with radiotherapy. And the rate of progression-free survival at 10 years was 46% with ADT alone, compared with 74% when radiotherapy was added, Dr. Mason and his associates said (J. Clin. Oncol. 2015 Feb. 17 [doi:10.1200/JCO.2014.57.7510]).
“The radiotherapy technique used in our trial reflects the prevailing treatment philosophies of the time. The study predated outcome data from randomized trials of dose-escalated radiotherapy, and the radiotherapy doses used here are modest by modern standards,” they noted.
The addition of radiotherapy had “a detectable, although modest” impact on toxicity, with small increases in the number of patients who experienced impotence, urinary frequency, ischemia, or hypertension. Bowel-related adverse effects were more frequent with the addition of radiotherapy, but most were grade 1 or 2. Any toxic effects of grade 3 or higher were of short duration, “and we would suggest that the toxicity of radiotherapy should not be regarded as a barrier to its routine use in this patient population,” the researchers said.
The National Cancer Institute, the Canadian Cancer Society Research Institute, the United Kingdom Medical Research Council, and the United Kingdom National Cancer Research Network supported the study. Dr. Mason disclosed ties to Sanofi and Dendreon; his associates reported ties to MIOT Institute Chennai, BiPar/Sanofi-Aventis, Novartis, Pfizer, Janssen Pharmaceuticals, Astellas Pharma, and B&C.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Clinicians should offer additional radiotherapy to ADT patients with “clinically node-negative, locally advanced prostate cancer.”
Major finding: Two hundred sixty deaths occurred among patients who received ADT alone, compared with 205 deaths for those who received ADT plus radiotherapy.
Data source: Final (8-year) follow-up of an international randomized controlled clinical trial comparing ADT alone (602 patients) against ADT plus radiotherapy (603 patients).
Disclosures: The National Cancer Institute, the Canadian Cancer Society Research Institute, the United Kingdom Medical Research Council, and the United Kingdom National Cancer Research Network supported the study. Dr. Mason disclosed ties with Sanofi and Dendreon; his associates reported ties to MIOT Institute Chennai, BiPar/Sanofi-Aventis, Novartis, Pfizer, Janssen Pharmaceuticals, Astellas Pharma, and B&C.

Low marks for bevacizumab in cost/benefit analysis
Adding bevacizumab to standard chemotherapy for metastatic colorectal cancer yields a minimal incremental benefit at high incremental cost per quality-adjusted life year, according to a report published online Feb. 16 in the Journal of Clinical Oncology.
In a cost-effectiveness analysis using computer modeling based on the results of the most recent randomized phase III clinical trials of the agent, even the best-case scenario produced an incremental cost-effectiveness ratio of more than $200,000 per QALY. Probabilistic sensitivity analyses also showed that the likelihood of bevacizumab being cost effective was 0% as either first- or second-line treatment for a willingness-to-pay threshold of $100,000 per QALY, said Dr. Daniel A. Goldstein of Emory University, Atlanta, and his associates.
At present, it is standard practice to add bevacizumab to chemotherapy for metastatic colorectal cancer in both first- and second-line settings, but the modest survival benefit conferred by the agent in this patient population, together with its very high cost, have raised concerns about its cost effectiveness.
Dr. Goldstein and his associates developed computer models of the clinical effectiveness and direct medical costs associated with bevacizumab using data from the most recent randomized controlled trials. They then performed both internal and external validation studies, which demonstrated that the survival curves generated by their models closely approximated those that actually occurred in other clinical trials of the agent.
As first-line therapy, FOLFOX alone provided 1.31 QALYs at a cost of $32,561, while FOLFOX plus bevacizumab provided 1.41 QALYs at a cost of $92,112. The incremental cost-effectiveness ratio for FOLFOX plus bevacizumab was $571,240 per QALY. As second-line therapy, FOLFIRI alone provided 0.64 QALYs at a cost of $7,443, while FOLFIRI plus bevacizumab provided 0.75 QALYs at a cost of $46,764. The incremental cost-effectiveness ratio for FOLFIRI plus bevacizumab was $364,083 per QALY. A threshold between $50,000 and $100,000 is usually used to define a cost-effective health intervention, the investigators said (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.58.4904]).
In sensitivity analyses that tested a broad range of variations in each parameter – such as different drug costs, physician fees, and treatment toxicities – the incremental cost-effectiveness ratio remained higher than $200,000 per QALY in every case, even in the “best case scenario” model.
These findings closely accord with those of similar studies in other countries, even though there were significant differences in modeling approaches, costs, and health care systems. In the United Kingdom, adding bevacizumab to FOLFIRI cost the equivalent of $102,000 U.S. per QALY. In Canada it cost the equivalent of $120,000 U.S. per QALY, and in Japan it cost the equivalent of $113,000 U.S. per QALY, Dr. Goldstein and his associates said.
Presumably, the poor cost/benefit ratio could be improved if an effective biomarker were developed and used to select only the patients most likely to benefit for bevacizumab therapy, they added.
No funding sources were reported for this study. Dr. Goldstein reported having no financial disclosures; his associates reported numerous ties to industry sources, mostly through research funding of their institutions.
The real problem with cost-effectiveness studies such as this one is that this type of research has become an academic exercise of no meaningful consequence since there is virtually no medical expense that we, as a society, have been willing to deny.
We can collectively wring our hands and agree that this agent is too expensive for what the available data indicate is the modest improvement in survival it confers, but that solves nothing. To make a meaningful change we must confront the data, acknowledge the limitations of this and other treatment “advances,” assign a realistic value to them, and either demand lower prices or refuse to use them.
Dr. Leonard B. Saltz is with Memorial Sloan Kettering Cancer Center, New York. His financial disclosures were not available at press time. Dr. Saltz made these remarks in an editorial accompanying Dr. Goldstein’s report (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.60.1401]).
The real problem with cost-effectiveness studies such as this one is that this type of research has become an academic exercise of no meaningful consequence since there is virtually no medical expense that we, as a society, have been willing to deny.
We can collectively wring our hands and agree that this agent is too expensive for what the available data indicate is the modest improvement in survival it confers, but that solves nothing. To make a meaningful change we must confront the data, acknowledge the limitations of this and other treatment “advances,” assign a realistic value to them, and either demand lower prices or refuse to use them.
Dr. Leonard B. Saltz is with Memorial Sloan Kettering Cancer Center, New York. His financial disclosures were not available at press time. Dr. Saltz made these remarks in an editorial accompanying Dr. Goldstein’s report (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.60.1401]).
The real problem with cost-effectiveness studies such as this one is that this type of research has become an academic exercise of no meaningful consequence since there is virtually no medical expense that we, as a society, have been willing to deny.
We can collectively wring our hands and agree that this agent is too expensive for what the available data indicate is the modest improvement in survival it confers, but that solves nothing. To make a meaningful change we must confront the data, acknowledge the limitations of this and other treatment “advances,” assign a realistic value to them, and either demand lower prices or refuse to use them.
Dr. Leonard B. Saltz is with Memorial Sloan Kettering Cancer Center, New York. His financial disclosures were not available at press time. Dr. Saltz made these remarks in an editorial accompanying Dr. Goldstein’s report (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.60.1401]).
Adding bevacizumab to standard chemotherapy for metastatic colorectal cancer yields a minimal incremental benefit at high incremental cost per quality-adjusted life year, according to a report published online Feb. 16 in the Journal of Clinical Oncology.
In a cost-effectiveness analysis using computer modeling based on the results of the most recent randomized phase III clinical trials of the agent, even the best-case scenario produced an incremental cost-effectiveness ratio of more than $200,000 per QALY. Probabilistic sensitivity analyses also showed that the likelihood of bevacizumab being cost effective was 0% as either first- or second-line treatment for a willingness-to-pay threshold of $100,000 per QALY, said Dr. Daniel A. Goldstein of Emory University, Atlanta, and his associates.
At present, it is standard practice to add bevacizumab to chemotherapy for metastatic colorectal cancer in both first- and second-line settings, but the modest survival benefit conferred by the agent in this patient population, together with its very high cost, have raised concerns about its cost effectiveness.
Dr. Goldstein and his associates developed computer models of the clinical effectiveness and direct medical costs associated with bevacizumab using data from the most recent randomized controlled trials. They then performed both internal and external validation studies, which demonstrated that the survival curves generated by their models closely approximated those that actually occurred in other clinical trials of the agent.
As first-line therapy, FOLFOX alone provided 1.31 QALYs at a cost of $32,561, while FOLFOX plus bevacizumab provided 1.41 QALYs at a cost of $92,112. The incremental cost-effectiveness ratio for FOLFOX plus bevacizumab was $571,240 per QALY. As second-line therapy, FOLFIRI alone provided 0.64 QALYs at a cost of $7,443, while FOLFIRI plus bevacizumab provided 0.75 QALYs at a cost of $46,764. The incremental cost-effectiveness ratio for FOLFIRI plus bevacizumab was $364,083 per QALY. A threshold between $50,000 and $100,000 is usually used to define a cost-effective health intervention, the investigators said (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.58.4904]).
In sensitivity analyses that tested a broad range of variations in each parameter – such as different drug costs, physician fees, and treatment toxicities – the incremental cost-effectiveness ratio remained higher than $200,000 per QALY in every case, even in the “best case scenario” model.
These findings closely accord with those of similar studies in other countries, even though there were significant differences in modeling approaches, costs, and health care systems. In the United Kingdom, adding bevacizumab to FOLFIRI cost the equivalent of $102,000 U.S. per QALY. In Canada it cost the equivalent of $120,000 U.S. per QALY, and in Japan it cost the equivalent of $113,000 U.S. per QALY, Dr. Goldstein and his associates said.
Presumably, the poor cost/benefit ratio could be improved if an effective biomarker were developed and used to select only the patients most likely to benefit for bevacizumab therapy, they added.
No funding sources were reported for this study. Dr. Goldstein reported having no financial disclosures; his associates reported numerous ties to industry sources, mostly through research funding of their institutions.
Adding bevacizumab to standard chemotherapy for metastatic colorectal cancer yields a minimal incremental benefit at high incremental cost per quality-adjusted life year, according to a report published online Feb. 16 in the Journal of Clinical Oncology.
In a cost-effectiveness analysis using computer modeling based on the results of the most recent randomized phase III clinical trials of the agent, even the best-case scenario produced an incremental cost-effectiveness ratio of more than $200,000 per QALY. Probabilistic sensitivity analyses also showed that the likelihood of bevacizumab being cost effective was 0% as either first- or second-line treatment for a willingness-to-pay threshold of $100,000 per QALY, said Dr. Daniel A. Goldstein of Emory University, Atlanta, and his associates.
At present, it is standard practice to add bevacizumab to chemotherapy for metastatic colorectal cancer in both first- and second-line settings, but the modest survival benefit conferred by the agent in this patient population, together with its very high cost, have raised concerns about its cost effectiveness.
Dr. Goldstein and his associates developed computer models of the clinical effectiveness and direct medical costs associated with bevacizumab using data from the most recent randomized controlled trials. They then performed both internal and external validation studies, which demonstrated that the survival curves generated by their models closely approximated those that actually occurred in other clinical trials of the agent.
As first-line therapy, FOLFOX alone provided 1.31 QALYs at a cost of $32,561, while FOLFOX plus bevacizumab provided 1.41 QALYs at a cost of $92,112. The incremental cost-effectiveness ratio for FOLFOX plus bevacizumab was $571,240 per QALY. As second-line therapy, FOLFIRI alone provided 0.64 QALYs at a cost of $7,443, while FOLFIRI plus bevacizumab provided 0.75 QALYs at a cost of $46,764. The incremental cost-effectiveness ratio for FOLFIRI plus bevacizumab was $364,083 per QALY. A threshold between $50,000 and $100,000 is usually used to define a cost-effective health intervention, the investigators said (J. Clin. Oncol. 2015 Feb. 16 [doi:10.1200/JCO.2014.58.4904]).
In sensitivity analyses that tested a broad range of variations in each parameter – such as different drug costs, physician fees, and treatment toxicities – the incremental cost-effectiveness ratio remained higher than $200,000 per QALY in every case, even in the “best case scenario” model.
These findings closely accord with those of similar studies in other countries, even though there were significant differences in modeling approaches, costs, and health care systems. In the United Kingdom, adding bevacizumab to FOLFIRI cost the equivalent of $102,000 U.S. per QALY. In Canada it cost the equivalent of $120,000 U.S. per QALY, and in Japan it cost the equivalent of $113,000 U.S. per QALY, Dr. Goldstein and his associates said.
Presumably, the poor cost/benefit ratio could be improved if an effective biomarker were developed and used to select only the patients most likely to benefit for bevacizumab therapy, they added.
No funding sources were reported for this study. Dr. Goldstein reported having no financial disclosures; his associates reported numerous ties to industry sources, mostly through research funding of their institutions.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Adding bevacizumab to standard chemotherapy for metastatic colorectal cancer yields minimal benefit at high cost.
Major finding: The incremental cost-effectiveness ratio for FOLFOX plus bevacizumab was $571,240 per QALY, and that for FOLFIRI plus bevacizumab was $364,083 per QALY.
Data source: A cost-effectiveness analysis of adding bevacizumab to standard chemotherapy using computer models based on recent clinical trial data.
Disclosures: No funding sources were reported for this study. Dr. Goldstein reported having no financial disclosures; his associates reported numerous ties to industry sources, mostly through research funding of their institutions.
Varenicline facilitates gradual smoking cessation
A 24-week course of varenicline improved quit rates among smokers who were unwilling or unable to stop smoking abruptly but preferred to gradually reduce their use of cigarettes, according to a report published online Feb. 17 in JAMA.
In an industry-sponsored, randomized, double-blind, controlled trial, participants who were given varenicline showed higher quit rates at the end of treatment as well as 1 year later, compared with those given placebo, said Dr. Jon O. Ebbert of the Mayo Clinic, Rochester, Minn., and his associates.

Current U.S. clinical practice guidelines recommend that smokers set an immediate quit date and quit abruptly, “even though only 8% of smokers report being ready to quit within the next month.” The findings of this study show that a more gradual, reduce-to-quit approach also can be effective, and “would be expected to be of interest to 14 million of the 42 million current smokers in this country,” the investigators noted.
The study was performed at 61 medical centers in 10 countries during a 2-year period. The 1,510 participants would not quit abruptly, as is recommended, but were willing to reduce their smoking and make a quit attempt within the next 3 months. They were asked to reduce their smoking rate by 50% or more by week 4, to further reduce it by 75% or more by week 8, and to quit altogether by week 12.
Study participants, who smoked an average of 10 or more cigarettes per day at baseline, were randomly assigned to receive varenicline (760 patients) or a matching placebo (750 patients) for 24 weeks. All also received written materials and smoking cessation counseling focused on reduction techniques, problem-solving, and skills training. This was provided during 18 clinic visits and 10 telephone sessions of about 10 minutes’ duration.
The primary efficacy endpoint was the continuous abstinence rate during weeks 15-24, which was self-reported by the participants and confirmed using exhaled carbon monoxide measurements. This rate was significantly higher for the varenicline group (32.1%) than for the placebo group (6.9%). The continuous abstinence rate remained significantly higher through 1 year of follow-up for varenicline (27%) than for placebo (9.9%).
In addition, the median time to abstinence was significantly shorter with varenicline (50 days) than with placebo (85 days). Results of sensitivity analyses confirmed those of the primary analysis, the investigators said (JAMA 2015 Feb. 17;313:687-94).
The percentage of participants who reported adverse events was higher with varenicline (82.3% vs. 72.5%), and the difference was largely accounted for by increases in nausea, abnormal dreams, insomnia, constipation, vomiting, and weight gain. Rates of serious adverse events were similar between the two study groups, as were rates of treatment discontinuation (8.4% for varenicline and 7% for placebo). In particular, rates of suicidal ideation or behavior and depression scores were not significantly higher with varenicline.
The study findings indicate that prescribing varenicline “with a recommendation to reduce the number of cigarettes smoked per day, with the eventual goal of quitting, could be a useful therapeutic option for this population of smokers,” Dr. Ebbert and his associates said.
One limitation of this study was that patients were excluded from participating if they had severe psychiatric, pulmonary, cardiovascular, or cerebrovascular disease, which hinders the generalizability of the results to a broader population of smokers. In addition, study participants received significant counseling support that would not necessarily be available to patients in real-world clinical practice, so quit rates would be expected to be lower in actual practice, they added.
This study was funded by Pfizer, maker of varenicline (Chantix). Pfizer also participated in the design and conduct of the study; the collection, analysis, and interpretation of the data; and manuscript preparation. Dr. Ebbert reported receiving grants from Pfizer, JHP Pharmaceuticals, and Orexigen, as well as personal fees from GlaxoSmithKline. His associates reported numerous ties to industry sources.
A 24-week course of varenicline improved quit rates among smokers who were unwilling or unable to stop smoking abruptly but preferred to gradually reduce their use of cigarettes, according to a report published online Feb. 17 in JAMA.
In an industry-sponsored, randomized, double-blind, controlled trial, participants who were given varenicline showed higher quit rates at the end of treatment as well as 1 year later, compared with those given placebo, said Dr. Jon O. Ebbert of the Mayo Clinic, Rochester, Minn., and his associates.
Current U.S. clinical practice guidelines recommend that smokers set an immediate quit date and quit abruptly, “even though only 8% of smokers report being ready to quit within the next month.” The findings of this study show that a more gradual, reduce-to-quit approach also can be effective, and “would be expected to be of interest to 14 million of the 42 million current smokers in this country,” the investigators noted.
The study was performed at 61 medical centers in 10 countries during a 2-year period. The 1,510 participants would not quit abruptly, as is recommended, but were willing to reduce their smoking and make a quit attempt within the next 3 months. They were asked to reduce their smoking rate by 50% or more by week 4, to further reduce it by 75% or more by week 8, and to quit altogether by week 12.
Study participants, who smoked an average of 10 or more cigarettes per day at baseline, were randomly assigned to receive varenicline (760 patients) or a matching placebo (750 patients) for 24 weeks. All also received written materials and smoking cessation counseling focused on reduction techniques, problem-solving, and skills training. This was provided during 18 clinic visits and 10 telephone sessions of about 10 minutes’ duration.
The primary efficacy endpoint was the continuous abstinence rate during weeks 15-24, which was self-reported by the participants and confirmed using exhaled carbon monoxide measurements. This rate was significantly higher for the varenicline group (32.1%) than for the placebo group (6.9%). The continuous abstinence rate remained significantly higher through 1 year of follow-up for varenicline (27%) than for placebo (9.9%).
In addition, the median time to abstinence was significantly shorter with varenicline (50 days) than with placebo (85 days). Results of sensitivity analyses confirmed those of the primary analysis, the investigators said (JAMA 2015 Feb. 17;313:687-94).
The percentage of participants who reported adverse events was higher with varenicline (82.3% vs. 72.5%), and the difference was largely accounted for by increases in nausea, abnormal dreams, insomnia, constipation, vomiting, and weight gain. Rates of serious adverse events were similar between the two study groups, as were rates of treatment discontinuation (8.4% for varenicline and 7% for placebo). In particular, rates of suicidal ideation or behavior and depression scores were not significantly higher with varenicline.
The study findings indicate that prescribing varenicline “with a recommendation to reduce the number of cigarettes smoked per day, with the eventual goal of quitting, could be a useful therapeutic option for this population of smokers,” Dr. Ebbert and his associates said.
One limitation of this study was that patients were excluded from participating if they had severe psychiatric, pulmonary, cardiovascular, or cerebrovascular disease, which hinders the generalizability of the results to a broader population of smokers. In addition, study participants received significant counseling support that would not necessarily be available to patients in real-world clinical practice, so quit rates would be expected to be lower in actual practice, they added.
This study was funded by Pfizer, maker of varenicline (Chantix). Pfizer also participated in the design and conduct of the study; the collection, analysis, and interpretation of the data; and manuscript preparation. Dr. Ebbert reported receiving grants from Pfizer, JHP Pharmaceuticals, and Orexigen, as well as personal fees from GlaxoSmithKline. His associates reported numerous ties to industry sources.
A 24-week course of varenicline improved quit rates among smokers who were unwilling or unable to stop smoking abruptly but preferred to gradually reduce their use of cigarettes, according to a report published online Feb. 17 in JAMA.
In an industry-sponsored, randomized, double-blind, controlled trial, participants who were given varenicline showed higher quit rates at the end of treatment as well as 1 year later, compared with those given placebo, said Dr. Jon O. Ebbert of the Mayo Clinic, Rochester, Minn., and his associates.
Current U.S. clinical practice guidelines recommend that smokers set an immediate quit date and quit abruptly, “even though only 8% of smokers report being ready to quit within the next month.” The findings of this study show that a more gradual, reduce-to-quit approach also can be effective, and “would be expected to be of interest to 14 million of the 42 million current smokers in this country,” the investigators noted.
The study was performed at 61 medical centers in 10 countries during a 2-year period. The 1,510 participants would not quit abruptly, as is recommended, but were willing to reduce their smoking and make a quit attempt within the next 3 months. They were asked to reduce their smoking rate by 50% or more by week 4, to further reduce it by 75% or more by week 8, and to quit altogether by week 12.
Study participants, who smoked an average of 10 or more cigarettes per day at baseline, were randomly assigned to receive varenicline (760 patients) or a matching placebo (750 patients) for 24 weeks. All also received written materials and smoking cessation counseling focused on reduction techniques, problem-solving, and skills training. This was provided during 18 clinic visits and 10 telephone sessions of about 10 minutes’ duration.
The primary efficacy endpoint was the continuous abstinence rate during weeks 15-24, which was self-reported by the participants and confirmed using exhaled carbon monoxide measurements. This rate was significantly higher for the varenicline group (32.1%) than for the placebo group (6.9%). The continuous abstinence rate remained significantly higher through 1 year of follow-up for varenicline (27%) than for placebo (9.9%).
In addition, the median time to abstinence was significantly shorter with varenicline (50 days) than with placebo (85 days). Results of sensitivity analyses confirmed those of the primary analysis, the investigators said (JAMA 2015 Feb. 17;313:687-94).
The percentage of participants who reported adverse events was higher with varenicline (82.3% vs. 72.5%), and the difference was largely accounted for by increases in nausea, abnormal dreams, insomnia, constipation, vomiting, and weight gain. Rates of serious adverse events were similar between the two study groups, as were rates of treatment discontinuation (8.4% for varenicline and 7% for placebo). In particular, rates of suicidal ideation or behavior and depression scores were not significantly higher with varenicline.
The study findings indicate that prescribing varenicline “with a recommendation to reduce the number of cigarettes smoked per day, with the eventual goal of quitting, could be a useful therapeutic option for this population of smokers,” Dr. Ebbert and his associates said.
One limitation of this study was that patients were excluded from participating if they had severe psychiatric, pulmonary, cardiovascular, or cerebrovascular disease, which hinders the generalizability of the results to a broader population of smokers. In addition, study participants received significant counseling support that would not necessarily be available to patients in real-world clinical practice, so quit rates would be expected to be lower in actual practice, they added.
This study was funded by Pfizer, maker of varenicline (Chantix). Pfizer also participated in the design and conduct of the study; the collection, analysis, and interpretation of the data; and manuscript preparation. Dr. Ebbert reported receiving grants from Pfizer, JHP Pharmaceuticals, and Orexigen, as well as personal fees from GlaxoSmithKline. His associates reported numerous ties to industry sources.
Key clinical point: Twenty-four weeks of varenicline therapy aimed at gradually cutting back on cigarette smoking can improve quit rates for smokers during treatment and at 1 year.
Major finding: The primary efficacy endpoint, the continuous abstinence rate during weeks 15-24 of treatment, was significantly higher for the varenicline group (32.1%) than for the placebo group (6.9%).
Data source: An industry-funded, international, randomized, double-blind, controlled trial involving 1,510 adult smokers treated for 24 weeks and followed for 1 year.
Disclosures: This study was funded by Pfizer, maker of varenicline (Chantix). Pfizer also participated in the design and conduct of the study; the collection, analysis, and interpretation of the data; and manuscript preparation. Dr. Ebbert reported receiving grants from Pfizer, JHP Pharmaceuticals, and Orexigen, as well as personal fees from GlaxoSmithKline. His associates reported numerous ties to industry sources.

Methylprednisolone cuts treatment failure in severe CAP
A 5-day course of methylprednisolone reduced the rate of treatment failure in adults with severe community-acquired pneumonia and a high initial inflammatory response, according to a report published online Feb. 17 in JAMA.
“If replicated, these findings would support the use of corticosteroids as adjunctive treatment in this clinical population,” said Dr. Antoni Torres of Institut Clinic del Torax, Hospital Clinic, Barcelona, and his associates.
The use of corticosteroids in community-acquired pneumonia is controversial. Some studies show that corticosteroids decrease radiographic progression of the disease, prevent shock, prevent respiratory failure, decrease length of stay, and reduce mortality. Other studies show no benefit. Dr. Torres and his colleagues studied the use of corticosteroids in the subgroup of patients who present with severe disease and a proinflammatory profile characterized by a serum CRP level over 150 mg/dL – the patients most likely to benefit from anti-inflammatories and least likely to be harmed by steroid-induced superinfection.
In a randomized double-blind trial at three teaching hospitals in Spain, 120 such patients were randomly assigned to receive 5 days of either IV methylprednisolone (61 patients) at a dose of 0.5 mg/kg every 12 hours or a matching placebo (59 patients), in addition to antibiotics. The most common cause of pneumonia in both study groups was Streptococcus pneumoniae, and the most frequent empiric antimicrobial treatment was a combination of ceftriaxone, levofloxacin, and azithromycin.
As expected, CRP and IL-10 levels decreased more in patients who received the corticosteroid than in those who received placebo.
The primary efficacy endpoint was the rate of treatment failure, both within 72 hours (early) and at 72-120 hours (late) after initiation of therapy. This rate was significantly lower in patients who received methylprednisolone (8 patients, or 13%) than in those who received placebo (18, or 31%). This reduction was largely attributed to the prevention of radiographic progression and late septic shock.
However, there were no significant differences in the secondary outcomes of time to clinical stabilization, length of ICU stay, length of hospitalization, and in-hospital mortality, the investigators said (JAMA 2015 Feb. 17 [doi:10.1001/jama.2015.88]). Adverse events were similar between the two study groups and included hyperglycemia (18% with methylprednisolone vs 12% with placebo) and acute kidney injury (13% vs. 14%). One patient taking methylprednisolone developed a superinfection. Other serious adverse events included one case of delirium and one of acute hepatic failure in the methylprednisolone group and one case of GI hemorrhage in the placebo group.
It is likely that the most feared adverse effect of corticosteroid therapy – immunosuppression leading to superinfection – wasn’t an issue in this study because of the short course of treatment and the relatively low dose of methylprednisolone used, the researchers said.
These findings are important because any efficacious adjunctive treatment may help reduce the high mortality associated with severe community-acquired pneumonia. It is estimated that despite effective antibiotic treatment, 12%-36% of patients admitted to an ICU with this disease will die within a short period, Dr. Torres and his associates noted.
They are conducting another study to confirm these results, in part because the small difference between the two study groups in the number of treatment failures – only 10 patients – indicates that replication is needed.
This study was supported by the Sociedad Españolade Neumologia, Societat Catalanade Pneumologia, Fundació Catalanade Pneumologia, Grup de Recercade Qualitatdela Generalitatde Catalunya, Fondode Investigación Sanitaria, Institut d’Investigacions Biomèdiques August Pii Sunyer, and Centrode Investigación Biomédica En Red-Enfermedades Respiratorias. Dr. Torres reported having no financial disclosures; his associates reported ties to GlaxoSmithKline, Dey Pharma, Pfizer, Boehringer Ingelheim, Bayer, Sherin Pharma, AstraZeneca, Cubist, Thermo Diagnostics, and Theravance.
These findings raise the intriguing possibility that corticosteroids block a Jarisch-Herxheimer–like reaction to the initiation of antibiotics in patients who have a high genomic bacterial load. Such a reaction is thought to result from high concentrations of cytokines observed shortly after antibiotics are first administered, possibly through the release of endotoxin or other bacterial mediators in patients with a high bacterial burden.
Richard G. Wunderink, M.D., is at Northwestern University, Chicago. He reported having no financial disclosures. Dr. Wunderink made these remarks in an editorial accompanying Dr. Torres’ report (JAMA 2015;313:673-4).
These findings raise the intriguing possibility that corticosteroids block a Jarisch-Herxheimer–like reaction to the initiation of antibiotics in patients who have a high genomic bacterial load. Such a reaction is thought to result from high concentrations of cytokines observed shortly after antibiotics are first administered, possibly through the release of endotoxin or other bacterial mediators in patients with a high bacterial burden.
Richard G. Wunderink, M.D., is at Northwestern University, Chicago. He reported having no financial disclosures. Dr. Wunderink made these remarks in an editorial accompanying Dr. Torres’ report (JAMA 2015;313:673-4).
These findings raise the intriguing possibility that corticosteroids block a Jarisch-Herxheimer–like reaction to the initiation of antibiotics in patients who have a high genomic bacterial load. Such a reaction is thought to result from high concentrations of cytokines observed shortly after antibiotics are first administered, possibly through the release of endotoxin or other bacterial mediators in patients with a high bacterial burden.
Richard G. Wunderink, M.D., is at Northwestern University, Chicago. He reported having no financial disclosures. Dr. Wunderink made these remarks in an editorial accompanying Dr. Torres’ report (JAMA 2015;313:673-4).
A 5-day course of methylprednisolone reduced the rate of treatment failure in adults with severe community-acquired pneumonia and a high initial inflammatory response, according to a report published online Feb. 17 in JAMA.
“If replicated, these findings would support the use of corticosteroids as adjunctive treatment in this clinical population,” said Dr. Antoni Torres of Institut Clinic del Torax, Hospital Clinic, Barcelona, and his associates.
The use of corticosteroids in community-acquired pneumonia is controversial. Some studies show that corticosteroids decrease radiographic progression of the disease, prevent shock, prevent respiratory failure, decrease length of stay, and reduce mortality. Other studies show no benefit. Dr. Torres and his colleagues studied the use of corticosteroids in the subgroup of patients who present with severe disease and a proinflammatory profile characterized by a serum CRP level over 150 mg/dL – the patients most likely to benefit from anti-inflammatories and least likely to be harmed by steroid-induced superinfection.
In a randomized double-blind trial at three teaching hospitals in Spain, 120 such patients were randomly assigned to receive 5 days of either IV methylprednisolone (61 patients) at a dose of 0.5 mg/kg every 12 hours or a matching placebo (59 patients), in addition to antibiotics. The most common cause of pneumonia in both study groups was Streptococcus pneumoniae, and the most frequent empiric antimicrobial treatment was a combination of ceftriaxone, levofloxacin, and azithromycin.
As expected, CRP and IL-10 levels decreased more in patients who received the corticosteroid than in those who received placebo.
The primary efficacy endpoint was the rate of treatment failure, both within 72 hours (early) and at 72-120 hours (late) after initiation of therapy. This rate was significantly lower in patients who received methylprednisolone (8 patients, or 13%) than in those who received placebo (18, or 31%). This reduction was largely attributed to the prevention of radiographic progression and late septic shock.
However, there were no significant differences in the secondary outcomes of time to clinical stabilization, length of ICU stay, length of hospitalization, and in-hospital mortality, the investigators said (JAMA 2015 Feb. 17 [doi:10.1001/jama.2015.88]). Adverse events were similar between the two study groups and included hyperglycemia (18% with methylprednisolone vs 12% with placebo) and acute kidney injury (13% vs. 14%). One patient taking methylprednisolone developed a superinfection. Other serious adverse events included one case of delirium and one of acute hepatic failure in the methylprednisolone group and one case of GI hemorrhage in the placebo group.
It is likely that the most feared adverse effect of corticosteroid therapy – immunosuppression leading to superinfection – wasn’t an issue in this study because of the short course of treatment and the relatively low dose of methylprednisolone used, the researchers said.
These findings are important because any efficacious adjunctive treatment may help reduce the high mortality associated with severe community-acquired pneumonia. It is estimated that despite effective antibiotic treatment, 12%-36% of patients admitted to an ICU with this disease will die within a short period, Dr. Torres and his associates noted.
They are conducting another study to confirm these results, in part because the small difference between the two study groups in the number of treatment failures – only 10 patients – indicates that replication is needed.
This study was supported by the Sociedad Españolade Neumologia, Societat Catalanade Pneumologia, Fundació Catalanade Pneumologia, Grup de Recercade Qualitatdela Generalitatde Catalunya, Fondode Investigación Sanitaria, Institut d’Investigacions Biomèdiques August Pii Sunyer, and Centrode Investigación Biomédica En Red-Enfermedades Respiratorias. Dr. Torres reported having no financial disclosures; his associates reported ties to GlaxoSmithKline, Dey Pharma, Pfizer, Boehringer Ingelheim, Bayer, Sherin Pharma, AstraZeneca, Cubist, Thermo Diagnostics, and Theravance.
A 5-day course of methylprednisolone reduced the rate of treatment failure in adults with severe community-acquired pneumonia and a high initial inflammatory response, according to a report published online Feb. 17 in JAMA.
“If replicated, these findings would support the use of corticosteroids as adjunctive treatment in this clinical population,” said Dr. Antoni Torres of Institut Clinic del Torax, Hospital Clinic, Barcelona, and his associates.
The use of corticosteroids in community-acquired pneumonia is controversial. Some studies show that corticosteroids decrease radiographic progression of the disease, prevent shock, prevent respiratory failure, decrease length of stay, and reduce mortality. Other studies show no benefit. Dr. Torres and his colleagues studied the use of corticosteroids in the subgroup of patients who present with severe disease and a proinflammatory profile characterized by a serum CRP level over 150 mg/dL – the patients most likely to benefit from anti-inflammatories and least likely to be harmed by steroid-induced superinfection.
In a randomized double-blind trial at three teaching hospitals in Spain, 120 such patients were randomly assigned to receive 5 days of either IV methylprednisolone (61 patients) at a dose of 0.5 mg/kg every 12 hours or a matching placebo (59 patients), in addition to antibiotics. The most common cause of pneumonia in both study groups was Streptococcus pneumoniae, and the most frequent empiric antimicrobial treatment was a combination of ceftriaxone, levofloxacin, and azithromycin.
As expected, CRP and IL-10 levels decreased more in patients who received the corticosteroid than in those who received placebo.
The primary efficacy endpoint was the rate of treatment failure, both within 72 hours (early) and at 72-120 hours (late) after initiation of therapy. This rate was significantly lower in patients who received methylprednisolone (8 patients, or 13%) than in those who received placebo (18, or 31%). This reduction was largely attributed to the prevention of radiographic progression and late septic shock.
However, there were no significant differences in the secondary outcomes of time to clinical stabilization, length of ICU stay, length of hospitalization, and in-hospital mortality, the investigators said (JAMA 2015 Feb. 17 [doi:10.1001/jama.2015.88]). Adverse events were similar between the two study groups and included hyperglycemia (18% with methylprednisolone vs 12% with placebo) and acute kidney injury (13% vs. 14%). One patient taking methylprednisolone developed a superinfection. Other serious adverse events included one case of delirium and one of acute hepatic failure in the methylprednisolone group and one case of GI hemorrhage in the placebo group.
It is likely that the most feared adverse effect of corticosteroid therapy – immunosuppression leading to superinfection – wasn’t an issue in this study because of the short course of treatment and the relatively low dose of methylprednisolone used, the researchers said.
These findings are important because any efficacious adjunctive treatment may help reduce the high mortality associated with severe community-acquired pneumonia. It is estimated that despite effective antibiotic treatment, 12%-36% of patients admitted to an ICU with this disease will die within a short period, Dr. Torres and his associates noted.
They are conducting another study to confirm these results, in part because the small difference between the two study groups in the number of treatment failures – only 10 patients – indicates that replication is needed.
This study was supported by the Sociedad Españolade Neumologia, Societat Catalanade Pneumologia, Fundació Catalanade Pneumologia, Grup de Recercade Qualitatdela Generalitatde Catalunya, Fondode Investigación Sanitaria, Institut d’Investigacions Biomèdiques August Pii Sunyer, and Centrode Investigación Biomédica En Red-Enfermedades Respiratorias. Dr. Torres reported having no financial disclosures; his associates reported ties to GlaxoSmithKline, Dey Pharma, Pfizer, Boehringer Ingelheim, Bayer, Sherin Pharma, AstraZeneca, Cubist, Thermo Diagnostics, and Theravance.
FROM JAMA
Key clinical point: Acute methylprednisolone decreased the rate of treatment failure in severe community-acquired pneumonia with a high initial inflammatory response.
Major finding: The primary efficacy endpoint – the rate of treatment failure – was significantly lower in patients who received methylprednisolone (8, or 13%) than in those who received placebo (18, or 31%).
Data source: A prospective randomized double-bind controlled trial involving 120 adults with severe community-acquired pneumonia treated at three hospitals in Spain.
Disclosures: This study was supported by the Sociedad Españolade Neumologia, Societat Catalanade Pneumologia, Fundació Catalanade Pneumologia, Grup de Recercade Qualitatdela Generalitatde Catalunya, Fondode Investigación Sanitaria, Institut d’Investigacions Biomèdiques August Pii Sunyer, and Centrode Investigación Biomédica En Red-Enfermedades Respiratorias. Dr. Torres reported having no financial disclosures; his associates reported ties to GlaxoSmithKline, Dey Pharma, Pfizer, Boehringer Ingelheim, Bayer, Sherin Pharma, AstraZeneca, Cubist, Thermo Diagnostics, and Theravance.
HPV-16 E6 seropositivity common before anal cancer develops
HPV-16-E6 seropositivity is relatively common among people who later develop anal cancer, and less common but still more frequent than normal among people who later develop cervical, penile, vaginal, or vulvar cancer, according to a report published online Feb. 9 in Journal of Clinical Oncology.
Recent research has suggested that HPV-16 E6 seropositivity may indicate a higher risk of developing oropharyngeal cancers, and case-control studies have linked this HPV-16 protein to anogenital cancers as well. To explore this association, investigators analyzed data from the European Prospective Investigation into Cancer and Nutrition (EPIC) study, which examined nutritional and lifestyle factors in more than 521,000 adults across 11 European countries in 1992-2000, said Aimée R. Kreimer, Ph.D., of the National Cancer Institute, Bethesda, Md, and her associates.
Using national cancer registries, Dr. Kreimer and her colleagues identified incident anogenital cancers that developed in study participants during the intervening decade. They then determined HPV-16 E6 seropositivity from blood samples drawn during the EPIC study for 273 patients with cervical cancer, 24 with anal cancer, 67 with vulvar cancer, 12 with vaginal cancer, and 24 with penile cancer, as well as for 718 matched but cancer-free controls. The median time between the blood draw and cancer diagnosis ranged between 7 and 8 years for all cancers except those of the cervix, for which the median lead time was only 3 years.
HPV-16 E6 seropositivity was relatively common only in the patients who developed anal cancer. It occurred in 7 of the 24 (29.2%), compared with only 4 of the control subjects (0.6%), for an odds ratio of 75.9. This means that the rate of anal cancer was 75 times higher among seropositive than seronegative participants. HPV-16 E6 seropositivity also was seen in 3.3% of patients with cervical cancer, 8.3% of those with penile cancer, 8.3% of those with vaginal cancer, and 1.5% of those with vulvar cancer, indicating a significantly elevated risk, but of a much smaller magnitude than found for anal cancer, the investigators said (J. Clin. Oncol. 2015 Feb. 9 [doi:10.1200/JCO.2014.57.8435]).
It is not yet clear why the strength of the associations between HPV-16 E6 and cancer differ by anatomic site, “but the immunobiology and proximity to the lymphatic system (and thus access to antigen-presenting cells) are likely important,” they said.
These results suggest that screening for anal cancer should be considered in the clinical work-up of people who test positive for HPV-16 E6. The main limitation of this study was that even using this large cohort, the number of cancers in every anatomic location except the cervix was quite small, “an inherent issue in studies of rare cancers,” the investigators added.
HPV-16-E6 seropositivity is relatively common among people who later develop anal cancer, and less common but still more frequent than normal among people who later develop cervical, penile, vaginal, or vulvar cancer, according to a report published online Feb. 9 in Journal of Clinical Oncology.
Recent research has suggested that HPV-16 E6 seropositivity may indicate a higher risk of developing oropharyngeal cancers, and case-control studies have linked this HPV-16 protein to anogenital cancers as well. To explore this association, investigators analyzed data from the European Prospective Investigation into Cancer and Nutrition (EPIC) study, which examined nutritional and lifestyle factors in more than 521,000 adults across 11 European countries in 1992-2000, said Aimée R. Kreimer, Ph.D., of the National Cancer Institute, Bethesda, Md, and her associates.
Using national cancer registries, Dr. Kreimer and her colleagues identified incident anogenital cancers that developed in study participants during the intervening decade. They then determined HPV-16 E6 seropositivity from blood samples drawn during the EPIC study for 273 patients with cervical cancer, 24 with anal cancer, 67 with vulvar cancer, 12 with vaginal cancer, and 24 with penile cancer, as well as for 718 matched but cancer-free controls. The median time between the blood draw and cancer diagnosis ranged between 7 and 8 years for all cancers except those of the cervix, for which the median lead time was only 3 years.
HPV-16 E6 seropositivity was relatively common only in the patients who developed anal cancer. It occurred in 7 of the 24 (29.2%), compared with only 4 of the control subjects (0.6%), for an odds ratio of 75.9. This means that the rate of anal cancer was 75 times higher among seropositive than seronegative participants. HPV-16 E6 seropositivity also was seen in 3.3% of patients with cervical cancer, 8.3% of those with penile cancer, 8.3% of those with vaginal cancer, and 1.5% of those with vulvar cancer, indicating a significantly elevated risk, but of a much smaller magnitude than found for anal cancer, the investigators said (J. Clin. Oncol. 2015 Feb. 9 [doi:10.1200/JCO.2014.57.8435]).
It is not yet clear why the strength of the associations between HPV-16 E6 and cancer differ by anatomic site, “but the immunobiology and proximity to the lymphatic system (and thus access to antigen-presenting cells) are likely important,” they said.
These results suggest that screening for anal cancer should be considered in the clinical work-up of people who test positive for HPV-16 E6. The main limitation of this study was that even using this large cohort, the number of cancers in every anatomic location except the cervix was quite small, “an inherent issue in studies of rare cancers,” the investigators added.
HPV-16-E6 seropositivity is relatively common among people who later develop anal cancer, and less common but still more frequent than normal among people who later develop cervical, penile, vaginal, or vulvar cancer, according to a report published online Feb. 9 in Journal of Clinical Oncology.
Recent research has suggested that HPV-16 E6 seropositivity may indicate a higher risk of developing oropharyngeal cancers, and case-control studies have linked this HPV-16 protein to anogenital cancers as well. To explore this association, investigators analyzed data from the European Prospective Investigation into Cancer and Nutrition (EPIC) study, which examined nutritional and lifestyle factors in more than 521,000 adults across 11 European countries in 1992-2000, said Aimée R. Kreimer, Ph.D., of the National Cancer Institute, Bethesda, Md, and her associates.
Using national cancer registries, Dr. Kreimer and her colleagues identified incident anogenital cancers that developed in study participants during the intervening decade. They then determined HPV-16 E6 seropositivity from blood samples drawn during the EPIC study for 273 patients with cervical cancer, 24 with anal cancer, 67 with vulvar cancer, 12 with vaginal cancer, and 24 with penile cancer, as well as for 718 matched but cancer-free controls. The median time between the blood draw and cancer diagnosis ranged between 7 and 8 years for all cancers except those of the cervix, for which the median lead time was only 3 years.
HPV-16 E6 seropositivity was relatively common only in the patients who developed anal cancer. It occurred in 7 of the 24 (29.2%), compared with only 4 of the control subjects (0.6%), for an odds ratio of 75.9. This means that the rate of anal cancer was 75 times higher among seropositive than seronegative participants. HPV-16 E6 seropositivity also was seen in 3.3% of patients with cervical cancer, 8.3% of those with penile cancer, 8.3% of those with vaginal cancer, and 1.5% of those with vulvar cancer, indicating a significantly elevated risk, but of a much smaller magnitude than found for anal cancer, the investigators said (J. Clin. Oncol. 2015 Feb. 9 [doi:10.1200/JCO.2014.57.8435]).
It is not yet clear why the strength of the associations between HPV-16 E6 and cancer differ by anatomic site, “but the immunobiology and proximity to the lymphatic system (and thus access to antigen-presenting cells) are likely important,” they said.
These results suggest that screening for anal cancer should be considered in the clinical work-up of people who test positive for HPV-16 E6. The main limitation of this study was that even using this large cohort, the number of cancers in every anatomic location except the cervix was quite small, “an inherent issue in studies of rare cancers,” the investigators added.
Key clinical point: HPV-16 E6 seropositivity was relatively common among people who went on to develop anal cancer, and less common but still elevated in people who went on to develop cervical, penile, vaginal, or vulvar cancer, compared with controls.
Major finding: HPV-16 E6 seropositivity occurred in 7 of 24 (29.2%) of patients who later developed anal cancer, compared with only 4 control subjects (0.6%), for an odds ratio of 75.9.
Data source: A secondary analysis of data from an international cohort study, involving 273 patients with cervical cancer, 24 with anal cancer, 67 with vulvar cancer, 12 with vaginal cancer, and 24 with penile cancer, as well as 718 matched controls.
Disclosures: This study was supported by the National Cancer Institute Intramural Research Program, the International Agency for Research on Cancer, the Associazione Italiana per la Ricerca sul Cancro, and the regional government of Asturias. Dr. Kreimer reported having no financial disclosures; her associates reported ties to multiple pharmaceutical companies.