User login
Hemophilia A drug appears safe, effective long-term
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Groups draft guidelines for acute leukemia

Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Mutations may contribute to CTCL

mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
Selinexor dose lowered due to sepsis in AML patients

Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Education doesn’t ensure appropriate use of VTE prophylaxis

Photo courtesy of the CDC
In a single-center study, researchers found that educating healthcare providers about the need for venous thromboembolism (VTE) prophylaxis did not ensure that patients received appropriate treatment.
Before the educational program was introduced, 36% of patients who were at risk of VTE were not receiving VTE prophylaxis. After the program, 26% of at-risk patients were not receiving prophylaxis.
The researchers reported these findings in the Canadian Journal of Cardiology.
The team carried out chart reviews of patients in a university-affiliated, tertiary care cardiology center, which included a clinical teaching unit and a coronary care unit.
Audits were conducted 3 and 5 months before the introduction of an educational program on VTE prophylaxis protocol, followed by a second series of audits 3 and 5 months after protocol initiation.
In each set of audits, conducted over 2 months, 3 independent groups consisting of a physician and a nonphysician healthcare provider (nursing, pharmacy) each reviewed the data. Discrepancies were settled by the senior investigators.
In the first set of audits, 173 charts for patients considered at high risk for VTE were evaluated. The second set of audits included 247 patients.
Prior to the educational program, including a guideline-based protocol, 36% of all patients who were considered at risk for VTE did not receive prophylaxis.
Three months after the program was initiated, 21% of patients were still not being treated according to the recommended guidelines, and that percentage rose to 28% at 5 months post-protocol.
“Awareness and education surrounding VTE prophylaxis is challenging in the inpatient teaching unit model due to a number of factors, including the high turnover of senior and junior physicians as well as nursing staff,” said study author Colette Seifer, of the University of Manitoba in Winnipeg, Manitoba, Canada.
“A single time point intervention is unlikely to result in a sustained improvement in VTE prophylaxis rates.”
However, Seifer and her colleagues believe that automated alerts and checklists incorporated into electronic patient records or used via innovative software programs have the potential to improve compliance rates. ![]()
Photo courtesy of the CDC
In a single-center study, researchers found that educating healthcare providers about the need for venous thromboembolism (VTE) prophylaxis did not ensure that patients received appropriate treatment.
Before the educational program was introduced, 36% of patients who were at risk of VTE were not receiving VTE prophylaxis. After the program, 26% of at-risk patients were not receiving prophylaxis.
The researchers reported these findings in the Canadian Journal of Cardiology.
The team carried out chart reviews of patients in a university-affiliated, tertiary care cardiology center, which included a clinical teaching unit and a coronary care unit.
Audits were conducted 3 and 5 months before the introduction of an educational program on VTE prophylaxis protocol, followed by a second series of audits 3 and 5 months after protocol initiation.
In each set of audits, conducted over 2 months, 3 independent groups consisting of a physician and a nonphysician healthcare provider (nursing, pharmacy) each reviewed the data. Discrepancies were settled by the senior investigators.
In the first set of audits, 173 charts for patients considered at high risk for VTE were evaluated. The second set of audits included 247 patients.
Prior to the educational program, including a guideline-based protocol, 36% of all patients who were considered at risk for VTE did not receive prophylaxis.
Three months after the program was initiated, 21% of patients were still not being treated according to the recommended guidelines, and that percentage rose to 28% at 5 months post-protocol.
“Awareness and education surrounding VTE prophylaxis is challenging in the inpatient teaching unit model due to a number of factors, including the high turnover of senior and junior physicians as well as nursing staff,” said study author Colette Seifer, of the University of Manitoba in Winnipeg, Manitoba, Canada.
“A single time point intervention is unlikely to result in a sustained improvement in VTE prophylaxis rates.”
However, Seifer and her colleagues believe that automated alerts and checklists incorporated into electronic patient records or used via innovative software programs have the potential to improve compliance rates. ![]()
Photo courtesy of the CDC
In a single-center study, researchers found that educating healthcare providers about the need for venous thromboembolism (VTE) prophylaxis did not ensure that patients received appropriate treatment.
Before the educational program was introduced, 36% of patients who were at risk of VTE were not receiving VTE prophylaxis. After the program, 26% of at-risk patients were not receiving prophylaxis.
The researchers reported these findings in the Canadian Journal of Cardiology.
The team carried out chart reviews of patients in a university-affiliated, tertiary care cardiology center, which included a clinical teaching unit and a coronary care unit.
Audits were conducted 3 and 5 months before the introduction of an educational program on VTE prophylaxis protocol, followed by a second series of audits 3 and 5 months after protocol initiation.
In each set of audits, conducted over 2 months, 3 independent groups consisting of a physician and a nonphysician healthcare provider (nursing, pharmacy) each reviewed the data. Discrepancies were settled by the senior investigators.
In the first set of audits, 173 charts for patients considered at high risk for VTE were evaluated. The second set of audits included 247 patients.
Prior to the educational program, including a guideline-based protocol, 36% of all patients who were considered at risk for VTE did not receive prophylaxis.
Three months after the program was initiated, 21% of patients were still not being treated according to the recommended guidelines, and that percentage rose to 28% at 5 months post-protocol.
“Awareness and education surrounding VTE prophylaxis is challenging in the inpatient teaching unit model due to a number of factors, including the high turnover of senior and junior physicians as well as nursing staff,” said study author Colette Seifer, of the University of Manitoba in Winnipeg, Manitoba, Canada.
“A single time point intervention is unlikely to result in a sustained improvement in VTE prophylaxis rates.”
However, Seifer and her colleagues believe that automated alerts and checklists incorporated into electronic patient records or used via innovative software programs have the potential to improve compliance rates. ![]()
How religion affects well-being in cancer patients

Photo by Petr Kratochvil
Three meta-analyses shed new light on the role religion and spirituality play in cancer patients’ mental, social, and physical well-being.
The analyses, published in Cancer, indicate that religion and spirituality have significant associations with patients’ health.
But investigators observed wide variability among studies with regard to how different dimensions of religion and spirituality relate to different aspects of health.
In the first analysis, the investigators focused on physical health. Patients reporting greater overall religiousness and spirituality reported better physical health, greater ability to perform their usual daily tasks, and fewer physical symptoms of cancer and treatment.
“These relationships were particularly strong in patients who experienced greater emotional aspects of religion and spirituality, including a sense of meaning and purpose in life as well as a connection to a source larger than oneself,” said study author Heather Jim, PhD, of the Moffitt Cancer Center in Tampa, Florida.
Dr Jim noted that patients who reported greater cognitive aspects of religion and spirituality, such as the ability to integrate the cancer into their religious or spiritual beliefs, also reported better physical health. However, physical health was not related to behavioral aspects of religion and spiritualty, such as church attendance, prayer, or meditation.
In the second analysis, the investigators examined patients’ mental health. The team discovered that emotional aspects of religion and spirituality were more strongly associated with positive mental health than behavioral or cognitive aspects of religion and spirituality.
“Spiritual well-being was, unsurprisingly, associated with less anxiety, depression, or distress,” said study author John Salsman, PhD, of Wake Forest School of Medicine in Winston-Salem, North Carolina.
“Also, greater levels of spiritual distress and a sense of disconnectedness with God or a religious community was associated with greater psychological distress or poorer emotional well-being.”
The third analysis pertained to social health, or patients’ capacity to retain social roles and relationships in the face of illness. Religion and spirituality, as well as each of its dimensions, had modest but reliable links with social health.
“When we took a closer look, we found that patients with stronger spiritual well-being, more benign images of God (such as perceptions of a benevolent God rather than an angry or distant God), or stronger beliefs (such as convictions that a personal God can be called upon for assistance) reported better social health,” said study author Allen Sherman, PhD, of the University of Arkansas for Medical Sciences in Little Rock. “In contrast, those who struggled with their faith fared more poorly.”
The investigators believe future research should focus on how relationships between religious or spiritual involvement and health change over time and whether support services designed to enhance particular aspects of religion and spirituality in interested patients might help improve their well-being.
“In addition, some patients struggle with the religious or spiritual significance of their cancer, which is normal,” Dr Jim said. “How they resolve their struggle may impact their health, but more research is needed to better understand and support these patients.” ![]()
Photo by Petr Kratochvil
Three meta-analyses shed new light on the role religion and spirituality play in cancer patients’ mental, social, and physical well-being.
The analyses, published in Cancer, indicate that religion and spirituality have significant associations with patients’ health.
But investigators observed wide variability among studies with regard to how different dimensions of religion and spirituality relate to different aspects of health.
In the first analysis, the investigators focused on physical health. Patients reporting greater overall religiousness and spirituality reported better physical health, greater ability to perform their usual daily tasks, and fewer physical symptoms of cancer and treatment.
“These relationships were particularly strong in patients who experienced greater emotional aspects of religion and spirituality, including a sense of meaning and purpose in life as well as a connection to a source larger than oneself,” said study author Heather Jim, PhD, of the Moffitt Cancer Center in Tampa, Florida.
Dr Jim noted that patients who reported greater cognitive aspects of religion and spirituality, such as the ability to integrate the cancer into their religious or spiritual beliefs, also reported better physical health. However, physical health was not related to behavioral aspects of religion and spiritualty, such as church attendance, prayer, or meditation.
In the second analysis, the investigators examined patients’ mental health. The team discovered that emotional aspects of religion and spirituality were more strongly associated with positive mental health than behavioral or cognitive aspects of religion and spirituality.
“Spiritual well-being was, unsurprisingly, associated with less anxiety, depression, or distress,” said study author John Salsman, PhD, of Wake Forest School of Medicine in Winston-Salem, North Carolina.
“Also, greater levels of spiritual distress and a sense of disconnectedness with God or a religious community was associated with greater psychological distress or poorer emotional well-being.”
The third analysis pertained to social health, or patients’ capacity to retain social roles and relationships in the face of illness. Religion and spirituality, as well as each of its dimensions, had modest but reliable links with social health.
“When we took a closer look, we found that patients with stronger spiritual well-being, more benign images of God (such as perceptions of a benevolent God rather than an angry or distant God), or stronger beliefs (such as convictions that a personal God can be called upon for assistance) reported better social health,” said study author Allen Sherman, PhD, of the University of Arkansas for Medical Sciences in Little Rock. “In contrast, those who struggled with their faith fared more poorly.”
The investigators believe future research should focus on how relationships between religious or spiritual involvement and health change over time and whether support services designed to enhance particular aspects of religion and spirituality in interested patients might help improve their well-being.
“In addition, some patients struggle with the religious or spiritual significance of their cancer, which is normal,” Dr Jim said. “How they resolve their struggle may impact their health, but more research is needed to better understand and support these patients.” ![]()
Photo by Petr Kratochvil
Three meta-analyses shed new light on the role religion and spirituality play in cancer patients’ mental, social, and physical well-being.
The analyses, published in Cancer, indicate that religion and spirituality have significant associations with patients’ health.
But investigators observed wide variability among studies with regard to how different dimensions of religion and spirituality relate to different aspects of health.
In the first analysis, the investigators focused on physical health. Patients reporting greater overall religiousness and spirituality reported better physical health, greater ability to perform their usual daily tasks, and fewer physical symptoms of cancer and treatment.
“These relationships were particularly strong in patients who experienced greater emotional aspects of religion and spirituality, including a sense of meaning and purpose in life as well as a connection to a source larger than oneself,” said study author Heather Jim, PhD, of the Moffitt Cancer Center in Tampa, Florida.
Dr Jim noted that patients who reported greater cognitive aspects of religion and spirituality, such as the ability to integrate the cancer into their religious or spiritual beliefs, also reported better physical health. However, physical health was not related to behavioral aspects of religion and spiritualty, such as church attendance, prayer, or meditation.
In the second analysis, the investigators examined patients’ mental health. The team discovered that emotional aspects of religion and spirituality were more strongly associated with positive mental health than behavioral or cognitive aspects of religion and spirituality.
“Spiritual well-being was, unsurprisingly, associated with less anxiety, depression, or distress,” said study author John Salsman, PhD, of Wake Forest School of Medicine in Winston-Salem, North Carolina.
“Also, greater levels of spiritual distress and a sense of disconnectedness with God or a religious community was associated with greater psychological distress or poorer emotional well-being.”
The third analysis pertained to social health, or patients’ capacity to retain social roles and relationships in the face of illness. Religion and spirituality, as well as each of its dimensions, had modest but reliable links with social health.
“When we took a closer look, we found that patients with stronger spiritual well-being, more benign images of God (such as perceptions of a benevolent God rather than an angry or distant God), or stronger beliefs (such as convictions that a personal God can be called upon for assistance) reported better social health,” said study author Allen Sherman, PhD, of the University of Arkansas for Medical Sciences in Little Rock. “In contrast, those who struggled with their faith fared more poorly.”
The investigators believe future research should focus on how relationships between religious or spiritual involvement and health change over time and whether support services designed to enhance particular aspects of religion and spirituality in interested patients might help improve their well-being.
“In addition, some patients struggle with the religious or spiritual significance of their cancer, which is normal,” Dr Jim said. “How they resolve their struggle may impact their health, but more research is needed to better understand and support these patients.” ![]()
Mutations linked to Fanconi anemia, repair pathway

Photo courtesy of NIGMS
Investigators say they have identified mutations that cause Fanconi anemia and, in the process, gained new insight into interstrand crosslink (ICL) repair.
The researchers studied two patients who had Fanconi anemia with no known genetic cause.
Genomic sequencing revealed that one patient had a mutation in RAD51, and the other had mutations in UBE2T.
These genes—and others linked to Fanconi anemia in previous studies—contribute to ICL repair, which fixes a misplaced attachment between two strands of DNA.
Caused by chemical agents, ICLs block the replication of DNA, making it impossible for cells to accurately copy their genomes as they divide. The ICL repair process uses multiple enzymes that cut away the connection between the DNA strands, freeing them up and allowing the cells to grow.
The genome is at constant risk of forming ICLs, and defects in the ICL repair pathway can produce a constellation of symptoms associated with Fanconi anemia—a predisposition to cancer, bone marrow failure, infertility, and developmental defects.
Via two different studies, Agata Smogorzewska, MD, PhD, of The Rockefeller University in New York, New York, and her colleagues unearthed new discoveries relating to the disease and the pathway.
“Our work began, as it often does, with samples and histories from patients,” Dr Smogorzewska said. “In these cases, we had two patients who each represented a sort of mystery. They had symptoms of Fanconi anemia but no genetic cause yet identified.”
With the RAD51 research, which was published in Molecular Cell, the investigators set out to determine the cause of the Fanconi anemia-like symptoms in a girl in the university’s International Fanconi Anemia Registry.
When they sequenced the protein-coding genes in her genome, the researchers found a mutation in one of two copies of the RAD51 gene.
The RAD1 protein was already known to be important for another DNA repair process—homologous recombination, in which a missing section of DNA is replaced using its sister strand as a template. Homologous recombination is thought to be used during the last step of ICL repair, after the crosslink has been cut.
Because only one copy of the RAD51 gene was partially defective, the patient’s cells could still perform homologous recombination but not ICL repair.
To show that the defective copy of the RAD51 gene was indeed responsible for the patient’s symptoms, the investigators genetically engineered the girl’s own cells to remove the defect, which restored their ability to fix ICLs.
Further experiments on the patient’s cells led the researchers to suspect that RAD51 plays a role outside of homologous recombination, by tamping down the activity of two enzymes that degrade the DNA at the ICL. When RAD51 is defective, these enzymes—DNA2 and WRN—become overly destructive.
With the UBE2T study, published in Cell Reports, the investigators analyzed genomic data from another patient in the International Fanconi Anemia Registry.
They found compound heterozygous mutations in UBE2T—a large genomic deletion in the paternally derived allele and a large duplication in the maternally derived allele.
While it was already known that UBE2T is involved in activating ICL repair, the researchers said the discovery that these mutations could produce Fanconi anemia revealed that UBE2T is an irreplaceable player in the pathway.
“Although we have discovered new causes for this devastating but very rare genetic disease, the implications of this work go much further,” Dr Smogorzewska said.
“By identifying new disruptions to this repair pathway, we can better understand the mechanisms of an event that is crucial to every cell division—a process that occurs constantly within the human body throughout a lifetime.” ![]()
Photo courtesy of NIGMS
Investigators say they have identified mutations that cause Fanconi anemia and, in the process, gained new insight into interstrand crosslink (ICL) repair.
The researchers studied two patients who had Fanconi anemia with no known genetic cause.
Genomic sequencing revealed that one patient had a mutation in RAD51, and the other had mutations in UBE2T.
These genes—and others linked to Fanconi anemia in previous studies—contribute to ICL repair, which fixes a misplaced attachment between two strands of DNA.
Caused by chemical agents, ICLs block the replication of DNA, making it impossible for cells to accurately copy their genomes as they divide. The ICL repair process uses multiple enzymes that cut away the connection between the DNA strands, freeing them up and allowing the cells to grow.
The genome is at constant risk of forming ICLs, and defects in the ICL repair pathway can produce a constellation of symptoms associated with Fanconi anemia—a predisposition to cancer, bone marrow failure, infertility, and developmental defects.
Via two different studies, Agata Smogorzewska, MD, PhD, of The Rockefeller University in New York, New York, and her colleagues unearthed new discoveries relating to the disease and the pathway.
“Our work began, as it often does, with samples and histories from patients,” Dr Smogorzewska said. “In these cases, we had two patients who each represented a sort of mystery. They had symptoms of Fanconi anemia but no genetic cause yet identified.”
With the RAD51 research, which was published in Molecular Cell, the investigators set out to determine the cause of the Fanconi anemia-like symptoms in a girl in the university’s International Fanconi Anemia Registry.
When they sequenced the protein-coding genes in her genome, the researchers found a mutation in one of two copies of the RAD51 gene.
The RAD1 protein was already known to be important for another DNA repair process—homologous recombination, in which a missing section of DNA is replaced using its sister strand as a template. Homologous recombination is thought to be used during the last step of ICL repair, after the crosslink has been cut.
Because only one copy of the RAD51 gene was partially defective, the patient’s cells could still perform homologous recombination but not ICL repair.
To show that the defective copy of the RAD51 gene was indeed responsible for the patient’s symptoms, the investigators genetically engineered the girl’s own cells to remove the defect, which restored their ability to fix ICLs.
Further experiments on the patient’s cells led the researchers to suspect that RAD51 plays a role outside of homologous recombination, by tamping down the activity of two enzymes that degrade the DNA at the ICL. When RAD51 is defective, these enzymes—DNA2 and WRN—become overly destructive.
With the UBE2T study, published in Cell Reports, the investigators analyzed genomic data from another patient in the International Fanconi Anemia Registry.
They found compound heterozygous mutations in UBE2T—a large genomic deletion in the paternally derived allele and a large duplication in the maternally derived allele.
While it was already known that UBE2T is involved in activating ICL repair, the researchers said the discovery that these mutations could produce Fanconi anemia revealed that UBE2T is an irreplaceable player in the pathway.
“Although we have discovered new causes for this devastating but very rare genetic disease, the implications of this work go much further,” Dr Smogorzewska said.
“By identifying new disruptions to this repair pathway, we can better understand the mechanisms of an event that is crucial to every cell division—a process that occurs constantly within the human body throughout a lifetime.” ![]()
Photo courtesy of NIGMS
Investigators say they have identified mutations that cause Fanconi anemia and, in the process, gained new insight into interstrand crosslink (ICL) repair.
The researchers studied two patients who had Fanconi anemia with no known genetic cause.
Genomic sequencing revealed that one patient had a mutation in RAD51, and the other had mutations in UBE2T.
These genes—and others linked to Fanconi anemia in previous studies—contribute to ICL repair, which fixes a misplaced attachment between two strands of DNA.
Caused by chemical agents, ICLs block the replication of DNA, making it impossible for cells to accurately copy their genomes as they divide. The ICL repair process uses multiple enzymes that cut away the connection between the DNA strands, freeing them up and allowing the cells to grow.
The genome is at constant risk of forming ICLs, and defects in the ICL repair pathway can produce a constellation of symptoms associated with Fanconi anemia—a predisposition to cancer, bone marrow failure, infertility, and developmental defects.
Via two different studies, Agata Smogorzewska, MD, PhD, of The Rockefeller University in New York, New York, and her colleagues unearthed new discoveries relating to the disease and the pathway.
“Our work began, as it often does, with samples and histories from patients,” Dr Smogorzewska said. “In these cases, we had two patients who each represented a sort of mystery. They had symptoms of Fanconi anemia but no genetic cause yet identified.”
With the RAD51 research, which was published in Molecular Cell, the investigators set out to determine the cause of the Fanconi anemia-like symptoms in a girl in the university’s International Fanconi Anemia Registry.
When they sequenced the protein-coding genes in her genome, the researchers found a mutation in one of two copies of the RAD51 gene.
The RAD1 protein was already known to be important for another DNA repair process—homologous recombination, in which a missing section of DNA is replaced using its sister strand as a template. Homologous recombination is thought to be used during the last step of ICL repair, after the crosslink has been cut.
Because only one copy of the RAD51 gene was partially defective, the patient’s cells could still perform homologous recombination but not ICL repair.
To show that the defective copy of the RAD51 gene was indeed responsible for the patient’s symptoms, the investigators genetically engineered the girl’s own cells to remove the defect, which restored their ability to fix ICLs.
Further experiments on the patient’s cells led the researchers to suspect that RAD51 plays a role outside of homologous recombination, by tamping down the activity of two enzymes that degrade the DNA at the ICL. When RAD51 is defective, these enzymes—DNA2 and WRN—become overly destructive.
With the UBE2T study, published in Cell Reports, the investigators analyzed genomic data from another patient in the International Fanconi Anemia Registry.
They found compound heterozygous mutations in UBE2T—a large genomic deletion in the paternally derived allele and a large duplication in the maternally derived allele.
While it was already known that UBE2T is involved in activating ICL repair, the researchers said the discovery that these mutations could produce Fanconi anemia revealed that UBE2T is an irreplaceable player in the pathway.
“Although we have discovered new causes for this devastating but very rare genetic disease, the implications of this work go much further,” Dr Smogorzewska said.
“By identifying new disruptions to this repair pathway, we can better understand the mechanisms of an event that is crucial to every cell division—a process that occurs constantly within the human body throughout a lifetime.”
Compound could aid fight against malaria
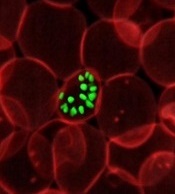
infecting an RBC
Image courtesy of St. Jude
Children’s Research Hospital
Luminol, the compound detectives spray at crime scenes to find trace amounts of blood, can help kill malaria parasites, according to preclinical research published in eLife.
Luminol glows blue when it encounters the hemoglobin in red blood cells (RBCs), and researchers have found they can trick malaria-infected RBCs into
building up a volatile chemical stockpile that can be set off by luminol’s glow.
To achieve this, the researchers exposed infected RBCs to an amino acid known as 5-aminolevulinic acid (ALA), luminol, and 4-iodophenol (a small-molecule that enhances the intensity and duration of luminol chemiluminescence).
This triggered buildup of the chemical, protoporphyrin IX (PPIX), which effectively killed the parasites. When the team substituted artemisinin for 4-iodophenol, they observed similar results.
“The light that luminol emits is enhanced by the antimalarial drug artemisinin,” said study author Daniel Goldberg, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“We think these agents could be combined to form an innovative treatment for malaria.”
The researchers believe this type of therapy would have an advantage over current malaria treatments, which have become less effective as the parasite mutates. That is because the new approach targets proteins made by human RBCs, which the parasite can’t mutate.
To uncover this approach, Dr Goldberg and his colleagues worked with human RBCs infected with Plasmodium falciparum. The team wanted to better understand how the parasite gets hold of heme, which is essential to the parasite’s survival.
They found that P falciparum opens an unnatural channel on the surface of RBCs. When the researchers put ALA (an ingredient of heme) into a solution containing infected RBCs, ALA entered the cells through the channel and started the heme-making process.
This led to a buildup of PPIX. When exposed to luminol and 4-iodophenol, PPIX emitted free radicals. This potently inhibited parasite growth, according to the researchers. And microscopic examination revealed widespread parasite death.
The ALA/luminol/4-iodophenol combination also worked in a parasite line that was resistant to antifolate and quinolone antibiotics, as well as one with a kelch-13 protein mutation, which confers artemisinin tolerance.
The researchers then wanted to determine if artemisinin would enhance their strategy. So they incubated malaria-infected RBCs with ALA, luminol, and/or sub-therapeutic doses of dihydroartemisinin.
Each of the components alone or 2 of them together had little effect, but all 3 in combination successfully ablated parasite growth.
The researchers are now planning to test this treatment approach in vivo.
“All of these agents—the amino acid, the luminol, and artemisinin—have been cleared for use in humans individually, so we are optimistic that they won’t present any safety problems together,” Dr Goldberg said. “This could be a promising new treatment for a devastating disease.”
infecting an RBC
Image courtesy of St. Jude
Children’s Research Hospital
Luminol, the compound detectives spray at crime scenes to find trace amounts of blood, can help kill malaria parasites, according to preclinical research published in eLife.
Luminol glows blue when it encounters the hemoglobin in red blood cells (RBCs), and researchers have found they can trick malaria-infected RBCs into
building up a volatile chemical stockpile that can be set off by luminol’s glow.
To achieve this, the researchers exposed infected RBCs to an amino acid known as 5-aminolevulinic acid (ALA), luminol, and 4-iodophenol (a small-molecule that enhances the intensity and duration of luminol chemiluminescence).
This triggered buildup of the chemical, protoporphyrin IX (PPIX), which effectively killed the parasites. When the team substituted artemisinin for 4-iodophenol, they observed similar results.
“The light that luminol emits is enhanced by the antimalarial drug artemisinin,” said study author Daniel Goldberg, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“We think these agents could be combined to form an innovative treatment for malaria.”
The researchers believe this type of therapy would have an advantage over current malaria treatments, which have become less effective as the parasite mutates. That is because the new approach targets proteins made by human RBCs, which the parasite can’t mutate.
To uncover this approach, Dr Goldberg and his colleagues worked with human RBCs infected with Plasmodium falciparum. The team wanted to better understand how the parasite gets hold of heme, which is essential to the parasite’s survival.
They found that P falciparum opens an unnatural channel on the surface of RBCs. When the researchers put ALA (an ingredient of heme) into a solution containing infected RBCs, ALA entered the cells through the channel and started the heme-making process.
This led to a buildup of PPIX. When exposed to luminol and 4-iodophenol, PPIX emitted free radicals. This potently inhibited parasite growth, according to the researchers. And microscopic examination revealed widespread parasite death.
The ALA/luminol/4-iodophenol combination also worked in a parasite line that was resistant to antifolate and quinolone antibiotics, as well as one with a kelch-13 protein mutation, which confers artemisinin tolerance.
The researchers then wanted to determine if artemisinin would enhance their strategy. So they incubated malaria-infected RBCs with ALA, luminol, and/or sub-therapeutic doses of dihydroartemisinin.
Each of the components alone or 2 of them together had little effect, but all 3 in combination successfully ablated parasite growth.
The researchers are now planning to test this treatment approach in vivo.
“All of these agents—the amino acid, the luminol, and artemisinin—have been cleared for use in humans individually, so we are optimistic that they won’t present any safety problems together,” Dr Goldberg said. “This could be a promising new treatment for a devastating disease.”
infecting an RBC
Image courtesy of St. Jude
Children’s Research Hospital
Luminol, the compound detectives spray at crime scenes to find trace amounts of blood, can help kill malaria parasites, according to preclinical research published in eLife.
Luminol glows blue when it encounters the hemoglobin in red blood cells (RBCs), and researchers have found they can trick malaria-infected RBCs into
building up a volatile chemical stockpile that can be set off by luminol’s glow.
To achieve this, the researchers exposed infected RBCs to an amino acid known as 5-aminolevulinic acid (ALA), luminol, and 4-iodophenol (a small-molecule that enhances the intensity and duration of luminol chemiluminescence).
This triggered buildup of the chemical, protoporphyrin IX (PPIX), which effectively killed the parasites. When the team substituted artemisinin for 4-iodophenol, they observed similar results.
“The light that luminol emits is enhanced by the antimalarial drug artemisinin,” said study author Daniel Goldberg, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“We think these agents could be combined to form an innovative treatment for malaria.”
The researchers believe this type of therapy would have an advantage over current malaria treatments, which have become less effective as the parasite mutates. That is because the new approach targets proteins made by human RBCs, which the parasite can’t mutate.
To uncover this approach, Dr Goldberg and his colleagues worked with human RBCs infected with Plasmodium falciparum. The team wanted to better understand how the parasite gets hold of heme, which is essential to the parasite’s survival.
They found that P falciparum opens an unnatural channel on the surface of RBCs. When the researchers put ALA (an ingredient of heme) into a solution containing infected RBCs, ALA entered the cells through the channel and started the heme-making process.
This led to a buildup of PPIX. When exposed to luminol and 4-iodophenol, PPIX emitted free radicals. This potently inhibited parasite growth, according to the researchers. And microscopic examination revealed widespread parasite death.
The ALA/luminol/4-iodophenol combination also worked in a parasite line that was resistant to antifolate and quinolone antibiotics, as well as one with a kelch-13 protein mutation, which confers artemisinin tolerance.
The researchers then wanted to determine if artemisinin would enhance their strategy. So they incubated malaria-infected RBCs with ALA, luminol, and/or sub-therapeutic doses of dihydroartemisinin.
Each of the components alone or 2 of them together had little effect, but all 3 in combination successfully ablated parasite growth.
The researchers are now planning to test this treatment approach in vivo.
“All of these agents—the amino acid, the luminol, and artemisinin—have been cleared for use in humans individually, so we are optimistic that they won’t present any safety problems together,” Dr Goldberg said. “This could be a promising new treatment for a devastating disease.”
Topical gel appears safe, effective in CTCL
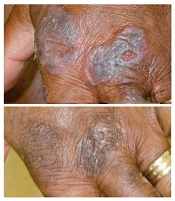
treatment (top) and 16 weeks
after treatment began
Photo from Penn Medicine
Results of a phase 1 trial suggest a topical gel can prompt regression of both treated and untreated lesions in patients with
early stage cutaneous T-cell lymphoma (CTCL).
Of the 12 patients who received the treatment, resiquimod gel, 75% had a significant improvement in treated lesions.
And 92% of patients had a more than 50% improvement in body surface area involvement, which included untreated lesions. Two patients experienced complete disease clearance.
Adverse events associated with resiquimod were largely limited to the skin, although 2 patients had transient, low-grade fever. Five patients developed superficial skin erosions that healed when treatment was stopped and did not reappear after treatment began again.
Alain Rook, MD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these results in Blood.
“The results of the trial suggest that resiquimod is safely and effectively absorbed into the skin and, beyond diminishing treated lesions, also enhances the immune response, leading to healing of even untreated lesions,” Dr Rook said.
“To our knowledge, this is the first topical therapy that can clear untreated lesions and lead to complete remission in some patients.”
Treatment and response
Dr Rook and his colleagues tested resiquimod gel in 12 patients who had previously undergone an average of 6 treatments for early stage CTCL.
The patients applied specified doses of resiquimod (0.03% or 0.06%) to select skin lesions for 16 weeks. Some patients using the 0.06% dose had complete clearance of all malignant cells after 8 weeks.
By the final evaluation, treated lesions had significantly improved in 75% of patients, and 30% of patients had complete clearance of all treated lesions.
Resiquimod also improved untreated lesions, resulting in more than 50% improvement in body surface area involvement for 92% of patients.
Two participants, one of whom had been living with CTCL for more than 15 years without responding to treatment, experienced complete eradication of the disease.
Malignant cell analysis
The researchers used high-throughput sequencing to detect malignant cells in patient samples. The technique could identify a single malignant cell among 100,000 healthy cells.
The team analyzed DNA from biopsies of the same lesion before treatment and 8 weeks after treatment began.
They observed a significant reduction of malignant T cells in 9 of 10 patients tested, 3 of whom had complete eradication of the malignant population and 1 of whom had a 99.6% reduction.
Adverse events
Adverse events associated with resiquimod were all grade 1 and were primarily related to local skin irritation. There were no serious adverse events.
Five of 8 patients receiving resiquimod at the 0.06% dose developed superficial skin erosions at some sites of treatment. These erosions healed completely within a week of stopping treatment and did not recur with re-initiation of treatment.
Two patients receiving the 0.06% dose experienced 2 days of low-grade fever (less than 100.50 F) when treatment began.
“Overall, lesions responded far better to topical resiquimod than they have with other topical therapies, including some potent topical steroids and topical chemotherapy, and [resiquimod] was extremely well tolerated by patients,” Dr Rook said.
“Building upon previous research, our study suggests resiquimod might be useful in combination with other therapies in the treatment of more advanced CTCL. Further research with larger participant populations is needed to determine the best approach and application for these patients.”
treatment (top) and 16 weeks
after treatment began
Photo from Penn Medicine
Results of a phase 1 trial suggest a topical gel can prompt regression of both treated and untreated lesions in patients with
early stage cutaneous T-cell lymphoma (CTCL).
Of the 12 patients who received the treatment, resiquimod gel, 75% had a significant improvement in treated lesions.
And 92% of patients had a more than 50% improvement in body surface area involvement, which included untreated lesions. Two patients experienced complete disease clearance.
Adverse events associated with resiquimod were largely limited to the skin, although 2 patients had transient, low-grade fever. Five patients developed superficial skin erosions that healed when treatment was stopped and did not reappear after treatment began again.
Alain Rook, MD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these results in Blood.
“The results of the trial suggest that resiquimod is safely and effectively absorbed into the skin and, beyond diminishing treated lesions, also enhances the immune response, leading to healing of even untreated lesions,” Dr Rook said.
“To our knowledge, this is the first topical therapy that can clear untreated lesions and lead to complete remission in some patients.”
Treatment and response
Dr Rook and his colleagues tested resiquimod gel in 12 patients who had previously undergone an average of 6 treatments for early stage CTCL.
The patients applied specified doses of resiquimod (0.03% or 0.06%) to select skin lesions for 16 weeks. Some patients using the 0.06% dose had complete clearance of all malignant cells after 8 weeks.
By the final evaluation, treated lesions had significantly improved in 75% of patients, and 30% of patients had complete clearance of all treated lesions.
Resiquimod also improved untreated lesions, resulting in more than 50% improvement in body surface area involvement for 92% of patients.
Two participants, one of whom had been living with CTCL for more than 15 years without responding to treatment, experienced complete eradication of the disease.
Malignant cell analysis
The researchers used high-throughput sequencing to detect malignant cells in patient samples. The technique could identify a single malignant cell among 100,000 healthy cells.
The team analyzed DNA from biopsies of the same lesion before treatment and 8 weeks after treatment began.
They observed a significant reduction of malignant T cells in 9 of 10 patients tested, 3 of whom had complete eradication of the malignant population and 1 of whom had a 99.6% reduction.
Adverse events
Adverse events associated with resiquimod were all grade 1 and were primarily related to local skin irritation. There were no serious adverse events.
Five of 8 patients receiving resiquimod at the 0.06% dose developed superficial skin erosions at some sites of treatment. These erosions healed completely within a week of stopping treatment and did not recur with re-initiation of treatment.
Two patients receiving the 0.06% dose experienced 2 days of low-grade fever (less than 100.50 F) when treatment began.
“Overall, lesions responded far better to topical resiquimod than they have with other topical therapies, including some potent topical steroids and topical chemotherapy, and [resiquimod] was extremely well tolerated by patients,” Dr Rook said.
“Building upon previous research, our study suggests resiquimod might be useful in combination with other therapies in the treatment of more advanced CTCL. Further research with larger participant populations is needed to determine the best approach and application for these patients.”
treatment (top) and 16 weeks
after treatment began
Photo from Penn Medicine
Results of a phase 1 trial suggest a topical gel can prompt regression of both treated and untreated lesions in patients with
early stage cutaneous T-cell lymphoma (CTCL).
Of the 12 patients who received the treatment, resiquimod gel, 75% had a significant improvement in treated lesions.
And 92% of patients had a more than 50% improvement in body surface area involvement, which included untreated lesions. Two patients experienced complete disease clearance.
Adverse events associated with resiquimod were largely limited to the skin, although 2 patients had transient, low-grade fever. Five patients developed superficial skin erosions that healed when treatment was stopped and did not reappear after treatment began again.
Alain Rook, MD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these results in Blood.
“The results of the trial suggest that resiquimod is safely and effectively absorbed into the skin and, beyond diminishing treated lesions, also enhances the immune response, leading to healing of even untreated lesions,” Dr Rook said.
“To our knowledge, this is the first topical therapy that can clear untreated lesions and lead to complete remission in some patients.”
Treatment and response
Dr Rook and his colleagues tested resiquimod gel in 12 patients who had previously undergone an average of 6 treatments for early stage CTCL.
The patients applied specified doses of resiquimod (0.03% or 0.06%) to select skin lesions for 16 weeks. Some patients using the 0.06% dose had complete clearance of all malignant cells after 8 weeks.
By the final evaluation, treated lesions had significantly improved in 75% of patients, and 30% of patients had complete clearance of all treated lesions.
Resiquimod also improved untreated lesions, resulting in more than 50% improvement in body surface area involvement for 92% of patients.
Two participants, one of whom had been living with CTCL for more than 15 years without responding to treatment, experienced complete eradication of the disease.
Malignant cell analysis
The researchers used high-throughput sequencing to detect malignant cells in patient samples. The technique could identify a single malignant cell among 100,000 healthy cells.
The team analyzed DNA from biopsies of the same lesion before treatment and 8 weeks after treatment began.
They observed a significant reduction of malignant T cells in 9 of 10 patients tested, 3 of whom had complete eradication of the malignant population and 1 of whom had a 99.6% reduction.
Adverse events
Adverse events associated with resiquimod were all grade 1 and were primarily related to local skin irritation. There were no serious adverse events.
Five of 8 patients receiving resiquimod at the 0.06% dose developed superficial skin erosions at some sites of treatment. These erosions healed completely within a week of stopping treatment and did not recur with re-initiation of treatment.
Two patients receiving the 0.06% dose experienced 2 days of low-grade fever (less than 100.50 F) when treatment began.
“Overall, lesions responded far better to topical resiquimod than they have with other topical therapies, including some potent topical steroids and topical chemotherapy, and [resiquimod] was extremely well tolerated by patients,” Dr Rook said.
“Building upon previous research, our study suggests resiquimod might be useful in combination with other therapies in the treatment of more advanced CTCL. Further research with larger participant populations is needed to determine the best approach and application for these patients.”
FDA approves IVIG product for kids

Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information.
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information.
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information.