User login
Drug gets orphan designation for CLL/SLL

The US Food and Drug Administration (FDA) has granted orphan designation for MOR208, an anti-CD19 monoclonal antibody, for the treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The European Medicines Agency (EMA) also announced a positive opinion of the orphan medicinal product application for MOR208 to treat patients with CLL/SLL.
Researchers said MOR208 showed promise in a phase 1 study of CLL/SLL patients.
Orphan drug and orphan medicinal product status are granted by the FDA and EMA to promote the development of promising therapeutics for the treatment of rare diseases affecting fewer than 200,000 people in the US annually and no more than 5 in 10,000 people in the European Union (EU).
Orphan drug designation includes benefits such as a 7-year period of marketing exclusivity in the US and 10 years of market exclusivity in the EU after approval. Other potential advantages come in the form of protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and fee waivers for the regulatory procedures.
MOR208 development, results
MOR208 is a humanized monoclonal antibody that targets the antigen CD19 for treatment of B-cell malignancies and autoimmune diseases. The development program for MOR208 is currently in phase 2 development in CLL, B-cell acute lymphoblastic leukemia, and non-Hodgkin lymphoma.
The drug is being developed by MorphoSys AG. The development program was in-licensed from Xencor in 2010.
Researchers evaluated MOR208 (formerly known as XmAb5574) in a phase 1 study of patients with CLL/SLL and reported their results at the 2012 ASH Annual Meeting (abstract 2894). The study was sponsored by Xencor.
The study included 27 patients with relapsed or refractory CLL/SLL. The median patient age was 66 years (range, 40-84), and patients were generally high-risk. The median number of prior therapies was 4 (range, 1-14).
Patients received MOR208 at a range of doses—0.3 mg/kg, 1 mg/kg, 3 mg/kg, 6 mg/kg, 9 mg/kg, and 12 mg/kg. MOR208 was administered as an intravenous infusion on days 1, 4, 8, 15, and 22 of cycle 1, and on days 1, 8, 15, and 22 of cycle 2.
Dose escalation continued without dose-limiting toxicities until the highest dose level. At this dose, 1 patient experienced grade 4 neutropenia associated with febrile neutropenia, and the patient was taken off treatment.
All 27 patients experienced adverse events, but the majority of them were grade 1-2. The most common events were infusion reactions. These occurred in 67% (n=18) of patients and were all grade 1 or 2.
Nineteen percent of patients (n=5) experienced grade 3-4 treatment-related adverse events, including neutropenia (n=3), thrombocytopenia (n=2), increased aspartate aminotransferase (n=1), febrile neutropenia (n=1), and tumor lysis syndrome (n=1). Four of these patients were on the 12 mg/kg dose, but 1 patient who experienced neutropenia was receiving 1 mg/kg.
Eleven percent of patients experienced a partial response according to IWCLL 2008 criteria. Responses occurred at the 6 mg/kg, 9 mg/kg, and 12 mg/kg dose levels.
All objective responses occurred in patients categorized as CLL as opposed to SLL, and none of the patients with lymph nodes greater than 5 cm responded. Two patients had progressed at the 8-week evaluation point. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for MOR208, an anti-CD19 monoclonal antibody, for the treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The European Medicines Agency (EMA) also announced a positive opinion of the orphan medicinal product application for MOR208 to treat patients with CLL/SLL.
Researchers said MOR208 showed promise in a phase 1 study of CLL/SLL patients.
Orphan drug and orphan medicinal product status are granted by the FDA and EMA to promote the development of promising therapeutics for the treatment of rare diseases affecting fewer than 200,000 people in the US annually and no more than 5 in 10,000 people in the European Union (EU).
Orphan drug designation includes benefits such as a 7-year period of marketing exclusivity in the US and 10 years of market exclusivity in the EU after approval. Other potential advantages come in the form of protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and fee waivers for the regulatory procedures.
MOR208 development, results
MOR208 is a humanized monoclonal antibody that targets the antigen CD19 for treatment of B-cell malignancies and autoimmune diseases. The development program for MOR208 is currently in phase 2 development in CLL, B-cell acute lymphoblastic leukemia, and non-Hodgkin lymphoma.
The drug is being developed by MorphoSys AG. The development program was in-licensed from Xencor in 2010.
Researchers evaluated MOR208 (formerly known as XmAb5574) in a phase 1 study of patients with CLL/SLL and reported their results at the 2012 ASH Annual Meeting (abstract 2894). The study was sponsored by Xencor.
The study included 27 patients with relapsed or refractory CLL/SLL. The median patient age was 66 years (range, 40-84), and patients were generally high-risk. The median number of prior therapies was 4 (range, 1-14).
Patients received MOR208 at a range of doses—0.3 mg/kg, 1 mg/kg, 3 mg/kg, 6 mg/kg, 9 mg/kg, and 12 mg/kg. MOR208 was administered as an intravenous infusion on days 1, 4, 8, 15, and 22 of cycle 1, and on days 1, 8, 15, and 22 of cycle 2.
Dose escalation continued without dose-limiting toxicities until the highest dose level. At this dose, 1 patient experienced grade 4 neutropenia associated with febrile neutropenia, and the patient was taken off treatment.
All 27 patients experienced adverse events, but the majority of them were grade 1-2. The most common events were infusion reactions. These occurred in 67% (n=18) of patients and were all grade 1 or 2.
Nineteen percent of patients (n=5) experienced grade 3-4 treatment-related adverse events, including neutropenia (n=3), thrombocytopenia (n=2), increased aspartate aminotransferase (n=1), febrile neutropenia (n=1), and tumor lysis syndrome (n=1). Four of these patients were on the 12 mg/kg dose, but 1 patient who experienced neutropenia was receiving 1 mg/kg.
Eleven percent of patients experienced a partial response according to IWCLL 2008 criteria. Responses occurred at the 6 mg/kg, 9 mg/kg, and 12 mg/kg dose levels.
All objective responses occurred in patients categorized as CLL as opposed to SLL, and none of the patients with lymph nodes greater than 5 cm responded. Two patients had progressed at the 8-week evaluation point. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for MOR208, an anti-CD19 monoclonal antibody, for the treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The European Medicines Agency (EMA) also announced a positive opinion of the orphan medicinal product application for MOR208 to treat patients with CLL/SLL.
Researchers said MOR208 showed promise in a phase 1 study of CLL/SLL patients.
Orphan drug and orphan medicinal product status are granted by the FDA and EMA to promote the development of promising therapeutics for the treatment of rare diseases affecting fewer than 200,000 people in the US annually and no more than 5 in 10,000 people in the European Union (EU).
Orphan drug designation includes benefits such as a 7-year period of marketing exclusivity in the US and 10 years of market exclusivity in the EU after approval. Other potential advantages come in the form of protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and fee waivers for the regulatory procedures.
MOR208 development, results
MOR208 is a humanized monoclonal antibody that targets the antigen CD19 for treatment of B-cell malignancies and autoimmune diseases. The development program for MOR208 is currently in phase 2 development in CLL, B-cell acute lymphoblastic leukemia, and non-Hodgkin lymphoma.
The drug is being developed by MorphoSys AG. The development program was in-licensed from Xencor in 2010.
Researchers evaluated MOR208 (formerly known as XmAb5574) in a phase 1 study of patients with CLL/SLL and reported their results at the 2012 ASH Annual Meeting (abstract 2894). The study was sponsored by Xencor.
The study included 27 patients with relapsed or refractory CLL/SLL. The median patient age was 66 years (range, 40-84), and patients were generally high-risk. The median number of prior therapies was 4 (range, 1-14).
Patients received MOR208 at a range of doses—0.3 mg/kg, 1 mg/kg, 3 mg/kg, 6 mg/kg, 9 mg/kg, and 12 mg/kg. MOR208 was administered as an intravenous infusion on days 1, 4, 8, 15, and 22 of cycle 1, and on days 1, 8, 15, and 22 of cycle 2.
Dose escalation continued without dose-limiting toxicities until the highest dose level. At this dose, 1 patient experienced grade 4 neutropenia associated with febrile neutropenia, and the patient was taken off treatment.
All 27 patients experienced adverse events, but the majority of them were grade 1-2. The most common events were infusion reactions. These occurred in 67% (n=18) of patients and were all grade 1 or 2.
Nineteen percent of patients (n=5) experienced grade 3-4 treatment-related adverse events, including neutropenia (n=3), thrombocytopenia (n=2), increased aspartate aminotransferase (n=1), febrile neutropenia (n=1), and tumor lysis syndrome (n=1). Four of these patients were on the 12 mg/kg dose, but 1 patient who experienced neutropenia was receiving 1 mg/kg.
Eleven percent of patients experienced a partial response according to IWCLL 2008 criteria. Responses occurred at the 6 mg/kg, 9 mg/kg, and 12 mg/kg dose levels.
All objective responses occurred in patients categorized as CLL as opposed to SLL, and none of the patients with lymph nodes greater than 5 cm responded. Two patients had progressed at the 8-week evaluation point. ![]()
FDA approves first molecular test for blood typing

Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Trapping parasites to fight malaria
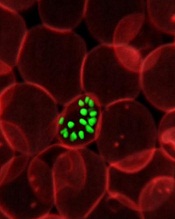
Credit: St Jude Children’s
Research Hospital
Investigators have identified antibodies that prevent malaria-causing parasites at the schizont stage from rupturing and spilling into the bloodstream.
These antibodies reduced loads of the parasite significantly in mice and humans, and they might one day be exploited to create a malaria vaccine, according to the researchers.
Jonathan Kurtis, MD, PhD, of Rhode Island Hospital in Providence, and his colleagues described the antibodies and their effects in Science.
The investigators studied the plasma of malaria-resistant 2-year-olds in Tanzania, where the disease is endemic. The team thought the naturally acquired immunity in these chronically exposed individuals provided a good model through which to identify vaccine antigens.
The analysis revealed that a particular Plasmodium falciparum antigen, known as P falciparum schizont egress antigen-1 (PfSEA-1), triggered antibodies in the children that, in turn, blocked replication of the parasite.
When the researchers vaccinated malaria-infected mice with the antigen or passively transferred PfSEA-1 antibodies to the rodents, they observed a 4-fold reduction of malaria parasites in the animals’ blood.
“When my post-doctoral fellow, Dipak Raj, discovered that antibodies to this protein, PfSEA-1, effectively trapped the malaria-causing parasite within the red blood cells, it was truly a moment of discovery,” Dr Kurtis said.
“Many researchers are trying to find ways to develop a malaria vaccine by preventing the parasite from entering the red blood cell, and, here, we found a way to block it from leaving the cell once it has entered. If it’s trapped in the red blood cell, it can’t go anywhere. It can’t do any further damage.”
The presence of PfSEA-1 antibodies also appeared to protect the Tanzanian study participants from severe cases of malaria. The investigators measured antibodies to PfSEA-1 in the entire cohort of 785 children and found that, among those with antibodies to PfSEA-1, there were no cases of severe malaria.
To generalize their results, the researchers then went back to serum samples they had collected from 140 children in Kenya in 1997. Analyses revealed that individuals with antibodies to PfSEA-1 had 50% lower parasitemia than individuals without these antibodies during a high-transmission season.
The investigators believe these findings could bring researchers a step closer to an effective malaria vaccine that targets parasites at multiple life stages.
“We still have additional trials ahead of us, first in another animal model, but we hope to begin phase 1 trials in humans very soon,” Dr Kurtis said.
“Our findings support PfSEA-1 as a potential vaccine candidate. And we are confident that, by partnering with our colleagues at the National Institutes of Health and other researchers focused on vaccines to prevent the parasites from entering red blood cells, we can approach the parasite from all angles, which could help us develop a truly effective vaccine to prevent this infectious disease that kills millions of children every year.” ![]()
Credit: St Jude Children’s
Research Hospital
Investigators have identified antibodies that prevent malaria-causing parasites at the schizont stage from rupturing and spilling into the bloodstream.
These antibodies reduced loads of the parasite significantly in mice and humans, and they might one day be exploited to create a malaria vaccine, according to the researchers.
Jonathan Kurtis, MD, PhD, of Rhode Island Hospital in Providence, and his colleagues described the antibodies and their effects in Science.
The investigators studied the plasma of malaria-resistant 2-year-olds in Tanzania, where the disease is endemic. The team thought the naturally acquired immunity in these chronically exposed individuals provided a good model through which to identify vaccine antigens.
The analysis revealed that a particular Plasmodium falciparum antigen, known as P falciparum schizont egress antigen-1 (PfSEA-1), triggered antibodies in the children that, in turn, blocked replication of the parasite.
When the researchers vaccinated malaria-infected mice with the antigen or passively transferred PfSEA-1 antibodies to the rodents, they observed a 4-fold reduction of malaria parasites in the animals’ blood.
“When my post-doctoral fellow, Dipak Raj, discovered that antibodies to this protein, PfSEA-1, effectively trapped the malaria-causing parasite within the red blood cells, it was truly a moment of discovery,” Dr Kurtis said.
“Many researchers are trying to find ways to develop a malaria vaccine by preventing the parasite from entering the red blood cell, and, here, we found a way to block it from leaving the cell once it has entered. If it’s trapped in the red blood cell, it can’t go anywhere. It can’t do any further damage.”
The presence of PfSEA-1 antibodies also appeared to protect the Tanzanian study participants from severe cases of malaria. The investigators measured antibodies to PfSEA-1 in the entire cohort of 785 children and found that, among those with antibodies to PfSEA-1, there were no cases of severe malaria.
To generalize their results, the researchers then went back to serum samples they had collected from 140 children in Kenya in 1997. Analyses revealed that individuals with antibodies to PfSEA-1 had 50% lower parasitemia than individuals without these antibodies during a high-transmission season.
The investigators believe these findings could bring researchers a step closer to an effective malaria vaccine that targets parasites at multiple life stages.
“We still have additional trials ahead of us, first in another animal model, but we hope to begin phase 1 trials in humans very soon,” Dr Kurtis said.
“Our findings support PfSEA-1 as a potential vaccine candidate. And we are confident that, by partnering with our colleagues at the National Institutes of Health and other researchers focused on vaccines to prevent the parasites from entering red blood cells, we can approach the parasite from all angles, which could help us develop a truly effective vaccine to prevent this infectious disease that kills millions of children every year.” ![]()
Credit: St Jude Children’s
Research Hospital
Investigators have identified antibodies that prevent malaria-causing parasites at the schizont stage from rupturing and spilling into the bloodstream.
These antibodies reduced loads of the parasite significantly in mice and humans, and they might one day be exploited to create a malaria vaccine, according to the researchers.
Jonathan Kurtis, MD, PhD, of Rhode Island Hospital in Providence, and his colleagues described the antibodies and their effects in Science.
The investigators studied the plasma of malaria-resistant 2-year-olds in Tanzania, where the disease is endemic. The team thought the naturally acquired immunity in these chronically exposed individuals provided a good model through which to identify vaccine antigens.
The analysis revealed that a particular Plasmodium falciparum antigen, known as P falciparum schizont egress antigen-1 (PfSEA-1), triggered antibodies in the children that, in turn, blocked replication of the parasite.
When the researchers vaccinated malaria-infected mice with the antigen or passively transferred PfSEA-1 antibodies to the rodents, they observed a 4-fold reduction of malaria parasites in the animals’ blood.
“When my post-doctoral fellow, Dipak Raj, discovered that antibodies to this protein, PfSEA-1, effectively trapped the malaria-causing parasite within the red blood cells, it was truly a moment of discovery,” Dr Kurtis said.
“Many researchers are trying to find ways to develop a malaria vaccine by preventing the parasite from entering the red blood cell, and, here, we found a way to block it from leaving the cell once it has entered. If it’s trapped in the red blood cell, it can’t go anywhere. It can’t do any further damage.”
The presence of PfSEA-1 antibodies also appeared to protect the Tanzanian study participants from severe cases of malaria. The investigators measured antibodies to PfSEA-1 in the entire cohort of 785 children and found that, among those with antibodies to PfSEA-1, there were no cases of severe malaria.
To generalize their results, the researchers then went back to serum samples they had collected from 140 children in Kenya in 1997. Analyses revealed that individuals with antibodies to PfSEA-1 had 50% lower parasitemia than individuals without these antibodies during a high-transmission season.
The investigators believe these findings could bring researchers a step closer to an effective malaria vaccine that targets parasites at multiple life stages.
“We still have additional trials ahead of us, first in another animal model, but we hope to begin phase 1 trials in humans very soon,” Dr Kurtis said.
“Our findings support PfSEA-1 as a potential vaccine candidate. And we are confident that, by partnering with our colleagues at the National Institutes of Health and other researchers focused on vaccines to prevent the parasites from entering red blood cells, we can approach the parasite from all angles, which could help us develop a truly effective vaccine to prevent this infectious disease that kills millions of children every year.” ![]()
Large-volume infusion pump recalled

Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
TPN calculation software recalled
The US Food and Drug Administration (FDA) has announced a Class I recall of Baxter Corporation Englewood’s ABACUS Total Parenteral Nutrition (TPN) Calculation Software, versions 3.1, 3.0, 2.1, and 2.0.
Baxter has received 2 reports of malfunctioning software and said errors with this software may cause adverse effects.
ABACUS TPN Calculation Software is a Windows-based software application used by pharmacists to calculate or order TPN formulas.
The errors explained
Due to software failures, the following errors may occur:
- ABACUS v3.1 may calculate quantities of electrolytes that are double the expected values during the creation of TPN orders.
- ABACUS v3.1 may automatically add additional sterile water to a formula equal to the volume of a premix, resulting in an over-dilution.
- All software versions of ABACUS software display the calcium phosphate curve points for Premasol incorrectly.
- All software versions of ABACUS may display an inaccurate estimation for calcium and phosphate precipitation in certain circumstances where multiple ingredients provide calcium.
If any of these failures occur, patients may be at risk of developing overdose symptoms. The symptoms are varied and depend on the type of software failure and composition of the fluid being compounded.
Symptoms may be non-specific and include nausea, vomiting, dizziness, or fatigue. Some more severe symptoms include cardiac arrhythmia, pulmonary edema, congestive heart failure, and seizures. A fatal outcome is possible, especially in the high-risk population.
Actions to take
Baxter is recommending that customers contact the company to ensure the ABACUS software is configured correctly.
Customers with a software version earlier than 3.1 will have software version 3.1 installed, which addresses the issues that prompted the recall. In addition, Baxter Support Services will schedule upgrades and assist customers with establishing the proper ABACUS configuration in the customers’ facilities.
Baxter has also requested that customers follow safe compounding practices. Namely, use the “Summary” button to verify the order against the calculated amounts prior to completing the order.
In addition, verify that the ordered ingredients and quantities displayed in the software and printed on the Bag label and the Solution Formula label match the PN prescription prior to preparation. And use a filter for administration of a PN bag.
For more information on the recall, see the FDA’s recall notice, or contact Baxter at 303-617-2242. For technical support, call 1-800-678-2292 or email COtechsupport@baxter.com.
To report adverse reactions or quality problems related to this product, visit the FDA’s MedWatch website. ![]()
The US Food and Drug Administration (FDA) has announced a Class I recall of Baxter Corporation Englewood’s ABACUS Total Parenteral Nutrition (TPN) Calculation Software, versions 3.1, 3.0, 2.1, and 2.0.
Baxter has received 2 reports of malfunctioning software and said errors with this software may cause adverse effects.
ABACUS TPN Calculation Software is a Windows-based software application used by pharmacists to calculate or order TPN formulas.
The errors explained
Due to software failures, the following errors may occur:
- ABACUS v3.1 may calculate quantities of electrolytes that are double the expected values during the creation of TPN orders.
- ABACUS v3.1 may automatically add additional sterile water to a formula equal to the volume of a premix, resulting in an over-dilution.
- All software versions of ABACUS software display the calcium phosphate curve points for Premasol incorrectly.
- All software versions of ABACUS may display an inaccurate estimation for calcium and phosphate precipitation in certain circumstances where multiple ingredients provide calcium.
If any of these failures occur, patients may be at risk of developing overdose symptoms. The symptoms are varied and depend on the type of software failure and composition of the fluid being compounded.
Symptoms may be non-specific and include nausea, vomiting, dizziness, or fatigue. Some more severe symptoms include cardiac arrhythmia, pulmonary edema, congestive heart failure, and seizures. A fatal outcome is possible, especially in the high-risk population.
Actions to take
Baxter is recommending that customers contact the company to ensure the ABACUS software is configured correctly.
Customers with a software version earlier than 3.1 will have software version 3.1 installed, which addresses the issues that prompted the recall. In addition, Baxter Support Services will schedule upgrades and assist customers with establishing the proper ABACUS configuration in the customers’ facilities.
Baxter has also requested that customers follow safe compounding practices. Namely, use the “Summary” button to verify the order against the calculated amounts prior to completing the order.
In addition, verify that the ordered ingredients and quantities displayed in the software and printed on the Bag label and the Solution Formula label match the PN prescription prior to preparation. And use a filter for administration of a PN bag.
For more information on the recall, see the FDA’s recall notice, or contact Baxter at 303-617-2242. For technical support, call 1-800-678-2292 or email COtechsupport@baxter.com.
To report adverse reactions or quality problems related to this product, visit the FDA’s MedWatch website. ![]()
The US Food and Drug Administration (FDA) has announced a Class I recall of Baxter Corporation Englewood’s ABACUS Total Parenteral Nutrition (TPN) Calculation Software, versions 3.1, 3.0, 2.1, and 2.0.
Baxter has received 2 reports of malfunctioning software and said errors with this software may cause adverse effects.
ABACUS TPN Calculation Software is a Windows-based software application used by pharmacists to calculate or order TPN formulas.
The errors explained
Due to software failures, the following errors may occur:
- ABACUS v3.1 may calculate quantities of electrolytes that are double the expected values during the creation of TPN orders.
- ABACUS v3.1 may automatically add additional sterile water to a formula equal to the volume of a premix, resulting in an over-dilution.
- All software versions of ABACUS software display the calcium phosphate curve points for Premasol incorrectly.
- All software versions of ABACUS may display an inaccurate estimation for calcium and phosphate precipitation in certain circumstances where multiple ingredients provide calcium.
If any of these failures occur, patients may be at risk of developing overdose symptoms. The symptoms are varied and depend on the type of software failure and composition of the fluid being compounded.
Symptoms may be non-specific and include nausea, vomiting, dizziness, or fatigue. Some more severe symptoms include cardiac arrhythmia, pulmonary edema, congestive heart failure, and seizures. A fatal outcome is possible, especially in the high-risk population.
Actions to take
Baxter is recommending that customers contact the company to ensure the ABACUS software is configured correctly.
Customers with a software version earlier than 3.1 will have software version 3.1 installed, which addresses the issues that prompted the recall. In addition, Baxter Support Services will schedule upgrades and assist customers with establishing the proper ABACUS configuration in the customers’ facilities.
Baxter has also requested that customers follow safe compounding practices. Namely, use the “Summary” button to verify the order against the calculated amounts prior to completing the order.
In addition, verify that the ordered ingredients and quantities displayed in the software and printed on the Bag label and the Solution Formula label match the PN prescription prior to preparation. And use a filter for administration of a PN bag.
For more information on the recall, see the FDA’s recall notice, or contact Baxter at 303-617-2242. For technical support, call 1-800-678-2292 or email COtechsupport@baxter.com.
To report adverse reactions or quality problems related to this product, visit the FDA’s MedWatch website. ![]()
Chemists discover true structure of anticancer agent

Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Drug gains orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.” ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.” ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.”
Cancer trial publications often omit minority accrual rates

Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology.
Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology.
Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology.
mAb gets breakthrough designation for MM

Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression.
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression.
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression.
Team reports way to reduce sickling, SCD progression

Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.”
Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.”
Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.”