User login
Supercomputer accelerates whole-genome analysis

Credit: NIGMS
The time needed to sequence an entire human genome has decreased greatly in recent years, but analyzing the resulting 3 billion base pairs of genetic information from a single genome can take many months.
Now, researchers have found they can accelerate whole-genome analysis using a Cray XE6 supercomputer.
The team found this computer could process many genomes at once and was able to analyze 240 full genomes in a little over 2 days.
The researchers reported these results in Bioinformatics.
The team used Beagle, a Cray XE6 supercomputer located at Argonne National Laboratory in Illinois, in an attempt to analyze multiple genomes concurrently.
Using publicly available software packages and one quarter of its total capacity, the computer was able to align and call variants on 240 whole genomes in approximately 50 hours.
But the computer did not only speed up whole-genome analysis. It also increased the usable sequences per genome.
“Improving analysis through both speed and accuracy reduces the price per genome,” said study author Elizabeth McNally, MD, PhD, of the University of Chicago.
“With this approach, the price for analyzing an entire genome is less than the cost of looking at just a fraction of the genome. New technology promises to bring the costs of sequencing down to around $1000 per genome. Our goal is get the cost of analysis down into that range.”
The findings of this research have immediate medical applications, according to Dr McNally. She noted that she and her colleagues must often sequence genes from an initial patient as well as multiple family members in order to better understand and either treat or prevent a disease.
“We start genetic testing with the patient,” she said. “But when we find a significant mutation, we have to think about testing the whole family to identify individuals at risk.”
Furthermore, the range of testable mutations has greatly increased in recent years.
“In the early days, we would test 1 to 3 genes,” Dr McNally said. “In 2007, we did our first 5-gene panel. Now, we order 50 to 70 genes at a time, which usually gets us an answer. At that point, it can be more useful and less expensive to sequence the whole genome.”
The information from these genomes combined with careful attention to patient and family histories adds to our knowledge about inherited disorders, according to Dr McNally.
“It can refine the classification of these disorders,” she said. “By paying close attention to family members with genes that place them at increased risk, but who do not yet show signs of disease, we can investigate early phases of a disorder.” ![]()
Credit: NIGMS
The time needed to sequence an entire human genome has decreased greatly in recent years, but analyzing the resulting 3 billion base pairs of genetic information from a single genome can take many months.
Now, researchers have found they can accelerate whole-genome analysis using a Cray XE6 supercomputer.
The team found this computer could process many genomes at once and was able to analyze 240 full genomes in a little over 2 days.
The researchers reported these results in Bioinformatics.
The team used Beagle, a Cray XE6 supercomputer located at Argonne National Laboratory in Illinois, in an attempt to analyze multiple genomes concurrently.
Using publicly available software packages and one quarter of its total capacity, the computer was able to align and call variants on 240 whole genomes in approximately 50 hours.
But the computer did not only speed up whole-genome analysis. It also increased the usable sequences per genome.
“Improving analysis through both speed and accuracy reduces the price per genome,” said study author Elizabeth McNally, MD, PhD, of the University of Chicago.
“With this approach, the price for analyzing an entire genome is less than the cost of looking at just a fraction of the genome. New technology promises to bring the costs of sequencing down to around $1000 per genome. Our goal is get the cost of analysis down into that range.”
The findings of this research have immediate medical applications, according to Dr McNally. She noted that she and her colleagues must often sequence genes from an initial patient as well as multiple family members in order to better understand and either treat or prevent a disease.
“We start genetic testing with the patient,” she said. “But when we find a significant mutation, we have to think about testing the whole family to identify individuals at risk.”
Furthermore, the range of testable mutations has greatly increased in recent years.
“In the early days, we would test 1 to 3 genes,” Dr McNally said. “In 2007, we did our first 5-gene panel. Now, we order 50 to 70 genes at a time, which usually gets us an answer. At that point, it can be more useful and less expensive to sequence the whole genome.”
The information from these genomes combined with careful attention to patient and family histories adds to our knowledge about inherited disorders, according to Dr McNally.
“It can refine the classification of these disorders,” she said. “By paying close attention to family members with genes that place them at increased risk, but who do not yet show signs of disease, we can investigate early phases of a disorder.” ![]()
Credit: NIGMS
The time needed to sequence an entire human genome has decreased greatly in recent years, but analyzing the resulting 3 billion base pairs of genetic information from a single genome can take many months.
Now, researchers have found they can accelerate whole-genome analysis using a Cray XE6 supercomputer.
The team found this computer could process many genomes at once and was able to analyze 240 full genomes in a little over 2 days.
The researchers reported these results in Bioinformatics.
The team used Beagle, a Cray XE6 supercomputer located at Argonne National Laboratory in Illinois, in an attempt to analyze multiple genomes concurrently.
Using publicly available software packages and one quarter of its total capacity, the computer was able to align and call variants on 240 whole genomes in approximately 50 hours.
But the computer did not only speed up whole-genome analysis. It also increased the usable sequences per genome.
“Improving analysis through both speed and accuracy reduces the price per genome,” said study author Elizabeth McNally, MD, PhD, of the University of Chicago.
“With this approach, the price for analyzing an entire genome is less than the cost of looking at just a fraction of the genome. New technology promises to bring the costs of sequencing down to around $1000 per genome. Our goal is get the cost of analysis down into that range.”
The findings of this research have immediate medical applications, according to Dr McNally. She noted that she and her colleagues must often sequence genes from an initial patient as well as multiple family members in order to better understand and either treat or prevent a disease.
“We start genetic testing with the patient,” she said. “But when we find a significant mutation, we have to think about testing the whole family to identify individuals at risk.”
Furthermore, the range of testable mutations has greatly increased in recent years.
“In the early days, we would test 1 to 3 genes,” Dr McNally said. “In 2007, we did our first 5-gene panel. Now, we order 50 to 70 genes at a time, which usually gets us an answer. At that point, it can be more useful and less expensive to sequence the whole genome.”
The information from these genomes combined with careful attention to patient and family histories adds to our knowledge about inherited disorders, according to Dr McNally.
“It can refine the classification of these disorders,” she said. “By paying close attention to family members with genes that place them at increased risk, but who do not yet show signs of disease, we can investigate early phases of a disorder.” ![]()
Mutations linked to blood vessel disorders
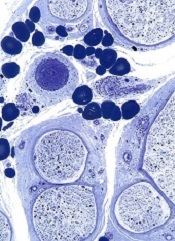
Two groups of researchers have shown that variants in the gene CECR1, which encodes for ADA2, are associated with blood vessel disorders.
One group found that mutations in CECR1 are associated with an ADA2 deficiency syndrome that is characterized by early onset, recurrent strokes and systemic vasculopathy.
The other group discovered that variants in CECR1 can cause polyarteritis nodosa vasculopathy.
Results of both studies appear in NEJM.
ADA2 deficiency syndrome
In the first study, Qing Zhou, PhD, of the National Human Genome Research Institute (NHGRI), and his colleagues used whole-exome sequencing to gain insight into the aforementioned syndrome, which was characterized by early onset lacunar strokes, systemic vasculopathy, and other symptoms.
The researchers performed whole-exome sequencing in 3 patients with the syndrome and their unaffected parents. In comparing 2 of the patients’ exomes to the exomes of their parents, the researchers located 2 variants in CECR1.
Sequencing a third patient revealed another harmful variant of CECR1, in addition to a small genomic deletion that shuts down the second copy of the gene.
The researchers needed only a sequence reading of that single gene to confirm that 3 other patients were affected by variants in CECR1. In all, these 6 patients were compound heterozygous for 8 mutations in CECR1.
The team discovered that these CECR1 mutations lead to the elimination of ADA2, which produces abnormalities and inflammation in blood vessel walls that ultimately result in the syndrome. So the team called the syndrome deficiency of ADA2 (DADA2).
To gain more insight into DADA2, the researchers induced ADA2 deficiency in a zebrafish model. They found that zebrafish embryos that produce less ADA2 than normal embryos have cerebral bleeds similar to those seen in some of the children with DADA2.
“Our study raises the possibility that the ADA2 pathway may contribute to susceptibility to stroke in the more general population,” said Daniel Kastner, MD, PhD, of NHGRI.
“This genome sequencing study expands what has previously been known about vascular biology. The role of ADA2 in such serious human disease is important and suggests that ADA2 variants may contribute to other, more common illnesses.”
Dr Kastner and his colleagues also sequenced the CECR1 gene in 3 patients from Turkey who had some of the symptoms of DADA2. These patients, who had polyarteritis nodosa (n=2) or small-vessel vasculitis (n=1), were homozygous for the p.Gly47Arg mutation.
ADA2 in polyarteritis nodosa
In the second study, researchers found the same mutation (p.Gly47Arg) in patients that emigrated to Israel from the country of Georgia.
“We now know that this mutation exists in the Middle East and in Pakistani populations and that it is not that uncommon,” said Ivona Aksentijevich, MD, of NHGRI. “This is the first time a single gene has been discovered that is involved in causing a system-wide form of vasculitis.”
To uncover this mutation, Paulina Navon-Elkan, MD, of the Shaare Zedek Medical Center in Jerusalem, and her colleagues sequenced 37 patients who had features of polyarteritis nodosa.
Nineteen patients were of Georgian Jewish ancestry. Sixteen of these patients came from 5 families, and 3 patients were unrelated. An additional 14 unrelated patients were of Turkish ancestry. And 4 German patients came from the same family.
The researchers found that, in all the families, vasculitis was a result of recessive mutations in CECR1.
All of the Georgian Jewish patients were homozygous for the p.Gly47Arg mutation. The German patients were compound heterozygous for Arg169Gln and Pro251Leu mutations. And 1 Turkish patient was compound heterozygous for Gly47Val and Trp264Ser mutations.
The researchers also analyzed serum specimens from the patients and found their ADA2 activity was significantly reduced.
Taking these results together, the team concluded that mutations in CECR1 cause loss of ADA2 function that can result in polyarteritis nodosa vasculopathy with a highly varied clinical expression. ![]()
Two groups of researchers have shown that variants in the gene CECR1, which encodes for ADA2, are associated with blood vessel disorders.
One group found that mutations in CECR1 are associated with an ADA2 deficiency syndrome that is characterized by early onset, recurrent strokes and systemic vasculopathy.
The other group discovered that variants in CECR1 can cause polyarteritis nodosa vasculopathy.
Results of both studies appear in NEJM.
ADA2 deficiency syndrome
In the first study, Qing Zhou, PhD, of the National Human Genome Research Institute (NHGRI), and his colleagues used whole-exome sequencing to gain insight into the aforementioned syndrome, which was characterized by early onset lacunar strokes, systemic vasculopathy, and other symptoms.
The researchers performed whole-exome sequencing in 3 patients with the syndrome and their unaffected parents. In comparing 2 of the patients’ exomes to the exomes of their parents, the researchers located 2 variants in CECR1.
Sequencing a third patient revealed another harmful variant of CECR1, in addition to a small genomic deletion that shuts down the second copy of the gene.
The researchers needed only a sequence reading of that single gene to confirm that 3 other patients were affected by variants in CECR1. In all, these 6 patients were compound heterozygous for 8 mutations in CECR1.
The team discovered that these CECR1 mutations lead to the elimination of ADA2, which produces abnormalities and inflammation in blood vessel walls that ultimately result in the syndrome. So the team called the syndrome deficiency of ADA2 (DADA2).
To gain more insight into DADA2, the researchers induced ADA2 deficiency in a zebrafish model. They found that zebrafish embryos that produce less ADA2 than normal embryos have cerebral bleeds similar to those seen in some of the children with DADA2.
“Our study raises the possibility that the ADA2 pathway may contribute to susceptibility to stroke in the more general population,” said Daniel Kastner, MD, PhD, of NHGRI.
“This genome sequencing study expands what has previously been known about vascular biology. The role of ADA2 in such serious human disease is important and suggests that ADA2 variants may contribute to other, more common illnesses.”
Dr Kastner and his colleagues also sequenced the CECR1 gene in 3 patients from Turkey who had some of the symptoms of DADA2. These patients, who had polyarteritis nodosa (n=2) or small-vessel vasculitis (n=1), were homozygous for the p.Gly47Arg mutation.
ADA2 in polyarteritis nodosa
In the second study, researchers found the same mutation (p.Gly47Arg) in patients that emigrated to Israel from the country of Georgia.
“We now know that this mutation exists in the Middle East and in Pakistani populations and that it is not that uncommon,” said Ivona Aksentijevich, MD, of NHGRI. “This is the first time a single gene has been discovered that is involved in causing a system-wide form of vasculitis.”
To uncover this mutation, Paulina Navon-Elkan, MD, of the Shaare Zedek Medical Center in Jerusalem, and her colleagues sequenced 37 patients who had features of polyarteritis nodosa.
Nineteen patients were of Georgian Jewish ancestry. Sixteen of these patients came from 5 families, and 3 patients were unrelated. An additional 14 unrelated patients were of Turkish ancestry. And 4 German patients came from the same family.
The researchers found that, in all the families, vasculitis was a result of recessive mutations in CECR1.
All of the Georgian Jewish patients were homozygous for the p.Gly47Arg mutation. The German patients were compound heterozygous for Arg169Gln and Pro251Leu mutations. And 1 Turkish patient was compound heterozygous for Gly47Val and Trp264Ser mutations.
The researchers also analyzed serum specimens from the patients and found their ADA2 activity was significantly reduced.
Taking these results together, the team concluded that mutations in CECR1 cause loss of ADA2 function that can result in polyarteritis nodosa vasculopathy with a highly varied clinical expression. ![]()
Two groups of researchers have shown that variants in the gene CECR1, which encodes for ADA2, are associated with blood vessel disorders.
One group found that mutations in CECR1 are associated with an ADA2 deficiency syndrome that is characterized by early onset, recurrent strokes and systemic vasculopathy.
The other group discovered that variants in CECR1 can cause polyarteritis nodosa vasculopathy.
Results of both studies appear in NEJM.
ADA2 deficiency syndrome
In the first study, Qing Zhou, PhD, of the National Human Genome Research Institute (NHGRI), and his colleagues used whole-exome sequencing to gain insight into the aforementioned syndrome, which was characterized by early onset lacunar strokes, systemic vasculopathy, and other symptoms.
The researchers performed whole-exome sequencing in 3 patients with the syndrome and their unaffected parents. In comparing 2 of the patients’ exomes to the exomes of their parents, the researchers located 2 variants in CECR1.
Sequencing a third patient revealed another harmful variant of CECR1, in addition to a small genomic deletion that shuts down the second copy of the gene.
The researchers needed only a sequence reading of that single gene to confirm that 3 other patients were affected by variants in CECR1. In all, these 6 patients were compound heterozygous for 8 mutations in CECR1.
The team discovered that these CECR1 mutations lead to the elimination of ADA2, which produces abnormalities and inflammation in blood vessel walls that ultimately result in the syndrome. So the team called the syndrome deficiency of ADA2 (DADA2).
To gain more insight into DADA2, the researchers induced ADA2 deficiency in a zebrafish model. They found that zebrafish embryos that produce less ADA2 than normal embryos have cerebral bleeds similar to those seen in some of the children with DADA2.
“Our study raises the possibility that the ADA2 pathway may contribute to susceptibility to stroke in the more general population,” said Daniel Kastner, MD, PhD, of NHGRI.
“This genome sequencing study expands what has previously been known about vascular biology. The role of ADA2 in such serious human disease is important and suggests that ADA2 variants may contribute to other, more common illnesses.”
Dr Kastner and his colleagues also sequenced the CECR1 gene in 3 patients from Turkey who had some of the symptoms of DADA2. These patients, who had polyarteritis nodosa (n=2) or small-vessel vasculitis (n=1), were homozygous for the p.Gly47Arg mutation.
ADA2 in polyarteritis nodosa
In the second study, researchers found the same mutation (p.Gly47Arg) in patients that emigrated to Israel from the country of Georgia.
“We now know that this mutation exists in the Middle East and in Pakistani populations and that it is not that uncommon,” said Ivona Aksentijevich, MD, of NHGRI. “This is the first time a single gene has been discovered that is involved in causing a system-wide form of vasculitis.”
To uncover this mutation, Paulina Navon-Elkan, MD, of the Shaare Zedek Medical Center in Jerusalem, and her colleagues sequenced 37 patients who had features of polyarteritis nodosa.
Nineteen patients were of Georgian Jewish ancestry. Sixteen of these patients came from 5 families, and 3 patients were unrelated. An additional 14 unrelated patients were of Turkish ancestry. And 4 German patients came from the same family.
The researchers found that, in all the families, vasculitis was a result of recessive mutations in CECR1.
All of the Georgian Jewish patients were homozygous for the p.Gly47Arg mutation. The German patients were compound heterozygous for Arg169Gln and Pro251Leu mutations. And 1 Turkish patient was compound heterozygous for Gly47Val and Trp264Ser mutations.
The researchers also analyzed serum specimens from the patients and found their ADA2 activity was significantly reduced.
Taking these results together, the team concluded that mutations in CECR1 cause loss of ADA2 function that can result in polyarteritis nodosa vasculopathy with a highly varied clinical expression. ![]()
Study reveals potential target for mucositis, GVHD prevention

Results of preclinical research point to a possible way of preventing mucositis, graft-vs-host disease, and other disorders associated with epithelial permeability.
Investigators created a mouse model of mucositis and discovered that interleukin-1 (IL-1) beta, a protein secreted by the stressed mucosa, played an important role in the condition.
But inhibiting IL-1 beta alleviated mucositis. So the researchers speculated that targeting IL-1 beta might prevent mucositis in humans.
Naama Kanarek, a doctoral student at Hebrew University Hadassah Medical School in Jerusalem, and her colleagues described this research in PNAS.
The investigators began by generating a mouse model deficient in a gene encoding the enzyme beta-TrCP. They chose this enzyme because it’s a major regulator of inflammatory cascades.
The team found that beta-TrCP deletion in the gut caused mucosal DNA damage in the mice, mimicking the effects of chemotherapy and irradiation. Similar to human patients, a severe mucositis reaction occurred in mice that were genetically engineered to be beta-TrCP-deficient.
Tracing the pathological basis of the mouse mucositis revealed that the source of the problem was IL-1 beta. IL-1 beta opened the gut lining, allowing gut bacteria to penetrate and destroy the gut interior.
To confirm this finding, the researchers treated mice with an antibody neutralizing IL-1 beta prior to deleting beta-TrCP. They found this prevented the onset of mucositis.
Therefore, the team has proposed that IL-1 receptor agonists should be tested as mucositis prophylaxis in humans. An example is anakinra (Kineret), which is used to treat chronic inflammatory conditions, such as rheumatoid arthritis and Crohn’s disease.
The investigators believe such treatments might also be used to prevent graft-vs-host disease, burn injuries, head and neck trauma, and other disorders associated with
epithelial permeability. ![]()
Results of preclinical research point to a possible way of preventing mucositis, graft-vs-host disease, and other disorders associated with epithelial permeability.
Investigators created a mouse model of mucositis and discovered that interleukin-1 (IL-1) beta, a protein secreted by the stressed mucosa, played an important role in the condition.
But inhibiting IL-1 beta alleviated mucositis. So the researchers speculated that targeting IL-1 beta might prevent mucositis in humans.
Naama Kanarek, a doctoral student at Hebrew University Hadassah Medical School in Jerusalem, and her colleagues described this research in PNAS.
The investigators began by generating a mouse model deficient in a gene encoding the enzyme beta-TrCP. They chose this enzyme because it’s a major regulator of inflammatory cascades.
The team found that beta-TrCP deletion in the gut caused mucosal DNA damage in the mice, mimicking the effects of chemotherapy and irradiation. Similar to human patients, a severe mucositis reaction occurred in mice that were genetically engineered to be beta-TrCP-deficient.
Tracing the pathological basis of the mouse mucositis revealed that the source of the problem was IL-1 beta. IL-1 beta opened the gut lining, allowing gut bacteria to penetrate and destroy the gut interior.
To confirm this finding, the researchers treated mice with an antibody neutralizing IL-1 beta prior to deleting beta-TrCP. They found this prevented the onset of mucositis.
Therefore, the team has proposed that IL-1 receptor agonists should be tested as mucositis prophylaxis in humans. An example is anakinra (Kineret), which is used to treat chronic inflammatory conditions, such as rheumatoid arthritis and Crohn’s disease.
The investigators believe such treatments might also be used to prevent graft-vs-host disease, burn injuries, head and neck trauma, and other disorders associated with
epithelial permeability. ![]()
Results of preclinical research point to a possible way of preventing mucositis, graft-vs-host disease, and other disorders associated with epithelial permeability.
Investigators created a mouse model of mucositis and discovered that interleukin-1 (IL-1) beta, a protein secreted by the stressed mucosa, played an important role in the condition.
But inhibiting IL-1 beta alleviated mucositis. So the researchers speculated that targeting IL-1 beta might prevent mucositis in humans.
Naama Kanarek, a doctoral student at Hebrew University Hadassah Medical School in Jerusalem, and her colleagues described this research in PNAS.
The investigators began by generating a mouse model deficient in a gene encoding the enzyme beta-TrCP. They chose this enzyme because it’s a major regulator of inflammatory cascades.
The team found that beta-TrCP deletion in the gut caused mucosal DNA damage in the mice, mimicking the effects of chemotherapy and irradiation. Similar to human patients, a severe mucositis reaction occurred in mice that were genetically engineered to be beta-TrCP-deficient.
Tracing the pathological basis of the mouse mucositis revealed that the source of the problem was IL-1 beta. IL-1 beta opened the gut lining, allowing gut bacteria to penetrate and destroy the gut interior.
To confirm this finding, the researchers treated mice with an antibody neutralizing IL-1 beta prior to deleting beta-TrCP. They found this prevented the onset of mucositis.
Therefore, the team has proposed that IL-1 receptor agonists should be tested as mucositis prophylaxis in humans. An example is anakinra (Kineret), which is used to treat chronic inflammatory conditions, such as rheumatoid arthritis and Crohn’s disease.
The investigators believe such treatments might also be used to prevent graft-vs-host disease, burn injuries, head and neck trauma, and other disorders associated with
epithelial permeability. ![]()
Investigating the cause of infant leukemias
Infants who develop leukemia during the first year of life inherit a combination of genetic variations that can make them highly susceptible to the disease, according to a study published in Leukemia.
Results of whole-exome sequencing suggested that infants with leukemia inherited genetic variants from both parents that, by themselves, would not cause leukemia but, in combination, put the infants at high risk of developing the disease.
“We sequenced every single gene and found that infants with leukemia were born with an excess of damaging changes in genes known to be linked to leukemia,” said study author Todd Druley, MD, PhD, of Washington University School of Medicine in St Louis, Missouri.
“For each child, both parents carried a few harmful genetic variations in their DNA, and, just by chance, their child inherited all of these changes.”
However, it’s unlikely that the inherited variations alone cause leukemia, Dr Druley said. The infants likely needed to accumulate a few additional variations.
To uncover these findings, Dr Druley and his colleagues performed whole-exome sequencing in infants with acute myeloid leukemia (AML), infants with acute lymphoblastic leukemia (ALL), and the mothers of these children. The researchers used the process of elimination to determine a father’s contribution to a child’s DNA.
Among the 23 families studied, there was no history of pediatric cancers. As a comparison, the researchers also sequenced the DNA of 25 healthy children.
The team found the average amount of congenital coding variations was higher in infants with leukemia than in their mothers or the control subjects. The average total variants per exome was 1264.4 in infants with ALL, 1112.6 in their mothers, 2549.9 in infants with AML, 1225.0 in their mothers, and 582.8 in healthy controls.
The researchers then decided to home in on variants that were likely to impart a functional effect associated with leukemia. Using the COSMIC database, the team identified 126 ALL-associated genes and 655 AML-associated genes.
They found an average of 12.1 variants per ALL patient in the ALL-associated genes and 163.4 variants per AML patient in the AML-associated genes. There were 6.4 ALL-associated variants in the ALL patients’ mothers, 132.5 AML-associated variants in the AML patients’ mothers, 1.9 ALL variants in controls, and 27.5 AML variants in controls.
To prioritize genes that might be most relevant to infant leukemia, the researchers looked for compound heterozygous genes and the genes that were most commonly variant in all patients.
All of the infants with AML and 50% of the infants with ALL were compound heterozygotes for MLL3. Sixty-seven percent of AML patients were compound heterozygotes for RYR1 and FLG, and 50% of ALL patients were compound heterozygotes for RBMX.
The most variant (but not necessarily compound heterozygous) AML-associated genes in infants with AML were TTN, MLL3, and FLG. But the ALL-associated genes MDN1, SYNE1, and MLL2 were frequently variable in AML patients as well.
For infants with ALL, MDN1 was the most variable ALL-associated gene. But these infants also had frequent variations in the AML-associated genes TTN, RBMX, and MLL3.
Dr Druley and his colleagues plan to study these variations in more detail to understand how they contribute to infant leukemia development. ![]()
Infants who develop leukemia during the first year of life inherit a combination of genetic variations that can make them highly susceptible to the disease, according to a study published in Leukemia.
Results of whole-exome sequencing suggested that infants with leukemia inherited genetic variants from both parents that, by themselves, would not cause leukemia but, in combination, put the infants at high risk of developing the disease.
“We sequenced every single gene and found that infants with leukemia were born with an excess of damaging changes in genes known to be linked to leukemia,” said study author Todd Druley, MD, PhD, of Washington University School of Medicine in St Louis, Missouri.
“For each child, both parents carried a few harmful genetic variations in their DNA, and, just by chance, their child inherited all of these changes.”
However, it’s unlikely that the inherited variations alone cause leukemia, Dr Druley said. The infants likely needed to accumulate a few additional variations.
To uncover these findings, Dr Druley and his colleagues performed whole-exome sequencing in infants with acute myeloid leukemia (AML), infants with acute lymphoblastic leukemia (ALL), and the mothers of these children. The researchers used the process of elimination to determine a father’s contribution to a child’s DNA.
Among the 23 families studied, there was no history of pediatric cancers. As a comparison, the researchers also sequenced the DNA of 25 healthy children.
The team found the average amount of congenital coding variations was higher in infants with leukemia than in their mothers or the control subjects. The average total variants per exome was 1264.4 in infants with ALL, 1112.6 in their mothers, 2549.9 in infants with AML, 1225.0 in their mothers, and 582.8 in healthy controls.
The researchers then decided to home in on variants that were likely to impart a functional effect associated with leukemia. Using the COSMIC database, the team identified 126 ALL-associated genes and 655 AML-associated genes.
They found an average of 12.1 variants per ALL patient in the ALL-associated genes and 163.4 variants per AML patient in the AML-associated genes. There were 6.4 ALL-associated variants in the ALL patients’ mothers, 132.5 AML-associated variants in the AML patients’ mothers, 1.9 ALL variants in controls, and 27.5 AML variants in controls.
To prioritize genes that might be most relevant to infant leukemia, the researchers looked for compound heterozygous genes and the genes that were most commonly variant in all patients.
All of the infants with AML and 50% of the infants with ALL were compound heterozygotes for MLL3. Sixty-seven percent of AML patients were compound heterozygotes for RYR1 and FLG, and 50% of ALL patients were compound heterozygotes for RBMX.
The most variant (but not necessarily compound heterozygous) AML-associated genes in infants with AML were TTN, MLL3, and FLG. But the ALL-associated genes MDN1, SYNE1, and MLL2 were frequently variable in AML patients as well.
For infants with ALL, MDN1 was the most variable ALL-associated gene. But these infants also had frequent variations in the AML-associated genes TTN, RBMX, and MLL3.
Dr Druley and his colleagues plan to study these variations in more detail to understand how they contribute to infant leukemia development. ![]()
Infants who develop leukemia during the first year of life inherit a combination of genetic variations that can make them highly susceptible to the disease, according to a study published in Leukemia.
Results of whole-exome sequencing suggested that infants with leukemia inherited genetic variants from both parents that, by themselves, would not cause leukemia but, in combination, put the infants at high risk of developing the disease.
“We sequenced every single gene and found that infants with leukemia were born with an excess of damaging changes in genes known to be linked to leukemia,” said study author Todd Druley, MD, PhD, of Washington University School of Medicine in St Louis, Missouri.
“For each child, both parents carried a few harmful genetic variations in their DNA, and, just by chance, their child inherited all of these changes.”
However, it’s unlikely that the inherited variations alone cause leukemia, Dr Druley said. The infants likely needed to accumulate a few additional variations.
To uncover these findings, Dr Druley and his colleagues performed whole-exome sequencing in infants with acute myeloid leukemia (AML), infants with acute lymphoblastic leukemia (ALL), and the mothers of these children. The researchers used the process of elimination to determine a father’s contribution to a child’s DNA.
Among the 23 families studied, there was no history of pediatric cancers. As a comparison, the researchers also sequenced the DNA of 25 healthy children.
The team found the average amount of congenital coding variations was higher in infants with leukemia than in their mothers or the control subjects. The average total variants per exome was 1264.4 in infants with ALL, 1112.6 in their mothers, 2549.9 in infants with AML, 1225.0 in their mothers, and 582.8 in healthy controls.
The researchers then decided to home in on variants that were likely to impart a functional effect associated with leukemia. Using the COSMIC database, the team identified 126 ALL-associated genes and 655 AML-associated genes.
They found an average of 12.1 variants per ALL patient in the ALL-associated genes and 163.4 variants per AML patient in the AML-associated genes. There were 6.4 ALL-associated variants in the ALL patients’ mothers, 132.5 AML-associated variants in the AML patients’ mothers, 1.9 ALL variants in controls, and 27.5 AML variants in controls.
To prioritize genes that might be most relevant to infant leukemia, the researchers looked for compound heterozygous genes and the genes that were most commonly variant in all patients.
All of the infants with AML and 50% of the infants with ALL were compound heterozygotes for MLL3. Sixty-seven percent of AML patients were compound heterozygotes for RYR1 and FLG, and 50% of ALL patients were compound heterozygotes for RBMX.
The most variant (but not necessarily compound heterozygous) AML-associated genes in infants with AML were TTN, MLL3, and FLG. But the ALL-associated genes MDN1, SYNE1, and MLL2 were frequently variable in AML patients as well.
For infants with ALL, MDN1 was the most variable ALL-associated gene. But these infants also had frequent variations in the AML-associated genes TTN, RBMX, and MLL3.
Dr Druley and his colleagues plan to study these variations in more detail to understand how they contribute to infant leukemia development. ![]()
Malaria maps show progress, room for improvement

Credit: CDC
Malaria prevalence maps indicate that, in 2010, nearly 184 million Africans were still living in areas where there is a high risk of contracting malaria, despite a decade of efforts to control the spread of the disease.
The maps showed that 40 African countries experienced reductions in childhood malaria transmission between 2000 and 2010.
Despite this progress, 57% of the population in malaria-endemic countries continued to live in areas of moderate to intense malaria transmission, with infection rates higher than 10%.
These findings are published in The Lancet.
Researchers compiled data from a collection of 26,746 community-based surveys of Plasmodium falciparum prevalence. The surveys covered 3,575,418 person observations from 44 malaria-endemic countries and territories in Africa since 1980.
“Health information systems in many African countries are weak, and it has been difficult to reliably estimate how many people get sick or die of malaria,” said study author Abdisalan Mohamed Noor, PhD, of the Kenya Medical Research Institute-Wellcome Trust Research Programme in Nairobi and the University of Oxford in the UK.
“The population surveys we used in this study are a more reliable indicator for tracking, and we hope our study will help countries assess their progress and adapt their strategies for more effective malaria control.”
Using model-based geostatistics, Dr Noor and his colleagues estimated the proportion of the population, aged 2 to 10 years old, infected with different levels of P falciparum across Africa in 2000 and 2010.
The researchers wanted to evaluate the effects of the Roll Back Malaria Partnership, which was launched in 2000 and resulted in a large increase in investments targeting malaria control.
The team found that the number of people living in high-risk areas, where more than 50% of the population is likely to carry infections, fell from 218.6 million in 2000 to 183.5 million in 2010—a 16% decrease.
But the population living in areas where the risk of infection is considered moderate to high grew from 178.6 million to 280.1 million—a 57% increase.
And the population living in areas where risk is regarded as very low grew from 78.2 million to 128.2 million—a 64% increase.
The researchers also discovered that 10 countries harbor 87% of the population remaining at high risk of malaria transmission. These countries are Guinea, Togo, Mali, Mozambique, Burkina Faso, Ghana, Côte d’Ivoire, Uganda, Nigeria, and the Democratic Republic of Congo.
On the other hand, the team noted that 7 countries have levels of malaria transmission so low that eliminating the disease is a realistic goal. These countries are Cape Verde, Eritrea, South Africa, Ethiopia, Swaziland, Djibouti, and Mayotte.
“The results of our analysis are pause for thought,” said study author Robert Snow, PhD, also of the Kenya Medical Research Institute-Wellcome Trust Research Programme and the University of Oxford.
“On the one hand, it’s a glass half full, with several countries showing significant reductions in malaria transmission. And on the other, it’s a glass half empty, where, despite a decade of massive investment in malaria control, the populations living in several African countries are as likely to be infected with malaria in 2000 as they were 10 years later.” ![]()
Credit: CDC
Malaria prevalence maps indicate that, in 2010, nearly 184 million Africans were still living in areas where there is a high risk of contracting malaria, despite a decade of efforts to control the spread of the disease.
The maps showed that 40 African countries experienced reductions in childhood malaria transmission between 2000 and 2010.
Despite this progress, 57% of the population in malaria-endemic countries continued to live in areas of moderate to intense malaria transmission, with infection rates higher than 10%.
These findings are published in The Lancet.
Researchers compiled data from a collection of 26,746 community-based surveys of Plasmodium falciparum prevalence. The surveys covered 3,575,418 person observations from 44 malaria-endemic countries and territories in Africa since 1980.
“Health information systems in many African countries are weak, and it has been difficult to reliably estimate how many people get sick or die of malaria,” said study author Abdisalan Mohamed Noor, PhD, of the Kenya Medical Research Institute-Wellcome Trust Research Programme in Nairobi and the University of Oxford in the UK.
“The population surveys we used in this study are a more reliable indicator for tracking, and we hope our study will help countries assess their progress and adapt their strategies for more effective malaria control.”
Using model-based geostatistics, Dr Noor and his colleagues estimated the proportion of the population, aged 2 to 10 years old, infected with different levels of P falciparum across Africa in 2000 and 2010.
The researchers wanted to evaluate the effects of the Roll Back Malaria Partnership, which was launched in 2000 and resulted in a large increase in investments targeting malaria control.
The team found that the number of people living in high-risk areas, where more than 50% of the population is likely to carry infections, fell from 218.6 million in 2000 to 183.5 million in 2010—a 16% decrease.
But the population living in areas where the risk of infection is considered moderate to high grew from 178.6 million to 280.1 million—a 57% increase.
And the population living in areas where risk is regarded as very low grew from 78.2 million to 128.2 million—a 64% increase.
The researchers also discovered that 10 countries harbor 87% of the population remaining at high risk of malaria transmission. These countries are Guinea, Togo, Mali, Mozambique, Burkina Faso, Ghana, Côte d’Ivoire, Uganda, Nigeria, and the Democratic Republic of Congo.
On the other hand, the team noted that 7 countries have levels of malaria transmission so low that eliminating the disease is a realistic goal. These countries are Cape Verde, Eritrea, South Africa, Ethiopia, Swaziland, Djibouti, and Mayotte.
“The results of our analysis are pause for thought,” said study author Robert Snow, PhD, also of the Kenya Medical Research Institute-Wellcome Trust Research Programme and the University of Oxford.
“On the one hand, it’s a glass half full, with several countries showing significant reductions in malaria transmission. And on the other, it’s a glass half empty, where, despite a decade of massive investment in malaria control, the populations living in several African countries are as likely to be infected with malaria in 2000 as they were 10 years later.” ![]()
Credit: CDC
Malaria prevalence maps indicate that, in 2010, nearly 184 million Africans were still living in areas where there is a high risk of contracting malaria, despite a decade of efforts to control the spread of the disease.
The maps showed that 40 African countries experienced reductions in childhood malaria transmission between 2000 and 2010.
Despite this progress, 57% of the population in malaria-endemic countries continued to live in areas of moderate to intense malaria transmission, with infection rates higher than 10%.
These findings are published in The Lancet.
Researchers compiled data from a collection of 26,746 community-based surveys of Plasmodium falciparum prevalence. The surveys covered 3,575,418 person observations from 44 malaria-endemic countries and territories in Africa since 1980.
“Health information systems in many African countries are weak, and it has been difficult to reliably estimate how many people get sick or die of malaria,” said study author Abdisalan Mohamed Noor, PhD, of the Kenya Medical Research Institute-Wellcome Trust Research Programme in Nairobi and the University of Oxford in the UK.
“The population surveys we used in this study are a more reliable indicator for tracking, and we hope our study will help countries assess their progress and adapt their strategies for more effective malaria control.”
Using model-based geostatistics, Dr Noor and his colleagues estimated the proportion of the population, aged 2 to 10 years old, infected with different levels of P falciparum across Africa in 2000 and 2010.
The researchers wanted to evaluate the effects of the Roll Back Malaria Partnership, which was launched in 2000 and resulted in a large increase in investments targeting malaria control.
The team found that the number of people living in high-risk areas, where more than 50% of the population is likely to carry infections, fell from 218.6 million in 2000 to 183.5 million in 2010—a 16% decrease.
But the population living in areas where the risk of infection is considered moderate to high grew from 178.6 million to 280.1 million—a 57% increase.
And the population living in areas where risk is regarded as very low grew from 78.2 million to 128.2 million—a 64% increase.
The researchers also discovered that 10 countries harbor 87% of the population remaining at high risk of malaria transmission. These countries are Guinea, Togo, Mali, Mozambique, Burkina Faso, Ghana, Côte d’Ivoire, Uganda, Nigeria, and the Democratic Republic of Congo.
On the other hand, the team noted that 7 countries have levels of malaria transmission so low that eliminating the disease is a realistic goal. These countries are Cape Verde, Eritrea, South Africa, Ethiopia, Swaziland, Djibouti, and Mayotte.
“The results of our analysis are pause for thought,” said study author Robert Snow, PhD, also of the Kenya Medical Research Institute-Wellcome Trust Research Programme and the University of Oxford.
“On the one hand, it’s a glass half full, with several countries showing significant reductions in malaria transmission. And on the other, it’s a glass half empty, where, despite a decade of massive investment in malaria control, the populations living in several African countries are as likely to be infected with malaria in 2000 as they were 10 years later.” ![]()
How Bcl-2 helps cancer cells survive treatment

Researchers believe they’ve discovered how the Bcl-2 protein helps leukemia and lymphoma cells survive anticancer treatment.
The team found that Bcl-2 alters the level of calcium ions in cancer cells, and this promotes the cells’ survival.
The group thinks these findings, published in PNAS, could help spur the development of drugs that effectively inhibit Bcl-2 and produce better outcomes for cancer patients.
“Since 1993, our team has been conducting research on key mechanisms by which the protein Bcl-2 keeps cancer cells alive,” said study author Clark W. Distelhorst, MD, of Case Western Reserve School of Medicine in Cleveland, Ohio.
“Now, for the first time, we have evidence of how Bcl-2 is promoting abnormally long survival of the cancer cells by regulating calcium levels within cells, and [we] will use the discovery and data to deliver therapies designed to attack the Bcl-2 protein and inhibit its impact.”
More than a decade ago, researchers in Dr Distelhorst’s lab discovered that Bcl-2 binds to the inositol 1,4,5-trisphosphate receptor (InsP3R) channel and regulates the release of calcium ions.
In the current study, the team found that when Bcl-2 binds to the InsP3R channel, it initiates a complex feedback mechanism that blocks the release of calcium ions intended to induce cell death. Instead of dying, the cancer cells continue to proliferate.
Specifically, the researchers discovered that Bcl-2 interacts with the Ca2+-activated protein phosphatase calcineurin (CaN) and dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a CaN-regulated inhibitor of protein phosphatase 1.
Bcl-2 docks DARPP-32 and CaN on the InsP3R, creating a negative feedback loop that responds to InsP3R-mediated Ca2+ release by inhibiting InsP3R phosphorylation at Ser1755. And this prevents the excessive Ca2+ elevation that induces cell death.
The team theorized that cancer cells overexpressing Bcl-2 may exploit this mechanism to prevent apoptosis. And experiments in chronic lymphocytic leukemia cells appeared to confirm this theory.
The researchers treated the cells with the peptide TAT-IDPDD/AA, which inhibits Bcl-2–InsP3R interaction. This increased P-Ser1755 InsP3R-1 levels and elevated Ca2+, which induced apoptosis.
“We have recognized for decades that cancer cells grow and forget to die,” said Stanton Gerson, MD, director of the Case Comprehensive Cancer Center, who was not involved in this study.
“[N]ow, we understand why. I predict that this work will focus the discovery of new drugs against the Bcl-2-calcium-flow system.” ![]()
Researchers believe they’ve discovered how the Bcl-2 protein helps leukemia and lymphoma cells survive anticancer treatment.
The team found that Bcl-2 alters the level of calcium ions in cancer cells, and this promotes the cells’ survival.
The group thinks these findings, published in PNAS, could help spur the development of drugs that effectively inhibit Bcl-2 and produce better outcomes for cancer patients.
“Since 1993, our team has been conducting research on key mechanisms by which the protein Bcl-2 keeps cancer cells alive,” said study author Clark W. Distelhorst, MD, of Case Western Reserve School of Medicine in Cleveland, Ohio.
“Now, for the first time, we have evidence of how Bcl-2 is promoting abnormally long survival of the cancer cells by regulating calcium levels within cells, and [we] will use the discovery and data to deliver therapies designed to attack the Bcl-2 protein and inhibit its impact.”
More than a decade ago, researchers in Dr Distelhorst’s lab discovered that Bcl-2 binds to the inositol 1,4,5-trisphosphate receptor (InsP3R) channel and regulates the release of calcium ions.
In the current study, the team found that when Bcl-2 binds to the InsP3R channel, it initiates a complex feedback mechanism that blocks the release of calcium ions intended to induce cell death. Instead of dying, the cancer cells continue to proliferate.
Specifically, the researchers discovered that Bcl-2 interacts with the Ca2+-activated protein phosphatase calcineurin (CaN) and dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a CaN-regulated inhibitor of protein phosphatase 1.
Bcl-2 docks DARPP-32 and CaN on the InsP3R, creating a negative feedback loop that responds to InsP3R-mediated Ca2+ release by inhibiting InsP3R phosphorylation at Ser1755. And this prevents the excessive Ca2+ elevation that induces cell death.
The team theorized that cancer cells overexpressing Bcl-2 may exploit this mechanism to prevent apoptosis. And experiments in chronic lymphocytic leukemia cells appeared to confirm this theory.
The researchers treated the cells with the peptide TAT-IDPDD/AA, which inhibits Bcl-2–InsP3R interaction. This increased P-Ser1755 InsP3R-1 levels and elevated Ca2+, which induced apoptosis.
“We have recognized for decades that cancer cells grow and forget to die,” said Stanton Gerson, MD, director of the Case Comprehensive Cancer Center, who was not involved in this study.
“[N]ow, we understand why. I predict that this work will focus the discovery of new drugs against the Bcl-2-calcium-flow system.” ![]()
Researchers believe they’ve discovered how the Bcl-2 protein helps leukemia and lymphoma cells survive anticancer treatment.
The team found that Bcl-2 alters the level of calcium ions in cancer cells, and this promotes the cells’ survival.
The group thinks these findings, published in PNAS, could help spur the development of drugs that effectively inhibit Bcl-2 and produce better outcomes for cancer patients.
“Since 1993, our team has been conducting research on key mechanisms by which the protein Bcl-2 keeps cancer cells alive,” said study author Clark W. Distelhorst, MD, of Case Western Reserve School of Medicine in Cleveland, Ohio.
“Now, for the first time, we have evidence of how Bcl-2 is promoting abnormally long survival of the cancer cells by regulating calcium levels within cells, and [we] will use the discovery and data to deliver therapies designed to attack the Bcl-2 protein and inhibit its impact.”
More than a decade ago, researchers in Dr Distelhorst’s lab discovered that Bcl-2 binds to the inositol 1,4,5-trisphosphate receptor (InsP3R) channel and regulates the release of calcium ions.
In the current study, the team found that when Bcl-2 binds to the InsP3R channel, it initiates a complex feedback mechanism that blocks the release of calcium ions intended to induce cell death. Instead of dying, the cancer cells continue to proliferate.
Specifically, the researchers discovered that Bcl-2 interacts with the Ca2+-activated protein phosphatase calcineurin (CaN) and dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), a CaN-regulated inhibitor of protein phosphatase 1.
Bcl-2 docks DARPP-32 and CaN on the InsP3R, creating a negative feedback loop that responds to InsP3R-mediated Ca2+ release by inhibiting InsP3R phosphorylation at Ser1755. And this prevents the excessive Ca2+ elevation that induces cell death.
The team theorized that cancer cells overexpressing Bcl-2 may exploit this mechanism to prevent apoptosis. And experiments in chronic lymphocytic leukemia cells appeared to confirm this theory.
The researchers treated the cells with the peptide TAT-IDPDD/AA, which inhibits Bcl-2–InsP3R interaction. This increased P-Ser1755 InsP3R-1 levels and elevated Ca2+, which induced apoptosis.
“We have recognized for decades that cancer cells grow and forget to die,” said Stanton Gerson, MD, director of the Case Comprehensive Cancer Center, who was not involved in this study.
“[N]ow, we understand why. I predict that this work will focus the discovery of new drugs against the Bcl-2-calcium-flow system.” ![]()
Platelets, iron may affect stroke risk in HHT patients

Credit: Andre E.X. Brown
New research suggests patients with hereditary hemorrhagic telangiectasia (HHT) have an increased risk of ischemic stroke if they are iron deficient, and this may be due to enhanced platelet aggregation.
In the last few years, several studies have shown that iron deficiency may be a risk factor for ischemic stroke, but exactly how this occurs has been unclear.
Previous research also revealed a possible link between iron deficiency and platelet aggregation, but it has been largely overlooked, until now.
Claire Shovlin, PhD, of Imperial College London in the UK, and her colleagues found that HHT patients who are iron deficient have both an increased risk of stroke and enhanced aggregation of circulating platelets.
The researchers reported these findings in PLOS ONE.
The team had studied 497 HHT patients who had enlarged blood vessels in the lungs known as pulmonary arteriovenous malformations.
Normally, the lungs’ blood vessels act as a filter to remove small clots before blood enters arteries. In patients with pulmonary arteriovenous malformations, blood is able to bypass the filter, so small blood clots can travel to the brain.
Dr Shovlin and her colleagues found that patients with iron deficiency had a greater risk of stroke than patients with normal iron levels. Even moderately low iron levels, around 6 μmol/L, approximately doubled the risk of stroke when compared with levels in the middle of the normal range (7 to 27 μmol/L).
The researchers evaluated platelet activity in 15 patients, dividing them into 2 groups according to serum ferritin. Iron-deficient patients (n=7) had ferritin levels ranging from 2 μg/L to 17 μg/L. Control subjects (n=8) had ferritin levels of 24 μg/L to 98 μg/L. (None of the patients had levels between 17μg/L and 24 μg/L.)
The team found that ADP induced similar, dose-dependent platelet aggregation in iron-deficient patients and control subjects. But iron-deficient patients exhibited enhanced total aggregation to 5HT over a 5-minute period. And the iron-deficient group displayed faster rates of aggregation in response to 5HT.
“Since platelets in the blood stick together more if you are short of iron, we think this may explain why being short of iron can lead to strokes, though much more research will be needed to prove this link,” Dr Shovlin said.
“The next step is to test whether we can reduce high-risk patients’ chances of having a stroke by treating their iron deficiency. We will be able to look at whether their platelets become less sticky.”
“There are many additional steps from a clot blocking a blood vessel to the final stroke developing, so it is still unclear just how important sticky platelets are to the overall process. We would certainly encourage more studies to investigate this link.” ![]()
Credit: Andre E.X. Brown
New research suggests patients with hereditary hemorrhagic telangiectasia (HHT) have an increased risk of ischemic stroke if they are iron deficient, and this may be due to enhanced platelet aggregation.
In the last few years, several studies have shown that iron deficiency may be a risk factor for ischemic stroke, but exactly how this occurs has been unclear.
Previous research also revealed a possible link between iron deficiency and platelet aggregation, but it has been largely overlooked, until now.
Claire Shovlin, PhD, of Imperial College London in the UK, and her colleagues found that HHT patients who are iron deficient have both an increased risk of stroke and enhanced aggregation of circulating platelets.
The researchers reported these findings in PLOS ONE.
The team had studied 497 HHT patients who had enlarged blood vessels in the lungs known as pulmonary arteriovenous malformations.
Normally, the lungs’ blood vessels act as a filter to remove small clots before blood enters arteries. In patients with pulmonary arteriovenous malformations, blood is able to bypass the filter, so small blood clots can travel to the brain.
Dr Shovlin and her colleagues found that patients with iron deficiency had a greater risk of stroke than patients with normal iron levels. Even moderately low iron levels, around 6 μmol/L, approximately doubled the risk of stroke when compared with levels in the middle of the normal range (7 to 27 μmol/L).
The researchers evaluated platelet activity in 15 patients, dividing them into 2 groups according to serum ferritin. Iron-deficient patients (n=7) had ferritin levels ranging from 2 μg/L to 17 μg/L. Control subjects (n=8) had ferritin levels of 24 μg/L to 98 μg/L. (None of the patients had levels between 17μg/L and 24 μg/L.)
The team found that ADP induced similar, dose-dependent platelet aggregation in iron-deficient patients and control subjects. But iron-deficient patients exhibited enhanced total aggregation to 5HT over a 5-minute period. And the iron-deficient group displayed faster rates of aggregation in response to 5HT.
“Since platelets in the blood stick together more if you are short of iron, we think this may explain why being short of iron can lead to strokes, though much more research will be needed to prove this link,” Dr Shovlin said.
“The next step is to test whether we can reduce high-risk patients’ chances of having a stroke by treating their iron deficiency. We will be able to look at whether their platelets become less sticky.”
“There are many additional steps from a clot blocking a blood vessel to the final stroke developing, so it is still unclear just how important sticky platelets are to the overall process. We would certainly encourage more studies to investigate this link.” ![]()
Credit: Andre E.X. Brown
New research suggests patients with hereditary hemorrhagic telangiectasia (HHT) have an increased risk of ischemic stroke if they are iron deficient, and this may be due to enhanced platelet aggregation.
In the last few years, several studies have shown that iron deficiency may be a risk factor for ischemic stroke, but exactly how this occurs has been unclear.
Previous research also revealed a possible link between iron deficiency and platelet aggregation, but it has been largely overlooked, until now.
Claire Shovlin, PhD, of Imperial College London in the UK, and her colleagues found that HHT patients who are iron deficient have both an increased risk of stroke and enhanced aggregation of circulating platelets.
The researchers reported these findings in PLOS ONE.
The team had studied 497 HHT patients who had enlarged blood vessels in the lungs known as pulmonary arteriovenous malformations.
Normally, the lungs’ blood vessels act as a filter to remove small clots before blood enters arteries. In patients with pulmonary arteriovenous malformations, blood is able to bypass the filter, so small blood clots can travel to the brain.
Dr Shovlin and her colleagues found that patients with iron deficiency had a greater risk of stroke than patients with normal iron levels. Even moderately low iron levels, around 6 μmol/L, approximately doubled the risk of stroke when compared with levels in the middle of the normal range (7 to 27 μmol/L).
The researchers evaluated platelet activity in 15 patients, dividing them into 2 groups according to serum ferritin. Iron-deficient patients (n=7) had ferritin levels ranging from 2 μg/L to 17 μg/L. Control subjects (n=8) had ferritin levels of 24 μg/L to 98 μg/L. (None of the patients had levels between 17μg/L and 24 μg/L.)
The team found that ADP induced similar, dose-dependent platelet aggregation in iron-deficient patients and control subjects. But iron-deficient patients exhibited enhanced total aggregation to 5HT over a 5-minute period. And the iron-deficient group displayed faster rates of aggregation in response to 5HT.
“Since platelets in the blood stick together more if you are short of iron, we think this may explain why being short of iron can lead to strokes, though much more research will be needed to prove this link,” Dr Shovlin said.
“The next step is to test whether we can reduce high-risk patients’ chances of having a stroke by treating their iron deficiency. We will be able to look at whether their platelets become less sticky.”
“There are many additional steps from a clot blocking a blood vessel to the final stroke developing, so it is still unclear just how important sticky platelets are to the overall process. We would certainly encourage more studies to investigate this link.”
Inhibitor shows preclinical promise in leukemia, MM

Credit: VCU
The experimental drug dinaciclib could potentially improve the treatment of multiple myeloma (MM) and myeloid leukemias, according to preclinical research published in Molecular Cancer Therapeutics.
The study showed that dinaciclib disrupts a cell survival mechanism known as the unfolded protein response (UPR).
And without the UPR, MM and myeloid leukemia cells were unable to combat damage caused by anticancer agents that induce stress in the endoplasmic reticulum (ER).
“Although dinaciclib has shown promising preclinical activity against a variety of tumor cells and is currently undergoing phase 1/2 clinical trials in several malignancies, the mechanisms responsible for its antitumor activity are not fully understood,” said study author Steven Grant, MD, of the Virginia Commonwealth University Massey Cancer Center.
“Our research highlights a potentially new mechanism of dinaciclib action and raises the possibility that this agent could be a useful addition to current multiple myeloma and myeloid leukemia therapies.”
Dinaciclib is a cyclin-dependent kinase (CDK) inhibitor. CDKs are overactive in many cancers, which results in unregulated proliferation of cancer cells.
Observations from this study suggest that 2 specific CDKs, CDK1 and CDK5, play key roles in regulating the UPR by helping to control the production and accumulation of X-box binding pretein-1 (XBP-1). The spliced form of XBP-1 (XBP-1s) helps regulate the expression of genes critical to cellular stress responses.
External stressors, including certain anticancer agents, can cause misfolded proteins to accumulate in the ER. These stressors can also cause XBP-1s to accumulate in the cell’s nucleus, which promotes the UPR and helps cells withstand the damaging effects of misfolded proteins.
This research showed that dinaciclib, by interfering with UPR activation, caused MM and myeloid leukemia cells to initiate apoptosis when exposed to thapsigargin and tunicamycin—2 agents that induce ER stress.
And single-agent dinaciclib treatment significantly decreased tumor growth in mouse models of MM, when compared to vehicle control.
“These findings build on a long history of work in our laboratory investigating mechanisms by which cancer cells respond to environmental stresses,” Dr Grant said.
“We intend to continue investigating ways in which dinaciclib and other CDK inhibitors might be used to disrupt the UPR and potentially improve the effectiveness of certain agents for the treatment of multiple myeloma or myeloid leukemia.”
Credit: VCU
The experimental drug dinaciclib could potentially improve the treatment of multiple myeloma (MM) and myeloid leukemias, according to preclinical research published in Molecular Cancer Therapeutics.
The study showed that dinaciclib disrupts a cell survival mechanism known as the unfolded protein response (UPR).
And without the UPR, MM and myeloid leukemia cells were unable to combat damage caused by anticancer agents that induce stress in the endoplasmic reticulum (ER).
“Although dinaciclib has shown promising preclinical activity against a variety of tumor cells and is currently undergoing phase 1/2 clinical trials in several malignancies, the mechanisms responsible for its antitumor activity are not fully understood,” said study author Steven Grant, MD, of the Virginia Commonwealth University Massey Cancer Center.
“Our research highlights a potentially new mechanism of dinaciclib action and raises the possibility that this agent could be a useful addition to current multiple myeloma and myeloid leukemia therapies.”
Dinaciclib is a cyclin-dependent kinase (CDK) inhibitor. CDKs are overactive in many cancers, which results in unregulated proliferation of cancer cells.
Observations from this study suggest that 2 specific CDKs, CDK1 and CDK5, play key roles in regulating the UPR by helping to control the production and accumulation of X-box binding pretein-1 (XBP-1). The spliced form of XBP-1 (XBP-1s) helps regulate the expression of genes critical to cellular stress responses.
External stressors, including certain anticancer agents, can cause misfolded proteins to accumulate in the ER. These stressors can also cause XBP-1s to accumulate in the cell’s nucleus, which promotes the UPR and helps cells withstand the damaging effects of misfolded proteins.
This research showed that dinaciclib, by interfering with UPR activation, caused MM and myeloid leukemia cells to initiate apoptosis when exposed to thapsigargin and tunicamycin—2 agents that induce ER stress.
And single-agent dinaciclib treatment significantly decreased tumor growth in mouse models of MM, when compared to vehicle control.
“These findings build on a long history of work in our laboratory investigating mechanisms by which cancer cells respond to environmental stresses,” Dr Grant said.
“We intend to continue investigating ways in which dinaciclib and other CDK inhibitors might be used to disrupt the UPR and potentially improve the effectiveness of certain agents for the treatment of multiple myeloma or myeloid leukemia.”
Credit: VCU
The experimental drug dinaciclib could potentially improve the treatment of multiple myeloma (MM) and myeloid leukemias, according to preclinical research published in Molecular Cancer Therapeutics.
The study showed that dinaciclib disrupts a cell survival mechanism known as the unfolded protein response (UPR).
And without the UPR, MM and myeloid leukemia cells were unable to combat damage caused by anticancer agents that induce stress in the endoplasmic reticulum (ER).
“Although dinaciclib has shown promising preclinical activity against a variety of tumor cells and is currently undergoing phase 1/2 clinical trials in several malignancies, the mechanisms responsible for its antitumor activity are not fully understood,” said study author Steven Grant, MD, of the Virginia Commonwealth University Massey Cancer Center.
“Our research highlights a potentially new mechanism of dinaciclib action and raises the possibility that this agent could be a useful addition to current multiple myeloma and myeloid leukemia therapies.”
Dinaciclib is a cyclin-dependent kinase (CDK) inhibitor. CDKs are overactive in many cancers, which results in unregulated proliferation of cancer cells.
Observations from this study suggest that 2 specific CDKs, CDK1 and CDK5, play key roles in regulating the UPR by helping to control the production and accumulation of X-box binding pretein-1 (XBP-1). The spliced form of XBP-1 (XBP-1s) helps regulate the expression of genes critical to cellular stress responses.
External stressors, including certain anticancer agents, can cause misfolded proteins to accumulate in the ER. These stressors can also cause XBP-1s to accumulate in the cell’s nucleus, which promotes the UPR and helps cells withstand the damaging effects of misfolded proteins.
This research showed that dinaciclib, by interfering with UPR activation, caused MM and myeloid leukemia cells to initiate apoptosis when exposed to thapsigargin and tunicamycin—2 agents that induce ER stress.
And single-agent dinaciclib treatment significantly decreased tumor growth in mouse models of MM, when compared to vehicle control.
“These findings build on a long history of work in our laboratory investigating mechanisms by which cancer cells respond to environmental stresses,” Dr Grant said.
“We intend to continue investigating ways in which dinaciclib and other CDK inhibitors might be used to disrupt the UPR and potentially improve the effectiveness of certain agents for the treatment of multiple myeloma or myeloid leukemia.”
FDA program aims to expedite drug importation

Credit: Steven Harbour
The US Food and Drug Administration (FDA) has launched a program that allows certain companies to expedite the importation of drugs and drug ingredients.
Thirteen companies have been selected to take part in this 2-year program, called the Secure Supply Chain Pilot Program.
As the companies meet certain criteria, they are eligible to receive expedited entry for up to 5 drug products. These products can enter the US after electronic screening without undergoing human examination.
The FDA said its goal with this program is to allow the agency to focus its imports surveillance resources on preventing the entry of drugs that are most likely to compromise the quality and safety of the US drug supply.
The companies that have been accepted into the program are:
- AbbVie Inc.
- Allergan, Inc.
- Astellas U.S. Technologies, Inc.
- Bristol-Myers Squibb Company
- Celgene Corporation
- GE Healthcare Inc.
- GlaxoSmithKline LLC
- Merck Sharp & Dohme Corporation
- Mylan Pharmaceuticals Inc.
- Novartis Pharmaceuticals Corporation
- Pfizer, Inc.
- Teva Pharmaceutcials USA, Inc.
- Watson Laboratories, Inc.
Each of these companies met the participation conditions, including:
- Committing to comply with requirements of the Food, Drug, and Cosmetics Act (FDCA)
- Having a validated, secure supply chain protocol per the US Customs and Border Protection’s Customs-Trade Partnership Against Terrorism (C-TPAT) program
- Having a plan in place to quickly correct potential problems the FDA identifies regarding importation of specific products
- Having effective recall and corrective action plans in place
- Maintaining control over their drugs from the time of manufacture abroad through entry into the US.
Over the next 2 years, the FDA will evaluate whether this program enhances imported drug compliance with FDA regulations and the security of the drug supply chain. If the FDA deems the program effective, a more permanent program may be established and possibly extended to additional companies.
For more information, see the FDA’s notice about the program, published in the Federal Register last August.
Credit: Steven Harbour
The US Food and Drug Administration (FDA) has launched a program that allows certain companies to expedite the importation of drugs and drug ingredients.
Thirteen companies have been selected to take part in this 2-year program, called the Secure Supply Chain Pilot Program.
As the companies meet certain criteria, they are eligible to receive expedited entry for up to 5 drug products. These products can enter the US after electronic screening without undergoing human examination.
The FDA said its goal with this program is to allow the agency to focus its imports surveillance resources on preventing the entry of drugs that are most likely to compromise the quality and safety of the US drug supply.
The companies that have been accepted into the program are:
- AbbVie Inc.
- Allergan, Inc.
- Astellas U.S. Technologies, Inc.
- Bristol-Myers Squibb Company
- Celgene Corporation
- GE Healthcare Inc.
- GlaxoSmithKline LLC
- Merck Sharp & Dohme Corporation
- Mylan Pharmaceuticals Inc.
- Novartis Pharmaceuticals Corporation
- Pfizer, Inc.
- Teva Pharmaceutcials USA, Inc.
- Watson Laboratories, Inc.
Each of these companies met the participation conditions, including:
- Committing to comply with requirements of the Food, Drug, and Cosmetics Act (FDCA)
- Having a validated, secure supply chain protocol per the US Customs and Border Protection’s Customs-Trade Partnership Against Terrorism (C-TPAT) program
- Having a plan in place to quickly correct potential problems the FDA identifies regarding importation of specific products
- Having effective recall and corrective action plans in place
- Maintaining control over their drugs from the time of manufacture abroad through entry into the US.
Over the next 2 years, the FDA will evaluate whether this program enhances imported drug compliance with FDA regulations and the security of the drug supply chain. If the FDA deems the program effective, a more permanent program may be established and possibly extended to additional companies.
For more information, see the FDA’s notice about the program, published in the Federal Register last August.
Credit: Steven Harbour
The US Food and Drug Administration (FDA) has launched a program that allows certain companies to expedite the importation of drugs and drug ingredients.
Thirteen companies have been selected to take part in this 2-year program, called the Secure Supply Chain Pilot Program.
As the companies meet certain criteria, they are eligible to receive expedited entry for up to 5 drug products. These products can enter the US after electronic screening without undergoing human examination.
The FDA said its goal with this program is to allow the agency to focus its imports surveillance resources on preventing the entry of drugs that are most likely to compromise the quality and safety of the US drug supply.
The companies that have been accepted into the program are:
- AbbVie Inc.
- Allergan, Inc.
- Astellas U.S. Technologies, Inc.
- Bristol-Myers Squibb Company
- Celgene Corporation
- GE Healthcare Inc.
- GlaxoSmithKline LLC
- Merck Sharp & Dohme Corporation
- Mylan Pharmaceuticals Inc.
- Novartis Pharmaceuticals Corporation
- Pfizer, Inc.
- Teva Pharmaceutcials USA, Inc.
- Watson Laboratories, Inc.
Each of these companies met the participation conditions, including:
- Committing to comply with requirements of the Food, Drug, and Cosmetics Act (FDCA)
- Having a validated, secure supply chain protocol per the US Customs and Border Protection’s Customs-Trade Partnership Against Terrorism (C-TPAT) program
- Having a plan in place to quickly correct potential problems the FDA identifies regarding importation of specific products
- Having effective recall and corrective action plans in place
- Maintaining control over their drugs from the time of manufacture abroad through entry into the US.
Over the next 2 years, the FDA will evaluate whether this program enhances imported drug compliance with FDA regulations and the security of the drug supply chain. If the FDA deems the program effective, a more permanent program may be established and possibly extended to additional companies.
For more information, see the FDA’s notice about the program, published in the Federal Register last August.
CAR T-cell therapy: The good and the bad

Credit: MSKCC
Several studies have shown that infusions of T cells modified with chimeric antigen receptors (CARs) can elicit complete responses in leukemia patients who have run out of treatment options.
However, the therapy also puts patients at risk of developing cytokine release syndrome (CRS).
With updated research, investigators have again shown that CAR T cells can produce complete responses in patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL), thereby allowing them to receive allogeneic stem cell transplant (allo-SCT).
But the researchers have also used this group of patients to define diagnostic criteria for severe CRS. And the team has discovered that measuring C-reactive protein levels can help predict the severity of CRS.
Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, and his colleagues described these findings in Science Translational Medicine.
Response, bridge to allo-SCT
Dr Sadelain and his colleagues previously reported results in 5 patients with relapsed/refractory B-ALL who received autologous T cells expressing a CD19-specific, CD28/CD3z CAR called 19-28z.
After receiving salvage chemotherapy and CAR T cells, all 5 patients were negative for minimal residual disease. And 4 of the patients went on to receive allo-SCT.
Now, the investigators have expanded upon these findings, reporting results in a total of 16 patients with relapsed/refractory B-ALL who received the 19-28z CAR T cells.
Forty-four percent of patients (n=7) had a complete response to the salvage chemotherapy, and 88% (n=14) had a complete response after CAR T-cell therapy (alghough some had incomplete count recovery). Sixty-three percent of patients (n=10) achieved a complete remission.
Of the 10 patients who were eligible for allo-SCT, 7 underwent the procedure, and all 7 remain free of relapse.
“These extraordinary results demonstrate that cell therapy is a powerful treatment for patients who have exhausted all conventional therapies,” Dr Sadelain said. “Our initial findings have held up in a larger cohort of patients, and we are already looking at new clinical studies to advance this novel therapeutic approach in fighting cancer.”
CRS diagnosis, stratification
In their analysis of 5 B-ALL patients, Dr Sadelain and his colleagues observed a correlation between cytokine elevation and tumor burden at the time of CAR T-cell administration. The team confirmed this correlation in the larger cohort of 16 patients and identified 7 cytokines whose elevation was correlated with pretreatment tumor burden and severe CRS.
Patients with CRS that required intensive medical intervention had a 75-fold increase over baseline levels in 2 of the 7 cytokines, which included IFN-γ, IL-5, IL-6, IL-10, Flt-3L, Fracktalkine, and GM-CSF. These patients also had at least 1 of the following: hypoxia, hypotension, and neurologic changes (such as delirium and seizure-like activity).
Taking these findings together, the researchers concluded that patients had severe CRS if they had persistent fevers (38°C) for more than 3 days, selected cytokine elevations, and additional clinical evidence of toxicity.
The investigators stressed that these patients should be closely monitored. Patients with severe CRS are more likely to need medical intervention than patients with mild CRS, which is characterized by low-grade fever and mild cytokine increases, or absent CRS, which is defined as no fevers and/or no significant cytokine elevations.
Finally, the researchers found that measuring C-reactive protein in serum samples could predict the severity of CRS. Only those patients who met the criteria for severe CRS had a C-reactive protein level of 20 mg/dL or higher.
Patients who had received high-dose steroids were excluded from this analysis, due to the inverse correlation between high-dose steroid treatment and serum C-reactive protein.
Incidentally, the investigators confirmed prior findings that the monoclonal antibody tocilizumab can ameliorate severe CRS as effectively as steroid treatment, without inhibiting the expansion of CAR T cells.
Credit: MSKCC
Several studies have shown that infusions of T cells modified with chimeric antigen receptors (CARs) can elicit complete responses in leukemia patients who have run out of treatment options.
However, the therapy also puts patients at risk of developing cytokine release syndrome (CRS).
With updated research, investigators have again shown that CAR T cells can produce complete responses in patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL), thereby allowing them to receive allogeneic stem cell transplant (allo-SCT).
But the researchers have also used this group of patients to define diagnostic criteria for severe CRS. And the team has discovered that measuring C-reactive protein levels can help predict the severity of CRS.
Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, and his colleagues described these findings in Science Translational Medicine.
Response, bridge to allo-SCT
Dr Sadelain and his colleagues previously reported results in 5 patients with relapsed/refractory B-ALL who received autologous T cells expressing a CD19-specific, CD28/CD3z CAR called 19-28z.
After receiving salvage chemotherapy and CAR T cells, all 5 patients were negative for minimal residual disease. And 4 of the patients went on to receive allo-SCT.
Now, the investigators have expanded upon these findings, reporting results in a total of 16 patients with relapsed/refractory B-ALL who received the 19-28z CAR T cells.
Forty-four percent of patients (n=7) had a complete response to the salvage chemotherapy, and 88% (n=14) had a complete response after CAR T-cell therapy (alghough some had incomplete count recovery). Sixty-three percent of patients (n=10) achieved a complete remission.
Of the 10 patients who were eligible for allo-SCT, 7 underwent the procedure, and all 7 remain free of relapse.
“These extraordinary results demonstrate that cell therapy is a powerful treatment for patients who have exhausted all conventional therapies,” Dr Sadelain said. “Our initial findings have held up in a larger cohort of patients, and we are already looking at new clinical studies to advance this novel therapeutic approach in fighting cancer.”
CRS diagnosis, stratification
In their analysis of 5 B-ALL patients, Dr Sadelain and his colleagues observed a correlation between cytokine elevation and tumor burden at the time of CAR T-cell administration. The team confirmed this correlation in the larger cohort of 16 patients and identified 7 cytokines whose elevation was correlated with pretreatment tumor burden and severe CRS.
Patients with CRS that required intensive medical intervention had a 75-fold increase over baseline levels in 2 of the 7 cytokines, which included IFN-γ, IL-5, IL-6, IL-10, Flt-3L, Fracktalkine, and GM-CSF. These patients also had at least 1 of the following: hypoxia, hypotension, and neurologic changes (such as delirium and seizure-like activity).
Taking these findings together, the researchers concluded that patients had severe CRS if they had persistent fevers (38°C) for more than 3 days, selected cytokine elevations, and additional clinical evidence of toxicity.
The investigators stressed that these patients should be closely monitored. Patients with severe CRS are more likely to need medical intervention than patients with mild CRS, which is characterized by low-grade fever and mild cytokine increases, or absent CRS, which is defined as no fevers and/or no significant cytokine elevations.
Finally, the researchers found that measuring C-reactive protein in serum samples could predict the severity of CRS. Only those patients who met the criteria for severe CRS had a C-reactive protein level of 20 mg/dL or higher.
Patients who had received high-dose steroids were excluded from this analysis, due to the inverse correlation between high-dose steroid treatment and serum C-reactive protein.
Incidentally, the investigators confirmed prior findings that the monoclonal antibody tocilizumab can ameliorate severe CRS as effectively as steroid treatment, without inhibiting the expansion of CAR T cells.
Credit: MSKCC
Several studies have shown that infusions of T cells modified with chimeric antigen receptors (CARs) can elicit complete responses in leukemia patients who have run out of treatment options.
However, the therapy also puts patients at risk of developing cytokine release syndrome (CRS).
With updated research, investigators have again shown that CAR T cells can produce complete responses in patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL), thereby allowing them to receive allogeneic stem cell transplant (allo-SCT).
But the researchers have also used this group of patients to define diagnostic criteria for severe CRS. And the team has discovered that measuring C-reactive protein levels can help predict the severity of CRS.
Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, and his colleagues described these findings in Science Translational Medicine.
Response, bridge to allo-SCT
Dr Sadelain and his colleagues previously reported results in 5 patients with relapsed/refractory B-ALL who received autologous T cells expressing a CD19-specific, CD28/CD3z CAR called 19-28z.
After receiving salvage chemotherapy and CAR T cells, all 5 patients were negative for minimal residual disease. And 4 of the patients went on to receive allo-SCT.
Now, the investigators have expanded upon these findings, reporting results in a total of 16 patients with relapsed/refractory B-ALL who received the 19-28z CAR T cells.
Forty-four percent of patients (n=7) had a complete response to the salvage chemotherapy, and 88% (n=14) had a complete response after CAR T-cell therapy (alghough some had incomplete count recovery). Sixty-three percent of patients (n=10) achieved a complete remission.
Of the 10 patients who were eligible for allo-SCT, 7 underwent the procedure, and all 7 remain free of relapse.
“These extraordinary results demonstrate that cell therapy is a powerful treatment for patients who have exhausted all conventional therapies,” Dr Sadelain said. “Our initial findings have held up in a larger cohort of patients, and we are already looking at new clinical studies to advance this novel therapeutic approach in fighting cancer.”
CRS diagnosis, stratification
In their analysis of 5 B-ALL patients, Dr Sadelain and his colleagues observed a correlation between cytokine elevation and tumor burden at the time of CAR T-cell administration. The team confirmed this correlation in the larger cohort of 16 patients and identified 7 cytokines whose elevation was correlated with pretreatment tumor burden and severe CRS.
Patients with CRS that required intensive medical intervention had a 75-fold increase over baseline levels in 2 of the 7 cytokines, which included IFN-γ, IL-5, IL-6, IL-10, Flt-3L, Fracktalkine, and GM-CSF. These patients also had at least 1 of the following: hypoxia, hypotension, and neurologic changes (such as delirium and seizure-like activity).
Taking these findings together, the researchers concluded that patients had severe CRS if they had persistent fevers (38°C) for more than 3 days, selected cytokine elevations, and additional clinical evidence of toxicity.
The investigators stressed that these patients should be closely monitored. Patients with severe CRS are more likely to need medical intervention than patients with mild CRS, which is characterized by low-grade fever and mild cytokine increases, or absent CRS, which is defined as no fevers and/or no significant cytokine elevations.
Finally, the researchers found that measuring C-reactive protein in serum samples could predict the severity of CRS. Only those patients who met the criteria for severe CRS had a C-reactive protein level of 20 mg/dL or higher.
Patients who had received high-dose steroids were excluded from this analysis, due to the inverse correlation between high-dose steroid treatment and serum C-reactive protein.
Incidentally, the investigators confirmed prior findings that the monoclonal antibody tocilizumab can ameliorate severe CRS as effectively as steroid treatment, without inhibiting the expansion of CAR T cells.