User login
Lens-free microscope a ‘milestone’
Credit: Максим Кукушкин
Researchers say they’ve developed a lens-free microscope that can detect the presence of cell-level abnormalities with the same accuracy as larger and more expensive optical microscopes.
The invention could lead to less expensive, more portable technology for performing examinations of tissue, blood, and other biomedical specimens, according to the researchers.
It may prove especially useful in remote areas and when large numbers of samples need to be examined quickly.
Aydogan Ozcan, PhD, of the University of California, Los Angeles, and his colleagues described their work with the microscope in Science Translational Medicine.
“This is a milestone in the work we’ve been doing,” Dr Ozcan said. “This is the first time tissue samples have been imaged in 3D using a lens-free, on-chip microscope.”
The device works by using a laser or light-emitting-diode to illuminate a tissue or blood sample that has been placed on a slide and inserted into the device. A sensor array on a microchip captures and records the pattern of shadows created by the sample.
The device processes these patterns as a series of holograms, forming 3-D images of the specimen and giving medical personnel a virtual depth-of-field view. An algorithm color codes the reconstructed images, making the contrasts in the samples more apparent than they would be in the holograms and making any abnormalities easier to detect.
Dr Ozcan’s team tested the device using blood samples containing sickle cell anemia, Pap smears that indicated cervical cancer, and tissue specimens containing cancerous breast cells.
In a blind test, a board-certified pathologist analyzed sets of specimen images that had been created by the lens-free technology and by conventional microscopes. The pathologist’s diagnoses using the lens-free microscopic images proved accurate 99% of the time.
Another benefit of the lens-free device, according to the researchers, is that it produces images that are several hundred times larger in area, or field of view, than those captured by conventional bright-field optical microscopes. This makes it possible to process specimens more quickly.
“While mobile healthcare has expanded rapidly with the growth of consumer electronics—cellphones in particular—pathology is still, by and large, constrained to advanced clinical laboratory settings,” Dr Ozcan said. “Accompanied by advances in its graphical user interface, this platform could scale up for use in clinical, biomedical, scientific, educational, and citizen-science applications, among others.” ![]()
Credit: Максим Кукушкин
Researchers say they’ve developed a lens-free microscope that can detect the presence of cell-level abnormalities with the same accuracy as larger and more expensive optical microscopes.
The invention could lead to less expensive, more portable technology for performing examinations of tissue, blood, and other biomedical specimens, according to the researchers.
It may prove especially useful in remote areas and when large numbers of samples need to be examined quickly.
Aydogan Ozcan, PhD, of the University of California, Los Angeles, and his colleagues described their work with the microscope in Science Translational Medicine.
“This is a milestone in the work we’ve been doing,” Dr Ozcan said. “This is the first time tissue samples have been imaged in 3D using a lens-free, on-chip microscope.”
The device works by using a laser or light-emitting-diode to illuminate a tissue or blood sample that has been placed on a slide and inserted into the device. A sensor array on a microchip captures and records the pattern of shadows created by the sample.
The device processes these patterns as a series of holograms, forming 3-D images of the specimen and giving medical personnel a virtual depth-of-field view. An algorithm color codes the reconstructed images, making the contrasts in the samples more apparent than they would be in the holograms and making any abnormalities easier to detect.
Dr Ozcan’s team tested the device using blood samples containing sickle cell anemia, Pap smears that indicated cervical cancer, and tissue specimens containing cancerous breast cells.
In a blind test, a board-certified pathologist analyzed sets of specimen images that had been created by the lens-free technology and by conventional microscopes. The pathologist’s diagnoses using the lens-free microscopic images proved accurate 99% of the time.
Another benefit of the lens-free device, according to the researchers, is that it produces images that are several hundred times larger in area, or field of view, than those captured by conventional bright-field optical microscopes. This makes it possible to process specimens more quickly.
“While mobile healthcare has expanded rapidly with the growth of consumer electronics—cellphones in particular—pathology is still, by and large, constrained to advanced clinical laboratory settings,” Dr Ozcan said. “Accompanied by advances in its graphical user interface, this platform could scale up for use in clinical, biomedical, scientific, educational, and citizen-science applications, among others.” ![]()
Credit: Максим Кукушкин
Researchers say they’ve developed a lens-free microscope that can detect the presence of cell-level abnormalities with the same accuracy as larger and more expensive optical microscopes.
The invention could lead to less expensive, more portable technology for performing examinations of tissue, blood, and other biomedical specimens, according to the researchers.
It may prove especially useful in remote areas and when large numbers of samples need to be examined quickly.
Aydogan Ozcan, PhD, of the University of California, Los Angeles, and his colleagues described their work with the microscope in Science Translational Medicine.
“This is a milestone in the work we’ve been doing,” Dr Ozcan said. “This is the first time tissue samples have been imaged in 3D using a lens-free, on-chip microscope.”
The device works by using a laser or light-emitting-diode to illuminate a tissue or blood sample that has been placed on a slide and inserted into the device. A sensor array on a microchip captures and records the pattern of shadows created by the sample.
The device processes these patterns as a series of holograms, forming 3-D images of the specimen and giving medical personnel a virtual depth-of-field view. An algorithm color codes the reconstructed images, making the contrasts in the samples more apparent than they would be in the holograms and making any abnormalities easier to detect.
Dr Ozcan’s team tested the device using blood samples containing sickle cell anemia, Pap smears that indicated cervical cancer, and tissue specimens containing cancerous breast cells.
In a blind test, a board-certified pathologist analyzed sets of specimen images that had been created by the lens-free technology and by conventional microscopes. The pathologist’s diagnoses using the lens-free microscopic images proved accurate 99% of the time.
Another benefit of the lens-free device, according to the researchers, is that it produces images that are several hundred times larger in area, or field of view, than those captured by conventional bright-field optical microscopes. This makes it possible to process specimens more quickly.
“While mobile healthcare has expanded rapidly with the growth of consumer electronics—cellphones in particular—pathology is still, by and large, constrained to advanced clinical laboratory settings,” Dr Ozcan said. “Accompanied by advances in its graphical user interface, this platform could scale up for use in clinical, biomedical, scientific, educational, and citizen-science applications, among others.” ![]()
Study shows higher risk of MDS, leukemia after breast cancer

chemotherapy
Credit: Rhoda Baer
The risk of developing myelodysplastic syndromes (MDS) or leukemias after treatment for early stage breast cancer is higher than previously reported, according to a study published in the Journal of Clinical Oncology.
Data from earlier studies showed that about 0.25% of breast cancer patients develop MDS or leukemia as a late effect of chemotherapy, said Judith Karp, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
But Dr Karp and her colleagues found the 10-year incidence of MDS and leukemia among breast cancer patients to be about 0.5%.
“[T]he cumulative risk over a decade is now shown to be twice as high as we thought it was, and that risk doesn’t seem to slow down 5 years after treatment,” Dr Karp said. “Most medical oncologists have come to think that the risk is early and short-lived. So this was a little bit of a wake-up call that we are not seeing any plateau of that risk, and it is higher.”
Dr Karp and her colleagues reviewed data on 20,063 breast cancer patients treated at 8 US cancer centers between 1998 and 2007 whose cancer recurrence and secondary cancer rates were recorded in a database kept by the National Comprehensive Cancer Network.
At a median follow-up of 5.1 years, 50 patients had developed a marrow neoplasm, including acute myeloid leukemia (n=24), MDS/acute myeloid leukemia (n=15), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=5), chronic myeloid leukemia (n=3), or acute lymphoblastic leukemia (n=3).
The risk of developing MDS/leukemia was about 7 times higher for patients who underwent surgery and received chemotherapy, compared to patients who did not receive chemotherapy. For patients who underwent surgery and received both chemotherapy and radiation, the risk was about 8 times higher.
The MDS/leukemia rates per 1000 person-years were 0.16 for surgery, 0.43 for surgery plus radiation, 0.46 for surgery plus chemotherapy, and 0.54 for all 3 modalities.
The cumulative incidence of MDS/leukemia doubled between years 5 and 10, rising from 0.24% to 0.48%. And only 9% of patients were alive at 10 years.
Antonio Wolff, MD, of the Johns Hopkins University School of Medicine, said this study could help early stage breast cancer patients and their physicians think more carefully about the use of preventive or adjuvant chemotherapy, especially when patients have a low risk of cancer recurrence.
“Our study provides useful information for physicians and patients to consider a potential downside of preventive or adjuvant chemotherapy in patients with very low risk of breast cancer recurrence,” he said.
“It could be a false and dangerous security blanket to some patients by exposing them to a small risk of serious late effects with little or no real benefit from the treatment.”
The researchers included a hypothetical case to put the risks of early stage breast cancer and its treatment in perspective. They described a 60-year-old woman in average health who was diagnosed with stage 1 breast cancer that was rapidly growing and estrogen receptor-positive.
The patient had an estimated 12.3% risk of dying of breast cancer after 10 years. She could improve her 10-year survival rate by 1.8% with 4 cycles of chemotherapy, but she would also increase her risk of MDS/leukemia over that same time by 0.5%.
Dr Wolff noted that it’s unclear whether the increased risk of MDS/leukemia after postsurgical chemotherapy applies to patients with other kinds of solid tumors, as the drug regimens used in breast cancer differ from those used for other cancers. ![]()
chemotherapy
Credit: Rhoda Baer
The risk of developing myelodysplastic syndromes (MDS) or leukemias after treatment for early stage breast cancer is higher than previously reported, according to a study published in the Journal of Clinical Oncology.
Data from earlier studies showed that about 0.25% of breast cancer patients develop MDS or leukemia as a late effect of chemotherapy, said Judith Karp, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
But Dr Karp and her colleagues found the 10-year incidence of MDS and leukemia among breast cancer patients to be about 0.5%.
“[T]he cumulative risk over a decade is now shown to be twice as high as we thought it was, and that risk doesn’t seem to slow down 5 years after treatment,” Dr Karp said. “Most medical oncologists have come to think that the risk is early and short-lived. So this was a little bit of a wake-up call that we are not seeing any plateau of that risk, and it is higher.”
Dr Karp and her colleagues reviewed data on 20,063 breast cancer patients treated at 8 US cancer centers between 1998 and 2007 whose cancer recurrence and secondary cancer rates were recorded in a database kept by the National Comprehensive Cancer Network.
At a median follow-up of 5.1 years, 50 patients had developed a marrow neoplasm, including acute myeloid leukemia (n=24), MDS/acute myeloid leukemia (n=15), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=5), chronic myeloid leukemia (n=3), or acute lymphoblastic leukemia (n=3).
The risk of developing MDS/leukemia was about 7 times higher for patients who underwent surgery and received chemotherapy, compared to patients who did not receive chemotherapy. For patients who underwent surgery and received both chemotherapy and radiation, the risk was about 8 times higher.
The MDS/leukemia rates per 1000 person-years were 0.16 for surgery, 0.43 for surgery plus radiation, 0.46 for surgery plus chemotherapy, and 0.54 for all 3 modalities.
The cumulative incidence of MDS/leukemia doubled between years 5 and 10, rising from 0.24% to 0.48%. And only 9% of patients were alive at 10 years.
Antonio Wolff, MD, of the Johns Hopkins University School of Medicine, said this study could help early stage breast cancer patients and their physicians think more carefully about the use of preventive or adjuvant chemotherapy, especially when patients have a low risk of cancer recurrence.
“Our study provides useful information for physicians and patients to consider a potential downside of preventive or adjuvant chemotherapy in patients with very low risk of breast cancer recurrence,” he said.
“It could be a false and dangerous security blanket to some patients by exposing them to a small risk of serious late effects with little or no real benefit from the treatment.”
The researchers included a hypothetical case to put the risks of early stage breast cancer and its treatment in perspective. They described a 60-year-old woman in average health who was diagnosed with stage 1 breast cancer that was rapidly growing and estrogen receptor-positive.
The patient had an estimated 12.3% risk of dying of breast cancer after 10 years. She could improve her 10-year survival rate by 1.8% with 4 cycles of chemotherapy, but she would also increase her risk of MDS/leukemia over that same time by 0.5%.
Dr Wolff noted that it’s unclear whether the increased risk of MDS/leukemia after postsurgical chemotherapy applies to patients with other kinds of solid tumors, as the drug regimens used in breast cancer differ from those used for other cancers. ![]()
chemotherapy
Credit: Rhoda Baer
The risk of developing myelodysplastic syndromes (MDS) or leukemias after treatment for early stage breast cancer is higher than previously reported, according to a study published in the Journal of Clinical Oncology.
Data from earlier studies showed that about 0.25% of breast cancer patients develop MDS or leukemia as a late effect of chemotherapy, said Judith Karp, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
But Dr Karp and her colleagues found the 10-year incidence of MDS and leukemia among breast cancer patients to be about 0.5%.
“[T]he cumulative risk over a decade is now shown to be twice as high as we thought it was, and that risk doesn’t seem to slow down 5 years after treatment,” Dr Karp said. “Most medical oncologists have come to think that the risk is early and short-lived. So this was a little bit of a wake-up call that we are not seeing any plateau of that risk, and it is higher.”
Dr Karp and her colleagues reviewed data on 20,063 breast cancer patients treated at 8 US cancer centers between 1998 and 2007 whose cancer recurrence and secondary cancer rates were recorded in a database kept by the National Comprehensive Cancer Network.
At a median follow-up of 5.1 years, 50 patients had developed a marrow neoplasm, including acute myeloid leukemia (n=24), MDS/acute myeloid leukemia (n=15), chronic lymphocytic leukemia/small lymphocytic lymphoma (n=5), chronic myeloid leukemia (n=3), or acute lymphoblastic leukemia (n=3).
The risk of developing MDS/leukemia was about 7 times higher for patients who underwent surgery and received chemotherapy, compared to patients who did not receive chemotherapy. For patients who underwent surgery and received both chemotherapy and radiation, the risk was about 8 times higher.
The MDS/leukemia rates per 1000 person-years were 0.16 for surgery, 0.43 for surgery plus radiation, 0.46 for surgery plus chemotherapy, and 0.54 for all 3 modalities.
The cumulative incidence of MDS/leukemia doubled between years 5 and 10, rising from 0.24% to 0.48%. And only 9% of patients were alive at 10 years.
Antonio Wolff, MD, of the Johns Hopkins University School of Medicine, said this study could help early stage breast cancer patients and their physicians think more carefully about the use of preventive or adjuvant chemotherapy, especially when patients have a low risk of cancer recurrence.
“Our study provides useful information for physicians and patients to consider a potential downside of preventive or adjuvant chemotherapy in patients with very low risk of breast cancer recurrence,” he said.
“It could be a false and dangerous security blanket to some patients by exposing them to a small risk of serious late effects with little or no real benefit from the treatment.”
The researchers included a hypothetical case to put the risks of early stage breast cancer and its treatment in perspective. They described a 60-year-old woman in average health who was diagnosed with stage 1 breast cancer that was rapidly growing and estrogen receptor-positive.
The patient had an estimated 12.3% risk of dying of breast cancer after 10 years. She could improve her 10-year survival rate by 1.8% with 4 cycles of chemotherapy, but she would also increase her risk of MDS/leukemia over that same time by 0.5%.
Dr Wolff noted that it’s unclear whether the increased risk of MDS/leukemia after postsurgical chemotherapy applies to patients with other kinds of solid tumors, as the drug regimens used in breast cancer differ from those used for other cancers. ![]()
How malaria parasites evade the immune system
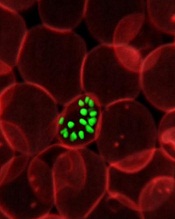
Credit: St Jude
Children’s Research Hospital
A new study has shown that malaria parasites can rapidly change proteins on the surface of human red blood cells (RBCs) during the course of a single infection, which helps the parasites evade the immune system.
The findings, which overturn previous thinking about the Plasmodium falciparum parasite’s lifecycle, could explain why so many attempts to create an effective malaria vaccine have failed and how the parasites are able to survive in the human body for such long periods of time.
Investigators described this research in PLOS Genetics.
The team kept P falciparum parasites dividing in human blood in the lab for over a year and sequenced the full parasite genome regularly. This provided snapshots of the parasite’s genome at multiple time points, allowing them to track evolution as it unfolded in the lab.
They found that the 60 or so genes that control proteins on the surface of infected human RBCs, known as var genes, swapped genetic information regularly, creating around a million new and unrecognizable surface proteins in every infected human every 2 days.
“These genes are like decks of cards constantly being shuffled,” explained study author William Hamilton, a graduate student at the Wellcome Trust Sanger Institute in Cambridge, UK.
“The use of whole-genome sequencing and the sheer number of samples we collected gave us a detailed picture of how the var gene repertoire changes continuously within red blood cells.”
The results showed, for the first time, that recombination does not occur when the malaria parasite is inside the mosquito, as previously thought. Instead, it occurs during the asexual stage of the parasite’s lifecycle inside human RBCs. This finding may help explain how chronic asymptomatic infection, a crucial problem for malaria elimination, is possible.
“It’s very likely that mosquitos are re-infected with Plasmodium falciparum parasites at the beginning of each wet season by biting humans who have carried the parasites, often asymptomatically, for up to 8 months during the dry season,” said study author Antoine Claessens, PhD, of the Wellcome Trust Sanger Institute.
“During those months, the parasite’s var genes are busy recombining to create millions of different versions—cunning disguises that mean they remain safe from the immune system and ready for the new malarial season.”
While further work will be required to fully understand the mechanism driving the recombination of P falciparum’s var genes, the investigators were able to calculate the rate at which it happens. They found that var gene recombination takes place in about 0.2% of parasites after each 48-hour life cycle in the RBC.
With about a billion parasites living inside a typical infected human, there is huge potential for the parasite to create new, recombined var genes inside each person with malaria. This pace of change far exceeds that of genes in any other region of the parasite’s genome.
“When you consider that 200 million people across the world are infected with malaria, and each of them is harboring parasites that are continually generating millions of antigenic variants, it becomes apparent why our fight against malaria is so challenging,” said study author Dominic Kwiatkowski, MBBS, of the Wellcome Trust Sanger Institute.
“This study is a great example of how genome sequence analysis is enriching our understanding of malaria biology. By learning the genetic tricks that the parasite uses to evade the human immune system, we will be in a much better position to eliminate this deadly disease.” ![]()
Credit: St Jude
Children’s Research Hospital
A new study has shown that malaria parasites can rapidly change proteins on the surface of human red blood cells (RBCs) during the course of a single infection, which helps the parasites evade the immune system.
The findings, which overturn previous thinking about the Plasmodium falciparum parasite’s lifecycle, could explain why so many attempts to create an effective malaria vaccine have failed and how the parasites are able to survive in the human body for such long periods of time.
Investigators described this research in PLOS Genetics.
The team kept P falciparum parasites dividing in human blood in the lab for over a year and sequenced the full parasite genome regularly. This provided snapshots of the parasite’s genome at multiple time points, allowing them to track evolution as it unfolded in the lab.
They found that the 60 or so genes that control proteins on the surface of infected human RBCs, known as var genes, swapped genetic information regularly, creating around a million new and unrecognizable surface proteins in every infected human every 2 days.
“These genes are like decks of cards constantly being shuffled,” explained study author William Hamilton, a graduate student at the Wellcome Trust Sanger Institute in Cambridge, UK.
“The use of whole-genome sequencing and the sheer number of samples we collected gave us a detailed picture of how the var gene repertoire changes continuously within red blood cells.”
The results showed, for the first time, that recombination does not occur when the malaria parasite is inside the mosquito, as previously thought. Instead, it occurs during the asexual stage of the parasite’s lifecycle inside human RBCs. This finding may help explain how chronic asymptomatic infection, a crucial problem for malaria elimination, is possible.
“It’s very likely that mosquitos are re-infected with Plasmodium falciparum parasites at the beginning of each wet season by biting humans who have carried the parasites, often asymptomatically, for up to 8 months during the dry season,” said study author Antoine Claessens, PhD, of the Wellcome Trust Sanger Institute.
“During those months, the parasite’s var genes are busy recombining to create millions of different versions—cunning disguises that mean they remain safe from the immune system and ready for the new malarial season.”
While further work will be required to fully understand the mechanism driving the recombination of P falciparum’s var genes, the investigators were able to calculate the rate at which it happens. They found that var gene recombination takes place in about 0.2% of parasites after each 48-hour life cycle in the RBC.
With about a billion parasites living inside a typical infected human, there is huge potential for the parasite to create new, recombined var genes inside each person with malaria. This pace of change far exceeds that of genes in any other region of the parasite’s genome.
“When you consider that 200 million people across the world are infected with malaria, and each of them is harboring parasites that are continually generating millions of antigenic variants, it becomes apparent why our fight against malaria is so challenging,” said study author Dominic Kwiatkowski, MBBS, of the Wellcome Trust Sanger Institute.
“This study is a great example of how genome sequence analysis is enriching our understanding of malaria biology. By learning the genetic tricks that the parasite uses to evade the human immune system, we will be in a much better position to eliminate this deadly disease.” ![]()
Credit: St Jude
Children’s Research Hospital
A new study has shown that malaria parasites can rapidly change proteins on the surface of human red blood cells (RBCs) during the course of a single infection, which helps the parasites evade the immune system.
The findings, which overturn previous thinking about the Plasmodium falciparum parasite’s lifecycle, could explain why so many attempts to create an effective malaria vaccine have failed and how the parasites are able to survive in the human body for such long periods of time.
Investigators described this research in PLOS Genetics.
The team kept P falciparum parasites dividing in human blood in the lab for over a year and sequenced the full parasite genome regularly. This provided snapshots of the parasite’s genome at multiple time points, allowing them to track evolution as it unfolded in the lab.
They found that the 60 or so genes that control proteins on the surface of infected human RBCs, known as var genes, swapped genetic information regularly, creating around a million new and unrecognizable surface proteins in every infected human every 2 days.
“These genes are like decks of cards constantly being shuffled,” explained study author William Hamilton, a graduate student at the Wellcome Trust Sanger Institute in Cambridge, UK.
“The use of whole-genome sequencing and the sheer number of samples we collected gave us a detailed picture of how the var gene repertoire changes continuously within red blood cells.”
The results showed, for the first time, that recombination does not occur when the malaria parasite is inside the mosquito, as previously thought. Instead, it occurs during the asexual stage of the parasite’s lifecycle inside human RBCs. This finding may help explain how chronic asymptomatic infection, a crucial problem for malaria elimination, is possible.
“It’s very likely that mosquitos are re-infected with Plasmodium falciparum parasites at the beginning of each wet season by biting humans who have carried the parasites, often asymptomatically, for up to 8 months during the dry season,” said study author Antoine Claessens, PhD, of the Wellcome Trust Sanger Institute.
“During those months, the parasite’s var genes are busy recombining to create millions of different versions—cunning disguises that mean they remain safe from the immune system and ready for the new malarial season.”
While further work will be required to fully understand the mechanism driving the recombination of P falciparum’s var genes, the investigators were able to calculate the rate at which it happens. They found that var gene recombination takes place in about 0.2% of parasites after each 48-hour life cycle in the RBC.
With about a billion parasites living inside a typical infected human, there is huge potential for the parasite to create new, recombined var genes inside each person with malaria. This pace of change far exceeds that of genes in any other region of the parasite’s genome.
“When you consider that 200 million people across the world are infected with malaria, and each of them is harboring parasites that are continually generating millions of antigenic variants, it becomes apparent why our fight against malaria is so challenging,” said study author Dominic Kwiatkowski, MBBS, of the Wellcome Trust Sanger Institute.
“This study is a great example of how genome sequence analysis is enriching our understanding of malaria biology. By learning the genetic tricks that the parasite uses to evade the human immune system, we will be in a much better position to eliminate this deadly disease.” ![]()
FDA recommends changing blood donor policy for MSM

Photo by Elisa Amendola
The US Food and Drug Administration (FDA) is recommending a change to the policy that prevents men who have sex with men (MSM) from donating blood, according to FDA Commissioner Margaret A. Hamburg.
The FDA would like to allow MSM to donate blood if they have abstained from sexual contact for 1 year.
The agency intends to issue a draft guidance recommending this policy change in 2015. The guidance will be open for public comment.
In a prepared statement, Hamburg said that, over the past few years, the FDA and other government agencies have carefully considered the scientific evidence relevant to the blood donor deferral policy for MSM.
This review, as well as the recommendations of advisory committees to the US Department of Health and Human Services (HHS) and the FDA, has prompted the FDA to recommend the change.
“This recommended change is consistent with the recommendation of an independent expert advisory panel, the HHS Advisory Committee on Blood and Tissue Safety and Availability, and will better align the deferral period with that of other men and women at increased risk for HIV infection,” Hamburg said.
“Additionally, in collaboration with the NIH’s National Heart Lung and Blood Institute (NHLBI), the FDA has already taken steps to implement a national blood surveillance system that will help the agency monitor the effect of a policy change and further help to ensure the continued safety of the blood supply.” ![]()
Photo by Elisa Amendola
The US Food and Drug Administration (FDA) is recommending a change to the policy that prevents men who have sex with men (MSM) from donating blood, according to FDA Commissioner Margaret A. Hamburg.
The FDA would like to allow MSM to donate blood if they have abstained from sexual contact for 1 year.
The agency intends to issue a draft guidance recommending this policy change in 2015. The guidance will be open for public comment.
In a prepared statement, Hamburg said that, over the past few years, the FDA and other government agencies have carefully considered the scientific evidence relevant to the blood donor deferral policy for MSM.
This review, as well as the recommendations of advisory committees to the US Department of Health and Human Services (HHS) and the FDA, has prompted the FDA to recommend the change.
“This recommended change is consistent with the recommendation of an independent expert advisory panel, the HHS Advisory Committee on Blood and Tissue Safety and Availability, and will better align the deferral period with that of other men and women at increased risk for HIV infection,” Hamburg said.
“Additionally, in collaboration with the NIH’s National Heart Lung and Blood Institute (NHLBI), the FDA has already taken steps to implement a national blood surveillance system that will help the agency monitor the effect of a policy change and further help to ensure the continued safety of the blood supply.” ![]()
Photo by Elisa Amendola
The US Food and Drug Administration (FDA) is recommending a change to the policy that prevents men who have sex with men (MSM) from donating blood, according to FDA Commissioner Margaret A. Hamburg.
The FDA would like to allow MSM to donate blood if they have abstained from sexual contact for 1 year.
The agency intends to issue a draft guidance recommending this policy change in 2015. The guidance will be open for public comment.
In a prepared statement, Hamburg said that, over the past few years, the FDA and other government agencies have carefully considered the scientific evidence relevant to the blood donor deferral policy for MSM.
This review, as well as the recommendations of advisory committees to the US Department of Health and Human Services (HHS) and the FDA, has prompted the FDA to recommend the change.
“This recommended change is consistent with the recommendation of an independent expert advisory panel, the HHS Advisory Committee on Blood and Tissue Safety and Availability, and will better align the deferral period with that of other men and women at increased risk for HIV infection,” Hamburg said.
“Additionally, in collaboration with the NIH’s National Heart Lung and Blood Institute (NHLBI), the FDA has already taken steps to implement a national blood surveillance system that will help the agency monitor the effect of a policy change and further help to ensure the continued safety of the blood supply.” ![]()
Mitoxantrone lots recalled worldwide

Credit: Bill Branson
Hospira, Inc. has initiated a worldwide, user-level recall of 10 lots of Mitoxantrone (both human and veterinary) due to confirmed subpotency and elevated impurity levels.
Drugs in the affected lots may exhibit decreased effectiveness, require additional dosing, or prompt cumulative impurity toxicity requiring medical intervention.
However, Hospira has not received reports of any adverse events associated with subpotency and impurities for these lots to date.
The lots were distributed to hospitals and veterinary clinics worldwide from February 2013 through November 2014.
The following lots are affected by the recall. (To ensure this list displays properly, click the “Hide” icon on the right side of this page to hide the “In this Section” column.)
United States
Product NDC Number Lot Expiration Date
MitoXANTRONE Injection, USP, 61703-343-18 Z054636AA December 2014
(concentrate) 20 mg/10 mL, A014636AA April 2015
2 mg/mL in 10 mL, 10 mL Vial, A024636AB July 2015
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-65 A014643AA April 2015
(concentrate) 25 mg/12.5 mL,
2 mg/mL in 12.5 mL, 12.5 mL Vial,
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-66 A014645AA November 2015
(concentrate) 30 mg/15 mL,
2 mg/mL in 15 mL, 15 mL Vial,
Multi Dose Vial
Australia and New Zealand
Product Product Code Batch Number Expiration Date
DBL™ MitoXANTRONE M4636A A024636AA July 2015
Hydrochloride Injection
(concentrate) 20mg/10mL
Injection Vial
United Kingdom, Ireland, Cyprus, Saudi Arabia, Qatar, Oman and Bahrain
Product List Number Lot Expiration Date
MitoXANTRONE 2 mg/mL; M4636AGB1 A014636AB April 2015
Concentrate for Infusion A024636AD July 2015
Z054636AB Dec 2014
Canada
Product List Number DIN Lot Expiration Date
MitoXANTRONE for
Injection 20mg /10mL USP 4636A001 02244614 A024636AC July 2015
Anyone with an existing inventory of the recalled lots should stop use and distribution, and quarantine the product immediately. This recall is being carried out to the user level (both human and veterinary).
Hospira has notified its direct customers via a recall letter and is arranging for impacted product to be returned to Stericycle in the US. For additional assistance in the US, call Stericycle at 1-844-265-7407 between the hours of 8 am and 5 pm ET, Monday through Friday. Customers outside the US should work with their local Hospira offices to return the product per local recall notifications.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or medcom@hospira.com (Available 24 hours a day/7 days per week).
To report adverse events or for product complaints, contact Hospira Global Complaint Management at 1-800-441-4100 (M-F, 8 am to 5 pm CT).
Adverse events or quality problems associated with Mitoxantrone can also be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Credit: Bill Branson
Hospira, Inc. has initiated a worldwide, user-level recall of 10 lots of Mitoxantrone (both human and veterinary) due to confirmed subpotency and elevated impurity levels.
Drugs in the affected lots may exhibit decreased effectiveness, require additional dosing, or prompt cumulative impurity toxicity requiring medical intervention.
However, Hospira has not received reports of any adverse events associated with subpotency and impurities for these lots to date.
The lots were distributed to hospitals and veterinary clinics worldwide from February 2013 through November 2014.
The following lots are affected by the recall. (To ensure this list displays properly, click the “Hide” icon on the right side of this page to hide the “In this Section” column.)
United States
Product NDC Number Lot Expiration Date
MitoXANTRONE Injection, USP, 61703-343-18 Z054636AA December 2014
(concentrate) 20 mg/10 mL, A014636AA April 2015
2 mg/mL in 10 mL, 10 mL Vial, A024636AB July 2015
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-65 A014643AA April 2015
(concentrate) 25 mg/12.5 mL,
2 mg/mL in 12.5 mL, 12.5 mL Vial,
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-66 A014645AA November 2015
(concentrate) 30 mg/15 mL,
2 mg/mL in 15 mL, 15 mL Vial,
Multi Dose Vial
Australia and New Zealand
Product Product Code Batch Number Expiration Date
DBL™ MitoXANTRONE M4636A A024636AA July 2015
Hydrochloride Injection
(concentrate) 20mg/10mL
Injection Vial
United Kingdom, Ireland, Cyprus, Saudi Arabia, Qatar, Oman and Bahrain
Product List Number Lot Expiration Date
MitoXANTRONE 2 mg/mL; M4636AGB1 A014636AB April 2015
Concentrate for Infusion A024636AD July 2015
Z054636AB Dec 2014
Canada
Product List Number DIN Lot Expiration Date
MitoXANTRONE for
Injection 20mg /10mL USP 4636A001 02244614 A024636AC July 2015
Anyone with an existing inventory of the recalled lots should stop use and distribution, and quarantine the product immediately. This recall is being carried out to the user level (both human and veterinary).
Hospira has notified its direct customers via a recall letter and is arranging for impacted product to be returned to Stericycle in the US. For additional assistance in the US, call Stericycle at 1-844-265-7407 between the hours of 8 am and 5 pm ET, Monday through Friday. Customers outside the US should work with their local Hospira offices to return the product per local recall notifications.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or medcom@hospira.com (Available 24 hours a day/7 days per week).
To report adverse events or for product complaints, contact Hospira Global Complaint Management at 1-800-441-4100 (M-F, 8 am to 5 pm CT).
Adverse events or quality problems associated with Mitoxantrone can also be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Credit: Bill Branson
Hospira, Inc. has initiated a worldwide, user-level recall of 10 lots of Mitoxantrone (both human and veterinary) due to confirmed subpotency and elevated impurity levels.
Drugs in the affected lots may exhibit decreased effectiveness, require additional dosing, or prompt cumulative impurity toxicity requiring medical intervention.
However, Hospira has not received reports of any adverse events associated with subpotency and impurities for these lots to date.
The lots were distributed to hospitals and veterinary clinics worldwide from February 2013 through November 2014.
The following lots are affected by the recall. (To ensure this list displays properly, click the “Hide” icon on the right side of this page to hide the “In this Section” column.)
United States
Product NDC Number Lot Expiration Date
MitoXANTRONE Injection, USP, 61703-343-18 Z054636AA December 2014
(concentrate) 20 mg/10 mL, A014636AA April 2015
2 mg/mL in 10 mL, 10 mL Vial, A024636AB July 2015
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-65 A014643AA April 2015
(concentrate) 25 mg/12.5 mL,
2 mg/mL in 12.5 mL, 12.5 mL Vial,
Multi Dose Vial
MitoXANTRONE Injection, USP, 61703-343-66 A014645AA November 2015
(concentrate) 30 mg/15 mL,
2 mg/mL in 15 mL, 15 mL Vial,
Multi Dose Vial
Australia and New Zealand
Product Product Code Batch Number Expiration Date
DBL™ MitoXANTRONE M4636A A024636AA July 2015
Hydrochloride Injection
(concentrate) 20mg/10mL
Injection Vial
United Kingdom, Ireland, Cyprus, Saudi Arabia, Qatar, Oman and Bahrain
Product List Number Lot Expiration Date
MitoXANTRONE 2 mg/mL; M4636AGB1 A014636AB April 2015
Concentrate for Infusion A024636AD July 2015
Z054636AB Dec 2014
Canada
Product List Number DIN Lot Expiration Date
MitoXANTRONE for
Injection 20mg /10mL USP 4636A001 02244614 A024636AC July 2015
Anyone with an existing inventory of the recalled lots should stop use and distribution, and quarantine the product immediately. This recall is being carried out to the user level (both human and veterinary).
Hospira has notified its direct customers via a recall letter and is arranging for impacted product to be returned to Stericycle in the US. For additional assistance in the US, call Stericycle at 1-844-265-7407 between the hours of 8 am and 5 pm ET, Monday through Friday. Customers outside the US should work with their local Hospira offices to return the product per local recall notifications.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or medcom@hospira.com (Available 24 hours a day/7 days per week).
To report adverse events or for product complaints, contact Hospira Global Complaint Management at 1-800-441-4100 (M-F, 8 am to 5 pm CT).
Adverse events or quality problems associated with Mitoxantrone can also be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Age-adjusted D-dimer is ‘probably safe,’ team says

Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
FDA approves new formulation of drug for ALL

The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()
The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()
The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()
CHMP supports expanding use of lenalidomide in MM

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval of continuous oral treatment with lenalidomide (Revlimid) in adults with previously untreated multiple myeloma (MM) who are ineligible for hematopoietic stem cell transplant (HSCT).
The European Commission, which generally follows the CHMP’s recommendations, is expected to make its final decision in about 2 months.
Lenalidomide is not currently approved to treat newly diagnosed MM in any country.
The drug is approved in the European Union (EU) for use in combination with dexamethasone to treat adults with MM who have received at least one prior therapy.
Lenalidomide is also approved in the EU to treat patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with 5q deletion when other therapeutic options are insufficient or inadequate.
The CHMP’s recommendation to extend the use of lenalidomide to HSCT-ineligible patients with newly diagnosed MM was based on the results of 2 studies: MM-015 and MM-020, also known as FIRST.
The FIRST trial
In the phase 3 FIRST trial, researchers enrolled 1623 patients who were newly diagnosed with MM and not eligible for HSCT.
Patients were randomized to receive lenalidomide and dexamethasone (Rd) in 28-day cycles until disease progression (n=535), 18 cycles of lenalidomide and dexamethasone (Rd18) for 72 weeks (n=541), or melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
Response rates were significantly better with continuous Rd (75%) and Rd18 (73%) than with MPT (62%, P<0.001 for both comparisons). Complete response rates were 15%, 14%, and 9%, respectively.
The median progression-free survival was 25.5 months with continuous Rd, 20.7 months with Rd18, and 21.2 months with MPT.
This resulted in a 28% reduction in the risk of progression or death for patients treated with continuous Rd compared with those treated with MPT (hazard ratio[HR]=0.72, P<0.001) and a 30% reduction compared with Rd18 (HR=0.70, P<0.001).
The pre-planned interim analysis of overall survival showed a 22% reduction in the risk of death for continuous Rd vs MPT (HR=0.78, P=0.02), but the difference did not cross the pre-specified superiority boundary (P<0.0096).
Adverse events reported in 20% or more of patients in the continuous Rd, Rd18, or MPT arms included diarrhea (45.5%, 38.5%, 16.5%), anemia (43.8%, 35.7%, 42.3%), neutropenia (35.0%, 33.0%, 60.6%), fatigue (32.5%, 32.8%, 28.5%), back pain (32.0%, 26.9%, 21.4%), insomnia (27.6%, 23.5%, 9.8%), asthenia (28.2%, 22.8%, 22.9%), rash (26.1%, 28.0%, 19.4%), decreased appetite (23.1%, 21.3%, 13.3%), cough (22.7%, 17.4%, 12.6%), pyrexia (21.4%, 18.9%, 14.0%), muscle spasms (20.5%, 18.9%, 11.3%) and abdominal pain (20.5%, 14.4%, 11.1%).
The incidence of invasive second primary malignancies was 3% in patients taking continuous Rd, 6% in patients taking Rd18, and 5% in those taking MPT. The overall incidence of solid tumors was identical in the continuous Rd and MPT arms (3%) and 5% in the Rd18 arm.
The MM-015 trial
In the phase 3 MM-015 study, researchers enrolled 459 patients who were 65 or older and newly diagnosed with MM. The team compared melphalan-prednisone-lenalidomide induction followed by lenalidomide maintenance (MPR-R) with melphalan-prednisone-lenalidomide (MPR) or melphalan-prednisone (MP) followed by placebo.
Patients who received MPR-R or MPR had significantly better response rates than patients who received MP, at 77%, 68%, and 50%, respectively (P<0.001 and P=0.002, respectively, for the comparison with MP).
And the median progression-free survival was significantly longer with MPR-R (31 months) than with MPR (14 months, HR=0.49, P<0.001) or MP (13 months, HR=0.40, P<0.001).
During induction, the most frequent adverse events were hematologic. Grade 4 neutropenia occurred in 35% of patients in the MPR-R arm, 32% in the MPR arm, and 8% in the MP arm. The 3-year rate of second primary malignancies was 7%, 7%, and 3%, respectively. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval of continuous oral treatment with lenalidomide (Revlimid) in adults with previously untreated multiple myeloma (MM) who are ineligible for hematopoietic stem cell transplant (HSCT).
The European Commission, which generally follows the CHMP’s recommendations, is expected to make its final decision in about 2 months.
Lenalidomide is not currently approved to treat newly diagnosed MM in any country.
The drug is approved in the European Union (EU) for use in combination with dexamethasone to treat adults with MM who have received at least one prior therapy.
Lenalidomide is also approved in the EU to treat patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with 5q deletion when other therapeutic options are insufficient or inadequate.
The CHMP’s recommendation to extend the use of lenalidomide to HSCT-ineligible patients with newly diagnosed MM was based on the results of 2 studies: MM-015 and MM-020, also known as FIRST.
The FIRST trial
In the phase 3 FIRST trial, researchers enrolled 1623 patients who were newly diagnosed with MM and not eligible for HSCT.
Patients were randomized to receive lenalidomide and dexamethasone (Rd) in 28-day cycles until disease progression (n=535), 18 cycles of lenalidomide and dexamethasone (Rd18) for 72 weeks (n=541), or melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
Response rates were significantly better with continuous Rd (75%) and Rd18 (73%) than with MPT (62%, P<0.001 for both comparisons). Complete response rates were 15%, 14%, and 9%, respectively.
The median progression-free survival was 25.5 months with continuous Rd, 20.7 months with Rd18, and 21.2 months with MPT.
This resulted in a 28% reduction in the risk of progression or death for patients treated with continuous Rd compared with those treated with MPT (hazard ratio[HR]=0.72, P<0.001) and a 30% reduction compared with Rd18 (HR=0.70, P<0.001).
The pre-planned interim analysis of overall survival showed a 22% reduction in the risk of death for continuous Rd vs MPT (HR=0.78, P=0.02), but the difference did not cross the pre-specified superiority boundary (P<0.0096).
Adverse events reported in 20% or more of patients in the continuous Rd, Rd18, or MPT arms included diarrhea (45.5%, 38.5%, 16.5%), anemia (43.8%, 35.7%, 42.3%), neutropenia (35.0%, 33.0%, 60.6%), fatigue (32.5%, 32.8%, 28.5%), back pain (32.0%, 26.9%, 21.4%), insomnia (27.6%, 23.5%, 9.8%), asthenia (28.2%, 22.8%, 22.9%), rash (26.1%, 28.0%, 19.4%), decreased appetite (23.1%, 21.3%, 13.3%), cough (22.7%, 17.4%, 12.6%), pyrexia (21.4%, 18.9%, 14.0%), muscle spasms (20.5%, 18.9%, 11.3%) and abdominal pain (20.5%, 14.4%, 11.1%).
The incidence of invasive second primary malignancies was 3% in patients taking continuous Rd, 6% in patients taking Rd18, and 5% in those taking MPT. The overall incidence of solid tumors was identical in the continuous Rd and MPT arms (3%) and 5% in the Rd18 arm.
The MM-015 trial
In the phase 3 MM-015 study, researchers enrolled 459 patients who were 65 or older and newly diagnosed with MM. The team compared melphalan-prednisone-lenalidomide induction followed by lenalidomide maintenance (MPR-R) with melphalan-prednisone-lenalidomide (MPR) or melphalan-prednisone (MP) followed by placebo.
Patients who received MPR-R or MPR had significantly better response rates than patients who received MP, at 77%, 68%, and 50%, respectively (P<0.001 and P=0.002, respectively, for the comparison with MP).
And the median progression-free survival was significantly longer with MPR-R (31 months) than with MPR (14 months, HR=0.49, P<0.001) or MP (13 months, HR=0.40, P<0.001).
During induction, the most frequent adverse events were hematologic. Grade 4 neutropenia occurred in 35% of patients in the MPR-R arm, 32% in the MPR arm, and 8% in the MP arm. The 3-year rate of second primary malignancies was 7%, 7%, and 3%, respectively. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval of continuous oral treatment with lenalidomide (Revlimid) in adults with previously untreated multiple myeloma (MM) who are ineligible for hematopoietic stem cell transplant (HSCT).
The European Commission, which generally follows the CHMP’s recommendations, is expected to make its final decision in about 2 months.
Lenalidomide is not currently approved to treat newly diagnosed MM in any country.
The drug is approved in the European Union (EU) for use in combination with dexamethasone to treat adults with MM who have received at least one prior therapy.
Lenalidomide is also approved in the EU to treat patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with 5q deletion when other therapeutic options are insufficient or inadequate.
The CHMP’s recommendation to extend the use of lenalidomide to HSCT-ineligible patients with newly diagnosed MM was based on the results of 2 studies: MM-015 and MM-020, also known as FIRST.
The FIRST trial
In the phase 3 FIRST trial, researchers enrolled 1623 patients who were newly diagnosed with MM and not eligible for HSCT.
Patients were randomized to receive lenalidomide and dexamethasone (Rd) in 28-day cycles until disease progression (n=535), 18 cycles of lenalidomide and dexamethasone (Rd18) for 72 weeks (n=541), or melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
Response rates were significantly better with continuous Rd (75%) and Rd18 (73%) than with MPT (62%, P<0.001 for both comparisons). Complete response rates were 15%, 14%, and 9%, respectively.
The median progression-free survival was 25.5 months with continuous Rd, 20.7 months with Rd18, and 21.2 months with MPT.
This resulted in a 28% reduction in the risk of progression or death for patients treated with continuous Rd compared with those treated with MPT (hazard ratio[HR]=0.72, P<0.001) and a 30% reduction compared with Rd18 (HR=0.70, P<0.001).
The pre-planned interim analysis of overall survival showed a 22% reduction in the risk of death for continuous Rd vs MPT (HR=0.78, P=0.02), but the difference did not cross the pre-specified superiority boundary (P<0.0096).
Adverse events reported in 20% or more of patients in the continuous Rd, Rd18, or MPT arms included diarrhea (45.5%, 38.5%, 16.5%), anemia (43.8%, 35.7%, 42.3%), neutropenia (35.0%, 33.0%, 60.6%), fatigue (32.5%, 32.8%, 28.5%), back pain (32.0%, 26.9%, 21.4%), insomnia (27.6%, 23.5%, 9.8%), asthenia (28.2%, 22.8%, 22.9%), rash (26.1%, 28.0%, 19.4%), decreased appetite (23.1%, 21.3%, 13.3%), cough (22.7%, 17.4%, 12.6%), pyrexia (21.4%, 18.9%, 14.0%), muscle spasms (20.5%, 18.9%, 11.3%) and abdominal pain (20.5%, 14.4%, 11.1%).
The incidence of invasive second primary malignancies was 3% in patients taking continuous Rd, 6% in patients taking Rd18, and 5% in those taking MPT. The overall incidence of solid tumors was identical in the continuous Rd and MPT arms (3%) and 5% in the Rd18 arm.
The MM-015 trial
In the phase 3 MM-015 study, researchers enrolled 459 patients who were 65 or older and newly diagnosed with MM. The team compared melphalan-prednisone-lenalidomide induction followed by lenalidomide maintenance (MPR-R) with melphalan-prednisone-lenalidomide (MPR) or melphalan-prednisone (MP) followed by placebo.
Patients who received MPR-R or MPR had significantly better response rates than patients who received MP, at 77%, 68%, and 50%, respectively (P<0.001 and P=0.002, respectively, for the comparison with MP).
And the median progression-free survival was significantly longer with MPR-R (31 months) than with MPR (14 months, HR=0.49, P<0.001) or MP (13 months, HR=0.40, P<0.001).
During induction, the most frequent adverse events were hematologic. Grade 4 neutropenia occurred in 35% of patients in the MPR-R arm, 32% in the MPR arm, and 8% in the MP arm. The 3-year rate of second primary malignancies was 7%, 7%, and 3%, respectively. ![]()
CHMP recommends bortezomib for MCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) who are unsuitable for hematopoietic stem cell transplant.
Bortezomib is already approved in the European Union to treat multiple myeloma, either as monotherapy or in combination with other treatment regimens.
The CHMP’s decision to expand the approved use of bortezomib is based on data from the phase 3 LYM-3002 study. Results from this trial were presented at the 2014 ASCO Annual Meeting (abstract 8500).
LYM-3002 included 487 patients newly diagnosed with MCL who were ineligible, or not considered, for transplant. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median PFS times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median PFS times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AE) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85%, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3%, respectively.
The CHMP’s positive opinion will be reviewed by the European Commission, which has the authority to grant a label extension for medicines in the European Economic Area. A final decision on the use of bortezomib in MCL is expected early next year. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) who are unsuitable for hematopoietic stem cell transplant.
Bortezomib is already approved in the European Union to treat multiple myeloma, either as monotherapy or in combination with other treatment regimens.
The CHMP’s decision to expand the approved use of bortezomib is based on data from the phase 3 LYM-3002 study. Results from this trial were presented at the 2014 ASCO Annual Meeting (abstract 8500).
LYM-3002 included 487 patients newly diagnosed with MCL who were ineligible, or not considered, for transplant. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median PFS times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median PFS times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AE) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85%, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3%, respectively.
The CHMP’s positive opinion will be reviewed by the European Commission, which has the authority to grant a label extension for medicines in the European Economic Area. A final decision on the use of bortezomib in MCL is expected early next year. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) who are unsuitable for hematopoietic stem cell transplant.
Bortezomib is already approved in the European Union to treat multiple myeloma, either as monotherapy or in combination with other treatment regimens.
The CHMP’s decision to expand the approved use of bortezomib is based on data from the phase 3 LYM-3002 study. Results from this trial were presented at the 2014 ASCO Annual Meeting (abstract 8500).
LYM-3002 included 487 patients newly diagnosed with MCL who were ineligible, or not considered, for transplant. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median PFS times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median PFS times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AE) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85%, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3%, respectively.
The CHMP’s positive opinion will be reviewed by the European Commission, which has the authority to grant a label extension for medicines in the European Economic Area. A final decision on the use of bortezomib in MCL is expected early next year.
Obokata fails to create STAP cells, resigns

Credit: Associated Press
After months of trying, Haruko Obokata, PhD, and a team of researchers at her institution, RIKEN, have failed to produce stimulus-triggered acquisition of pluripotency (STAP) cells.
Officials from RIKEN said they have accepted Dr Obokata’s resignation, and the institution has decided to end its efforts to recreate the STAP cell phenomenon.
Dr Obokata and her colleagues initially reported the creation of STAP cells in an article and a letter published in Nature last January. The researchers said they had induced pluripotency in somatic cells by exposing the cells to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
In August, the group reported initial results, saying their attempts had failed, but they would continue trying to create STAP cells until March 2015. Meanwhile, Dr Obokata was trying to recreate the STAP cell phenomenon on her own, under supervision.
Shinichi Aizawa, PhD, the leader of RIKEN’s team, explained the final results of their experiments, as well as Dr Obokata’s, in a press conference in Tokyo on Friday.
Dr Obokata was able to show a fluorescent phenomenon that indicates the possibility of pluripotency in cells, albeit at a very low rate. However, she could not confirm the pluripotency of STAP cells in mice.
The RIKEN team had similar results. So they have decided not to continue with the experiments.
RIKEN accepted Dr Obokata’s resignation, and a disciplinary committee has been discussing how they will reprimand her for research misconduct. RIKEN officials said they will make an announcement once the decision has been made.
Credit: Associated Press
After months of trying, Haruko Obokata, PhD, and a team of researchers at her institution, RIKEN, have failed to produce stimulus-triggered acquisition of pluripotency (STAP) cells.
Officials from RIKEN said they have accepted Dr Obokata’s resignation, and the institution has decided to end its efforts to recreate the STAP cell phenomenon.
Dr Obokata and her colleagues initially reported the creation of STAP cells in an article and a letter published in Nature last January. The researchers said they had induced pluripotency in somatic cells by exposing the cells to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
In August, the group reported initial results, saying their attempts had failed, but they would continue trying to create STAP cells until March 2015. Meanwhile, Dr Obokata was trying to recreate the STAP cell phenomenon on her own, under supervision.
Shinichi Aizawa, PhD, the leader of RIKEN’s team, explained the final results of their experiments, as well as Dr Obokata’s, in a press conference in Tokyo on Friday.
Dr Obokata was able to show a fluorescent phenomenon that indicates the possibility of pluripotency in cells, albeit at a very low rate. However, she could not confirm the pluripotency of STAP cells in mice.
The RIKEN team had similar results. So they have decided not to continue with the experiments.
RIKEN accepted Dr Obokata’s resignation, and a disciplinary committee has been discussing how they will reprimand her for research misconduct. RIKEN officials said they will make an announcement once the decision has been made.
Credit: Associated Press
After months of trying, Haruko Obokata, PhD, and a team of researchers at her institution, RIKEN, have failed to produce stimulus-triggered acquisition of pluripotency (STAP) cells.
Officials from RIKEN said they have accepted Dr Obokata’s resignation, and the institution has decided to end its efforts to recreate the STAP cell phenomenon.
Dr Obokata and her colleagues initially reported the creation of STAP cells in an article and a letter published in Nature last January. The researchers said they had induced pluripotency in somatic cells by exposing the cells to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
In August, the group reported initial results, saying their attempts had failed, but they would continue trying to create STAP cells until March 2015. Meanwhile, Dr Obokata was trying to recreate the STAP cell phenomenon on her own, under supervision.
Shinichi Aizawa, PhD, the leader of RIKEN’s team, explained the final results of their experiments, as well as Dr Obokata’s, in a press conference in Tokyo on Friday.
Dr Obokata was able to show a fluorescent phenomenon that indicates the possibility of pluripotency in cells, albeit at a very low rate. However, she could not confirm the pluripotency of STAP cells in mice.
The RIKEN team had similar results. So they have decided not to continue with the experiments.
RIKEN accepted Dr Obokata’s resignation, and a disciplinary committee has been discussing how they will reprimand her for research misconduct. RIKEN officials said they will make an announcement once the decision has been made.