User login
Inhibitor gets breakthrough designation for HL
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
Method can help predict utility of tPA
Credit: Lucien Monfils
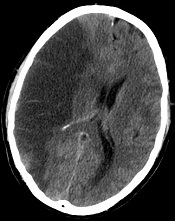
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke. ![]()
Credit: Lucien Monfils
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke. ![]()
Credit: Lucien Monfils
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke. ![]()
Drug granted orphan designation for AML

The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia. ![]()
C difficile vaccine generates immune response
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013. ![]()
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013. ![]()
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013. ![]()
Olive oil may protect against adverse vascular effects
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.” ![]()
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.” ![]()
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.” ![]()
Chip may allow for early cancer detection

Institute of Photonic Sciences
Scientists say they’ve developed a lab-on-a-chip device capable of detecting protein markers for cancer.
The device can detect very low concentrations of protein markers in the blood, enabling cancer diagnosis in its earliest stages, the team says.
Romain Quidant, PhD, of The Institute of Photonic Sciences in Barcelona, Spain, and his colleagues described the device in Nano Letters.
The lab on a chip hosts 32 sensing sites distributed across a network of 8 fluidic microchannels that enables it to conduct multiple analyses.
Gold nanoparticles lie on the surface of the chip and are chemically programed with an antibody receptor in such a way that they are capable of specifically attracting the protein markers circulating in blood.
When a drop of blood is injected into the chip, it circulates through the microchannels, and, if cancer markers are present in the blood, they will stick to the nanoparticles located on the microchannels as they pass by, setting off changes in what is known as the plasmonic resonance.
The device monitors these changes, the magnitude of which is directly related to the concentration/number of markers in the patient’s blood. In this way, it provides a direct assessment of the patient’s risk of developing cancer.
“The most fascinating finding is that we are capable of detecting extremely low concentrations of this protein in a matter of minutes, making this device an ultra-high-sensitivity, state-of-the-art, powerful instrument that will benefit early detection and treatment monitoring of cancer,” Dr Quidant said. ![]()
Institute of Photonic Sciences
Scientists say they’ve developed a lab-on-a-chip device capable of detecting protein markers for cancer.
The device can detect very low concentrations of protein markers in the blood, enabling cancer diagnosis in its earliest stages, the team says.
Romain Quidant, PhD, of The Institute of Photonic Sciences in Barcelona, Spain, and his colleagues described the device in Nano Letters.
The lab on a chip hosts 32 sensing sites distributed across a network of 8 fluidic microchannels that enables it to conduct multiple analyses.
Gold nanoparticles lie on the surface of the chip and are chemically programed with an antibody receptor in such a way that they are capable of specifically attracting the protein markers circulating in blood.
When a drop of blood is injected into the chip, it circulates through the microchannels, and, if cancer markers are present in the blood, they will stick to the nanoparticles located on the microchannels as they pass by, setting off changes in what is known as the plasmonic resonance.
The device monitors these changes, the magnitude of which is directly related to the concentration/number of markers in the patient’s blood. In this way, it provides a direct assessment of the patient’s risk of developing cancer.
“The most fascinating finding is that we are capable of detecting extremely low concentrations of this protein in a matter of minutes, making this device an ultra-high-sensitivity, state-of-the-art, powerful instrument that will benefit early detection and treatment monitoring of cancer,” Dr Quidant said. ![]()
Institute of Photonic Sciences
Scientists say they’ve developed a lab-on-a-chip device capable of detecting protein markers for cancer.
The device can detect very low concentrations of protein markers in the blood, enabling cancer diagnosis in its earliest stages, the team says.
Romain Quidant, PhD, of The Institute of Photonic Sciences in Barcelona, Spain, and his colleagues described the device in Nano Letters.
The lab on a chip hosts 32 sensing sites distributed across a network of 8 fluidic microchannels that enables it to conduct multiple analyses.
Gold nanoparticles lie on the surface of the chip and are chemically programed with an antibody receptor in such a way that they are capable of specifically attracting the protein markers circulating in blood.
When a drop of blood is injected into the chip, it circulates through the microchannels, and, if cancer markers are present in the blood, they will stick to the nanoparticles located on the microchannels as they pass by, setting off changes in what is known as the plasmonic resonance.
The device monitors these changes, the magnitude of which is directly related to the concentration/number of markers in the patient’s blood. In this way, it provides a direct assessment of the patient’s risk of developing cancer.
“The most fascinating finding is that we are capable of detecting extremely low concentrations of this protein in a matter of minutes, making this device an ultra-high-sensitivity, state-of-the-art, powerful instrument that will benefit early detection and treatment monitoring of cancer,” Dr Quidant said. ![]()
CHF screening guidelines need another look, group says

patient and her father
Credit: Rhoda Baer
New research suggests a need to revisit cardiac screening guidelines for survivors of childhood cancers.
The study indicates that less frequent screening for early signs of impending congestive heart failure (CHF) may yield a similar clinical benefit as current screening recommendations.
Furthermore, some survivors might be better served by a different method of screening than the one currently used. And early treatment of patients at high risk of CHF may be beneficial.
The researchers reported these findings in the Annals of Internal Medicine.
Current CHF screening guidelines recommend that childhood cancer survivors treated with chemotherapeutic agents known to affect long-term heart health be screened as often as every year, with a schedule dependent on their level of CHF risk.
The Children’s Oncology Group (COG) recommends that survivors undergo screening by echocardiography for asymptomatic left ventricular dysfunction (ALVD). If left untreated, this clinically silent condition can progress to CHF, so clinicians typically prescribe beta blockers and ACE inhibitors to patients with signs of ALVD.
The COG recommends that patients at high risk of developing CHF be screened every year or 2 and those at low risk be screened every 2 or 5 years
“It is important to monitor survivors so we can reduce the late effects of treatment whenever possible, but we may be asking them to be tested too often, which burdens both individuals and the healthcare system,” said study author Lisa Diller, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts. “We think it is worthwhile to review the current CHF screening guidelines.”
To estimate the clinical benefits and cost-effectiveness of the current heart screening guidelines, Dr Diller and her colleagues constructed a computer model of a virtual cohort of 15-year-olds who had survived cancer at least 5 years.
Using data from the Childhood Cancer Survivors Study and the Framingham Heart Study, the researchers modeled the cohort’s CHF risk and clinical progression over the course of survivors’ lifetimes. Results suggested that routine screening may prevent as many as 1 in 12 cases of CHF.
The team then used Medicare data to estimate the costs and value (expressed in cost per quality-adjusted life-year [QALY]) of different screening schedules—every 1, 2, 5, or 10 years—and methods—echocardiography vs cardiac magnetic resonance imaging (cMRI)—for the different CHF risk groups.
At a cost-effectiveness threshold of $100,000/QALY, the model’s results indicated that echocardiographic screening might not be the best value for resources invested to reduce lifetime CHF risk among survivors at low risk of developing the disease.
On the other hand, the data suggested that biennial echocardiography screening may be a high-value strategy for high-risk survivors.
The simulation’s data also suggested that cMRI may be preferable to echocardiography as a screening method, with cMRI’s greater cost per test balanced by its greater sensitivity. According to the model, cMRI-based screening of low-risk survivors every 10 years and high-risk survivors every 5 years was more cost-effective than any echocardiography-based schedule.
Lastly, the data suggested it may be most beneficial to treat high-risk survivors before signs of ALVD even appear. For instance, proactively treating all high-risk patients in the virtual cohort with ACE inhibitors and beta blockers reduced their lifetime CHF risk more than if they received an echocardiograph every 2 years.
The researchers relied on simulation modeling using the best available clinical and epidemiologic data because of the logistical obstacles to conducting a prospective, randomized, clinical trial.
They said enrolling the number of survivors needed for such a study would be challenging, given how rare childhood cancers are. Yet guidance on the health benefits associated with current recommendations is needed.
“Our findings suggest that there is a long-term benefit in screening survivors at elevated risk for CHF,” said study author Jennifer Yeh, PhD, of the Harvard School of Public Health in Boston.
“Yet less frequent screening than currently recommended may be reasonable when other factors are considered. We hope these results can help inform the ongoing discussion about screening childhood cancer survivors.” ![]()
patient and her father
Credit: Rhoda Baer
New research suggests a need to revisit cardiac screening guidelines for survivors of childhood cancers.
The study indicates that less frequent screening for early signs of impending congestive heart failure (CHF) may yield a similar clinical benefit as current screening recommendations.
Furthermore, some survivors might be better served by a different method of screening than the one currently used. And early treatment of patients at high risk of CHF may be beneficial.
The researchers reported these findings in the Annals of Internal Medicine.
Current CHF screening guidelines recommend that childhood cancer survivors treated with chemotherapeutic agents known to affect long-term heart health be screened as often as every year, with a schedule dependent on their level of CHF risk.
The Children’s Oncology Group (COG) recommends that survivors undergo screening by echocardiography for asymptomatic left ventricular dysfunction (ALVD). If left untreated, this clinically silent condition can progress to CHF, so clinicians typically prescribe beta blockers and ACE inhibitors to patients with signs of ALVD.
The COG recommends that patients at high risk of developing CHF be screened every year or 2 and those at low risk be screened every 2 or 5 years
“It is important to monitor survivors so we can reduce the late effects of treatment whenever possible, but we may be asking them to be tested too often, which burdens both individuals and the healthcare system,” said study author Lisa Diller, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts. “We think it is worthwhile to review the current CHF screening guidelines.”
To estimate the clinical benefits and cost-effectiveness of the current heart screening guidelines, Dr Diller and her colleagues constructed a computer model of a virtual cohort of 15-year-olds who had survived cancer at least 5 years.
Using data from the Childhood Cancer Survivors Study and the Framingham Heart Study, the researchers modeled the cohort’s CHF risk and clinical progression over the course of survivors’ lifetimes. Results suggested that routine screening may prevent as many as 1 in 12 cases of CHF.
The team then used Medicare data to estimate the costs and value (expressed in cost per quality-adjusted life-year [QALY]) of different screening schedules—every 1, 2, 5, or 10 years—and methods—echocardiography vs cardiac magnetic resonance imaging (cMRI)—for the different CHF risk groups.
At a cost-effectiveness threshold of $100,000/QALY, the model’s results indicated that echocardiographic screening might not be the best value for resources invested to reduce lifetime CHF risk among survivors at low risk of developing the disease.
On the other hand, the data suggested that biennial echocardiography screening may be a high-value strategy for high-risk survivors.
The simulation’s data also suggested that cMRI may be preferable to echocardiography as a screening method, with cMRI’s greater cost per test balanced by its greater sensitivity. According to the model, cMRI-based screening of low-risk survivors every 10 years and high-risk survivors every 5 years was more cost-effective than any echocardiography-based schedule.
Lastly, the data suggested it may be most beneficial to treat high-risk survivors before signs of ALVD even appear. For instance, proactively treating all high-risk patients in the virtual cohort with ACE inhibitors and beta blockers reduced their lifetime CHF risk more than if they received an echocardiograph every 2 years.
The researchers relied on simulation modeling using the best available clinical and epidemiologic data because of the logistical obstacles to conducting a prospective, randomized, clinical trial.
They said enrolling the number of survivors needed for such a study would be challenging, given how rare childhood cancers are. Yet guidance on the health benefits associated with current recommendations is needed.
“Our findings suggest that there is a long-term benefit in screening survivors at elevated risk for CHF,” said study author Jennifer Yeh, PhD, of the Harvard School of Public Health in Boston.
“Yet less frequent screening than currently recommended may be reasonable when other factors are considered. We hope these results can help inform the ongoing discussion about screening childhood cancer survivors.” ![]()
patient and her father
Credit: Rhoda Baer
New research suggests a need to revisit cardiac screening guidelines for survivors of childhood cancers.
The study indicates that less frequent screening for early signs of impending congestive heart failure (CHF) may yield a similar clinical benefit as current screening recommendations.
Furthermore, some survivors might be better served by a different method of screening than the one currently used. And early treatment of patients at high risk of CHF may be beneficial.
The researchers reported these findings in the Annals of Internal Medicine.
Current CHF screening guidelines recommend that childhood cancer survivors treated with chemotherapeutic agents known to affect long-term heart health be screened as often as every year, with a schedule dependent on their level of CHF risk.
The Children’s Oncology Group (COG) recommends that survivors undergo screening by echocardiography for asymptomatic left ventricular dysfunction (ALVD). If left untreated, this clinically silent condition can progress to CHF, so clinicians typically prescribe beta blockers and ACE inhibitors to patients with signs of ALVD.
The COG recommends that patients at high risk of developing CHF be screened every year or 2 and those at low risk be screened every 2 or 5 years
“It is important to monitor survivors so we can reduce the late effects of treatment whenever possible, but we may be asking them to be tested too often, which burdens both individuals and the healthcare system,” said study author Lisa Diller, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts. “We think it is worthwhile to review the current CHF screening guidelines.”
To estimate the clinical benefits and cost-effectiveness of the current heart screening guidelines, Dr Diller and her colleagues constructed a computer model of a virtual cohort of 15-year-olds who had survived cancer at least 5 years.
Using data from the Childhood Cancer Survivors Study and the Framingham Heart Study, the researchers modeled the cohort’s CHF risk and clinical progression over the course of survivors’ lifetimes. Results suggested that routine screening may prevent as many as 1 in 12 cases of CHF.
The team then used Medicare data to estimate the costs and value (expressed in cost per quality-adjusted life-year [QALY]) of different screening schedules—every 1, 2, 5, or 10 years—and methods—echocardiography vs cardiac magnetic resonance imaging (cMRI)—for the different CHF risk groups.
At a cost-effectiveness threshold of $100,000/QALY, the model’s results indicated that echocardiographic screening might not be the best value for resources invested to reduce lifetime CHF risk among survivors at low risk of developing the disease.
On the other hand, the data suggested that biennial echocardiography screening may be a high-value strategy for high-risk survivors.
The simulation’s data also suggested that cMRI may be preferable to echocardiography as a screening method, with cMRI’s greater cost per test balanced by its greater sensitivity. According to the model, cMRI-based screening of low-risk survivors every 10 years and high-risk survivors every 5 years was more cost-effective than any echocardiography-based schedule.
Lastly, the data suggested it may be most beneficial to treat high-risk survivors before signs of ALVD even appear. For instance, proactively treating all high-risk patients in the virtual cohort with ACE inhibitors and beta blockers reduced their lifetime CHF risk more than if they received an echocardiograph every 2 years.
The researchers relied on simulation modeling using the best available clinical and epidemiologic data because of the logistical obstacles to conducting a prospective, randomized, clinical trial.
They said enrolling the number of survivors needed for such a study would be challenging, given how rare childhood cancers are. Yet guidance on the health benefits associated with current recommendations is needed.
“Our findings suggest that there is a long-term benefit in screening survivors at elevated risk for CHF,” said study author Jennifer Yeh, PhD, of the Harvard School of Public Health in Boston.
“Yet less frequent screening than currently recommended may be reasonable when other factors are considered. We hope these results can help inform the ongoing discussion about screening childhood cancer survivors.”
Test detects PE with greater specificity than D-dimer

Credit: Andre E.X. Brown
SAN DIEGO—New research suggests a urine test can be used to detect a pulmonary embolism (PE), providing similar sensitivity and greater specificity than the D-dimer test.
The urine test measures levels of fibrinopeptide B (FPB), a peptide released when a thrombus forms.
“The urine FPB test offers advantages over other screening methods because it doesn’t require blood to be drawn, and it can provide more accurate results than the D-dimer test,” said Timothy Fernandes, MD, of the University of California, San Diego.
Dr Fernandes and his colleagues presented results observed with the FPB test at the American Thoracic Society’s 2014 International Conference (abstract 53841).
The researchers tested samples taken from 344 patients who participated in the Pulmonary Embolism Diagnosis Study (PEDS), a multicenter study of patients considered likely to have an acute PE. Sixty-one of these patients (18%) had a confirmed PE, and 283 (83%) did not.
For all urine samples, the researchers measured the FPB concentration and evaluated the sensitivity and specificity of the test at various cut-off points in relation to its ability to predict the presence of PE.
The team found that, at concentrations of 2.5 ng/mL, urine FPB demonstrated sensitivity comparable to previously published values for plasma latex and whole-blood D-dimer levels, but with greater specificity.
Specifically, at a threshold of 2.5 ng/mL, urine FPB could predict PE with sensitivity of 75.4%, specificity of 28.9%, and a negative likelihood ratio of 0.18. Sensitivity was lower at thresholds of 5 ng/mL and 7.5 ng/mL, at 55.7% and 42.6%, respectively.
The researchers noted that the FPB test has the potential for greater specificity than the D-dimer test because FPB can reflect ongoing clot activity, while D-dimer can only be measured once a thrombus has already become degraded.
“The results of our study indicate that urine FPB tests may be a useful complement to current biomarkers such as D-dimer to measure for the
presence and activity of venous thromboembolism,” Dr Fernandes said.
He and his colleagues are now planning to develop a urine dipstick test for FBP. The patent for the urine FPB test is held by the University of California Board of Regents.
The researchers are also planning studies to assess urine FPB in other settings where D-dimer is used, including the use of urine FPB after anticoagulation to determine the risk of recurrent venous thromboembolism.
Credit: Andre E.X. Brown
SAN DIEGO—New research suggests a urine test can be used to detect a pulmonary embolism (PE), providing similar sensitivity and greater specificity than the D-dimer test.
The urine test measures levels of fibrinopeptide B (FPB), a peptide released when a thrombus forms.
“The urine FPB test offers advantages over other screening methods because it doesn’t require blood to be drawn, and it can provide more accurate results than the D-dimer test,” said Timothy Fernandes, MD, of the University of California, San Diego.
Dr Fernandes and his colleagues presented results observed with the FPB test at the American Thoracic Society’s 2014 International Conference (abstract 53841).
The researchers tested samples taken from 344 patients who participated in the Pulmonary Embolism Diagnosis Study (PEDS), a multicenter study of patients considered likely to have an acute PE. Sixty-one of these patients (18%) had a confirmed PE, and 283 (83%) did not.
For all urine samples, the researchers measured the FPB concentration and evaluated the sensitivity and specificity of the test at various cut-off points in relation to its ability to predict the presence of PE.
The team found that, at concentrations of 2.5 ng/mL, urine FPB demonstrated sensitivity comparable to previously published values for plasma latex and whole-blood D-dimer levels, but with greater specificity.
Specifically, at a threshold of 2.5 ng/mL, urine FPB could predict PE with sensitivity of 75.4%, specificity of 28.9%, and a negative likelihood ratio of 0.18. Sensitivity was lower at thresholds of 5 ng/mL and 7.5 ng/mL, at 55.7% and 42.6%, respectively.
The researchers noted that the FPB test has the potential for greater specificity than the D-dimer test because FPB can reflect ongoing clot activity, while D-dimer can only be measured once a thrombus has already become degraded.
“The results of our study indicate that urine FPB tests may be a useful complement to current biomarkers such as D-dimer to measure for the
presence and activity of venous thromboembolism,” Dr Fernandes said.
He and his colleagues are now planning to develop a urine dipstick test for FBP. The patent for the urine FPB test is held by the University of California Board of Regents.
The researchers are also planning studies to assess urine FPB in other settings where D-dimer is used, including the use of urine FPB after anticoagulation to determine the risk of recurrent venous thromboembolism.
Credit: Andre E.X. Brown
SAN DIEGO—New research suggests a urine test can be used to detect a pulmonary embolism (PE), providing similar sensitivity and greater specificity than the D-dimer test.
The urine test measures levels of fibrinopeptide B (FPB), a peptide released when a thrombus forms.
“The urine FPB test offers advantages over other screening methods because it doesn’t require blood to be drawn, and it can provide more accurate results than the D-dimer test,” said Timothy Fernandes, MD, of the University of California, San Diego.
Dr Fernandes and his colleagues presented results observed with the FPB test at the American Thoracic Society’s 2014 International Conference (abstract 53841).
The researchers tested samples taken from 344 patients who participated in the Pulmonary Embolism Diagnosis Study (PEDS), a multicenter study of patients considered likely to have an acute PE. Sixty-one of these patients (18%) had a confirmed PE, and 283 (83%) did not.
For all urine samples, the researchers measured the FPB concentration and evaluated the sensitivity and specificity of the test at various cut-off points in relation to its ability to predict the presence of PE.
The team found that, at concentrations of 2.5 ng/mL, urine FPB demonstrated sensitivity comparable to previously published values for plasma latex and whole-blood D-dimer levels, but with greater specificity.
Specifically, at a threshold of 2.5 ng/mL, urine FPB could predict PE with sensitivity of 75.4%, specificity of 28.9%, and a negative likelihood ratio of 0.18. Sensitivity was lower at thresholds of 5 ng/mL and 7.5 ng/mL, at 55.7% and 42.6%, respectively.
The researchers noted that the FPB test has the potential for greater specificity than the D-dimer test because FPB can reflect ongoing clot activity, while D-dimer can only be measured once a thrombus has already become degraded.
“The results of our study indicate that urine FPB tests may be a useful complement to current biomarkers such as D-dimer to measure for the
presence and activity of venous thromboembolism,” Dr Fernandes said.
He and his colleagues are now planning to develop a urine dipstick test for FBP. The patent for the urine FPB test is held by the University of California Board of Regents.
The researchers are also planning studies to assess urine FPB in other settings where D-dimer is used, including the use of urine FPB after anticoagulation to determine the risk of recurrent venous thromboembolism.
Team unearths surprising sepsis finding
Credit: NHLBI
SAN DIEGO—Results of a retrospective study suggest sepsis may contribute to as many as half of all hospital deaths in the US.
Researchers analyzed information from 6.5 million hospital discharge records and found that, of all hospital deaths, as many as 52% occurred in patients with sepsis.
“We were surprised to find that as many as 1 in 2 patients dying in US hospitals had sepsis,” said Vincent Liu, MD, of the Kaiser Permanente Northern California Division of Research.
He and his colleagues presented this finding at the American Thoracic Society’s 2014 International Conference (abstract 50626). The results have also been published in JAMA.
The researchers analyzed data from 6.5 million hospital discharge records derived from the Healthcare Cost and Utilization Project Nationwide Inpatient Sample (NIS) in 2010. The NIS is the largest publicly available, all-payer inpatient database in the US, containing data from 100% of hospital discharges from a stratified sample of 1051 community hospitals.
The team also evaluated records for 482,828 adult patients admitted to 21 Kaiser Permanente Northern California (KPNC) hospitals.
Using diagnosis and procedure codes, the researchers identified hospital admissions and deaths of patients with sepsis and estimated the percentage of total hospital charges associated with sepsis hospitalizations.
They used 2 approaches to identify patients with sepsis from International Statistical Classification of Diseases, Ninth Revision, Clinical Modification codes. The explicit approach identified patients with codes 038 (septicemia), 995.91 (sepsis), 995.92 (severe sepsis), or 785.52 (septic shock). The team also used an implicit approach, which included patients with evidence of both infection and acute organ failure.
In the NIS cohort, there were 280,663 explicit and 717,718 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 4.3% (explicit) to 10.9% (implicit) of all hospitalizations.
There were 143,312 deaths in the NIS cohort, and 34.7% (explicit) to 52.0% (implicit) occurred among patients with sepsis.
In the KPNC cohort, there were 55,008 explicit and 80,678 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 11.4% (explicit) to 16.7% (implicit) of all hospitalizations.
There were 14,206 inpatient deaths in this cohort, and 36.9% (explicit) to 55.9% (implicit) occurred among patients with sepsis.
“[W]e found that most patients already had sepsis at the time of hospital admission,” Dr Liu said. “There was also a large number of patients with less severe sepsis, a group for whom treatment guidelines are less well-defined. The results of our study suggest that improved care for sepsis patients of all severity levels and in all hospital settings could result in many future lives saved.”
Credit: NHLBI
SAN DIEGO—Results of a retrospective study suggest sepsis may contribute to as many as half of all hospital deaths in the US.
Researchers analyzed information from 6.5 million hospital discharge records and found that, of all hospital deaths, as many as 52% occurred in patients with sepsis.
“We were surprised to find that as many as 1 in 2 patients dying in US hospitals had sepsis,” said Vincent Liu, MD, of the Kaiser Permanente Northern California Division of Research.
He and his colleagues presented this finding at the American Thoracic Society’s 2014 International Conference (abstract 50626). The results have also been published in JAMA.
The researchers analyzed data from 6.5 million hospital discharge records derived from the Healthcare Cost and Utilization Project Nationwide Inpatient Sample (NIS) in 2010. The NIS is the largest publicly available, all-payer inpatient database in the US, containing data from 100% of hospital discharges from a stratified sample of 1051 community hospitals.
The team also evaluated records for 482,828 adult patients admitted to 21 Kaiser Permanente Northern California (KPNC) hospitals.
Using diagnosis and procedure codes, the researchers identified hospital admissions and deaths of patients with sepsis and estimated the percentage of total hospital charges associated with sepsis hospitalizations.
They used 2 approaches to identify patients with sepsis from International Statistical Classification of Diseases, Ninth Revision, Clinical Modification codes. The explicit approach identified patients with codes 038 (septicemia), 995.91 (sepsis), 995.92 (severe sepsis), or 785.52 (septic shock). The team also used an implicit approach, which included patients with evidence of both infection and acute organ failure.
In the NIS cohort, there were 280,663 explicit and 717,718 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 4.3% (explicit) to 10.9% (implicit) of all hospitalizations.
There were 143,312 deaths in the NIS cohort, and 34.7% (explicit) to 52.0% (implicit) occurred among patients with sepsis.
In the KPNC cohort, there were 55,008 explicit and 80,678 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 11.4% (explicit) to 16.7% (implicit) of all hospitalizations.
There were 14,206 inpatient deaths in this cohort, and 36.9% (explicit) to 55.9% (implicit) occurred among patients with sepsis.
“[W]e found that most patients already had sepsis at the time of hospital admission,” Dr Liu said. “There was also a large number of patients with less severe sepsis, a group for whom treatment guidelines are less well-defined. The results of our study suggest that improved care for sepsis patients of all severity levels and in all hospital settings could result in many future lives saved.”
Credit: NHLBI
SAN DIEGO—Results of a retrospective study suggest sepsis may contribute to as many as half of all hospital deaths in the US.
Researchers analyzed information from 6.5 million hospital discharge records and found that, of all hospital deaths, as many as 52% occurred in patients with sepsis.
“We were surprised to find that as many as 1 in 2 patients dying in US hospitals had sepsis,” said Vincent Liu, MD, of the Kaiser Permanente Northern California Division of Research.
He and his colleagues presented this finding at the American Thoracic Society’s 2014 International Conference (abstract 50626). The results have also been published in JAMA.
The researchers analyzed data from 6.5 million hospital discharge records derived from the Healthcare Cost and Utilization Project Nationwide Inpatient Sample (NIS) in 2010. The NIS is the largest publicly available, all-payer inpatient database in the US, containing data from 100% of hospital discharges from a stratified sample of 1051 community hospitals.
The team also evaluated records for 482,828 adult patients admitted to 21 Kaiser Permanente Northern California (KPNC) hospitals.
Using diagnosis and procedure codes, the researchers identified hospital admissions and deaths of patients with sepsis and estimated the percentage of total hospital charges associated with sepsis hospitalizations.
They used 2 approaches to identify patients with sepsis from International Statistical Classification of Diseases, Ninth Revision, Clinical Modification codes. The explicit approach identified patients with codes 038 (septicemia), 995.91 (sepsis), 995.92 (severe sepsis), or 785.52 (septic shock). The team also used an implicit approach, which included patients with evidence of both infection and acute organ failure.
In the NIS cohort, there were 280,663 explicit and 717,718 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 4.3% (explicit) to 10.9% (implicit) of all hospitalizations.
There were 143,312 deaths in the NIS cohort, and 34.7% (explicit) to 52.0% (implicit) occurred among patients with sepsis.
In the KPNC cohort, there were 55,008 explicit and 80,678 implicit sepsis hospitalizations. Sepsis hospitalizations accounted for 11.4% (explicit) to 16.7% (implicit) of all hospitalizations.
There were 14,206 inpatient deaths in this cohort, and 36.9% (explicit) to 55.9% (implicit) occurred among patients with sepsis.
“[W]e found that most patients already had sepsis at the time of hospital admission,” Dr Liu said. “There was also a large number of patients with less severe sepsis, a group for whom treatment guidelines are less well-defined. The results of our study suggest that improved care for sepsis patients of all severity levels and in all hospital settings could result in many future lives saved.”
Protein may be therapeutic target for AML

Credit: Lance Liotta
Researchers have found evidence to suggest that the phosphoinositide (PI) modulator PIP4K2A plays a key role in acute myeloid leukemia (AML).
PIs appear to control the transformation of hematopoietic stem cells into leukemic cells.
Some of these PIs switch on specific cell-signaling pathways, resulting in rapid growth and enhanced survival. Regulation of the PIs is carried out by a variety of proteins known as PI modulators.
“Little is known about the role of PI modulators in leukemia,” said Tim Somervaille, PhD, of The University of Manchester in the UK.
“We wanted to find out which ones were responsible for cell growth or survival in acute myeloid leukemia.”
He and his colleagues described this research in Oncogene.
The researchers performed a targeted knockdown screen of PI modulator genes in human AML cells, looking for genes required to sustain proliferation or prevent apoptosis.
They found that one PI modulator, PIP4K2A, was essential for the growth of leukemia. PIP4K2A knockdown resulted in leukemic cell death. This occurred in both murine MLL-AF9 AML cells and primary human AML cells.
Additional investigation showed that PIP4K2A knockdown resulted in the accumulation of the cyclin-dependent kinase inhibitors CDKN1A and CDKN1B, as well as G1 cell-cycle arrest and apoptosis. CDKN1A accumulation and apoptosis were partially dependent on activation of the mTOR pathway.
Fortunately, PIP4K2A knockdown did not adversely affect normal hematopoietic stem and progenitor cells from mice or human subjects. Neither clonogenic nor multilineage differentiation potential was affected.
“This makes [PIP4K2A] an ideal target for future drug development in leukemia,” Dr Somervaille concluded.
Credit: Lance Liotta
Researchers have found evidence to suggest that the phosphoinositide (PI) modulator PIP4K2A plays a key role in acute myeloid leukemia (AML).
PIs appear to control the transformation of hematopoietic stem cells into leukemic cells.
Some of these PIs switch on specific cell-signaling pathways, resulting in rapid growth and enhanced survival. Regulation of the PIs is carried out by a variety of proteins known as PI modulators.
“Little is known about the role of PI modulators in leukemia,” said Tim Somervaille, PhD, of The University of Manchester in the UK.
“We wanted to find out which ones were responsible for cell growth or survival in acute myeloid leukemia.”
He and his colleagues described this research in Oncogene.
The researchers performed a targeted knockdown screen of PI modulator genes in human AML cells, looking for genes required to sustain proliferation or prevent apoptosis.
They found that one PI modulator, PIP4K2A, was essential for the growth of leukemia. PIP4K2A knockdown resulted in leukemic cell death. This occurred in both murine MLL-AF9 AML cells and primary human AML cells.
Additional investigation showed that PIP4K2A knockdown resulted in the accumulation of the cyclin-dependent kinase inhibitors CDKN1A and CDKN1B, as well as G1 cell-cycle arrest and apoptosis. CDKN1A accumulation and apoptosis were partially dependent on activation of the mTOR pathway.
Fortunately, PIP4K2A knockdown did not adversely affect normal hematopoietic stem and progenitor cells from mice or human subjects. Neither clonogenic nor multilineage differentiation potential was affected.
“This makes [PIP4K2A] an ideal target for future drug development in leukemia,” Dr Somervaille concluded.
Credit: Lance Liotta
Researchers have found evidence to suggest that the phosphoinositide (PI) modulator PIP4K2A plays a key role in acute myeloid leukemia (AML).
PIs appear to control the transformation of hematopoietic stem cells into leukemic cells.
Some of these PIs switch on specific cell-signaling pathways, resulting in rapid growth and enhanced survival. Regulation of the PIs is carried out by a variety of proteins known as PI modulators.
“Little is known about the role of PI modulators in leukemia,” said Tim Somervaille, PhD, of The University of Manchester in the UK.
“We wanted to find out which ones were responsible for cell growth or survival in acute myeloid leukemia.”
He and his colleagues described this research in Oncogene.
The researchers performed a targeted knockdown screen of PI modulator genes in human AML cells, looking for genes required to sustain proliferation or prevent apoptosis.
They found that one PI modulator, PIP4K2A, was essential for the growth of leukemia. PIP4K2A knockdown resulted in leukemic cell death. This occurred in both murine MLL-AF9 AML cells and primary human AML cells.
Additional investigation showed that PIP4K2A knockdown resulted in the accumulation of the cyclin-dependent kinase inhibitors CDKN1A and CDKN1B, as well as G1 cell-cycle arrest and apoptosis. CDKN1A accumulation and apoptosis were partially dependent on activation of the mTOR pathway.
Fortunately, PIP4K2A knockdown did not adversely affect normal hematopoietic stem and progenitor cells from mice or human subjects. Neither clonogenic nor multilineage differentiation potential was affected.
“This makes [PIP4K2A] an ideal target for future drug development in leukemia,” Dr Somervaille concluded.