User login
Panobinostat combos can treat rel/ref MM

©ASCO/Rodney White
CHICAGO—Combination regimens including the histone deacetylase inhibitor panobinostat can produce durable responses and prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma (MM), according to research presented at the 2015 ASCO Annual Meeting.
In a phase 2 trial, panobinostat plus lenalidomide and dexamethasone produced durable responses, even in high-risk, lenalidomide-refractory MM patients.
In a phase 3 trial, panobinostat in combination with bortezomib and dexamethasone led to a 7.8-month improvement in median PFS over placebo-bortezomib-dexamethasone in patients with relapsed or relapsed and refractory MM who had received 2 or more prior regimens.
Both studies were sponsored by Novartis, the company developing panobinostat.
PANORAMA-1 substudy
PANORAMA-1 was a phase 3, randomized, double-blind, placebo-controlled trial of 768 MM patients. Overall, panobinostat in combination with bortezomib and dexamethasone led to a clinically relevant and statistically significant increase in PFS of about 4 months compared to placebo-bortezomib-dexamethasone.
At ASCO, Jesús San Miguel, MD, of Clínica Universidad de Navarra in Pamplona, Spain, presented the results of an exploratory analysis of 147 patients in this trial (abstract 8526*).
The patients had relapsed or relapsed and refractory MM and had received 2 or more prior regimens, including bortezomib and an immunomodulatory agent (IMiD).
Disease and treatment characteristics were as follows:
| Panobinostat
(n=73) |
Placebo (n=74) | |
| Disease
characteristics, n (%) |
||
| Relapsed | 39 (53) | 30 (41) |
| Relapsed/refractory | 34 (47) | 43 (58) |
| Prior
therapies, n (%) |
||
| Bortezomib | 73 (100) | 74 (100) |
| Lenalidomide | 28 (38) | 37 (50) |
| Thalidomide | 63 (86) | 50 (68) |
| Bortezomib
+ lenalidomide |
28 (38) | 37 (50) |
| Bortezomib
+ dexamethasone |
69 (95) | 74 (100) |
| Prior
autologous transplant, n (%) |
54 (74) | 47 (64) |
| Median
prior lines of therapy (range) |
3 (2-4) | 3 (2-3) |
The median PFS was 12.5 months in the panobinostat arm, compared to 4.7 months in the placebo arm. Treatment with panobinostat also led to an increase in complete/near complete response rates (21.9% vs 8.1%) and overall response rate (58.9% vs 39.2%).
Common grade 3/4 non-hematologic adverse events in the panobinostat arm and placebo arm, respectively, included diarrhea (33.3% vs 15.1%), asthenia/fatigue (26.4% vs 13.7%), and peripheral neuropathy (16.7% vs 6.8%).
The most common grade 3/4 hematologic abnormalities in the panobinostat arm and placebo arm, respectively, were thrombocytopenia (68.1% vs 44.4%), lymphopenia (48.6% vs 49.3%), and neutropenia (40.3% vs 16.4%).
The percentage of on-treatment deaths was similar between the treatment arms (6.9% vs 6.8%).
“These data provide physicians with a better understanding of the clinical use of panobinostat, an HDAC inhibitor, a promising new drug class for this difficult-to-treat patient population with a high unmet need,” Dr San Miguel said.
Phase 2 trial
Ajai Chari, MD, of Mount Sinai Medical Center in New York, presented the results of a phase 2 study of panobinostat with lenalidomide and weekly dexamethasone in patients with relapsed/refractory MM (abstract 8528*).
There were 20 evaluable patients with a median age of 64 (range, 51-75). They had received a median of 3 prior therapies (range, 1-10). Prior regimens were as follows:
| Prior
therapy |
Exposed/Refractory, n (%) |
| Dexamethasone | 20 (100)/9
(45) |
| Thalidomide | 6 (30)/2
(10) |
| Lenalidomide |
20 (100)/15 (75) |
| Pomalidomide | 7 (35)/7
(35) |
| Bortezomib | 20 (100)/9
(45) |
| Carfilzomib | 6 (30)/6
(30) |
| Autologous
transplant |
15 (75) |
For this study, patients received panobinostat (20 mg on days 1, 3, 5, 15, 17, and 19), lenalidomide (25 mg on days 1-21), and dexamethasone (40 mg on days 1, 8, and 15).
The overall response rate was 45%. This included 1 complete response, 3 very good partial responses, 5 partial responses, and 8 minimal responses. Two patients had stable disease, and 1 progressed.
Among lenalidomide-refractory patients (n=16), the overall response rate was 38%. This included 3 very good partial responses, 3 partial responses, and 7 minimal responses. Two patients had stable disease, and 1 progressed.
The median PFS was 6.5 months overall and among lenalidomide-refractory patients.
Grade 3/4 toxicities were primarily hematologic, including neutropenia (55%), thrombocytopenia (40%), and anemia (5%). Grade 3/4 non-hematologic adverse events included infections (n=4), diarrhea (n=3), pulmonary emboli (n=2), neck pain (n=1), QTc prolongation (n=1), fatigue (n=1), and weight loss (n=1).
“In relapsed/refractory MM patients, panobinostat in combination with lenalidomide and dexamethasone demonstrated durable responses comparable to other recently approved agents, even in lenalidomide-refractory patients with high-risk molecular findings,” Dr Chari said.
“In notable contrast to PANORAMA-1 results, this completely oral regimen is well-tolerated, with no grade 3/4 [gastrointestinal] toxicities and primarily expected hematologic toxicities.” ![]()
*Information in the abstract differs from that presented at the meeting.
©ASCO/Rodney White
CHICAGO—Combination regimens including the histone deacetylase inhibitor panobinostat can produce durable responses and prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma (MM), according to research presented at the 2015 ASCO Annual Meeting.
In a phase 2 trial, panobinostat plus lenalidomide and dexamethasone produced durable responses, even in high-risk, lenalidomide-refractory MM patients.
In a phase 3 trial, panobinostat in combination with bortezomib and dexamethasone led to a 7.8-month improvement in median PFS over placebo-bortezomib-dexamethasone in patients with relapsed or relapsed and refractory MM who had received 2 or more prior regimens.
Both studies were sponsored by Novartis, the company developing panobinostat.
PANORAMA-1 substudy
PANORAMA-1 was a phase 3, randomized, double-blind, placebo-controlled trial of 768 MM patients. Overall, panobinostat in combination with bortezomib and dexamethasone led to a clinically relevant and statistically significant increase in PFS of about 4 months compared to placebo-bortezomib-dexamethasone.
At ASCO, Jesús San Miguel, MD, of Clínica Universidad de Navarra in Pamplona, Spain, presented the results of an exploratory analysis of 147 patients in this trial (abstract 8526*).
The patients had relapsed or relapsed and refractory MM and had received 2 or more prior regimens, including bortezomib and an immunomodulatory agent (IMiD).
Disease and treatment characteristics were as follows:
| Panobinostat
(n=73) |
Placebo (n=74) | |
| Disease
characteristics, n (%) |
||
| Relapsed | 39 (53) | 30 (41) |
| Relapsed/refractory | 34 (47) | 43 (58) |
| Prior
therapies, n (%) |
||
| Bortezomib | 73 (100) | 74 (100) |
| Lenalidomide | 28 (38) | 37 (50) |
| Thalidomide | 63 (86) | 50 (68) |
| Bortezomib
+ lenalidomide |
28 (38) | 37 (50) |
| Bortezomib
+ dexamethasone |
69 (95) | 74 (100) |
| Prior
autologous transplant, n (%) |
54 (74) | 47 (64) |
| Median
prior lines of therapy (range) |
3 (2-4) | 3 (2-3) |
The median PFS was 12.5 months in the panobinostat arm, compared to 4.7 months in the placebo arm. Treatment with panobinostat also led to an increase in complete/near complete response rates (21.9% vs 8.1%) and overall response rate (58.9% vs 39.2%).
Common grade 3/4 non-hematologic adverse events in the panobinostat arm and placebo arm, respectively, included diarrhea (33.3% vs 15.1%), asthenia/fatigue (26.4% vs 13.7%), and peripheral neuropathy (16.7% vs 6.8%).
The most common grade 3/4 hematologic abnormalities in the panobinostat arm and placebo arm, respectively, were thrombocytopenia (68.1% vs 44.4%), lymphopenia (48.6% vs 49.3%), and neutropenia (40.3% vs 16.4%).
The percentage of on-treatment deaths was similar between the treatment arms (6.9% vs 6.8%).
“These data provide physicians with a better understanding of the clinical use of panobinostat, an HDAC inhibitor, a promising new drug class for this difficult-to-treat patient population with a high unmet need,” Dr San Miguel said.
Phase 2 trial
Ajai Chari, MD, of Mount Sinai Medical Center in New York, presented the results of a phase 2 study of panobinostat with lenalidomide and weekly dexamethasone in patients with relapsed/refractory MM (abstract 8528*).
There were 20 evaluable patients with a median age of 64 (range, 51-75). They had received a median of 3 prior therapies (range, 1-10). Prior regimens were as follows:
| Prior
therapy |
Exposed/Refractory, n (%) |
| Dexamethasone | 20 (100)/9
(45) |
| Thalidomide | 6 (30)/2
(10) |
| Lenalidomide |
20 (100)/15 (75) |
| Pomalidomide | 7 (35)/7
(35) |
| Bortezomib | 20 (100)/9
(45) |
| Carfilzomib | 6 (30)/6
(30) |
| Autologous
transplant |
15 (75) |
For this study, patients received panobinostat (20 mg on days 1, 3, 5, 15, 17, and 19), lenalidomide (25 mg on days 1-21), and dexamethasone (40 mg on days 1, 8, and 15).
The overall response rate was 45%. This included 1 complete response, 3 very good partial responses, 5 partial responses, and 8 minimal responses. Two patients had stable disease, and 1 progressed.
Among lenalidomide-refractory patients (n=16), the overall response rate was 38%. This included 3 very good partial responses, 3 partial responses, and 7 minimal responses. Two patients had stable disease, and 1 progressed.
The median PFS was 6.5 months overall and among lenalidomide-refractory patients.
Grade 3/4 toxicities were primarily hematologic, including neutropenia (55%), thrombocytopenia (40%), and anemia (5%). Grade 3/4 non-hematologic adverse events included infections (n=4), diarrhea (n=3), pulmonary emboli (n=2), neck pain (n=1), QTc prolongation (n=1), fatigue (n=1), and weight loss (n=1).
“In relapsed/refractory MM patients, panobinostat in combination with lenalidomide and dexamethasone demonstrated durable responses comparable to other recently approved agents, even in lenalidomide-refractory patients with high-risk molecular findings,” Dr Chari said.
“In notable contrast to PANORAMA-1 results, this completely oral regimen is well-tolerated, with no grade 3/4 [gastrointestinal] toxicities and primarily expected hematologic toxicities.” ![]()
*Information in the abstract differs from that presented at the meeting.
©ASCO/Rodney White
CHICAGO—Combination regimens including the histone deacetylase inhibitor panobinostat can produce durable responses and prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma (MM), according to research presented at the 2015 ASCO Annual Meeting.
In a phase 2 trial, panobinostat plus lenalidomide and dexamethasone produced durable responses, even in high-risk, lenalidomide-refractory MM patients.
In a phase 3 trial, panobinostat in combination with bortezomib and dexamethasone led to a 7.8-month improvement in median PFS over placebo-bortezomib-dexamethasone in patients with relapsed or relapsed and refractory MM who had received 2 or more prior regimens.
Both studies were sponsored by Novartis, the company developing panobinostat.
PANORAMA-1 substudy
PANORAMA-1 was a phase 3, randomized, double-blind, placebo-controlled trial of 768 MM patients. Overall, panobinostat in combination with bortezomib and dexamethasone led to a clinically relevant and statistically significant increase in PFS of about 4 months compared to placebo-bortezomib-dexamethasone.
At ASCO, Jesús San Miguel, MD, of Clínica Universidad de Navarra in Pamplona, Spain, presented the results of an exploratory analysis of 147 patients in this trial (abstract 8526*).
The patients had relapsed or relapsed and refractory MM and had received 2 or more prior regimens, including bortezomib and an immunomodulatory agent (IMiD).
Disease and treatment characteristics were as follows:
| Panobinostat
(n=73) |
Placebo (n=74) | |
| Disease
characteristics, n (%) |
||
| Relapsed | 39 (53) | 30 (41) |
| Relapsed/refractory | 34 (47) | 43 (58) |
| Prior
therapies, n (%) |
||
| Bortezomib | 73 (100) | 74 (100) |
| Lenalidomide | 28 (38) | 37 (50) |
| Thalidomide | 63 (86) | 50 (68) |
| Bortezomib
+ lenalidomide |
28 (38) | 37 (50) |
| Bortezomib
+ dexamethasone |
69 (95) | 74 (100) |
| Prior
autologous transplant, n (%) |
54 (74) | 47 (64) |
| Median
prior lines of therapy (range) |
3 (2-4) | 3 (2-3) |
The median PFS was 12.5 months in the panobinostat arm, compared to 4.7 months in the placebo arm. Treatment with panobinostat also led to an increase in complete/near complete response rates (21.9% vs 8.1%) and overall response rate (58.9% vs 39.2%).
Common grade 3/4 non-hematologic adverse events in the panobinostat arm and placebo arm, respectively, included diarrhea (33.3% vs 15.1%), asthenia/fatigue (26.4% vs 13.7%), and peripheral neuropathy (16.7% vs 6.8%).
The most common grade 3/4 hematologic abnormalities in the panobinostat arm and placebo arm, respectively, were thrombocytopenia (68.1% vs 44.4%), lymphopenia (48.6% vs 49.3%), and neutropenia (40.3% vs 16.4%).
The percentage of on-treatment deaths was similar between the treatment arms (6.9% vs 6.8%).
“These data provide physicians with a better understanding of the clinical use of panobinostat, an HDAC inhibitor, a promising new drug class for this difficult-to-treat patient population with a high unmet need,” Dr San Miguel said.
Phase 2 trial
Ajai Chari, MD, of Mount Sinai Medical Center in New York, presented the results of a phase 2 study of panobinostat with lenalidomide and weekly dexamethasone in patients with relapsed/refractory MM (abstract 8528*).
There were 20 evaluable patients with a median age of 64 (range, 51-75). They had received a median of 3 prior therapies (range, 1-10). Prior regimens were as follows:
| Prior
therapy |
Exposed/Refractory, n (%) |
| Dexamethasone | 20 (100)/9
(45) |
| Thalidomide | 6 (30)/2
(10) |
| Lenalidomide |
20 (100)/15 (75) |
| Pomalidomide | 7 (35)/7
(35) |
| Bortezomib | 20 (100)/9
(45) |
| Carfilzomib | 6 (30)/6
(30) |
| Autologous
transplant |
15 (75) |
For this study, patients received panobinostat (20 mg on days 1, 3, 5, 15, 17, and 19), lenalidomide (25 mg on days 1-21), and dexamethasone (40 mg on days 1, 8, and 15).
The overall response rate was 45%. This included 1 complete response, 3 very good partial responses, 5 partial responses, and 8 minimal responses. Two patients had stable disease, and 1 progressed.
Among lenalidomide-refractory patients (n=16), the overall response rate was 38%. This included 3 very good partial responses, 3 partial responses, and 7 minimal responses. Two patients had stable disease, and 1 progressed.
The median PFS was 6.5 months overall and among lenalidomide-refractory patients.
Grade 3/4 toxicities were primarily hematologic, including neutropenia (55%), thrombocytopenia (40%), and anemia (5%). Grade 3/4 non-hematologic adverse events included infections (n=4), diarrhea (n=3), pulmonary emboli (n=2), neck pain (n=1), QTc prolongation (n=1), fatigue (n=1), and weight loss (n=1).
“In relapsed/refractory MM patients, panobinostat in combination with lenalidomide and dexamethasone demonstrated durable responses comparable to other recently approved agents, even in lenalidomide-refractory patients with high-risk molecular findings,” Dr Chari said.
“In notable contrast to PANORAMA-1 results, this completely oral regimen is well-tolerated, with no grade 3/4 [gastrointestinal] toxicities and primarily expected hematologic toxicities.” ![]()
*Information in the abstract differs from that presented at the meeting.
FDA extends approval of ITP drug to kids

Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
IDH1 inhibitor gets orphan designation for AML

The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
There may be room for improvement with VTE prophylaxis, team says

Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Drug granted orphan status for hemophilia A

The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
Pathway may play key role in FLT3-ITD AML

Results of preclinical research suggest the Hedgehog signaling pathway works with FLT3-ITD mutations to accelerate the development of acute myeloid leukemia (AML).
This finding prompted investigators to look at Hedgehog signaling as a possible second target for treatment in FLT3-ITD AML.
By combining the FLT3 inhibitor sorafenib with the Hedgehog pathway inhibitor IPI-926 (saridegib), the team found they could limit AML growth in vitro and in vivo.
William Matsui, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues conducted this research and disclosed the results in Science Translational Medicine.
The investigators began this work with the goal of discovering why FLT3 inhibitors have fallen short in treating AML. They analyzed gene expression profiles from AML patients and found abnormally enhanced activity in the Hedgehog signaling pathway.
This pathway, which normally regulates embryonic development, is known to fuel many cancer types, but its role in AML has, thus far, remained unresolved.
In experiments with mice, the investigators found the Hedgehog pathway seemed to work together with FLT3 to accelerate AML development and earlier death.
When the team activated Hedgehog signaling in mice expressing FLT3-ITD, they observed enhanced STAT5 signaling, myeloid progenitor proliferation, and AML development.
“From our data, it appears that Hedgehog signaling is like an accelerator,” Dr Matsui said. “It facilitates the cellular events that lead to cancer, but it itself is not the driver of the whole process.”
The investigators also found that mice with both the FLT3 mutation and Hedgehog activity had a significantly shorter lifespan than mice with FLT3-ITD only—an average of 12 weeks and 40 weeks, respectively.
To build upon these findings, the team tested IPI-926 and sorafenib, both alone and in combination, in mice with AML. They found that mice treated with the combination had significantly fewer leukemic cells in the blood and bone marrow than mice treated with either drug alone.
In addition, 3 of the 5 mice treated with the combination survived past the 16 days of the experiment without any further treatment, but none of the mice treated with sorafenib or IPI-926 alone survived that long.
“When we treat mice that have leukemia with both drugs, they live longer than with either drug alone, and there is a portion of them that don’t die at all,” Dr Matsui noted.
He said it’s likely that Hedgehog signaling is involved in the progression of a number of cancers, and “this study brings home the idea that, in treating these cancers, clinicians may need to inhibit Hedgehog along with specific gene mutations.”
Dr Matsui and his colleagues have begun investigating how Hedgehog inhibitors work when combined with newer drugs that target FLT3 more precisely than sorafenib. If these tests continue to show signs that the two types of inhibitors can fight AML, the investigators think such combination therapy could move on to clinical trials. ![]()
Results of preclinical research suggest the Hedgehog signaling pathway works with FLT3-ITD mutations to accelerate the development of acute myeloid leukemia (AML).
This finding prompted investigators to look at Hedgehog signaling as a possible second target for treatment in FLT3-ITD AML.
By combining the FLT3 inhibitor sorafenib with the Hedgehog pathway inhibitor IPI-926 (saridegib), the team found they could limit AML growth in vitro and in vivo.
William Matsui, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues conducted this research and disclosed the results in Science Translational Medicine.
The investigators began this work with the goal of discovering why FLT3 inhibitors have fallen short in treating AML. They analyzed gene expression profiles from AML patients and found abnormally enhanced activity in the Hedgehog signaling pathway.
This pathway, which normally regulates embryonic development, is known to fuel many cancer types, but its role in AML has, thus far, remained unresolved.
In experiments with mice, the investigators found the Hedgehog pathway seemed to work together with FLT3 to accelerate AML development and earlier death.
When the team activated Hedgehog signaling in mice expressing FLT3-ITD, they observed enhanced STAT5 signaling, myeloid progenitor proliferation, and AML development.
“From our data, it appears that Hedgehog signaling is like an accelerator,” Dr Matsui said. “It facilitates the cellular events that lead to cancer, but it itself is not the driver of the whole process.”
The investigators also found that mice with both the FLT3 mutation and Hedgehog activity had a significantly shorter lifespan than mice with FLT3-ITD only—an average of 12 weeks and 40 weeks, respectively.
To build upon these findings, the team tested IPI-926 and sorafenib, both alone and in combination, in mice with AML. They found that mice treated with the combination had significantly fewer leukemic cells in the blood and bone marrow than mice treated with either drug alone.
In addition, 3 of the 5 mice treated with the combination survived past the 16 days of the experiment without any further treatment, but none of the mice treated with sorafenib or IPI-926 alone survived that long.
“When we treat mice that have leukemia with both drugs, they live longer than with either drug alone, and there is a portion of them that don’t die at all,” Dr Matsui noted.
He said it’s likely that Hedgehog signaling is involved in the progression of a number of cancers, and “this study brings home the idea that, in treating these cancers, clinicians may need to inhibit Hedgehog along with specific gene mutations.”
Dr Matsui and his colleagues have begun investigating how Hedgehog inhibitors work when combined with newer drugs that target FLT3 more precisely than sorafenib. If these tests continue to show signs that the two types of inhibitors can fight AML, the investigators think such combination therapy could move on to clinical trials. ![]()
Results of preclinical research suggest the Hedgehog signaling pathway works with FLT3-ITD mutations to accelerate the development of acute myeloid leukemia (AML).
This finding prompted investigators to look at Hedgehog signaling as a possible second target for treatment in FLT3-ITD AML.
By combining the FLT3 inhibitor sorafenib with the Hedgehog pathway inhibitor IPI-926 (saridegib), the team found they could limit AML growth in vitro and in vivo.
William Matsui, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues conducted this research and disclosed the results in Science Translational Medicine.
The investigators began this work with the goal of discovering why FLT3 inhibitors have fallen short in treating AML. They analyzed gene expression profiles from AML patients and found abnormally enhanced activity in the Hedgehog signaling pathway.
This pathway, which normally regulates embryonic development, is known to fuel many cancer types, but its role in AML has, thus far, remained unresolved.
In experiments with mice, the investigators found the Hedgehog pathway seemed to work together with FLT3 to accelerate AML development and earlier death.
When the team activated Hedgehog signaling in mice expressing FLT3-ITD, they observed enhanced STAT5 signaling, myeloid progenitor proliferation, and AML development.
“From our data, it appears that Hedgehog signaling is like an accelerator,” Dr Matsui said. “It facilitates the cellular events that lead to cancer, but it itself is not the driver of the whole process.”
The investigators also found that mice with both the FLT3 mutation and Hedgehog activity had a significantly shorter lifespan than mice with FLT3-ITD only—an average of 12 weeks and 40 weeks, respectively.
To build upon these findings, the team tested IPI-926 and sorafenib, both alone and in combination, in mice with AML. They found that mice treated with the combination had significantly fewer leukemic cells in the blood and bone marrow than mice treated with either drug alone.
In addition, 3 of the 5 mice treated with the combination survived past the 16 days of the experiment without any further treatment, but none of the mice treated with sorafenib or IPI-926 alone survived that long.
“When we treat mice that have leukemia with both drugs, they live longer than with either drug alone, and there is a portion of them that don’t die at all,” Dr Matsui noted.
He said it’s likely that Hedgehog signaling is involved in the progression of a number of cancers, and “this study brings home the idea that, in treating these cancers, clinicians may need to inhibit Hedgehog along with specific gene mutations.”
Dr Matsui and his colleagues have begun investigating how Hedgehog inhibitors work when combined with newer drugs that target FLT3 more precisely than sorafenib. If these tests continue to show signs that the two types of inhibitors can fight AML, the investigators think such combination therapy could move on to clinical trials. ![]()
CAR T-cell therapy seems feasible for NHL, MM

©ASCO/Rodney White
CHICAGO—The CD19-directed chimeric antigen receptor (CAR) T-cell therapy CTL019 has shown promise for treating non-Hodgkin lymphoma (NHL) and may be a feasible treatment option for multiple myeloma (MM) as well, according to researchers.
In an ongoing phase 2 trial, CTL019 has produced durable responses in patients with relapsed or refractory NHL.
And early results of a phase 1 trial suggest CTL019 can provide clinical benefit in heavily pretreated patients with MM.
Both studies were presented at the 2015 ASCO Annual Meeting. The University of Pennsylvania and Novartis have an exclusive global collaboration to research, develop, and commercialize CTL019.
CTL019 in NHL
Stephen Schuster, MD, of the Abramson Cancer Center of the University of Pennsylvania in Philadelphia, presented results of the phase 2 NHL trial (abstract 8516*).
The trial included 20 evaluable patients, 13 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma (FL). At the time of presentation, the median follow-up was 274 days for the patients with DLBCL and 290 days for those with FL.
The overall response rate was 100% in patients with FL and 50% in those with DLBCL. Thirteen patients responded to the therapy, including 11 who achieved a complete response and 2 who experienced a partial response.
Six patients with a partial response to treatment at 3 months achieved a complete response by 6 months. Two patients with a partial response experienced disease progression at 6 and 12 months after treatment.
The researchers said toxicity appeared to be acceptable, with primarily grade 2 cytokine release syndrome (CRS). Two patients developed CRS of grade 3 or higher at peak T-cell expansion. There were no deaths from CRS.
“The results from this ongoing study of CTL019 are encouraging, as we now have data through 6 months showing that patients may have achieved durable overall response rates,” Dr Schuster said. “These data support our ongoing efforts to determine the potential role of CTL019 in improving outcomes for patients with certain types of B-cell lymphomas.”
CTL019 in MM
Alfred Garfall, MD, of the Abramson Cancer Center, presented preliminary results of an ongoing phase 1 study investigating CTL019 in patients with MM (abstract 8517*).
Dr Garfall and his colleagues hypothesized that CTL019 would exhibit efficacy in MM due to low-level CD19 expression on MM plasma cells or CD19 expression in drug-resistant, disease-propagating subsets of the MM clone.
The study included 5 patients who experienced disease progression within a year of a prior autologous stem cell transplant and were medically fit to undergo a second autologous transplant. The patients had received a median of 7.5 prior lines of therapy.
“We found potential evidence of clinical benefit in 3 of 4 patients with more than 100 days of follow-up,” Dr Garfall said.
Two patients experienced longer, deeper responses, and 1 patient experienced CRS.
The data suggest “it is safe and feasible to manufacture and administered CTL019 to refractory multiple myeloma patients,” Dr Garfall said.
*Information in the abstract differs from that presented at the meeting.
©ASCO/Rodney White
CHICAGO—The CD19-directed chimeric antigen receptor (CAR) T-cell therapy CTL019 has shown promise for treating non-Hodgkin lymphoma (NHL) and may be a feasible treatment option for multiple myeloma (MM) as well, according to researchers.
In an ongoing phase 2 trial, CTL019 has produced durable responses in patients with relapsed or refractory NHL.
And early results of a phase 1 trial suggest CTL019 can provide clinical benefit in heavily pretreated patients with MM.
Both studies were presented at the 2015 ASCO Annual Meeting. The University of Pennsylvania and Novartis have an exclusive global collaboration to research, develop, and commercialize CTL019.
CTL019 in NHL
Stephen Schuster, MD, of the Abramson Cancer Center of the University of Pennsylvania in Philadelphia, presented results of the phase 2 NHL trial (abstract 8516*).
The trial included 20 evaluable patients, 13 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma (FL). At the time of presentation, the median follow-up was 274 days for the patients with DLBCL and 290 days for those with FL.
The overall response rate was 100% in patients with FL and 50% in those with DLBCL. Thirteen patients responded to the therapy, including 11 who achieved a complete response and 2 who experienced a partial response.
Six patients with a partial response to treatment at 3 months achieved a complete response by 6 months. Two patients with a partial response experienced disease progression at 6 and 12 months after treatment.
The researchers said toxicity appeared to be acceptable, with primarily grade 2 cytokine release syndrome (CRS). Two patients developed CRS of grade 3 or higher at peak T-cell expansion. There were no deaths from CRS.
“The results from this ongoing study of CTL019 are encouraging, as we now have data through 6 months showing that patients may have achieved durable overall response rates,” Dr Schuster said. “These data support our ongoing efforts to determine the potential role of CTL019 in improving outcomes for patients with certain types of B-cell lymphomas.”
CTL019 in MM
Alfred Garfall, MD, of the Abramson Cancer Center, presented preliminary results of an ongoing phase 1 study investigating CTL019 in patients with MM (abstract 8517*).
Dr Garfall and his colleagues hypothesized that CTL019 would exhibit efficacy in MM due to low-level CD19 expression on MM plasma cells or CD19 expression in drug-resistant, disease-propagating subsets of the MM clone.
The study included 5 patients who experienced disease progression within a year of a prior autologous stem cell transplant and were medically fit to undergo a second autologous transplant. The patients had received a median of 7.5 prior lines of therapy.
“We found potential evidence of clinical benefit in 3 of 4 patients with more than 100 days of follow-up,” Dr Garfall said.
Two patients experienced longer, deeper responses, and 1 patient experienced CRS.
The data suggest “it is safe and feasible to manufacture and administered CTL019 to refractory multiple myeloma patients,” Dr Garfall said.
*Information in the abstract differs from that presented at the meeting.
©ASCO/Rodney White
CHICAGO—The CD19-directed chimeric antigen receptor (CAR) T-cell therapy CTL019 has shown promise for treating non-Hodgkin lymphoma (NHL) and may be a feasible treatment option for multiple myeloma (MM) as well, according to researchers.
In an ongoing phase 2 trial, CTL019 has produced durable responses in patients with relapsed or refractory NHL.
And early results of a phase 1 trial suggest CTL019 can provide clinical benefit in heavily pretreated patients with MM.
Both studies were presented at the 2015 ASCO Annual Meeting. The University of Pennsylvania and Novartis have an exclusive global collaboration to research, develop, and commercialize CTL019.
CTL019 in NHL
Stephen Schuster, MD, of the Abramson Cancer Center of the University of Pennsylvania in Philadelphia, presented results of the phase 2 NHL trial (abstract 8516*).
The trial included 20 evaluable patients, 13 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma (FL). At the time of presentation, the median follow-up was 274 days for the patients with DLBCL and 290 days for those with FL.
The overall response rate was 100% in patients with FL and 50% in those with DLBCL. Thirteen patients responded to the therapy, including 11 who achieved a complete response and 2 who experienced a partial response.
Six patients with a partial response to treatment at 3 months achieved a complete response by 6 months. Two patients with a partial response experienced disease progression at 6 and 12 months after treatment.
The researchers said toxicity appeared to be acceptable, with primarily grade 2 cytokine release syndrome (CRS). Two patients developed CRS of grade 3 or higher at peak T-cell expansion. There were no deaths from CRS.
“The results from this ongoing study of CTL019 are encouraging, as we now have data through 6 months showing that patients may have achieved durable overall response rates,” Dr Schuster said. “These data support our ongoing efforts to determine the potential role of CTL019 in improving outcomes for patients with certain types of B-cell lymphomas.”
CTL019 in MM
Alfred Garfall, MD, of the Abramson Cancer Center, presented preliminary results of an ongoing phase 1 study investigating CTL019 in patients with MM (abstract 8517*).
Dr Garfall and his colleagues hypothesized that CTL019 would exhibit efficacy in MM due to low-level CD19 expression on MM plasma cells or CD19 expression in drug-resistant, disease-propagating subsets of the MM clone.
The study included 5 patients who experienced disease progression within a year of a prior autologous stem cell transplant and were medically fit to undergo a second autologous transplant. The patients had received a median of 7.5 prior lines of therapy.
“We found potential evidence of clinical benefit in 3 of 4 patients with more than 100 days of follow-up,” Dr Garfall said.
Two patients experienced longer, deeper responses, and 1 patient experienced CRS.
The data suggest “it is safe and feasible to manufacture and administered CTL019 to refractory multiple myeloma patients,” Dr Garfall said.
*Information in the abstract differs from that presented at the meeting.
Establishing cause of death in HSCT recipients

With a new study, researchers hope to establish definitions for specific causes of death in patients who undergo allogeneic hematopoietic stem cell transplant (HSCT).
The group noted that scientists conducting clinical trials commonly use committees to review and define endpoints.
However, investigators conducting genome-wide association studies rarely do, relying instead on center-reported outcomes, which are variable.
“It is important that we accurately define outcomes in these types of genomic studies as precisely as possible,” said Lara Sucheston-Campbell, PhD, of Roswell Park Cancer Institute in Buffalo, New York.
To that end, she and her colleagues convened a consensus panel to review specific causes of death in HSCT recipients. The panel evaluated outcome data for 1484 patients who had been treated for acute leukemia or myelodysplasia at 1 of 11 US transplant centers and died within a year of undergoing HSCT.
Dr Sucheston-Campbell and her colleagues reported the results of this evaluation in Biology of Blood and Marrow Transplantation.
In the cases where the transplant center reported disease-related mortality, the researchers found almost perfect agreement between the consensus panel and the transplant center, in terms of how those deaths were classified. The panel agreed with more than 99% of deaths that were reported as disease-related.
There was less agreement for transplant-related mortality, however. The panel agreed with about 80% of deaths reported to be transplant-related.
And the level of agreement/discordance varied depending on the specific cause of death. For example, the panel agreed with transplant centers on most of the deaths that were reportedly caused by graft-vs-host disease.
But the panel reclassified many of the deaths that reportedly resulted from organ failure, saying these deaths should be considered disease-related, a result of graft-vs-host disease, infection-related, or due to “other” causes.
These results indicate that transplant-related mortality needs to be better defined, the researchers said. They believe this study provides a mechanism for prioritizing those HSCT cases that should be reviewed.
“We need to make sure that patients who experienced similar clinical events after a transplant were consistently defined regardless of where they were treated,” said study author Theresa Hahn, PhD, also of Roswell Park Cancer Center.
“This is a difficult topic to consider, but we can’t make progress to improve transplant outcomes without these discussions.” ![]()
With a new study, researchers hope to establish definitions for specific causes of death in patients who undergo allogeneic hematopoietic stem cell transplant (HSCT).
The group noted that scientists conducting clinical trials commonly use committees to review and define endpoints.
However, investigators conducting genome-wide association studies rarely do, relying instead on center-reported outcomes, which are variable.
“It is important that we accurately define outcomes in these types of genomic studies as precisely as possible,” said Lara Sucheston-Campbell, PhD, of Roswell Park Cancer Institute in Buffalo, New York.
To that end, she and her colleagues convened a consensus panel to review specific causes of death in HSCT recipients. The panel evaluated outcome data for 1484 patients who had been treated for acute leukemia or myelodysplasia at 1 of 11 US transplant centers and died within a year of undergoing HSCT.
Dr Sucheston-Campbell and her colleagues reported the results of this evaluation in Biology of Blood and Marrow Transplantation.
In the cases where the transplant center reported disease-related mortality, the researchers found almost perfect agreement between the consensus panel and the transplant center, in terms of how those deaths were classified. The panel agreed with more than 99% of deaths that were reported as disease-related.
There was less agreement for transplant-related mortality, however. The panel agreed with about 80% of deaths reported to be transplant-related.
And the level of agreement/discordance varied depending on the specific cause of death. For example, the panel agreed with transplant centers on most of the deaths that were reportedly caused by graft-vs-host disease.
But the panel reclassified many of the deaths that reportedly resulted from organ failure, saying these deaths should be considered disease-related, a result of graft-vs-host disease, infection-related, or due to “other” causes.
These results indicate that transplant-related mortality needs to be better defined, the researchers said. They believe this study provides a mechanism for prioritizing those HSCT cases that should be reviewed.
“We need to make sure that patients who experienced similar clinical events after a transplant were consistently defined regardless of where they were treated,” said study author Theresa Hahn, PhD, also of Roswell Park Cancer Center.
“This is a difficult topic to consider, but we can’t make progress to improve transplant outcomes without these discussions.” ![]()
With a new study, researchers hope to establish definitions for specific causes of death in patients who undergo allogeneic hematopoietic stem cell transplant (HSCT).
The group noted that scientists conducting clinical trials commonly use committees to review and define endpoints.
However, investigators conducting genome-wide association studies rarely do, relying instead on center-reported outcomes, which are variable.
“It is important that we accurately define outcomes in these types of genomic studies as precisely as possible,” said Lara Sucheston-Campbell, PhD, of Roswell Park Cancer Institute in Buffalo, New York.
To that end, she and her colleagues convened a consensus panel to review specific causes of death in HSCT recipients. The panel evaluated outcome data for 1484 patients who had been treated for acute leukemia or myelodysplasia at 1 of 11 US transplant centers and died within a year of undergoing HSCT.
Dr Sucheston-Campbell and her colleagues reported the results of this evaluation in Biology of Blood and Marrow Transplantation.
In the cases where the transplant center reported disease-related mortality, the researchers found almost perfect agreement between the consensus panel and the transplant center, in terms of how those deaths were classified. The panel agreed with more than 99% of deaths that were reported as disease-related.
There was less agreement for transplant-related mortality, however. The panel agreed with about 80% of deaths reported to be transplant-related.
And the level of agreement/discordance varied depending on the specific cause of death. For example, the panel agreed with transplant centers on most of the deaths that were reportedly caused by graft-vs-host disease.
But the panel reclassified many of the deaths that reportedly resulted from organ failure, saying these deaths should be considered disease-related, a result of graft-vs-host disease, infection-related, or due to “other” causes.
These results indicate that transplant-related mortality needs to be better defined, the researchers said. They believe this study provides a mechanism for prioritizing those HSCT cases that should be reviewed.
“We need to make sure that patients who experienced similar clinical events after a transplant were consistently defined regardless of where they were treated,” said study author Theresa Hahn, PhD, also of Roswell Park Cancer Center.
“This is a difficult topic to consider, but we can’t make progress to improve transplant outcomes without these discussions.”
Harnessing immune defense to treat Candida infection
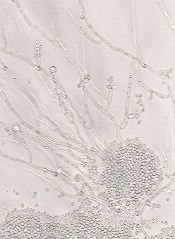
An upset in the body’s natural balance of gut bacteria that may lead to life-threatening bloodstream infections can be reversed by enhancing an
immune response, according to research published in Nature Medicine.
Researchers found that a transcription factor known as HIF-1α works with LL-37, a naturally occurring antibiotic, to kill the infection-causing fungi Candida albicans.
And this response can be enhanced with a drug called L-mimosine.
The researchers noted that Candida albicans can be lethal if it invades the bloodstream from the gut. And stem cell transplant recipients and immunosuppressed cancer patients have a high risk for this type of infection.
“For a cancer patient with a Candida bloodstream infection, the fatality rate is about 30%, [and] Candida is the number 1 fungal pathogen,” said study author Andrew Koh, MD, of the University of Texas Southwestern Medical Center in Dallas.
With that in mind, he and his colleagues set out to determine how the body’s natural immune defense system might be enhanced to fight a Candida infection. By studying how mice infected with Candida responded in different scenarios, the team found their answer.
“The commensal bacteria stimulate gut tissue to make a transcription factor and a natural antibiotic, which then kills the Candida fungus,” Dr Koh explained.
“When we gave the mice a pharmacologic agent called L-mimosine that stimulates the transcription factor, the agent knocked down Candida 100-fold, which translated into a 50% reduction in mortality from invasive Candida infection.”
Specifically, the researchers found that enhancing the transcription factor HIF-1α with L-mimosine led to increased production of the natural antibiotic peptide LL-37, which, in turn, killed the fungi. L-mimosine is a natural product derived from seeds of the koa haole tree that is known to boost HIF-1α activity.
The study also suggested that certain gut bacteria—Clostridial Firmicutes and Bacteroidetes—may be important in producing short-chain fatty acids that help fight infection.
The researchers said more work is needed to pinpoint the optimal method of inducing the body’s gut defense system, whether through use of an agent like L-mimosine or by administering short-chain fatty acids such as vinegar.
“Can we modulate the gut system to maintain balance so that it never gets to the point of pathogens invading the bloodstream?” Dr Koh asked. “Boosting [gastrointestinal] mucosal immune effectors to reduce fungal burden may be the key to tipping the balance back toward normal and preventing invasive fungal disease.”
An upset in the body’s natural balance of gut bacteria that may lead to life-threatening bloodstream infections can be reversed by enhancing an
immune response, according to research published in Nature Medicine.
Researchers found that a transcription factor known as HIF-1α works with LL-37, a naturally occurring antibiotic, to kill the infection-causing fungi Candida albicans.
And this response can be enhanced with a drug called L-mimosine.
The researchers noted that Candida albicans can be lethal if it invades the bloodstream from the gut. And stem cell transplant recipients and immunosuppressed cancer patients have a high risk for this type of infection.
“For a cancer patient with a Candida bloodstream infection, the fatality rate is about 30%, [and] Candida is the number 1 fungal pathogen,” said study author Andrew Koh, MD, of the University of Texas Southwestern Medical Center in Dallas.
With that in mind, he and his colleagues set out to determine how the body’s natural immune defense system might be enhanced to fight a Candida infection. By studying how mice infected with Candida responded in different scenarios, the team found their answer.
“The commensal bacteria stimulate gut tissue to make a transcription factor and a natural antibiotic, which then kills the Candida fungus,” Dr Koh explained.
“When we gave the mice a pharmacologic agent called L-mimosine that stimulates the transcription factor, the agent knocked down Candida 100-fold, which translated into a 50% reduction in mortality from invasive Candida infection.”
Specifically, the researchers found that enhancing the transcription factor HIF-1α with L-mimosine led to increased production of the natural antibiotic peptide LL-37, which, in turn, killed the fungi. L-mimosine is a natural product derived from seeds of the koa haole tree that is known to boost HIF-1α activity.
The study also suggested that certain gut bacteria—Clostridial Firmicutes and Bacteroidetes—may be important in producing short-chain fatty acids that help fight infection.
The researchers said more work is needed to pinpoint the optimal method of inducing the body’s gut defense system, whether through use of an agent like L-mimosine or by administering short-chain fatty acids such as vinegar.
“Can we modulate the gut system to maintain balance so that it never gets to the point of pathogens invading the bloodstream?” Dr Koh asked. “Boosting [gastrointestinal] mucosal immune effectors to reduce fungal burden may be the key to tipping the balance back toward normal and preventing invasive fungal disease.”
An upset in the body’s natural balance of gut bacteria that may lead to life-threatening bloodstream infections can be reversed by enhancing an
immune response, according to research published in Nature Medicine.
Researchers found that a transcription factor known as HIF-1α works with LL-37, a naturally occurring antibiotic, to kill the infection-causing fungi Candida albicans.
And this response can be enhanced with a drug called L-mimosine.
The researchers noted that Candida albicans can be lethal if it invades the bloodstream from the gut. And stem cell transplant recipients and immunosuppressed cancer patients have a high risk for this type of infection.
“For a cancer patient with a Candida bloodstream infection, the fatality rate is about 30%, [and] Candida is the number 1 fungal pathogen,” said study author Andrew Koh, MD, of the University of Texas Southwestern Medical Center in Dallas.
With that in mind, he and his colleagues set out to determine how the body’s natural immune defense system might be enhanced to fight a Candida infection. By studying how mice infected with Candida responded in different scenarios, the team found their answer.
“The commensal bacteria stimulate gut tissue to make a transcription factor and a natural antibiotic, which then kills the Candida fungus,” Dr Koh explained.
“When we gave the mice a pharmacologic agent called L-mimosine that stimulates the transcription factor, the agent knocked down Candida 100-fold, which translated into a 50% reduction in mortality from invasive Candida infection.”
Specifically, the researchers found that enhancing the transcription factor HIF-1α with L-mimosine led to increased production of the natural antibiotic peptide LL-37, which, in turn, killed the fungi. L-mimosine is a natural product derived from seeds of the koa haole tree that is known to boost HIF-1α activity.
The study also suggested that certain gut bacteria—Clostridial Firmicutes and Bacteroidetes—may be important in producing short-chain fatty acids that help fight infection.
The researchers said more work is needed to pinpoint the optimal method of inducing the body’s gut defense system, whether through use of an agent like L-mimosine or by administering short-chain fatty acids such as vinegar.
“Can we modulate the gut system to maintain balance so that it never gets to the point of pathogens invading the bloodstream?” Dr Koh asked. “Boosting [gastrointestinal] mucosal immune effectors to reduce fungal burden may be the key to tipping the balance back toward normal and preventing invasive fungal disease.”
Study suggests need for change in matching criteria
![]()
Photo by Chad McNeeley
A donor’s CD8 cell count can affect the outcome of hematopoietic stem cell transplant (HSCT) in older patients, according to a study published in the Journal of Clinical Oncology.
Older patients who received stem cells from unrelated donors with higher CD8 counts had a significantly lower risk of relapse and higher rate of
survival than patients who received grafts with lower CD8 counts, even if those grafts were from matched sibling donors.
This suggests that screening for donor T-cell characteristics could optimize donor selection and ultimately lead to more successful transplants in an older population, study investigators said.
“Developing better tools to identify ideal donors is an exciting prospect and fundamental to improving transplant outcomes,” said Ran Reshef, MD, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
“There may be suitable donors out there who are overlooked because they are considered a poorer match by today’s donor selection algorithms. Refining the screening method could greatly increase the chances of finding the most appropriate donor, one that will induce the most potent graft-vs-tumor response.”
For this study, Dr Reshef and his colleagues retrospectively evaluated associations between a graft’s T-cell dose and outcomes in 200 patients undergoing HSCT to treat acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin lymphoma, and other hematologic disorders.
The investigators looked specifically at CD4 and CD8 counts and found the number of CD8 cells in the graft had an impact on survival. They also found that high CD8 cell counts were much more common among young donors.
The 4-year overall survival rates were 59% for patients who received grafts from younger (<50 years), unrelated donors with high CD8 counts; 18% for those who received grafts from younger, unrelated donors with low CD8 counts; and 33% for patients who received grafts from older (>50 years), HLA-matched sibling donors.
In multivariable analysis, CD8 count was an independent predictor of relapse (adjusted hazard ratio [aHR]=0.43; P=0.009), relapse-free survival (aHR=0.50; P=0.006), and overall survival (aHR=0.57; P=0.04).
The investigators also assessed the effect of CD8 count on survival using a cutoff of 0.72 x 108 CD8 cells/kg. They found that patients with CD8 doses above this cutoff had significantly better relapse-free survival (P=0.005) and overall survival (P=0.007) rates than patients with CD8 doses below this cutoff.
These results suggest it would be better to use a younger, unrelated donor graft with a high CD8 cell count rather than an older sibling donor, the investigators said. However, this strategy should be tested in a prospective trial.
“This is a method deserving additional investigation,” Dr Reshef said, “which could refine the standardized matching system used by registries, such as Be the Match and others, and ultimately optimize the donor pool for older patients undergoing these transplants.”
![]()
Photo by Chad McNeeley
A donor’s CD8 cell count can affect the outcome of hematopoietic stem cell transplant (HSCT) in older patients, according to a study published in the Journal of Clinical Oncology.
Older patients who received stem cells from unrelated donors with higher CD8 counts had a significantly lower risk of relapse and higher rate of
survival than patients who received grafts with lower CD8 counts, even if those grafts were from matched sibling donors.
This suggests that screening for donor T-cell characteristics could optimize donor selection and ultimately lead to more successful transplants in an older population, study investigators said.
“Developing better tools to identify ideal donors is an exciting prospect and fundamental to improving transplant outcomes,” said Ran Reshef, MD, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
“There may be suitable donors out there who are overlooked because they are considered a poorer match by today’s donor selection algorithms. Refining the screening method could greatly increase the chances of finding the most appropriate donor, one that will induce the most potent graft-vs-tumor response.”
For this study, Dr Reshef and his colleagues retrospectively evaluated associations between a graft’s T-cell dose and outcomes in 200 patients undergoing HSCT to treat acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin lymphoma, and other hematologic disorders.
The investigators looked specifically at CD4 and CD8 counts and found the number of CD8 cells in the graft had an impact on survival. They also found that high CD8 cell counts were much more common among young donors.
The 4-year overall survival rates were 59% for patients who received grafts from younger (<50 years), unrelated donors with high CD8 counts; 18% for those who received grafts from younger, unrelated donors with low CD8 counts; and 33% for patients who received grafts from older (>50 years), HLA-matched sibling donors.
In multivariable analysis, CD8 count was an independent predictor of relapse (adjusted hazard ratio [aHR]=0.43; P=0.009), relapse-free survival (aHR=0.50; P=0.006), and overall survival (aHR=0.57; P=0.04).
The investigators also assessed the effect of CD8 count on survival using a cutoff of 0.72 x 108 CD8 cells/kg. They found that patients with CD8 doses above this cutoff had significantly better relapse-free survival (P=0.005) and overall survival (P=0.007) rates than patients with CD8 doses below this cutoff.
These results suggest it would be better to use a younger, unrelated donor graft with a high CD8 cell count rather than an older sibling donor, the investigators said. However, this strategy should be tested in a prospective trial.
“This is a method deserving additional investigation,” Dr Reshef said, “which could refine the standardized matching system used by registries, such as Be the Match and others, and ultimately optimize the donor pool for older patients undergoing these transplants.”
![]()
Photo by Chad McNeeley
A donor’s CD8 cell count can affect the outcome of hematopoietic stem cell transplant (HSCT) in older patients, according to a study published in the Journal of Clinical Oncology.
Older patients who received stem cells from unrelated donors with higher CD8 counts had a significantly lower risk of relapse and higher rate of
survival than patients who received grafts with lower CD8 counts, even if those grafts were from matched sibling donors.
This suggests that screening for donor T-cell characteristics could optimize donor selection and ultimately lead to more successful transplants in an older population, study investigators said.
“Developing better tools to identify ideal donors is an exciting prospect and fundamental to improving transplant outcomes,” said Ran Reshef, MD, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
“There may be suitable donors out there who are overlooked because they are considered a poorer match by today’s donor selection algorithms. Refining the screening method could greatly increase the chances of finding the most appropriate donor, one that will induce the most potent graft-vs-tumor response.”
For this study, Dr Reshef and his colleagues retrospectively evaluated associations between a graft’s T-cell dose and outcomes in 200 patients undergoing HSCT to treat acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin lymphoma, and other hematologic disorders.
The investigators looked specifically at CD4 and CD8 counts and found the number of CD8 cells in the graft had an impact on survival. They also found that high CD8 cell counts were much more common among young donors.
The 4-year overall survival rates were 59% for patients who received grafts from younger (<50 years), unrelated donors with high CD8 counts; 18% for those who received grafts from younger, unrelated donors with low CD8 counts; and 33% for patients who received grafts from older (>50 years), HLA-matched sibling donors.
In multivariable analysis, CD8 count was an independent predictor of relapse (adjusted hazard ratio [aHR]=0.43; P=0.009), relapse-free survival (aHR=0.50; P=0.006), and overall survival (aHR=0.57; P=0.04).
The investigators also assessed the effect of CD8 count on survival using a cutoff of 0.72 x 108 CD8 cells/kg. They found that patients with CD8 doses above this cutoff had significantly better relapse-free survival (P=0.005) and overall survival (P=0.007) rates than patients with CD8 doses below this cutoff.
These results suggest it would be better to use a younger, unrelated donor graft with a high CD8 cell count rather than an older sibling donor, the investigators said. However, this strategy should be tested in a prospective trial.
“This is a method deserving additional investigation,” Dr Reshef said, “which could refine the standardized matching system used by registries, such as Be the Match and others, and ultimately optimize the donor pool for older patients undergoing these transplants.”