User login
Protein helps HSP90 inhibitors fight cancers

Credit: PNAS
Researchers have discovered how a molecule called CUL5 helps HSP90 inhibitors kill cancer cells, according to a study published in Proceedings of the National Academy of Sciences.
The team found that CUL5 is required for the degradation of proteins that promote cancer cell proliferation, and CUL5 works in opposition to HSP90.
When cancer cells are treated with HSP90 inhibitors, CUL5 immediately steps in to help dispose of the proliferation-promoting proteins.
Based on these findings, the researchers speculate that some patients may be resistant to HSP90 inhibitors if their cancer cells have lower amounts of CUL5. And conversely, the drugs may work better in patients with higher CUL5 levels.
Paul Workman, PhD, of The Institute of Cancer Research in London, UK, and his colleagues conducted this research in cell lines of melanoma, as well as colon, breast, and lung cancers.
They first tested the HSP90 inhibitor 17-AAG in HT29 cells and found that CUL5 is involved in the drug-induced degradation of several protein kinase clients of HSP90.
Then, the researchers assessed the effects of silencing CUL5 and discovered that it delays the abrogation of protein signaling caused by an HSP90 inhibitor.
Furthermore, silencing CUL5 reduced cellular sensitivity to 3 different HSP90 inhibitors across the 4 different cancer types studied, which, as the researchers pointed out, are driven by different protein kinases.
So the team believes this research could apply to a number of different cancers. HSP90 inhibitors have proven effective against a range of malignancies, including leukemias, lymphomas, and multiple myeloma.
“We’ve known for some time that drugs that block HSP90 have great potential as treatments for cancers . . . , and we had an initial clue that the protein CUL5 may be involved in some way in how these drugs work,” Dr Workman said.
“Our new research shows that CUL5 is not only vital in the response of cancer cells to HSP90 inhibitors but also reveals surprising insights into precisely how it works by acting at several different levels. What also surprised us was that CUL5 gets rid of many more of the cancer-causing proteins than we’d previously imagined and that it’s effective across several types of tumor.”
“This suggests that a test for CUL5 in patients could help us tell whether they might respond to HSP90-blocking drugs, as well as pointing to new targets to develop more effective drugs.” ![]()
Credit: PNAS
Researchers have discovered how a molecule called CUL5 helps HSP90 inhibitors kill cancer cells, according to a study published in Proceedings of the National Academy of Sciences.
The team found that CUL5 is required for the degradation of proteins that promote cancer cell proliferation, and CUL5 works in opposition to HSP90.
When cancer cells are treated with HSP90 inhibitors, CUL5 immediately steps in to help dispose of the proliferation-promoting proteins.
Based on these findings, the researchers speculate that some patients may be resistant to HSP90 inhibitors if their cancer cells have lower amounts of CUL5. And conversely, the drugs may work better in patients with higher CUL5 levels.
Paul Workman, PhD, of The Institute of Cancer Research in London, UK, and his colleagues conducted this research in cell lines of melanoma, as well as colon, breast, and lung cancers.
They first tested the HSP90 inhibitor 17-AAG in HT29 cells and found that CUL5 is involved in the drug-induced degradation of several protein kinase clients of HSP90.
Then, the researchers assessed the effects of silencing CUL5 and discovered that it delays the abrogation of protein signaling caused by an HSP90 inhibitor.
Furthermore, silencing CUL5 reduced cellular sensitivity to 3 different HSP90 inhibitors across the 4 different cancer types studied, which, as the researchers pointed out, are driven by different protein kinases.
So the team believes this research could apply to a number of different cancers. HSP90 inhibitors have proven effective against a range of malignancies, including leukemias, lymphomas, and multiple myeloma.
“We’ve known for some time that drugs that block HSP90 have great potential as treatments for cancers . . . , and we had an initial clue that the protein CUL5 may be involved in some way in how these drugs work,” Dr Workman said.
“Our new research shows that CUL5 is not only vital in the response of cancer cells to HSP90 inhibitors but also reveals surprising insights into precisely how it works by acting at several different levels. What also surprised us was that CUL5 gets rid of many more of the cancer-causing proteins than we’d previously imagined and that it’s effective across several types of tumor.”
“This suggests that a test for CUL5 in patients could help us tell whether they might respond to HSP90-blocking drugs, as well as pointing to new targets to develop more effective drugs.” ![]()
Credit: PNAS
Researchers have discovered how a molecule called CUL5 helps HSP90 inhibitors kill cancer cells, according to a study published in Proceedings of the National Academy of Sciences.
The team found that CUL5 is required for the degradation of proteins that promote cancer cell proliferation, and CUL5 works in opposition to HSP90.
When cancer cells are treated with HSP90 inhibitors, CUL5 immediately steps in to help dispose of the proliferation-promoting proteins.
Based on these findings, the researchers speculate that some patients may be resistant to HSP90 inhibitors if their cancer cells have lower amounts of CUL5. And conversely, the drugs may work better in patients with higher CUL5 levels.
Paul Workman, PhD, of The Institute of Cancer Research in London, UK, and his colleagues conducted this research in cell lines of melanoma, as well as colon, breast, and lung cancers.
They first tested the HSP90 inhibitor 17-AAG in HT29 cells and found that CUL5 is involved in the drug-induced degradation of several protein kinase clients of HSP90.
Then, the researchers assessed the effects of silencing CUL5 and discovered that it delays the abrogation of protein signaling caused by an HSP90 inhibitor.
Furthermore, silencing CUL5 reduced cellular sensitivity to 3 different HSP90 inhibitors across the 4 different cancer types studied, which, as the researchers pointed out, are driven by different protein kinases.
So the team believes this research could apply to a number of different cancers. HSP90 inhibitors have proven effective against a range of malignancies, including leukemias, lymphomas, and multiple myeloma.
“We’ve known for some time that drugs that block HSP90 have great potential as treatments for cancers . . . , and we had an initial clue that the protein CUL5 may be involved in some way in how these drugs work,” Dr Workman said.
“Our new research shows that CUL5 is not only vital in the response of cancer cells to HSP90 inhibitors but also reveals surprising insights into precisely how it works by acting at several different levels. What also surprised us was that CUL5 gets rid of many more of the cancer-causing proteins than we’d previously imagined and that it’s effective across several types of tumor.”
“This suggests that a test for CUL5 in patients could help us tell whether they might respond to HSP90-blocking drugs, as well as pointing to new targets to develop more effective drugs.” ![]()
Drug gets orphan designation for AML

Credit: NIH
The US Food and Drug Administration and the European Commission have granted volasertib orphan designation for the treatment of acute myeloid leukemia (AML).
Volasertib, an investigational inhibitor of polo-like kinase 1 (Plk1), works by arresting the cell cycle and inducing apoptosis.
The drug is under evaluation as a potential treatment for patients aged 65 or older with previously untreated AML who are ineligible for intensive remission induction therapy.
In both the US and the European Union, orphan designation is awarded for drugs intended to treat rare conditions for which no authorized treatment exists. The designation gives the company developing volasertib, Boehringer Ingelheim, regulatory support and incentives to help the development and authorization process.
Volasertib has already been tested in a phase 1/2 trial of patients with newly diagnosed AML who were considered ineligible for intensive remission induction therapy. The results were presented at the 2012 ASH Annual Meeting as abstract 411.
In this study, volasertib in combination with low-dose cytarabine (LDAC) elicited higher rates of objective response and an improvement in event-free survival, when compared to LDAC alone.
Eighty-seven AML patients were assigned to receive volasertib + LDAC (n=42) or LDAC alone (n=45). Patient characteristics were similar between the 2 groups.
The objective response rate was 31% among patients who received volasertib + LDAC and 11.1% in those who received LDAC alone. The complete response rates were 16.7% and 6.7%, respectively.
The median event-free survival was 169 days for patients who received volasertib + LDAC and 69 days for patients who received LDAC alone.
Grade 3 or higher adverse events were more common in the volasertib + LDAC arm than the LDAC-alone arm—95.2% vs 68.9%.
The most frequent adverse events of any grade occurring in the volasertib + LDAC arm were febrile neutropenia (50%), constipation (45.2%), nausea (40.5%), and anemia (40.5%).
In the LDAC-alone arm, the most common adverse events were nausea (33.3%), anemia (28.9%), pyrexia (28.9%), constipation (26.7%), asthenia (26.7%), and diarrhea (26.7%).
Based on these results, researchers initiated a phase 3 study, called POLO-AML-2, comparing volasertib plus LDAC to LDAC plus placebo in older AML patients. ![]()
Credit: NIH
The US Food and Drug Administration and the European Commission have granted volasertib orphan designation for the treatment of acute myeloid leukemia (AML).
Volasertib, an investigational inhibitor of polo-like kinase 1 (Plk1), works by arresting the cell cycle and inducing apoptosis.
The drug is under evaluation as a potential treatment for patients aged 65 or older with previously untreated AML who are ineligible for intensive remission induction therapy.
In both the US and the European Union, orphan designation is awarded for drugs intended to treat rare conditions for which no authorized treatment exists. The designation gives the company developing volasertib, Boehringer Ingelheim, regulatory support and incentives to help the development and authorization process.
Volasertib has already been tested in a phase 1/2 trial of patients with newly diagnosed AML who were considered ineligible for intensive remission induction therapy. The results were presented at the 2012 ASH Annual Meeting as abstract 411.
In this study, volasertib in combination with low-dose cytarabine (LDAC) elicited higher rates of objective response and an improvement in event-free survival, when compared to LDAC alone.
Eighty-seven AML patients were assigned to receive volasertib + LDAC (n=42) or LDAC alone (n=45). Patient characteristics were similar between the 2 groups.
The objective response rate was 31% among patients who received volasertib + LDAC and 11.1% in those who received LDAC alone. The complete response rates were 16.7% and 6.7%, respectively.
The median event-free survival was 169 days for patients who received volasertib + LDAC and 69 days for patients who received LDAC alone.
Grade 3 or higher adverse events were more common in the volasertib + LDAC arm than the LDAC-alone arm—95.2% vs 68.9%.
The most frequent adverse events of any grade occurring in the volasertib + LDAC arm were febrile neutropenia (50%), constipation (45.2%), nausea (40.5%), and anemia (40.5%).
In the LDAC-alone arm, the most common adverse events were nausea (33.3%), anemia (28.9%), pyrexia (28.9%), constipation (26.7%), asthenia (26.7%), and diarrhea (26.7%).
Based on these results, researchers initiated a phase 3 study, called POLO-AML-2, comparing volasertib plus LDAC to LDAC plus placebo in older AML patients. ![]()
Credit: NIH
The US Food and Drug Administration and the European Commission have granted volasertib orphan designation for the treatment of acute myeloid leukemia (AML).
Volasertib, an investigational inhibitor of polo-like kinase 1 (Plk1), works by arresting the cell cycle and inducing apoptosis.
The drug is under evaluation as a potential treatment for patients aged 65 or older with previously untreated AML who are ineligible for intensive remission induction therapy.
In both the US and the European Union, orphan designation is awarded for drugs intended to treat rare conditions for which no authorized treatment exists. The designation gives the company developing volasertib, Boehringer Ingelheim, regulatory support and incentives to help the development and authorization process.
Volasertib has already been tested in a phase 1/2 trial of patients with newly diagnosed AML who were considered ineligible for intensive remission induction therapy. The results were presented at the 2012 ASH Annual Meeting as abstract 411.
In this study, volasertib in combination with low-dose cytarabine (LDAC) elicited higher rates of objective response and an improvement in event-free survival, when compared to LDAC alone.
Eighty-seven AML patients were assigned to receive volasertib + LDAC (n=42) or LDAC alone (n=45). Patient characteristics were similar between the 2 groups.
The objective response rate was 31% among patients who received volasertib + LDAC and 11.1% in those who received LDAC alone. The complete response rates were 16.7% and 6.7%, respectively.
The median event-free survival was 169 days for patients who received volasertib + LDAC and 69 days for patients who received LDAC alone.
Grade 3 or higher adverse events were more common in the volasertib + LDAC arm than the LDAC-alone arm—95.2% vs 68.9%.
The most frequent adverse events of any grade occurring in the volasertib + LDAC arm were febrile neutropenia (50%), constipation (45.2%), nausea (40.5%), and anemia (40.5%).
In the LDAC-alone arm, the most common adverse events were nausea (33.3%), anemia (28.9%), pyrexia (28.9%), constipation (26.7%), asthenia (26.7%), and diarrhea (26.7%).
Based on these results, researchers initiated a phase 3 study, called POLO-AML-2, comparing volasertib plus LDAC to LDAC plus placebo in older AML patients. ![]()
Protein discovery could aid antimalarial development
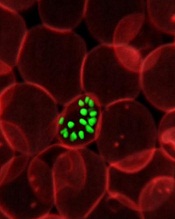
infecting a red blood cell
Credit: St Jude Hospital
High-resolution technology has revealed the structure of actin proteins in the malaria parasite Plasmodium falciparum.
Researchers found the 2 versions of the protein differ from each other more than actin proteins in any other organism.
And they were able to identify areas within these proteins that cause their different behaviors.
The team believes this discovery, published in PLOS Pathogens, may contribute to the development of tailor-made drugs against malaria.
Malaria parasites use actin to enter human cells and leave them again. Inside cells, actin confers stability, allows for cell division, and makes the movement of single cells possible.
The dynamic behavior needed for these processes is enabled by individual globular actin molecules assembling together to form thread-like structures called filaments.
The malaria parasite has 2 versions of actin, actin I and actin II, which differ substantially from each other. These structural proteins are crucial for the pathogen’s infectivity, but researchers have not been able to demonstrate filament formation in the parasite, until now.
Inari Kursula, PhD, of the University of Oulu in Finland, and her colleagues have succeeded in detecting filament assembly of the parasite actin II proteins. For this, they used electron microscopy, which overcomes the resolution limit of classical light microscopy.
Male malaria parasites from which the researchers had deleted actin II were not able to form mature germ cells and, consequently, could not reproduce and propagate.
The researchers therefore speculated that having only 1 actin variant is not sufficient for this process, but they wondered why the 2 proteins show such different behavior. To answer this question, the team deciphered the structure of the globular actin proteins using X-radiation.
“We were able to determine the structures of actin I and actin II at very high resolutions–down to 1.3 and 2.2 Ångström, respectively,” Dr Kursula said.
“With this, we are in the range of single atoms. The structures show us that the 2 variants differ more from each other than actins in any other known living organism do.”
The high resolution enabled the researchers to identify areas within the proteins that cause the different behavior.
“We now understand that Plasmodium actin filaments are very different from other actin filaments—like, for example, from those found in humans—and that they are assembled in a very different manner,” Dr Kursula said.
“Now that we know the structural basis for this, we can look for ways to specifically interfere with the parasite cytoskeleton.”
This knowledge might, in the future, aid the design of tailor-made antimalarial drugs. ![]()
infecting a red blood cell
Credit: St Jude Hospital
High-resolution technology has revealed the structure of actin proteins in the malaria parasite Plasmodium falciparum.
Researchers found the 2 versions of the protein differ from each other more than actin proteins in any other organism.
And they were able to identify areas within these proteins that cause their different behaviors.
The team believes this discovery, published in PLOS Pathogens, may contribute to the development of tailor-made drugs against malaria.
Malaria parasites use actin to enter human cells and leave them again. Inside cells, actin confers stability, allows for cell division, and makes the movement of single cells possible.
The dynamic behavior needed for these processes is enabled by individual globular actin molecules assembling together to form thread-like structures called filaments.
The malaria parasite has 2 versions of actin, actin I and actin II, which differ substantially from each other. These structural proteins are crucial for the pathogen’s infectivity, but researchers have not been able to demonstrate filament formation in the parasite, until now.
Inari Kursula, PhD, of the University of Oulu in Finland, and her colleagues have succeeded in detecting filament assembly of the parasite actin II proteins. For this, they used electron microscopy, which overcomes the resolution limit of classical light microscopy.
Male malaria parasites from which the researchers had deleted actin II were not able to form mature germ cells and, consequently, could not reproduce and propagate.
The researchers therefore speculated that having only 1 actin variant is not sufficient for this process, but they wondered why the 2 proteins show such different behavior. To answer this question, the team deciphered the structure of the globular actin proteins using X-radiation.
“We were able to determine the structures of actin I and actin II at very high resolutions–down to 1.3 and 2.2 Ångström, respectively,” Dr Kursula said.
“With this, we are in the range of single atoms. The structures show us that the 2 variants differ more from each other than actins in any other known living organism do.”
The high resolution enabled the researchers to identify areas within the proteins that cause the different behavior.
“We now understand that Plasmodium actin filaments are very different from other actin filaments—like, for example, from those found in humans—and that they are assembled in a very different manner,” Dr Kursula said.
“Now that we know the structural basis for this, we can look for ways to specifically interfere with the parasite cytoskeleton.”
This knowledge might, in the future, aid the design of tailor-made antimalarial drugs. ![]()
infecting a red blood cell
Credit: St Jude Hospital
High-resolution technology has revealed the structure of actin proteins in the malaria parasite Plasmodium falciparum.
Researchers found the 2 versions of the protein differ from each other more than actin proteins in any other organism.
And they were able to identify areas within these proteins that cause their different behaviors.
The team believes this discovery, published in PLOS Pathogens, may contribute to the development of tailor-made drugs against malaria.
Malaria parasites use actin to enter human cells and leave them again. Inside cells, actin confers stability, allows for cell division, and makes the movement of single cells possible.
The dynamic behavior needed for these processes is enabled by individual globular actin molecules assembling together to form thread-like structures called filaments.
The malaria parasite has 2 versions of actin, actin I and actin II, which differ substantially from each other. These structural proteins are crucial for the pathogen’s infectivity, but researchers have not been able to demonstrate filament formation in the parasite, until now.
Inari Kursula, PhD, of the University of Oulu in Finland, and her colleagues have succeeded in detecting filament assembly of the parasite actin II proteins. For this, they used electron microscopy, which overcomes the resolution limit of classical light microscopy.
Male malaria parasites from which the researchers had deleted actin II were not able to form mature germ cells and, consequently, could not reproduce and propagate.
The researchers therefore speculated that having only 1 actin variant is not sufficient for this process, but they wondered why the 2 proteins show such different behavior. To answer this question, the team deciphered the structure of the globular actin proteins using X-radiation.
“We were able to determine the structures of actin I and actin II at very high resolutions–down to 1.3 and 2.2 Ångström, respectively,” Dr Kursula said.
“With this, we are in the range of single atoms. The structures show us that the 2 variants differ more from each other than actins in any other known living organism do.”
The high resolution enabled the researchers to identify areas within the proteins that cause the different behavior.
“We now understand that Plasmodium actin filaments are very different from other actin filaments—like, for example, from those found in humans—and that they are assembled in a very different manner,” Dr Kursula said.
“Now that we know the structural basis for this, we can look for ways to specifically interfere with the parasite cytoskeleton.”
This knowledge might, in the future, aid the design of tailor-made antimalarial drugs. ![]()
FDA lifts clinical hold on CAR T-cell study

Credit: MSKCC
The US Food and Drug Administration (FDA) has lifted a clinical hold placed on a trial of chimeric antigen receptor (CAR) T-cell therapy, according to one of the study’s investigators.
The study is an evaluation of 19-28z CAR T cells in patients with B-cell acute lymphoblastic leukemia.
Trial enrollment was halted after 2 patients died from complications related to cytokine release syndrome, within 2 weeks of receiving CAR T-cell therapy.
Investigator Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center (MSKCC) in New York, said these deaths have been reviewed, trial enrollment criteria have been changed, and the clinical hold has been lifted.
The FDA put the trial on hold last month, after researchers at MSKCC notified the agency of the deaths and had decided to halt trial enrollment themselves.
Though several other patients have died on this study, only 2 of the deaths had unexpected causes and prompted additional investigation. One of these patients died of cardiovascular disease, and the other died following “persistent seizure activity.”
So MSKCC conducted a review of these cases. And the results prompted them to amend trial enrollment criteria and dosing recommendations. Now, patients with cardiac disease are ineligible to receive 19-28z CAR T cells.
And the T-cell dose a patient receives will depend on the extent of his or her disease. The hope is that this will reduce the risk of cytokine release syndrome and any resulting seizures.
The researchers also noted that the monoclonal antibody tocilizumab has proven effective in treating cytokine release syndrome. ![]()
Credit: MSKCC
The US Food and Drug Administration (FDA) has lifted a clinical hold placed on a trial of chimeric antigen receptor (CAR) T-cell therapy, according to one of the study’s investigators.
The study is an evaluation of 19-28z CAR T cells in patients with B-cell acute lymphoblastic leukemia.
Trial enrollment was halted after 2 patients died from complications related to cytokine release syndrome, within 2 weeks of receiving CAR T-cell therapy.
Investigator Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center (MSKCC) in New York, said these deaths have been reviewed, trial enrollment criteria have been changed, and the clinical hold has been lifted.
The FDA put the trial on hold last month, after researchers at MSKCC notified the agency of the deaths and had decided to halt trial enrollment themselves.
Though several other patients have died on this study, only 2 of the deaths had unexpected causes and prompted additional investigation. One of these patients died of cardiovascular disease, and the other died following “persistent seizure activity.”
So MSKCC conducted a review of these cases. And the results prompted them to amend trial enrollment criteria and dosing recommendations. Now, patients with cardiac disease are ineligible to receive 19-28z CAR T cells.
And the T-cell dose a patient receives will depend on the extent of his or her disease. The hope is that this will reduce the risk of cytokine release syndrome and any resulting seizures.
The researchers also noted that the monoclonal antibody tocilizumab has proven effective in treating cytokine release syndrome. ![]()
Credit: MSKCC
The US Food and Drug Administration (FDA) has lifted a clinical hold placed on a trial of chimeric antigen receptor (CAR) T-cell therapy, according to one of the study’s investigators.
The study is an evaluation of 19-28z CAR T cells in patients with B-cell acute lymphoblastic leukemia.
Trial enrollment was halted after 2 patients died from complications related to cytokine release syndrome, within 2 weeks of receiving CAR T-cell therapy.
Investigator Michel Sadelain, MD, PhD, of Memorial Sloan-Kettering Cancer Center (MSKCC) in New York, said these deaths have been reviewed, trial enrollment criteria have been changed, and the clinical hold has been lifted.
The FDA put the trial on hold last month, after researchers at MSKCC notified the agency of the deaths and had decided to halt trial enrollment themselves.
Though several other patients have died on this study, only 2 of the deaths had unexpected causes and prompted additional investigation. One of these patients died of cardiovascular disease, and the other died following “persistent seizure activity.”
So MSKCC conducted a review of these cases. And the results prompted them to amend trial enrollment criteria and dosing recommendations. Now, patients with cardiac disease are ineligible to receive 19-28z CAR T cells.
And the T-cell dose a patient receives will depend on the extent of his or her disease. The hope is that this will reduce the risk of cytokine release syndrome and any resulting seizures.
The researchers also noted that the monoclonal antibody tocilizumab has proven effective in treating cytokine release syndrome. ![]()
Explaining the link between Down syndrome and B-ALL

Credit: Aaron Logan
Investigators believe they’ve uncovered information that explains the connection between Down syndrome and B-cell acute lymphoblastic leukemia (B-ALL).
In a letter to Nature Genetics, the team described how they tracked the chain of events that links a chromosomal abnormality in Down syndrome to the cellular havoc that occurs in B-cell ALL.
Experiments in mice and patient samples revealed that the gene HMGN1 turns off the function of PRC2, which prompts B-cell proliferation.
“For 80 years, it hasn’t been clear why children with Down syndrome face a sharply elevated risk of ALL,” said the study’s lead author, Andrew Lane, MD, PhD, of Dana-Farber Cancer Institute in Boston.
“Advances in technology—which make it possible to study blood cells and leukemias that model Down syndrome in the laboratory—have enabled us to make that link.”
To trace the link between Down syndrome and B-ALL, the investigators acquired a strain of mice that carry an extra copy of 31 genes found on chromosome 21. B cells from these mice were abnormal and grew uncontrollably, just as they do in B-ALL patients.
The team set out to characterize the pattern of gene activity that distinguishes these abnormal B cells from normal B cells. They found the chief difference was that, in the abnormal cells, PRC2 proteins did not function. Somehow, the loss of PRC2 was spurring the B cells to divide and proliferate before they were fully mature.
To confirm that a shutdown of PRC2 is critical to the formation of B-ALL in Down syndrome patients, the investigators focused on the genes controlled by PRC2. Using 2 sets of B-ALL cell samples—1 from patients with Down syndrome and 1 from patients without it—they measured the activity of thousands of different genes, looking for differences between the 2 sets.
About 100 genes were much more active in the Down syndrome group, and all of them were under the control of PRC2. When PRC2 is silenced, those 100 genes respond with a burst of activity, driving cell growth and division.
The investigators then wondered what gene or group of genes was stifling PRC2 in Down syndrome patients’ B cells. Using cells from the mouse models, the team systematically switched off each of the 31 genes to determine its effect on the cells. When they turned off HMGN1, the cells stopped growing and died.
“We concluded that the extra copy of HMGN1 is important for turning off PRC2, and that, in turn, increases the cell proliferation,” Dr Lane said. “This provides the long-sought-after molecular link between Down syndrome and the development of B-cell ALL.”
Although there are currently no drugs that target HMGN1, the investigators suggest HDAC inhibitors that switch on PRC2 could have an antileukemic effect in patients with Down syndrome. Work is under way to improve these drugs so they can be tested in preclinical experiments.
As other forms of B-ALL also have the same 100-gene signature as the one discovered for B-ALL associated with Down syndrome, agents that target PRC2 might be effective in those cancers as well, Dr Lane noted. ![]()
Credit: Aaron Logan
Investigators believe they’ve uncovered information that explains the connection between Down syndrome and B-cell acute lymphoblastic leukemia (B-ALL).
In a letter to Nature Genetics, the team described how they tracked the chain of events that links a chromosomal abnormality in Down syndrome to the cellular havoc that occurs in B-cell ALL.
Experiments in mice and patient samples revealed that the gene HMGN1 turns off the function of PRC2, which prompts B-cell proliferation.
“For 80 years, it hasn’t been clear why children with Down syndrome face a sharply elevated risk of ALL,” said the study’s lead author, Andrew Lane, MD, PhD, of Dana-Farber Cancer Institute in Boston.
“Advances in technology—which make it possible to study blood cells and leukemias that model Down syndrome in the laboratory—have enabled us to make that link.”
To trace the link between Down syndrome and B-ALL, the investigators acquired a strain of mice that carry an extra copy of 31 genes found on chromosome 21. B cells from these mice were abnormal and grew uncontrollably, just as they do in B-ALL patients.
The team set out to characterize the pattern of gene activity that distinguishes these abnormal B cells from normal B cells. They found the chief difference was that, in the abnormal cells, PRC2 proteins did not function. Somehow, the loss of PRC2 was spurring the B cells to divide and proliferate before they were fully mature.
To confirm that a shutdown of PRC2 is critical to the formation of B-ALL in Down syndrome patients, the investigators focused on the genes controlled by PRC2. Using 2 sets of B-ALL cell samples—1 from patients with Down syndrome and 1 from patients without it—they measured the activity of thousands of different genes, looking for differences between the 2 sets.
About 100 genes were much more active in the Down syndrome group, and all of them were under the control of PRC2. When PRC2 is silenced, those 100 genes respond with a burst of activity, driving cell growth and division.
The investigators then wondered what gene or group of genes was stifling PRC2 in Down syndrome patients’ B cells. Using cells from the mouse models, the team systematically switched off each of the 31 genes to determine its effect on the cells. When they turned off HMGN1, the cells stopped growing and died.
“We concluded that the extra copy of HMGN1 is important for turning off PRC2, and that, in turn, increases the cell proliferation,” Dr Lane said. “This provides the long-sought-after molecular link between Down syndrome and the development of B-cell ALL.”
Although there are currently no drugs that target HMGN1, the investigators suggest HDAC inhibitors that switch on PRC2 could have an antileukemic effect in patients with Down syndrome. Work is under way to improve these drugs so they can be tested in preclinical experiments.
As other forms of B-ALL also have the same 100-gene signature as the one discovered for B-ALL associated with Down syndrome, agents that target PRC2 might be effective in those cancers as well, Dr Lane noted. ![]()
Credit: Aaron Logan
Investigators believe they’ve uncovered information that explains the connection between Down syndrome and B-cell acute lymphoblastic leukemia (B-ALL).
In a letter to Nature Genetics, the team described how they tracked the chain of events that links a chromosomal abnormality in Down syndrome to the cellular havoc that occurs in B-cell ALL.
Experiments in mice and patient samples revealed that the gene HMGN1 turns off the function of PRC2, which prompts B-cell proliferation.
“For 80 years, it hasn’t been clear why children with Down syndrome face a sharply elevated risk of ALL,” said the study’s lead author, Andrew Lane, MD, PhD, of Dana-Farber Cancer Institute in Boston.
“Advances in technology—which make it possible to study blood cells and leukemias that model Down syndrome in the laboratory—have enabled us to make that link.”
To trace the link between Down syndrome and B-ALL, the investigators acquired a strain of mice that carry an extra copy of 31 genes found on chromosome 21. B cells from these mice were abnormal and grew uncontrollably, just as they do in B-ALL patients.
The team set out to characterize the pattern of gene activity that distinguishes these abnormal B cells from normal B cells. They found the chief difference was that, in the abnormal cells, PRC2 proteins did not function. Somehow, the loss of PRC2 was spurring the B cells to divide and proliferate before they were fully mature.
To confirm that a shutdown of PRC2 is critical to the formation of B-ALL in Down syndrome patients, the investigators focused on the genes controlled by PRC2. Using 2 sets of B-ALL cell samples—1 from patients with Down syndrome and 1 from patients without it—they measured the activity of thousands of different genes, looking for differences between the 2 sets.
About 100 genes were much more active in the Down syndrome group, and all of them were under the control of PRC2. When PRC2 is silenced, those 100 genes respond with a burst of activity, driving cell growth and division.
The investigators then wondered what gene or group of genes was stifling PRC2 in Down syndrome patients’ B cells. Using cells from the mouse models, the team systematically switched off each of the 31 genes to determine its effect on the cells. When they turned off HMGN1, the cells stopped growing and died.
“We concluded that the extra copy of HMGN1 is important for turning off PRC2, and that, in turn, increases the cell proliferation,” Dr Lane said. “This provides the long-sought-after molecular link between Down syndrome and the development of B-cell ALL.”
Although there are currently no drugs that target HMGN1, the investigators suggest HDAC inhibitors that switch on PRC2 could have an antileukemic effect in patients with Down syndrome. Work is under way to improve these drugs so they can be tested in preclinical experiments.
As other forms of B-ALL also have the same 100-gene signature as the one discovered for B-ALL associated with Down syndrome, agents that target PRC2 might be effective in those cancers as well, Dr Lane noted. ![]()
Method speeds up analysis of RNA-seq data

Credit: Darren Baker
Researchers say they’ve developed a computational method that dramatically speeds up estimates of gene activity from RNA sequencing (RNA-seq) data.
With the new method, called Sailfish, estimates of gene expression that previously took hours can be completed in a few minutes.
And the researchers say the accuracy of Sailfish equals or exceeds that of previous methods.
The team described the Sailfish method in Nature Biotechnology.
They noted that gigantic repositories of RNA-seq data now exist, making it possible to re-analyze experiments in light of new discoveries.
“But 15 hours a pop really starts to add up, particularly if you want to look at 100 experiments,” said study author Carl Kingsford, PhD, of Carnegie Mellon University in Pittsburg, Pennsylvania.
“With Sailfish, we can give researchers everything they got from previous methods, but faster.”
With previous methods, the RNA molecules from which read sequences originated could be identified and measured only by mapping the reads to their original positions in the larger molecules—a time-consuming process.
Dr Kingsford and his colleagues found this step can actually be eliminated. The team discovered they could allocate parts of the reads to different types of RNA molecules, much as if each read acted as several “votes” for one molecule or another.
Without the mapping step, Sailfish can complete its RNA analysis 20 to 30 times faster than previous methods.
Dr Kingsford also said the Sailfish method is more robust than previous methods. It’s better able to tolerate errors in the reads or differences between individuals’ genomes.
These errors can prevent some reads from being mapped, he explained. But the Sailfish method can make use of all the RNA read “votes,” which improves the method’s accuracy.
For more information and to download the Sailfish code, visit: http://www.cs.cmu.edu/~ckingsf/software/sailfish/. ![]()
Credit: Darren Baker
Researchers say they’ve developed a computational method that dramatically speeds up estimates of gene activity from RNA sequencing (RNA-seq) data.
With the new method, called Sailfish, estimates of gene expression that previously took hours can be completed in a few minutes.
And the researchers say the accuracy of Sailfish equals or exceeds that of previous methods.
The team described the Sailfish method in Nature Biotechnology.
They noted that gigantic repositories of RNA-seq data now exist, making it possible to re-analyze experiments in light of new discoveries.
“But 15 hours a pop really starts to add up, particularly if you want to look at 100 experiments,” said study author Carl Kingsford, PhD, of Carnegie Mellon University in Pittsburg, Pennsylvania.
“With Sailfish, we can give researchers everything they got from previous methods, but faster.”
With previous methods, the RNA molecules from which read sequences originated could be identified and measured only by mapping the reads to their original positions in the larger molecules—a time-consuming process.
Dr Kingsford and his colleagues found this step can actually be eliminated. The team discovered they could allocate parts of the reads to different types of RNA molecules, much as if each read acted as several “votes” for one molecule or another.
Without the mapping step, Sailfish can complete its RNA analysis 20 to 30 times faster than previous methods.
Dr Kingsford also said the Sailfish method is more robust than previous methods. It’s better able to tolerate errors in the reads or differences between individuals’ genomes.
These errors can prevent some reads from being mapped, he explained. But the Sailfish method can make use of all the RNA read “votes,” which improves the method’s accuracy.
For more information and to download the Sailfish code, visit: http://www.cs.cmu.edu/~ckingsf/software/sailfish/. ![]()
Credit: Darren Baker
Researchers say they’ve developed a computational method that dramatically speeds up estimates of gene activity from RNA sequencing (RNA-seq) data.
With the new method, called Sailfish, estimates of gene expression that previously took hours can be completed in a few minutes.
And the researchers say the accuracy of Sailfish equals or exceeds that of previous methods.
The team described the Sailfish method in Nature Biotechnology.
They noted that gigantic repositories of RNA-seq data now exist, making it possible to re-analyze experiments in light of new discoveries.
“But 15 hours a pop really starts to add up, particularly if you want to look at 100 experiments,” said study author Carl Kingsford, PhD, of Carnegie Mellon University in Pittsburg, Pennsylvania.
“With Sailfish, we can give researchers everything they got from previous methods, but faster.”
With previous methods, the RNA molecules from which read sequences originated could be identified and measured only by mapping the reads to their original positions in the larger molecules—a time-consuming process.
Dr Kingsford and his colleagues found this step can actually be eliminated. The team discovered they could allocate parts of the reads to different types of RNA molecules, much as if each read acted as several “votes” for one molecule or another.
Without the mapping step, Sailfish can complete its RNA analysis 20 to 30 times faster than previous methods.
Dr Kingsford also said the Sailfish method is more robust than previous methods. It’s better able to tolerate errors in the reads or differences between individuals’ genomes.
These errors can prevent some reads from being mapped, he explained. But the Sailfish method can make use of all the RNA read “votes,” which improves the method’s accuracy.
For more information and to download the Sailfish code, visit: http://www.cs.cmu.edu/~ckingsf/software/sailfish/. ![]()
Molecule shows preclinical activity in leukemias, lymphomas

SAN DIEGO—A small molecule that has previously proven effective against solid tumors exhibits activity against leukemias and lymphomas, preclinical research suggests.
The molecule, LOR-253, showed antiproliferative activity in a range of leukemia and lymphoma cell lines, induced apoptosis in acute myeloid leukemia (AML) in vitro, and demonstrated synergy with chemotherapeutic agents.
Ronnie Lum, PhD, and colleagues at Lorus Therapeutics, Inc., the Toronto, Canada-based company developing LOR-253, presented these results at the AACR Annual Meeting 2014 (abstract 4544).
LOR-253 acts through induction of the innate tumor suppressor KLF4. Recent research has suggested that upregulation of the transcription factor CDX2 drives the development or progression of leukemic disease. And CDX2 has been shown to silence KLF4, which is reported to be a critical oncogenic event in AML.
Wih this in mind, the researchers decided to test LOR-253’s activity against AML and other hematologic malignancies in vitro.
Experiments revealed that LOR-253 exerts antiproliferative activity against a range of leukemia and lymphoma cell lines, including Ramos, Raji, K-562, Jurkat, MOLT-4, CCRF-CEM, HEL92.1.7, MOLM-13, THP-1, MV411, NB4, HL-60, KG-1, NOMO-1, SKM-1, OCI-AML-2, EOL-1, and Kasumi-1.
IC50 values were substantially lower in these cell lines than in melanoma cell lines, as well as lines of lung, bladder, colon, prostate, and breast cancers.
The researchers also found that LOR-253 induces KLF4 mRNA expression in the AML cell lines HL60 and THP1. This prompts increased expression of p21, a cyclin-dependent kinase inhibitor that is transcriptionally regulated by KLF4.
Consistent with these results, LOR-253 induced cell-cycle arrest and apoptosis in the AML cell lines, which suggests the molecule acted through its intended mechanism of action.
LOR-253 also showed “strong anticancer synergy” in HL60 cells when delivered in combination with daunorubicin, azacitidine, decitabine, or cytarabine.
When LOR-253 was delivered concurrently with chemotherapy, cell viability decreased the most with cytarabine, followed by decitabine, azacitidine, and daunorubicin. With sequential treatment, cell viability decreased the most when LOR-253 was delivered with decitabine, followed by azacitidine, cytarabine, and daunorubicin.
The researchers said these results suggest LOR-253 could provide a new approach to treat AML and, possibly, other hematologic malignancies.
They are now conducting studies to further characterize the pathway that mediates KLF4 induction by LOR-253, to characterize the effects of LOR-253 in combination with approved chemotherapies for AML, and to assess the efficacy of LOR-253 in animal models of AML.
Lorus Therapeutics is also planning a dose-escalating, phase 1b trial of LOR-253 as monotherapy in AML, myelodysplastic syndromes, and other hematologic malignancies. The company expects to begin the trial this summer. ![]()
SAN DIEGO—A small molecule that has previously proven effective against solid tumors exhibits activity against leukemias and lymphomas, preclinical research suggests.
The molecule, LOR-253, showed antiproliferative activity in a range of leukemia and lymphoma cell lines, induced apoptosis in acute myeloid leukemia (AML) in vitro, and demonstrated synergy with chemotherapeutic agents.
Ronnie Lum, PhD, and colleagues at Lorus Therapeutics, Inc., the Toronto, Canada-based company developing LOR-253, presented these results at the AACR Annual Meeting 2014 (abstract 4544).
LOR-253 acts through induction of the innate tumor suppressor KLF4. Recent research has suggested that upregulation of the transcription factor CDX2 drives the development or progression of leukemic disease. And CDX2 has been shown to silence KLF4, which is reported to be a critical oncogenic event in AML.
Wih this in mind, the researchers decided to test LOR-253’s activity against AML and other hematologic malignancies in vitro.
Experiments revealed that LOR-253 exerts antiproliferative activity against a range of leukemia and lymphoma cell lines, including Ramos, Raji, K-562, Jurkat, MOLT-4, CCRF-CEM, HEL92.1.7, MOLM-13, THP-1, MV411, NB4, HL-60, KG-1, NOMO-1, SKM-1, OCI-AML-2, EOL-1, and Kasumi-1.
IC50 values were substantially lower in these cell lines than in melanoma cell lines, as well as lines of lung, bladder, colon, prostate, and breast cancers.
The researchers also found that LOR-253 induces KLF4 mRNA expression in the AML cell lines HL60 and THP1. This prompts increased expression of p21, a cyclin-dependent kinase inhibitor that is transcriptionally regulated by KLF4.
Consistent with these results, LOR-253 induced cell-cycle arrest and apoptosis in the AML cell lines, which suggests the molecule acted through its intended mechanism of action.
LOR-253 also showed “strong anticancer synergy” in HL60 cells when delivered in combination with daunorubicin, azacitidine, decitabine, or cytarabine.
When LOR-253 was delivered concurrently with chemotherapy, cell viability decreased the most with cytarabine, followed by decitabine, azacitidine, and daunorubicin. With sequential treatment, cell viability decreased the most when LOR-253 was delivered with decitabine, followed by azacitidine, cytarabine, and daunorubicin.
The researchers said these results suggest LOR-253 could provide a new approach to treat AML and, possibly, other hematologic malignancies.
They are now conducting studies to further characterize the pathway that mediates KLF4 induction by LOR-253, to characterize the effects of LOR-253 in combination with approved chemotherapies for AML, and to assess the efficacy of LOR-253 in animal models of AML.
Lorus Therapeutics is also planning a dose-escalating, phase 1b trial of LOR-253 as monotherapy in AML, myelodysplastic syndromes, and other hematologic malignancies. The company expects to begin the trial this summer. ![]()
SAN DIEGO—A small molecule that has previously proven effective against solid tumors exhibits activity against leukemias and lymphomas, preclinical research suggests.
The molecule, LOR-253, showed antiproliferative activity in a range of leukemia and lymphoma cell lines, induced apoptosis in acute myeloid leukemia (AML) in vitro, and demonstrated synergy with chemotherapeutic agents.
Ronnie Lum, PhD, and colleagues at Lorus Therapeutics, Inc., the Toronto, Canada-based company developing LOR-253, presented these results at the AACR Annual Meeting 2014 (abstract 4544).
LOR-253 acts through induction of the innate tumor suppressor KLF4. Recent research has suggested that upregulation of the transcription factor CDX2 drives the development or progression of leukemic disease. And CDX2 has been shown to silence KLF4, which is reported to be a critical oncogenic event in AML.
Wih this in mind, the researchers decided to test LOR-253’s activity against AML and other hematologic malignancies in vitro.
Experiments revealed that LOR-253 exerts antiproliferative activity against a range of leukemia and lymphoma cell lines, including Ramos, Raji, K-562, Jurkat, MOLT-4, CCRF-CEM, HEL92.1.7, MOLM-13, THP-1, MV411, NB4, HL-60, KG-1, NOMO-1, SKM-1, OCI-AML-2, EOL-1, and Kasumi-1.
IC50 values were substantially lower in these cell lines than in melanoma cell lines, as well as lines of lung, bladder, colon, prostate, and breast cancers.
The researchers also found that LOR-253 induces KLF4 mRNA expression in the AML cell lines HL60 and THP1. This prompts increased expression of p21, a cyclin-dependent kinase inhibitor that is transcriptionally regulated by KLF4.
Consistent with these results, LOR-253 induced cell-cycle arrest and apoptosis in the AML cell lines, which suggests the molecule acted through its intended mechanism of action.
LOR-253 also showed “strong anticancer synergy” in HL60 cells when delivered in combination with daunorubicin, azacitidine, decitabine, or cytarabine.
When LOR-253 was delivered concurrently with chemotherapy, cell viability decreased the most with cytarabine, followed by decitabine, azacitidine, and daunorubicin. With sequential treatment, cell viability decreased the most when LOR-253 was delivered with decitabine, followed by azacitidine, cytarabine, and daunorubicin.
The researchers said these results suggest LOR-253 could provide a new approach to treat AML and, possibly, other hematologic malignancies.
They are now conducting studies to further characterize the pathway that mediates KLF4 induction by LOR-253, to characterize the effects of LOR-253 in combination with approved chemotherapies for AML, and to assess the efficacy of LOR-253 in animal models of AML.
Lorus Therapeutics is also planning a dose-escalating, phase 1b trial of LOR-253 as monotherapy in AML, myelodysplastic syndromes, and other hematologic malignancies. The company expects to begin the trial this summer.
Study may explain how CSCs survive treatment

Credit: Andre Karwath
Experiments conducted in fruit flies showed that when researchers eliminated a type of stem cell, a group of non-stem cells stepped in to replace them.
The team said this discovery sheds new light on stem cell niches and may help explain how cancer stem cells (CSCs) replenish themselves after exposure to radiation and chemotherapy.
Erika Matunis, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues detailed these findings in Cell Reports.
The researchers used the fruit fly as a model to examine stem cells in their natural state, studying stem cell niches in Drosophila testes.
In these niches are 3 kinds of cells: germ-line stem cells, which divide to produce sperm; somatic cyst stem cells, which make cyst cells; and hub cells, which produce signals that keep these 2 cell types going.
The hub cells have settled on their final form and are incapable of dividing further or changing their function—or so everyone thought.
In a bid to determine what happens when the somatic cyst stem cells are killed off, the researchers tried to figure out how to best do away with them. They thought the task would be straightforward, but it took many combinations of different genes working together to kill the somatic cyst cells.
“When we finally figured out a way to kill all of the somatic stem cells, we thought that the rest of the tissue would probably just empty out,” Dr Matunis said.
In 35% of testes, that’s just what happened. But in the rest, the somatic stem cells grew back.
This was a surprise, Dr Matunis said, and it raised the question of where these new stem cells originated.
The answer was another surprise: the hub cells. When the somatic stem cells were destroyed, the hub cells ramped up their machinery for cell division.
The team did several experiments to confirm the hub cells were involved, including one in which they genetically marked the hub cells and saw the mark appear in the newly formed somatic stem cells—a clear sign that hub cells had divided to make new stem cells.
Dr Matunis noted, however, that the new stem cells created by the hub cells weren’t exactly the same as the old ones. Sometimes, the new cells made molecules that only hub cells normally make.
As the researchers looked closer, they realized the damaged and recovered testes were making new niches. Instead of just one pocket of stem cells, a damaged testis might have 2 or 3.
The researchers have not determined how the new niches are formed, but they speculate that the original niche gets bigger as the new cells divide, then splits. The group is now conducting more experiments aimed at explaining the basics of how niches work, according to Dr Matunis.
She said this research may be useful for understanding CSCs. Knowing how tumor niches support the continued growth and division of CSCs might one day offer new targets for controlling such growth.
Credit: Andre Karwath
Experiments conducted in fruit flies showed that when researchers eliminated a type of stem cell, a group of non-stem cells stepped in to replace them.
The team said this discovery sheds new light on stem cell niches and may help explain how cancer stem cells (CSCs) replenish themselves after exposure to radiation and chemotherapy.
Erika Matunis, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues detailed these findings in Cell Reports.
The researchers used the fruit fly as a model to examine stem cells in their natural state, studying stem cell niches in Drosophila testes.
In these niches are 3 kinds of cells: germ-line stem cells, which divide to produce sperm; somatic cyst stem cells, which make cyst cells; and hub cells, which produce signals that keep these 2 cell types going.
The hub cells have settled on their final form and are incapable of dividing further or changing their function—or so everyone thought.
In a bid to determine what happens when the somatic cyst stem cells are killed off, the researchers tried to figure out how to best do away with them. They thought the task would be straightforward, but it took many combinations of different genes working together to kill the somatic cyst cells.
“When we finally figured out a way to kill all of the somatic stem cells, we thought that the rest of the tissue would probably just empty out,” Dr Matunis said.
In 35% of testes, that’s just what happened. But in the rest, the somatic stem cells grew back.
This was a surprise, Dr Matunis said, and it raised the question of where these new stem cells originated.
The answer was another surprise: the hub cells. When the somatic stem cells were destroyed, the hub cells ramped up their machinery for cell division.
The team did several experiments to confirm the hub cells were involved, including one in which they genetically marked the hub cells and saw the mark appear in the newly formed somatic stem cells—a clear sign that hub cells had divided to make new stem cells.
Dr Matunis noted, however, that the new stem cells created by the hub cells weren’t exactly the same as the old ones. Sometimes, the new cells made molecules that only hub cells normally make.
As the researchers looked closer, they realized the damaged and recovered testes were making new niches. Instead of just one pocket of stem cells, a damaged testis might have 2 or 3.
The researchers have not determined how the new niches are formed, but they speculate that the original niche gets bigger as the new cells divide, then splits. The group is now conducting more experiments aimed at explaining the basics of how niches work, according to Dr Matunis.
She said this research may be useful for understanding CSCs. Knowing how tumor niches support the continued growth and division of CSCs might one day offer new targets for controlling such growth.
Credit: Andre Karwath
Experiments conducted in fruit flies showed that when researchers eliminated a type of stem cell, a group of non-stem cells stepped in to replace them.
The team said this discovery sheds new light on stem cell niches and may help explain how cancer stem cells (CSCs) replenish themselves after exposure to radiation and chemotherapy.
Erika Matunis, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues detailed these findings in Cell Reports.
The researchers used the fruit fly as a model to examine stem cells in their natural state, studying stem cell niches in Drosophila testes.
In these niches are 3 kinds of cells: germ-line stem cells, which divide to produce sperm; somatic cyst stem cells, which make cyst cells; and hub cells, which produce signals that keep these 2 cell types going.
The hub cells have settled on their final form and are incapable of dividing further or changing their function—or so everyone thought.
In a bid to determine what happens when the somatic cyst stem cells are killed off, the researchers tried to figure out how to best do away with them. They thought the task would be straightforward, but it took many combinations of different genes working together to kill the somatic cyst cells.
“When we finally figured out a way to kill all of the somatic stem cells, we thought that the rest of the tissue would probably just empty out,” Dr Matunis said.
In 35% of testes, that’s just what happened. But in the rest, the somatic stem cells grew back.
This was a surprise, Dr Matunis said, and it raised the question of where these new stem cells originated.
The answer was another surprise: the hub cells. When the somatic stem cells were destroyed, the hub cells ramped up their machinery for cell division.
The team did several experiments to confirm the hub cells were involved, including one in which they genetically marked the hub cells and saw the mark appear in the newly formed somatic stem cells—a clear sign that hub cells had divided to make new stem cells.
Dr Matunis noted, however, that the new stem cells created by the hub cells weren’t exactly the same as the old ones. Sometimes, the new cells made molecules that only hub cells normally make.
As the researchers looked closer, they realized the damaged and recovered testes were making new niches. Instead of just one pocket of stem cells, a damaged testis might have 2 or 3.
The researchers have not determined how the new niches are formed, but they speculate that the original niche gets bigger as the new cells divide, then splits. The group is now conducting more experiments aimed at explaining the basics of how niches work, according to Dr Matunis.
She said this research may be useful for understanding CSCs. Knowing how tumor niches support the continued growth and division of CSCs might one day offer new targets for controlling such growth.
Compound targets mutated DLBCL, WM cells
SAN DIEGO—A Toll-like receptor (TLR) antagonist can target B-cell lymphoma cells harboring the MYD88 L265P mutation, preclinical research suggests.
The compound, IMO-8400, decreased the viability of mutated diffuse large B-cell lymphoma (DLBCL) cells and Waldenström’s macroglobulinemia (WM) cells in vitro.
IMO-8400 also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Lakshmi Bhagat, PhD, and colleagues from the Cambridge, Massachusetts-based Idera Pharmaceuticals, Inc.—the company developing IMO-8400—presented these results at the AACR Annual Meeting 2014 (abstract 2570).
The researchers said their data provide additional evidence that the MYD88 L265P mutation results in over-activation of TLR7- and TLR9-mediated signaling, and blocking these TLRs leads to tumor cell death. IMO-8400 is an oligonucleotide-based antagonist of TLRs 7, 8, and 9.
In experiments with OCI‐Ly10 cells (DLBCL cells harboring the MYD88 L265P mutation), IMO-8400 prompted cell death and decreased proliferative cell signaling. But the compound did not produce these effects in SU-DHL-6 cells (DLBCL cells without the MYD88 L265P mutation).
In OCI‐Ly10 cells, IMO-8400 inhibited the IRAK-1, IRAK-4, BTK, STAT-3, Ik-Ba, and NF-κB pathways. The compound did not affect signaling pathways in SU-DHL-6 cells.
IMO-8400 also inhibited tumor growth in a mouse model of MYD88 L265P-positive, activated B-cell-like DLBCL. This inhibition was linked to the suppression of tumor-associated cytokines, including human IL-10, IL-2R, IP-10, and MIG.
Treated mice had significantly longer survival than controls, and the effect was dose-dependent. When IMO-8400 was given at 12.5 mg/kg, the P value was 0.0002. At 25 mg/kg, the P value was less than 0.0002. And at 50 mg/kg, the P value was less than 0.0001.
The researchers also found that IMO‐8400 inhibited cell viability, cytokine production, and signaling pathways in MYD88 L265P-positive WM cells. They observed these effects in the MWCL‐1 cell line and in cells from WM patients.
The team said these results provide a “strong foundation” for accelerating the clinical development of IMO-8400 in patients with B-cell lymphomas harboring the MYD88 L265P mutation.
To that end, Idera has opened enrollment in a phase 1/2 trial of IMO-8400 in WM patients who are refractory to prior therapies. The company has also submitted a protocol to the US Food and Drug Administration to conduct a phase 1/2 trial in patients with MYD88 L265P-positive DLBCL.
SAN DIEGO—A Toll-like receptor (TLR) antagonist can target B-cell lymphoma cells harboring the MYD88 L265P mutation, preclinical research suggests.
The compound, IMO-8400, decreased the viability of mutated diffuse large B-cell lymphoma (DLBCL) cells and Waldenström’s macroglobulinemia (WM) cells in vitro.
IMO-8400 also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Lakshmi Bhagat, PhD, and colleagues from the Cambridge, Massachusetts-based Idera Pharmaceuticals, Inc.—the company developing IMO-8400—presented these results at the AACR Annual Meeting 2014 (abstract 2570).
The researchers said their data provide additional evidence that the MYD88 L265P mutation results in over-activation of TLR7- and TLR9-mediated signaling, and blocking these TLRs leads to tumor cell death. IMO-8400 is an oligonucleotide-based antagonist of TLRs 7, 8, and 9.
In experiments with OCI‐Ly10 cells (DLBCL cells harboring the MYD88 L265P mutation), IMO-8400 prompted cell death and decreased proliferative cell signaling. But the compound did not produce these effects in SU-DHL-6 cells (DLBCL cells without the MYD88 L265P mutation).
In OCI‐Ly10 cells, IMO-8400 inhibited the IRAK-1, IRAK-4, BTK, STAT-3, Ik-Ba, and NF-κB pathways. The compound did not affect signaling pathways in SU-DHL-6 cells.
IMO-8400 also inhibited tumor growth in a mouse model of MYD88 L265P-positive, activated B-cell-like DLBCL. This inhibition was linked to the suppression of tumor-associated cytokines, including human IL-10, IL-2R, IP-10, and MIG.
Treated mice had significantly longer survival than controls, and the effect was dose-dependent. When IMO-8400 was given at 12.5 mg/kg, the P value was 0.0002. At 25 mg/kg, the P value was less than 0.0002. And at 50 mg/kg, the P value was less than 0.0001.
The researchers also found that IMO‐8400 inhibited cell viability, cytokine production, and signaling pathways in MYD88 L265P-positive WM cells. They observed these effects in the MWCL‐1 cell line and in cells from WM patients.
The team said these results provide a “strong foundation” for accelerating the clinical development of IMO-8400 in patients with B-cell lymphomas harboring the MYD88 L265P mutation.
To that end, Idera has opened enrollment in a phase 1/2 trial of IMO-8400 in WM patients who are refractory to prior therapies. The company has also submitted a protocol to the US Food and Drug Administration to conduct a phase 1/2 trial in patients with MYD88 L265P-positive DLBCL.
SAN DIEGO—A Toll-like receptor (TLR) antagonist can target B-cell lymphoma cells harboring the MYD88 L265P mutation, preclinical research suggests.
The compound, IMO-8400, decreased the viability of mutated diffuse large B-cell lymphoma (DLBCL) cells and Waldenström’s macroglobulinemia (WM) cells in vitro.
IMO-8400 also decreased tumor growth and prolonged survival in mice with MYD88 L265P-positive DLBCL.
Lakshmi Bhagat, PhD, and colleagues from the Cambridge, Massachusetts-based Idera Pharmaceuticals, Inc.—the company developing IMO-8400—presented these results at the AACR Annual Meeting 2014 (abstract 2570).
The researchers said their data provide additional evidence that the MYD88 L265P mutation results in over-activation of TLR7- and TLR9-mediated signaling, and blocking these TLRs leads to tumor cell death. IMO-8400 is an oligonucleotide-based antagonist of TLRs 7, 8, and 9.
In experiments with OCI‐Ly10 cells (DLBCL cells harboring the MYD88 L265P mutation), IMO-8400 prompted cell death and decreased proliferative cell signaling. But the compound did not produce these effects in SU-DHL-6 cells (DLBCL cells without the MYD88 L265P mutation).
In OCI‐Ly10 cells, IMO-8400 inhibited the IRAK-1, IRAK-4, BTK, STAT-3, Ik-Ba, and NF-κB pathways. The compound did not affect signaling pathways in SU-DHL-6 cells.
IMO-8400 also inhibited tumor growth in a mouse model of MYD88 L265P-positive, activated B-cell-like DLBCL. This inhibition was linked to the suppression of tumor-associated cytokines, including human IL-10, IL-2R, IP-10, and MIG.
Treated mice had significantly longer survival than controls, and the effect was dose-dependent. When IMO-8400 was given at 12.5 mg/kg, the P value was 0.0002. At 25 mg/kg, the P value was less than 0.0002. And at 50 mg/kg, the P value was less than 0.0001.
The researchers also found that IMO‐8400 inhibited cell viability, cytokine production, and signaling pathways in MYD88 L265P-positive WM cells. They observed these effects in the MWCL‐1 cell line and in cells from WM patients.
The team said these results provide a “strong foundation” for accelerating the clinical development of IMO-8400 in patients with B-cell lymphomas harboring the MYD88 L265P mutation.
To that end, Idera has opened enrollment in a phase 1/2 trial of IMO-8400 in WM patients who are refractory to prior therapies. The company has also submitted a protocol to the US Food and Drug Administration to conduct a phase 1/2 trial in patients with MYD88 L265P-positive DLBCL.
FDA approves ofatumumab in combination for CLL

Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has approved ofatumumab (Arzerra) in combination with chlorambucil for previously untreated patients with chronic lymphocytic leukemia (CLL) who should not receive fludarabine-based therapy.
Ofatumumab, a CD20-directed monoclonal antibody, is already FDA-approved as monotherapy for CLL patients who are refractory to fludarabine and alemtuzumab.
The latest approval was based on results of the phase 3 COMPLEMENT 1 trial.
In this randomized trial, researchers compared single-agent chlorambucil to chlorambucil plus ofatumumab. They enrolled 447 patients for whom fludarabine-based therapy was considered inappropriate (due to factors such as advanced age or comorbidities).
In the overall trial population, the median age was 69 years (range, 35 to 92). Seventy-two percent of patients had 2 or more comorbidities, and 48% of patients had a creatinine clearance of less than 70 mL/min.
The researchers randomized 221 patients to receive chlorambucil plus ofatumumab and 226 patients to receive chlorambucil alone.
Patients in the ofatumumab arm received the drug as an intravenous infusion according to the following schedule: 300 mg in cycle 1 on day 1, 1000 mg in cycle 1 on day 8, and 1000 mg administered on day 1 of all subsequent 28-day cycles.
In both arms, patients received chlorambucil at a dose of 10 mg/m2 orally on days 1 to 7, every 28 days.
Prior to each infusion of ofatumumab, patients received acetaminophen, an antihistamine, and a glucocorticoid.
The primary endpoint of the trial was progression-free survival, as assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The median progression-free survival was 22.4 months for patients receiving ofatumumab and chlorambucil, compared to 13.1 months for patients receiving chlorambucil alone. The hazard ratio was 0.57 (P<0.001).
The most common adverse reactions observed in patients receiving ofatumumab and chlorambucil (at least 2% more than in the control arm) were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain.
Overall, 67% of patients who received ofatumumab experienced 1 or more symptoms of infusion reaction. Ten percent of patients experienced a grade 3 or greater infusion reaction.
The drug’s label carries a boxed warning detailing the risk of hepatitis B virus reactivation—which can result in fulminant hepatitis, hepatic failure, and death—as well as the risk of progressive multifocal leukoencephalopathy—which can result in death.
The recommended dose and schedule for ofatumumab in previously untreated CLL is 300 mg on day 1, followed 1 week later by 1000 mg on day 8 (cycle 1), followed by 1000 mg on day 1 of subsequent 28-day cycles, for a minimum of 3 cycles until best response or a maximum of 12 cycles.
Ofatumumab is under development by GlaxoSmithKline and GenMab. For more details on the drug, see the full prescribing information.
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has approved ofatumumab (Arzerra) in combination with chlorambucil for previously untreated patients with chronic lymphocytic leukemia (CLL) who should not receive fludarabine-based therapy.
Ofatumumab, a CD20-directed monoclonal antibody, is already FDA-approved as monotherapy for CLL patients who are refractory to fludarabine and alemtuzumab.
The latest approval was based on results of the phase 3 COMPLEMENT 1 trial.
In this randomized trial, researchers compared single-agent chlorambucil to chlorambucil plus ofatumumab. They enrolled 447 patients for whom fludarabine-based therapy was considered inappropriate (due to factors such as advanced age or comorbidities).
In the overall trial population, the median age was 69 years (range, 35 to 92). Seventy-two percent of patients had 2 or more comorbidities, and 48% of patients had a creatinine clearance of less than 70 mL/min.
The researchers randomized 221 patients to receive chlorambucil plus ofatumumab and 226 patients to receive chlorambucil alone.
Patients in the ofatumumab arm received the drug as an intravenous infusion according to the following schedule: 300 mg in cycle 1 on day 1, 1000 mg in cycle 1 on day 8, and 1000 mg administered on day 1 of all subsequent 28-day cycles.
In both arms, patients received chlorambucil at a dose of 10 mg/m2 orally on days 1 to 7, every 28 days.
Prior to each infusion of ofatumumab, patients received acetaminophen, an antihistamine, and a glucocorticoid.
The primary endpoint of the trial was progression-free survival, as assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The median progression-free survival was 22.4 months for patients receiving ofatumumab and chlorambucil, compared to 13.1 months for patients receiving chlorambucil alone. The hazard ratio was 0.57 (P<0.001).
The most common adverse reactions observed in patients receiving ofatumumab and chlorambucil (at least 2% more than in the control arm) were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain.
Overall, 67% of patients who received ofatumumab experienced 1 or more symptoms of infusion reaction. Ten percent of patients experienced a grade 3 or greater infusion reaction.
The drug’s label carries a boxed warning detailing the risk of hepatitis B virus reactivation—which can result in fulminant hepatitis, hepatic failure, and death—as well as the risk of progressive multifocal leukoencephalopathy—which can result in death.
The recommended dose and schedule for ofatumumab in previously untreated CLL is 300 mg on day 1, followed 1 week later by 1000 mg on day 8 (cycle 1), followed by 1000 mg on day 1 of subsequent 28-day cycles, for a minimum of 3 cycles until best response or a maximum of 12 cycles.
Ofatumumab is under development by GlaxoSmithKline and GenMab. For more details on the drug, see the full prescribing information.
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has approved ofatumumab (Arzerra) in combination with chlorambucil for previously untreated patients with chronic lymphocytic leukemia (CLL) who should not receive fludarabine-based therapy.
Ofatumumab, a CD20-directed monoclonal antibody, is already FDA-approved as monotherapy for CLL patients who are refractory to fludarabine and alemtuzumab.
The latest approval was based on results of the phase 3 COMPLEMENT 1 trial.
In this randomized trial, researchers compared single-agent chlorambucil to chlorambucil plus ofatumumab. They enrolled 447 patients for whom fludarabine-based therapy was considered inappropriate (due to factors such as advanced age or comorbidities).
In the overall trial population, the median age was 69 years (range, 35 to 92). Seventy-two percent of patients had 2 or more comorbidities, and 48% of patients had a creatinine clearance of less than 70 mL/min.
The researchers randomized 221 patients to receive chlorambucil plus ofatumumab and 226 patients to receive chlorambucil alone.
Patients in the ofatumumab arm received the drug as an intravenous infusion according to the following schedule: 300 mg in cycle 1 on day 1, 1000 mg in cycle 1 on day 8, and 1000 mg administered on day 1 of all subsequent 28-day cycles.
In both arms, patients received chlorambucil at a dose of 10 mg/m2 orally on days 1 to 7, every 28 days.
Prior to each infusion of ofatumumab, patients received acetaminophen, an antihistamine, and a glucocorticoid.
The primary endpoint of the trial was progression-free survival, as assessed by a blinded independent review committee using the 2008 International Workshop on Chronic Lymphocytic Leukemia update of the National Cancer Institute Working Group guidelines.
The median progression-free survival was 22.4 months for patients receiving ofatumumab and chlorambucil, compared to 13.1 months for patients receiving chlorambucil alone. The hazard ratio was 0.57 (P<0.001).
The most common adverse reactions observed in patients receiving ofatumumab and chlorambucil (at least 2% more than in the control arm) were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain.
Overall, 67% of patients who received ofatumumab experienced 1 or more symptoms of infusion reaction. Ten percent of patients experienced a grade 3 or greater infusion reaction.
The drug’s label carries a boxed warning detailing the risk of hepatitis B virus reactivation—which can result in fulminant hepatitis, hepatic failure, and death—as well as the risk of progressive multifocal leukoencephalopathy—which can result in death.
The recommended dose and schedule for ofatumumab in previously untreated CLL is 300 mg on day 1, followed 1 week later by 1000 mg on day 8 (cycle 1), followed by 1000 mg on day 1 of subsequent 28-day cycles, for a minimum of 3 cycles until best response or a maximum of 12 cycles.
Ofatumumab is under development by GlaxoSmithKline and GenMab. For more details on the drug, see the full prescribing information.