User login
System can detect sepsis early

Credit: CDC
An early warning and response system can predict a patient’s likelihood of developing sepsis, according to research published in the Journal of Hospital Medicine.
The system uses lab and vital-sign data in the electronic health record of hospital inpatients to identify those at risk for sepsis.
In a multi-hospital study, the system allowed for a marked increase in sepsis identification and care, transfer to the intensive care unit (ICU), and an indication of fewer deaths due to sepsis.
Craig A. Umscheid, MD, of Penn Medicine in Philadelphia, and his colleagues developed the system using 4575 patients admitted to the University of Pennsylvania Health System in October 2011.
The system monitored lab values and vital signs in real time. If a patient had 4 or more predefined abnormalities at any single time, an electronic communication was sent to the provider, nurse, and rapid response coordinator, who performed an immediate bedside patient evaluation.
The researchers validated the effectiveness of the system during a pre-implementation period from June to September 2012, when data on admitted patients was evaluated and alerts triggered in a database, but no notifications were sent to providers on the ground.
Outcomes in that control period were then compared to a post-implementation period from June to September 2013. The total number of patients included in both periods was 31,093.
In the pre- and post-implementation periods, 4% of patient visits triggered the alert. Analysis revealed that 90% of those patients received bedside evaluations by the care team within 30 minutes of the alert being issued.
The system resulted in a 2- to 3-fold increase in orders for tests that could help identify the presence of sepsis and a 1.5- to 2-fold increase in the administration of antibiotics and intravenous fluids.
The system prompted an increase of more than 50% in the proportion of patients quickly transferred to the ICU and a 50% increase in documentation of sepsis in the patients’ electronic health record.
There was a lower death rate from sepsis and an increase in the number of patients successfully discharged home in the post-implementation period. But these rates were not significantly different from those in the pre-implementation period.
“Our study is the first we’re aware of that was implemented throughout a multihospital health system,” Dr Umscheid said.
“Previous studies that have examined the impact of sepsis prediction tools at other institutions have only taken place on a limited number of inpatient wards. The varied patient populations, clinical staffing, practice models, and practice cultures across our health system increases the generalizability of our findings to other healthcare settings.”
Dr Umscheid also noted that the system could help triage patients for suitability of ICU transfer.
“By better identifying those with sepsis requiring advanced care,” he said, “the tool can help screen out patients not needing the inevitably limited number of ICU beds.” ![]()
Credit: CDC
An early warning and response system can predict a patient’s likelihood of developing sepsis, according to research published in the Journal of Hospital Medicine.
The system uses lab and vital-sign data in the electronic health record of hospital inpatients to identify those at risk for sepsis.
In a multi-hospital study, the system allowed for a marked increase in sepsis identification and care, transfer to the intensive care unit (ICU), and an indication of fewer deaths due to sepsis.
Craig A. Umscheid, MD, of Penn Medicine in Philadelphia, and his colleagues developed the system using 4575 patients admitted to the University of Pennsylvania Health System in October 2011.
The system monitored lab values and vital signs in real time. If a patient had 4 or more predefined abnormalities at any single time, an electronic communication was sent to the provider, nurse, and rapid response coordinator, who performed an immediate bedside patient evaluation.
The researchers validated the effectiveness of the system during a pre-implementation period from June to September 2012, when data on admitted patients was evaluated and alerts triggered in a database, but no notifications were sent to providers on the ground.
Outcomes in that control period were then compared to a post-implementation period from June to September 2013. The total number of patients included in both periods was 31,093.
In the pre- and post-implementation periods, 4% of patient visits triggered the alert. Analysis revealed that 90% of those patients received bedside evaluations by the care team within 30 minutes of the alert being issued.
The system resulted in a 2- to 3-fold increase in orders for tests that could help identify the presence of sepsis and a 1.5- to 2-fold increase in the administration of antibiotics and intravenous fluids.
The system prompted an increase of more than 50% in the proportion of patients quickly transferred to the ICU and a 50% increase in documentation of sepsis in the patients’ electronic health record.
There was a lower death rate from sepsis and an increase in the number of patients successfully discharged home in the post-implementation period. But these rates were not significantly different from those in the pre-implementation period.
“Our study is the first we’re aware of that was implemented throughout a multihospital health system,” Dr Umscheid said.
“Previous studies that have examined the impact of sepsis prediction tools at other institutions have only taken place on a limited number of inpatient wards. The varied patient populations, clinical staffing, practice models, and practice cultures across our health system increases the generalizability of our findings to other healthcare settings.”
Dr Umscheid also noted that the system could help triage patients for suitability of ICU transfer.
“By better identifying those with sepsis requiring advanced care,” he said, “the tool can help screen out patients not needing the inevitably limited number of ICU beds.” ![]()
Credit: CDC
An early warning and response system can predict a patient’s likelihood of developing sepsis, according to research published in the Journal of Hospital Medicine.
The system uses lab and vital-sign data in the electronic health record of hospital inpatients to identify those at risk for sepsis.
In a multi-hospital study, the system allowed for a marked increase in sepsis identification and care, transfer to the intensive care unit (ICU), and an indication of fewer deaths due to sepsis.
Craig A. Umscheid, MD, of Penn Medicine in Philadelphia, and his colleagues developed the system using 4575 patients admitted to the University of Pennsylvania Health System in October 2011.
The system monitored lab values and vital signs in real time. If a patient had 4 or more predefined abnormalities at any single time, an electronic communication was sent to the provider, nurse, and rapid response coordinator, who performed an immediate bedside patient evaluation.
The researchers validated the effectiveness of the system during a pre-implementation period from June to September 2012, when data on admitted patients was evaluated and alerts triggered in a database, but no notifications were sent to providers on the ground.
Outcomes in that control period were then compared to a post-implementation period from June to September 2013. The total number of patients included in both periods was 31,093.
In the pre- and post-implementation periods, 4% of patient visits triggered the alert. Analysis revealed that 90% of those patients received bedside evaluations by the care team within 30 minutes of the alert being issued.
The system resulted in a 2- to 3-fold increase in orders for tests that could help identify the presence of sepsis and a 1.5- to 2-fold increase in the administration of antibiotics and intravenous fluids.
The system prompted an increase of more than 50% in the proportion of patients quickly transferred to the ICU and a 50% increase in documentation of sepsis in the patients’ electronic health record.
There was a lower death rate from sepsis and an increase in the number of patients successfully discharged home in the post-implementation period. But these rates were not significantly different from those in the pre-implementation period.
“Our study is the first we’re aware of that was implemented throughout a multihospital health system,” Dr Umscheid said.
“Previous studies that have examined the impact of sepsis prediction tools at other institutions have only taken place on a limited number of inpatient wards. The varied patient populations, clinical staffing, practice models, and practice cultures across our health system increases the generalizability of our findings to other healthcare settings.”
Dr Umscheid also noted that the system could help triage patients for suitability of ICU transfer.
“By better identifying those with sepsis requiring advanced care,” he said, “the tool can help screen out patients not needing the inevitably limited number of ICU beds.” ![]()
FDA approves drug for nausea, vomiting
The US Food and Drug Administration (FDA) has approved Akynzeo to treat nausea and vomiting in cancer patients undergoing chemotherapy. Akynzeo is a capsule consisting of two drugs, netupitant and palonosetron.
Palonosetron prevents nausea and vomiting in the acute phase—within the first 24 hours of chemotherapy initiation.
Netupitant prevents nausea and vomiting in the acute phase and the delayed phase—25 to 120 hours after chemotherapy began.
Akynzeo’s effectiveness was established in two clinical trials of 1720 cancer patients receiving chemotherapy. Patients were randomized to receive Akynzeo or oral palonosetron.
The trials were designed to measure whether the drugs prevented any vomiting episodes in the acute, delayed, and overall phases after the start of chemotherapy.
Most Akynzeo-treated patients did not experience any vomiting or require rescue medication for nausea during the acute (98.5%), delayed (90.4%), and overall phases (89.6%).
The same was true for patients who received palonosetron, although percentages were lower—89.7%, 80.1%, and 76.5%, respectively.
The second trial showed similar results.
Common side effects of Akynzeo in the clinical trials were headache, asthenia, fatigue, dyspepsia, and constipation.
Akynzeo is distributed and marketed by Eisai Inc., under license from Helsinn Healthcare S.A. ![]()
The US Food and Drug Administration (FDA) has approved Akynzeo to treat nausea and vomiting in cancer patients undergoing chemotherapy. Akynzeo is a capsule consisting of two drugs, netupitant and palonosetron.
Palonosetron prevents nausea and vomiting in the acute phase—within the first 24 hours of chemotherapy initiation.
Netupitant prevents nausea and vomiting in the acute phase and the delayed phase—25 to 120 hours after chemotherapy began.
Akynzeo’s effectiveness was established in two clinical trials of 1720 cancer patients receiving chemotherapy. Patients were randomized to receive Akynzeo or oral palonosetron.
The trials were designed to measure whether the drugs prevented any vomiting episodes in the acute, delayed, and overall phases after the start of chemotherapy.
Most Akynzeo-treated patients did not experience any vomiting or require rescue medication for nausea during the acute (98.5%), delayed (90.4%), and overall phases (89.6%).
The same was true for patients who received palonosetron, although percentages were lower—89.7%, 80.1%, and 76.5%, respectively.
The second trial showed similar results.
Common side effects of Akynzeo in the clinical trials were headache, asthenia, fatigue, dyspepsia, and constipation.
Akynzeo is distributed and marketed by Eisai Inc., under license from Helsinn Healthcare S.A. ![]()
The US Food and Drug Administration (FDA) has approved Akynzeo to treat nausea and vomiting in cancer patients undergoing chemotherapy. Akynzeo is a capsule consisting of two drugs, netupitant and palonosetron.
Palonosetron prevents nausea and vomiting in the acute phase—within the first 24 hours of chemotherapy initiation.
Netupitant prevents nausea and vomiting in the acute phase and the delayed phase—25 to 120 hours after chemotherapy began.
Akynzeo’s effectiveness was established in two clinical trials of 1720 cancer patients receiving chemotherapy. Patients were randomized to receive Akynzeo or oral palonosetron.
The trials were designed to measure whether the drugs prevented any vomiting episodes in the acute, delayed, and overall phases after the start of chemotherapy.
Most Akynzeo-treated patients did not experience any vomiting or require rescue medication for nausea during the acute (98.5%), delayed (90.4%), and overall phases (89.6%).
The same was true for patients who received palonosetron, although percentages were lower—89.7%, 80.1%, and 76.5%, respectively.
The second trial showed similar results.
Common side effects of Akynzeo in the clinical trials were headache, asthenia, fatigue, dyspepsia, and constipation.
Akynzeo is distributed and marketed by Eisai Inc., under license from Helsinn Healthcare S.A. ![]()
Physical changes in RBCs remain a mystery

During their approximately 100-day lifespan in the bloodstream, red blood cells (RBCs) lose membrane surface area, volume, and hemoglobin content.
A study published in PLOS Computational Biology shows that, of these 3 changes, only surface-area loss can be explained by RBCs shedding small, hemoglobin-containing vesicles budding off their cells’ membrane.
Therefore, an unknown process must be primarily responsible for loss of RBC volume and hemoglobin reduction.
Roy Malka, PhD, of Massachusetts General Hospital in Boston, and his colleagues noted that variations in RBCs’ mean volume and hemoglobin content are associated with important clinical conditions, but the mechanisms controlling these physical characteristics are not well understood.
Vesicle shedding was thought to be the most important mechanism, but the researchers found evidence to suggest that a dominant role for vesicle shedding would violate empirical geometric and biophysical constraints.
So they concluded that an unknown mechanism must control loss of RBC volume and hemoglobin reduction. And this mechanism is likely responsible for 60% to 90% of volume loss and hemoglobin reduction.
The group’s work combined mathematical modeling of the mechanism that changes the physical properties of RBCs, clinical measurements of both cellular volume and hemoglobin content, and data from a new system for characterizing the non-water cellular mass of individual cells.
The researchers said the quantitative characterization of RBC loss processes will help focus future research into the molecular mechanisms of RBC maturation. And it may ultimately help in the early detection of clinical conditions where the RBC maturation pattern is altered. ![]()
During their approximately 100-day lifespan in the bloodstream, red blood cells (RBCs) lose membrane surface area, volume, and hemoglobin content.
A study published in PLOS Computational Biology shows that, of these 3 changes, only surface-area loss can be explained by RBCs shedding small, hemoglobin-containing vesicles budding off their cells’ membrane.
Therefore, an unknown process must be primarily responsible for loss of RBC volume and hemoglobin reduction.
Roy Malka, PhD, of Massachusetts General Hospital in Boston, and his colleagues noted that variations in RBCs’ mean volume and hemoglobin content are associated with important clinical conditions, but the mechanisms controlling these physical characteristics are not well understood.
Vesicle shedding was thought to be the most important mechanism, but the researchers found evidence to suggest that a dominant role for vesicle shedding would violate empirical geometric and biophysical constraints.
So they concluded that an unknown mechanism must control loss of RBC volume and hemoglobin reduction. And this mechanism is likely responsible for 60% to 90% of volume loss and hemoglobin reduction.
The group’s work combined mathematical modeling of the mechanism that changes the physical properties of RBCs, clinical measurements of both cellular volume and hemoglobin content, and data from a new system for characterizing the non-water cellular mass of individual cells.
The researchers said the quantitative characterization of RBC loss processes will help focus future research into the molecular mechanisms of RBC maturation. And it may ultimately help in the early detection of clinical conditions where the RBC maturation pattern is altered. ![]()
During their approximately 100-day lifespan in the bloodstream, red blood cells (RBCs) lose membrane surface area, volume, and hemoglobin content.
A study published in PLOS Computational Biology shows that, of these 3 changes, only surface-area loss can be explained by RBCs shedding small, hemoglobin-containing vesicles budding off their cells’ membrane.
Therefore, an unknown process must be primarily responsible for loss of RBC volume and hemoglobin reduction.
Roy Malka, PhD, of Massachusetts General Hospital in Boston, and his colleagues noted that variations in RBCs’ mean volume and hemoglobin content are associated with important clinical conditions, but the mechanisms controlling these physical characteristics are not well understood.
Vesicle shedding was thought to be the most important mechanism, but the researchers found evidence to suggest that a dominant role for vesicle shedding would violate empirical geometric and biophysical constraints.
So they concluded that an unknown mechanism must control loss of RBC volume and hemoglobin reduction. And this mechanism is likely responsible for 60% to 90% of volume loss and hemoglobin reduction.
The group’s work combined mathematical modeling of the mechanism that changes the physical properties of RBCs, clinical measurements of both cellular volume and hemoglobin content, and data from a new system for characterizing the non-water cellular mass of individual cells.
The researchers said the quantitative characterization of RBC loss processes will help focus future research into the molecular mechanisms of RBC maturation. And it may ultimately help in the early detection of clinical conditions where the RBC maturation pattern is altered. ![]()
FDA approves drug for untreated MCL

The US Food and Drug Administration (FDA) has approved bortezomib (Velcade) for use in previously untreated patients with mantle cell
lymphoma (MCL).
This is the first drug to be approved in the US for previously untreated patients with MCL.
The approval extends the utility of bortezomib beyond relapsed or refractory MCL, for which it has been approved since 2006.
The new approval is based on results of a phase 3 trial.
The study was a comparison of VcR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone) and R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in 487 patients newly diagnosed with stage II, III, or IV MCL.
Survival and response
VcR-CAP demonstrated a 59% relative improvement in the study’s primary endpoint of progression-free survival. At a median follow-up of 40 months, the median progression-free survival was 25 months in the VcR-CAP arm and 14 months in the R-CHOP arm (hazard ratio [HR]=0.63, P<0.001).
However, there was no significant improvement in overall survival. The median overall survival was not reached in the VcR-CAP arm and was 56.3 months in the R-CHOP arm (HR=0.80; P=0.173).
Patients in the VcR-CAP arm had a higher rate of complete response/unconfirmed complete response than those in the R-CHOP arm—53% and 42%, respectively (P=0.007). But there was no significant difference in overall response—92% and 90%, respectively (P=0.275).
The time to progression was significantly longer in the VcR-CAP arm—30.5 months, compared to 16.1 months in the R-CHOP arm (HR=0.58; P<0.001). And the median time to subsequent treatment was significantly longer in the VcR-CAP arm—44.5 months vs 24.8 months (HR 0.50; P<0.001).
Adverse events
VcR-CAP was associated with additional but manageable toxicity compared to R-CHOP.
Serious adverse events were reported in 38% of patients in the VcR-CAP arm and 30% in the R-CHOP arm. Grade 3 or higher adverse events were reported in 93% and 85%, respectively.
There were similar rates of all-grade peripheral neuropathy between the VcR-CAP arm and the R-CHOP arm—30% and 29%, respectively. But the rate of grade 3 or higher peripheral neuropathy was significantly higher in the VcR-CAP arm—7.5% vs 4.1%.
The incidence of all-grade thrombocytopenia was substantially higher in the VcR-CAP arm than the R-CHOP arm—72% and 19%, respectively. But there was no significant difference in bleeding events—6% and 5%, respectively.
The incidence of all-grade neutropenia was 88% in the VcR-CAP arm and 74% in the R-CHOP arm. The rate of grade 3 or higher febrile neutropenia was 14% and 15%, respectively, and the rate of infection was 60% and 46%, respectively.
These data were presented at ASCO 2014 as abstract 8500.
Bortezomib is marketed as Velcade by Millennium/Takeda and Janssen Pharmaceutical Companies. Millennium is responsible for commercialization in the US, and Janssen Pharmaceutical Companies are responsible for commercialization in the rest of the world.
For more details on the drug, visit www.velcade.com. ![]()
The US Food and Drug Administration (FDA) has approved bortezomib (Velcade) for use in previously untreated patients with mantle cell
lymphoma (MCL).
This is the first drug to be approved in the US for previously untreated patients with MCL.
The approval extends the utility of bortezomib beyond relapsed or refractory MCL, for which it has been approved since 2006.
The new approval is based on results of a phase 3 trial.
The study was a comparison of VcR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone) and R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in 487 patients newly diagnosed with stage II, III, or IV MCL.
Survival and response
VcR-CAP demonstrated a 59% relative improvement in the study’s primary endpoint of progression-free survival. At a median follow-up of 40 months, the median progression-free survival was 25 months in the VcR-CAP arm and 14 months in the R-CHOP arm (hazard ratio [HR]=0.63, P<0.001).
However, there was no significant improvement in overall survival. The median overall survival was not reached in the VcR-CAP arm and was 56.3 months in the R-CHOP arm (HR=0.80; P=0.173).
Patients in the VcR-CAP arm had a higher rate of complete response/unconfirmed complete response than those in the R-CHOP arm—53% and 42%, respectively (P=0.007). But there was no significant difference in overall response—92% and 90%, respectively (P=0.275).
The time to progression was significantly longer in the VcR-CAP arm—30.5 months, compared to 16.1 months in the R-CHOP arm (HR=0.58; P<0.001). And the median time to subsequent treatment was significantly longer in the VcR-CAP arm—44.5 months vs 24.8 months (HR 0.50; P<0.001).
Adverse events
VcR-CAP was associated with additional but manageable toxicity compared to R-CHOP.
Serious adverse events were reported in 38% of patients in the VcR-CAP arm and 30% in the R-CHOP arm. Grade 3 or higher adverse events were reported in 93% and 85%, respectively.
There were similar rates of all-grade peripheral neuropathy between the VcR-CAP arm and the R-CHOP arm—30% and 29%, respectively. But the rate of grade 3 or higher peripheral neuropathy was significantly higher in the VcR-CAP arm—7.5% vs 4.1%.
The incidence of all-grade thrombocytopenia was substantially higher in the VcR-CAP arm than the R-CHOP arm—72% and 19%, respectively. But there was no significant difference in bleeding events—6% and 5%, respectively.
The incidence of all-grade neutropenia was 88% in the VcR-CAP arm and 74% in the R-CHOP arm. The rate of grade 3 or higher febrile neutropenia was 14% and 15%, respectively, and the rate of infection was 60% and 46%, respectively.
These data were presented at ASCO 2014 as abstract 8500.
Bortezomib is marketed as Velcade by Millennium/Takeda and Janssen Pharmaceutical Companies. Millennium is responsible for commercialization in the US, and Janssen Pharmaceutical Companies are responsible for commercialization in the rest of the world.
For more details on the drug, visit www.velcade.com. ![]()
The US Food and Drug Administration (FDA) has approved bortezomib (Velcade) for use in previously untreated patients with mantle cell
lymphoma (MCL).
This is the first drug to be approved in the US for previously untreated patients with MCL.
The approval extends the utility of bortezomib beyond relapsed or refractory MCL, for which it has been approved since 2006.
The new approval is based on results of a phase 3 trial.
The study was a comparison of VcR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone) and R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in 487 patients newly diagnosed with stage II, III, or IV MCL.
Survival and response
VcR-CAP demonstrated a 59% relative improvement in the study’s primary endpoint of progression-free survival. At a median follow-up of 40 months, the median progression-free survival was 25 months in the VcR-CAP arm and 14 months in the R-CHOP arm (hazard ratio [HR]=0.63, P<0.001).
However, there was no significant improvement in overall survival. The median overall survival was not reached in the VcR-CAP arm and was 56.3 months in the R-CHOP arm (HR=0.80; P=0.173).
Patients in the VcR-CAP arm had a higher rate of complete response/unconfirmed complete response than those in the R-CHOP arm—53% and 42%, respectively (P=0.007). But there was no significant difference in overall response—92% and 90%, respectively (P=0.275).
The time to progression was significantly longer in the VcR-CAP arm—30.5 months, compared to 16.1 months in the R-CHOP arm (HR=0.58; P<0.001). And the median time to subsequent treatment was significantly longer in the VcR-CAP arm—44.5 months vs 24.8 months (HR 0.50; P<0.001).
Adverse events
VcR-CAP was associated with additional but manageable toxicity compared to R-CHOP.
Serious adverse events were reported in 38% of patients in the VcR-CAP arm and 30% in the R-CHOP arm. Grade 3 or higher adverse events were reported in 93% and 85%, respectively.
There were similar rates of all-grade peripheral neuropathy between the VcR-CAP arm and the R-CHOP arm—30% and 29%, respectively. But the rate of grade 3 or higher peripheral neuropathy was significantly higher in the VcR-CAP arm—7.5% vs 4.1%.
The incidence of all-grade thrombocytopenia was substantially higher in the VcR-CAP arm than the R-CHOP arm—72% and 19%, respectively. But there was no significant difference in bleeding events—6% and 5%, respectively.
The incidence of all-grade neutropenia was 88% in the VcR-CAP arm and 74% in the R-CHOP arm. The rate of grade 3 or higher febrile neutropenia was 14% and 15%, respectively, and the rate of infection was 60% and 46%, respectively.
These data were presented at ASCO 2014 as abstract 8500.
Bortezomib is marketed as Velcade by Millennium/Takeda and Janssen Pharmaceutical Companies. Millennium is responsible for commercialization in the US, and Janssen Pharmaceutical Companies are responsible for commercialization in the rest of the world.
For more details on the drug, visit www.velcade.com. ![]()
No need to switch antibiotics, study shows
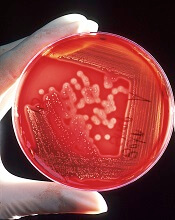
Staphylococcus
infectionCredit: Bill Branson
New research suggests the antibiotic vancomycin is still effective in treating Staphylococcus aureus bloodstream (SAB) infections, despite increases in minimum inhibitory concentration (MIC) values.
Researchers found no difference in mortality between patients with low-vancomycin MIC and those with high-vancomycin MIC.
So it seems physicians can continue using vancomycin when MIC values are elevated but within the susceptible range, rather than
switching to newer antibiotics.
Andre Kalil, MD, of the University of Nebraska Medical Center in Omaha, and his colleagues described this research in JAMA.
In recent years, physicians treating Staphylococcus infections with vancomycin have seen an increase in the MIC, the lowest concentration of an antimicrobial agent that inhibits the growth of a microorganism.
MIC values lower than 4 mg/L suggest Staphylococcus is susceptible to vancomycin. However, when the MIC value exceeds 1.5 mg/L, some physicians have taken it as an indication that vancomycin may not be working at maximum effectiveness.
Some reports have suggested that elevations in vancomycin MIC values may be associated with increased treatment failure and death.
To determine the effectiveness of vancomycin, Dr Kalil and his colleagues analyzed data from 38 studies covering 8291 episodes of SAB infection.
The team evaluated the association between vancomycin MIC elevation and mortality. Among all SAB infections studied, the overall mortality was 26.1%.
The adjusted absolute risk of mortality did not differ significantly between patients with high-vancomycin MIC and those with low-vancomycin MIC—26.8% and 25.8%, respectively.
In studies that included only methicillin-resistant Staphylococcus aureus infections, the mortality among SAB episodes in patients with high-vancomycin MIC was 27.6%, compared with a mortality of 27.4% among patients with low-vancomycin MIC.
“The study provides strong evidence that vancomycin remains highly useful,” Dr Kalil said. “Even though vancomycin is an older drug, it is still killing staph very efficiently. There are newer antibiotics available to treat Staphylococcus aureus infections, but this study demonstrates that physicians don’t necessarily need to switch to these new drugs when the MIC is increased but still within the susceptible range.”
“The prevention of a rapid switch to newer drugs has another great benefit to our patients—less unnecessary exposure to these drugs, which will translate into less development of antibiotic resistance.”
Dr Kalil said the study may have implications for clinical practice and public health.
The results suggest standards for vancomycin MIC likely do not need to be lowered, routine differentiation of MIC values between 1 mg/L and 2 mg/L appears unnecessary, and the use of alternative drugs may not be required for Staphylococcus aureus isolates with elevated but susceptible vancomycin MIC values. ![]()
Staphylococcus
infectionCredit: Bill Branson
New research suggests the antibiotic vancomycin is still effective in treating Staphylococcus aureus bloodstream (SAB) infections, despite increases in minimum inhibitory concentration (MIC) values.
Researchers found no difference in mortality between patients with low-vancomycin MIC and those with high-vancomycin MIC.
So it seems physicians can continue using vancomycin when MIC values are elevated but within the susceptible range, rather than
switching to newer antibiotics.
Andre Kalil, MD, of the University of Nebraska Medical Center in Omaha, and his colleagues described this research in JAMA.
In recent years, physicians treating Staphylococcus infections with vancomycin have seen an increase in the MIC, the lowest concentration of an antimicrobial agent that inhibits the growth of a microorganism.
MIC values lower than 4 mg/L suggest Staphylococcus is susceptible to vancomycin. However, when the MIC value exceeds 1.5 mg/L, some physicians have taken it as an indication that vancomycin may not be working at maximum effectiveness.
Some reports have suggested that elevations in vancomycin MIC values may be associated with increased treatment failure and death.
To determine the effectiveness of vancomycin, Dr Kalil and his colleagues analyzed data from 38 studies covering 8291 episodes of SAB infection.
The team evaluated the association between vancomycin MIC elevation and mortality. Among all SAB infections studied, the overall mortality was 26.1%.
The adjusted absolute risk of mortality did not differ significantly between patients with high-vancomycin MIC and those with low-vancomycin MIC—26.8% and 25.8%, respectively.
In studies that included only methicillin-resistant Staphylococcus aureus infections, the mortality among SAB episodes in patients with high-vancomycin MIC was 27.6%, compared with a mortality of 27.4% among patients with low-vancomycin MIC.
“The study provides strong evidence that vancomycin remains highly useful,” Dr Kalil said. “Even though vancomycin is an older drug, it is still killing staph very efficiently. There are newer antibiotics available to treat Staphylococcus aureus infections, but this study demonstrates that physicians don’t necessarily need to switch to these new drugs when the MIC is increased but still within the susceptible range.”
“The prevention of a rapid switch to newer drugs has another great benefit to our patients—less unnecessary exposure to these drugs, which will translate into less development of antibiotic resistance.”
Dr Kalil said the study may have implications for clinical practice and public health.
The results suggest standards for vancomycin MIC likely do not need to be lowered, routine differentiation of MIC values between 1 mg/L and 2 mg/L appears unnecessary, and the use of alternative drugs may not be required for Staphylococcus aureus isolates with elevated but susceptible vancomycin MIC values. ![]()
Staphylococcus
infectionCredit: Bill Branson
New research suggests the antibiotic vancomycin is still effective in treating Staphylococcus aureus bloodstream (SAB) infections, despite increases in minimum inhibitory concentration (MIC) values.
Researchers found no difference in mortality between patients with low-vancomycin MIC and those with high-vancomycin MIC.
So it seems physicians can continue using vancomycin when MIC values are elevated but within the susceptible range, rather than
switching to newer antibiotics.
Andre Kalil, MD, of the University of Nebraska Medical Center in Omaha, and his colleagues described this research in JAMA.
In recent years, physicians treating Staphylococcus infections with vancomycin have seen an increase in the MIC, the lowest concentration of an antimicrobial agent that inhibits the growth of a microorganism.
MIC values lower than 4 mg/L suggest Staphylococcus is susceptible to vancomycin. However, when the MIC value exceeds 1.5 mg/L, some physicians have taken it as an indication that vancomycin may not be working at maximum effectiveness.
Some reports have suggested that elevations in vancomycin MIC values may be associated with increased treatment failure and death.
To determine the effectiveness of vancomycin, Dr Kalil and his colleagues analyzed data from 38 studies covering 8291 episodes of SAB infection.
The team evaluated the association between vancomycin MIC elevation and mortality. Among all SAB infections studied, the overall mortality was 26.1%.
The adjusted absolute risk of mortality did not differ significantly between patients with high-vancomycin MIC and those with low-vancomycin MIC—26.8% and 25.8%, respectively.
In studies that included only methicillin-resistant Staphylococcus aureus infections, the mortality among SAB episodes in patients with high-vancomycin MIC was 27.6%, compared with a mortality of 27.4% among patients with low-vancomycin MIC.
“The study provides strong evidence that vancomycin remains highly useful,” Dr Kalil said. “Even though vancomycin is an older drug, it is still killing staph very efficiently. There are newer antibiotics available to treat Staphylococcus aureus infections, but this study demonstrates that physicians don’t necessarily need to switch to these new drugs when the MIC is increased but still within the susceptible range.”
“The prevention of a rapid switch to newer drugs has another great benefit to our patients—less unnecessary exposure to these drugs, which will translate into less development of antibiotic resistance.”
Dr Kalil said the study may have implications for clinical practice and public health.
The results suggest standards for vancomycin MIC likely do not need to be lowered, routine differentiation of MIC values between 1 mg/L and 2 mg/L appears unnecessary, and the use of alternative drugs may not be required for Staphylococcus aureus isolates with elevated but susceptible vancomycin MIC values. ![]()
New insight into T-cell development

Credit: NIAID
A fundamental theory about how the thymus “educates” T cells appears to be wrong, according to research published in Nature Communications.
It’s known that stem cells leave the bone marrow and travel to the thymus to become one of two CD4 T-cell types: effector cells or regulatory T cells (Tregs).
One widely held concept of why the cells become one type or the other is that they are exposed to different ligands in the thymus, said Leszek Ignatowicz, PhD, of Georgia Regents University in Augusta.
But when he and his colleagues limited the cells’ exposure to only one ligand, the same mix of T cells still emerged.
“We asked a simple question, ‘Is it going to affect their development,’ and the answer was ‘no,’” Dr Ignatowicz said. “The cells still mature in the thymus, so something else must be determining it.”
The finding provides more insight into immunity that could one day enable a new approach to vaccines that steer the thymus to produce more of whatever T-cell type a patient needs: more effector cells if they have an infection or cancer, more Tregs if they have autoimmune diseases such as arthritis and multiple sclerosis.
“We could help steer the education process in the desired direction,” Dr Ignatowicz said.
To uncover their findings, he and his colleagues studied two types of mice, each expressing a single ligand in the thymus. The researchers thought one would prompt strong ligand binding and favor Treg development, and the other would favor a weaker ligand bond and effector cell development.
The mix of resulting T cells was the same as if both were exposed to the usual thousands of ligands, although the experiment did reveal one difference.
Ligands—and, eventually, bacteria and other invaders—get the attention of T cells by activating their receptors. Both CD4 T-cell types generally have the same receptors, but they are organized differently.
The researchers found that as long as the binding was weak, as it was in the first mouse, there was a lot of overlap in the receptors the ligand bound to in both T-cell types. However, in the second mouse, where the ligand prompted strong binding, there was far less overlap.
“We are now trying to find what causes that difference,” Dr Ignatowicz said. ![]()
Credit: NIAID
A fundamental theory about how the thymus “educates” T cells appears to be wrong, according to research published in Nature Communications.
It’s known that stem cells leave the bone marrow and travel to the thymus to become one of two CD4 T-cell types: effector cells or regulatory T cells (Tregs).
One widely held concept of why the cells become one type or the other is that they are exposed to different ligands in the thymus, said Leszek Ignatowicz, PhD, of Georgia Regents University in Augusta.
But when he and his colleagues limited the cells’ exposure to only one ligand, the same mix of T cells still emerged.
“We asked a simple question, ‘Is it going to affect their development,’ and the answer was ‘no,’” Dr Ignatowicz said. “The cells still mature in the thymus, so something else must be determining it.”
The finding provides more insight into immunity that could one day enable a new approach to vaccines that steer the thymus to produce more of whatever T-cell type a patient needs: more effector cells if they have an infection or cancer, more Tregs if they have autoimmune diseases such as arthritis and multiple sclerosis.
“We could help steer the education process in the desired direction,” Dr Ignatowicz said.
To uncover their findings, he and his colleagues studied two types of mice, each expressing a single ligand in the thymus. The researchers thought one would prompt strong ligand binding and favor Treg development, and the other would favor a weaker ligand bond and effector cell development.
The mix of resulting T cells was the same as if both were exposed to the usual thousands of ligands, although the experiment did reveal one difference.
Ligands—and, eventually, bacteria and other invaders—get the attention of T cells by activating their receptors. Both CD4 T-cell types generally have the same receptors, but they are organized differently.
The researchers found that as long as the binding was weak, as it was in the first mouse, there was a lot of overlap in the receptors the ligand bound to in both T-cell types. However, in the second mouse, where the ligand prompted strong binding, there was far less overlap.
“We are now trying to find what causes that difference,” Dr Ignatowicz said. ![]()
Credit: NIAID
A fundamental theory about how the thymus “educates” T cells appears to be wrong, according to research published in Nature Communications.
It’s known that stem cells leave the bone marrow and travel to the thymus to become one of two CD4 T-cell types: effector cells or regulatory T cells (Tregs).
One widely held concept of why the cells become one type or the other is that they are exposed to different ligands in the thymus, said Leszek Ignatowicz, PhD, of Georgia Regents University in Augusta.
But when he and his colleagues limited the cells’ exposure to only one ligand, the same mix of T cells still emerged.
“We asked a simple question, ‘Is it going to affect their development,’ and the answer was ‘no,’” Dr Ignatowicz said. “The cells still mature in the thymus, so something else must be determining it.”
The finding provides more insight into immunity that could one day enable a new approach to vaccines that steer the thymus to produce more of whatever T-cell type a patient needs: more effector cells if they have an infection or cancer, more Tregs if they have autoimmune diseases such as arthritis and multiple sclerosis.
“We could help steer the education process in the desired direction,” Dr Ignatowicz said.
To uncover their findings, he and his colleagues studied two types of mice, each expressing a single ligand in the thymus. The researchers thought one would prompt strong ligand binding and favor Treg development, and the other would favor a weaker ligand bond and effector cell development.
The mix of resulting T cells was the same as if both were exposed to the usual thousands of ligands, although the experiment did reveal one difference.
Ligands—and, eventually, bacteria and other invaders—get the attention of T cells by activating their receptors. Both CD4 T-cell types generally have the same receptors, but they are organized differently.
The researchers found that as long as the binding was weak, as it was in the first mouse, there was a lot of overlap in the receptors the ligand bound to in both T-cell types. However, in the second mouse, where the ligand prompted strong binding, there was far less overlap.
“We are now trying to find what causes that difference,” Dr Ignatowicz said. ![]()
Shorter regimen can prevent GVHD
![]()
Credit: Chad McNeeley
Combining a couple of “promising” treatment approaches can prevent graft-vs-host disease (GVHD) as well as conventional therapy, researchers have reported in the Journal of Clinical Oncology.
They combined a 4-day myeloablative conditioning regimen of busulfan and fludarabine with 2 days of high-dose cyclophosphamide after transplant.
Typically, patients receive 6 months of immunosuppressive therapy to reduce their risk of GVHD.
The conditioning regimen and post-transplant cyclophosphamide have been tested separately in other studies and have good track records in controlling cancer and preventing severe GVHD.
Those successes led Leo Luznik, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues to combine the 2 therapies.
The team tested the combination in 92 patients with high-risk hematologic malignancies. Diagnoses included acute and chronic leukemias, multiple myeloma, non-Hodgkin lymphoma, and myelodysplastic syndromes. Patients had a median age of 49 (range, 21-65).
All patients received 40 mg/m2/day of intravenous (IV) fludarabine immediately before busulfan on all 4 days of conditioning. The busulfan dose of 130 mg/m2 IV daily was adjusted based on pharmacokinetics.
One or 2 days of rest were allowed before patients received a T-cell-replete bone marrow allograft. Forty-five patients had a matched, related donor, and 47 had a matched, unrelated donor.
Patients received 50 mg/kg/day of IV cyclophosphamide for 2 days, with the first dose starting 62 to 72 hours after the start of allograft infusion.
At 100 days after transplant, 51% of patients had developed grade 2-4 acute GVHD, and 15% had grade 3-4 acute GVHD. Fourteen percent of patients developed chronic GVHD.
The 2-year overall survival rate was 67%, and 2-year event-free survival was 62%.
Dr Luznik said he was encouraged by the low rate of chronic GVHD with the regimen. And he noted that percentages of acute GVHD are similar to those seen with the standard 6-month regimen of immunosuppressive drugs.
Reducing the post-transplant treatment to 2 days with cyclophosphamide, he said, “also allows for the earlier integration of other treatments.”
For example, immunotherapies used to eradicate any remaining cancer could be started much sooner with this regimen, said study author Christopher Kanakry, MD, of the Sidney Kimmel Cancer Center at Johns Hopkins.
“If you give patients immune cells to eradicate any remaining cancer cells that might be present,” he said, “those immune cells would not be prevented from doing their job by ongoing immune suppression drugs that are being used in patients treated with conventional transplant approaches.”
Dr Luznik said the researchers’ next step will be a phase 3 trial comparing this regimen to another experimental approach to prevent GVHD or to the more traditional 6-month immunosuppressive therapy.
Funding for this study was provided by Otsuka Pharmaceutical Co., Ltd. and the National Institutes of Health. ![]()
![]()
Credit: Chad McNeeley
Combining a couple of “promising” treatment approaches can prevent graft-vs-host disease (GVHD) as well as conventional therapy, researchers have reported in the Journal of Clinical Oncology.
They combined a 4-day myeloablative conditioning regimen of busulfan and fludarabine with 2 days of high-dose cyclophosphamide after transplant.
Typically, patients receive 6 months of immunosuppressive therapy to reduce their risk of GVHD.
The conditioning regimen and post-transplant cyclophosphamide have been tested separately in other studies and have good track records in controlling cancer and preventing severe GVHD.
Those successes led Leo Luznik, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues to combine the 2 therapies.
The team tested the combination in 92 patients with high-risk hematologic malignancies. Diagnoses included acute and chronic leukemias, multiple myeloma, non-Hodgkin lymphoma, and myelodysplastic syndromes. Patients had a median age of 49 (range, 21-65).
All patients received 40 mg/m2/day of intravenous (IV) fludarabine immediately before busulfan on all 4 days of conditioning. The busulfan dose of 130 mg/m2 IV daily was adjusted based on pharmacokinetics.
One or 2 days of rest were allowed before patients received a T-cell-replete bone marrow allograft. Forty-five patients had a matched, related donor, and 47 had a matched, unrelated donor.
Patients received 50 mg/kg/day of IV cyclophosphamide for 2 days, with the first dose starting 62 to 72 hours after the start of allograft infusion.
At 100 days after transplant, 51% of patients had developed grade 2-4 acute GVHD, and 15% had grade 3-4 acute GVHD. Fourteen percent of patients developed chronic GVHD.
The 2-year overall survival rate was 67%, and 2-year event-free survival was 62%.
Dr Luznik said he was encouraged by the low rate of chronic GVHD with the regimen. And he noted that percentages of acute GVHD are similar to those seen with the standard 6-month regimen of immunosuppressive drugs.
Reducing the post-transplant treatment to 2 days with cyclophosphamide, he said, “also allows for the earlier integration of other treatments.”
For example, immunotherapies used to eradicate any remaining cancer could be started much sooner with this regimen, said study author Christopher Kanakry, MD, of the Sidney Kimmel Cancer Center at Johns Hopkins.
“If you give patients immune cells to eradicate any remaining cancer cells that might be present,” he said, “those immune cells would not be prevented from doing their job by ongoing immune suppression drugs that are being used in patients treated with conventional transplant approaches.”
Dr Luznik said the researchers’ next step will be a phase 3 trial comparing this regimen to another experimental approach to prevent GVHD or to the more traditional 6-month immunosuppressive therapy.
Funding for this study was provided by Otsuka Pharmaceutical Co., Ltd. and the National Institutes of Health. ![]()
![]()
Credit: Chad McNeeley
Combining a couple of “promising” treatment approaches can prevent graft-vs-host disease (GVHD) as well as conventional therapy, researchers have reported in the Journal of Clinical Oncology.
They combined a 4-day myeloablative conditioning regimen of busulfan and fludarabine with 2 days of high-dose cyclophosphamide after transplant.
Typically, patients receive 6 months of immunosuppressive therapy to reduce their risk of GVHD.
The conditioning regimen and post-transplant cyclophosphamide have been tested separately in other studies and have good track records in controlling cancer and preventing severe GVHD.
Those successes led Leo Luznik, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues to combine the 2 therapies.
The team tested the combination in 92 patients with high-risk hematologic malignancies. Diagnoses included acute and chronic leukemias, multiple myeloma, non-Hodgkin lymphoma, and myelodysplastic syndromes. Patients had a median age of 49 (range, 21-65).
All patients received 40 mg/m2/day of intravenous (IV) fludarabine immediately before busulfan on all 4 days of conditioning. The busulfan dose of 130 mg/m2 IV daily was adjusted based on pharmacokinetics.
One or 2 days of rest were allowed before patients received a T-cell-replete bone marrow allograft. Forty-five patients had a matched, related donor, and 47 had a matched, unrelated donor.
Patients received 50 mg/kg/day of IV cyclophosphamide for 2 days, with the first dose starting 62 to 72 hours after the start of allograft infusion.
At 100 days after transplant, 51% of patients had developed grade 2-4 acute GVHD, and 15% had grade 3-4 acute GVHD. Fourteen percent of patients developed chronic GVHD.
The 2-year overall survival rate was 67%, and 2-year event-free survival was 62%.
Dr Luznik said he was encouraged by the low rate of chronic GVHD with the regimen. And he noted that percentages of acute GVHD are similar to those seen with the standard 6-month regimen of immunosuppressive drugs.
Reducing the post-transplant treatment to 2 days with cyclophosphamide, he said, “also allows for the earlier integration of other treatments.”
For example, immunotherapies used to eradicate any remaining cancer could be started much sooner with this regimen, said study author Christopher Kanakry, MD, of the Sidney Kimmel Cancer Center at Johns Hopkins.
“If you give patients immune cells to eradicate any remaining cancer cells that might be present,” he said, “those immune cells would not be prevented from doing their job by ongoing immune suppression drugs that are being used in patients treated with conventional transplant approaches.”
Dr Luznik said the researchers’ next step will be a phase 3 trial comparing this regimen to another experimental approach to prevent GVHD or to the more traditional 6-month immunosuppressive therapy.
Funding for this study was provided by Otsuka Pharmaceutical Co., Ltd. and the National Institutes of Health.
A safer gene therapy for SCID-X1?

Credit: Chad McNeeley
A new gene therapy appears safe and effective for boys with X-linked severe combined immunodeficiency syndrome (SCID-X1).
Early data from a small trial suggest the treatment may not pose a risk of leukemia, which was seen in previous trials of gene therapy in SCID-X1 patients.
The new therapy is a self-inactivating γ-retrovirus designed to deliver its payload while minimizing the chance of inadvertently activating
oncogenes that could lead to leukemia.
Researchers described results with this therapy in The New England Journal of Medicine.
The team enrolled 9 boys with SCID-X1. They received bone marrow-derived CD34+ cells transduced with the self-inactivating γ-chain vector, without preparative conditioning.
Eight boys are still alive after 12.1 to 38.7 months of follow-up, with no SCID-X1-associated infections. One child died of an overwhelming infection that was present at the time gene therapy began.
Gene therapy alone generated functioning immune systems in 7 of the patients.
Genetic studies of the boys’ new T cells revealed that the viral vector did not lead to an expansion of cells with vector insertions near known oncogenes, raising cautious hopes about the vector’s long-term safety.
The researchers said they will continue to monitor the patients for any signs of treatment-related leukemia for 15 years.
In prior European trials, which were the first to demonstrate gene therapy’s potential to successfully cure a disease, leukemia appeared 2 to 5 years after treatment. This outcome was one of several events that, together, slowed clinical progress in gene therapy for many years.
“Our goal was to take the molecular data from the prior trial and use it to produce a vector that would remain effective and, at the same time, reduce the risk of leukemia,” said David A. Williams, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts.
“The efficacy data from our study is clear: The vector does work to correct the disease. And by a surrogate endpoint, we have improved the treatment’s safety, although it’s too early to say that we’ve completely eliminated the long-term risk of leukemia.”
After a single round of treatment, 6 of 7 boys for whom the gene therapy was successful had achieved the trial’s primary efficacy endpoints—a T-cell count greater than 300 cells per microliter of blood and T-cell proliferation in response to stimulation with phytohemagglutinin.
The seventh boy received a second round of gene therapy and remains healthy despite having relatively low T-cell counts. The eighth surviving patient was successfully treated with a conventional hematopoietic stem cell transplant after gene therapy failed to stimulate T-cell production.
Credit: Chad McNeeley
A new gene therapy appears safe and effective for boys with X-linked severe combined immunodeficiency syndrome (SCID-X1).
Early data from a small trial suggest the treatment may not pose a risk of leukemia, which was seen in previous trials of gene therapy in SCID-X1 patients.
The new therapy is a self-inactivating γ-retrovirus designed to deliver its payload while minimizing the chance of inadvertently activating
oncogenes that could lead to leukemia.
Researchers described results with this therapy in The New England Journal of Medicine.
The team enrolled 9 boys with SCID-X1. They received bone marrow-derived CD34+ cells transduced with the self-inactivating γ-chain vector, without preparative conditioning.
Eight boys are still alive after 12.1 to 38.7 months of follow-up, with no SCID-X1-associated infections. One child died of an overwhelming infection that was present at the time gene therapy began.
Gene therapy alone generated functioning immune systems in 7 of the patients.
Genetic studies of the boys’ new T cells revealed that the viral vector did not lead to an expansion of cells with vector insertions near known oncogenes, raising cautious hopes about the vector’s long-term safety.
The researchers said they will continue to monitor the patients for any signs of treatment-related leukemia for 15 years.
In prior European trials, which were the first to demonstrate gene therapy’s potential to successfully cure a disease, leukemia appeared 2 to 5 years after treatment. This outcome was one of several events that, together, slowed clinical progress in gene therapy for many years.
“Our goal was to take the molecular data from the prior trial and use it to produce a vector that would remain effective and, at the same time, reduce the risk of leukemia,” said David A. Williams, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts.
“The efficacy data from our study is clear: The vector does work to correct the disease. And by a surrogate endpoint, we have improved the treatment’s safety, although it’s too early to say that we’ve completely eliminated the long-term risk of leukemia.”
After a single round of treatment, 6 of 7 boys for whom the gene therapy was successful had achieved the trial’s primary efficacy endpoints—a T-cell count greater than 300 cells per microliter of blood and T-cell proliferation in response to stimulation with phytohemagglutinin.
The seventh boy received a second round of gene therapy and remains healthy despite having relatively low T-cell counts. The eighth surviving patient was successfully treated with a conventional hematopoietic stem cell transplant after gene therapy failed to stimulate T-cell production.
Credit: Chad McNeeley
A new gene therapy appears safe and effective for boys with X-linked severe combined immunodeficiency syndrome (SCID-X1).
Early data from a small trial suggest the treatment may not pose a risk of leukemia, which was seen in previous trials of gene therapy in SCID-X1 patients.
The new therapy is a self-inactivating γ-retrovirus designed to deliver its payload while minimizing the chance of inadvertently activating
oncogenes that could lead to leukemia.
Researchers described results with this therapy in The New England Journal of Medicine.
The team enrolled 9 boys with SCID-X1. They received bone marrow-derived CD34+ cells transduced with the self-inactivating γ-chain vector, without preparative conditioning.
Eight boys are still alive after 12.1 to 38.7 months of follow-up, with no SCID-X1-associated infections. One child died of an overwhelming infection that was present at the time gene therapy began.
Gene therapy alone generated functioning immune systems in 7 of the patients.
Genetic studies of the boys’ new T cells revealed that the viral vector did not lead to an expansion of cells with vector insertions near known oncogenes, raising cautious hopes about the vector’s long-term safety.
The researchers said they will continue to monitor the patients for any signs of treatment-related leukemia for 15 years.
In prior European trials, which were the first to demonstrate gene therapy’s potential to successfully cure a disease, leukemia appeared 2 to 5 years after treatment. This outcome was one of several events that, together, slowed clinical progress in gene therapy for many years.
“Our goal was to take the molecular data from the prior trial and use it to produce a vector that would remain effective and, at the same time, reduce the risk of leukemia,” said David A. Williams, MD, of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center in Massachusetts.
“The efficacy data from our study is clear: The vector does work to correct the disease. And by a surrogate endpoint, we have improved the treatment’s safety, although it’s too early to say that we’ve completely eliminated the long-term risk of leukemia.”
After a single round of treatment, 6 of 7 boys for whom the gene therapy was successful had achieved the trial’s primary efficacy endpoints—a T-cell count greater than 300 cells per microliter of blood and T-cell proliferation in response to stimulation with phytohemagglutinin.
The seventh boy received a second round of gene therapy and remains healthy despite having relatively low T-cell counts. The eighth surviving patient was successfully treated with a conventional hematopoietic stem cell transplant after gene therapy failed to stimulate T-cell production.
Microscopy advances net Nobel Prize

Credit: Max Planck Institute
for Biophysical Chemistry
Three scientists have received the 2014 Nobel Prize in Chemistry for aiding the development of super-resolved fluorescence microscopy.
For a long time, optical microscopy was held back by a presumed limitation: that it would never obtain a better resolution than half the wavelength
of light.
Working separately, this year’s Nobel Laureates in Chemistry circumvented this limitation and brought optical microscopy into the nanodimension.
Now, scientists can monitor the interplay between individual molecules inside cells, watch disease-related proteins aggregate, and track cell division at the nanolevel.
For enabling these advances, Eric Betzig, PhD, of the Howard Hughes Medical Institute in Ashburn, Virginia; Stefan W. Hell, PhD, of the Max Planck Institute for Biophysical Chemistry in Göttingen, Germany; and William E. Moerner, PhD, of Stanford University in California, received the prize. The prize amount was SEK 8 million, to be shared equally among the Laureates.
The work in brief
In 1873, the microscopist Ernst Abbe stipulated a physical limit for the maximum resolution of traditional optical microscopy—0.2 micrometers. Drs Moerner, Hell, and Betzig were able to bypass this limit.
Dr Hell developed stimulated emission depletion (STED) microscopy. This method employs 2 laser beams. One stimulates fluorescent molecules to glow, and another cancels out all fluorescence except for that in a nanometer-sized volume.
Scanning over the sample, nanometer for nanometer, yields an image with a resolution better than Abbe’s stipulated limit.
Drs Betzig and Moerner laid the foundation for another method, single-molecule microscopy. This method relies upon the possibility to turn the fluorescence of individual molecules on and off.
Scientists image the same area multiple times, letting just a few interspersed molecules glow each time. Superimposing these images yields a dense super-image resolved at the nanolevel. In 2006, Dr Betzig used this method for the first time.
For more details on the Nobel Laureates and their work, visit Nobelprize.org.
Credit: Max Planck Institute
for Biophysical Chemistry
Three scientists have received the 2014 Nobel Prize in Chemistry for aiding the development of super-resolved fluorescence microscopy.
For a long time, optical microscopy was held back by a presumed limitation: that it would never obtain a better resolution than half the wavelength
of light.
Working separately, this year’s Nobel Laureates in Chemistry circumvented this limitation and brought optical microscopy into the nanodimension.
Now, scientists can monitor the interplay between individual molecules inside cells, watch disease-related proteins aggregate, and track cell division at the nanolevel.
For enabling these advances, Eric Betzig, PhD, of the Howard Hughes Medical Institute in Ashburn, Virginia; Stefan W. Hell, PhD, of the Max Planck Institute for Biophysical Chemistry in Göttingen, Germany; and William E. Moerner, PhD, of Stanford University in California, received the prize. The prize amount was SEK 8 million, to be shared equally among the Laureates.
The work in brief
In 1873, the microscopist Ernst Abbe stipulated a physical limit for the maximum resolution of traditional optical microscopy—0.2 micrometers. Drs Moerner, Hell, and Betzig were able to bypass this limit.
Dr Hell developed stimulated emission depletion (STED) microscopy. This method employs 2 laser beams. One stimulates fluorescent molecules to glow, and another cancels out all fluorescence except for that in a nanometer-sized volume.
Scanning over the sample, nanometer for nanometer, yields an image with a resolution better than Abbe’s stipulated limit.
Drs Betzig and Moerner laid the foundation for another method, single-molecule microscopy. This method relies upon the possibility to turn the fluorescence of individual molecules on and off.
Scientists image the same area multiple times, letting just a few interspersed molecules glow each time. Superimposing these images yields a dense super-image resolved at the nanolevel. In 2006, Dr Betzig used this method for the first time.
For more details on the Nobel Laureates and their work, visit Nobelprize.org.
Credit: Max Planck Institute
for Biophysical Chemistry
Three scientists have received the 2014 Nobel Prize in Chemistry for aiding the development of super-resolved fluorescence microscopy.
For a long time, optical microscopy was held back by a presumed limitation: that it would never obtain a better resolution than half the wavelength
of light.
Working separately, this year’s Nobel Laureates in Chemistry circumvented this limitation and brought optical microscopy into the nanodimension.
Now, scientists can monitor the interplay between individual molecules inside cells, watch disease-related proteins aggregate, and track cell division at the nanolevel.
For enabling these advances, Eric Betzig, PhD, of the Howard Hughes Medical Institute in Ashburn, Virginia; Stefan W. Hell, PhD, of the Max Planck Institute for Biophysical Chemistry in Göttingen, Germany; and William E. Moerner, PhD, of Stanford University in California, received the prize. The prize amount was SEK 8 million, to be shared equally among the Laureates.
The work in brief
In 1873, the microscopist Ernst Abbe stipulated a physical limit for the maximum resolution of traditional optical microscopy—0.2 micrometers. Drs Moerner, Hell, and Betzig were able to bypass this limit.
Dr Hell developed stimulated emission depletion (STED) microscopy. This method employs 2 laser beams. One stimulates fluorescent molecules to glow, and another cancels out all fluorescence except for that in a nanometer-sized volume.
Scanning over the sample, nanometer for nanometer, yields an image with a resolution better than Abbe’s stipulated limit.
Drs Betzig and Moerner laid the foundation for another method, single-molecule microscopy. This method relies upon the possibility to turn the fluorescence of individual molecules on and off.
Scientists image the same area multiple times, letting just a few interspersed molecules glow each time. Superimposing these images yields a dense super-image resolved at the nanolevel. In 2006, Dr Betzig used this method for the first time.
For more details on the Nobel Laureates and their work, visit Nobelprize.org.
Price increases drive spending on cancer drugs

Credit: Steven Harbour
The recent surge in spending on oral anticancer drugs in the US exceeds the increase in use of these drugs, new research shows.
Average quarterly national spending on oral oncologics increased 37% during the period studied, from $940 million in the first quarter of 2006 to $1.4 billion in the third quarter of 2011.
But the average quarterly use of these drugs in the same time period increased by only 10%.
This suggests price increases are a significant driver of spending trends.
Rena M. Conti, PhD, of the University of Chicago in Illinois, and her colleagues examined recent trends in spending and use of oral oncologics and disclosed their findings in Health Affairs.
Of the 47 drugs analyzed, most were targeted agents (30%), hormonal agents (26%), and alkylating agents (19%).
The researchers observed a significant increase in national spending on oral oncologics from 2006 to 2011—an estimated average quarterly increase of $20 million.
This was driven by brand-name, patent-protected drugs, but the use of these drugs climbed a comparatively small amount. Average quarterly spending of patent-protected drugs increased 61%, and average quarterly use increased 30% between 2006 and the period from September 2010 to September 2011.
“This is an exciting time, an era of breakthrough cancer drugs,” Dr Conti said. “Some of these medications have extended the lives of many people with certain types of cancer. However, spending on these brand-name oral oncologics is outstripping national spending on all pharmaceuticals and all medical care spending generally.”
The researchers also discovered that when oncologics lose patent protection, spending takes a nosedive. The use of newly off-patent drugs increased by 16%, but average quarterly spending on those drugs fell by 65%.
Another finding was that US spending on targeted anticancer agents increased from 35% of all oral cancer drugs in 2006 to nearly 60% in 2011.
Meanwhile, spending on hormonal agents decreased from 42% of total spending to 19%, spending on antimetabolites increased from 11% to 12%, and spending on alkylating agents decreased from 10% to 8%.
Credit: Steven Harbour
The recent surge in spending on oral anticancer drugs in the US exceeds the increase in use of these drugs, new research shows.
Average quarterly national spending on oral oncologics increased 37% during the period studied, from $940 million in the first quarter of 2006 to $1.4 billion in the third quarter of 2011.
But the average quarterly use of these drugs in the same time period increased by only 10%.
This suggests price increases are a significant driver of spending trends.
Rena M. Conti, PhD, of the University of Chicago in Illinois, and her colleagues examined recent trends in spending and use of oral oncologics and disclosed their findings in Health Affairs.
Of the 47 drugs analyzed, most were targeted agents (30%), hormonal agents (26%), and alkylating agents (19%).
The researchers observed a significant increase in national spending on oral oncologics from 2006 to 2011—an estimated average quarterly increase of $20 million.
This was driven by brand-name, patent-protected drugs, but the use of these drugs climbed a comparatively small amount. Average quarterly spending of patent-protected drugs increased 61%, and average quarterly use increased 30% between 2006 and the period from September 2010 to September 2011.
“This is an exciting time, an era of breakthrough cancer drugs,” Dr Conti said. “Some of these medications have extended the lives of many people with certain types of cancer. However, spending on these brand-name oral oncologics is outstripping national spending on all pharmaceuticals and all medical care spending generally.”
The researchers also discovered that when oncologics lose patent protection, spending takes a nosedive. The use of newly off-patent drugs increased by 16%, but average quarterly spending on those drugs fell by 65%.
Another finding was that US spending on targeted anticancer agents increased from 35% of all oral cancer drugs in 2006 to nearly 60% in 2011.
Meanwhile, spending on hormonal agents decreased from 42% of total spending to 19%, spending on antimetabolites increased from 11% to 12%, and spending on alkylating agents decreased from 10% to 8%.
Credit: Steven Harbour
The recent surge in spending on oral anticancer drugs in the US exceeds the increase in use of these drugs, new research shows.
Average quarterly national spending on oral oncologics increased 37% during the period studied, from $940 million in the first quarter of 2006 to $1.4 billion in the third quarter of 2011.
But the average quarterly use of these drugs in the same time period increased by only 10%.
This suggests price increases are a significant driver of spending trends.
Rena M. Conti, PhD, of the University of Chicago in Illinois, and her colleagues examined recent trends in spending and use of oral oncologics and disclosed their findings in Health Affairs.
Of the 47 drugs analyzed, most were targeted agents (30%), hormonal agents (26%), and alkylating agents (19%).
The researchers observed a significant increase in national spending on oral oncologics from 2006 to 2011—an estimated average quarterly increase of $20 million.
This was driven by brand-name, patent-protected drugs, but the use of these drugs climbed a comparatively small amount. Average quarterly spending of patent-protected drugs increased 61%, and average quarterly use increased 30% between 2006 and the period from September 2010 to September 2011.
“This is an exciting time, an era of breakthrough cancer drugs,” Dr Conti said. “Some of these medications have extended the lives of many people with certain types of cancer. However, spending on these brand-name oral oncologics is outstripping national spending on all pharmaceuticals and all medical care spending generally.”
The researchers also discovered that when oncologics lose patent protection, spending takes a nosedive. The use of newly off-patent drugs increased by 16%, but average quarterly spending on those drugs fell by 65%.
Another finding was that US spending on targeted anticancer agents increased from 35% of all oral cancer drugs in 2006 to nearly 60% in 2011.
Meanwhile, spending on hormonal agents decreased from 42% of total spending to 19%, spending on antimetabolites increased from 11% to 12%, and spending on alkylating agents decreased from 10% to 8%.