User login
FDA Approves Test for HER2 Gene in Breast Ca
A test that measures the number of copies of the HER2 gene in breast tumor tissue has been approved by the Food and Drug Administration.
If the Inform Dual ISH test is positive, then the patient is a candidate for treatment with trastuzumab, the recombinant monoclonal antibody directed against HER2 that is marketed as Herceptin by Genentech for the treatment of HER2 overexpressing breast cancer. The test is manufactured by Tucson, Ariz.—based Ventana Medical Systems, a member of the Roche group, as is Genentech.
“When used with other clinical information and laboratory tests, this test can provide health care professionals with additional insight on treatment decisions for patients with breast cancer,” Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostic Device Evaluation and Safety in the FDA's Center for Devices and Radiological Health, said in the statement announcing the approval. The test makes it possible “to see and count copies of chromosome 17 and HER2 genes on the same slide, similar to HER2 amplification measurements that have traditionally only been available using fluorescence microscopes,” the statement said.
But the new test, “allows lab staff to see the HER2 and chromosome 17 signals directly under a microscope, for longer periods of time.”
Approval is based on a study conducted in the United States that evaluated the test in 510 women with breast cancer. The test confirmed that the tumor sample contained more than the normal number of copies of the HER2 gene, located on chromosome 17, in 96% of the HER2 positive samples, according to the statement.
About 20% of women diagnosed with breast cancer are HER2-positive, according to the FDA.
A test that measures the number of copies of the HER2 gene in breast tumor tissue has been approved by the Food and Drug Administration.
If the Inform Dual ISH test is positive, then the patient is a candidate for treatment with trastuzumab, the recombinant monoclonal antibody directed against HER2 that is marketed as Herceptin by Genentech for the treatment of HER2 overexpressing breast cancer. The test is manufactured by Tucson, Ariz.—based Ventana Medical Systems, a member of the Roche group, as is Genentech.
“When used with other clinical information and laboratory tests, this test can provide health care professionals with additional insight on treatment decisions for patients with breast cancer,” Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostic Device Evaluation and Safety in the FDA's Center for Devices and Radiological Health, said in the statement announcing the approval. The test makes it possible “to see and count copies of chromosome 17 and HER2 genes on the same slide, similar to HER2 amplification measurements that have traditionally only been available using fluorescence microscopes,” the statement said.
But the new test, “allows lab staff to see the HER2 and chromosome 17 signals directly under a microscope, for longer periods of time.”
Approval is based on a study conducted in the United States that evaluated the test in 510 women with breast cancer. The test confirmed that the tumor sample contained more than the normal number of copies of the HER2 gene, located on chromosome 17, in 96% of the HER2 positive samples, according to the statement.
About 20% of women diagnosed with breast cancer are HER2-positive, according to the FDA.
A test that measures the number of copies of the HER2 gene in breast tumor tissue has been approved by the Food and Drug Administration.
If the Inform Dual ISH test is positive, then the patient is a candidate for treatment with trastuzumab, the recombinant monoclonal antibody directed against HER2 that is marketed as Herceptin by Genentech for the treatment of HER2 overexpressing breast cancer. The test is manufactured by Tucson, Ariz.—based Ventana Medical Systems, a member of the Roche group, as is Genentech.
“When used with other clinical information and laboratory tests, this test can provide health care professionals with additional insight on treatment decisions for patients with breast cancer,” Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostic Device Evaluation and Safety in the FDA's Center for Devices and Radiological Health, said in the statement announcing the approval. The test makes it possible “to see and count copies of chromosome 17 and HER2 genes on the same slide, similar to HER2 amplification measurements that have traditionally only been available using fluorescence microscopes,” the statement said.
But the new test, “allows lab staff to see the HER2 and chromosome 17 signals directly under a microscope, for longer periods of time.”
Approval is based on a study conducted in the United States that evaluated the test in 510 women with breast cancer. The test confirmed that the tumor sample contained more than the normal number of copies of the HER2 gene, located on chromosome 17, in 96% of the HER2 positive samples, according to the statement.
About 20% of women diagnosed with breast cancer are HER2-positive, according to the FDA.
Denosumab Approved for Bone Loss From Hormone Ablation Therapy
The Prolia brand of denosumab has been approved as a treatment for bone loss in people receiving hormone ablation therapy for prostate or breast cancer, according to a statement from Amgen, the manufacturer.
The approved indications are for increasing bone mass in women at high risk for fracture on adjuvant aromatase inhibitor therapy for breast cancer and in men at high risk for fracture on androgen deprivation therapy (ADT) for nonmetastatic prostate cancer, the company's statement said.
Denosumab, a RANK ligand inhibitor, was first approved in June 2010 for treating postmenopausal women with osteoporosis at high risk of fracture and is marketed as Prolia for this indication. Prolia is administered in a subcutaneous injection once every 6 months, at a dose of 60 mg.
In November 2010, denosumab was also approved for preventing skeletal-related events in patients with bone metastases from solid tumors. It is marketed as Xgeva and administered more frequently at a higher dose for this indication.
Approval of the new indications for Prolia was based on phase III studies of these two groups of patients, according to Amgen. In an international study of almost 1,500 men with nonmetastatic prostate cancer who were being treated with ADT, bone mineral density at the lumbar spine after 2 years of treatment was significantly higher among men who had received denosumab compared with those who received placebo. After 3 years of treatment, the incidence of new vertebral fractures was 1.5% among those treated with denosumab, compared with 3.9% for those on placebo, a risk reduction of 62%, the company said.
And in a study of 252 postmenopausal women with breast cancer under treatment with an aromatase inhibitor, bone mineral density at the lumbar spine was significantly higher among those treated with denosumab compared with those on placebo after 12 months of treatment, according to the statement.
Arthralgia and back pain were the most common adverse events associated with treatment in these two groups of patients. Hypocalcemia was also reported; denosumab is contraindicated in people with hypocalcemia. Treatment was associated with cataract events in men in the prostate cancer trial.
In the European Union, denosumab is approved for the osteoporosis indication as well as the prostate cancer indication just approved in the United States, according to Amgen.
The Prolia brand of denosumab has been approved as a treatment for bone loss in people receiving hormone ablation therapy for prostate or breast cancer, according to a statement from Amgen, the manufacturer.
The approved indications are for increasing bone mass in women at high risk for fracture on adjuvant aromatase inhibitor therapy for breast cancer and in men at high risk for fracture on androgen deprivation therapy (ADT) for nonmetastatic prostate cancer, the company's statement said.
Denosumab, a RANK ligand inhibitor, was first approved in June 2010 for treating postmenopausal women with osteoporosis at high risk of fracture and is marketed as Prolia for this indication. Prolia is administered in a subcutaneous injection once every 6 months, at a dose of 60 mg.
In November 2010, denosumab was also approved for preventing skeletal-related events in patients with bone metastases from solid tumors. It is marketed as Xgeva and administered more frequently at a higher dose for this indication.
Approval of the new indications for Prolia was based on phase III studies of these two groups of patients, according to Amgen. In an international study of almost 1,500 men with nonmetastatic prostate cancer who were being treated with ADT, bone mineral density at the lumbar spine after 2 years of treatment was significantly higher among men who had received denosumab compared with those who received placebo. After 3 years of treatment, the incidence of new vertebral fractures was 1.5% among those treated with denosumab, compared with 3.9% for those on placebo, a risk reduction of 62%, the company said.
And in a study of 252 postmenopausal women with breast cancer under treatment with an aromatase inhibitor, bone mineral density at the lumbar spine was significantly higher among those treated with denosumab compared with those on placebo after 12 months of treatment, according to the statement.
Arthralgia and back pain were the most common adverse events associated with treatment in these two groups of patients. Hypocalcemia was also reported; denosumab is contraindicated in people with hypocalcemia. Treatment was associated with cataract events in men in the prostate cancer trial.
In the European Union, denosumab is approved for the osteoporosis indication as well as the prostate cancer indication just approved in the United States, according to Amgen.
The Prolia brand of denosumab has been approved as a treatment for bone loss in people receiving hormone ablation therapy for prostate or breast cancer, according to a statement from Amgen, the manufacturer.
The approved indications are for increasing bone mass in women at high risk for fracture on adjuvant aromatase inhibitor therapy for breast cancer and in men at high risk for fracture on androgen deprivation therapy (ADT) for nonmetastatic prostate cancer, the company's statement said.
Denosumab, a RANK ligand inhibitor, was first approved in June 2010 for treating postmenopausal women with osteoporosis at high risk of fracture and is marketed as Prolia for this indication. Prolia is administered in a subcutaneous injection once every 6 months, at a dose of 60 mg.
In November 2010, denosumab was also approved for preventing skeletal-related events in patients with bone metastases from solid tumors. It is marketed as Xgeva and administered more frequently at a higher dose for this indication.
Approval of the new indications for Prolia was based on phase III studies of these two groups of patients, according to Amgen. In an international study of almost 1,500 men with nonmetastatic prostate cancer who were being treated with ADT, bone mineral density at the lumbar spine after 2 years of treatment was significantly higher among men who had received denosumab compared with those who received placebo. After 3 years of treatment, the incidence of new vertebral fractures was 1.5% among those treated with denosumab, compared with 3.9% for those on placebo, a risk reduction of 62%, the company said.
And in a study of 252 postmenopausal women with breast cancer under treatment with an aromatase inhibitor, bone mineral density at the lumbar spine was significantly higher among those treated with denosumab compared with those on placebo after 12 months of treatment, according to the statement.
Arthralgia and back pain were the most common adverse events associated with treatment in these two groups of patients. Hypocalcemia was also reported; denosumab is contraindicated in people with hypocalcemia. Treatment was associated with cataract events in men in the prostate cancer trial.
In the European Union, denosumab is approved for the osteoporosis indication as well as the prostate cancer indication just approved in the United States, according to Amgen.
Arrhythmia Risk Leads to Ondansetron Label Changes
A review of the potential for prolongation of the QT interval associated with ondansetron has resulted in some "interim" changes to the drug’s label, the Food and Drug Administration announced on Sept. 15.
In a drug safety announcement, the FDA said that the agency conducted a review of all the available information on the risk of QT prolongation with ondansetron and is making the changes, which include recommending against the use of the antinausea drug in patients with congenital long QT syndrome.
Used to prevent nausea and vomiting caused by cancer chemotherapy, radiation therapy, and surgery, ondansetron is a 5-hydroxytryptamine-3 (5-HT3) receptor antagonist marketed as Zofran by GlaxoSmithKline, and is available in generic formulations. Information on the potential for QT prolongation is already included in the label, and QT interval prolongation with ondansetron has been reported in the medical literature, according to the announcement.
But the label will now also recommend ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias, and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. QT prolongation can result in the abnormal and potentially fatal heart rhythm torsades de pointes, which has also been reported in patients taking ondansetron, the FDA statement points out.
Patients taking ondansetron should also be advised to contact a health care professional immediately, if they develop signs and symptoms of an abnormal heart rate or rhythm, the statement says.
GlaxoSmithKline is being required by the FDA to conduct a study that will evaluate the potential for ondansetron to prolong the QT interval; results are expected in the summer of 2012. The safety review is ongoing, and "the FDA will continue to assess all available data supporting the safety and effectiveness of ondansetron and will update the public when more information becomes available," the statement said.
The notice is available online. Serious adverse events associated with ondansetron should be reported to the FDA’s MedWatch program at 800-332-1088.
A review of the potential for prolongation of the QT interval associated with ondansetron has resulted in some "interim" changes to the drug’s label, the Food and Drug Administration announced on Sept. 15.
In a drug safety announcement, the FDA said that the agency conducted a review of all the available information on the risk of QT prolongation with ondansetron and is making the changes, which include recommending against the use of the antinausea drug in patients with congenital long QT syndrome.
Used to prevent nausea and vomiting caused by cancer chemotherapy, radiation therapy, and surgery, ondansetron is a 5-hydroxytryptamine-3 (5-HT3) receptor antagonist marketed as Zofran by GlaxoSmithKline, and is available in generic formulations. Information on the potential for QT prolongation is already included in the label, and QT interval prolongation with ondansetron has been reported in the medical literature, according to the announcement.
But the label will now also recommend ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias, and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. QT prolongation can result in the abnormal and potentially fatal heart rhythm torsades de pointes, which has also been reported in patients taking ondansetron, the FDA statement points out.
Patients taking ondansetron should also be advised to contact a health care professional immediately, if they develop signs and symptoms of an abnormal heart rate or rhythm, the statement says.
GlaxoSmithKline is being required by the FDA to conduct a study that will evaluate the potential for ondansetron to prolong the QT interval; results are expected in the summer of 2012. The safety review is ongoing, and "the FDA will continue to assess all available data supporting the safety and effectiveness of ondansetron and will update the public when more information becomes available," the statement said.
The notice is available online. Serious adverse events associated with ondansetron should be reported to the FDA’s MedWatch program at 800-332-1088.
A review of the potential for prolongation of the QT interval associated with ondansetron has resulted in some "interim" changes to the drug’s label, the Food and Drug Administration announced on Sept. 15.
In a drug safety announcement, the FDA said that the agency conducted a review of all the available information on the risk of QT prolongation with ondansetron and is making the changes, which include recommending against the use of the antinausea drug in patients with congenital long QT syndrome.
Used to prevent nausea and vomiting caused by cancer chemotherapy, radiation therapy, and surgery, ondansetron is a 5-hydroxytryptamine-3 (5-HT3) receptor antagonist marketed as Zofran by GlaxoSmithKline, and is available in generic formulations. Information on the potential for QT prolongation is already included in the label, and QT interval prolongation with ondansetron has been reported in the medical literature, according to the announcement.
But the label will now also recommend ECG monitoring in patients who are treated with ondansetron and have an electrolyte abnormality, congestive heart failure, or bradyarrhythmias, and in patients taking other medications that prolong the QT interval – who are at an increased risk of developing torsades de pointes. QT prolongation can result in the abnormal and potentially fatal heart rhythm torsades de pointes, which has also been reported in patients taking ondansetron, the FDA statement points out.
Patients taking ondansetron should also be advised to contact a health care professional immediately, if they develop signs and symptoms of an abnormal heart rate or rhythm, the statement says.
GlaxoSmithKline is being required by the FDA to conduct a study that will evaluate the potential for ondansetron to prolong the QT interval; results are expected in the summer of 2012. The safety review is ongoing, and "the FDA will continue to assess all available data supporting the safety and effectiveness of ondansetron and will update the public when more information becomes available," the statement said.
The notice is available online. Serious adverse events associated with ondansetron should be reported to the FDA’s MedWatch program at 800-332-1088.
FDA Panel Supports Deferiprone for Iron Overload
SILVER SPRING, MD. – The majority of a Food and Drug Administration Advisory panel on Sept. 14 supported the approval of deferiprone as a second-line treatment for patients with transfusional iron overload.
At the meeting of the FDA’s Oncologic Drugs Advisory Committee, the panel voted 10-2 that treatment with deferiprone had a favorable benefit-risk profile for the proposed indication: treatment of patients with transfusional iron overload when current chelation therapy is inadequate. If approved, deferiprone will be marketed as Ferriprox by ApoPharma Inc. and would be the third chelating treatment approved by the FDA for transfusional iron overload. (In the United States, ApoPharma is represented by CATO Research Ltd., the company’s authorized U.S. agent.)
Approved in Europe in 1999, deferiprone, an oral therapy, is indicated for treating iron overload in patients with thalassemia major when deferoxamine treatment "is contraindicated or inadequate." Deferiprone is now approved in 61 countries. In the United States, deferoxamine (Desferal), an iron chelator administered via a subcutaneous infusion pump (usually over 6 nights a week), was approved in 1968, and deferasirox (Exjade), an oral chelator, was approved in 2005.
ApoPharma presented data from two studies, composed mostly of patients with beta-thalassemia. In one retrospective uncontrolled analysis of about 260 patients from different studies who had failed previous treatment with an iron chelator, 52% of patients had at least a 20% drop in serum ferritin over 1 year of treatment, the primary end point. In the second study, a randomized controlled study of 61 patients treated with deferiprone or deferoxamine for 12 months, deferiprone was superior in removing excess cardiac iron but the effects of the two treatments on serum ferritin and liver iron concentrations were not significantly different, according to the company.
The most common adverse events were mild to moderate and were transient, the company said. Agranulocytosis is the most serious adverse event; in the retrospective studies, the overall rate of agranulocytosis was 1.7%, usually occurring in the first year of treatment. Agranulocytosis appears to be reversible if the drug is discontinued, according to the FDA reviewer.
The panelists who voted in favor of the benefit-risk profile cited the unmet need for chelating treatments in the United States and agreed that, despite some limitations of the data, including retrospective data, deferiprone had been shown to be effective and reasonably safe. But they stressed that there was a need for studies in patients with sickle cell anemia, the population that would be more likely to be treated with the drug in the United States.
There are no data in this population. If deferiprone is approved, ApoPharma plans to conduct a study of the agent in patients with sickle cell disease.
The FDA usually follows the recommendations of its advisory panels. Members of the panel had no conflicts.
The FDA is expected to make a decision on approval by Oct. 14.
SILVER SPRING, MD. – The majority of a Food and Drug Administration Advisory panel on Sept. 14 supported the approval of deferiprone as a second-line treatment for patients with transfusional iron overload.
At the meeting of the FDA’s Oncologic Drugs Advisory Committee, the panel voted 10-2 that treatment with deferiprone had a favorable benefit-risk profile for the proposed indication: treatment of patients with transfusional iron overload when current chelation therapy is inadequate. If approved, deferiprone will be marketed as Ferriprox by ApoPharma Inc. and would be the third chelating treatment approved by the FDA for transfusional iron overload. (In the United States, ApoPharma is represented by CATO Research Ltd., the company’s authorized U.S. agent.)
Approved in Europe in 1999, deferiprone, an oral therapy, is indicated for treating iron overload in patients with thalassemia major when deferoxamine treatment "is contraindicated or inadequate." Deferiprone is now approved in 61 countries. In the United States, deferoxamine (Desferal), an iron chelator administered via a subcutaneous infusion pump (usually over 6 nights a week), was approved in 1968, and deferasirox (Exjade), an oral chelator, was approved in 2005.
ApoPharma presented data from two studies, composed mostly of patients with beta-thalassemia. In one retrospective uncontrolled analysis of about 260 patients from different studies who had failed previous treatment with an iron chelator, 52% of patients had at least a 20% drop in serum ferritin over 1 year of treatment, the primary end point. In the second study, a randomized controlled study of 61 patients treated with deferiprone or deferoxamine for 12 months, deferiprone was superior in removing excess cardiac iron but the effects of the two treatments on serum ferritin and liver iron concentrations were not significantly different, according to the company.
The most common adverse events were mild to moderate and were transient, the company said. Agranulocytosis is the most serious adverse event; in the retrospective studies, the overall rate of agranulocytosis was 1.7%, usually occurring in the first year of treatment. Agranulocytosis appears to be reversible if the drug is discontinued, according to the FDA reviewer.
The panelists who voted in favor of the benefit-risk profile cited the unmet need for chelating treatments in the United States and agreed that, despite some limitations of the data, including retrospective data, deferiprone had been shown to be effective and reasonably safe. But they stressed that there was a need for studies in patients with sickle cell anemia, the population that would be more likely to be treated with the drug in the United States.
There are no data in this population. If deferiprone is approved, ApoPharma plans to conduct a study of the agent in patients with sickle cell disease.
The FDA usually follows the recommendations of its advisory panels. Members of the panel had no conflicts.
The FDA is expected to make a decision on approval by Oct. 14.
SILVER SPRING, MD. – The majority of a Food and Drug Administration Advisory panel on Sept. 14 supported the approval of deferiprone as a second-line treatment for patients with transfusional iron overload.
At the meeting of the FDA’s Oncologic Drugs Advisory Committee, the panel voted 10-2 that treatment with deferiprone had a favorable benefit-risk profile for the proposed indication: treatment of patients with transfusional iron overload when current chelation therapy is inadequate. If approved, deferiprone will be marketed as Ferriprox by ApoPharma Inc. and would be the third chelating treatment approved by the FDA for transfusional iron overload. (In the United States, ApoPharma is represented by CATO Research Ltd., the company’s authorized U.S. agent.)
Approved in Europe in 1999, deferiprone, an oral therapy, is indicated for treating iron overload in patients with thalassemia major when deferoxamine treatment "is contraindicated or inadequate." Deferiprone is now approved in 61 countries. In the United States, deferoxamine (Desferal), an iron chelator administered via a subcutaneous infusion pump (usually over 6 nights a week), was approved in 1968, and deferasirox (Exjade), an oral chelator, was approved in 2005.
ApoPharma presented data from two studies, composed mostly of patients with beta-thalassemia. In one retrospective uncontrolled analysis of about 260 patients from different studies who had failed previous treatment with an iron chelator, 52% of patients had at least a 20% drop in serum ferritin over 1 year of treatment, the primary end point. In the second study, a randomized controlled study of 61 patients treated with deferiprone or deferoxamine for 12 months, deferiprone was superior in removing excess cardiac iron but the effects of the two treatments on serum ferritin and liver iron concentrations were not significantly different, according to the company.
The most common adverse events were mild to moderate and were transient, the company said. Agranulocytosis is the most serious adverse event; in the retrospective studies, the overall rate of agranulocytosis was 1.7%, usually occurring in the first year of treatment. Agranulocytosis appears to be reversible if the drug is discontinued, according to the FDA reviewer.
The panelists who voted in favor of the benefit-risk profile cited the unmet need for chelating treatments in the United States and agreed that, despite some limitations of the data, including retrospective data, deferiprone had been shown to be effective and reasonably safe. But they stressed that there was a need for studies in patients with sickle cell anemia, the population that would be more likely to be treated with the drug in the United States.
There are no data in this population. If deferiprone is approved, ApoPharma plans to conduct a study of the agent in patients with sickle cell disease.
The FDA usually follows the recommendations of its advisory panels. Members of the panel had no conflicts.
The FDA is expected to make a decision on approval by Oct. 14.
FROM THE FDA'S ONCOLOGIC DRUGS ADVISORY COMMITTEE
FDA Guidance on Biosimilars Expected by End of 2011
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA’s summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson noted that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA’s Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8.)
The coauthors of the article, titled "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for rheumatoid arthritis and other inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA’s discretion. FDA’s regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.
The Importance of Cheaper, Reliable Biologics
In an interview, Dr. Karen S. Kolba, chair of the American College of Rheumatology’s committee on rheumatologic care, said that the issue of more affordable generic biologics is as critical as ever for rheumatologists. It is important that cheaper versions of these products, with reliable clinical effects and acceptable safety profiles, become available for patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, she said. Many patients who could benefit from these products do not receive them because of their cost, a problem in the United States and worldwide, noted Dr. Kolba, a rheumatologist in private practice in Santa Maria, Calif.
While these products are not without risks, she said, with such huge profit margins at stake, "there’s no reason it can’t be done in a safe responsible way."
Currently, Medicare covers the costs of infliximab (Remicade), rituximab (Rituxan), and tocilizumab (Actemra) under Part B, because they are administered as infusions in the office. But just because they are covered does not make the high cost of these drugs acceptable, she said. Her patients who self-administered adalimumab (Humira) or etanercept (Enbrel) must still pay copayments that are high even when the patients qualify for company assistance programs. These patients also would benefit from cheaper versions.
An example of the perspective of the biologic manufacturers is provided in a statement on biosimilars on the Genentech website, which states that Genentech, a member of the Roche group, which manufactures rituximab and other biologics, believes that indication-specific clinical trials should be used to establish every indication for a biosimilar to ensure the product’s safety and efficacy. In addition, "biosimilars should only be substituted for an innovator biologic if comparative clinical trials demonstrate that substitution is appropriate," and that they should be "uniquely identified and should be traceable to ensure patient safety."
The company also states that biologics, especially large-molecule biologics, "tend to be produced as mixtures of molecules that differ very slightly from one another, which make them difficult to characterize."
Dr. Kolba pointed out that some pharmaceutical companies started to manufacture generic formulations of their own blockbuster trade-name drugs once the patents expired. Current manufacturers of biologics could do the same. Reducing the price of their products to 10% plus cost would solve the problem of equivalence and alleviate concerns about safety and substitution while dramatically reducing the cost to patients, she said.
At least one manufacture of biologics has plans to do so: Amgen, the manufacturer of etanercept, recently announced its intentions to manufacture biosimilars, an Amgen spokesperson said.
In the dermatology arena, biosimilars would also be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis. Dr. Kolba disclosed that she has been a past consultant or speaker to Abbott, Bristol-Myers Squibb, Genentech and UCB; and is currently an investigator in trials sponsored by Abbott, Amgen, Lilly, Pfizer, Sanofi-Aventis, and UCB.
More on the BPCI Act is available on the FDA website.
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA’s summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson noted that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA’s Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8.)
The coauthors of the article, titled "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for rheumatoid arthritis and other inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA’s discretion. FDA’s regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.
The Importance of Cheaper, Reliable Biologics
In an interview, Dr. Karen S. Kolba, chair of the American College of Rheumatology’s committee on rheumatologic care, said that the issue of more affordable generic biologics is as critical as ever for rheumatologists. It is important that cheaper versions of these products, with reliable clinical effects and acceptable safety profiles, become available for patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, she said. Many patients who could benefit from these products do not receive them because of their cost, a problem in the United States and worldwide, noted Dr. Kolba, a rheumatologist in private practice in Santa Maria, Calif.
While these products are not without risks, she said, with such huge profit margins at stake, "there’s no reason it can’t be done in a safe responsible way."
Currently, Medicare covers the costs of infliximab (Remicade), rituximab (Rituxan), and tocilizumab (Actemra) under Part B, because they are administered as infusions in the office. But just because they are covered does not make the high cost of these drugs acceptable, she said. Her patients who self-administered adalimumab (Humira) or etanercept (Enbrel) must still pay copayments that are high even when the patients qualify for company assistance programs. These patients also would benefit from cheaper versions.
An example of the perspective of the biologic manufacturers is provided in a statement on biosimilars on the Genentech website, which states that Genentech, a member of the Roche group, which manufactures rituximab and other biologics, believes that indication-specific clinical trials should be used to establish every indication for a biosimilar to ensure the product’s safety and efficacy. In addition, "biosimilars should only be substituted for an innovator biologic if comparative clinical trials demonstrate that substitution is appropriate," and that they should be "uniquely identified and should be traceable to ensure patient safety."
The company also states that biologics, especially large-molecule biologics, "tend to be produced as mixtures of molecules that differ very slightly from one another, which make them difficult to characterize."
Dr. Kolba pointed out that some pharmaceutical companies started to manufacture generic formulations of their own blockbuster trade-name drugs once the patents expired. Current manufacturers of biologics could do the same. Reducing the price of their products to 10% plus cost would solve the problem of equivalence and alleviate concerns about safety and substitution while dramatically reducing the cost to patients, she said.
At least one manufacture of biologics has plans to do so: Amgen, the manufacturer of etanercept, recently announced its intentions to manufacture biosimilars, an Amgen spokesperson said.
In the dermatology arena, biosimilars would also be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis. Dr. Kolba disclosed that she has been a past consultant or speaker to Abbott, Bristol-Myers Squibb, Genentech and UCB; and is currently an investigator in trials sponsored by Abbott, Amgen, Lilly, Pfizer, Sanofi-Aventis, and UCB.
More on the BPCI Act is available on the FDA website.
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA’s summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson noted that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA’s Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8.)
The coauthors of the article, titled "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for rheumatoid arthritis and other inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA’s discretion. FDA’s regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.
The Importance of Cheaper, Reliable Biologics
In an interview, Dr. Karen S. Kolba, chair of the American College of Rheumatology’s committee on rheumatologic care, said that the issue of more affordable generic biologics is as critical as ever for rheumatologists. It is important that cheaper versions of these products, with reliable clinical effects and acceptable safety profiles, become available for patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, she said. Many patients who could benefit from these products do not receive them because of their cost, a problem in the United States and worldwide, noted Dr. Kolba, a rheumatologist in private practice in Santa Maria, Calif.
While these products are not without risks, she said, with such huge profit margins at stake, "there’s no reason it can’t be done in a safe responsible way."
Currently, Medicare covers the costs of infliximab (Remicade), rituximab (Rituxan), and tocilizumab (Actemra) under Part B, because they are administered as infusions in the office. But just because they are covered does not make the high cost of these drugs acceptable, she said. Her patients who self-administered adalimumab (Humira) or etanercept (Enbrel) must still pay copayments that are high even when the patients qualify for company assistance programs. These patients also would benefit from cheaper versions.
An example of the perspective of the biologic manufacturers is provided in a statement on biosimilars on the Genentech website, which states that Genentech, a member of the Roche group, which manufactures rituximab and other biologics, believes that indication-specific clinical trials should be used to establish every indication for a biosimilar to ensure the product’s safety and efficacy. In addition, "biosimilars should only be substituted for an innovator biologic if comparative clinical trials demonstrate that substitution is appropriate," and that they should be "uniquely identified and should be traceable to ensure patient safety."
The company also states that biologics, especially large-molecule biologics, "tend to be produced as mixtures of molecules that differ very slightly from one another, which make them difficult to characterize."
Dr. Kolba pointed out that some pharmaceutical companies started to manufacture generic formulations of their own blockbuster trade-name drugs once the patents expired. Current manufacturers of biologics could do the same. Reducing the price of their products to 10% plus cost would solve the problem of equivalence and alleviate concerns about safety and substitution while dramatically reducing the cost to patients, she said.
At least one manufacture of biologics has plans to do so: Amgen, the manufacturer of etanercept, recently announced its intentions to manufacture biosimilars, an Amgen spokesperson said.
In the dermatology arena, biosimilars would also be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis. Dr. Kolba disclosed that she has been a past consultant or speaker to Abbott, Bristol-Myers Squibb, Genentech and UCB; and is currently an investigator in trials sponsored by Abbott, Amgen, Lilly, Pfizer, Sanofi-Aventis, and UCB.
More on the BPCI Act is available on the FDA website.
FDA Guidance on Biosimilars Expected by End of Year
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA's summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson said that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA's Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8).
The coauthors of the article, "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA's Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA's discretion. FDA's regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.

In the dermatology arena, biosimilars would be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis.
More on the BPCI Act is available on the FDA website.
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA's summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson said that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA's Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8).
The coauthors of the article, "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA's Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA's discretion. FDA's regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.
In the dermatology arena, biosimilars would be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis.
More on the BPCI Act is available on the FDA website.
The Food and Drug Administration expects to issue its guidance on biosimilars by the end of 2011, more than a year after legislation designed to make cheaper generic versions of therapeutic biologics available was incorporated into the Patient Protection and Affordable Care Act of 2010.
A year earlier, the Biologics Price Competition and Innovation Act of 2009 (BPCI) established an abbreviated approval pathway for biological products that are shown to be "highly similar" or "biosimilar" to, or "interchangeable" with, an FDA-approved biological product, according to the FDA's summary of the legislation.
The complex issues related to its implementation are still being worked out by a special committee at the agency, which met in November 2010 to discuss the issues and challenges of implementing the BPCI Act. Manufacturers of biologic therapeutics, generic pharmaceutical companies, patient groups, and other interested parties provided their input at that meeting.
An FDA spokesperson said that the agency "continues to carefully review and consider all comments from the November 2010 hearing and from the docket as we move forward in our implementation" of the statute. Biosimilars are also referred to as "follow-on biologics."
Of the major biologics, etanercept (Enbrel) will be the first to lose its patent, in 2012.
Dr. Steven Kozlowski, director of the FDA's Office of Biotechnology Products, and other FDA officials noted that access to expensive biologic therapies "may be limited, not infrequently because of their cost" and emphasized the challenges the agency faced in implementing the act in a "Perspective" article published online on Aug. 4 in the New England Journal of Medicine (2011;365:385-8).
The coauthors of the article, "Developing the Nation's Biosimilar Program," were Dr. Janet Woodcock, director of the FDA's Center for Drug Evaluation and Research; Dr. Karen Midthun, director of the Center for Biologics Evaluation and Research; and Dr. Rachel Behrman Sherman, director of the Office of Medical Policy.
"Reconciling the science of biosimilar development with the new regulatory framework required by the BPCI Act presents the FDA with numerous challenges," primarily, establishing scientific criteria "that address the key question: how similar is similar enough when it comes to the substitution of complex biologic drug products in clinical practice?" They cited immunogenicity as "a critical factor" when evaluating biosimilars, and noted that product-specific safety monitoring should be included in the process.
For now, it remains difficult to predict when these more affordable generic formulations of the expensive biologic treatments for inflammatory diseases will become available.
A spokesperson for the Generic Pharmaceutical Association (GPhA) said that the legislation left a lot to the FDA's discretion. FDA's regulations must not be burdensome and should not create barriers to making these products available, he said. "Ultimately, we want patients to have access to these drugs at affordable prices." A representative of the GPhA made a presentation at the November 2010 meeting and submitted formal comments to the docket in December.
In the dermatology arena, biosimilars would be welcomed primarily because of cost savings for patients and increased access to these effective treatments, said Dr. Mark G. Lebwohl, professor and chairman of the department of dermatology at Mount Sinai School of Medicine, N.Y.
Biosimilars will not be identical to the drugs they replace, "and when tested, they will have to come into a certain narrow window of similarity in terms of both safety and side effects," he said in an interview. The dilemma with the side effect profile is that it may not be fully characterized for years and, in terms of effectiveness, "the dilemma is that they have to be approximately equal to the drug that it is replacing."
For example, a biosimilar for etanercept that is 10% less effective than the original product could make a difference clinically. A biosimilar could conceivably work better than the biologic drug it is replacing, but "if it is too much more effective, it’s not a biosimilar and will have to through a new drug application." Moreover, if it is more effective, it might be more immunosuppressive, raising concerns about more side effects, Dr. Lebwohl pointed out.
Dr. Lebwohl disclosed that he has been an investigator for and the department of dermatology at Mount Sinai has received funds from most of the manufacturers of biologics for psoriasis.
More on the BPCI Act is available on the FDA website.
FDA Approves Icatibant for Hereditary Angioedema Attacks
The Food and Drug Administration on Aug. 25 approved icatibant for the treatment of acute attacks of hereditary angioedema in adults aged 18 years and older.
Icatibant is administered subcutaneously at the onset of attack symptoms, which can include rapid swelling of different body parts – usually the face, GI tract, extremities, or genitals – with airway swelling that can increase the risk of suffocation.
"Because it can be self-administered through an injection in the abdominal area, patients can treat themselves upon recognition of an HAE attack," Dr. Curtis Rosebraugh, director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement announcing the approval.
Icatibant, a selective bradykinin B2 receptor antagonist, inhibits the effects of bradykinin, thought to be the cause of HAE symptoms, according to Shire Human Genetic Therapies, which will market the drug as Firazyr. One syringe costs $6,800 (average wholesale price), according to a company spokesperson. The company has a program to help eligible patients help pay for copays as well as a program to help uninsured patients or those patients whose insurance does not cover the treatment.
This is the third drug approved by the FDA to treat attacks of HAE, a rare genetic disorder that affects fewer than 30,000 people in the United States: a C1 esterase inhibitor (Berinert) was approved in October 2009, to treat facial and abdominal attacks of HAE, and ecallantide (Kalbitor) was approved in December 2009 to treat acute attacks of HAE in patients aged 16 years and older.
In three controlled studies, with open-label extension periods, 225 patients were treated with 1,076 icatibant injections. Those treated with icatibant started to experience relief of symptoms in a median of 2 hours after the injection, compared with almost 20 hours among those who received a placebo injection. The most common side effects associated with icatibant were injection site reactions, fever, elevated liver enzymes, dizziness, and rash, according to the FDA.
Icatibant, which is launching in the United States on Aug. 26, is now approved in 38 countries, including those in the European Union, according to Shire.
The Food and Drug Administration on Aug. 25 approved icatibant for the treatment of acute attacks of hereditary angioedema in adults aged 18 years and older.
Icatibant is administered subcutaneously at the onset of attack symptoms, which can include rapid swelling of different body parts – usually the face, GI tract, extremities, or genitals – with airway swelling that can increase the risk of suffocation.
"Because it can be self-administered through an injection in the abdominal area, patients can treat themselves upon recognition of an HAE attack," Dr. Curtis Rosebraugh, director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement announcing the approval.
Icatibant, a selective bradykinin B2 receptor antagonist, inhibits the effects of bradykinin, thought to be the cause of HAE symptoms, according to Shire Human Genetic Therapies, which will market the drug as Firazyr. One syringe costs $6,800 (average wholesale price), according to a company spokesperson. The company has a program to help eligible patients help pay for copays as well as a program to help uninsured patients or those patients whose insurance does not cover the treatment.
This is the third drug approved by the FDA to treat attacks of HAE, a rare genetic disorder that affects fewer than 30,000 people in the United States: a C1 esterase inhibitor (Berinert) was approved in October 2009, to treat facial and abdominal attacks of HAE, and ecallantide (Kalbitor) was approved in December 2009 to treat acute attacks of HAE in patients aged 16 years and older.
In three controlled studies, with open-label extension periods, 225 patients were treated with 1,076 icatibant injections. Those treated with icatibant started to experience relief of symptoms in a median of 2 hours after the injection, compared with almost 20 hours among those who received a placebo injection. The most common side effects associated with icatibant were injection site reactions, fever, elevated liver enzymes, dizziness, and rash, according to the FDA.
Icatibant, which is launching in the United States on Aug. 26, is now approved in 38 countries, including those in the European Union, according to Shire.
The Food and Drug Administration on Aug. 25 approved icatibant for the treatment of acute attacks of hereditary angioedema in adults aged 18 years and older.
Icatibant is administered subcutaneously at the onset of attack symptoms, which can include rapid swelling of different body parts – usually the face, GI tract, extremities, or genitals – with airway swelling that can increase the risk of suffocation.
"Because it can be self-administered through an injection in the abdominal area, patients can treat themselves upon recognition of an HAE attack," Dr. Curtis Rosebraugh, director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement announcing the approval.
Icatibant, a selective bradykinin B2 receptor antagonist, inhibits the effects of bradykinin, thought to be the cause of HAE symptoms, according to Shire Human Genetic Therapies, which will market the drug as Firazyr. One syringe costs $6,800 (average wholesale price), according to a company spokesperson. The company has a program to help eligible patients help pay for copays as well as a program to help uninsured patients or those patients whose insurance does not cover the treatment.
This is the third drug approved by the FDA to treat attacks of HAE, a rare genetic disorder that affects fewer than 30,000 people in the United States: a C1 esterase inhibitor (Berinert) was approved in October 2009, to treat facial and abdominal attacks of HAE, and ecallantide (Kalbitor) was approved in December 2009 to treat acute attacks of HAE in patients aged 16 years and older.
In three controlled studies, with open-label extension periods, 225 patients were treated with 1,076 icatibant injections. Those treated with icatibant started to experience relief of symptoms in a median of 2 hours after the injection, compared with almost 20 hours among those who received a placebo injection. The most common side effects associated with icatibant were injection site reactions, fever, elevated liver enzymes, dizziness, and rash, according to the FDA.
Icatibant, which is launching in the United States on Aug. 26, is now approved in 38 countries, including those in the European Union, according to Shire.
FROM THE FDA
Variant Creutzfeldt-Jakob Blood Test on the Horizon?
The prospects for a practical blood test that quickly detects the presence of prions in blood, organs, and tissues became more encouraging with the results of two studies published earlier this year.
But until such screening and diagnostic tests become available, the greatest concern for U.S. officials is the deferment of blood and tissue donors who have spent substantial time in countries in which variant Creutzfeldt-Jakob disease (vCJD) is most prevalent, according to speakers at a meeting of the Food and Drug Administration’s Transmissible Spongiform Encephalopathies Advisory Committee in August.
One test from researchers at the National Institute of Allergy and Infectious Diseases uses an antibody-based technique to isolate abnormal prion proteins and detect them at levels 10,000 times more sensitive than in previous tests for detecting vCJD. Another test from researchers at University College London Institute of Neurology and the National Hospital for Neurology and Neurosurgery in London was able to detect vCJD prions in the presence of the normal prion protein with a sensitivity of 71% and specificity of 100%.
The safety of the blood supply is a concern, especially in places like the United Kingdom, because of five documented cases of transmission of vCJD via blood transfusions from donors with subclinical vCJD, NIAID study investigator Byron Caughey, Ph.D., said in an interview.
"The ability of [this assay] to detect prions in plasma samples raises the possibility that this assay could be used to improve prion disease diagnosis in humans and animals and to screen the blood supply for prion contamination," wrote Dr. Caughey and his coauthors at NIAID’s Rocky Mountain Laboratories in Hamilton, Mont. (mBio 2011 [doi:10.1128/mBio.00078-11]). Besides its sensitivity for detecting prions in human plasma spiked with brain tissue from patients with vCJD, the assay, called eQuIC, also accurately distinguished between hamsters infected with scrapie – including some that were in early preclinical phases of infection – and uninfected hamsters.
However, they pointed out that the test has not been studied in plasma or CSF from vCJD patients and that "further studies will be required to assess the diagnostic utility of eQuIC based on the detection of vCJD seeding activity that is endogenous to these and other specimens."
In addition to helping prevent infections through the screening of blood, blood products, and transplanted tissues, if the sensitivity is dramatically improved, the test might be used to catch the disease as early as possible, before irreversible brain damage occurs; at which point treatments, as they become available, could be used. Such a test might also be used to test medical instruments such as electrodes to prevent iatrogenic transmission, as well as to detect low levels of contamination in the food supply, such as bovine spongiform encephalopathy (BSE) in cattle and scrapie in sheep, Dr. Caughey said.
The test developed by English researchers was positive in 15 of 21 blood samples from patients with clinical vCJD and was negative in blood from 169 patients without vCJD. The authors said that the use of the test in the differential diagnosis of suspected vCJD would be studied further in large studies (Lancet 2011;377:487-93).
The Implications of a vCJD Blood Test
Dr. Brian Appleby, a neuropsychiatrist at the Cleveland Clinic Lou Ruvo Center for Brain Health, speculated that a blood test could have "huge implications" for screening the blood supply, largely in the United Kingdom, and for use in checking deer and elk for chronic wasting disease, the main acquired prion disease seen in the United States.
A better test could also help to expedite the diagnosis of vCJD in people, who often get diagnosed very late in the course of the disease – if at all – and usually survive for less than a month after diagnosis, he said in an interview. Currently, the only forms of Creutzfeldt-Jakob disease seen in the United States are the sporadic and familial types.
Of all the prion diseases, a better diagnostic test is needed for sporadic CJD, which accounts for about 85% of all human prion diseases, but there is no evidence that sporadic CJD is in the blood, Dr. Appleby said. vCJD, however, is present not only in the blood but also in lymphoreticular tissues such as the tonsils.
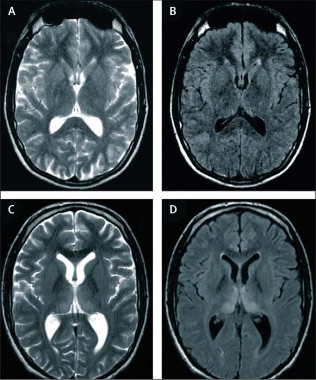
He added that sporadic CJD cases are being identified much earlier now with brain MRI, which has increasingly been used and has become very useful in making and expediting the diagnosis. Unique to vCJD is the "hockey stick" sign on brain MRI, which is a symmetrical hyperintensity in the pulvinar (posterior) nucleiof the thalamus.
Dr. Appleby is a member of the FDA’s advisory committee that in August discussed the implications of three cases of vCJD diagnosed in North America on current recommendations for vCJD screening of donors of blood, cell, and tissue-based products.
The three individuals were long-time residents of Saudi Arabia who were likely infected with the BSE agent in beef imported from the United Kingdom to Saudi Arabia during 1980-1996. After hearing data that included detailed descriptions of the cases from public health officials, the majority of the panel voted that the available data supported the FDA’s consideration to recommend deferring donors who had spent a cumulative of 6 months or more during 1980-1996 in Saudi Arabia as U.S. military personnel or who had spent more than 5 years cumulatively in Saudi Arabia during the same time period.
Having lived in the United Kingdom for at least 3 months during 1980-1996 (considered the highest risk period for dietary exposure to BSE) is among the reasons for donor deferral under current U.S. and Canadian blood donor deferral policies. Luisa Gregori, Ph.D., of the FDA’s division of emerging and transfusion-transmitted diseases said at the meeting that donor deferral is currently the only way to protect the U.S. blood supply from transfusion-transmitted CJD and vCJD and that the current donor deferral policy in the United States would not have deferred these three individuals.
The risk of CJD transmission via a blood transfusion is theoretical, but the risk of transfusion transmission of vCJD has been demonstrated, Dr. Gregori noted.
None of the sources for this story had relevant financial disclosures.
The prospects for a practical blood test that quickly detects the presence of prions in blood, organs, and tissues became more encouraging with the results of two studies published earlier this year.
But until such screening and diagnostic tests become available, the greatest concern for U.S. officials is the deferment of blood and tissue donors who have spent substantial time in countries in which variant Creutzfeldt-Jakob disease (vCJD) is most prevalent, according to speakers at a meeting of the Food and Drug Administration’s Transmissible Spongiform Encephalopathies Advisory Committee in August.
One test from researchers at the National Institute of Allergy and Infectious Diseases uses an antibody-based technique to isolate abnormal prion proteins and detect them at levels 10,000 times more sensitive than in previous tests for detecting vCJD. Another test from researchers at University College London Institute of Neurology and the National Hospital for Neurology and Neurosurgery in London was able to detect vCJD prions in the presence of the normal prion protein with a sensitivity of 71% and specificity of 100%.
The safety of the blood supply is a concern, especially in places like the United Kingdom, because of five documented cases of transmission of vCJD via blood transfusions from donors with subclinical vCJD, NIAID study investigator Byron Caughey, Ph.D., said in an interview.
"The ability of [this assay] to detect prions in plasma samples raises the possibility that this assay could be used to improve prion disease diagnosis in humans and animals and to screen the blood supply for prion contamination," wrote Dr. Caughey and his coauthors at NIAID’s Rocky Mountain Laboratories in Hamilton, Mont. (mBio 2011 [doi:10.1128/mBio.00078-11]). Besides its sensitivity for detecting prions in human plasma spiked with brain tissue from patients with vCJD, the assay, called eQuIC, also accurately distinguished between hamsters infected with scrapie – including some that were in early preclinical phases of infection – and uninfected hamsters.
However, they pointed out that the test has not been studied in plasma or CSF from vCJD patients and that "further studies will be required to assess the diagnostic utility of eQuIC based on the detection of vCJD seeding activity that is endogenous to these and other specimens."
In addition to helping prevent infections through the screening of blood, blood products, and transplanted tissues, if the sensitivity is dramatically improved, the test might be used to catch the disease as early as possible, before irreversible brain damage occurs; at which point treatments, as they become available, could be used. Such a test might also be used to test medical instruments such as electrodes to prevent iatrogenic transmission, as well as to detect low levels of contamination in the food supply, such as bovine spongiform encephalopathy (BSE) in cattle and scrapie in sheep, Dr. Caughey said.
The test developed by English researchers was positive in 15 of 21 blood samples from patients with clinical vCJD and was negative in blood from 169 patients without vCJD. The authors said that the use of the test in the differential diagnosis of suspected vCJD would be studied further in large studies (Lancet 2011;377:487-93).
The Implications of a vCJD Blood Test
Dr. Brian Appleby, a neuropsychiatrist at the Cleveland Clinic Lou Ruvo Center for Brain Health, speculated that a blood test could have "huge implications" for screening the blood supply, largely in the United Kingdom, and for use in checking deer and elk for chronic wasting disease, the main acquired prion disease seen in the United States.
A better test could also help to expedite the diagnosis of vCJD in people, who often get diagnosed very late in the course of the disease – if at all – and usually survive for less than a month after diagnosis, he said in an interview. Currently, the only forms of Creutzfeldt-Jakob disease seen in the United States are the sporadic and familial types.
Of all the prion diseases, a better diagnostic test is needed for sporadic CJD, which accounts for about 85% of all human prion diseases, but there is no evidence that sporadic CJD is in the blood, Dr. Appleby said. vCJD, however, is present not only in the blood but also in lymphoreticular tissues such as the tonsils.
He added that sporadic CJD cases are being identified much earlier now with brain MRI, which has increasingly been used and has become very useful in making and expediting the diagnosis. Unique to vCJD is the "hockey stick" sign on brain MRI, which is a symmetrical hyperintensity in the pulvinar (posterior) nucleiof the thalamus.
Dr. Appleby is a member of the FDA’s advisory committee that in August discussed the implications of three cases of vCJD diagnosed in North America on current recommendations for vCJD screening of donors of blood, cell, and tissue-based products.
The three individuals were long-time residents of Saudi Arabia who were likely infected with the BSE agent in beef imported from the United Kingdom to Saudi Arabia during 1980-1996. After hearing data that included detailed descriptions of the cases from public health officials, the majority of the panel voted that the available data supported the FDA’s consideration to recommend deferring donors who had spent a cumulative of 6 months or more during 1980-1996 in Saudi Arabia as U.S. military personnel or who had spent more than 5 years cumulatively in Saudi Arabia during the same time period.
Having lived in the United Kingdom for at least 3 months during 1980-1996 (considered the highest risk period for dietary exposure to BSE) is among the reasons for donor deferral under current U.S. and Canadian blood donor deferral policies. Luisa Gregori, Ph.D., of the FDA’s division of emerging and transfusion-transmitted diseases said at the meeting that donor deferral is currently the only way to protect the U.S. blood supply from transfusion-transmitted CJD and vCJD and that the current donor deferral policy in the United States would not have deferred these three individuals.
The risk of CJD transmission via a blood transfusion is theoretical, but the risk of transfusion transmission of vCJD has been demonstrated, Dr. Gregori noted.
None of the sources for this story had relevant financial disclosures.
The prospects for a practical blood test that quickly detects the presence of prions in blood, organs, and tissues became more encouraging with the results of two studies published earlier this year.
But until such screening and diagnostic tests become available, the greatest concern for U.S. officials is the deferment of blood and tissue donors who have spent substantial time in countries in which variant Creutzfeldt-Jakob disease (vCJD) is most prevalent, according to speakers at a meeting of the Food and Drug Administration’s Transmissible Spongiform Encephalopathies Advisory Committee in August.
One test from researchers at the National Institute of Allergy and Infectious Diseases uses an antibody-based technique to isolate abnormal prion proteins and detect them at levels 10,000 times more sensitive than in previous tests for detecting vCJD. Another test from researchers at University College London Institute of Neurology and the National Hospital for Neurology and Neurosurgery in London was able to detect vCJD prions in the presence of the normal prion protein with a sensitivity of 71% and specificity of 100%.
The safety of the blood supply is a concern, especially in places like the United Kingdom, because of five documented cases of transmission of vCJD via blood transfusions from donors with subclinical vCJD, NIAID study investigator Byron Caughey, Ph.D., said in an interview.
"The ability of [this assay] to detect prions in plasma samples raises the possibility that this assay could be used to improve prion disease diagnosis in humans and animals and to screen the blood supply for prion contamination," wrote Dr. Caughey and his coauthors at NIAID’s Rocky Mountain Laboratories in Hamilton, Mont. (mBio 2011 [doi:10.1128/mBio.00078-11]). Besides its sensitivity for detecting prions in human plasma spiked with brain tissue from patients with vCJD, the assay, called eQuIC, also accurately distinguished between hamsters infected with scrapie – including some that were in early preclinical phases of infection – and uninfected hamsters.
However, they pointed out that the test has not been studied in plasma or CSF from vCJD patients and that "further studies will be required to assess the diagnostic utility of eQuIC based on the detection of vCJD seeding activity that is endogenous to these and other specimens."
In addition to helping prevent infections through the screening of blood, blood products, and transplanted tissues, if the sensitivity is dramatically improved, the test might be used to catch the disease as early as possible, before irreversible brain damage occurs; at which point treatments, as they become available, could be used. Such a test might also be used to test medical instruments such as electrodes to prevent iatrogenic transmission, as well as to detect low levels of contamination in the food supply, such as bovine spongiform encephalopathy (BSE) in cattle and scrapie in sheep, Dr. Caughey said.
The test developed by English researchers was positive in 15 of 21 blood samples from patients with clinical vCJD and was negative in blood from 169 patients without vCJD. The authors said that the use of the test in the differential diagnosis of suspected vCJD would be studied further in large studies (Lancet 2011;377:487-93).
The Implications of a vCJD Blood Test
Dr. Brian Appleby, a neuropsychiatrist at the Cleveland Clinic Lou Ruvo Center for Brain Health, speculated that a blood test could have "huge implications" for screening the blood supply, largely in the United Kingdom, and for use in checking deer and elk for chronic wasting disease, the main acquired prion disease seen in the United States.
A better test could also help to expedite the diagnosis of vCJD in people, who often get diagnosed very late in the course of the disease – if at all – and usually survive for less than a month after diagnosis, he said in an interview. Currently, the only forms of Creutzfeldt-Jakob disease seen in the United States are the sporadic and familial types.
Of all the prion diseases, a better diagnostic test is needed for sporadic CJD, which accounts for about 85% of all human prion diseases, but there is no evidence that sporadic CJD is in the blood, Dr. Appleby said. vCJD, however, is present not only in the blood but also in lymphoreticular tissues such as the tonsils.
He added that sporadic CJD cases are being identified much earlier now with brain MRI, which has increasingly been used and has become very useful in making and expediting the diagnosis. Unique to vCJD is the "hockey stick" sign on brain MRI, which is a symmetrical hyperintensity in the pulvinar (posterior) nucleiof the thalamus.
Dr. Appleby is a member of the FDA’s advisory committee that in August discussed the implications of three cases of vCJD diagnosed in North America on current recommendations for vCJD screening of donors of blood, cell, and tissue-based products.
The three individuals were long-time residents of Saudi Arabia who were likely infected with the BSE agent in beef imported from the United Kingdom to Saudi Arabia during 1980-1996. After hearing data that included detailed descriptions of the cases from public health officials, the majority of the panel voted that the available data supported the FDA’s consideration to recommend deferring donors who had spent a cumulative of 6 months or more during 1980-1996 in Saudi Arabia as U.S. military personnel or who had spent more than 5 years cumulatively in Saudi Arabia during the same time period.
Having lived in the United Kingdom for at least 3 months during 1980-1996 (considered the highest risk period for dietary exposure to BSE) is among the reasons for donor deferral under current U.S. and Canadian blood donor deferral policies. Luisa Gregori, Ph.D., of the FDA’s division of emerging and transfusion-transmitted diseases said at the meeting that donor deferral is currently the only way to protect the U.S. blood supply from transfusion-transmitted CJD and vCJD and that the current donor deferral policy in the United States would not have deferred these three individuals.
The risk of CJD transmission via a blood transfusion is theoretical, but the risk of transfusion transmission of vCJD has been demonstrated, Dr. Gregori noted.
None of the sources for this story had relevant financial disclosures.
FDA Approves Brentuximab for Two Lymphomas
The Food and Drug Administration on Aug. 19 approved brentuximab, a CD30-directed antibody drug-conjugate, for the treatment of Hodgkin’s lymphoma and systemic anaplastic large-cell lymphoma, after other treatments have failed.
Specifically, brentuximab was approved for the treatment of patients with Hodgkin’s lymphoma (HL) after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates. It is the first new treatment approved for HL by the FDA since 1977, according to the FDA statement. The indications are based on response rates, and there are no available data that show "improvement in patient reported outcomes or survival" with treatment, according to the prescribing information.
The FDA also approved brentuximab for patients with systemic anaplastic large-cell lymphoma (ALCL), a rare form of lymphoma, after failure of at least one prior multiagent chemotherapy regimen. It is the first treatment approved for this rare lymphoma; approximately 2,000 new cases are diagnosed yearly in the United States.
Both types of lymphomas express the CD30 antigen. Brentuximab is a combination of an anti-CD30 antibody and a drug, monomethyl auristatin E (MMAE), a microtubule disrupting agent. The antibody directs the drug to C30-expressing tumor cells, where it is released.
Brentuximab is administered intravenously every 3 weeks; it will be marketed in the United States as Adcetris by Seattle Genetics. It will be available the week of Aug. 22, according to a company spokesperson.
The product was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug for a serious disease based on data showing the drug is effective on a surrogate end point that is "reasonably likely" to predict clinical benefit. Accelerated approvals provide patients with access to promising treatments, but companies are required to provide confirmatory clinical data in order for the drug to be converted to a full approval and remain on the market.
"Early clinical data suggest that patients who received Adcetris for Hodgkin’s lymphoma and systemic anaplastic lymphoma experienced a significant response to the therapy," Dr. Richard Pazdur, director of the Office of Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
In a study of 102 patients with HL treated with brentuximab, 73% of the patients had either a complete or partial response to treatment. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response to treatment. Neutropenia, peripheral sensory neuropathy, fatigue, nausea, anemia, upper-respiratory infection, diarrhea, fever, cough, vomiting, and thrombocytopenia were among the most common adverse effects associated with treatment, according to the FDA.
At a meeting in July, an FDA advisory panel recommended the accelerated approval of both indications.
Brentuximab is under review for the same indications in Europe; a decision on approval is expected in the first half of 2012, according to a spokesperson for Millennium: the Takeda Oncology Co., which has rights to commercialize brentuximab outside the United States and Canada.
The Food and Drug Administration on Aug. 19 approved brentuximab, a CD30-directed antibody drug-conjugate, for the treatment of Hodgkin’s lymphoma and systemic anaplastic large-cell lymphoma, after other treatments have failed.
Specifically, brentuximab was approved for the treatment of patients with Hodgkin’s lymphoma (HL) after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates. It is the first new treatment approved for HL by the FDA since 1977, according to the FDA statement. The indications are based on response rates, and there are no available data that show "improvement in patient reported outcomes or survival" with treatment, according to the prescribing information.
The FDA also approved brentuximab for patients with systemic anaplastic large-cell lymphoma (ALCL), a rare form of lymphoma, after failure of at least one prior multiagent chemotherapy regimen. It is the first treatment approved for this rare lymphoma; approximately 2,000 new cases are diagnosed yearly in the United States.
Both types of lymphomas express the CD30 antigen. Brentuximab is a combination of an anti-CD30 antibody and a drug, monomethyl auristatin E (MMAE), a microtubule disrupting agent. The antibody directs the drug to C30-expressing tumor cells, where it is released.
Brentuximab is administered intravenously every 3 weeks; it will be marketed in the United States as Adcetris by Seattle Genetics. It will be available the week of Aug. 22, according to a company spokesperson.
The product was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug for a serious disease based on data showing the drug is effective on a surrogate end point that is "reasonably likely" to predict clinical benefit. Accelerated approvals provide patients with access to promising treatments, but companies are required to provide confirmatory clinical data in order for the drug to be converted to a full approval and remain on the market.
"Early clinical data suggest that patients who received Adcetris for Hodgkin’s lymphoma and systemic anaplastic lymphoma experienced a significant response to the therapy," Dr. Richard Pazdur, director of the Office of Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
In a study of 102 patients with HL treated with brentuximab, 73% of the patients had either a complete or partial response to treatment. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response to treatment. Neutropenia, peripheral sensory neuropathy, fatigue, nausea, anemia, upper-respiratory infection, diarrhea, fever, cough, vomiting, and thrombocytopenia were among the most common adverse effects associated with treatment, according to the FDA.
At a meeting in July, an FDA advisory panel recommended the accelerated approval of both indications.
Brentuximab is under review for the same indications in Europe; a decision on approval is expected in the first half of 2012, according to a spokesperson for Millennium: the Takeda Oncology Co., which has rights to commercialize brentuximab outside the United States and Canada.
The Food and Drug Administration on Aug. 19 approved brentuximab, a CD30-directed antibody drug-conjugate, for the treatment of Hodgkin’s lymphoma and systemic anaplastic large-cell lymphoma, after other treatments have failed.
Specifically, brentuximab was approved for the treatment of patients with Hodgkin’s lymphoma (HL) after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multiagent chemotherapy regimens in patients who are not ASCT candidates. It is the first new treatment approved for HL by the FDA since 1977, according to the FDA statement. The indications are based on response rates, and there are no available data that show "improvement in patient reported outcomes or survival" with treatment, according to the prescribing information.
The FDA also approved brentuximab for patients with systemic anaplastic large-cell lymphoma (ALCL), a rare form of lymphoma, after failure of at least one prior multiagent chemotherapy regimen. It is the first treatment approved for this rare lymphoma; approximately 2,000 new cases are diagnosed yearly in the United States.
Both types of lymphomas express the CD30 antigen. Brentuximab is a combination of an anti-CD30 antibody and a drug, monomethyl auristatin E (MMAE), a microtubule disrupting agent. The antibody directs the drug to C30-expressing tumor cells, where it is released.
Brentuximab is administered intravenously every 3 weeks; it will be marketed in the United States as Adcetris by Seattle Genetics. It will be available the week of Aug. 22, according to a company spokesperson.
The product was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug for a serious disease based on data showing the drug is effective on a surrogate end point that is "reasonably likely" to predict clinical benefit. Accelerated approvals provide patients with access to promising treatments, but companies are required to provide confirmatory clinical data in order for the drug to be converted to a full approval and remain on the market.
"Early clinical data suggest that patients who received Adcetris for Hodgkin’s lymphoma and systemic anaplastic lymphoma experienced a significant response to the therapy," Dr. Richard Pazdur, director of the Office of Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
In a study of 102 patients with HL treated with brentuximab, 73% of the patients had either a complete or partial response to treatment. In a similarly designed study of 58 patients with ALCL, 86% achieved a partial or complete response to treatment. Neutropenia, peripheral sensory neuropathy, fatigue, nausea, anemia, upper-respiratory infection, diarrhea, fever, cough, vomiting, and thrombocytopenia were among the most common adverse effects associated with treatment, according to the FDA.
At a meeting in July, an FDA advisory panel recommended the accelerated approval of both indications.
Brentuximab is under review for the same indications in Europe; a decision on approval is expected in the first half of 2012, according to a spokesperson for Millennium: the Takeda Oncology Co., which has rights to commercialize brentuximab outside the United States and Canada.
FROM THE FOOD AND DRUG ADMINISTRATION
Liraglutide Risks Revisited in Letter From Drug Maker
The manufacturer of liraglutide has issued a letter to health care professionals reminding them about the increased risk of pancreatitis associated with treatment in patients as well as informing them about the development of thyroid tumors in rodents exposed to clinically relevant doses of liraglutide, the Food and Drug Administration announced.
The information is not new, but the letter is being sent to clinicians because “a recent assessment of healthcare providers showed that some primary care providers are not fully aware of the serious risks associated with the use of Victoza,” according to the FDA statement. Victoza is the trade name for liraglutide (rDNA origin) injection, which was approved by the FDA in 2010 as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
The letter describes the “potential risk” of thyroid C-cell tumors, including medullary carcinoma, which may be associated with liraglutide. Inbrats and mice, liraglutide causesdohyroid C-cell tumors at clinically relevant exposures, but the relevance to humans cannot be ruled out by clinical or nonclinical studies, according to the letter. In addition, pancreatitis was more commonamn patients treated with liraglutide, compared with comparators in clinical trials, so the drug may increase the risk of acute pancreatitis.
Because of these risks, liraglutide is not recommended as first-line therapy, and patients should be observed closely for signs and symptoms of pancreatitis after starting treatment and after dose increases. The company is monitoring cases of medullary thyroid cancer registry cases to determine whether there was an increase in cases associated with the availability of liraglutide in the United States.
More information including a link to the letter is available in the FDA statement at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm258826.htmwww.fda.gov/medwatch
The manufacturer of liraglutide has issued a letter to health care professionals reminding them about the increased risk of pancreatitis associated with treatment in patients as well as informing them about the development of thyroid tumors in rodents exposed to clinically relevant doses of liraglutide, the Food and Drug Administration announced.
The information is not new, but the letter is being sent to clinicians because “a recent assessment of healthcare providers showed that some primary care providers are not fully aware of the serious risks associated with the use of Victoza,” according to the FDA statement. Victoza is the trade name for liraglutide (rDNA origin) injection, which was approved by the FDA in 2010 as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
The letter describes the “potential risk” of thyroid C-cell tumors, including medullary carcinoma, which may be associated with liraglutide. Inbrats and mice, liraglutide causesdohyroid C-cell tumors at clinically relevant exposures, but the relevance to humans cannot be ruled out by clinical or nonclinical studies, according to the letter. In addition, pancreatitis was more commonamn patients treated with liraglutide, compared with comparators in clinical trials, so the drug may increase the risk of acute pancreatitis.
Because of these risks, liraglutide is not recommended as first-line therapy, and patients should be observed closely for signs and symptoms of pancreatitis after starting treatment and after dose increases. The company is monitoring cases of medullary thyroid cancer registry cases to determine whether there was an increase in cases associated with the availability of liraglutide in the United States.
More information including a link to the letter is available in the FDA statement at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm258826.htmwww.fda.gov/medwatch
The manufacturer of liraglutide has issued a letter to health care professionals reminding them about the increased risk of pancreatitis associated with treatment in patients as well as informing them about the development of thyroid tumors in rodents exposed to clinically relevant doses of liraglutide, the Food and Drug Administration announced.
The information is not new, but the letter is being sent to clinicians because “a recent assessment of healthcare providers showed that some primary care providers are not fully aware of the serious risks associated with the use of Victoza,” according to the FDA statement. Victoza is the trade name for liraglutide (rDNA origin) injection, which was approved by the FDA in 2010 as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
The letter describes the “potential risk” of thyroid C-cell tumors, including medullary carcinoma, which may be associated with liraglutide. Inbrats and mice, liraglutide causesdohyroid C-cell tumors at clinically relevant exposures, but the relevance to humans cannot be ruled out by clinical or nonclinical studies, according to the letter. In addition, pancreatitis was more commonamn patients treated with liraglutide, compared with comparators in clinical trials, so the drug may increase the risk of acute pancreatitis.
Because of these risks, liraglutide is not recommended as first-line therapy, and patients should be observed closely for signs and symptoms of pancreatitis after starting treatment and after dose increases. The company is monitoring cases of medullary thyroid cancer registry cases to determine whether there was an increase in cases associated with the availability of liraglutide in the United States.
More information including a link to the letter is available in the FDA statement at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm258826.htmwww.fda.gov/medwatch
From the Food and Drug Administration