User login
Armored CAR T cells next on the production line

NEW YORK—Chimeric antigen receptor (CAR) T cells have “remarkable” activity, according to a speaker at the NCCN 9th Annual Congress: Hematologic Malignancies.
“[T]his chimera binds like an antibody, but it acts like a T cell, so it combines the best of both worlds,” said Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York.
He then traced the evolution of CAR T-cell design, discussed clinical trials using CD19-targed T cells, and described how investigators are working at building a better T cell.
Researchers found that T-cell activation and proliferation require signaling through a costimulatory receptor, such as CD28, 4-1BB, or OX-40. Without costimulation, the T cell becomes unresponsive or undergoes apoptosis.
So based on this observation, Dr Park said, several research groups created second- and third-generation CARs to incorporate the costimulatory signal.
The first generation was typically fused to the CD8 domain. Second-generation CARs include a costimulatory signaling domain, such as CD28, 4-1BB, or OX40. And the third generation contains signaling domains from 2 costimulatory receptors, CD28 with 4-1BB and CD28 with OX40.
The built-in costimulatory signal proved superior to the first-generation CAR T cells.
In NOD/SCID mice inoculated with NALM-6 lymphoma cells, Dr Park said, about 50% more were “cured,” in terms of survival, using a CD80 costimulatory ligand with CD19-targeted T cells compared to those without the ligand.
Clinical trials
Clinical trials using second-generation CD19-targeted T cells in relapsed B-cell acute lymphoblastic leukemia (ALL) at MSKCC produced an overall complete response (CR) rate of 88% in a median of 22.1 days. And 72% of the CRs were minimal residual disease (MRD) negative.
So the CAR T cells produce a “very rapid and deep remission,” Dr Park said.
CAR T-cell therapy, however, comes with adverse events, most notably, cytokine release syndrome (CRS), which results from T-cell activation. CRS causes fevers, hypotension, and neurologic toxicities including mental status changes, obtundation, and seizures.
“CRS is not unique to CAR T-cell therapy,” Dr Park said. “Any therapy that activates T cells can have this type of side effect.”
Dr Park noted that CRS is associated with disease burden at the time of treatment. “The larger the disease burden pre T-cell therapy,” he said, “the more likely [patients are] to develop CRS.”
In the MSKCC trial, no patient with very low disease burden—5% blasts in the bone marrow—developed CRS.
However, there is also a correlation between tumor burden and T-cell expansion, he added. T cells expand much better with a larger disease burden, because there is a greater antigen load.
The investigators found that serum C-reactive protein can serve as a surrogate marker for the severity of CRS. Patients with levels above 20 mg/dL are more likely to experience CRS.
And Dr Park pointed out that CRS symptoms respond pretty rapidly to steroids or interleukin-6 receptor blockade.
CAR T-cell therapy has also been used to treat chronic lymphocytic leukemia, but with much more modest response rates than in ALL. Both University of Pennsylvania and MSKCC trials in CLL have produced overall response rates around 40%.
Building a better T cell
Dr Park described efforts underway to develop the fourth-generation “armored” CAR T cells to overcome the hostile tumor microenvironment, which contains multiple inhibitory factors designed to suppress effector T cells.
Armored T cells can actually secrete some of the inflammatory cytokines to change the tumor microenvironment and overcome the inhibitory effect.
Dr Park described a potential scenario: The armored CAR T cells secrete IL-12, enhance the central memory phenotype, enhance cytotoxicity, enhance persistence, modify the endogenous immune system and T-cell activation, and reactivate tumor-infiltrating lymphocytes.
He said future studies will focus on translation of these armored CAR T cells to the clinical setting in both hematologic and solid tumor malignancies. ![]()
NEW YORK—Chimeric antigen receptor (CAR) T cells have “remarkable” activity, according to a speaker at the NCCN 9th Annual Congress: Hematologic Malignancies.
“[T]his chimera binds like an antibody, but it acts like a T cell, so it combines the best of both worlds,” said Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York.
He then traced the evolution of CAR T-cell design, discussed clinical trials using CD19-targed T cells, and described how investigators are working at building a better T cell.
Researchers found that T-cell activation and proliferation require signaling through a costimulatory receptor, such as CD28, 4-1BB, or OX-40. Without costimulation, the T cell becomes unresponsive or undergoes apoptosis.
So based on this observation, Dr Park said, several research groups created second- and third-generation CARs to incorporate the costimulatory signal.
The first generation was typically fused to the CD8 domain. Second-generation CARs include a costimulatory signaling domain, such as CD28, 4-1BB, or OX40. And the third generation contains signaling domains from 2 costimulatory receptors, CD28 with 4-1BB and CD28 with OX40.
The built-in costimulatory signal proved superior to the first-generation CAR T cells.
In NOD/SCID mice inoculated with NALM-6 lymphoma cells, Dr Park said, about 50% more were “cured,” in terms of survival, using a CD80 costimulatory ligand with CD19-targeted T cells compared to those without the ligand.
Clinical trials
Clinical trials using second-generation CD19-targeted T cells in relapsed B-cell acute lymphoblastic leukemia (ALL) at MSKCC produced an overall complete response (CR) rate of 88% in a median of 22.1 days. And 72% of the CRs were minimal residual disease (MRD) negative.
So the CAR T cells produce a “very rapid and deep remission,” Dr Park said.
CAR T-cell therapy, however, comes with adverse events, most notably, cytokine release syndrome (CRS), which results from T-cell activation. CRS causes fevers, hypotension, and neurologic toxicities including mental status changes, obtundation, and seizures.
“CRS is not unique to CAR T-cell therapy,” Dr Park said. “Any therapy that activates T cells can have this type of side effect.”
Dr Park noted that CRS is associated with disease burden at the time of treatment. “The larger the disease burden pre T-cell therapy,” he said, “the more likely [patients are] to develop CRS.”
In the MSKCC trial, no patient with very low disease burden—5% blasts in the bone marrow—developed CRS.
However, there is also a correlation between tumor burden and T-cell expansion, he added. T cells expand much better with a larger disease burden, because there is a greater antigen load.
The investigators found that serum C-reactive protein can serve as a surrogate marker for the severity of CRS. Patients with levels above 20 mg/dL are more likely to experience CRS.
And Dr Park pointed out that CRS symptoms respond pretty rapidly to steroids or interleukin-6 receptor blockade.
CAR T-cell therapy has also been used to treat chronic lymphocytic leukemia, but with much more modest response rates than in ALL. Both University of Pennsylvania and MSKCC trials in CLL have produced overall response rates around 40%.
Building a better T cell
Dr Park described efforts underway to develop the fourth-generation “armored” CAR T cells to overcome the hostile tumor microenvironment, which contains multiple inhibitory factors designed to suppress effector T cells.
Armored T cells can actually secrete some of the inflammatory cytokines to change the tumor microenvironment and overcome the inhibitory effect.
Dr Park described a potential scenario: The armored CAR T cells secrete IL-12, enhance the central memory phenotype, enhance cytotoxicity, enhance persistence, modify the endogenous immune system and T-cell activation, and reactivate tumor-infiltrating lymphocytes.
He said future studies will focus on translation of these armored CAR T cells to the clinical setting in both hematologic and solid tumor malignancies. ![]()
NEW YORK—Chimeric antigen receptor (CAR) T cells have “remarkable” activity, according to a speaker at the NCCN 9th Annual Congress: Hematologic Malignancies.
“[T]his chimera binds like an antibody, but it acts like a T cell, so it combines the best of both worlds,” said Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York.
He then traced the evolution of CAR T-cell design, discussed clinical trials using CD19-targed T cells, and described how investigators are working at building a better T cell.
Researchers found that T-cell activation and proliferation require signaling through a costimulatory receptor, such as CD28, 4-1BB, or OX-40. Without costimulation, the T cell becomes unresponsive or undergoes apoptosis.
So based on this observation, Dr Park said, several research groups created second- and third-generation CARs to incorporate the costimulatory signal.
The first generation was typically fused to the CD8 domain. Second-generation CARs include a costimulatory signaling domain, such as CD28, 4-1BB, or OX40. And the third generation contains signaling domains from 2 costimulatory receptors, CD28 with 4-1BB and CD28 with OX40.
The built-in costimulatory signal proved superior to the first-generation CAR T cells.
In NOD/SCID mice inoculated with NALM-6 lymphoma cells, Dr Park said, about 50% more were “cured,” in terms of survival, using a CD80 costimulatory ligand with CD19-targeted T cells compared to those without the ligand.
Clinical trials
Clinical trials using second-generation CD19-targeted T cells in relapsed B-cell acute lymphoblastic leukemia (ALL) at MSKCC produced an overall complete response (CR) rate of 88% in a median of 22.1 days. And 72% of the CRs were minimal residual disease (MRD) negative.
So the CAR T cells produce a “very rapid and deep remission,” Dr Park said.
CAR T-cell therapy, however, comes with adverse events, most notably, cytokine release syndrome (CRS), which results from T-cell activation. CRS causes fevers, hypotension, and neurologic toxicities including mental status changes, obtundation, and seizures.
“CRS is not unique to CAR T-cell therapy,” Dr Park said. “Any therapy that activates T cells can have this type of side effect.”
Dr Park noted that CRS is associated with disease burden at the time of treatment. “The larger the disease burden pre T-cell therapy,” he said, “the more likely [patients are] to develop CRS.”
In the MSKCC trial, no patient with very low disease burden—5% blasts in the bone marrow—developed CRS.
However, there is also a correlation between tumor burden and T-cell expansion, he added. T cells expand much better with a larger disease burden, because there is a greater antigen load.
The investigators found that serum C-reactive protein can serve as a surrogate marker for the severity of CRS. Patients with levels above 20 mg/dL are more likely to experience CRS.
And Dr Park pointed out that CRS symptoms respond pretty rapidly to steroids or interleukin-6 receptor blockade.
CAR T-cell therapy has also been used to treat chronic lymphocytic leukemia, but with much more modest response rates than in ALL. Both University of Pennsylvania and MSKCC trials in CLL have produced overall response rates around 40%.
Building a better T cell
Dr Park described efforts underway to develop the fourth-generation “armored” CAR T cells to overcome the hostile tumor microenvironment, which contains multiple inhibitory factors designed to suppress effector T cells.
Armored T cells can actually secrete some of the inflammatory cytokines to change the tumor microenvironment and overcome the inhibitory effect.
Dr Park described a potential scenario: The armored CAR T cells secrete IL-12, enhance the central memory phenotype, enhance cytotoxicity, enhance persistence, modify the endogenous immune system and T-cell activation, and reactivate tumor-infiltrating lymphocytes.
He said future studies will focus on translation of these armored CAR T cells to the clinical setting in both hematologic and solid tumor malignancies. ![]()
Bacterium could help control malaria, dengue

Credit: CDC
A bacterium isolated from a mosquito’s gut could aid the fight against malaria and dengue, according to a study published in PLOS Pathogens.
With previous research, scientists isolated Csp_P, a member of the family of chromobacteria, from the gut of Aedes aegypti mosquitoes.
Now, the team has found that Csp_P can directly inhibit malaria and dengue pathogens in vitro and shorten the life span of the mosquitoes that transmit both diseases.
George Dimopoulos, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues examined Csp_P’s actions on both mosquitoes and pathogens, and the results suggest that Csp_P might help to fight malaria and dengue at different levels.
The researchers added Csp_P to sugar water fed to mosquitoes and found that the bacteria are able to quickly colonize the gut of the two most important mosquito disease vectors—Aedes aegypti and Anopheles gambiae.
Moreover, the presence of Csp_P in the gut reduced the susceptibility of the respective mosquitoes to infection with the malaria parasite Plasmodium falciparum or with dengue virus.
Even without gut colonization, exposure to Csp_P through food or breeding water shortened the lifespan of adult mosquitoes and mosquito larvae of both species.
When the researchers tested whether Csp_P could act against the malaria or dengue pathogens directly, they found that the bacterium, likely through the production of toxic metabolites, can inhibit the growth of Plasmodium at various stages during the parasite’s life cycle and also abolish dengue virus infectivity.
The team said these toxic metabolites could potentially be developed into drugs to treat malaria and dengue.
Overall, the researchers concluded that Csp_P’s broad-spectrum antipathogen properties and ability to kill mosquitoes make it a good candidate for the development of novel control strategies for malaria and dengue, so it warrants further study. ![]()
Credit: CDC
A bacterium isolated from a mosquito’s gut could aid the fight against malaria and dengue, according to a study published in PLOS Pathogens.
With previous research, scientists isolated Csp_P, a member of the family of chromobacteria, from the gut of Aedes aegypti mosquitoes.
Now, the team has found that Csp_P can directly inhibit malaria and dengue pathogens in vitro and shorten the life span of the mosquitoes that transmit both diseases.
George Dimopoulos, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues examined Csp_P’s actions on both mosquitoes and pathogens, and the results suggest that Csp_P might help to fight malaria and dengue at different levels.
The researchers added Csp_P to sugar water fed to mosquitoes and found that the bacteria are able to quickly colonize the gut of the two most important mosquito disease vectors—Aedes aegypti and Anopheles gambiae.
Moreover, the presence of Csp_P in the gut reduced the susceptibility of the respective mosquitoes to infection with the malaria parasite Plasmodium falciparum or with dengue virus.
Even without gut colonization, exposure to Csp_P through food or breeding water shortened the lifespan of adult mosquitoes and mosquito larvae of both species.
When the researchers tested whether Csp_P could act against the malaria or dengue pathogens directly, they found that the bacterium, likely through the production of toxic metabolites, can inhibit the growth of Plasmodium at various stages during the parasite’s life cycle and also abolish dengue virus infectivity.
The team said these toxic metabolites could potentially be developed into drugs to treat malaria and dengue.
Overall, the researchers concluded that Csp_P’s broad-spectrum antipathogen properties and ability to kill mosquitoes make it a good candidate for the development of novel control strategies for malaria and dengue, so it warrants further study. ![]()
Credit: CDC
A bacterium isolated from a mosquito’s gut could aid the fight against malaria and dengue, according to a study published in PLOS Pathogens.
With previous research, scientists isolated Csp_P, a member of the family of chromobacteria, from the gut of Aedes aegypti mosquitoes.
Now, the team has found that Csp_P can directly inhibit malaria and dengue pathogens in vitro and shorten the life span of the mosquitoes that transmit both diseases.
George Dimopoulos, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues examined Csp_P’s actions on both mosquitoes and pathogens, and the results suggest that Csp_P might help to fight malaria and dengue at different levels.
The researchers added Csp_P to sugar water fed to mosquitoes and found that the bacteria are able to quickly colonize the gut of the two most important mosquito disease vectors—Aedes aegypti and Anopheles gambiae.
Moreover, the presence of Csp_P in the gut reduced the susceptibility of the respective mosquitoes to infection with the malaria parasite Plasmodium falciparum or with dengue virus.
Even without gut colonization, exposure to Csp_P through food or breeding water shortened the lifespan of adult mosquitoes and mosquito larvae of both species.
When the researchers tested whether Csp_P could act against the malaria or dengue pathogens directly, they found that the bacterium, likely through the production of toxic metabolites, can inhibit the growth of Plasmodium at various stages during the parasite’s life cycle and also abolish dengue virus infectivity.
The team said these toxic metabolites could potentially be developed into drugs to treat malaria and dengue.
Overall, the researchers concluded that Csp_P’s broad-spectrum antipathogen properties and ability to kill mosquitoes make it a good candidate for the development of novel control strategies for malaria and dengue, so it warrants further study. ![]()
Bortezomib can treat chronic GVHD, study shows
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Team creates functional vascular grafts in a week
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
CHMP says ponatinib’s benefits outweigh risks

Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
FDA approves treatment for acquired hemophilia A

The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()
The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()
The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()
Insured cancer patients forgo care, scrimp to get by

cancer patient and her father
Credit: Rhoda Baer
Despite having insurance, many US cancer patients may forgo medical care and change their lifestyles due to treatment-related financial burdens, a new survey suggests.
Nearly 40% of the 174 patients surveyed adopted medical care-altering strategies, such as skipping recommended testing and treatment.
And nearly 9 out of 10 patients made at least one lifestyle change, such as spending less money on food and clothing or selling their possessions.
Prior research has suggested that about 13% of patients suffer from high out-of-pocket financial burden after they are diagnosed with cancer. According to the American Cancer Society, as many as 20% of Americans with cancer spend their life savings to pay for their care.
“We need a better, more open dialogue between patients and providers about the financial burden associated with cancer care costs,” said lead study author Ryan Nipp, MD, of Dana-Farber Cancer Institute in Boston.
“We found that people use a range of different cost-coping strategies, and we need to engage with patients on their choices and develop screening tools to identify patients who are likely to make potentially harmful decisions about their treatment.”
Dr Nipp presented these findings (abstract 161*) in a presscast in advance of the 2014 Palliative Care in Oncology Symposium, which is taking place October 24-25 at the Westin Boston Waterfront in Boston.
The researchers surveyed 174 patients (median age 67, 96% female, 83% white) undergoing treatment for cancer (85% breast, 4% colorectal, 11% other solid tumors). All patients were insured and had requested financial assistance through a national copay assistance program.
Overall, 89% of participants used at least one lifestyle-altering strategy, and 39% used at least one medical care-altering strategy.
The most common medical care-altering strategies were not filling a prescription (28%) and taking less medication than prescribed (22%). Patients also reported skipping tests (10%), forgoing procedures (8%), and missing doctor’s appointments (6%).
Lifestyle-altering strategies included spending less on leisure activities (78%), spending less on basics like food and clothing (57%), borrowing money (54%), spending savings (50%), selling possessions (18%), and having family members work more (15%).
Younger age, higher education, and shorter time on chemotherapy were all associated with a greater likelihood of adopting lifestyle coping strategies compared to older age, lower education, and longer time on chemotherapy.
Younger patients were also more likely to use care-altering strategies compared to older patients. And lower-income patients used more care-altering strategies than higher-income patients. ![]()
*Data in the abstract differ from data presented.
cancer patient and her father
Credit: Rhoda Baer
Despite having insurance, many US cancer patients may forgo medical care and change their lifestyles due to treatment-related financial burdens, a new survey suggests.
Nearly 40% of the 174 patients surveyed adopted medical care-altering strategies, such as skipping recommended testing and treatment.
And nearly 9 out of 10 patients made at least one lifestyle change, such as spending less money on food and clothing or selling their possessions.
Prior research has suggested that about 13% of patients suffer from high out-of-pocket financial burden after they are diagnosed with cancer. According to the American Cancer Society, as many as 20% of Americans with cancer spend their life savings to pay for their care.
“We need a better, more open dialogue between patients and providers about the financial burden associated with cancer care costs,” said lead study author Ryan Nipp, MD, of Dana-Farber Cancer Institute in Boston.
“We found that people use a range of different cost-coping strategies, and we need to engage with patients on their choices and develop screening tools to identify patients who are likely to make potentially harmful decisions about their treatment.”
Dr Nipp presented these findings (abstract 161*) in a presscast in advance of the 2014 Palliative Care in Oncology Symposium, which is taking place October 24-25 at the Westin Boston Waterfront in Boston.
The researchers surveyed 174 patients (median age 67, 96% female, 83% white) undergoing treatment for cancer (85% breast, 4% colorectal, 11% other solid tumors). All patients were insured and had requested financial assistance through a national copay assistance program.
Overall, 89% of participants used at least one lifestyle-altering strategy, and 39% used at least one medical care-altering strategy.
The most common medical care-altering strategies were not filling a prescription (28%) and taking less medication than prescribed (22%). Patients also reported skipping tests (10%), forgoing procedures (8%), and missing doctor’s appointments (6%).
Lifestyle-altering strategies included spending less on leisure activities (78%), spending less on basics like food and clothing (57%), borrowing money (54%), spending savings (50%), selling possessions (18%), and having family members work more (15%).
Younger age, higher education, and shorter time on chemotherapy were all associated with a greater likelihood of adopting lifestyle coping strategies compared to older age, lower education, and longer time on chemotherapy.
Younger patients were also more likely to use care-altering strategies compared to older patients. And lower-income patients used more care-altering strategies than higher-income patients. ![]()
*Data in the abstract differ from data presented.
cancer patient and her father
Credit: Rhoda Baer
Despite having insurance, many US cancer patients may forgo medical care and change their lifestyles due to treatment-related financial burdens, a new survey suggests.
Nearly 40% of the 174 patients surveyed adopted medical care-altering strategies, such as skipping recommended testing and treatment.
And nearly 9 out of 10 patients made at least one lifestyle change, such as spending less money on food and clothing or selling their possessions.
Prior research has suggested that about 13% of patients suffer from high out-of-pocket financial burden after they are diagnosed with cancer. According to the American Cancer Society, as many as 20% of Americans with cancer spend their life savings to pay for their care.
“We need a better, more open dialogue between patients and providers about the financial burden associated with cancer care costs,” said lead study author Ryan Nipp, MD, of Dana-Farber Cancer Institute in Boston.
“We found that people use a range of different cost-coping strategies, and we need to engage with patients on their choices and develop screening tools to identify patients who are likely to make potentially harmful decisions about their treatment.”
Dr Nipp presented these findings (abstract 161*) in a presscast in advance of the 2014 Palliative Care in Oncology Symposium, which is taking place October 24-25 at the Westin Boston Waterfront in Boston.
The researchers surveyed 174 patients (median age 67, 96% female, 83% white) undergoing treatment for cancer (85% breast, 4% colorectal, 11% other solid tumors). All patients were insured and had requested financial assistance through a national copay assistance program.
Overall, 89% of participants used at least one lifestyle-altering strategy, and 39% used at least one medical care-altering strategy.
The most common medical care-altering strategies were not filling a prescription (28%) and taking less medication than prescribed (22%). Patients also reported skipping tests (10%), forgoing procedures (8%), and missing doctor’s appointments (6%).
Lifestyle-altering strategies included spending less on leisure activities (78%), spending less on basics like food and clothing (57%), borrowing money (54%), spending savings (50%), selling possessions (18%), and having family members work more (15%).
Younger age, higher education, and shorter time on chemotherapy were all associated with a greater likelihood of adopting lifestyle coping strategies compared to older age, lower education, and longer time on chemotherapy.
Younger patients were also more likely to use care-altering strategies compared to older patients. And lower-income patients used more care-altering strategies than higher-income patients.
*Data in the abstract differ from data presented.
NICE supports use of rivaroxaban in ACS

Credit: Andre E.X. Brown
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending rivaroxaban (Xarelto) as an option for preventing atherothrombotic events in patients with acute coronary syndrome (ACS).
The agency is recommending rivaroxaban in combination with aspirin plus clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the draft guidance.
Because rivaroxaban increases the risk of bleeding, NICE recommends that clinicians undertake a careful assessment of a patient’s bleeding risk prior to treatment and ensure patients understand the benefits and risks associated with rivaroxaban.
Furthermore, clinicians should reassess the relative benefits and risks of continuing rivaroxaban treatment no later than 12 months after starting treatment.
Clinical and cost-effectiveness
An independent appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
“The committee therefore recommended rivaroxaban as a cost-effective use of [National Health Service] resources,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
The committee noted that, according to Bayer Healthcare (makers of rivaroxaban), the base case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base case estimate was £5622 per QALY gained.
The committee acknowledged that there is uncertainty about the validity of the results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts.
The draft guidance for rivaroxaban in ACS can be found on the NICE website. The closing date for comments is November 13, 2014.
Credit: Andre E.X. Brown
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending rivaroxaban (Xarelto) as an option for preventing atherothrombotic events in patients with acute coronary syndrome (ACS).
The agency is recommending rivaroxaban in combination with aspirin plus clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the draft guidance.
Because rivaroxaban increases the risk of bleeding, NICE recommends that clinicians undertake a careful assessment of a patient’s bleeding risk prior to treatment and ensure patients understand the benefits and risks associated with rivaroxaban.
Furthermore, clinicians should reassess the relative benefits and risks of continuing rivaroxaban treatment no later than 12 months after starting treatment.
Clinical and cost-effectiveness
An independent appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
“The committee therefore recommended rivaroxaban as a cost-effective use of [National Health Service] resources,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
The committee noted that, according to Bayer Healthcare (makers of rivaroxaban), the base case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base case estimate was £5622 per QALY gained.
The committee acknowledged that there is uncertainty about the validity of the results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts.
The draft guidance for rivaroxaban in ACS can be found on the NICE website. The closing date for comments is November 13, 2014.
Credit: Andre E.X. Brown
The UK’s National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending rivaroxaban (Xarelto) as an option for preventing atherothrombotic events in patients with acute coronary syndrome (ACS).
The agency is recommending rivaroxaban in combination with aspirin plus clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the draft guidance.
Because rivaroxaban increases the risk of bleeding, NICE recommends that clinicians undertake a careful assessment of a patient’s bleeding risk prior to treatment and ensure patients understand the benefits and risks associated with rivaroxaban.
Furthermore, clinicians should reassess the relative benefits and risks of continuing rivaroxaban treatment no later than 12 months after starting treatment.
Clinical and cost-effectiveness
An independent appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
“The committee therefore recommended rivaroxaban as a cost-effective use of [National Health Service] resources,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
The committee noted that, according to Bayer Healthcare (makers of rivaroxaban), the base case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base case estimate was £5622 per QALY gained.
The committee acknowledged that there is uncertainty about the validity of the results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts.
The draft guidance for rivaroxaban in ACS can be found on the NICE website. The closing date for comments is November 13, 2014.
Managing CNS relapse in cardiac lymphoma
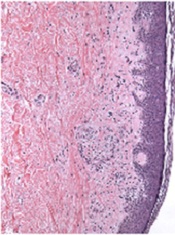
Two cases of central nervous system (CNS) relapse in patients with primary cardiac lymphoma underline the importance of CNS evaluation in these patients, according to researchers.
Cardiac lymphoma is a rare condition, and doctors often overlook the potential of metastasis to the CNS, said study author Niccolò Frungillo, MD, of the European Institute of Oncology in Milan, Italy.
“In my opinion, it is very important to identify prognostic factors that predict the brain relapse of lymphoma,” he said. “It’s a rare—but often fatal—complication.”
In ecancermedicalscience, Dr Frungillo and his colleagues described the diagnosis and treatment of two women with cardiac lymphoma who achieved remission and later presented with isolated CNS relapse.
Doctors diagnosed the patients via endomyocardial biopsy, and results were consistent with diffuse large B-cell lymphoma in both cases. Immunochemotherapy produced complete remissions (CRs) in both women.
The patients then experienced isolated CNS relapses, one 8 weeks after achieving a CR, and one 5 weeks after CR.
In the first patient, MRI and cerebrospinal fluid confirmed her relapse, after she presented with a 3-day history of headache and vomiting.
The patient then received high-dose methotrexate and high-dose cytarabine, which prompted a second CR. She went on to receive an autologous stem cell transplant but died before engraftment due to a pulmonary infection.
The second patient received systemic CNS prophylaxis with high-dose methotrexate prior to her relapse. Nevertheless, one week after she was declared lymphoma-free, she presented with headache and a deterioration of motor skills.
MRI and lumbar puncture confirmed her relapse, and she received induction chemotherapy with high-dose methotrexate. Her disease progressed after two courses of treatment, so she was placed on salvage therapy with cytarabine and high-dose methotrexate.
Whole-brain radiotherapy prompted a CR, and the patient went on to receive an autologous stem cell transplant.
The researchers noted that isolated CNS relapse is very uncommon in cardiac lymphoma, but CNS evaluation should be considered. Additional studies are needed to determine the appropriate management of CNS relapse in cardiac lymphoma.
Two cases of central nervous system (CNS) relapse in patients with primary cardiac lymphoma underline the importance of CNS evaluation in these patients, according to researchers.
Cardiac lymphoma is a rare condition, and doctors often overlook the potential of metastasis to the CNS, said study author Niccolò Frungillo, MD, of the European Institute of Oncology in Milan, Italy.
“In my opinion, it is very important to identify prognostic factors that predict the brain relapse of lymphoma,” he said. “It’s a rare—but often fatal—complication.”
In ecancermedicalscience, Dr Frungillo and his colleagues described the diagnosis and treatment of two women with cardiac lymphoma who achieved remission and later presented with isolated CNS relapse.
Doctors diagnosed the patients via endomyocardial biopsy, and results were consistent with diffuse large B-cell lymphoma in both cases. Immunochemotherapy produced complete remissions (CRs) in both women.
The patients then experienced isolated CNS relapses, one 8 weeks after achieving a CR, and one 5 weeks after CR.
In the first patient, MRI and cerebrospinal fluid confirmed her relapse, after she presented with a 3-day history of headache and vomiting.
The patient then received high-dose methotrexate and high-dose cytarabine, which prompted a second CR. She went on to receive an autologous stem cell transplant but died before engraftment due to a pulmonary infection.
The second patient received systemic CNS prophylaxis with high-dose methotrexate prior to her relapse. Nevertheless, one week after she was declared lymphoma-free, she presented with headache and a deterioration of motor skills.
MRI and lumbar puncture confirmed her relapse, and she received induction chemotherapy with high-dose methotrexate. Her disease progressed after two courses of treatment, so she was placed on salvage therapy with cytarabine and high-dose methotrexate.
Whole-brain radiotherapy prompted a CR, and the patient went on to receive an autologous stem cell transplant.
The researchers noted that isolated CNS relapse is very uncommon in cardiac lymphoma, but CNS evaluation should be considered. Additional studies are needed to determine the appropriate management of CNS relapse in cardiac lymphoma.
Two cases of central nervous system (CNS) relapse in patients with primary cardiac lymphoma underline the importance of CNS evaluation in these patients, according to researchers.
Cardiac lymphoma is a rare condition, and doctors often overlook the potential of metastasis to the CNS, said study author Niccolò Frungillo, MD, of the European Institute of Oncology in Milan, Italy.
“In my opinion, it is very important to identify prognostic factors that predict the brain relapse of lymphoma,” he said. “It’s a rare—but often fatal—complication.”
In ecancermedicalscience, Dr Frungillo and his colleagues described the diagnosis and treatment of two women with cardiac lymphoma who achieved remission and later presented with isolated CNS relapse.
Doctors diagnosed the patients via endomyocardial biopsy, and results were consistent with diffuse large B-cell lymphoma in both cases. Immunochemotherapy produced complete remissions (CRs) in both women.
The patients then experienced isolated CNS relapses, one 8 weeks after achieving a CR, and one 5 weeks after CR.
In the first patient, MRI and cerebrospinal fluid confirmed her relapse, after she presented with a 3-day history of headache and vomiting.
The patient then received high-dose methotrexate and high-dose cytarabine, which prompted a second CR. She went on to receive an autologous stem cell transplant but died before engraftment due to a pulmonary infection.
The second patient received systemic CNS prophylaxis with high-dose methotrexate prior to her relapse. Nevertheless, one week after she was declared lymphoma-free, she presented with headache and a deterioration of motor skills.
MRI and lumbar puncture confirmed her relapse, and she received induction chemotherapy with high-dose methotrexate. Her disease progressed after two courses of treatment, so she was placed on salvage therapy with cytarabine and high-dose methotrexate.
Whole-brain radiotherapy prompted a CR, and the patient went on to receive an autologous stem cell transplant.
The researchers noted that isolated CNS relapse is very uncommon in cardiac lymphoma, but CNS evaluation should be considered. Additional studies are needed to determine the appropriate management of CNS relapse in cardiac lymphoma.
Patients in GDR trials were not properly informed

Credit: Esther Dyson
New research has shown that clinical trials carried out in the German Democratic Republic (GDR) in the second half of the 20th century were not always conducted with the full knowledge or understanding of participants.
A review of documents from that time suggests that, although questionable practices took place, the GDR attempted to conduct trials according to international ethical standards.
And there was no evidence to suggest that trial investigators intentionally hurt patients.
Nevertheless, these trials were hidden from the public, there was no record of patient consent, and some documents suggest patients did not receive adequate information.
Dr Rainer Erices, of Friedrich-Alexander-Universität Erlangen-Nuernberg in Germany, and his colleagues detailed these findings in the Journal of Medical Ethics.
The GDR, also known as East Germany, was a state within the Eastern Bloc during the Cold War period and between 1949 and 1990. Since the 1990s, the media has reported that unofficial clinical trials were conducted by Western pharmaceutical companies in East Germany from the 1960s onward.
Reports have suggested the GDR “sold” its patients as “guinea pigs” for experiments in exchange for hard currency; for example, for tests on doping effects in premature babies and on treating seriously ill patients with placebo instead of actual medicine.
However, there is still a lack of reliable data about the extent of studies taking place then, the contracts, the amount of money paid, and more moral issues, such as patient education and informed consent.
Dr Erices and his colleagues set out to uncover more information by evaluating the clinical trials based on archival material from the health system and the secret service.
The team found documents related to 220 trials carried out between 1983 and 1990, which involved more than 14,000 patients and 68 Western pharmaceutical companies.
However, there was no record of patient information forms or systematic documentation regarding the provision of patient consent.
A range of drugs were tested in these studies, including chemotherapeutic agents, heparin, insulin, anti-depressants, anti-allergy drugs, contrast agents, and toothpastes.
Between 1983 and 1990, the GDR’s health system received approximately DM 16.5 million for the trials, which were cost-effective for the drug firms, according to the researchers. The team also noted that the GDR agreed to these trials due to its desperate need for hard currency (impending bankruptcy).
Overall, the files the researchers studied suggested that the GDR attempted to conduct trials according to international ethical standards.
However, the trials were concealed from the public. And state legislation stipulated that patients had to consent to the trials, but no evidence was found to suggest that patients were systematically informed.
Some documents suggested that at least some of the trials were carried out without patients having a comprehensive understanding of what the trial involved. And it was unclear whether the patients themselves knew that they were participating in trials and were aware of all the risks.
The researchers concluded that further investigation of these trials is needed, and specific trials should be studied separately.
Credit: Esther Dyson
New research has shown that clinical trials carried out in the German Democratic Republic (GDR) in the second half of the 20th century were not always conducted with the full knowledge or understanding of participants.
A review of documents from that time suggests that, although questionable practices took place, the GDR attempted to conduct trials according to international ethical standards.
And there was no evidence to suggest that trial investigators intentionally hurt patients.
Nevertheless, these trials were hidden from the public, there was no record of patient consent, and some documents suggest patients did not receive adequate information.
Dr Rainer Erices, of Friedrich-Alexander-Universität Erlangen-Nuernberg in Germany, and his colleagues detailed these findings in the Journal of Medical Ethics.
The GDR, also known as East Germany, was a state within the Eastern Bloc during the Cold War period and between 1949 and 1990. Since the 1990s, the media has reported that unofficial clinical trials were conducted by Western pharmaceutical companies in East Germany from the 1960s onward.
Reports have suggested the GDR “sold” its patients as “guinea pigs” for experiments in exchange for hard currency; for example, for tests on doping effects in premature babies and on treating seriously ill patients with placebo instead of actual medicine.
However, there is still a lack of reliable data about the extent of studies taking place then, the contracts, the amount of money paid, and more moral issues, such as patient education and informed consent.
Dr Erices and his colleagues set out to uncover more information by evaluating the clinical trials based on archival material from the health system and the secret service.
The team found documents related to 220 trials carried out between 1983 and 1990, which involved more than 14,000 patients and 68 Western pharmaceutical companies.
However, there was no record of patient information forms or systematic documentation regarding the provision of patient consent.
A range of drugs were tested in these studies, including chemotherapeutic agents, heparin, insulin, anti-depressants, anti-allergy drugs, contrast agents, and toothpastes.
Between 1983 and 1990, the GDR’s health system received approximately DM 16.5 million for the trials, which were cost-effective for the drug firms, according to the researchers. The team also noted that the GDR agreed to these trials due to its desperate need for hard currency (impending bankruptcy).
Overall, the files the researchers studied suggested that the GDR attempted to conduct trials according to international ethical standards.
However, the trials were concealed from the public. And state legislation stipulated that patients had to consent to the trials, but no evidence was found to suggest that patients were systematically informed.
Some documents suggested that at least some of the trials were carried out without patients having a comprehensive understanding of what the trial involved. And it was unclear whether the patients themselves knew that they were participating in trials and were aware of all the risks.
The researchers concluded that further investigation of these trials is needed, and specific trials should be studied separately.
Credit: Esther Dyson
New research has shown that clinical trials carried out in the German Democratic Republic (GDR) in the second half of the 20th century were not always conducted with the full knowledge or understanding of participants.
A review of documents from that time suggests that, although questionable practices took place, the GDR attempted to conduct trials according to international ethical standards.
And there was no evidence to suggest that trial investigators intentionally hurt patients.
Nevertheless, these trials were hidden from the public, there was no record of patient consent, and some documents suggest patients did not receive adequate information.
Dr Rainer Erices, of Friedrich-Alexander-Universität Erlangen-Nuernberg in Germany, and his colleagues detailed these findings in the Journal of Medical Ethics.
The GDR, also known as East Germany, was a state within the Eastern Bloc during the Cold War period and between 1949 and 1990. Since the 1990s, the media has reported that unofficial clinical trials were conducted by Western pharmaceutical companies in East Germany from the 1960s onward.
Reports have suggested the GDR “sold” its patients as “guinea pigs” for experiments in exchange for hard currency; for example, for tests on doping effects in premature babies and on treating seriously ill patients with placebo instead of actual medicine.
However, there is still a lack of reliable data about the extent of studies taking place then, the contracts, the amount of money paid, and more moral issues, such as patient education and informed consent.
Dr Erices and his colleagues set out to uncover more information by evaluating the clinical trials based on archival material from the health system and the secret service.
The team found documents related to 220 trials carried out between 1983 and 1990, which involved more than 14,000 patients and 68 Western pharmaceutical companies.
However, there was no record of patient information forms or systematic documentation regarding the provision of patient consent.
A range of drugs were tested in these studies, including chemotherapeutic agents, heparin, insulin, anti-depressants, anti-allergy drugs, contrast agents, and toothpastes.
Between 1983 and 1990, the GDR’s health system received approximately DM 16.5 million for the trials, which were cost-effective for the drug firms, according to the researchers. The team also noted that the GDR agreed to these trials due to its desperate need for hard currency (impending bankruptcy).
Overall, the files the researchers studied suggested that the GDR attempted to conduct trials according to international ethical standards.
However, the trials were concealed from the public. And state legislation stipulated that patients had to consent to the trials, but no evidence was found to suggest that patients were systematically informed.
Some documents suggested that at least some of the trials were carried out without patients having a comprehensive understanding of what the trial involved. And it was unclear whether the patients themselves knew that they were participating in trials and were aware of all the risks.
The researchers concluded that further investigation of these trials is needed, and specific trials should be studied separately.