User login
Combo shows promise for treating DLBCL

The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
Study: CMV doesn’t lower risk of relapse, death
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Dementia isn’t passed on via transfusion, team says

Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Artemisinin resistance confined to Asia, study shows
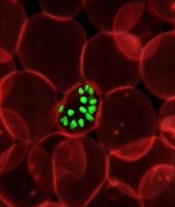
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.” ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.” ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.” ![]()
Agreements may constrain publication of trial results

for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted. ![]()
for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted. ![]()
for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted. ![]()
Team maps chromatin landscape in CLL

Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.” ![]()
Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.” ![]()
Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.” ![]()
EBV-CTL product classified as ATMP
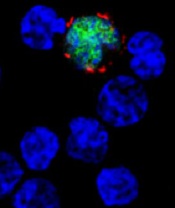
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US. ![]()
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US. ![]()
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US.
P vivax evolving differently in different regions
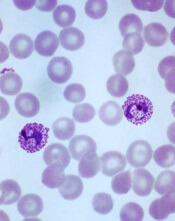
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.”
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.”
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.”
Immunotherapy drugs linked to rheumatic diseases

Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications.
Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications.
Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications.
Study explains how a mutation spurs AML development
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML.
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML.
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML.