User login
Group calls for standardized data collection practices across cancer centers

Credit: CDC
Researchers have identified significant variations in how cancer centers gather data, particularly that pertaining to racial and ethnic minorities.
Although racial and ethnic categories were similar across the centers, those categories were defined differently.
And the centers’ definitions of “catchment area,” the geographic region they expect to influence with their programs, differed widely.
This research, published in Cancer, was part of a national effort to recruit more racial/ethnic minorities into clinical trials and, ultimately, reduce the disproportional incidence of many cancers among those populations.
Five National Cancer Institute-designated comprehensive cancer centers participated in the endeavor, known as EMPaCT—Enhancing Minority Participation in Clinical Trials. They were:
- University of Minnesota, Minneapolis, which represents the Midwest and targets the accrual of Native Americans and African Americans
- University of Alabama, Birmingham, representing the Southeast, targeting African Americans
- Johns Hopkins University, representing the East, targeting African Americans
- University of Texas MD Anderson, Houston, representing the Southwest, targeting Latinos
- University of California, Davis, representing the West, targeting Asian Americans.
Ernest T. Hawk, MD, of the MD Anderson Cancer Center, and his colleagues reviewed the collection and reporting of patient data and other practices by these 5 centers.
This revealed significant variation in the centers’ methods of data collection. For example, patients’ insurance status was routinely documented at 2 centers, collected for non-research patients only at a third center, collected for billing of researcher enrollees at a fourth center, and not documented at all at a fifth center.
There were differences in data collection according to race/ethnicity as well. Racial/ethnic categories were generally similar across the centers—white, black/African American, Asian, Native American, Hispanic/Latino, and “other/unknown.”
However, the means of race/ethnicity data collection differed. Each center collected self-reported data on race/ethnicity, but 2 centers included data from staff observations.
Two centers compared the proportions of racial/ethnic groups enrolled in trials with those of their catchment area(s). But the others did not.
The centers also differed in how they defined their patient catchment area, in terms of their cancer patient-vs-general-population specificity, levels of specificity, and geographic coverage.
That merits notice, according to the researchers, because National Cancer Institute cancer centers are required to accrue women and minorities to clinical trials in rough proportion to the cancer patient population of the center’s primary catchment area.
Given these findings, the researchers recommended better standardization of data definition, collection, and reporting as an essential first step toward expanding minority participation in clinical trials.
The team also advised that cancer centers collect socioeconomic data, including a patient’s income and education levels, given past evidence of the strong link between socioeconomic status and cancer outcomes.
Finally, the group recommended collecting patient zip codes and insurance status to allow researchers to assess differences in access to clinical trials that may be related to geography and the availability of health insurance coverage. ![]()
Credit: CDC
Researchers have identified significant variations in how cancer centers gather data, particularly that pertaining to racial and ethnic minorities.
Although racial and ethnic categories were similar across the centers, those categories were defined differently.
And the centers’ definitions of “catchment area,” the geographic region they expect to influence with their programs, differed widely.
This research, published in Cancer, was part of a national effort to recruit more racial/ethnic minorities into clinical trials and, ultimately, reduce the disproportional incidence of many cancers among those populations.
Five National Cancer Institute-designated comprehensive cancer centers participated in the endeavor, known as EMPaCT—Enhancing Minority Participation in Clinical Trials. They were:
- University of Minnesota, Minneapolis, which represents the Midwest and targets the accrual of Native Americans and African Americans
- University of Alabama, Birmingham, representing the Southeast, targeting African Americans
- Johns Hopkins University, representing the East, targeting African Americans
- University of Texas MD Anderson, Houston, representing the Southwest, targeting Latinos
- University of California, Davis, representing the West, targeting Asian Americans.
Ernest T. Hawk, MD, of the MD Anderson Cancer Center, and his colleagues reviewed the collection and reporting of patient data and other practices by these 5 centers.
This revealed significant variation in the centers’ methods of data collection. For example, patients’ insurance status was routinely documented at 2 centers, collected for non-research patients only at a third center, collected for billing of researcher enrollees at a fourth center, and not documented at all at a fifth center.
There were differences in data collection according to race/ethnicity as well. Racial/ethnic categories were generally similar across the centers—white, black/African American, Asian, Native American, Hispanic/Latino, and “other/unknown.”
However, the means of race/ethnicity data collection differed. Each center collected self-reported data on race/ethnicity, but 2 centers included data from staff observations.
Two centers compared the proportions of racial/ethnic groups enrolled in trials with those of their catchment area(s). But the others did not.
The centers also differed in how they defined their patient catchment area, in terms of their cancer patient-vs-general-population specificity, levels of specificity, and geographic coverage.
That merits notice, according to the researchers, because National Cancer Institute cancer centers are required to accrue women and minorities to clinical trials in rough proportion to the cancer patient population of the center’s primary catchment area.
Given these findings, the researchers recommended better standardization of data definition, collection, and reporting as an essential first step toward expanding minority participation in clinical trials.
The team also advised that cancer centers collect socioeconomic data, including a patient’s income and education levels, given past evidence of the strong link between socioeconomic status and cancer outcomes.
Finally, the group recommended collecting patient zip codes and insurance status to allow researchers to assess differences in access to clinical trials that may be related to geography and the availability of health insurance coverage. ![]()
Credit: CDC
Researchers have identified significant variations in how cancer centers gather data, particularly that pertaining to racial and ethnic minorities.
Although racial and ethnic categories were similar across the centers, those categories were defined differently.
And the centers’ definitions of “catchment area,” the geographic region they expect to influence with their programs, differed widely.
This research, published in Cancer, was part of a national effort to recruit more racial/ethnic minorities into clinical trials and, ultimately, reduce the disproportional incidence of many cancers among those populations.
Five National Cancer Institute-designated comprehensive cancer centers participated in the endeavor, known as EMPaCT—Enhancing Minority Participation in Clinical Trials. They were:
- University of Minnesota, Minneapolis, which represents the Midwest and targets the accrual of Native Americans and African Americans
- University of Alabama, Birmingham, representing the Southeast, targeting African Americans
- Johns Hopkins University, representing the East, targeting African Americans
- University of Texas MD Anderson, Houston, representing the Southwest, targeting Latinos
- University of California, Davis, representing the West, targeting Asian Americans.
Ernest T. Hawk, MD, of the MD Anderson Cancer Center, and his colleagues reviewed the collection and reporting of patient data and other practices by these 5 centers.
This revealed significant variation in the centers’ methods of data collection. For example, patients’ insurance status was routinely documented at 2 centers, collected for non-research patients only at a third center, collected for billing of researcher enrollees at a fourth center, and not documented at all at a fifth center.
There were differences in data collection according to race/ethnicity as well. Racial/ethnic categories were generally similar across the centers—white, black/African American, Asian, Native American, Hispanic/Latino, and “other/unknown.”
However, the means of race/ethnicity data collection differed. Each center collected self-reported data on race/ethnicity, but 2 centers included data from staff observations.
Two centers compared the proportions of racial/ethnic groups enrolled in trials with those of their catchment area(s). But the others did not.
The centers also differed in how they defined their patient catchment area, in terms of their cancer patient-vs-general-population specificity, levels of specificity, and geographic coverage.
That merits notice, according to the researchers, because National Cancer Institute cancer centers are required to accrue women and minorities to clinical trials in rough proportion to the cancer patient population of the center’s primary catchment area.
Given these findings, the researchers recommended better standardization of data definition, collection, and reporting as an essential first step toward expanding minority participation in clinical trials.
The team also advised that cancer centers collect socioeconomic data, including a patient’s income and education levels, given past evidence of the strong link between socioeconomic status and cancer outcomes.
Finally, the group recommended collecting patient zip codes and insurance status to allow researchers to assess differences in access to clinical trials that may be related to geography and the availability of health insurance coverage. ![]()
Cancer survivors’ risk of health problems increases with age

cancer patient and her father
Credit: Rhoda Baer
The “health gap” between childhood cancer survivors and their siblings widens with age, according to a study published in the Journal of Clinical Oncology.
Cancer survivors aged 20 to 34 years old were 3.8 times more likely than siblings of the same age to develop new cancers and other serious health conditions.
By age 35 and beyond, survivors had a 5-fold greater risk.
“Survivors remain at risk for serious health problems into their 40s and 50s, decades after they have completed treatment for childhood cancer,” said study author Gregory Armstrong, MD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
“In fact, for survivors, the risk of illness and death increases significantly beyond the age of 35. Their siblings don’t share these same risks.”
Dr Armstrong and his colleagues uncovered these results by analyzing data from the Childhood Cancer Survivor Study, which included 14,359 survivors and 4301 healthy siblings.
The patients had been diagnosed with leukemias, lymphomas, and other pediatric cancers before age 21 and were followed for a median of 24.5 years (range, 5 to 39.3 years).
The researchers compared survivors to age-matched siblings, evaluating the incidence of severe, disabling, life-threatening, or fatal health conditions. This included new malignancies as well as diseases of the heart, lungs, liver, kidneys, and hormones.
The team found a heightened risk of these health conditions among cancer survivors. And that risk increased as the survivors aged.
At 20 years of age, 16% of survivors had serious health conditions, compared to 3.3% of siblings. But by age 50, the incidence had increased to 53.6% among survivors and 19.8% among siblings. At 50, 22.5% of survivors had at least 2 serious health problems, and 10.1% had 3 or more.
In a multivariate analysis, the hazard ratio for developing serious health conditions was significantly higher among survivors aged 35 and older than for those aged 20 to 34 (P=0.03).
Among survivors who reached age 35 without serious health problems, 25.9% developed a significant health problem in the next decade. In comparison, 6% of siblings developed their first serious health condition between the ages of 35 and 45.
In addition to showing a health gap between childhood cancer survivors and their siblings, this research adds to evidence that survivors experience accelerated aging. The 24-year-old cancer survivors had roughly the same cumulative incidence of grade 3 to 5 health conditions (19.6%) as the 50-year-old siblings (19.8%).
Overall, these findings highlight the importance of lifelong, risk-based healthcare for childhood cancer survivors, Dr Armstrong said. Depending on their cancer treatment and other risk factors, follow-up care may include performing health checks at a younger age than is recommended for the general public.
This study involved survivors whose cancer was diagnosed between 1970 and 1986. The researchers are now studying the health of adult cancer survivors from a more recent treatment era. ![]()
cancer patient and her father
Credit: Rhoda Baer
The “health gap” between childhood cancer survivors and their siblings widens with age, according to a study published in the Journal of Clinical Oncology.
Cancer survivors aged 20 to 34 years old were 3.8 times more likely than siblings of the same age to develop new cancers and other serious health conditions.
By age 35 and beyond, survivors had a 5-fold greater risk.
“Survivors remain at risk for serious health problems into their 40s and 50s, decades after they have completed treatment for childhood cancer,” said study author Gregory Armstrong, MD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
“In fact, for survivors, the risk of illness and death increases significantly beyond the age of 35. Their siblings don’t share these same risks.”
Dr Armstrong and his colleagues uncovered these results by analyzing data from the Childhood Cancer Survivor Study, which included 14,359 survivors and 4301 healthy siblings.
The patients had been diagnosed with leukemias, lymphomas, and other pediatric cancers before age 21 and were followed for a median of 24.5 years (range, 5 to 39.3 years).
The researchers compared survivors to age-matched siblings, evaluating the incidence of severe, disabling, life-threatening, or fatal health conditions. This included new malignancies as well as diseases of the heart, lungs, liver, kidneys, and hormones.
The team found a heightened risk of these health conditions among cancer survivors. And that risk increased as the survivors aged.
At 20 years of age, 16% of survivors had serious health conditions, compared to 3.3% of siblings. But by age 50, the incidence had increased to 53.6% among survivors and 19.8% among siblings. At 50, 22.5% of survivors had at least 2 serious health problems, and 10.1% had 3 or more.
In a multivariate analysis, the hazard ratio for developing serious health conditions was significantly higher among survivors aged 35 and older than for those aged 20 to 34 (P=0.03).
Among survivors who reached age 35 without serious health problems, 25.9% developed a significant health problem in the next decade. In comparison, 6% of siblings developed their first serious health condition between the ages of 35 and 45.
In addition to showing a health gap between childhood cancer survivors and their siblings, this research adds to evidence that survivors experience accelerated aging. The 24-year-old cancer survivors had roughly the same cumulative incidence of grade 3 to 5 health conditions (19.6%) as the 50-year-old siblings (19.8%).
Overall, these findings highlight the importance of lifelong, risk-based healthcare for childhood cancer survivors, Dr Armstrong said. Depending on their cancer treatment and other risk factors, follow-up care may include performing health checks at a younger age than is recommended for the general public.
This study involved survivors whose cancer was diagnosed between 1970 and 1986. The researchers are now studying the health of adult cancer survivors from a more recent treatment era. ![]()
cancer patient and her father
Credit: Rhoda Baer
The “health gap” between childhood cancer survivors and their siblings widens with age, according to a study published in the Journal of Clinical Oncology.
Cancer survivors aged 20 to 34 years old were 3.8 times more likely than siblings of the same age to develop new cancers and other serious health conditions.
By age 35 and beyond, survivors had a 5-fold greater risk.
“Survivors remain at risk for serious health problems into their 40s and 50s, decades after they have completed treatment for childhood cancer,” said study author Gregory Armstrong, MD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
“In fact, for survivors, the risk of illness and death increases significantly beyond the age of 35. Their siblings don’t share these same risks.”
Dr Armstrong and his colleagues uncovered these results by analyzing data from the Childhood Cancer Survivor Study, which included 14,359 survivors and 4301 healthy siblings.
The patients had been diagnosed with leukemias, lymphomas, and other pediatric cancers before age 21 and were followed for a median of 24.5 years (range, 5 to 39.3 years).
The researchers compared survivors to age-matched siblings, evaluating the incidence of severe, disabling, life-threatening, or fatal health conditions. This included new malignancies as well as diseases of the heart, lungs, liver, kidneys, and hormones.
The team found a heightened risk of these health conditions among cancer survivors. And that risk increased as the survivors aged.
At 20 years of age, 16% of survivors had serious health conditions, compared to 3.3% of siblings. But by age 50, the incidence had increased to 53.6% among survivors and 19.8% among siblings. At 50, 22.5% of survivors had at least 2 serious health problems, and 10.1% had 3 or more.
In a multivariate analysis, the hazard ratio for developing serious health conditions was significantly higher among survivors aged 35 and older than for those aged 20 to 34 (P=0.03).
Among survivors who reached age 35 without serious health problems, 25.9% developed a significant health problem in the next decade. In comparison, 6% of siblings developed their first serious health condition between the ages of 35 and 45.
In addition to showing a health gap between childhood cancer survivors and their siblings, this research adds to evidence that survivors experience accelerated aging. The 24-year-old cancer survivors had roughly the same cumulative incidence of grade 3 to 5 health conditions (19.6%) as the 50-year-old siblings (19.8%).
Overall, these findings highlight the importance of lifelong, risk-based healthcare for childhood cancer survivors, Dr Armstrong said. Depending on their cancer treatment and other risk factors, follow-up care may include performing health checks at a younger age than is recommended for the general public.
This study involved survivors whose cancer was diagnosed between 1970 and 1986. The researchers are now studying the health of adult cancer survivors from a more recent treatment era. ![]()
Adult minorities underrepresented in cancer trials
Credit: Rhoda Baer
New research indicates that less than 2% of trials funded by the National Cancer Institute focus on racial and ethnic minorities, and minority participation in adult cancer trials is not representative of the US population.
The researchers said these findings suggest we must do more to promote minority-focused research and clinical trial recruitment, beyond the National Institutes of Health (NIH) Revitalization Act of 1993, which mandated the appropriate inclusion of minorities in all NIH-funded research.
“What is needed is deliberate effort,” said study author Moon Chen, Jr, PhD, of the University of California, Davis. “Minorities are not hard to reach. They are hardly reached.”
To assess minority inclusion in clinical trials, Dr Chen and his colleagues searched ClinicalTrials.gov, looking for trials sponsored by the National Cancer Institute that were available in January 2013.
They searched using terms for different minority groups, then counted the number of clinical trials with a primary focus on a particular ethnic or minority population. Roughly 150 trials out of 10,000—or less than 2%—met the criteria.
The researchers also reviewed abstracts and articles accessed from January through March 2013 on PubMed to find those that specifically examined minority accrual in clinical trials.
Of the 42 citations found, 5 included reports explicitly discussing participation levels by race and ethnicity. Those reports revealed an “encouraging but less than optimal” increase in specification of race or ethnicity in published results of clinical trials.
Dr Chen and his colleagues also reported that participation of adult minorities is not proportional to their representation in the US population.
For example, African Americans experience the highest cancer incidence of any racial group (593.7 cases per 100,000), but they have the lowest rates of cancer trial participation (tied with Hispanics), at 1.3%. It’s important to note, however, that clinical trial participation is low for all adult cancer patients, at 3% to 5%.
In contrast, the researchers pointed out that 60% of all patients under age 15 are enrolled in clinical trials. And minority representation among children is excellent, either equal to or greater than their proportion of the population.
To put the adult population on par with the pediatric population, researchers should design trials to include and focus on specific populations, Dr Chen said. Furthermore, scientific journals should insist on appropriate representation and analyses of NIH research by race and ethnicity.
“Whatever happens in the laboratory or in the clinic needs to be applied to solving real-world problems,” Dr Chen said. “And those relate to the disproportionate effects of cancer and other diseases on racial and ethnic minorities.”
Dr Chen and his colleagues reported this research in Cancer. ![]()
Credit: Rhoda Baer
New research indicates that less than 2% of trials funded by the National Cancer Institute focus on racial and ethnic minorities, and minority participation in adult cancer trials is not representative of the US population.
The researchers said these findings suggest we must do more to promote minority-focused research and clinical trial recruitment, beyond the National Institutes of Health (NIH) Revitalization Act of 1993, which mandated the appropriate inclusion of minorities in all NIH-funded research.
“What is needed is deliberate effort,” said study author Moon Chen, Jr, PhD, of the University of California, Davis. “Minorities are not hard to reach. They are hardly reached.”
To assess minority inclusion in clinical trials, Dr Chen and his colleagues searched ClinicalTrials.gov, looking for trials sponsored by the National Cancer Institute that were available in January 2013.
They searched using terms for different minority groups, then counted the number of clinical trials with a primary focus on a particular ethnic or minority population. Roughly 150 trials out of 10,000—or less than 2%—met the criteria.
The researchers also reviewed abstracts and articles accessed from January through March 2013 on PubMed to find those that specifically examined minority accrual in clinical trials.
Of the 42 citations found, 5 included reports explicitly discussing participation levels by race and ethnicity. Those reports revealed an “encouraging but less than optimal” increase in specification of race or ethnicity in published results of clinical trials.
Dr Chen and his colleagues also reported that participation of adult minorities is not proportional to their representation in the US population.
For example, African Americans experience the highest cancer incidence of any racial group (593.7 cases per 100,000), but they have the lowest rates of cancer trial participation (tied with Hispanics), at 1.3%. It’s important to note, however, that clinical trial participation is low for all adult cancer patients, at 3% to 5%.
In contrast, the researchers pointed out that 60% of all patients under age 15 are enrolled in clinical trials. And minority representation among children is excellent, either equal to or greater than their proportion of the population.
To put the adult population on par with the pediatric population, researchers should design trials to include and focus on specific populations, Dr Chen said. Furthermore, scientific journals should insist on appropriate representation and analyses of NIH research by race and ethnicity.
“Whatever happens in the laboratory or in the clinic needs to be applied to solving real-world problems,” Dr Chen said. “And those relate to the disproportionate effects of cancer and other diseases on racial and ethnic minorities.”
Dr Chen and his colleagues reported this research in Cancer. ![]()
Credit: Rhoda Baer
New research indicates that less than 2% of trials funded by the National Cancer Institute focus on racial and ethnic minorities, and minority participation in adult cancer trials is not representative of the US population.
The researchers said these findings suggest we must do more to promote minority-focused research and clinical trial recruitment, beyond the National Institutes of Health (NIH) Revitalization Act of 1993, which mandated the appropriate inclusion of minorities in all NIH-funded research.
“What is needed is deliberate effort,” said study author Moon Chen, Jr, PhD, of the University of California, Davis. “Minorities are not hard to reach. They are hardly reached.”
To assess minority inclusion in clinical trials, Dr Chen and his colleagues searched ClinicalTrials.gov, looking for trials sponsored by the National Cancer Institute that were available in January 2013.
They searched using terms for different minority groups, then counted the number of clinical trials with a primary focus on a particular ethnic or minority population. Roughly 150 trials out of 10,000—or less than 2%—met the criteria.
The researchers also reviewed abstracts and articles accessed from January through March 2013 on PubMed to find those that specifically examined minority accrual in clinical trials.
Of the 42 citations found, 5 included reports explicitly discussing participation levels by race and ethnicity. Those reports revealed an “encouraging but less than optimal” increase in specification of race or ethnicity in published results of clinical trials.
Dr Chen and his colleagues also reported that participation of adult minorities is not proportional to their representation in the US population.
For example, African Americans experience the highest cancer incidence of any racial group (593.7 cases per 100,000), but they have the lowest rates of cancer trial participation (tied with Hispanics), at 1.3%. It’s important to note, however, that clinical trial participation is low for all adult cancer patients, at 3% to 5%.
In contrast, the researchers pointed out that 60% of all patients under age 15 are enrolled in clinical trials. And minority representation among children is excellent, either equal to or greater than their proportion of the population.
To put the adult population on par with the pediatric population, researchers should design trials to include and focus on specific populations, Dr Chen said. Furthermore, scientific journals should insist on appropriate representation and analyses of NIH research by race and ethnicity.
“Whatever happens in the laboratory or in the clinic needs to be applied to solving real-world problems,” Dr Chen said. “And those relate to the disproportionate effects of cancer and other diseases on racial and ethnic minorities.”
Dr Chen and his colleagues reported this research in Cancer. ![]()
Company issues nationwide recall of blood sets

Credit: Elise Amendola
Hospira, Inc. has announced a nationwide recall of 2 lots of Hemoset Dual Channel Plum Sets, which are used to administer blood products.
The affected lots—28005-5H and 34100-5H (list number 11241-03)—contain an incorrect component.
Using these sets, which were distributed across the US, could result in the over-delivery of blood products.
However, Hospira has not received any reports of adverse events associated with the sets. The recall is a precautionary measure.
Possible risk associated with the sets
The Hemostat Dual Channel Plum Set is designed to administer blood and blood products via the Plum infusion pump. If the Plum infusion pump is used with one of the sets being recalled, the blood product will be delivered at its intended dosage.
However, if one of the affected sets is removed from the Plum infusion pump and used in a gravity infusion, there is a risk of over-delivering blood products, due to the incorrect component—a lower lid.
In a gravity delivery, the correct lower lid dispenses 15 drops per mL. But the incorrect lower lid dispenses 10 drops per mL. If a caregiver does not realize that each drop contains more volume, over-delivery could occur.
Over-delivery of blood products in the populations at greatest risk (eg, neonates and patients with heart and/or kidney failure) may result in injuries that require medical intervention. These injuries are expected to fully resolve with medical intervention.
Steps to take
The sets impacted by the recall were distributed to US healthcare and veterinary facilities from May 2013 through December 2013.
Customers should check their inventory and immediately quarantine any affected sets. They should also inform individuals who might use the sets about the recall.
The affected sets should be returned to Stericycle. To do so, call 1-888-240-4282, Monday through Friday between 8 am and 5 pm Eastern Time.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187.
Adverse reactions or quality problems associated with the use of these sets can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Credit: Elise Amendola
Hospira, Inc. has announced a nationwide recall of 2 lots of Hemoset Dual Channel Plum Sets, which are used to administer blood products.
The affected lots—28005-5H and 34100-5H (list number 11241-03)—contain an incorrect component.
Using these sets, which were distributed across the US, could result in the over-delivery of blood products.
However, Hospira has not received any reports of adverse events associated with the sets. The recall is a precautionary measure.
Possible risk associated with the sets
The Hemostat Dual Channel Plum Set is designed to administer blood and blood products via the Plum infusion pump. If the Plum infusion pump is used with one of the sets being recalled, the blood product will be delivered at its intended dosage.
However, if one of the affected sets is removed from the Plum infusion pump and used in a gravity infusion, there is a risk of over-delivering blood products, due to the incorrect component—a lower lid.
In a gravity delivery, the correct lower lid dispenses 15 drops per mL. But the incorrect lower lid dispenses 10 drops per mL. If a caregiver does not realize that each drop contains more volume, over-delivery could occur.
Over-delivery of blood products in the populations at greatest risk (eg, neonates and patients with heart and/or kidney failure) may result in injuries that require medical intervention. These injuries are expected to fully resolve with medical intervention.
Steps to take
The sets impacted by the recall were distributed to US healthcare and veterinary facilities from May 2013 through December 2013.
Customers should check their inventory and immediately quarantine any affected sets. They should also inform individuals who might use the sets about the recall.
The affected sets should be returned to Stericycle. To do so, call 1-888-240-4282, Monday through Friday between 8 am and 5 pm Eastern Time.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187.
Adverse reactions or quality problems associated with the use of these sets can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Credit: Elise Amendola
Hospira, Inc. has announced a nationwide recall of 2 lots of Hemoset Dual Channel Plum Sets, which are used to administer blood products.
The affected lots—28005-5H and 34100-5H (list number 11241-03)—contain an incorrect component.
Using these sets, which were distributed across the US, could result in the over-delivery of blood products.
However, Hospira has not received any reports of adverse events associated with the sets. The recall is a precautionary measure.
Possible risk associated with the sets
The Hemostat Dual Channel Plum Set is designed to administer blood and blood products via the Plum infusion pump. If the Plum infusion pump is used with one of the sets being recalled, the blood product will be delivered at its intended dosage.
However, if one of the affected sets is removed from the Plum infusion pump and used in a gravity infusion, there is a risk of over-delivering blood products, due to the incorrect component—a lower lid.
In a gravity delivery, the correct lower lid dispenses 15 drops per mL. But the incorrect lower lid dispenses 10 drops per mL. If a caregiver does not realize that each drop contains more volume, over-delivery could occur.
Over-delivery of blood products in the populations at greatest risk (eg, neonates and patients with heart and/or kidney failure) may result in injuries that require medical intervention. These injuries are expected to fully resolve with medical intervention.
Steps to take
The sets impacted by the recall were distributed to US healthcare and veterinary facilities from May 2013 through December 2013.
Customers should check their inventory and immediately quarantine any affected sets. They should also inform individuals who might use the sets about the recall.
The affected sets should be returned to Stericycle. To do so, call 1-888-240-4282, Monday through Friday between 8 am and 5 pm Eastern Time.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187.
Adverse reactions or quality problems associated with the use of these sets can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Electronics workers may have elevated risk of death from NHL

Researchers have found evidence suggesting that men who work in microelectronics and business machine facilities may have an increased risk of dying from certain cancers, including non-Hodgkin lymphoma (NHL).
Their study, published in the American Journal of Industrial Medicine, was designed to assess the effects chemical exposure might have on the incidence of diseases and worker mortality.
The results showed that hourly male workers, who were more likely than other employees to be exposed to the chemicals studied, had a 1.5-fold increased risk of death from NHL.
However, the investigators did not observe a significant relationship between NHL and any of the chemicals studied.
This research originated from concerns about the release of trichloroethylene (TCE), perchlorethylene (PCE), and other industrial chemicals through groundwater and air emissions from several industrial facilities in a town in upstate New York.
Previous studies suggested the chemicals were associated with increases in the incidence of kidney, lung, and testicular cancer in the community. So researchers initiated a study of current and former workers of the local microelectronics and business machine facility.
Patient population
Sharon R. Silver, of the National Institute for Occupational Safety and Health in Cincinnati, Ohio, and her colleagues examined health outcomes among 34,494 former workers employed at the facility for at least 91 days between 1969 and 2001.
Machining workers were exposed to dust, noise, solvents, and metals. And “wet” process workers were exposed to chemical solutions used in manufacturing circuit boards and their substrates. The facility also had employees in non-production roles, including sales and office support, as well as computer programming.
The researchers evaluated the relationship between health outcomes and the estimated cumulative extent of potential chemical exposures, stratified according to gender and pay code.
Of the 34,494 workers, 69.7% were male. Among males, 15,447 were hourly workers, and 8590 were salaried. Among females, 8934 were hourly workers, and 1523 were salaried.
Chemical exposure
A previous study of this population revealed the use of 6 chemical agents (fiberglass, lead, methylene chloride, methyl chloroform, PCE, and TCE), 6 chemical classes (acid-base, aromatic hydrocarbons, chlorinated hydrocarbons, other hydrocarbons, chlorofluorocarbons, and metals), and general chemicals (including unspecified).
The potential for exposure to a chemical agent or class was much more common among hourly workers than salaried workers. Among males, 65.7% of hourly workers and 20% of salaried workers were exposed to at least 1 of the chemicals studied. Among females, exposure rates were 58.5% and 13.9%, respectively.
“Other hydrocarbons” was the chemical class that male hourly workers were potentially exposed to most often (60.5%). At least one-third of workers in this group had potential exposure to chlorinated hydrocarbons, lead, and acids and bases. TCE and PCE were the least common exposure agents among male hourly workers, with 13.9% and 15.1% exposed, respectively.
Cancer mortality, incidence
The investigators used mortality rates from the US population, as well as New York State (excluding New York City), to calculate the number of expected deaths among study participants. The standardized mortality ratio (SMR) is the ratio of observed to expected deaths.
The average follow-up was 25.7 years. By the study end date, 5966 workers (17.3%) had died. Workers employed less than a year at the facility (n=8397) comprised 363 of these deaths.
Both all-cause mortality (SMR=0.67) and all-cancer mortality (SMR=0.74) showed a statistically significant deficit for the entire workforce. Most of the individual cancers and other conditions studied were not associated with an increased risk of death.
There were significant increases in death for certain cancers among males, but there was no significant increase in a specific cause of death among females belonging to either pay code.
There was an increased risk of death from NHL among male hourly workers but not salaried workers, with SMRs of 1.49 and 0.68, respectively. The same pattern occurred for rectal cancer, with SMRs of 1.71 and 0.71, respectively.
The study also revealed an elevated incidence of pleural cancers in salaried males, mesothelioma in hourly workers, and testicular cancer in salaried males.
The increase in mesothelioma and pleural cancers was seen only in workers hired before 1969, which would support a link between the cancers and asbestos exposure. However, the researchers could find no evidence that asbestos was used in manufacturing at the facility.
Similarly, the investigators found no significant link between exposure to specific chemicals and the increased mortality from NHL or rectal cancer. And there was no significant link between exposure and testicular cancer.
Although these results do not suggest a strong role for occupational chemical exposures in cancer incidence and mortality, the researchers said risks from occupational exposures cannot be ruled out due to limitations of this study and the relative youth of this patient cohort. ![]()
Researchers have found evidence suggesting that men who work in microelectronics and business machine facilities may have an increased risk of dying from certain cancers, including non-Hodgkin lymphoma (NHL).
Their study, published in the American Journal of Industrial Medicine, was designed to assess the effects chemical exposure might have on the incidence of diseases and worker mortality.
The results showed that hourly male workers, who were more likely than other employees to be exposed to the chemicals studied, had a 1.5-fold increased risk of death from NHL.
However, the investigators did not observe a significant relationship between NHL and any of the chemicals studied.
This research originated from concerns about the release of trichloroethylene (TCE), perchlorethylene (PCE), and other industrial chemicals through groundwater and air emissions from several industrial facilities in a town in upstate New York.
Previous studies suggested the chemicals were associated with increases in the incidence of kidney, lung, and testicular cancer in the community. So researchers initiated a study of current and former workers of the local microelectronics and business machine facility.
Patient population
Sharon R. Silver, of the National Institute for Occupational Safety and Health in Cincinnati, Ohio, and her colleagues examined health outcomes among 34,494 former workers employed at the facility for at least 91 days between 1969 and 2001.
Machining workers were exposed to dust, noise, solvents, and metals. And “wet” process workers were exposed to chemical solutions used in manufacturing circuit boards and their substrates. The facility also had employees in non-production roles, including sales and office support, as well as computer programming.
The researchers evaluated the relationship between health outcomes and the estimated cumulative extent of potential chemical exposures, stratified according to gender and pay code.
Of the 34,494 workers, 69.7% were male. Among males, 15,447 were hourly workers, and 8590 were salaried. Among females, 8934 were hourly workers, and 1523 were salaried.
Chemical exposure
A previous study of this population revealed the use of 6 chemical agents (fiberglass, lead, methylene chloride, methyl chloroform, PCE, and TCE), 6 chemical classes (acid-base, aromatic hydrocarbons, chlorinated hydrocarbons, other hydrocarbons, chlorofluorocarbons, and metals), and general chemicals (including unspecified).
The potential for exposure to a chemical agent or class was much more common among hourly workers than salaried workers. Among males, 65.7% of hourly workers and 20% of salaried workers were exposed to at least 1 of the chemicals studied. Among females, exposure rates were 58.5% and 13.9%, respectively.
“Other hydrocarbons” was the chemical class that male hourly workers were potentially exposed to most often (60.5%). At least one-third of workers in this group had potential exposure to chlorinated hydrocarbons, lead, and acids and bases. TCE and PCE were the least common exposure agents among male hourly workers, with 13.9% and 15.1% exposed, respectively.
Cancer mortality, incidence
The investigators used mortality rates from the US population, as well as New York State (excluding New York City), to calculate the number of expected deaths among study participants. The standardized mortality ratio (SMR) is the ratio of observed to expected deaths.
The average follow-up was 25.7 years. By the study end date, 5966 workers (17.3%) had died. Workers employed less than a year at the facility (n=8397) comprised 363 of these deaths.
Both all-cause mortality (SMR=0.67) and all-cancer mortality (SMR=0.74) showed a statistically significant deficit for the entire workforce. Most of the individual cancers and other conditions studied were not associated with an increased risk of death.
There were significant increases in death for certain cancers among males, but there was no significant increase in a specific cause of death among females belonging to either pay code.
There was an increased risk of death from NHL among male hourly workers but not salaried workers, with SMRs of 1.49 and 0.68, respectively. The same pattern occurred for rectal cancer, with SMRs of 1.71 and 0.71, respectively.
The study also revealed an elevated incidence of pleural cancers in salaried males, mesothelioma in hourly workers, and testicular cancer in salaried males.
The increase in mesothelioma and pleural cancers was seen only in workers hired before 1969, which would support a link between the cancers and asbestos exposure. However, the researchers could find no evidence that asbestos was used in manufacturing at the facility.
Similarly, the investigators found no significant link between exposure to specific chemicals and the increased mortality from NHL or rectal cancer. And there was no significant link between exposure and testicular cancer.
Although these results do not suggest a strong role for occupational chemical exposures in cancer incidence and mortality, the researchers said risks from occupational exposures cannot be ruled out due to limitations of this study and the relative youth of this patient cohort. ![]()
Researchers have found evidence suggesting that men who work in microelectronics and business machine facilities may have an increased risk of dying from certain cancers, including non-Hodgkin lymphoma (NHL).
Their study, published in the American Journal of Industrial Medicine, was designed to assess the effects chemical exposure might have on the incidence of diseases and worker mortality.
The results showed that hourly male workers, who were more likely than other employees to be exposed to the chemicals studied, had a 1.5-fold increased risk of death from NHL.
However, the investigators did not observe a significant relationship between NHL and any of the chemicals studied.
This research originated from concerns about the release of trichloroethylene (TCE), perchlorethylene (PCE), and other industrial chemicals through groundwater and air emissions from several industrial facilities in a town in upstate New York.
Previous studies suggested the chemicals were associated with increases in the incidence of kidney, lung, and testicular cancer in the community. So researchers initiated a study of current and former workers of the local microelectronics and business machine facility.
Patient population
Sharon R. Silver, of the National Institute for Occupational Safety and Health in Cincinnati, Ohio, and her colleagues examined health outcomes among 34,494 former workers employed at the facility for at least 91 days between 1969 and 2001.
Machining workers were exposed to dust, noise, solvents, and metals. And “wet” process workers were exposed to chemical solutions used in manufacturing circuit boards and their substrates. The facility also had employees in non-production roles, including sales and office support, as well as computer programming.
The researchers evaluated the relationship between health outcomes and the estimated cumulative extent of potential chemical exposures, stratified according to gender and pay code.
Of the 34,494 workers, 69.7% were male. Among males, 15,447 were hourly workers, and 8590 were salaried. Among females, 8934 were hourly workers, and 1523 were salaried.
Chemical exposure
A previous study of this population revealed the use of 6 chemical agents (fiberglass, lead, methylene chloride, methyl chloroform, PCE, and TCE), 6 chemical classes (acid-base, aromatic hydrocarbons, chlorinated hydrocarbons, other hydrocarbons, chlorofluorocarbons, and metals), and general chemicals (including unspecified).
The potential for exposure to a chemical agent or class was much more common among hourly workers than salaried workers. Among males, 65.7% of hourly workers and 20% of salaried workers were exposed to at least 1 of the chemicals studied. Among females, exposure rates were 58.5% and 13.9%, respectively.
“Other hydrocarbons” was the chemical class that male hourly workers were potentially exposed to most often (60.5%). At least one-third of workers in this group had potential exposure to chlorinated hydrocarbons, lead, and acids and bases. TCE and PCE were the least common exposure agents among male hourly workers, with 13.9% and 15.1% exposed, respectively.
Cancer mortality, incidence
The investigators used mortality rates from the US population, as well as New York State (excluding New York City), to calculate the number of expected deaths among study participants. The standardized mortality ratio (SMR) is the ratio of observed to expected deaths.
The average follow-up was 25.7 years. By the study end date, 5966 workers (17.3%) had died. Workers employed less than a year at the facility (n=8397) comprised 363 of these deaths.
Both all-cause mortality (SMR=0.67) and all-cancer mortality (SMR=0.74) showed a statistically significant deficit for the entire workforce. Most of the individual cancers and other conditions studied were not associated with an increased risk of death.
There were significant increases in death for certain cancers among males, but there was no significant increase in a specific cause of death among females belonging to either pay code.
There was an increased risk of death from NHL among male hourly workers but not salaried workers, with SMRs of 1.49 and 0.68, respectively. The same pattern occurred for rectal cancer, with SMRs of 1.71 and 0.71, respectively.
The study also revealed an elevated incidence of pleural cancers in salaried males, mesothelioma in hourly workers, and testicular cancer in salaried males.
The increase in mesothelioma and pleural cancers was seen only in workers hired before 1969, which would support a link between the cancers and asbestos exposure. However, the researchers could find no evidence that asbestos was used in manufacturing at the facility.
Similarly, the investigators found no significant link between exposure to specific chemicals and the increased mortality from NHL or rectal cancer. And there was no significant link between exposure and testicular cancer.
Although these results do not suggest a strong role for occupational chemical exposures in cancer incidence and mortality, the researchers said risks from occupational exposures cannot be ruled out due to limitations of this study and the relative youth of this patient cohort. ![]()
Age-adjusted D-dimer cutoff appears safe, effective

embolism; Credit: Medical
College of Georgia
Incorporating age into D-dimer test results can help clinicians more accurately diagnose pulmonary embolism (PE) in older patients, according to a new study.
Adjusting the D-dimer cutoff between abnormal and normal results according to a patient’s age allowed researchers to better differentiate healthy patients from those with PE.
The method did prove ineffective in 1 patient, who was ultimately diagnosed with venous thromboembolism (VTE).
But the remaining 300 patients who had D-dimer levels above the standard cutoff and below their age-adjusted cutoff were VTE-free during follow-up.
And among patients aged 75 and older, the age-adjusted cutoff safely excluded PE in about 30% of patients, whereas the standard cutoff excluded PE in about 6%.
Marc Righini, MD, of Geneva University Hospital in Switzerland, and his colleagues conducted this research and described their results in JAMA.
Previous studies showed that D-dimer levels increase with age. So the proportion of healthy patients with abnormal test results (above 500 µg/L for most tests) increases with age, and this limits the test’s utility in older patients.
Therefore, Dr Righini and his colleagues decided to determine whether an age-adjusted D-dimer threshold could safely exclude the diagnosis of PE in older patients. The team changed the cutoff between abnormal and normal results by multiplying the patient’s age by 10 in those 50 years or older.
The study included 3324 patients with suspected PE who first underwent a clinical probability assessment using either the simplified, revised Geneva score or the 2-level Wells score for PE.
If patients had a high clinical probability of PE (n=426), they underwent computed tomography pulmonary angiography (CTPA) and received treatment according to the results.
If PE was deemed unlikely, patients had a D-dimer test. Of these 2898 patients, 817 had normal results (< 500 μg/L), 337 had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, and 1744 had results higher than their age-adjusted cutoff.
Those 1744 patients joined the group of 426 who underwent CTPA (n=2170), and 631 of them were diagnosed with PE.
Of the 1539 patients who were not diagnosed with PE, 1481 completed the 3-month follow-up without receiving anticoagulant therapy. During that time, 7 of those patients had a confirmed VTE.
Of the 337 patients who had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, 331 completed follow-up without anticoagulation. And 1 of those patients was diagnosed with a VTE during that time.
Similarly, of the 817 patients who had normal D-dimer results, 810 completed follow-up without anticoagulation, and 1 patient was diagnosed with VTE.
So the age-adjusted D-dimer test and clinical probability assessment proved largely effective in identifying those patients at risk of VTE. But it also increased the proportion of elderly patients in whom PE could be safely excluded without further imaging.
Among the 766 patients aged 75 and older, 673 did not have a high clinical probability of PE. And using the age-adjusted cutoff instead of the 500 μg/L cutoff increased the proportion of patients in whom PE could be excluded from 6.4% (43/673) to 29.7% (200/673), without any additional false-negative findings.
The researchers said future studies should assess the utility of the age-adjusted D-dimer cutoff in clinical practice. It remains to be seen whether this method can decrease costs or improve the quality of care. ![]()
embolism; Credit: Medical
College of Georgia
Incorporating age into D-dimer test results can help clinicians more accurately diagnose pulmonary embolism (PE) in older patients, according to a new study.
Adjusting the D-dimer cutoff between abnormal and normal results according to a patient’s age allowed researchers to better differentiate healthy patients from those with PE.
The method did prove ineffective in 1 patient, who was ultimately diagnosed with venous thromboembolism (VTE).
But the remaining 300 patients who had D-dimer levels above the standard cutoff and below their age-adjusted cutoff were VTE-free during follow-up.
And among patients aged 75 and older, the age-adjusted cutoff safely excluded PE in about 30% of patients, whereas the standard cutoff excluded PE in about 6%.
Marc Righini, MD, of Geneva University Hospital in Switzerland, and his colleagues conducted this research and described their results in JAMA.
Previous studies showed that D-dimer levels increase with age. So the proportion of healthy patients with abnormal test results (above 500 µg/L for most tests) increases with age, and this limits the test’s utility in older patients.
Therefore, Dr Righini and his colleagues decided to determine whether an age-adjusted D-dimer threshold could safely exclude the diagnosis of PE in older patients. The team changed the cutoff between abnormal and normal results by multiplying the patient’s age by 10 in those 50 years or older.
The study included 3324 patients with suspected PE who first underwent a clinical probability assessment using either the simplified, revised Geneva score or the 2-level Wells score for PE.
If patients had a high clinical probability of PE (n=426), they underwent computed tomography pulmonary angiography (CTPA) and received treatment according to the results.
If PE was deemed unlikely, patients had a D-dimer test. Of these 2898 patients, 817 had normal results (< 500 μg/L), 337 had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, and 1744 had results higher than their age-adjusted cutoff.
Those 1744 patients joined the group of 426 who underwent CTPA (n=2170), and 631 of them were diagnosed with PE.
Of the 1539 patients who were not diagnosed with PE, 1481 completed the 3-month follow-up without receiving anticoagulant therapy. During that time, 7 of those patients had a confirmed VTE.
Of the 337 patients who had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, 331 completed follow-up without anticoagulation. And 1 of those patients was diagnosed with a VTE during that time.
Similarly, of the 817 patients who had normal D-dimer results, 810 completed follow-up without anticoagulation, and 1 patient was diagnosed with VTE.
So the age-adjusted D-dimer test and clinical probability assessment proved largely effective in identifying those patients at risk of VTE. But it also increased the proportion of elderly patients in whom PE could be safely excluded without further imaging.
Among the 766 patients aged 75 and older, 673 did not have a high clinical probability of PE. And using the age-adjusted cutoff instead of the 500 μg/L cutoff increased the proportion of patients in whom PE could be excluded from 6.4% (43/673) to 29.7% (200/673), without any additional false-negative findings.
The researchers said future studies should assess the utility of the age-adjusted D-dimer cutoff in clinical practice. It remains to be seen whether this method can decrease costs or improve the quality of care. ![]()
embolism; Credit: Medical
College of Georgia
Incorporating age into D-dimer test results can help clinicians more accurately diagnose pulmonary embolism (PE) in older patients, according to a new study.
Adjusting the D-dimer cutoff between abnormal and normal results according to a patient’s age allowed researchers to better differentiate healthy patients from those with PE.
The method did prove ineffective in 1 patient, who was ultimately diagnosed with venous thromboembolism (VTE).
But the remaining 300 patients who had D-dimer levels above the standard cutoff and below their age-adjusted cutoff were VTE-free during follow-up.
And among patients aged 75 and older, the age-adjusted cutoff safely excluded PE in about 30% of patients, whereas the standard cutoff excluded PE in about 6%.
Marc Righini, MD, of Geneva University Hospital in Switzerland, and his colleagues conducted this research and described their results in JAMA.
Previous studies showed that D-dimer levels increase with age. So the proportion of healthy patients with abnormal test results (above 500 µg/L for most tests) increases with age, and this limits the test’s utility in older patients.
Therefore, Dr Righini and his colleagues decided to determine whether an age-adjusted D-dimer threshold could safely exclude the diagnosis of PE in older patients. The team changed the cutoff between abnormal and normal results by multiplying the patient’s age by 10 in those 50 years or older.
The study included 3324 patients with suspected PE who first underwent a clinical probability assessment using either the simplified, revised Geneva score or the 2-level Wells score for PE.
If patients had a high clinical probability of PE (n=426), they underwent computed tomography pulmonary angiography (CTPA) and received treatment according to the results.
If PE was deemed unlikely, patients had a D-dimer test. Of these 2898 patients, 817 had normal results (< 500 μg/L), 337 had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, and 1744 had results higher than their age-adjusted cutoff.
Those 1744 patients joined the group of 426 who underwent CTPA (n=2170), and 631 of them were diagnosed with PE.
Of the 1539 patients who were not diagnosed with PE, 1481 completed the 3-month follow-up without receiving anticoagulant therapy. During that time, 7 of those patients had a confirmed VTE.
Of the 337 patients who had D-dimer levels ≥ 500 μg/L but below their age-adjusted cutoff, 331 completed follow-up without anticoagulation. And 1 of those patients was diagnosed with a VTE during that time.
Similarly, of the 817 patients who had normal D-dimer results, 810 completed follow-up without anticoagulation, and 1 patient was diagnosed with VTE.
So the age-adjusted D-dimer test and clinical probability assessment proved largely effective in identifying those patients at risk of VTE. But it also increased the proportion of elderly patients in whom PE could be safely excluded without further imaging.
Among the 766 patients aged 75 and older, 673 did not have a high clinical probability of PE. And using the age-adjusted cutoff instead of the 500 μg/L cutoff increased the proportion of patients in whom PE could be excluded from 6.4% (43/673) to 29.7% (200/673), without any additional false-negative findings.
The researchers said future studies should assess the utility of the age-adjusted D-dimer cutoff in clinical practice. It remains to be seen whether this method can decrease costs or improve the quality of care. ![]()
NICE wants more info on lenalidomide in MM

Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations. ![]()
Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations. ![]()
Credit: CDC
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of lenalidomide (Revlimid) in multiple myeloma (MM) patients who have previously received bortezomib.
Based on current information, NICE has said it cannot recommend the drug for patients who have received bortezomib once and are unable to receive thalidomide or undergo hematopoietic stem cell transplant.
NICE’s previous recommendation regarding lenalidomide in MM has not changed. The drug will still be available through the National Health Service (NHS) for MM patients who have received 2 or more prior therapies.
“We are now looking specifically at how well lenalidomide works after someone has received bortezomib, and whether it provides value for money,” said Sir Andrew Dillon, NICE Chief Executive.
“However, from the information provided by the manufacturer, it was unclear if lenalidomide was as effective as re-treatment with bortezomib, and the manufacturer’s own economic model showed that the drug would not be cost effective at this stage.”
“Because of this, we are unable to recommend the drug in preliminary recommendations. We hope that the manufacturer, Celgene, will look again at their submission.”
NICE also pointed out that, since the organization recommended lenalidomide for MM in 2009, there have been no studies comparing lenalidomide to other treatments in these patients. Celgene has only provided data comparing lenalidomide to placebo.
In addition, for the 2009 guidance, Celgene submitted a patient access scheme, where they bear the costs of the drug beyond 26 cycles (normally for a period of 2 years). And this enabled NICE to recommend the drug. But Celgene has not submitted a patient access scheme for the current appraisal.
NICE considered all the cost effectiveness models Celgene submitted to be fundamentally flawed.
NICE concluded that the most plausible costs per quality-adjusted life-year for lenalidomide compared with bortezomib or standard chemotherapies were more than £30,000, whether bortezomib re-treatment was appropriate or not.
Lenalidomide is available as a 21-capsule pack. The cost per pack varies according to capsule size: £3570 (5 mg), £3780 (10 mg), £3969 (15 mg), and £4368 (25 mg). The recommended starting dose is 25 mg orally, once daily on days 1-21 of repeated 28-day cycles.
The draft guidance is open for public comment until April 4. Until a final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance, it replaces local recommendations.
FDA approves drug for infantile hemangioma
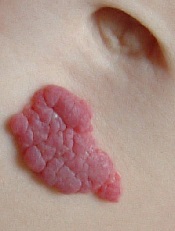
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients.
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients.
infant’s stomach
The US Food and Drug Administration has approved oral propranolol hydrochloride (Hemangeol) to treat proliferating infantile hemangiomas that require systemic therapy.
The drug, which is also under review in the European Union, will be available in the US in June.
Infantile hemangioma is the most common vascular benign tumor of infancy, affecting 3% to 10% of newborns. The lesions are rarely detectable at birth and start growing noticeably in the first 4 to 6 weeks of life.
While most infantile hemangiomas do not require treatment, approximately 12% do. Depending upon their location, infantile hemangiomas might impair breathing, eating, or vision, or become life-threatening.
Propranolol has long been used in cardiology, but its use in infantile hemangiomas is relatively new. In 2007, Christine Léauté-Labreze, MD, a dermatologist at the Bordeaux University Hospital in France, discovered that propranolol could treat infantile hemangiomas.
Since then, the drug has been used for this indication off-label. And in 2009, Pierre Fabre Dermatologie began developing propranolol hydrochloride for use in infantile hemangiomas.
In a study of 32 children, propranolol slowed the growth of infantile hemangiomas in 100% of patients. Patients had received propranolol at 2 to 3 mg/kg per day for a median of 6.1 months. Side effects were “limited and mild,” according to researchers (V Sans et al. Pediatrics 2009).
Researchers also conducted a randomized, controlled trial of the drug in infants 5 weeks to 5 months old at therapy initiation. The team compared 4 propranolol treatment protocols (1 or 3 mg/kg/day for 3 or 6 months) to placebo.
Propranolol at a daily dose of 3 mg/kg for 6 months had a 60.4% success rate, compared to 3.6% in the placebo group (P<0.0001). Success was defined as complete or nearly complete resolution of the target hemangioma. However, 11.4% of patients needed to be re-treated after stopping propranolol.
The drug is contraindicated in premature infants who have a corrected age of less than 5 weeks, weight less than 2 kg, known hypersensitivity to propranolol or any of its excipients, asthma or a history of bronchospasm, pheochromocytoma, blood pressure less than 50/30 mmHg, a heart rate less than 80 beats per minute, greater than first degree heart block, or decompensated heart failure.
Propranolol hydrochloride can cause serious side effects, including hypoglycemia, bradycardia, hypotension, and bronchospasm. The drug can worsen congestive heart failure and may increase the risk of stroke in children with PHACE syndrome.
The most frequently reported adverse reactions, occurring in more than 10% of infants receiving propranolol hydrochloride, were sleep disorders, diarrhea, vomiting, and aggravated respiratory tract infections, such as bronchitis and bronchiolitis associated with cough and fever. Adverse reactions led to treatment discontinuation in fewer than 2% of treated patients.
FDA approves IV formulation of antifungal agent
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck.
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck.
The US Food and Drug Administration has approved an intravenous formulation of posaconazole (Noxafil), which is expected to be available at wholesalers in mid-April.
The antifungal agent is already available as delayed-release tablets and in an oral suspension formulation.
In any formulation, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in immunocompromised patients who are at high risk of developing these infections.
This includes patients who have developed graft-vs-host disease after hematopoietic stem cell transplant and patients with hematologic malignancies who have prolonged neutropenia resulting from chemotherapy.
Posaconazole injection is indicated for use in patients 18 years of age and older. The delayed-release tablets and oral suspension are indicated for patients 13 years of age and older.
Posaconazole injection is administered with a loading dose of 300 mg (one 300 mg vial) twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by slow intravenous infusion over approximately 90 minutes.
Once combined with a mixture of intravenous solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole injection should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2-8 degrees C (36-46 degrees F).
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
In clinical trials, the adverse reactions reported for posaconazole injection were generally similar to those reported in trials of posaconazole oral suspension. The most frequently reported adverse reactions with an onset during the posaconazole intravenous phase of dosing 300 mg once-daily therapy were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
Patients who are allergic to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given along with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
For more details, see the complete prescribing information. Posaconazole is marketed as Noxafil by Merck.
High cost of eculizumab needs explaining, NICE says

Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients).
Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients).
Credit: Bill Branson
The UK’s National Institute for Health and Care Excellence (NICE) has asked the manufacturer of eculizumab (Soliris) to explain the high cost of the drug.
Research has suggested that eculizumab can be effective against atypical hemolytic uremic syndrome (aHUS), a rare disease that often proves difficult to treat.
So the National Health Service (NHS) has made eculizumab available for these patients on an interim basis, pending NICE appraisal.
However, an advisory committee for NICE has estimated that routine use of eculizumab would cost the NHS about £58 million in the first year, and costs would exceed £80 million in 5 years.
Therefore, in its draft guidance for eculizumab, the committee has asked the drug’s manufacturer, Alexion Pharma, to explain its costs.
“[The committee has] asked for clarification from the company on aspects of the manufacturing, research, and development costs of a medicinal product for the treatment of a very rare condition,” said Sir Andrew Dillon, Chief Executive at NICE.
“It has also asked NHS England for clarification on treatment costs for a highly specialized technology in the context of a highly specialized service. The information provided will be considered at the next meeting of the evaluation committee in April.”
The committee will also consider comments on its draft guidance at the meeting. The guidance is available for public comment until midday on March 25.
About aHUS
Estimated to affect more than 200 people in England, aHUS is a chronic condition that causes severe inflammation of blood vessels and thrombus formation in small blood vessels throughout the body.
Patients with aHUS can experience significant kidney impairment, thrombosis, heart failure, and brain injury. In about 70% of patients, aHUS is associated with an underlying genetic or acquired abnormality of proteins in the complement immune system.
Before eculizumab became available, plasma therapy (infusion and/or exchange) was the main treatment for aHUS. However, not all patients with aHUS respond to plasma therapy. And up to 40% of patients may die or progress to end-stage renal failure and require dialysis with the first clinical aHUS manifestation, despite the use of plasma therapy.
Some patients may be eligible for a kidney or combined kidney-liver transplantation. However, there is a high risk of organ rejection following recurrent disease.
Eculizumab in aHUS: Treatment and cost
Eculizumab inhibits the disease process by blocking pro-thrombotic and pro-inflammatory processes that can lead to cellular damage in small blood vessels throughout the body, renal failure, and damage to other organs.
Eculizumab is given intravenously in adults as initial treatment at a dose of 900 mg for 4 weeks, then as maintenance treatment at a dose of 1200 mg on week 5 and then every 12 to 16 days. The summary of product characteristics for eculizumab states that treatment should be continued for the patient’s lifetime, unless discontinuation is clinically indicated.
Eculizumab costs £3150 per 30 mL vial, excluding tax, according to the British National Formulary.
“Alexion insisted that its information about the overall cost of eculizumab be kept confidential, and so NICE is unable to share these details of the Alexion submission with stakeholders,” Dillon said.
However, to allow consultees and commentators to properly engage in the consultation process, NICE has prepared an estimate of the possible budget impact eculizumab might have, using information available in the public domain.
This is based on a treatment cost of £340,200 per adult patient in the first year (based on the acquisition cost of the drug and the recommended dosing for an adult), and assumes a patient cohort of 170, as estimated by NHS England in its interim commissioning policy.
Assuming all of these patients receive eculizumab, the budget impact for the first year would be £57.8 million. If an additional 20 new patients are treated the following year (based on a worldwide incidence of 0.4 million), the budget impact will rise to £62.5 million. That is assuming all new patients are treated and all existing patients continue to be treated at the maintenance cost of £327,600 per year.
Using the same assumptions, the budget impact will rise to £69 million in year 3 (190 existing and 20 new patients), £75 million in year 4 (210 existing and 20 new patients) and £82 million in year 5 (230 existing and 20 new patients).