User login
HSC engraftment across the species barrier

Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant. ![]()
Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant. ![]()
Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant. ![]()
Survival differences in blood cancers across Europe

Credit: Rhoda Baer
Differences in treatment access and quality may explain why survival rates vary widely for European patients with hematologic malignancies, researchers have reported in The Lancet Oncology.
“The good news is that 5-year survival for most cancers of the blood has increased over the past 11 years, most likely reflecting the approval of new targeted drugs in the early 2000s . . . ,” said Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy.
“But there continue to be persistent differences between regions. For example, the uptake and use of new technologies and effective treatments has been far slower in eastern Europe than other regions. This might have contributed to the large differences in the management and outcomes of patients.”
Dr Sant and her colleagues uncovered these differences by analyzing data from 30 cancer registries covering all patients diagnosed in 20 European countries.*
The researchers compared changes in 5-year survival for 560,444 adults (aged 15 years and older) who were diagnosed with 11 lymphoid and myeloid cancers between 1997 and 2008, and followed up to the end of 2008.
Some cancers have shown particularly large increases in survival between 1997-1999 and 2006-2008, such as follicular lymphoma (59% to 74%), diffuse large B-cell lymphoma (42% to 55%), chronic myeloid leukemia (32% to 54%), and acute promyelocytic leukemia (50% to 62%).
The greatest improvements in survival have been in northern, central, and eastern Europe, even though adults in eastern Europe (where survival in 1997 was the lowest) continue to have lower survival for most hematologic malignancies than elsewhere.
Survival gains have been lower in southern Europe and the UK. For example, improvements in 5-year chronic myeloid leukemia survival in northern Europe (29% to 60%) and central Europe (34% to 65%) have been persistently higher than in the UK (35% to 56%) and southern Europe (37% to 55%).
Overall, the risk of death within 5 years from diagnosis fell significantly for all malignancies except myelodysplastic syndromes. But not all regions have seen such improvements.
For example, compared with the UK, the excess risk of death was significantly higher in eastern Europe than in other regions for most of the cancers investigated, but significantly lower in northern Europe.
The researchers said the most likely reasons for continuing geographical differences in survival are inequalities in the provision of care and in the availability and use of new treatments.
“We know that rituximab, imatinib, thalidomide, and bortezomib were first made available for general use in Europe in 1997, 2001, 1998, and 2003, respectively,” the researchers wrote.
“The years following general release of these drugs coincided with large increases in survival for chronic myeloid leukemia, diffuse large B-cell lymphoma, and follicular lymphoma, with a smaller but still significant survival increase for multiple myeloma plasmacytoma.”
However, they pointed out that the uptake and use of these drugs has not been uniform across Europe. For example, market uptake of rituximab, imatinib, and bortezomib was lower in eastern Europe than elsewhere and might explain the consistently lower survival in this region.
Writing in a linked comment article, Alastair Munro, MD, of the University of Dundee Medical School in Scotland, questioned whether improvements in survival can be attributed to drugs alone.
He said that better understanding of the conclusions from this study (called EUROCARE-5) requires additional information about changes affecting survival according to disease categories, the distribution of histological subtypes and their relation with the age distribution of the population, the distribution of stages at diagnosis, and the timing of active intervention for indolent tumors. ![]()
*The areas included in the study were northern Europe (Denmark, Iceland, and Norway), the UK (England, Northern Ireland, Scotland, and Wales), central Europe (Austria, France, Germany, Switzerland, and The Netherlands), eastern Europe (Bulgaria, Estonia, Lithuania, Poland, and Slovakia), and southern Europe (Italy, Malta, and Slovenia).
Credit: Rhoda Baer
Differences in treatment access and quality may explain why survival rates vary widely for European patients with hematologic malignancies, researchers have reported in The Lancet Oncology.
“The good news is that 5-year survival for most cancers of the blood has increased over the past 11 years, most likely reflecting the approval of new targeted drugs in the early 2000s . . . ,” said Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy.
“But there continue to be persistent differences between regions. For example, the uptake and use of new technologies and effective treatments has been far slower in eastern Europe than other regions. This might have contributed to the large differences in the management and outcomes of patients.”
Dr Sant and her colleagues uncovered these differences by analyzing data from 30 cancer registries covering all patients diagnosed in 20 European countries.*
The researchers compared changes in 5-year survival for 560,444 adults (aged 15 years and older) who were diagnosed with 11 lymphoid and myeloid cancers between 1997 and 2008, and followed up to the end of 2008.
Some cancers have shown particularly large increases in survival between 1997-1999 and 2006-2008, such as follicular lymphoma (59% to 74%), diffuse large B-cell lymphoma (42% to 55%), chronic myeloid leukemia (32% to 54%), and acute promyelocytic leukemia (50% to 62%).
The greatest improvements in survival have been in northern, central, and eastern Europe, even though adults in eastern Europe (where survival in 1997 was the lowest) continue to have lower survival for most hematologic malignancies than elsewhere.
Survival gains have been lower in southern Europe and the UK. For example, improvements in 5-year chronic myeloid leukemia survival in northern Europe (29% to 60%) and central Europe (34% to 65%) have been persistently higher than in the UK (35% to 56%) and southern Europe (37% to 55%).
Overall, the risk of death within 5 years from diagnosis fell significantly for all malignancies except myelodysplastic syndromes. But not all regions have seen such improvements.
For example, compared with the UK, the excess risk of death was significantly higher in eastern Europe than in other regions for most of the cancers investigated, but significantly lower in northern Europe.
The researchers said the most likely reasons for continuing geographical differences in survival are inequalities in the provision of care and in the availability and use of new treatments.
“We know that rituximab, imatinib, thalidomide, and bortezomib were first made available for general use in Europe in 1997, 2001, 1998, and 2003, respectively,” the researchers wrote.
“The years following general release of these drugs coincided with large increases in survival for chronic myeloid leukemia, diffuse large B-cell lymphoma, and follicular lymphoma, with a smaller but still significant survival increase for multiple myeloma plasmacytoma.”
However, they pointed out that the uptake and use of these drugs has not been uniform across Europe. For example, market uptake of rituximab, imatinib, and bortezomib was lower in eastern Europe than elsewhere and might explain the consistently lower survival in this region.
Writing in a linked comment article, Alastair Munro, MD, of the University of Dundee Medical School in Scotland, questioned whether improvements in survival can be attributed to drugs alone.
He said that better understanding of the conclusions from this study (called EUROCARE-5) requires additional information about changes affecting survival according to disease categories, the distribution of histological subtypes and their relation with the age distribution of the population, the distribution of stages at diagnosis, and the timing of active intervention for indolent tumors. ![]()
*The areas included in the study were northern Europe (Denmark, Iceland, and Norway), the UK (England, Northern Ireland, Scotland, and Wales), central Europe (Austria, France, Germany, Switzerland, and The Netherlands), eastern Europe (Bulgaria, Estonia, Lithuania, Poland, and Slovakia), and southern Europe (Italy, Malta, and Slovenia).
Credit: Rhoda Baer
Differences in treatment access and quality may explain why survival rates vary widely for European patients with hematologic malignancies, researchers have reported in The Lancet Oncology.
“The good news is that 5-year survival for most cancers of the blood has increased over the past 11 years, most likely reflecting the approval of new targeted drugs in the early 2000s . . . ,” said Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy.
“But there continue to be persistent differences between regions. For example, the uptake and use of new technologies and effective treatments has been far slower in eastern Europe than other regions. This might have contributed to the large differences in the management and outcomes of patients.”
Dr Sant and her colleagues uncovered these differences by analyzing data from 30 cancer registries covering all patients diagnosed in 20 European countries.*
The researchers compared changes in 5-year survival for 560,444 adults (aged 15 years and older) who were diagnosed with 11 lymphoid and myeloid cancers between 1997 and 2008, and followed up to the end of 2008.
Some cancers have shown particularly large increases in survival between 1997-1999 and 2006-2008, such as follicular lymphoma (59% to 74%), diffuse large B-cell lymphoma (42% to 55%), chronic myeloid leukemia (32% to 54%), and acute promyelocytic leukemia (50% to 62%).
The greatest improvements in survival have been in northern, central, and eastern Europe, even though adults in eastern Europe (where survival in 1997 was the lowest) continue to have lower survival for most hematologic malignancies than elsewhere.
Survival gains have been lower in southern Europe and the UK. For example, improvements in 5-year chronic myeloid leukemia survival in northern Europe (29% to 60%) and central Europe (34% to 65%) have been persistently higher than in the UK (35% to 56%) and southern Europe (37% to 55%).
Overall, the risk of death within 5 years from diagnosis fell significantly for all malignancies except myelodysplastic syndromes. But not all regions have seen such improvements.
For example, compared with the UK, the excess risk of death was significantly higher in eastern Europe than in other regions for most of the cancers investigated, but significantly lower in northern Europe.
The researchers said the most likely reasons for continuing geographical differences in survival are inequalities in the provision of care and in the availability and use of new treatments.
“We know that rituximab, imatinib, thalidomide, and bortezomib were first made available for general use in Europe in 1997, 2001, 1998, and 2003, respectively,” the researchers wrote.
“The years following general release of these drugs coincided with large increases in survival for chronic myeloid leukemia, diffuse large B-cell lymphoma, and follicular lymphoma, with a smaller but still significant survival increase for multiple myeloma plasmacytoma.”
However, they pointed out that the uptake and use of these drugs has not been uniform across Europe. For example, market uptake of rituximab, imatinib, and bortezomib was lower in eastern Europe than elsewhere and might explain the consistently lower survival in this region.
Writing in a linked comment article, Alastair Munro, MD, of the University of Dundee Medical School in Scotland, questioned whether improvements in survival can be attributed to drugs alone.
He said that better understanding of the conclusions from this study (called EUROCARE-5) requires additional information about changes affecting survival according to disease categories, the distribution of histological subtypes and their relation with the age distribution of the population, the distribution of stages at diagnosis, and the timing of active intervention for indolent tumors. ![]()
*The areas included in the study were northern Europe (Denmark, Iceland, and Norway), the UK (England, Northern Ireland, Scotland, and Wales), central Europe (Austria, France, Germany, Switzerland, and The Netherlands), eastern Europe (Bulgaria, Estonia, Lithuania, Poland, and Slovakia), and southern Europe (Italy, Malta, and Slovenia).
FDA puts clinical hold on REGULATE-PCI trial

Credit: Kevin MacKenzie
The US Food and Drug Administration (FDA) has placed a hold on patient enrollment and dosing of drugs under investigation in the phase 3 REGULATE-PCI trial.
The trial is a comparison of the reversible thrombin inhibitor bivalirudin and the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation Factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing percutaneous coronary intervention (PCI).
The company sponsoring the REGULATE-PCI trial, Regado Biosciences, had voluntarily halted enrollment prior to the FDA’s announcement. The FDA’s clinical hold formalizes its involvement in any decision to re-initiate enrollment and dosing in the trial in the future.
Regado stopped enrollment after its Data Safety Monitoring Board (DSMB) started an unplanned review of the study data. The board has said the review is focusing on serious adverse events and allergic reactions.
“[W]e remain blinded to REGULATE-PCI study data and are awaiting the outcome of the full safety and efficacy analysis, including an analysis of benefit/risk ratio, being performed by our DSMB,” said David J Mazzo, PhD, CEO of Regado.
“Any recommendation to re-initiate patient enrollment in REGULATE-PCI will be based on the DSMB’s conclusions and would always be implemented in agreement with FDA.”
The REGULATE-PCI trial is a comparison of the Revolixys Kit (formerly known as REG-1) with bivalirudin (Angiomax) in patients undergoing PCI. The goal is to enroll 13,200 patients, and 3234 have been enrolled to date.
Eligible patients are those receiving PCI electively or for the treatment of unstable angina or non-ST elevated myocardial infarction.
Patients randomized to the Revolixys arm receive pegnivacogin at a dose of 1 mg/kg along with an 80% reversal dose of anivamersen. The timing of the remaining 20% is at the discretion of the treating physician.
The trial is powered to show superiority in efficacy and noninferiority in safety of the Revolixys Kit compared to bivalirudin. The study’s primary endpoint is a composite of death, nonfatal myocardial infarction, nonfatal stroke, and urgent target lesion revascularization through day 3 post-PCI.
For more details, visit clinicaltrials.gov. ![]()
Credit: Kevin MacKenzie
The US Food and Drug Administration (FDA) has placed a hold on patient enrollment and dosing of drugs under investigation in the phase 3 REGULATE-PCI trial.
The trial is a comparison of the reversible thrombin inhibitor bivalirudin and the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation Factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing percutaneous coronary intervention (PCI).
The company sponsoring the REGULATE-PCI trial, Regado Biosciences, had voluntarily halted enrollment prior to the FDA’s announcement. The FDA’s clinical hold formalizes its involvement in any decision to re-initiate enrollment and dosing in the trial in the future.
Regado stopped enrollment after its Data Safety Monitoring Board (DSMB) started an unplanned review of the study data. The board has said the review is focusing on serious adverse events and allergic reactions.
“[W]e remain blinded to REGULATE-PCI study data and are awaiting the outcome of the full safety and efficacy analysis, including an analysis of benefit/risk ratio, being performed by our DSMB,” said David J Mazzo, PhD, CEO of Regado.
“Any recommendation to re-initiate patient enrollment in REGULATE-PCI will be based on the DSMB’s conclusions and would always be implemented in agreement with FDA.”
The REGULATE-PCI trial is a comparison of the Revolixys Kit (formerly known as REG-1) with bivalirudin (Angiomax) in patients undergoing PCI. The goal is to enroll 13,200 patients, and 3234 have been enrolled to date.
Eligible patients are those receiving PCI electively or for the treatment of unstable angina or non-ST elevated myocardial infarction.
Patients randomized to the Revolixys arm receive pegnivacogin at a dose of 1 mg/kg along with an 80% reversal dose of anivamersen. The timing of the remaining 20% is at the discretion of the treating physician.
The trial is powered to show superiority in efficacy and noninferiority in safety of the Revolixys Kit compared to bivalirudin. The study’s primary endpoint is a composite of death, nonfatal myocardial infarction, nonfatal stroke, and urgent target lesion revascularization through day 3 post-PCI.
For more details, visit clinicaltrials.gov. ![]()
Credit: Kevin MacKenzie
The US Food and Drug Administration (FDA) has placed a hold on patient enrollment and dosing of drugs under investigation in the phase 3 REGULATE-PCI trial.
The trial is a comparison of the reversible thrombin inhibitor bivalirudin and the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation Factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing percutaneous coronary intervention (PCI).
The company sponsoring the REGULATE-PCI trial, Regado Biosciences, had voluntarily halted enrollment prior to the FDA’s announcement. The FDA’s clinical hold formalizes its involvement in any decision to re-initiate enrollment and dosing in the trial in the future.
Regado stopped enrollment after its Data Safety Monitoring Board (DSMB) started an unplanned review of the study data. The board has said the review is focusing on serious adverse events and allergic reactions.
“[W]e remain blinded to REGULATE-PCI study data and are awaiting the outcome of the full safety and efficacy analysis, including an analysis of benefit/risk ratio, being performed by our DSMB,” said David J Mazzo, PhD, CEO of Regado.
“Any recommendation to re-initiate patient enrollment in REGULATE-PCI will be based on the DSMB’s conclusions and would always be implemented in agreement with FDA.”
The REGULATE-PCI trial is a comparison of the Revolixys Kit (formerly known as REG-1) with bivalirudin (Angiomax) in patients undergoing PCI. The goal is to enroll 13,200 patients, and 3234 have been enrolled to date.
Eligible patients are those receiving PCI electively or for the treatment of unstable angina or non-ST elevated myocardial infarction.
Patients randomized to the Revolixys arm receive pegnivacogin at a dose of 1 mg/kg along with an 80% reversal dose of anivamersen. The timing of the remaining 20% is at the discretion of the treating physician.
The trial is powered to show superiority in efficacy and noninferiority in safety of the Revolixys Kit compared to bivalirudin. The study’s primary endpoint is a composite of death, nonfatal myocardial infarction, nonfatal stroke, and urgent target lesion revascularization through day 3 post-PCI.
For more details, visit clinicaltrials.gov. ![]()
Pair details ‘promise and perils’ of antioxidants

Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets. ![]()
Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets. ![]()
Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets. ![]()
Product recalled due to mold contamination
Image courtesy of Hospira
Hospira, Inc., has announced a US-wide, user-level recall of one lot of Lactated Ringers and 5% Dextrose Injection, USP, 1000 mL, Flexible Container (NDC 0409-7929-09, Lot 35-118-JT).
The company confirmed a report of particulate within the solution of the primary container. It was a filamentous-like structured particulate indicative of mold.
An analysis of the primary container and overwrap revealed a puncture that had caused the container to leak.
Intravenous administration of a non-sterile product can result in infections that may be life-threatening and could result in prolonged hospitalization or organ failure.
However, Hospira has not received reports of any adverse events associated with this issue for this lot and has not identified any quality issues with retention samples for this lot.
Lactated Ringers and 5% Dextrose Injection is indicated for parenteral replacement of extracellular losses of fluid and electrolytes, with or without minimal carbohydrate calories, as required by the clinical condition of the patient.
The product is packaged in 1000mL flexible containers, 1 container per overwrap, and 12 overwrapped containers in each case. The lot number is located in the upper left hand side of the primary container.
This lot (35-118-JT) was distributed nationwide—from December 2013 through February 2014—to hospitals, clinics, wholesalers, and distributors.
Anyone with an existing inventory should stop use and distribution, quarantine the product immediately, and call Stericycle at 1-888-912-8457 (8am to 5pm EST, Monday through Friday) to arrange for the return of the product.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 (available 24 hours a day, 7 days per week) or medcom@hospira.com.
To report adverse events or product complaints, contact Hospira Global
Complaint Management at 1-800-441-4100 (8am to 5pm CT, Monday through Friday) or
medcom@hospira.com.
Adverse events or quality problems can also be reported to the Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Image courtesy of Hospira
Hospira, Inc., has announced a US-wide, user-level recall of one lot of Lactated Ringers and 5% Dextrose Injection, USP, 1000 mL, Flexible Container (NDC 0409-7929-09, Lot 35-118-JT).
The company confirmed a report of particulate within the solution of the primary container. It was a filamentous-like structured particulate indicative of mold.
An analysis of the primary container and overwrap revealed a puncture that had caused the container to leak.
Intravenous administration of a non-sterile product can result in infections that may be life-threatening and could result in prolonged hospitalization or organ failure.
However, Hospira has not received reports of any adverse events associated with this issue for this lot and has not identified any quality issues with retention samples for this lot.
Lactated Ringers and 5% Dextrose Injection is indicated for parenteral replacement of extracellular losses of fluid and electrolytes, with or without minimal carbohydrate calories, as required by the clinical condition of the patient.
The product is packaged in 1000mL flexible containers, 1 container per overwrap, and 12 overwrapped containers in each case. The lot number is located in the upper left hand side of the primary container.
This lot (35-118-JT) was distributed nationwide—from December 2013 through February 2014—to hospitals, clinics, wholesalers, and distributors.
Anyone with an existing inventory should stop use and distribution, quarantine the product immediately, and call Stericycle at 1-888-912-8457 (8am to 5pm EST, Monday through Friday) to arrange for the return of the product.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 (available 24 hours a day, 7 days per week) or medcom@hospira.com.
To report adverse events or product complaints, contact Hospira Global
Complaint Management at 1-800-441-4100 (8am to 5pm CT, Monday through Friday) or
medcom@hospira.com.
Adverse events or quality problems can also be reported to the Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Image courtesy of Hospira
Hospira, Inc., has announced a US-wide, user-level recall of one lot of Lactated Ringers and 5% Dextrose Injection, USP, 1000 mL, Flexible Container (NDC 0409-7929-09, Lot 35-118-JT).
The company confirmed a report of particulate within the solution of the primary container. It was a filamentous-like structured particulate indicative of mold.
An analysis of the primary container and overwrap revealed a puncture that had caused the container to leak.
Intravenous administration of a non-sterile product can result in infections that may be life-threatening and could result in prolonged hospitalization or organ failure.
However, Hospira has not received reports of any adverse events associated with this issue for this lot and has not identified any quality issues with retention samples for this lot.
Lactated Ringers and 5% Dextrose Injection is indicated for parenteral replacement of extracellular losses of fluid and electrolytes, with or without minimal carbohydrate calories, as required by the clinical condition of the patient.
The product is packaged in 1000mL flexible containers, 1 container per overwrap, and 12 overwrapped containers in each case. The lot number is located in the upper left hand side of the primary container.
This lot (35-118-JT) was distributed nationwide—from December 2013 through February 2014—to hospitals, clinics, wholesalers, and distributors.
Anyone with an existing inventory should stop use and distribution, quarantine the product immediately, and call Stericycle at 1-888-912-8457 (8am to 5pm EST, Monday through Friday) to arrange for the return of the product.
For medical inquiries, contact Hospira Medical Communications at 1-800-615-0187 (available 24 hours a day, 7 days per week) or medcom@hospira.com.
To report adverse events or product complaints, contact Hospira Global
Complaint Management at 1-800-441-4100 (8am to 5pm CT, Monday through Friday) or
medcom@hospira.com.
Adverse events or quality problems can also be reported to the Food and Drug Administration’s MedWatch Adverse Event Reporting Program. ![]()
Gene editing doesn’t increase mutations in iPSCs
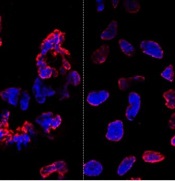
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
Telomeres can help predict prognosis in CLL
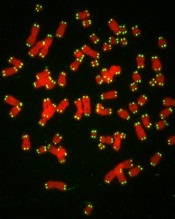
with telomeres in green
Credit: Claus Azzalin
Measuring the length and function of telomeres can help us predict prognosis in patients with chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
Investigators found that CLL patients with short, dysfunctional telomeres had a considerably poorer clinical outcome than those with long, functional telomeres.
“For the first time, confident predictions of clinical outcome can be made for individual CLL patients at diagnosis based on accurate analysis of the length of telomeres in cancer cells,” said Chris Pepper, PhD, who led the research at Cardiff University’s School of Medicine in the UK.
“This should prove enormously valuable to doctors, patients, and their families, and there is no reason why there should not be widespread implementation of this powerful prognostic tool in the near future.”
CLL progression is known to be sped up by the loss of telomeres, which cap the ends of chromosomes and protect them from damage when a cell divides. Every time a cell divides, telomeres get shorter.
When they become too short in a healthy cell, signals are sent to instruct the cell to stop dividing and die. But this “safety check” does not occur in CLL cells. Telomeres become so short that chromosomes are left exposed and are prone to fusing together during cell division, causing even larger DNA faults and even greater instability.
So Dr Pepper and his colleagues set out to identify the telomere length at which fusions start to occur in CLL patients.
The team measured telomeres in patient samples using single telomere length analysis (STELA) along with an experimentally derived definition of telomere dysfunction. They defined the upper telomere length threshold at which telomere fusions occur and used the mean of the telomere “fusogenic” range as a prognostic tool.
The researchers first analyzed samples from 200 CLL patients and found that patients with telomeres below the fusogenic mean had significantly shorter overall survival than patients with telomeres above the fusogenic mean (hazard ratio [HR]=13.2, P<0.0001). This was also true for patients with early stage disease (HR=19.3, P<0.0001).
The investigators confirmed this association by analyzing samples from an additional 121 CLL patients. The prognostic impact of telomere dysfunction was evident in the entire cohort (HR=7.4, P<0.0001) and among patients classified as Binet stage A (HR=8.9, P<0.0001).
The researchers also found they could use telomere dysfunction to accurately classify Binet stage A patients into an indolent disease group and a poor prognostic group. At 10 years, the survival rate was 91% in the favorable prognostic group and 13% in the poor prognostic group.
Of note, patients with telomeres above the fusogenic mean had superior prognosis regardless of their IGHV mutation status or cytogenetic risk group. And in a multivariate analysis, the telomere fusogenic mean was associated with the highest hazard of progression and death, independent of all other biomarkers.
“The accuracy of this test in predicting how a person’s disease will develop is unprecedented and, if confirmed in clinical trials, would help doctors decide on the best treatment courses for individual CLL patients,” said Matt Kaiser, PhD, Head of Research at Leukaemia & Lymphoma Research, which funded this study.
“Telomeres are known to play a part in the progress of other forms of cancer, so this type of testing could have far-reaching benefits.” ![]()
with telomeres in green
Credit: Claus Azzalin
Measuring the length and function of telomeres can help us predict prognosis in patients with chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
Investigators found that CLL patients with short, dysfunctional telomeres had a considerably poorer clinical outcome than those with long, functional telomeres.
“For the first time, confident predictions of clinical outcome can be made for individual CLL patients at diagnosis based on accurate analysis of the length of telomeres in cancer cells,” said Chris Pepper, PhD, who led the research at Cardiff University’s School of Medicine in the UK.
“This should prove enormously valuable to doctors, patients, and their families, and there is no reason why there should not be widespread implementation of this powerful prognostic tool in the near future.”
CLL progression is known to be sped up by the loss of telomeres, which cap the ends of chromosomes and protect them from damage when a cell divides. Every time a cell divides, telomeres get shorter.
When they become too short in a healthy cell, signals are sent to instruct the cell to stop dividing and die. But this “safety check” does not occur in CLL cells. Telomeres become so short that chromosomes are left exposed and are prone to fusing together during cell division, causing even larger DNA faults and even greater instability.
So Dr Pepper and his colleagues set out to identify the telomere length at which fusions start to occur in CLL patients.
The team measured telomeres in patient samples using single telomere length analysis (STELA) along with an experimentally derived definition of telomere dysfunction. They defined the upper telomere length threshold at which telomere fusions occur and used the mean of the telomere “fusogenic” range as a prognostic tool.
The researchers first analyzed samples from 200 CLL patients and found that patients with telomeres below the fusogenic mean had significantly shorter overall survival than patients with telomeres above the fusogenic mean (hazard ratio [HR]=13.2, P<0.0001). This was also true for patients with early stage disease (HR=19.3, P<0.0001).
The investigators confirmed this association by analyzing samples from an additional 121 CLL patients. The prognostic impact of telomere dysfunction was evident in the entire cohort (HR=7.4, P<0.0001) and among patients classified as Binet stage A (HR=8.9, P<0.0001).
The researchers also found they could use telomere dysfunction to accurately classify Binet stage A patients into an indolent disease group and a poor prognostic group. At 10 years, the survival rate was 91% in the favorable prognostic group and 13% in the poor prognostic group.
Of note, patients with telomeres above the fusogenic mean had superior prognosis regardless of their IGHV mutation status or cytogenetic risk group. And in a multivariate analysis, the telomere fusogenic mean was associated with the highest hazard of progression and death, independent of all other biomarkers.
“The accuracy of this test in predicting how a person’s disease will develop is unprecedented and, if confirmed in clinical trials, would help doctors decide on the best treatment courses for individual CLL patients,” said Matt Kaiser, PhD, Head of Research at Leukaemia & Lymphoma Research, which funded this study.
“Telomeres are known to play a part in the progress of other forms of cancer, so this type of testing could have far-reaching benefits.” ![]()
with telomeres in green
Credit: Claus Azzalin
Measuring the length and function of telomeres can help us predict prognosis in patients with chronic lymphocytic leukemia (CLL), according to research published in the British Journal of Haematology.
Investigators found that CLL patients with short, dysfunctional telomeres had a considerably poorer clinical outcome than those with long, functional telomeres.
“For the first time, confident predictions of clinical outcome can be made for individual CLL patients at diagnosis based on accurate analysis of the length of telomeres in cancer cells,” said Chris Pepper, PhD, who led the research at Cardiff University’s School of Medicine in the UK.
“This should prove enormously valuable to doctors, patients, and their families, and there is no reason why there should not be widespread implementation of this powerful prognostic tool in the near future.”
CLL progression is known to be sped up by the loss of telomeres, which cap the ends of chromosomes and protect them from damage when a cell divides. Every time a cell divides, telomeres get shorter.
When they become too short in a healthy cell, signals are sent to instruct the cell to stop dividing and die. But this “safety check” does not occur in CLL cells. Telomeres become so short that chromosomes are left exposed and are prone to fusing together during cell division, causing even larger DNA faults and even greater instability.
So Dr Pepper and his colleagues set out to identify the telomere length at which fusions start to occur in CLL patients.
The team measured telomeres in patient samples using single telomere length analysis (STELA) along with an experimentally derived definition of telomere dysfunction. They defined the upper telomere length threshold at which telomere fusions occur and used the mean of the telomere “fusogenic” range as a prognostic tool.
The researchers first analyzed samples from 200 CLL patients and found that patients with telomeres below the fusogenic mean had significantly shorter overall survival than patients with telomeres above the fusogenic mean (hazard ratio [HR]=13.2, P<0.0001). This was also true for patients with early stage disease (HR=19.3, P<0.0001).
The investigators confirmed this association by analyzing samples from an additional 121 CLL patients. The prognostic impact of telomere dysfunction was evident in the entire cohort (HR=7.4, P<0.0001) and among patients classified as Binet stage A (HR=8.9, P<0.0001).
The researchers also found they could use telomere dysfunction to accurately classify Binet stage A patients into an indolent disease group and a poor prognostic group. At 10 years, the survival rate was 91% in the favorable prognostic group and 13% in the poor prognostic group.
Of note, patients with telomeres above the fusogenic mean had superior prognosis regardless of their IGHV mutation status or cytogenetic risk group. And in a multivariate analysis, the telomere fusogenic mean was associated with the highest hazard of progression and death, independent of all other biomarkers.
“The accuracy of this test in predicting how a person’s disease will develop is unprecedented and, if confirmed in clinical trials, would help doctors decide on the best treatment courses for individual CLL patients,” said Matt Kaiser, PhD, Head of Research at Leukaemia & Lymphoma Research, which funded this study.
“Telomeres are known to play a part in the progress of other forms of cancer, so this type of testing could have far-reaching benefits.” ![]()
Immune function after trauma and transfusion

Credit: Graham Colm
An immune marker may help predict which child trauma patients are likely to develop a hospital-acquired infection, and it may also provide new insight into immune response following transfusion.
In a small study, blood samples from critically ill children showed decreased production of TNF-alpha, a cytokine that’s part of the first line of defense in the innate immune system, when compared to samples from healthy control children.
In addition, TNF-alpha production was lower among children who received transfusions with older blood, compared to children who received fresher blood.
Mark W. Hall, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and his colleagues reported these findings in Shock.
The researchers had collected blood samples from 21 healthy children and 76 critically injured children aged 18 years or younger. The team then exposed each sample to lipopolysaccharide (LPS), a known stimulant of the immune response. When healthy cells are exposed to LPS, it prompts the production of TNF-alpha.
When they analyzed the immune response, the researchers found that blood samples from the healthy children responded normally to LPS, producing high levels of TNF-alpha.
Samples from the patients with critical injuries all showed at least a moderate decrease in the production of TNF-alpha. But the children who went on to develop an infection showed a much more severe and persistent drop in TNF-alpha following injury.
While the findings strongly suggest that infection risk is associated with immune system function after critical injury, they don’t explain what’s causing the malfunction. Dr Hall’s team is investigating that question now.
Transfusion implications
The research also highlighted another issue that may affect the immune response in critical illness. The team found that patients who received a transfusion of blood stored for more than 2 weeks had a lower level of TNF-alpha production than kids whose transfused blood was less than 2 weeks old, regardless of the severity of their original injury.
This supports a study published by Dr Hall and his colleagues in 2012 in Transfusion. The study showed the same immunosuppressive effect in a human cell culture model.
Dr Hall plans to look into this further through his work with a multi-institutional effort called The Pediatric Critical Care Blood Research Network (BloodNet). The group is studying, among other things, what impact blood transfusions have on immune function.
“There’s a whole line of research in which we’re involved that is dedicated to understanding the effects of transfusion in critical illness,” he said. “It’s not clear yet if blood transfusions are immunosuppressive, but our work so far suggests that blood becomes more immunosuppressive the longer it sits on the shelf.”
Reversing immunosuppression
Yet another element to the study involves reversing the immunosuppression that follows critical injury or illness. The researchers took 3 blood samples in which TNF-alpha production was decreased and cultured them with GM-CSF.
Once treated, the cells began to produce normal levels of TNF-alpha—an indication that the immunosuppression had been reversed.
Dr Hall is now leading a phase 4 clinical trial of GM-CSF to reverse immunosuppression in critically injured patients aged 1 to 21 years old.
Although findings from that project won’t be ready for another year or so, the results in the Shock article seem to offer yet another weapon in physicians’ arsenal when caring for critically ill and injured children, Dr Hall said.
“We have certainly made headway in reducing preventable infections through programs such as our own Zero Hero initiative,” he noted.
“But what this paper suggests is that it’s also important to consider the patient’s immune system and how well they are able to fight off infection. We believe that critical illness- and injury-related immune suppression may be reversible with beneficial effects on clinical outcomes.” ![]()
Credit: Graham Colm
An immune marker may help predict which child trauma patients are likely to develop a hospital-acquired infection, and it may also provide new insight into immune response following transfusion.
In a small study, blood samples from critically ill children showed decreased production of TNF-alpha, a cytokine that’s part of the first line of defense in the innate immune system, when compared to samples from healthy control children.
In addition, TNF-alpha production was lower among children who received transfusions with older blood, compared to children who received fresher blood.
Mark W. Hall, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and his colleagues reported these findings in Shock.
The researchers had collected blood samples from 21 healthy children and 76 critically injured children aged 18 years or younger. The team then exposed each sample to lipopolysaccharide (LPS), a known stimulant of the immune response. When healthy cells are exposed to LPS, it prompts the production of TNF-alpha.
When they analyzed the immune response, the researchers found that blood samples from the healthy children responded normally to LPS, producing high levels of TNF-alpha.
Samples from the patients with critical injuries all showed at least a moderate decrease in the production of TNF-alpha. But the children who went on to develop an infection showed a much more severe and persistent drop in TNF-alpha following injury.
While the findings strongly suggest that infection risk is associated with immune system function after critical injury, they don’t explain what’s causing the malfunction. Dr Hall’s team is investigating that question now.
Transfusion implications
The research also highlighted another issue that may affect the immune response in critical illness. The team found that patients who received a transfusion of blood stored for more than 2 weeks had a lower level of TNF-alpha production than kids whose transfused blood was less than 2 weeks old, regardless of the severity of their original injury.
This supports a study published by Dr Hall and his colleagues in 2012 in Transfusion. The study showed the same immunosuppressive effect in a human cell culture model.
Dr Hall plans to look into this further through his work with a multi-institutional effort called The Pediatric Critical Care Blood Research Network (BloodNet). The group is studying, among other things, what impact blood transfusions have on immune function.
“There’s a whole line of research in which we’re involved that is dedicated to understanding the effects of transfusion in critical illness,” he said. “It’s not clear yet if blood transfusions are immunosuppressive, but our work so far suggests that blood becomes more immunosuppressive the longer it sits on the shelf.”
Reversing immunosuppression
Yet another element to the study involves reversing the immunosuppression that follows critical injury or illness. The researchers took 3 blood samples in which TNF-alpha production was decreased and cultured them with GM-CSF.
Once treated, the cells began to produce normal levels of TNF-alpha—an indication that the immunosuppression had been reversed.
Dr Hall is now leading a phase 4 clinical trial of GM-CSF to reverse immunosuppression in critically injured patients aged 1 to 21 years old.
Although findings from that project won’t be ready for another year or so, the results in the Shock article seem to offer yet another weapon in physicians’ arsenal when caring for critically ill and injured children, Dr Hall said.
“We have certainly made headway in reducing preventable infections through programs such as our own Zero Hero initiative,” he noted.
“But what this paper suggests is that it’s also important to consider the patient’s immune system and how well they are able to fight off infection. We believe that critical illness- and injury-related immune suppression may be reversible with beneficial effects on clinical outcomes.” ![]()
Credit: Graham Colm
An immune marker may help predict which child trauma patients are likely to develop a hospital-acquired infection, and it may also provide new insight into immune response following transfusion.
In a small study, blood samples from critically ill children showed decreased production of TNF-alpha, a cytokine that’s part of the first line of defense in the innate immune system, when compared to samples from healthy control children.
In addition, TNF-alpha production was lower among children who received transfusions with older blood, compared to children who received fresher blood.
Mark W. Hall, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and his colleagues reported these findings in Shock.
The researchers had collected blood samples from 21 healthy children and 76 critically injured children aged 18 years or younger. The team then exposed each sample to lipopolysaccharide (LPS), a known stimulant of the immune response. When healthy cells are exposed to LPS, it prompts the production of TNF-alpha.
When they analyzed the immune response, the researchers found that blood samples from the healthy children responded normally to LPS, producing high levels of TNF-alpha.
Samples from the patients with critical injuries all showed at least a moderate decrease in the production of TNF-alpha. But the children who went on to develop an infection showed a much more severe and persistent drop in TNF-alpha following injury.
While the findings strongly suggest that infection risk is associated with immune system function after critical injury, they don’t explain what’s causing the malfunction. Dr Hall’s team is investigating that question now.
Transfusion implications
The research also highlighted another issue that may affect the immune response in critical illness. The team found that patients who received a transfusion of blood stored for more than 2 weeks had a lower level of TNF-alpha production than kids whose transfused blood was less than 2 weeks old, regardless of the severity of their original injury.
This supports a study published by Dr Hall and his colleagues in 2012 in Transfusion. The study showed the same immunosuppressive effect in a human cell culture model.
Dr Hall plans to look into this further through his work with a multi-institutional effort called The Pediatric Critical Care Blood Research Network (BloodNet). The group is studying, among other things, what impact blood transfusions have on immune function.
“There’s a whole line of research in which we’re involved that is dedicated to understanding the effects of transfusion in critical illness,” he said. “It’s not clear yet if blood transfusions are immunosuppressive, but our work so far suggests that blood becomes more immunosuppressive the longer it sits on the shelf.”
Reversing immunosuppression
Yet another element to the study involves reversing the immunosuppression that follows critical injury or illness. The researchers took 3 blood samples in which TNF-alpha production was decreased and cultured them with GM-CSF.
Once treated, the cells began to produce normal levels of TNF-alpha—an indication that the immunosuppression had been reversed.
Dr Hall is now leading a phase 4 clinical trial of GM-CSF to reverse immunosuppression in critically injured patients aged 1 to 21 years old.
Although findings from that project won’t be ready for another year or so, the results in the Shock article seem to offer yet another weapon in physicians’ arsenal when caring for critically ill and injured children, Dr Hall said.
“We have certainly made headway in reducing preventable infections through programs such as our own Zero Hero initiative,” he noted.
“But what this paper suggests is that it’s also important to consider the patient’s immune system and how well they are able to fight off infection. We believe that critical illness- and injury-related immune suppression may be reversible with beneficial effects on clinical outcomes.”
New formulation improves chemo drug

Credit: Larry Ostby
A new formulation of the chemotherapy drug cisplatin can significantly increase the drug’s ability to target and destroy cancer cells, a new study suggests.
Scientists constructed a modified version of cisplatin called Platin-M, which is designed to overcome treatment resistance by attacking mitochondria within cancer cells.
“You can think of mitochondria as a kind of powerhouse for the cell, generating the energy it needs to grow and reproduce,” said Shanta Dhar, PhD, of the University of Georgia in Athens, Georgia.
“This prodrug delivers cisplatin directly to the mitochondria in cancerous cells. Without that essential powerhouse, the cell cannot survive.”
Dr Dhar and her colleagues described the creation of this prodrug in the Proceedings of the National Academy of Sciences.
Sean Marrache, a graduate student in Dr Dhar’s lab, entrapped Platin-M in a specially designed nanoparticle that seeks out the mitochondria and releases the drug. Once inside, Platin-M interferes with the mitochondria’s DNA, triggering cell death.
The researchers tested Platin-M on neuroblastoma cells. In experiments using a cisplatin-resistant cell culture, Platin-M nanoparticles were roughly 17 times more active than cisplatin alone.
“This technique could become a treatment for a number of cancers, but it may prove most useful for more aggressive forms of cancer that are resistant to current therapies,” said Rakesh Pathak, PhD, a postdoctoral researcher in Dr Dhar’s lab.
However, the researchers cautioned that these results are preliminary, and more work is necessary before Platin-M enters clinical trials. Still, their early results in mouse models are encouraging, and they are currently developing safety trials in larger animals.
“Cisplatin is a well-studied chemotherapy, so we hope our unique formulation will enhance its efficacy,” Dr Dhar said. “We are excited about these early results, which look very promising.”
Credit: Larry Ostby
A new formulation of the chemotherapy drug cisplatin can significantly increase the drug’s ability to target and destroy cancer cells, a new study suggests.
Scientists constructed a modified version of cisplatin called Platin-M, which is designed to overcome treatment resistance by attacking mitochondria within cancer cells.
“You can think of mitochondria as a kind of powerhouse for the cell, generating the energy it needs to grow and reproduce,” said Shanta Dhar, PhD, of the University of Georgia in Athens, Georgia.
“This prodrug delivers cisplatin directly to the mitochondria in cancerous cells. Without that essential powerhouse, the cell cannot survive.”
Dr Dhar and her colleagues described the creation of this prodrug in the Proceedings of the National Academy of Sciences.
Sean Marrache, a graduate student in Dr Dhar’s lab, entrapped Platin-M in a specially designed nanoparticle that seeks out the mitochondria and releases the drug. Once inside, Platin-M interferes with the mitochondria’s DNA, triggering cell death.
The researchers tested Platin-M on neuroblastoma cells. In experiments using a cisplatin-resistant cell culture, Platin-M nanoparticles were roughly 17 times more active than cisplatin alone.
“This technique could become a treatment for a number of cancers, but it may prove most useful for more aggressive forms of cancer that are resistant to current therapies,” said Rakesh Pathak, PhD, a postdoctoral researcher in Dr Dhar’s lab.
However, the researchers cautioned that these results are preliminary, and more work is necessary before Platin-M enters clinical trials. Still, their early results in mouse models are encouraging, and they are currently developing safety trials in larger animals.
“Cisplatin is a well-studied chemotherapy, so we hope our unique formulation will enhance its efficacy,” Dr Dhar said. “We are excited about these early results, which look very promising.”
Credit: Larry Ostby
A new formulation of the chemotherapy drug cisplatin can significantly increase the drug’s ability to target and destroy cancer cells, a new study suggests.
Scientists constructed a modified version of cisplatin called Platin-M, which is designed to overcome treatment resistance by attacking mitochondria within cancer cells.
“You can think of mitochondria as a kind of powerhouse for the cell, generating the energy it needs to grow and reproduce,” said Shanta Dhar, PhD, of the University of Georgia in Athens, Georgia.
“This prodrug delivers cisplatin directly to the mitochondria in cancerous cells. Without that essential powerhouse, the cell cannot survive.”
Dr Dhar and her colleagues described the creation of this prodrug in the Proceedings of the National Academy of Sciences.
Sean Marrache, a graduate student in Dr Dhar’s lab, entrapped Platin-M in a specially designed nanoparticle that seeks out the mitochondria and releases the drug. Once inside, Platin-M interferes with the mitochondria’s DNA, triggering cell death.
The researchers tested Platin-M on neuroblastoma cells. In experiments using a cisplatin-resistant cell culture, Platin-M nanoparticles were roughly 17 times more active than cisplatin alone.
“This technique could become a treatment for a number of cancers, but it may prove most useful for more aggressive forms of cancer that are resistant to current therapies,” said Rakesh Pathak, PhD, a postdoctoral researcher in Dr Dhar’s lab.
However, the researchers cautioned that these results are preliminary, and more work is necessary before Platin-M enters clinical trials. Still, their early results in mouse models are encouraging, and they are currently developing safety trials in larger animals.
“Cisplatin is a well-studied chemotherapy, so we hope our unique formulation will enhance its efficacy,” Dr Dhar said. “We are excited about these early results, which look very promising.”
CAR T cells may fight fungal infections
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”