User login
Less competition equals higher healthcare costs

Credit: Petr Kratochvil
Medical practices in less competitive markets charge more for their services, according to a study published in JAMA.
The study, based on US healthcare data from 2010, provides new information about the effects of competition on prices for office visits paid by preferred provider organizations (PPOs).
“The research comes out of trying to understand some dramatic changes that have occurred in the healthcare system over a couple of decades,” said Laurence Baker, PhD, of the Stanford University School of Medicine in California.
One such change is the shift from practices with one or two doctors toward larger, more complex organizations with many physicians. An impact of this can be reduced competition among physician practices.
Dr Baker and his colleagues sought to understand how variation in the amount of competition within a region affects the amounts doctors are paid.
The researchers assessed the relationship between competition and prices paid by PPOs for the most commonly billed services within 10 prominent physician specialties—internal medicine, family practice, cardiology, dermatology, gastroenterology, neurology, general surgery, orthopedics, urology, and otolaryngology.
To establish what prices various medical practices were paid for services, the team used Truven Analytics MarketScan Commercial Claims and Encounters database, which contains the prices paid to physicians for more than 49 million privately insured people from all over the US. They obtained the number of claims and the mean price paid for each service in 1058 counties representing all 50 states.
To measure competitiveness, the researchers drew inspiration from the business world. Using Medicare data, they adapted a standard economic competition measure to track physician practice competition for different US regions.
The Hirschman-Herfindahl Index (HHI) uses the relative sizes of practices to measure market concentration. A higher HHI indicates a less competitive market, and a lower HHI indicates higher competition.
Less competition, higher prices
The researchers found that less competition among physician practices was associated with higher prices paid by private PPOs for office visits.
Across 10 types of office visits, the difference in the HHI was associated with average prices for office visits 8.3% to 16.1% higher. In a more conservative model, the difference in the HHI was associated with 3.5% to 5.4% higher average prices.
The researchers pointed out that, in 2011, privately insured individuals in the US spent nearly $250 billion on physician services. In that context, these small percentage increases could translate to tens of billions of dollars in extra spending.
The team also found that, between 2003 and 2010, prices increased more rapidly in areas that were less competitive. Even when there is no change in HHI, practices in less competitive areas could continue to drive up prices.
“These larger organizations might have better processes in place to optimize care,” said Kate Bundorf, PhD, also of the Stanford University School of Medicine.
“But our research also points out [that we] have to think about the effect on prices and try to balance those two things when we think about how to form policy about these organizations.”
Dr Baker echoed that sentiment.
“Sometimes, it can be tempting to say our goals for the healthcare system should be only about taking care of patients and doing it as well as possible,” he said. “We don’t want to worry about the economics. But the truth is, we do have to worry about the prices because the bill does come, even if you wish it wouldn’t.” ![]()
Credit: Petr Kratochvil
Medical practices in less competitive markets charge more for their services, according to a study published in JAMA.
The study, based on US healthcare data from 2010, provides new information about the effects of competition on prices for office visits paid by preferred provider organizations (PPOs).
“The research comes out of trying to understand some dramatic changes that have occurred in the healthcare system over a couple of decades,” said Laurence Baker, PhD, of the Stanford University School of Medicine in California.
One such change is the shift from practices with one or two doctors toward larger, more complex organizations with many physicians. An impact of this can be reduced competition among physician practices.
Dr Baker and his colleagues sought to understand how variation in the amount of competition within a region affects the amounts doctors are paid.
The researchers assessed the relationship between competition and prices paid by PPOs for the most commonly billed services within 10 prominent physician specialties—internal medicine, family practice, cardiology, dermatology, gastroenterology, neurology, general surgery, orthopedics, urology, and otolaryngology.
To establish what prices various medical practices were paid for services, the team used Truven Analytics MarketScan Commercial Claims and Encounters database, which contains the prices paid to physicians for more than 49 million privately insured people from all over the US. They obtained the number of claims and the mean price paid for each service in 1058 counties representing all 50 states.
To measure competitiveness, the researchers drew inspiration from the business world. Using Medicare data, they adapted a standard economic competition measure to track physician practice competition for different US regions.
The Hirschman-Herfindahl Index (HHI) uses the relative sizes of practices to measure market concentration. A higher HHI indicates a less competitive market, and a lower HHI indicates higher competition.
Less competition, higher prices
The researchers found that less competition among physician practices was associated with higher prices paid by private PPOs for office visits.
Across 10 types of office visits, the difference in the HHI was associated with average prices for office visits 8.3% to 16.1% higher. In a more conservative model, the difference in the HHI was associated with 3.5% to 5.4% higher average prices.
The researchers pointed out that, in 2011, privately insured individuals in the US spent nearly $250 billion on physician services. In that context, these small percentage increases could translate to tens of billions of dollars in extra spending.
The team also found that, between 2003 and 2010, prices increased more rapidly in areas that were less competitive. Even when there is no change in HHI, practices in less competitive areas could continue to drive up prices.
“These larger organizations might have better processes in place to optimize care,” said Kate Bundorf, PhD, also of the Stanford University School of Medicine.
“But our research also points out [that we] have to think about the effect on prices and try to balance those two things when we think about how to form policy about these organizations.”
Dr Baker echoed that sentiment.
“Sometimes, it can be tempting to say our goals for the healthcare system should be only about taking care of patients and doing it as well as possible,” he said. “We don’t want to worry about the economics. But the truth is, we do have to worry about the prices because the bill does come, even if you wish it wouldn’t.” ![]()
Credit: Petr Kratochvil
Medical practices in less competitive markets charge more for their services, according to a study published in JAMA.
The study, based on US healthcare data from 2010, provides new information about the effects of competition on prices for office visits paid by preferred provider organizations (PPOs).
“The research comes out of trying to understand some dramatic changes that have occurred in the healthcare system over a couple of decades,” said Laurence Baker, PhD, of the Stanford University School of Medicine in California.
One such change is the shift from practices with one or two doctors toward larger, more complex organizations with many physicians. An impact of this can be reduced competition among physician practices.
Dr Baker and his colleagues sought to understand how variation in the amount of competition within a region affects the amounts doctors are paid.
The researchers assessed the relationship between competition and prices paid by PPOs for the most commonly billed services within 10 prominent physician specialties—internal medicine, family practice, cardiology, dermatology, gastroenterology, neurology, general surgery, orthopedics, urology, and otolaryngology.
To establish what prices various medical practices were paid for services, the team used Truven Analytics MarketScan Commercial Claims and Encounters database, which contains the prices paid to physicians for more than 49 million privately insured people from all over the US. They obtained the number of claims and the mean price paid for each service in 1058 counties representing all 50 states.
To measure competitiveness, the researchers drew inspiration from the business world. Using Medicare data, they adapted a standard economic competition measure to track physician practice competition for different US regions.
The Hirschman-Herfindahl Index (HHI) uses the relative sizes of practices to measure market concentration. A higher HHI indicates a less competitive market, and a lower HHI indicates higher competition.
Less competition, higher prices
The researchers found that less competition among physician practices was associated with higher prices paid by private PPOs for office visits.
Across 10 types of office visits, the difference in the HHI was associated with average prices for office visits 8.3% to 16.1% higher. In a more conservative model, the difference in the HHI was associated with 3.5% to 5.4% higher average prices.
The researchers pointed out that, in 2011, privately insured individuals in the US spent nearly $250 billion on physician services. In that context, these small percentage increases could translate to tens of billions of dollars in extra spending.
The team also found that, between 2003 and 2010, prices increased more rapidly in areas that were less competitive. Even when there is no change in HHI, practices in less competitive areas could continue to drive up prices.
“These larger organizations might have better processes in place to optimize care,” said Kate Bundorf, PhD, also of the Stanford University School of Medicine.
“But our research also points out [that we] have to think about the effect on prices and try to balance those two things when we think about how to form policy about these organizations.”
Dr Baker echoed that sentiment.
“Sometimes, it can be tempting to say our goals for the healthcare system should be only about taking care of patients and doing it as well as possible,” he said. “We don’t want to worry about the economics. But the truth is, we do have to worry about the prices because the bill does come, even if you wish it wouldn’t.” ![]()
Docs often don’t know about patients’ CVCs
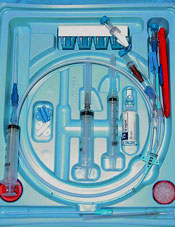
A multicenter study showed that roughly 1 in 5 physicians did not know when a hospitalized patient had a central venous catheter (CVC) in place.
Hospitalists were the least likely to know of a CVC’s presence, followed by general medicine teaching attendings, interns, and residents.
These findings raise questions about patient safety, as CVCs pose a risk of bloodstream infection and thrombosis, both of which can be prevented by removing catheters that are no longer necessary.
“We know that central venous catheters are invaluable for the safe and comprehensive care of some hospitalized patients, but just as they are helpful, they can be harmful,” said study author Vineet Chopra, MD, of the University of Michigan Health System in Ann Arbor.
“The key to preventing complications is to remove devices when they are no longer necessary, but that requires knowing they are there in the first place. Our findings suggest that patient safety may be jeopardized if medical providers don’t improve their practices regarding device awareness.”
Dr Chopra and his colleagues reported their findings in the Annals of Internal Medicine.
The study included 990 patients and 1881 clinical assessments at 3 academic medical centers in the US. Responses from interns (n=454), residents and physician extenders (n=513), general medicine teaching attendings (n=245), subspecialty attendings (n=176), intensivists (n=95), and hospitalists (n=398) were included.
The overall prevalence of CVCs was 21.1%, 60.3% of which were peripherally inserted central catheters (PICCs). The remaining CVCs were triple-lumen catheters inserted in the neck (19.6%), chest (11.5%), or groin (8.6%).
In all, 21.2% of clinicians interviewed did not know when a patient had a CVC in place. And 5.6% of clinicians said a patient had a CVC when there was no device in place.
Hospitalists were most likely to be unaware of a CVC (30.5%), followed by general medicine teaching attendings (25.8%), interns (19.1%), and residents (13.8%).
When assessed by service, critical care physicians were the least likely to be unaware of a CVC (12.6%), compared to general medicine teaching attendings/hospitalists (26.2%) and clinicians in other subspecialties (22.5%). Awareness was best among specialties that used CVCs often, such as cardiology and oncology.
Lack of awareness was greatest for PICCs; 25.1% of clinicians were unaware of a PICC’s presence, and 15.6% were unaware of a triple lumen catheter’s presence.
“These findings have important patient safety and policy implications for health systems nationwide,” Dr Chopra said, “because they suggest that removal of clinically unnecessary catheters may be limited by lack of awareness by providers, especially in non-intensive care settings.” ![]()
A multicenter study showed that roughly 1 in 5 physicians did not know when a hospitalized patient had a central venous catheter (CVC) in place.
Hospitalists were the least likely to know of a CVC’s presence, followed by general medicine teaching attendings, interns, and residents.
These findings raise questions about patient safety, as CVCs pose a risk of bloodstream infection and thrombosis, both of which can be prevented by removing catheters that are no longer necessary.
“We know that central venous catheters are invaluable for the safe and comprehensive care of some hospitalized patients, but just as they are helpful, they can be harmful,” said study author Vineet Chopra, MD, of the University of Michigan Health System in Ann Arbor.
“The key to preventing complications is to remove devices when they are no longer necessary, but that requires knowing they are there in the first place. Our findings suggest that patient safety may be jeopardized if medical providers don’t improve their practices regarding device awareness.”
Dr Chopra and his colleagues reported their findings in the Annals of Internal Medicine.
The study included 990 patients and 1881 clinical assessments at 3 academic medical centers in the US. Responses from interns (n=454), residents and physician extenders (n=513), general medicine teaching attendings (n=245), subspecialty attendings (n=176), intensivists (n=95), and hospitalists (n=398) were included.
The overall prevalence of CVCs was 21.1%, 60.3% of which were peripherally inserted central catheters (PICCs). The remaining CVCs were triple-lumen catheters inserted in the neck (19.6%), chest (11.5%), or groin (8.6%).
In all, 21.2% of clinicians interviewed did not know when a patient had a CVC in place. And 5.6% of clinicians said a patient had a CVC when there was no device in place.
Hospitalists were most likely to be unaware of a CVC (30.5%), followed by general medicine teaching attendings (25.8%), interns (19.1%), and residents (13.8%).
When assessed by service, critical care physicians were the least likely to be unaware of a CVC (12.6%), compared to general medicine teaching attendings/hospitalists (26.2%) and clinicians in other subspecialties (22.5%). Awareness was best among specialties that used CVCs often, such as cardiology and oncology.
Lack of awareness was greatest for PICCs; 25.1% of clinicians were unaware of a PICC’s presence, and 15.6% were unaware of a triple lumen catheter’s presence.
“These findings have important patient safety and policy implications for health systems nationwide,” Dr Chopra said, “because they suggest that removal of clinically unnecessary catheters may be limited by lack of awareness by providers, especially in non-intensive care settings.” ![]()
A multicenter study showed that roughly 1 in 5 physicians did not know when a hospitalized patient had a central venous catheter (CVC) in place.
Hospitalists were the least likely to know of a CVC’s presence, followed by general medicine teaching attendings, interns, and residents.
These findings raise questions about patient safety, as CVCs pose a risk of bloodstream infection and thrombosis, both of which can be prevented by removing catheters that are no longer necessary.
“We know that central venous catheters are invaluable for the safe and comprehensive care of some hospitalized patients, but just as they are helpful, they can be harmful,” said study author Vineet Chopra, MD, of the University of Michigan Health System in Ann Arbor.
“The key to preventing complications is to remove devices when they are no longer necessary, but that requires knowing they are there in the first place. Our findings suggest that patient safety may be jeopardized if medical providers don’t improve their practices regarding device awareness.”
Dr Chopra and his colleagues reported their findings in the Annals of Internal Medicine.
The study included 990 patients and 1881 clinical assessments at 3 academic medical centers in the US. Responses from interns (n=454), residents and physician extenders (n=513), general medicine teaching attendings (n=245), subspecialty attendings (n=176), intensivists (n=95), and hospitalists (n=398) were included.
The overall prevalence of CVCs was 21.1%, 60.3% of which were peripherally inserted central catheters (PICCs). The remaining CVCs were triple-lumen catheters inserted in the neck (19.6%), chest (11.5%), or groin (8.6%).
In all, 21.2% of clinicians interviewed did not know when a patient had a CVC in place. And 5.6% of clinicians said a patient had a CVC when there was no device in place.
Hospitalists were most likely to be unaware of a CVC (30.5%), followed by general medicine teaching attendings (25.8%), interns (19.1%), and residents (13.8%).
When assessed by service, critical care physicians were the least likely to be unaware of a CVC (12.6%), compared to general medicine teaching attendings/hospitalists (26.2%) and clinicians in other subspecialties (22.5%). Awareness was best among specialties that used CVCs often, such as cardiology and oncology.
Lack of awareness was greatest for PICCs; 25.1% of clinicians were unaware of a PICC’s presence, and 15.6% were unaware of a triple lumen catheter’s presence.
“These findings have important patient safety and policy implications for health systems nationwide,” Dr Chopra said, “because they suggest that removal of clinically unnecessary catheters may be limited by lack of awareness by providers, especially in non-intensive care settings.” ![]()
Enzyme ‘switch’ is key to new treatment strategy for T-ALL

Credit: Thomas Semkow
Blocking the action of an enzyme “switch” needed to activate tumor growth may be a practical strategy for treating T-cell acute lymphoblastic leukemia (T-ALL), new research suggests.
The study showed that this enzyme, JMJD3, acts as a cancer “on” switch by splitting off a chemical methyl group of another protein that is usually methylated by the tumor-suppressing enzyme PRC2.
PRC2 acts, in turn, as an “off” switch for cancer cell proliferation.
The researchers previously showed that this destabilizing and cutting loose of PRC2 leads to activation of the NOTCH1 pathway, a process common to many cancers but especially active in at least half of all people with T-ALL.
The team said the drug manufacturer GlaxoSmithKline is already developing an investigational compound called GSKJ4, whose treatment path follows the biological road map revealed in this research.
“Our investigations are showing incredible promise in fighting this disease at the transcriptional level,” said Iannis Aifantis, PhD, of NYU Langone Medical Center in New York.
“We are blocking the action of enzymes controlling the transcription of proteins involved in leukemia, rather than attempting to directly suppress cancer genes.”
Dr Aifantis and his colleagues described this approach in a letter to Nature.
The group’s findings are the culmination of several years of research to unravel precisely how PRC2 suppresses tumor growth since the team first reported the phenomenon in leukemia.
For the current study, the researchers investigated precisely how demethylation triggers the chain of events that evicts PRC2 from cells, thereby removing PRC2 suppression of NOTCH1, which directly binds to and activates cancer-causing genes.
Specifically, they focused on a protein controlled and methylated by PRC2 called H3K27, as well as two other enzymes closely tied to H3K27—JMJD3 and UTX.
The study showed that JMJD3 was highly active in both mice and human leukemia cells at all stages of tumor growth and development. By contrast, UTX was not overexpressed in leukemia, but it was highly active in noncancerous mouse and human cells.
When mice and human leukemia cells were treated with the experimental drug GSKJ4, JMJD3 activity stopped, and all cancer cells eventually died.
Subsequent experiments showed that, in leukemic JMJD3 knockout mice, NOTCH1 activity declined, while UTX activity remained the same.
The disease also progressed much faster in mice bred without UTX, while mice lived longer if they produced UTX. These findings suggest that UTX production controls several tumor-suppressing genes.
To further confirm their findings, the researchers screened more than 200 blood samples from children and adults with T-ALL, revealing several common mutations in UTX.
Dr Aifantis said plans are underway to test GSKJ4 against human leukemia cells transplanted in mice. Other experiments will use the drug in combination with standard chemotherapy in animals with leukemia.
“Our report serves as a valuable reminder of just how complex cancers like T-cell acute lymphoblastic leukemia can be,” Dr Aifantis said, “and that enzymes can play many, even opposing, roles in both tumor growth and suppression.” ![]()
Credit: Thomas Semkow
Blocking the action of an enzyme “switch” needed to activate tumor growth may be a practical strategy for treating T-cell acute lymphoblastic leukemia (T-ALL), new research suggests.
The study showed that this enzyme, JMJD3, acts as a cancer “on” switch by splitting off a chemical methyl group of another protein that is usually methylated by the tumor-suppressing enzyme PRC2.
PRC2 acts, in turn, as an “off” switch for cancer cell proliferation.
The researchers previously showed that this destabilizing and cutting loose of PRC2 leads to activation of the NOTCH1 pathway, a process common to many cancers but especially active in at least half of all people with T-ALL.
The team said the drug manufacturer GlaxoSmithKline is already developing an investigational compound called GSKJ4, whose treatment path follows the biological road map revealed in this research.
“Our investigations are showing incredible promise in fighting this disease at the transcriptional level,” said Iannis Aifantis, PhD, of NYU Langone Medical Center in New York.
“We are blocking the action of enzymes controlling the transcription of proteins involved in leukemia, rather than attempting to directly suppress cancer genes.”
Dr Aifantis and his colleagues described this approach in a letter to Nature.
The group’s findings are the culmination of several years of research to unravel precisely how PRC2 suppresses tumor growth since the team first reported the phenomenon in leukemia.
For the current study, the researchers investigated precisely how demethylation triggers the chain of events that evicts PRC2 from cells, thereby removing PRC2 suppression of NOTCH1, which directly binds to and activates cancer-causing genes.
Specifically, they focused on a protein controlled and methylated by PRC2 called H3K27, as well as two other enzymes closely tied to H3K27—JMJD3 and UTX.
The study showed that JMJD3 was highly active in both mice and human leukemia cells at all stages of tumor growth and development. By contrast, UTX was not overexpressed in leukemia, but it was highly active in noncancerous mouse and human cells.
When mice and human leukemia cells were treated with the experimental drug GSKJ4, JMJD3 activity stopped, and all cancer cells eventually died.
Subsequent experiments showed that, in leukemic JMJD3 knockout mice, NOTCH1 activity declined, while UTX activity remained the same.
The disease also progressed much faster in mice bred without UTX, while mice lived longer if they produced UTX. These findings suggest that UTX production controls several tumor-suppressing genes.
To further confirm their findings, the researchers screened more than 200 blood samples from children and adults with T-ALL, revealing several common mutations in UTX.
Dr Aifantis said plans are underway to test GSKJ4 against human leukemia cells transplanted in mice. Other experiments will use the drug in combination with standard chemotherapy in animals with leukemia.
“Our report serves as a valuable reminder of just how complex cancers like T-cell acute lymphoblastic leukemia can be,” Dr Aifantis said, “and that enzymes can play many, even opposing, roles in both tumor growth and suppression.” ![]()
Credit: Thomas Semkow
Blocking the action of an enzyme “switch” needed to activate tumor growth may be a practical strategy for treating T-cell acute lymphoblastic leukemia (T-ALL), new research suggests.
The study showed that this enzyme, JMJD3, acts as a cancer “on” switch by splitting off a chemical methyl group of another protein that is usually methylated by the tumor-suppressing enzyme PRC2.
PRC2 acts, in turn, as an “off” switch for cancer cell proliferation.
The researchers previously showed that this destabilizing and cutting loose of PRC2 leads to activation of the NOTCH1 pathway, a process common to many cancers but especially active in at least half of all people with T-ALL.
The team said the drug manufacturer GlaxoSmithKline is already developing an investigational compound called GSKJ4, whose treatment path follows the biological road map revealed in this research.
“Our investigations are showing incredible promise in fighting this disease at the transcriptional level,” said Iannis Aifantis, PhD, of NYU Langone Medical Center in New York.
“We are blocking the action of enzymes controlling the transcription of proteins involved in leukemia, rather than attempting to directly suppress cancer genes.”
Dr Aifantis and his colleagues described this approach in a letter to Nature.
The group’s findings are the culmination of several years of research to unravel precisely how PRC2 suppresses tumor growth since the team first reported the phenomenon in leukemia.
For the current study, the researchers investigated precisely how demethylation triggers the chain of events that evicts PRC2 from cells, thereby removing PRC2 suppression of NOTCH1, which directly binds to and activates cancer-causing genes.
Specifically, they focused on a protein controlled and methylated by PRC2 called H3K27, as well as two other enzymes closely tied to H3K27—JMJD3 and UTX.
The study showed that JMJD3 was highly active in both mice and human leukemia cells at all stages of tumor growth and development. By contrast, UTX was not overexpressed in leukemia, but it was highly active in noncancerous mouse and human cells.
When mice and human leukemia cells were treated with the experimental drug GSKJ4, JMJD3 activity stopped, and all cancer cells eventually died.
Subsequent experiments showed that, in leukemic JMJD3 knockout mice, NOTCH1 activity declined, while UTX activity remained the same.
The disease also progressed much faster in mice bred without UTX, while mice lived longer if they produced UTX. These findings suggest that UTX production controls several tumor-suppressing genes.
To further confirm their findings, the researchers screened more than 200 blood samples from children and adults with T-ALL, revealing several common mutations in UTX.
Dr Aifantis said plans are underway to test GSKJ4 against human leukemia cells transplanted in mice. Other experiments will use the drug in combination with standard chemotherapy in animals with leukemia.
“Our report serves as a valuable reminder of just how complex cancers like T-cell acute lymphoblastic leukemia can be,” Dr Aifantis said, “and that enzymes can play many, even opposing, roles in both tumor growth and suppression.” ![]()
Study reveals incidence of mutations linked to leukemia, lymphoma
At least 2% of people over the age of 40 and 5% over age 70 have mutations linked to leukemia and lymphoma, according to research published in Nature Medicine.
The findings, based on blood samples from nearly 3000 patients, don’t necessarily mean that people with these mutations will develop leukemia or lymphoma.
They may have a higher-than-normal risk of developing these malignancies, but more research is needed to determine the risk.
“We would not want anyone to think they should be screened for these mutations to understand their risk of leukemia or lymphoma,” said Timothy Ley, MD, of the Washington University School of Medicine in St Louis, Missouri.
“The ability to understand how mutations in these genes increase a person’s risk of blood cancers is a long way off, and genetic testing would be of no benefit at this time.”
Dr Ley and his colleagues analyzed blood samples from people enrolled in The Cancer Genome Atlas project. The patients had been diagnosed with cancer but were not known to have leukemia, lymphoma, or a blood disease.
They ranged in age from 10 to 90 at the time of diagnosis and had donated blood and tumor samples before starting cancer treatment. Therefore, any mutations the researchers identified would not have been associated with chemotherapy or radiation.
The team looked closely at 556 known cancer genes. In 341 patients ages 40 to 49, fewer than 1% had mutations in 19 leukemia- or lymphoma-related genes.
But among 475 people ages 70 to 79, more than 5% did. And more than 6% of the 132 people ages 80 to 89 had mutations in these genes.
The researchers noted that 9 of the 19 genes were mutated repeatedly, an indicator that the changes drive or initiate the expansion of blood cells.
This expansion of cells is clearly not leukemia or lymphoma, the researchers said. It may be a precursor to hematologic malignancies in a small subset of patients, but the study was not designed to predict the future risk of developing these diseases.
The researchers also said this study likely underestimates the percentage of people with mutations in leukemia and lymphoma genes because the team was only able to identify small mutations, not large structural variations or the insertions and deletions of chunks of genetic material. ![]()
At least 2% of people over the age of 40 and 5% over age 70 have mutations linked to leukemia and lymphoma, according to research published in Nature Medicine.
The findings, based on blood samples from nearly 3000 patients, don’t necessarily mean that people with these mutations will develop leukemia or lymphoma.
They may have a higher-than-normal risk of developing these malignancies, but more research is needed to determine the risk.
“We would not want anyone to think they should be screened for these mutations to understand their risk of leukemia or lymphoma,” said Timothy Ley, MD, of the Washington University School of Medicine in St Louis, Missouri.
“The ability to understand how mutations in these genes increase a person’s risk of blood cancers is a long way off, and genetic testing would be of no benefit at this time.”
Dr Ley and his colleagues analyzed blood samples from people enrolled in The Cancer Genome Atlas project. The patients had been diagnosed with cancer but were not known to have leukemia, lymphoma, or a blood disease.
They ranged in age from 10 to 90 at the time of diagnosis and had donated blood and tumor samples before starting cancer treatment. Therefore, any mutations the researchers identified would not have been associated with chemotherapy or radiation.
The team looked closely at 556 known cancer genes. In 341 patients ages 40 to 49, fewer than 1% had mutations in 19 leukemia- or lymphoma-related genes.
But among 475 people ages 70 to 79, more than 5% did. And more than 6% of the 132 people ages 80 to 89 had mutations in these genes.
The researchers noted that 9 of the 19 genes were mutated repeatedly, an indicator that the changes drive or initiate the expansion of blood cells.
This expansion of cells is clearly not leukemia or lymphoma, the researchers said. It may be a precursor to hematologic malignancies in a small subset of patients, but the study was not designed to predict the future risk of developing these diseases.
The researchers also said this study likely underestimates the percentage of people with mutations in leukemia and lymphoma genes because the team was only able to identify small mutations, not large structural variations or the insertions and deletions of chunks of genetic material. ![]()
At least 2% of people over the age of 40 and 5% over age 70 have mutations linked to leukemia and lymphoma, according to research published in Nature Medicine.
The findings, based on blood samples from nearly 3000 patients, don’t necessarily mean that people with these mutations will develop leukemia or lymphoma.
They may have a higher-than-normal risk of developing these malignancies, but more research is needed to determine the risk.
“We would not want anyone to think they should be screened for these mutations to understand their risk of leukemia or lymphoma,” said Timothy Ley, MD, of the Washington University School of Medicine in St Louis, Missouri.
“The ability to understand how mutations in these genes increase a person’s risk of blood cancers is a long way off, and genetic testing would be of no benefit at this time.”
Dr Ley and his colleagues analyzed blood samples from people enrolled in The Cancer Genome Atlas project. The patients had been diagnosed with cancer but were not known to have leukemia, lymphoma, or a blood disease.
They ranged in age from 10 to 90 at the time of diagnosis and had donated blood and tumor samples before starting cancer treatment. Therefore, any mutations the researchers identified would not have been associated with chemotherapy or radiation.
The team looked closely at 556 known cancer genes. In 341 patients ages 40 to 49, fewer than 1% had mutations in 19 leukemia- or lymphoma-related genes.
But among 475 people ages 70 to 79, more than 5% did. And more than 6% of the 132 people ages 80 to 89 had mutations in these genes.
The researchers noted that 9 of the 19 genes were mutated repeatedly, an indicator that the changes drive or initiate the expansion of blood cells.
This expansion of cells is clearly not leukemia or lymphoma, the researchers said. It may be a precursor to hematologic malignancies in a small subset of patients, but the study was not designed to predict the future risk of developing these diseases.
The researchers also said this study likely underestimates the percentage of people with mutations in leukemia and lymphoma genes because the team was only able to identify small mutations, not large structural variations or the insertions and deletions of chunks of genetic material. ![]()
Hemorrhage control system gets expanded approval

Control System
The US Food and Drug Administration has expanded the indication for the iTClamp® Hemorrhage Control System.
It is now approved to provide temporary control of severe bleeding of the neck. The product was already approved for use on the extremities, axilla, inguinal areas, and the scalp.
The iTClamp is a temporary wound closure device designed to control severe bleeding in seconds.
It seals the edges of a wound closed to create a temporary pool of blood under pressure. This forms a stable clot that mitigates further blood
loss until the wound can be surgically repaired.
Each iTClamp measures less than 2 by 2 inches and weighs less than 3 ounces. It requires only minimal training and gross motor skills to use, according to iTraumaCare, the company that makes the product.
“Addressing difficult-to-control hemorrhage in the neck has been a consistent problem with few solutions,” said Dennis Filips, MD, founder and chief medical officer of iTraumaCare.
“This expanded indication for the iTClamp will allow first responders, medical professionals, and tactical and battlefield medics to use the device in even more meaningful ways to improve patient care.” ![]()
Control System
The US Food and Drug Administration has expanded the indication for the iTClamp® Hemorrhage Control System.
It is now approved to provide temporary control of severe bleeding of the neck. The product was already approved for use on the extremities, axilla, inguinal areas, and the scalp.
The iTClamp is a temporary wound closure device designed to control severe bleeding in seconds.
It seals the edges of a wound closed to create a temporary pool of blood under pressure. This forms a stable clot that mitigates further blood
loss until the wound can be surgically repaired.
Each iTClamp measures less than 2 by 2 inches and weighs less than 3 ounces. It requires only minimal training and gross motor skills to use, according to iTraumaCare, the company that makes the product.
“Addressing difficult-to-control hemorrhage in the neck has been a consistent problem with few solutions,” said Dennis Filips, MD, founder and chief medical officer of iTraumaCare.
“This expanded indication for the iTClamp will allow first responders, medical professionals, and tactical and battlefield medics to use the device in even more meaningful ways to improve patient care.” ![]()
Control System
The US Food and Drug Administration has expanded the indication for the iTClamp® Hemorrhage Control System.
It is now approved to provide temporary control of severe bleeding of the neck. The product was already approved for use on the extremities, axilla, inguinal areas, and the scalp.
The iTClamp is a temporary wound closure device designed to control severe bleeding in seconds.
It seals the edges of a wound closed to create a temporary pool of blood under pressure. This forms a stable clot that mitigates further blood
loss until the wound can be surgically repaired.
Each iTClamp measures less than 2 by 2 inches and weighs less than 3 ounces. It requires only minimal training and gross motor skills to use, according to iTraumaCare, the company that makes the product.
“Addressing difficult-to-control hemorrhage in the neck has been a consistent problem with few solutions,” said Dennis Filips, MD, founder and chief medical officer of iTraumaCare.
“This expanded indication for the iTClamp will allow first responders, medical professionals, and tactical and battlefield medics to use the device in even more meaningful ways to improve patient care.” ![]()
Group creates universal platelets using iPSCs

Credit: Salk Institute
Researchers say they can use induced pluripotent stem cells (iPSCs) to produce large-scale quantities of universal donor platelets.
The team generated megakaryocytes and platelets from iPSCs under feeder-free conditions.
They were able to produce universal platelets by removing a gene essential to expression of the major histocompatibility antigens.
The resulting platelets were functional and behaved like normal human platelets.
The researchers described this method of platelet production, owned by Advanced Cell Technology, Inc., in Stem Cell Reports.
“Unlike other sources of platelets, human induced pluripotent stem cells can be propagated indefinitely, providing a potentially unlimited source of cells for therapeutic purposes,” said Robert Lanza, MD, Chief Scientific Officer at Advanced Cell Technology.
“This study shows that platelets may be produced from [iPSCs] without the need for serum and feeders and, thus, removes potential risks associated with contaminants and pathogens.”
Dr Lanza and his colleagues used a 3-step protocol to differentiate human iPSCs into megakaryocytes and functional platelets in less than 20 days. The method incorporates several discrete intermediate cells, including proprietary hemogenic endothelium-like cells.
The technique allows for long-term storage of megakaryocyte progenitors so they can be available within a few days when needed to produce large quantities of platelets for transfusion.
In addition, by knocking out the β2-microglobulin gene, the researchers were able to generate platelets that are negative for the major histocompatibility antigens.
This suggests the platelets could be transfused into almost any patient, and the method might even prevent platelet refractoriness, according to the researchers.
The team found no major differences in the iPSC platelets and normal human platelets. The iPSC platelets formed aggregates, lamellipodia, and filopodia after activation, just like normal platelets.
Also like normal platelets, the iPSC platelets circulated for at least 8 hours in macrophage-depleted NOD/SCID mice, with a time to reach maximal accumulation of 30 minutes to an hour.
In another murine experiment, iPSC platelets incorporated into a growing thrombus just like normal human platelets, with an average number of 9.0 ± 1.8 platelets per thrombus.
“The platelets generated with our technology are functional and behave like normal human platelets,” Dr Lanza said. “This technology and these results represent an important step towards generating unlimited supplies of universal donor platelets for transfusion.” ![]()
Credit: Salk Institute
Researchers say they can use induced pluripotent stem cells (iPSCs) to produce large-scale quantities of universal donor platelets.
The team generated megakaryocytes and platelets from iPSCs under feeder-free conditions.
They were able to produce universal platelets by removing a gene essential to expression of the major histocompatibility antigens.
The resulting platelets were functional and behaved like normal human platelets.
The researchers described this method of platelet production, owned by Advanced Cell Technology, Inc., in Stem Cell Reports.
“Unlike other sources of platelets, human induced pluripotent stem cells can be propagated indefinitely, providing a potentially unlimited source of cells for therapeutic purposes,” said Robert Lanza, MD, Chief Scientific Officer at Advanced Cell Technology.
“This study shows that platelets may be produced from [iPSCs] without the need for serum and feeders and, thus, removes potential risks associated with contaminants and pathogens.”
Dr Lanza and his colleagues used a 3-step protocol to differentiate human iPSCs into megakaryocytes and functional platelets in less than 20 days. The method incorporates several discrete intermediate cells, including proprietary hemogenic endothelium-like cells.
The technique allows for long-term storage of megakaryocyte progenitors so they can be available within a few days when needed to produce large quantities of platelets for transfusion.
In addition, by knocking out the β2-microglobulin gene, the researchers were able to generate platelets that are negative for the major histocompatibility antigens.
This suggests the platelets could be transfused into almost any patient, and the method might even prevent platelet refractoriness, according to the researchers.
The team found no major differences in the iPSC platelets and normal human platelets. The iPSC platelets formed aggregates, lamellipodia, and filopodia after activation, just like normal platelets.
Also like normal platelets, the iPSC platelets circulated for at least 8 hours in macrophage-depleted NOD/SCID mice, with a time to reach maximal accumulation of 30 minutes to an hour.
In another murine experiment, iPSC platelets incorporated into a growing thrombus just like normal human platelets, with an average number of 9.0 ± 1.8 platelets per thrombus.
“The platelets generated with our technology are functional and behave like normal human platelets,” Dr Lanza said. “This technology and these results represent an important step towards generating unlimited supplies of universal donor platelets for transfusion.” ![]()
Credit: Salk Institute
Researchers say they can use induced pluripotent stem cells (iPSCs) to produce large-scale quantities of universal donor platelets.
The team generated megakaryocytes and platelets from iPSCs under feeder-free conditions.
They were able to produce universal platelets by removing a gene essential to expression of the major histocompatibility antigens.
The resulting platelets were functional and behaved like normal human platelets.
The researchers described this method of platelet production, owned by Advanced Cell Technology, Inc., in Stem Cell Reports.
“Unlike other sources of platelets, human induced pluripotent stem cells can be propagated indefinitely, providing a potentially unlimited source of cells for therapeutic purposes,” said Robert Lanza, MD, Chief Scientific Officer at Advanced Cell Technology.
“This study shows that platelets may be produced from [iPSCs] without the need for serum and feeders and, thus, removes potential risks associated with contaminants and pathogens.”
Dr Lanza and his colleagues used a 3-step protocol to differentiate human iPSCs into megakaryocytes and functional platelets in less than 20 days. The method incorporates several discrete intermediate cells, including proprietary hemogenic endothelium-like cells.
The technique allows for long-term storage of megakaryocyte progenitors so they can be available within a few days when needed to produce large quantities of platelets for transfusion.
In addition, by knocking out the β2-microglobulin gene, the researchers were able to generate platelets that are negative for the major histocompatibility antigens.
This suggests the platelets could be transfused into almost any patient, and the method might even prevent platelet refractoriness, according to the researchers.
The team found no major differences in the iPSC platelets and normal human platelets. The iPSC platelets formed aggregates, lamellipodia, and filopodia after activation, just like normal platelets.
Also like normal platelets, the iPSC platelets circulated for at least 8 hours in macrophage-depleted NOD/SCID mice, with a time to reach maximal accumulation of 30 minutes to an hour.
In another murine experiment, iPSC platelets incorporated into a growing thrombus just like normal human platelets, with an average number of 9.0 ± 1.8 platelets per thrombus.
“The platelets generated with our technology are functional and behave like normal human platelets,” Dr Lanza said. “This technology and these results represent an important step towards generating unlimited supplies of universal donor platelets for transfusion.” ![]()
Megakaryocytes can control HSCs, team finds

to a megakaryocyte (red)
Credit: Meng Zhao
For the first time, researchers have shown that hematopoietic stem cells (HSCs) can be directly controlled by their own progeny, megakaryocytes.
Preclinical experiments revealed that megakaryocytes maintain HSC quiescence during homeostasis and promote HSC regeneration after chemotherapeutic stress.
The discovery suggests megakaryocytes might be used to treat patients with low blood cell counts and to expand HSCs for transplant.
The researchers described these findings in Nature Medicine.
The team examined the relationship between megakaryocytes and HSCs in mouse bone marrow. And they discovered that, as a terminally differentiated progeny, megakaryocytes regulate HSCs by performing two previously unknown functions.
“Megakaryocytes can directly regulate the amount of hematopoietic stem cells by telling the cells when they need to keep in the quiescent stage and when they need to start proliferating to meet increased demand,” said study author Meng Zhao, PhD, of the Stowers Institute for Medical Research in Kansas City, Missouri.
The researchers found that the protein transforming growth factor B1 (TGF-B1), contained in megakaryocytes, signaled quiescence.
And, when under stress from chemotherapy, megakaryocytes signaled fibroblast growth factor 1 (FGF1), to stimulate HSC proliferation.
“Our findings suggest that megakaryocytes are required for the recovery of hematopoietic stem cells post-chemotherapy,” said Linheng Li, PhD, also of the Stowers Institute.
The discovery could provide insight for using megakaryocyte-derived factors, such as TGF-B1 and FGF1, clinically to facilitate the regeneration of HSCs, he added.
Engineering a megakaryocyte niche that supports the growth of HSCs in culture is the next step for the researchers. They are also investigating whether a megakaryocyte niche can be used to help expand human HSCs in vitro for transplant.
These findings are supported by similar research also reported in Nature Medicine. ![]()
to a megakaryocyte (red)
Credit: Meng Zhao
For the first time, researchers have shown that hematopoietic stem cells (HSCs) can be directly controlled by their own progeny, megakaryocytes.
Preclinical experiments revealed that megakaryocytes maintain HSC quiescence during homeostasis and promote HSC regeneration after chemotherapeutic stress.
The discovery suggests megakaryocytes might be used to treat patients with low blood cell counts and to expand HSCs for transplant.
The researchers described these findings in Nature Medicine.
The team examined the relationship between megakaryocytes and HSCs in mouse bone marrow. And they discovered that, as a terminally differentiated progeny, megakaryocytes regulate HSCs by performing two previously unknown functions.
“Megakaryocytes can directly regulate the amount of hematopoietic stem cells by telling the cells when they need to keep in the quiescent stage and when they need to start proliferating to meet increased demand,” said study author Meng Zhao, PhD, of the Stowers Institute for Medical Research in Kansas City, Missouri.
The researchers found that the protein transforming growth factor B1 (TGF-B1), contained in megakaryocytes, signaled quiescence.
And, when under stress from chemotherapy, megakaryocytes signaled fibroblast growth factor 1 (FGF1), to stimulate HSC proliferation.
“Our findings suggest that megakaryocytes are required for the recovery of hematopoietic stem cells post-chemotherapy,” said Linheng Li, PhD, also of the Stowers Institute.
The discovery could provide insight for using megakaryocyte-derived factors, such as TGF-B1 and FGF1, clinically to facilitate the regeneration of HSCs, he added.
Engineering a megakaryocyte niche that supports the growth of HSCs in culture is the next step for the researchers. They are also investigating whether a megakaryocyte niche can be used to help expand human HSCs in vitro for transplant.
These findings are supported by similar research also reported in Nature Medicine. ![]()
to a megakaryocyte (red)
Credit: Meng Zhao
For the first time, researchers have shown that hematopoietic stem cells (HSCs) can be directly controlled by their own progeny, megakaryocytes.
Preclinical experiments revealed that megakaryocytes maintain HSC quiescence during homeostasis and promote HSC regeneration after chemotherapeutic stress.
The discovery suggests megakaryocytes might be used to treat patients with low blood cell counts and to expand HSCs for transplant.
The researchers described these findings in Nature Medicine.
The team examined the relationship between megakaryocytes and HSCs in mouse bone marrow. And they discovered that, as a terminally differentiated progeny, megakaryocytes regulate HSCs by performing two previously unknown functions.
“Megakaryocytes can directly regulate the amount of hematopoietic stem cells by telling the cells when they need to keep in the quiescent stage and when they need to start proliferating to meet increased demand,” said study author Meng Zhao, PhD, of the Stowers Institute for Medical Research in Kansas City, Missouri.
The researchers found that the protein transforming growth factor B1 (TGF-B1), contained in megakaryocytes, signaled quiescence.
And, when under stress from chemotherapy, megakaryocytes signaled fibroblast growth factor 1 (FGF1), to stimulate HSC proliferation.
“Our findings suggest that megakaryocytes are required for the recovery of hematopoietic stem cells post-chemotherapy,” said Linheng Li, PhD, also of the Stowers Institute.
The discovery could provide insight for using megakaryocyte-derived factors, such as TGF-B1 and FGF1, clinically to facilitate the regeneration of HSCs, he added.
Engineering a megakaryocyte niche that supports the growth of HSCs in culture is the next step for the researchers. They are also investigating whether a megakaryocyte niche can be used to help expand human HSCs in vitro for transplant.
These findings are supported by similar research also reported in Nature Medicine.
Trio-CES produces higher molecular diagnostic yield

Calvin, who was diagnosed
with Pitt-Hopkins Syndrome
via trio-CES
Credit: Lapidus family
A 3-pronged approach to clinical exome sequencing (CES) can provide a higher diagnostic yield than traditional molecular diagnostic methods, results of a new study suggest.
Investigators found that sequencing a patient’s exome together with his or her parents’—a method known as trio-CES—greatly improved the ability to reach a firm diagnosis in children with suspected genetic conditions.
This research was published in JAMA. It was released to coincide with a presentation at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers performed CES on 814 patients with undiagnosed, suspected genetic conditions between January 2012 and August 2014. Sequencing was conducted as trio-CES or as proband-CES (only the affected individual sequenced) when parental samples were not available.
The team funneled the raw data through an informatics pipeline to identify variants from the standard human genome. Next, they applied a series of filters to the data based on the patient’s family history and other relevant aspects of his or her condition.
The investigators then hunted for all genes and mutations linked by medical literature to the patient’s symptoms. And a multidisciplinary team of experts reviewed the findings to reach a diagnosis.
Overall, 26% of patients (213/814) received a molecular diagnosis, with the causative variant(s) identified in a well-established clinical gene.
There was a significantly higher molecular diagnostic yield from cases performed as trio-CES relative to proband-CES—31% (127/410) and 22% (74/338), respectively.
In cases of developmental delay in children younger than 5 years (n=138), the molecular diagnosis rate was 41% (45/ 109) for trio-CES cases and 9% (2/23) for proband-CES cases.
The typical turnaround time for exome sequencing is less than 8 weeks, though test results have been returned to physicians within 10 days in medically urgent situations.
With preauthorization, many insurance providers cover the cost to sequence a child and both parents. If not, the out-of-pocket fee is about $6650.
“All families deserve a clear diagnosis of their child’s condition,” said study author Wayne Grody, MD, PhD, of the University of California, Los Angeles.
“Exome sequencing plays an important role in identifying the precise cause of a child’s illness. This is immediately useful to families and physicians in understanding how the disease occurred, preventing unnecessary testing, and developing the best strategies to treat it.”
The researchers noted, however, that the clinical implications of their findings should be better understood before trio- or proband-CES are routinely adopted.
Calvin, who was diagnosed
with Pitt-Hopkins Syndrome
via trio-CES
Credit: Lapidus family
A 3-pronged approach to clinical exome sequencing (CES) can provide a higher diagnostic yield than traditional molecular diagnostic methods, results of a new study suggest.
Investigators found that sequencing a patient’s exome together with his or her parents’—a method known as trio-CES—greatly improved the ability to reach a firm diagnosis in children with suspected genetic conditions.
This research was published in JAMA. It was released to coincide with a presentation at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers performed CES on 814 patients with undiagnosed, suspected genetic conditions between January 2012 and August 2014. Sequencing was conducted as trio-CES or as proband-CES (only the affected individual sequenced) when parental samples were not available.
The team funneled the raw data through an informatics pipeline to identify variants from the standard human genome. Next, they applied a series of filters to the data based on the patient’s family history and other relevant aspects of his or her condition.
The investigators then hunted for all genes and mutations linked by medical literature to the patient’s symptoms. And a multidisciplinary team of experts reviewed the findings to reach a diagnosis.
Overall, 26% of patients (213/814) received a molecular diagnosis, with the causative variant(s) identified in a well-established clinical gene.
There was a significantly higher molecular diagnostic yield from cases performed as trio-CES relative to proband-CES—31% (127/410) and 22% (74/338), respectively.
In cases of developmental delay in children younger than 5 years (n=138), the molecular diagnosis rate was 41% (45/ 109) for trio-CES cases and 9% (2/23) for proband-CES cases.
The typical turnaround time for exome sequencing is less than 8 weeks, though test results have been returned to physicians within 10 days in medically urgent situations.
With preauthorization, many insurance providers cover the cost to sequence a child and both parents. If not, the out-of-pocket fee is about $6650.
“All families deserve a clear diagnosis of their child’s condition,” said study author Wayne Grody, MD, PhD, of the University of California, Los Angeles.
“Exome sequencing plays an important role in identifying the precise cause of a child’s illness. This is immediately useful to families and physicians in understanding how the disease occurred, preventing unnecessary testing, and developing the best strategies to treat it.”
The researchers noted, however, that the clinical implications of their findings should be better understood before trio- or proband-CES are routinely adopted.
Calvin, who was diagnosed
with Pitt-Hopkins Syndrome
via trio-CES
Credit: Lapidus family
A 3-pronged approach to clinical exome sequencing (CES) can provide a higher diagnostic yield than traditional molecular diagnostic methods, results of a new study suggest.
Investigators found that sequencing a patient’s exome together with his or her parents’—a method known as trio-CES—greatly improved the ability to reach a firm diagnosis in children with suspected genetic conditions.
This research was published in JAMA. It was released to coincide with a presentation at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers performed CES on 814 patients with undiagnosed, suspected genetic conditions between January 2012 and August 2014. Sequencing was conducted as trio-CES or as proband-CES (only the affected individual sequenced) when parental samples were not available.
The team funneled the raw data through an informatics pipeline to identify variants from the standard human genome. Next, they applied a series of filters to the data based on the patient’s family history and other relevant aspects of his or her condition.
The investigators then hunted for all genes and mutations linked by medical literature to the patient’s symptoms. And a multidisciplinary team of experts reviewed the findings to reach a diagnosis.
Overall, 26% of patients (213/814) received a molecular diagnosis, with the causative variant(s) identified in a well-established clinical gene.
There was a significantly higher molecular diagnostic yield from cases performed as trio-CES relative to proband-CES—31% (127/410) and 22% (74/338), respectively.
In cases of developmental delay in children younger than 5 years (n=138), the molecular diagnosis rate was 41% (45/ 109) for trio-CES cases and 9% (2/23) for proband-CES cases.
The typical turnaround time for exome sequencing is less than 8 weeks, though test results have been returned to physicians within 10 days in medically urgent situations.
With preauthorization, many insurance providers cover the cost to sequence a child and both parents. If not, the out-of-pocket fee is about $6650.
“All families deserve a clear diagnosis of their child’s condition,” said study author Wayne Grody, MD, PhD, of the University of California, Los Angeles.
“Exome sequencing plays an important role in identifying the precise cause of a child’s illness. This is immediately useful to families and physicians in understanding how the disease occurred, preventing unnecessary testing, and developing the best strategies to treat it.”
The researchers noted, however, that the clinical implications of their findings should be better understood before trio- or proband-CES are routinely adopted.
Exome sequencing shows potential as diagnostic tool

Credit: Graham Colm
In a large study, whole-exome sequencing provided 25% of patients with a diagnosis related to a known genetic disease, giving young
patients and their parents some long-sought answers.
The technology was able to detect a number of rare genetic events and new mutations contributing to disease.
Among the medically actionable findings were mutations related to Fanconi anemia, erythrocytosis, hemolytic anemia, and von Willebrand disease.
“The findings in this report, I believe, will forever change the future practice of pediatrics and medicine as a whole,” said study author James R. Lupski, MD, PhD, of the Baylor College of Medicine in Houston.
“It is just a matter of time before genomics moves up on the physician’s list of things to do and is ordered before formulating a differential diagnosis. It will be the new ‘family history’ that, better yet, gets you both the important variants inherited from each parent and the new mutations that contribute to disease susceptibility.”
This research was published in JAMA and is set to be presented on October 21 at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers had previously conducted a pilot study of whole-exome sequencing that included 250 patients and revealed a 25% molecular diagnostic rate.
This current study included 2000 patients (88% pediatric) with clinical whole-exome sequencing analyzed between June 2012 and August 2014. The majority of patients—87.8%—had neurological disorders or a developmental delay, and 12.2% had non-neurological disorders.
The researchers collected peripheral blood, tissue, or extracted DNA samples from patients and their parents. The team sequenced patients’ DNA and compared those results to the normal reference. Any disease-associated mutations were then compared with the parent’s DNA to determine if the child inherited it from one or both parents.
In all, 504 patients (25.2%) received a molecular diagnosis, and 58% of the diagnostic mutations had not previously been reported. Two hundred and eighty patients had a single mutation that caused disease, 181 were autosomal recessive, 65 were X-linked, and 1 was presumed inherited through the mitochondria.
In 5 cases, the patient inherited 2 copies of the mutated gene from the same parent. Of the dominant mutations, 208 were de novo mutations not inherited from either parent, 32 were inherited, and 40 were not determined because parental samples were not available.
Among the de novo mutations, 5 demonstrated mosaicism, which suggested the mutation occurred after fertilization.
The researchers found 708 presumptive causative variant alleles in the 504 cases. Almost 30% of the diagnoses occurred in disease genes only identified by researchers in the last 3 years. In 65 cases, there was no available genetic test other than whole-exome sequencing to find the mutated gene at the time the test was ordered.
Twenty-three patients (about 5%) had mutations in 2 different genes, which could account for various aspects of the patient’s medical condition.
“Doctors generally try to find one diagnosis that explains all the issues a patient may have,” said study author Christine Eng, MD, of the Baylor College of Medicine.
“We have found that, in some cases, a patient may have a blended phenotype of 2 different conditions. That patients may have 2 different rare genetic diseases to explain their condition was an unexpected finding prior to the use of whole-exome sequencing.”
In the 2000 cases, incidental findings of medically actionable results that could result in early diagnosis, screening, or treatment were found in 92 patients. Three patients had more than 1 finding.
“Clinical exome sequencing can assist diagnosis in a wide range of disorders that are diagnostic dilemmas,” Dr Lupski said. “Rare variants and Mendelian disease are important contributors to disease populations. This is in sharp contrast to the thinking of population geneticists, who investigate how common variants contribute to disease susceptibility.”
“We find ‘rare variants’ in aggregate actually contribute to disease susceptibility in a big way. The individual diseases may be rare, but there are thousands of such diseases and many more being defined through genomics.”
Credit: Graham Colm
In a large study, whole-exome sequencing provided 25% of patients with a diagnosis related to a known genetic disease, giving young
patients and their parents some long-sought answers.
The technology was able to detect a number of rare genetic events and new mutations contributing to disease.
Among the medically actionable findings were mutations related to Fanconi anemia, erythrocytosis, hemolytic anemia, and von Willebrand disease.
“The findings in this report, I believe, will forever change the future practice of pediatrics and medicine as a whole,” said study author James R. Lupski, MD, PhD, of the Baylor College of Medicine in Houston.
“It is just a matter of time before genomics moves up on the physician’s list of things to do and is ordered before formulating a differential diagnosis. It will be the new ‘family history’ that, better yet, gets you both the important variants inherited from each parent and the new mutations that contribute to disease susceptibility.”
This research was published in JAMA and is set to be presented on October 21 at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers had previously conducted a pilot study of whole-exome sequencing that included 250 patients and revealed a 25% molecular diagnostic rate.
This current study included 2000 patients (88% pediatric) with clinical whole-exome sequencing analyzed between June 2012 and August 2014. The majority of patients—87.8%—had neurological disorders or a developmental delay, and 12.2% had non-neurological disorders.
The researchers collected peripheral blood, tissue, or extracted DNA samples from patients and their parents. The team sequenced patients’ DNA and compared those results to the normal reference. Any disease-associated mutations were then compared with the parent’s DNA to determine if the child inherited it from one or both parents.
In all, 504 patients (25.2%) received a molecular diagnosis, and 58% of the diagnostic mutations had not previously been reported. Two hundred and eighty patients had a single mutation that caused disease, 181 were autosomal recessive, 65 were X-linked, and 1 was presumed inherited through the mitochondria.
In 5 cases, the patient inherited 2 copies of the mutated gene from the same parent. Of the dominant mutations, 208 were de novo mutations not inherited from either parent, 32 were inherited, and 40 were not determined because parental samples were not available.
Among the de novo mutations, 5 demonstrated mosaicism, which suggested the mutation occurred after fertilization.
The researchers found 708 presumptive causative variant alleles in the 504 cases. Almost 30% of the diagnoses occurred in disease genes only identified by researchers in the last 3 years. In 65 cases, there was no available genetic test other than whole-exome sequencing to find the mutated gene at the time the test was ordered.
Twenty-three patients (about 5%) had mutations in 2 different genes, which could account for various aspects of the patient’s medical condition.
“Doctors generally try to find one diagnosis that explains all the issues a patient may have,” said study author Christine Eng, MD, of the Baylor College of Medicine.
“We have found that, in some cases, a patient may have a blended phenotype of 2 different conditions. That patients may have 2 different rare genetic diseases to explain their condition was an unexpected finding prior to the use of whole-exome sequencing.”
In the 2000 cases, incidental findings of medically actionable results that could result in early diagnosis, screening, or treatment were found in 92 patients. Three patients had more than 1 finding.
“Clinical exome sequencing can assist diagnosis in a wide range of disorders that are diagnostic dilemmas,” Dr Lupski said. “Rare variants and Mendelian disease are important contributors to disease populations. This is in sharp contrast to the thinking of population geneticists, who investigate how common variants contribute to disease susceptibility.”
“We find ‘rare variants’ in aggregate actually contribute to disease susceptibility in a big way. The individual diseases may be rare, but there are thousands of such diseases and many more being defined through genomics.”
Credit: Graham Colm
In a large study, whole-exome sequencing provided 25% of patients with a diagnosis related to a known genetic disease, giving young
patients and their parents some long-sought answers.
The technology was able to detect a number of rare genetic events and new mutations contributing to disease.
Among the medically actionable findings were mutations related to Fanconi anemia, erythrocytosis, hemolytic anemia, and von Willebrand disease.
“The findings in this report, I believe, will forever change the future practice of pediatrics and medicine as a whole,” said study author James R. Lupski, MD, PhD, of the Baylor College of Medicine in Houston.
“It is just a matter of time before genomics moves up on the physician’s list of things to do and is ordered before formulating a differential diagnosis. It will be the new ‘family history’ that, better yet, gets you both the important variants inherited from each parent and the new mutations that contribute to disease susceptibility.”
This research was published in JAMA and is set to be presented on October 21 at the American Society of Human Genetics Annual Meeting in San Diego.
The researchers had previously conducted a pilot study of whole-exome sequencing that included 250 patients and revealed a 25% molecular diagnostic rate.
This current study included 2000 patients (88% pediatric) with clinical whole-exome sequencing analyzed between June 2012 and August 2014. The majority of patients—87.8%—had neurological disorders or a developmental delay, and 12.2% had non-neurological disorders.
The researchers collected peripheral blood, tissue, or extracted DNA samples from patients and their parents. The team sequenced patients’ DNA and compared those results to the normal reference. Any disease-associated mutations were then compared with the parent’s DNA to determine if the child inherited it from one or both parents.
In all, 504 patients (25.2%) received a molecular diagnosis, and 58% of the diagnostic mutations had not previously been reported. Two hundred and eighty patients had a single mutation that caused disease, 181 were autosomal recessive, 65 were X-linked, and 1 was presumed inherited through the mitochondria.
In 5 cases, the patient inherited 2 copies of the mutated gene from the same parent. Of the dominant mutations, 208 were de novo mutations not inherited from either parent, 32 were inherited, and 40 were not determined because parental samples were not available.
Among the de novo mutations, 5 demonstrated mosaicism, which suggested the mutation occurred after fertilization.
The researchers found 708 presumptive causative variant alleles in the 504 cases. Almost 30% of the diagnoses occurred in disease genes only identified by researchers in the last 3 years. In 65 cases, there was no available genetic test other than whole-exome sequencing to find the mutated gene at the time the test was ordered.
Twenty-three patients (about 5%) had mutations in 2 different genes, which could account for various aspects of the patient’s medical condition.
“Doctors generally try to find one diagnosis that explains all the issues a patient may have,” said study author Christine Eng, MD, of the Baylor College of Medicine.
“We have found that, in some cases, a patient may have a blended phenotype of 2 different conditions. That patients may have 2 different rare genetic diseases to explain their condition was an unexpected finding prior to the use of whole-exome sequencing.”
In the 2000 cases, incidental findings of medically actionable results that could result in early diagnosis, screening, or treatment were found in 92 patients. Three patients had more than 1 finding.
“Clinical exome sequencing can assist diagnosis in a wide range of disorders that are diagnostic dilemmas,” Dr Lupski said. “Rare variants and Mendelian disease are important contributors to disease populations. This is in sharp contrast to the thinking of population geneticists, who investigate how common variants contribute to disease susceptibility.”
“We find ‘rare variants’ in aggregate actually contribute to disease susceptibility in a big way. The individual diseases may be rare, but there are thousands of such diseases and many more being defined through genomics.”
Ibrutinib gets EU approval for CLL, MCL

Credit: Steven Harbour
The European Commission has granted marketing approval for the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica) in the European Union (EU).
The drug is now approved to treat adult patients with relapsed or refractory mantle cell lymphoma (MCL), adults with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy, and first-line CLL patients who have 17p deletion or TP53 mutation and are unsuitable for chemotherapy.
In the EU and all other countries except the US, ibrutinib is marketed by Janssen Pharmaceutical Companies. In the US, the drug is being jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc.
The EU approval of ibrutinib was based on data from a phase 2 study (PCYC-1104) in patients with MCL, the phase 3 RESONATE trial (PCYC-1112-CA) in CLL and small lymphocytic lymphoma (SLL), and a phase 1b/2 study (PCYC-1102) in CLL/SLL.
PCYC-1104: Ibrutinib in MCL
Results of this trial were presented at ASH 2012 and published in NEJM in 2013. The NEJM data included 111 patients who received ibrutinib at 560 mg daily in continuous, 28-day cycles until disease progression.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With an estimated median follow-up of 15.3 months, the estimated median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
RESONATE: Ibrutinib in CLL/SLL
Results of the RESONATE trial were reported at EHA 2014 and published in NEJM in July. This trial was stopped early after an interim analysis showed that ibrutinib-treated patients experienced a 78% reduction in the risk of disease progression or death.
The trial included 391 patients with relapsed or refractory CLL or SLL who were randomized to receive ibrutinib (n=195) or ofatumumab (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57). The median time on study was 9.4 months.
The best overall response rate was higher in the ibrutinib arm than the ofatumumab arm, at 78% and 11%, respectively. And ibrutinib significantly prolonged progression-free survival. The median was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001).
Ibrutinib significantly prolonged overall survival as well. The median overall survival was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049).
Adverse events occurred in 99% of patients in the ibrutinib arm and 98% of those in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation, bleeding-related events, diarrhea, and arthralgia were more common in the ibrutinib arm. Infusion-related reactions, peripheral sensory neuropathy, urticaria, night sweats, and pruritus were more common in the ofatumumab arm.
PCYC-1102: Ibrutinib in CLL/SLL
Results of this phase 1b/2 trial were published in The Lancet Oncology in January. The trial enrolled 29 patients with previously untreated CLL and 2 with SLL.
They received 28-day cycles of once-daily ibrutinib at 420 mg or 840 mg. The 840 mg dose was discontinued after enrollment had begun because the doses showed comparable activity.
After a median follow-up of 22.1 months, 71% of patients achieved an objective response. Four patients (13%) had a complete response. The median time to response was 1.9 months.
Study investigators did not establish whether ibrutinib confers improvements in survival or disease-related symptoms.
Common adverse events included diarrhea (68%), nausea (48%), fatigue (32%), peripheral edema (29%), hypertension (29%), dizziness (26%), dyspepsia (26%), upper respiratory tract infection (26%), arthralgia (23%), constipation (23%), urinary tract infection (23%), and vomiting (23%).
Grade 3 adverse events included diarrhea (13%), fatigue (3%), hypertension (6%), dizziness (3%), urinary tract infection (3%), headache (3%), back pain (3%), and neutropenia (3%). One patient (3%) had grade 4 thrombocytopenia.
Credit: Steven Harbour
The European Commission has granted marketing approval for the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica) in the European Union (EU).
The drug is now approved to treat adult patients with relapsed or refractory mantle cell lymphoma (MCL), adults with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy, and first-line CLL patients who have 17p deletion or TP53 mutation and are unsuitable for chemotherapy.
In the EU and all other countries except the US, ibrutinib is marketed by Janssen Pharmaceutical Companies. In the US, the drug is being jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc.
The EU approval of ibrutinib was based on data from a phase 2 study (PCYC-1104) in patients with MCL, the phase 3 RESONATE trial (PCYC-1112-CA) in CLL and small lymphocytic lymphoma (SLL), and a phase 1b/2 study (PCYC-1102) in CLL/SLL.
PCYC-1104: Ibrutinib in MCL
Results of this trial were presented at ASH 2012 and published in NEJM in 2013. The NEJM data included 111 patients who received ibrutinib at 560 mg daily in continuous, 28-day cycles until disease progression.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With an estimated median follow-up of 15.3 months, the estimated median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
RESONATE: Ibrutinib in CLL/SLL
Results of the RESONATE trial were reported at EHA 2014 and published in NEJM in July. This trial was stopped early after an interim analysis showed that ibrutinib-treated patients experienced a 78% reduction in the risk of disease progression or death.
The trial included 391 patients with relapsed or refractory CLL or SLL who were randomized to receive ibrutinib (n=195) or ofatumumab (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57). The median time on study was 9.4 months.
The best overall response rate was higher in the ibrutinib arm than the ofatumumab arm, at 78% and 11%, respectively. And ibrutinib significantly prolonged progression-free survival. The median was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001).
Ibrutinib significantly prolonged overall survival as well. The median overall survival was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049).
Adverse events occurred in 99% of patients in the ibrutinib arm and 98% of those in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation, bleeding-related events, diarrhea, and arthralgia were more common in the ibrutinib arm. Infusion-related reactions, peripheral sensory neuropathy, urticaria, night sweats, and pruritus were more common in the ofatumumab arm.
PCYC-1102: Ibrutinib in CLL/SLL
Results of this phase 1b/2 trial were published in The Lancet Oncology in January. The trial enrolled 29 patients with previously untreated CLL and 2 with SLL.
They received 28-day cycles of once-daily ibrutinib at 420 mg or 840 mg. The 840 mg dose was discontinued after enrollment had begun because the doses showed comparable activity.
After a median follow-up of 22.1 months, 71% of patients achieved an objective response. Four patients (13%) had a complete response. The median time to response was 1.9 months.
Study investigators did not establish whether ibrutinib confers improvements in survival or disease-related symptoms.
Common adverse events included diarrhea (68%), nausea (48%), fatigue (32%), peripheral edema (29%), hypertension (29%), dizziness (26%), dyspepsia (26%), upper respiratory tract infection (26%), arthralgia (23%), constipation (23%), urinary tract infection (23%), and vomiting (23%).
Grade 3 adverse events included diarrhea (13%), fatigue (3%), hypertension (6%), dizziness (3%), urinary tract infection (3%), headache (3%), back pain (3%), and neutropenia (3%). One patient (3%) had grade 4 thrombocytopenia.
Credit: Steven Harbour
The European Commission has granted marketing approval for the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica) in the European Union (EU).
The drug is now approved to treat adult patients with relapsed or refractory mantle cell lymphoma (MCL), adults with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy, and first-line CLL patients who have 17p deletion or TP53 mutation and are unsuitable for chemotherapy.
In the EU and all other countries except the US, ibrutinib is marketed by Janssen Pharmaceutical Companies. In the US, the drug is being jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc.
The EU approval of ibrutinib was based on data from a phase 2 study (PCYC-1104) in patients with MCL, the phase 3 RESONATE trial (PCYC-1112-CA) in CLL and small lymphocytic lymphoma (SLL), and a phase 1b/2 study (PCYC-1102) in CLL/SLL.
PCYC-1104: Ibrutinib in MCL
Results of this trial were presented at ASH 2012 and published in NEJM in 2013. The NEJM data included 111 patients who received ibrutinib at 560 mg daily in continuous, 28-day cycles until disease progression.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With an estimated median follow-up of 15.3 months, the estimated median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
RESONATE: Ibrutinib in CLL/SLL
Results of the RESONATE trial were reported at EHA 2014 and published in NEJM in July. This trial was stopped early after an interim analysis showed that ibrutinib-treated patients experienced a 78% reduction in the risk of disease progression or death.
The trial included 391 patients with relapsed or refractory CLL or SLL who were randomized to receive ibrutinib (n=195) or ofatumumab (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57). The median time on study was 9.4 months.
The best overall response rate was higher in the ibrutinib arm than the ofatumumab arm, at 78% and 11%, respectively. And ibrutinib significantly prolonged progression-free survival. The median was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001).
Ibrutinib significantly prolonged overall survival as well. The median overall survival was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049).
Adverse events occurred in 99% of patients in the ibrutinib arm and 98% of those in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation, bleeding-related events, diarrhea, and arthralgia were more common in the ibrutinib arm. Infusion-related reactions, peripheral sensory neuropathy, urticaria, night sweats, and pruritus were more common in the ofatumumab arm.
PCYC-1102: Ibrutinib in CLL/SLL
Results of this phase 1b/2 trial were published in The Lancet Oncology in January. The trial enrolled 29 patients with previously untreated CLL and 2 with SLL.
They received 28-day cycles of once-daily ibrutinib at 420 mg or 840 mg. The 840 mg dose was discontinued after enrollment had begun because the doses showed comparable activity.
After a median follow-up of 22.1 months, 71% of patients achieved an objective response. Four patients (13%) had a complete response. The median time to response was 1.9 months.
Study investigators did not establish whether ibrutinib confers improvements in survival or disease-related symptoms.
Common adverse events included diarrhea (68%), nausea (48%), fatigue (32%), peripheral edema (29%), hypertension (29%), dizziness (26%), dyspepsia (26%), upper respiratory tract infection (26%), arthralgia (23%), constipation (23%), urinary tract infection (23%), and vomiting (23%).
Grade 3 adverse events included diarrhea (13%), fatigue (3%), hypertension (6%), dizziness (3%), urinary tract infection (3%), headache (3%), back pain (3%), and neutropenia (3%). One patient (3%) had grade 4 thrombocytopenia.