User login
NICE recommends approval for bosutinib

Photo courtesy of CDC
The National Institute for Health and Care Excellence (NICE) has issued a final draft guidance recommending approval for bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML).
NICE is recommending that bosutinib be made available through normal National Health Service (NHS) funding channels so patients don’t have to apply to the Cancer Drugs Fund (CDF) to obtain it.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.
“People with this type of chronic myeloid leukemia, who haven’t responded to first- and second-line treatment or who experience severe side effects, have few or no treatment options left,” said Carole Longson, director of the Centre for Health Technology Evaluation at NICE.
“New patients who need this drug can be reassured that bosutinib should be made available for routine use within the NHS.”
The current list price of bosutinib is £45,000 per patient per year. However, the NHS has been offered a discount by Pfizer, the drug’s manufacturer.
NICE previously looked at bosutinib in 2013 but did not recommend the drug for use on the NHS at that time, saying the drug was not cost-effective. Bosutinib was then made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
“The company positively engaged with our CDF reconsideration process and demonstrated that their drug can be cost-effective, which resulted in a positive recommendation,” Longson said. “This decision, when implemented, frees up funding in the CDF, which can be spent on other new and innovative cancer treatments.”
NICE’s final draft guidance is now with consultees who have the opportunity to appeal against the decision or notify NICE of any factual errors. The appeal period will close at 5 pm on July 21, 2016.
Until the final decision is published, bosutinib will still be available to new and existing patients through the old CDF. ![]()
Photo courtesy of CDC
The National Institute for Health and Care Excellence (NICE) has issued a final draft guidance recommending approval for bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML).
NICE is recommending that bosutinib be made available through normal National Health Service (NHS) funding channels so patients don’t have to apply to the Cancer Drugs Fund (CDF) to obtain it.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.
“People with this type of chronic myeloid leukemia, who haven’t responded to first- and second-line treatment or who experience severe side effects, have few or no treatment options left,” said Carole Longson, director of the Centre for Health Technology Evaluation at NICE.
“New patients who need this drug can be reassured that bosutinib should be made available for routine use within the NHS.”
The current list price of bosutinib is £45,000 per patient per year. However, the NHS has been offered a discount by Pfizer, the drug’s manufacturer.
NICE previously looked at bosutinib in 2013 but did not recommend the drug for use on the NHS at that time, saying the drug was not cost-effective. Bosutinib was then made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
“The company positively engaged with our CDF reconsideration process and demonstrated that their drug can be cost-effective, which resulted in a positive recommendation,” Longson said. “This decision, when implemented, frees up funding in the CDF, which can be spent on other new and innovative cancer treatments.”
NICE’s final draft guidance is now with consultees who have the opportunity to appeal against the decision or notify NICE of any factual errors. The appeal period will close at 5 pm on July 21, 2016.
Until the final decision is published, bosutinib will still be available to new and existing patients through the old CDF. ![]()
Photo courtesy of CDC
The National Institute for Health and Care Excellence (NICE) has issued a final draft guidance recommending approval for bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML).
NICE is recommending that bosutinib be made available through normal National Health Service (NHS) funding channels so patients don’t have to apply to the Cancer Drugs Fund (CDF) to obtain it.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.
“People with this type of chronic myeloid leukemia, who haven’t responded to first- and second-line treatment or who experience severe side effects, have few or no treatment options left,” said Carole Longson, director of the Centre for Health Technology Evaluation at NICE.
“New patients who need this drug can be reassured that bosutinib should be made available for routine use within the NHS.”
The current list price of bosutinib is £45,000 per patient per year. However, the NHS has been offered a discount by Pfizer, the drug’s manufacturer.
NICE previously looked at bosutinib in 2013 but did not recommend the drug for use on the NHS at that time, saying the drug was not cost-effective. Bosutinib was then made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
“The company positively engaged with our CDF reconsideration process and demonstrated that their drug can be cost-effective, which resulted in a positive recommendation,” Longson said. “This decision, when implemented, frees up funding in the CDF, which can be spent on other new and innovative cancer treatments.”
NICE’s final draft guidance is now with consultees who have the opportunity to appeal against the decision or notify NICE of any factual errors. The appeal period will close at 5 pm on July 21, 2016.
Until the final decision is published, bosutinib will still be available to new and existing patients through the old CDF. ![]()
EC extends marketing authorization for brentuximab vedotin

Photo from Business Wire
The European Commission (EC) has extended the current conditional marketing authorization of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) who are at an increased risk of relapse or progression following autologous stem cell transplant (ASCT).
Conditional marketing authorizations are valid for 1 year and are reviewed annually.
The company developing the drug is required to provide comprehensive data confirming the drug’s benefit-risk balance is positive. Once these data are available, the marketing authorization may be converted into a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The EC’s decision to extend the conditional marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The European Commission (EC) has extended the current conditional marketing authorization of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) who are at an increased risk of relapse or progression following autologous stem cell transplant (ASCT).
Conditional marketing authorizations are valid for 1 year and are reviewed annually.
The company developing the drug is required to provide comprehensive data confirming the drug’s benefit-risk balance is positive. Once these data are available, the marketing authorization may be converted into a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The EC’s decision to extend the conditional marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The European Commission (EC) has extended the current conditional marketing authorization of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) who are at an increased risk of relapse or progression following autologous stem cell transplant (ASCT).
Conditional marketing authorizations are valid for 1 year and are reviewed annually.
The company developing the drug is required to provide comprehensive data confirming the drug’s benefit-risk balance is positive. Once these data are available, the marketing authorization may be converted into a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The EC’s decision to extend the conditional marketing authorization of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
NHL patients may have higher risk of second cancer

Photo courtesy of NIH
Compared to patients with other common cancers, patients with non-Hodgkin lymphoma (NHL) have a higher risk of developing a second, unrelated malignancy, according to a new study.
Researchers looked at data on more than 2.1 million patients with 10 of the most common cancers and found that patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The researchers reported these findings in Cancer.
For this study, Karim Chamie, MD, of the University of California, Los Angeles, and his colleagues looked at data from Surveillance, Epidemiology, and End Results database.
The team identified patients age 18 and older who were diagnosed with one of the 10 most common cancers—NHL, melanoma, and prostate, breast, lung, colon, rectal, bladder, uterine, and kidney cancers—between 1992 and 2008.
Of the 2,116,163 patients identified, 170,865 (8.1%) developed a second primary malignancy.
In multivariable analysis, patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The hazard ratios for patients with NHL were 2.70 for men and 2.88 for women. The hazard ratios for bladder cancer were 1.88 for men and 1.66 for women.
Lung cancer was a common second malignancy for both NHL and bladder cancer patients. NHL patients also tended to develop prostate and breast cancer.
Among patients with 2 incident cancers, 13% died of their initial cancer, and 55% died of their second primary malignancy. Lung cancer was the cause of death in 12% of the patients.
“As clinicians, we can become so focused on surveilling our patients to see if a primary cancer recurs that we sometimes may not be aware that patients can be at risk of developing a second, unrelated cancer,” Dr Chamie said.
He and his colleagues believe this study makes a case for monitoring cancer patients for second malignancies. ![]()
Photo courtesy of NIH
Compared to patients with other common cancers, patients with non-Hodgkin lymphoma (NHL) have a higher risk of developing a second, unrelated malignancy, according to a new study.
Researchers looked at data on more than 2.1 million patients with 10 of the most common cancers and found that patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The researchers reported these findings in Cancer.
For this study, Karim Chamie, MD, of the University of California, Los Angeles, and his colleagues looked at data from Surveillance, Epidemiology, and End Results database.
The team identified patients age 18 and older who were diagnosed with one of the 10 most common cancers—NHL, melanoma, and prostate, breast, lung, colon, rectal, bladder, uterine, and kidney cancers—between 1992 and 2008.
Of the 2,116,163 patients identified, 170,865 (8.1%) developed a second primary malignancy.
In multivariable analysis, patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The hazard ratios for patients with NHL were 2.70 for men and 2.88 for women. The hazard ratios for bladder cancer were 1.88 for men and 1.66 for women.
Lung cancer was a common second malignancy for both NHL and bladder cancer patients. NHL patients also tended to develop prostate and breast cancer.
Among patients with 2 incident cancers, 13% died of their initial cancer, and 55% died of their second primary malignancy. Lung cancer was the cause of death in 12% of the patients.
“As clinicians, we can become so focused on surveilling our patients to see if a primary cancer recurs that we sometimes may not be aware that patients can be at risk of developing a second, unrelated cancer,” Dr Chamie said.
He and his colleagues believe this study makes a case for monitoring cancer patients for second malignancies. ![]()
Photo courtesy of NIH
Compared to patients with other common cancers, patients with non-Hodgkin lymphoma (NHL) have a higher risk of developing a second, unrelated malignancy, according to a new study.
Researchers looked at data on more than 2.1 million patients with 10 of the most common cancers and found that patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The researchers reported these findings in Cancer.
For this study, Karim Chamie, MD, of the University of California, Los Angeles, and his colleagues looked at data from Surveillance, Epidemiology, and End Results database.
The team identified patients age 18 and older who were diagnosed with one of the 10 most common cancers—NHL, melanoma, and prostate, breast, lung, colon, rectal, bladder, uterine, and kidney cancers—between 1992 and 2008.
Of the 2,116,163 patients identified, 170,865 (8.1%) developed a second primary malignancy.
In multivariable analysis, patients with NHL or bladder cancer had the highest risk of developing a second malignancy.
The hazard ratios for patients with NHL were 2.70 for men and 2.88 for women. The hazard ratios for bladder cancer were 1.88 for men and 1.66 for women.
Lung cancer was a common second malignancy for both NHL and bladder cancer patients. NHL patients also tended to develop prostate and breast cancer.
Among patients with 2 incident cancers, 13% died of their initial cancer, and 55% died of their second primary malignancy. Lung cancer was the cause of death in 12% of the patients.
“As clinicians, we can become so focused on surveilling our patients to see if a primary cancer recurs that we sometimes may not be aware that patients can be at risk of developing a second, unrelated cancer,” Dr Chamie said.
He and his colleagues believe this study makes a case for monitoring cancer patients for second malignancies. ![]()
Study reveals global variations of P vivax
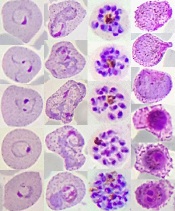
from patients in Thailand
Image by Wanlapa Roobsoong
Researchers say they have uncovered the global, evolving, and historic make-up of the malaria parasite Plasmodium vivax.
The group’s study revealed 4 genetically distinct populations of P vivax that provide insight into the movement of the parasite over time and
show how it is still adapting to regional variations in the mosquitoes that transmit P vivax, the humans infected with the parasite, and the drugs used to fight it.
“Our findings show it is evolving in response to antimalarial drugs and adapting to regional differences, indicating a wide range of approaches will likely be necessary to eliminate it globally,” said Jane Carlton, PhD, of New York University in New York, New York.
Dr Carlton and her colleagues reported these findings in Nature Genetics.
The researchers sequenced 182 DNA samples of P vivax collected from patients in 11 countries. The team said this provided new insights into the nature of P vivax as it exists today and also served as a “genetic history book” of the studied regions.
“The DNA data show that P vivax has clearly had a different history of association with global human populations than other malaria parasites, indicating that unique aspects of its biology may have influenced the ways in which it spread around the world,” said Daniel Neafsey, PhD, of the Broad Institute in Cambridge, Massachusetts.
Specifically, the researchers found that Central and South American P vivax populations are genetically diverse and distinct from all other contemporary P vivax populations. The team said this suggests that New World parasites may have been introduced by colonial seafarers and represent a now-eliminated European parasite population.
The researchers also found that contemporary African and South Asian P vivax populations are genetically similar. They said this suggests that South Asian P vivax populations may have genetically mingled with European lineages during the colonial era, or it may reflect ancient connections between human populations in the Eastern Mediterranean, Middle East, and Indian subcontinent.
Another finding was the relatively homogeneous genetic makeup of P vivax in Mexico, which reflects a steady decline of the disease in this country over the last decade.
By contrast, the Papua New Guinea population of P vivax was shown to be very diverse relative to other P vivax populations.
A similar study, which also illustrated the global variations of P vivax, was recently published in Nature Genetics in as well. ![]()
from patients in Thailand
Image by Wanlapa Roobsoong
Researchers say they have uncovered the global, evolving, and historic make-up of the malaria parasite Plasmodium vivax.
The group’s study revealed 4 genetically distinct populations of P vivax that provide insight into the movement of the parasite over time and
show how it is still adapting to regional variations in the mosquitoes that transmit P vivax, the humans infected with the parasite, and the drugs used to fight it.
“Our findings show it is evolving in response to antimalarial drugs and adapting to regional differences, indicating a wide range of approaches will likely be necessary to eliminate it globally,” said Jane Carlton, PhD, of New York University in New York, New York.
Dr Carlton and her colleagues reported these findings in Nature Genetics.
The researchers sequenced 182 DNA samples of P vivax collected from patients in 11 countries. The team said this provided new insights into the nature of P vivax as it exists today and also served as a “genetic history book” of the studied regions.
“The DNA data show that P vivax has clearly had a different history of association with global human populations than other malaria parasites, indicating that unique aspects of its biology may have influenced the ways in which it spread around the world,” said Daniel Neafsey, PhD, of the Broad Institute in Cambridge, Massachusetts.
Specifically, the researchers found that Central and South American P vivax populations are genetically diverse and distinct from all other contemporary P vivax populations. The team said this suggests that New World parasites may have been introduced by colonial seafarers and represent a now-eliminated European parasite population.
The researchers also found that contemporary African and South Asian P vivax populations are genetically similar. They said this suggests that South Asian P vivax populations may have genetically mingled with European lineages during the colonial era, or it may reflect ancient connections between human populations in the Eastern Mediterranean, Middle East, and Indian subcontinent.
Another finding was the relatively homogeneous genetic makeup of P vivax in Mexico, which reflects a steady decline of the disease in this country over the last decade.
By contrast, the Papua New Guinea population of P vivax was shown to be very diverse relative to other P vivax populations.
A similar study, which also illustrated the global variations of P vivax, was recently published in Nature Genetics in as well. ![]()
from patients in Thailand
Image by Wanlapa Roobsoong
Researchers say they have uncovered the global, evolving, and historic make-up of the malaria parasite Plasmodium vivax.
The group’s study revealed 4 genetically distinct populations of P vivax that provide insight into the movement of the parasite over time and
show how it is still adapting to regional variations in the mosquitoes that transmit P vivax, the humans infected with the parasite, and the drugs used to fight it.
“Our findings show it is evolving in response to antimalarial drugs and adapting to regional differences, indicating a wide range of approaches will likely be necessary to eliminate it globally,” said Jane Carlton, PhD, of New York University in New York, New York.
Dr Carlton and her colleagues reported these findings in Nature Genetics.
The researchers sequenced 182 DNA samples of P vivax collected from patients in 11 countries. The team said this provided new insights into the nature of P vivax as it exists today and also served as a “genetic history book” of the studied regions.
“The DNA data show that P vivax has clearly had a different history of association with global human populations than other malaria parasites, indicating that unique aspects of its biology may have influenced the ways in which it spread around the world,” said Daniel Neafsey, PhD, of the Broad Institute in Cambridge, Massachusetts.
Specifically, the researchers found that Central and South American P vivax populations are genetically diverse and distinct from all other contemporary P vivax populations. The team said this suggests that New World parasites may have been introduced by colonial seafarers and represent a now-eliminated European parasite population.
The researchers also found that contemporary African and South Asian P vivax populations are genetically similar. They said this suggests that South Asian P vivax populations may have genetically mingled with European lineages during the colonial era, or it may reflect ancient connections between human populations in the Eastern Mediterranean, Middle East, and Indian subcontinent.
Another finding was the relatively homogeneous genetic makeup of P vivax in Mexico, which reflects a steady decline of the disease in this country over the last decade.
By contrast, the Papua New Guinea population of P vivax was shown to be very diverse relative to other P vivax populations.
A similar study, which also illustrated the global variations of P vivax, was recently published in Nature Genetics in as well. ![]()
Results support INR self-monitoring/management

Results of a systematic review support self-monitoring and self-management for patients on long-term oral anticoagulation therapy.
Researchers found that both self-monitoring and self-management were associated with a reduction in thromboembolic events when compared to standard monitoring.
Self-management, but not self-monitoring, was associated with a reduction in all-cause mortality.
And neither practice appeared to have an effect on major bleeding.
These results were published in the Cochrane Database of Systematic Reviews.
Point-of-care testing has made it possible for patients on long-term oral anticoagulation therapy to monitor their own international normalized ratio (INR).
Patients can either adjust their own medication according to a pre-determined dose-INR schedule, which is known as self-management, or they can call into a clinic to be told the appropriate dose adjustment, which is known as self-monitoring.
For the current review, Carl Heneghan, BM BCh, DPhil, of the University of Oxford in the UK, and his colleagues evaluated data from 28 randomized trials including 8950 patients. The trials compared self-monitoring and self-management with standard monitoring.
The researchers said pooled estimates showed a reduction in thromboembolic events with both self-management and self-monitoring, compared to standard monitoring.
Eleven studies including 3497 patients showed the relative risk (RR) of thromboembolic events was 0.47 (95% CI 0.31 to 0.70) for self-management compared to standard monitoring.
Seven studies including 4097 patients showed the RR of thromboembolic events was 0.69 (95% CI 0.49 to 0.97) for self-monitoring compared to standard monitoring.
Eight studies including 3058 patients suggested that self-management caused a reduction in all-cause mortality (RR=0.55, 95% CI 0.36 to 0.84) when compared to standard monitoring.
But 3 studies of 3300 patients indicated that self-monitoring did not reduce all-cause mortality (RR=0.94, 95% CI 0.78 to 1.15).
In 20 trials of 8018 patients, neither self-monitoring nor self-management reduced major hemorrhage (RR=0.95, 95% CI, 0.80 to 1.12) when compared to standard monitoring.
For all of these analyses, the quality of evidence was moderate.
“Our review of the latest research finds that self-monitoring alone does indeed result in a statistically significant reduction in thromboembolic events, whereas our previous review did not find this effect,” Dr Heneghan said.
“Suitable patients still need to be identified and educated for self-monitoring as it is not feasible for everyone, but the evidence clearly demonstrates that self-monitoring can improve the quality of oral anticoagulation therapy and adds weight to the argument that more patients should be given the opportunity to benefit from this treatment approach.”
The current review is an update on a previous review carried out by the same researchers in 2010. It includes 10 new studies of 4227 participants. ![]()
Results of a systematic review support self-monitoring and self-management for patients on long-term oral anticoagulation therapy.
Researchers found that both self-monitoring and self-management were associated with a reduction in thromboembolic events when compared to standard monitoring.
Self-management, but not self-monitoring, was associated with a reduction in all-cause mortality.
And neither practice appeared to have an effect on major bleeding.
These results were published in the Cochrane Database of Systematic Reviews.
Point-of-care testing has made it possible for patients on long-term oral anticoagulation therapy to monitor their own international normalized ratio (INR).
Patients can either adjust their own medication according to a pre-determined dose-INR schedule, which is known as self-management, or they can call into a clinic to be told the appropriate dose adjustment, which is known as self-monitoring.
For the current review, Carl Heneghan, BM BCh, DPhil, of the University of Oxford in the UK, and his colleagues evaluated data from 28 randomized trials including 8950 patients. The trials compared self-monitoring and self-management with standard monitoring.
The researchers said pooled estimates showed a reduction in thromboembolic events with both self-management and self-monitoring, compared to standard monitoring.
Eleven studies including 3497 patients showed the relative risk (RR) of thromboembolic events was 0.47 (95% CI 0.31 to 0.70) for self-management compared to standard monitoring.
Seven studies including 4097 patients showed the RR of thromboembolic events was 0.69 (95% CI 0.49 to 0.97) for self-monitoring compared to standard monitoring.
Eight studies including 3058 patients suggested that self-management caused a reduction in all-cause mortality (RR=0.55, 95% CI 0.36 to 0.84) when compared to standard monitoring.
But 3 studies of 3300 patients indicated that self-monitoring did not reduce all-cause mortality (RR=0.94, 95% CI 0.78 to 1.15).
In 20 trials of 8018 patients, neither self-monitoring nor self-management reduced major hemorrhage (RR=0.95, 95% CI, 0.80 to 1.12) when compared to standard monitoring.
For all of these analyses, the quality of evidence was moderate.
“Our review of the latest research finds that self-monitoring alone does indeed result in a statistically significant reduction in thromboembolic events, whereas our previous review did not find this effect,” Dr Heneghan said.
“Suitable patients still need to be identified and educated for self-monitoring as it is not feasible for everyone, but the evidence clearly demonstrates that self-monitoring can improve the quality of oral anticoagulation therapy and adds weight to the argument that more patients should be given the opportunity to benefit from this treatment approach.”
The current review is an update on a previous review carried out by the same researchers in 2010. It includes 10 new studies of 4227 participants. ![]()
Results of a systematic review support self-monitoring and self-management for patients on long-term oral anticoagulation therapy.
Researchers found that both self-monitoring and self-management were associated with a reduction in thromboembolic events when compared to standard monitoring.
Self-management, but not self-monitoring, was associated with a reduction in all-cause mortality.
And neither practice appeared to have an effect on major bleeding.
These results were published in the Cochrane Database of Systematic Reviews.
Point-of-care testing has made it possible for patients on long-term oral anticoagulation therapy to monitor their own international normalized ratio (INR).
Patients can either adjust their own medication according to a pre-determined dose-INR schedule, which is known as self-management, or they can call into a clinic to be told the appropriate dose adjustment, which is known as self-monitoring.
For the current review, Carl Heneghan, BM BCh, DPhil, of the University of Oxford in the UK, and his colleagues evaluated data from 28 randomized trials including 8950 patients. The trials compared self-monitoring and self-management with standard monitoring.
The researchers said pooled estimates showed a reduction in thromboembolic events with both self-management and self-monitoring, compared to standard monitoring.
Eleven studies including 3497 patients showed the relative risk (RR) of thromboembolic events was 0.47 (95% CI 0.31 to 0.70) for self-management compared to standard monitoring.
Seven studies including 4097 patients showed the RR of thromboembolic events was 0.69 (95% CI 0.49 to 0.97) for self-monitoring compared to standard monitoring.
Eight studies including 3058 patients suggested that self-management caused a reduction in all-cause mortality (RR=0.55, 95% CI 0.36 to 0.84) when compared to standard monitoring.
But 3 studies of 3300 patients indicated that self-monitoring did not reduce all-cause mortality (RR=0.94, 95% CI 0.78 to 1.15).
In 20 trials of 8018 patients, neither self-monitoring nor self-management reduced major hemorrhage (RR=0.95, 95% CI, 0.80 to 1.12) when compared to standard monitoring.
For all of these analyses, the quality of evidence was moderate.
“Our review of the latest research finds that self-monitoring alone does indeed result in a statistically significant reduction in thromboembolic events, whereas our previous review did not find this effect,” Dr Heneghan said.
“Suitable patients still need to be identified and educated for self-monitoring as it is not feasible for everyone, but the evidence clearly demonstrates that self-monitoring can improve the quality of oral anticoagulation therapy and adds weight to the argument that more patients should be given the opportunity to benefit from this treatment approach.”
The current review is an update on a previous review carried out by the same researchers in 2010. It includes 10 new studies of 4227 participants. ![]()
Drugs produce comparable results in CP-CML

Long-term results from the DASISION trial suggest that dasatinib and imatinib produce similar outcomes in patients with newly diagnosed chronic phase chronic myeloid leukemia (CP-CML).
Although patients who received dasatinib experienced faster and deeper molecular responses than patients who received imatinib, the overall survival and progression-free survival rates were similar between the treatment arms.
Overall, adverse events (AEs) were similar between the arms as well.
Researchers said these results suggest that dasatinib should continue to be considered an option for patients with newly diagnosed CP-CML.
The team reported the results of this study in the Journal of Clinical Oncology. The research was sponsored by Bristol-Myers Squibb.
The trial enrolled 519 patients with newly diagnosed CP-CML. They were randomized to receive dasatinib at 100 mg once daily (n=259) or imatinib at 400 mg once daily (n=260). Baseline characteristics were well-balanced between the arms.
At 5 years of follow-up, 61% of patients in the dasatinib arm and 63% of patients in the imatinib arm remained on treatment.
Response and survival
The cumulative 5-year rate of major molecular response was 76% in the dasatinib arm and 64% in the imatinib arm (P=0.0022). The rates of MR4.5 were 42% and 33%, respectively (P=0.0251).
The estimated 5-year overall survival was 91% in the dasatinib arm and 90% in the imatinib arm (hazard ratio=1.01; 95% CI, 0.58 to 1.73).
The estimated 5-year progression-free survival was 85% and 86%, respectively (hazard ratio=1.06; 95% CI, 0.68 to 1.66).
Safety
In both treatment arms, most AEs were grade 1 or 2. Grade 3/4 AEs occurred in 15% of patients in the dasatinib arm and 11% of patients in the imatinib arm.
Rates of grade 3/4 hematologic AEs tended to be higher in the dasatinib arm than the imatinib arm.
But the rates of most drug-related, nonhematologic AEs were lower in the dasatinib arm than the imatinib arm or were comparable between the arms.
The exception was drug-related pleural effusion, which was more common with dasatinib (28%) than with imatinib (0.8%).
Drug-related AEs were largely manageable, although they led to treatment discontinuation in 16% of dasatinib-treated patients and 7% of imatinib-treated patients.
By 5 years, 26 patients (10%) in each treatment arm had died. Nine patients in the dasatinib arm died of disease progression, as did 17 patients in the imatinib arm. ![]()
Long-term results from the DASISION trial suggest that dasatinib and imatinib produce similar outcomes in patients with newly diagnosed chronic phase chronic myeloid leukemia (CP-CML).
Although patients who received dasatinib experienced faster and deeper molecular responses than patients who received imatinib, the overall survival and progression-free survival rates were similar between the treatment arms.
Overall, adverse events (AEs) were similar between the arms as well.
Researchers said these results suggest that dasatinib should continue to be considered an option for patients with newly diagnosed CP-CML.
The team reported the results of this study in the Journal of Clinical Oncology. The research was sponsored by Bristol-Myers Squibb.
The trial enrolled 519 patients with newly diagnosed CP-CML. They were randomized to receive dasatinib at 100 mg once daily (n=259) or imatinib at 400 mg once daily (n=260). Baseline characteristics were well-balanced between the arms.
At 5 years of follow-up, 61% of patients in the dasatinib arm and 63% of patients in the imatinib arm remained on treatment.
Response and survival
The cumulative 5-year rate of major molecular response was 76% in the dasatinib arm and 64% in the imatinib arm (P=0.0022). The rates of MR4.5 were 42% and 33%, respectively (P=0.0251).
The estimated 5-year overall survival was 91% in the dasatinib arm and 90% in the imatinib arm (hazard ratio=1.01; 95% CI, 0.58 to 1.73).
The estimated 5-year progression-free survival was 85% and 86%, respectively (hazard ratio=1.06; 95% CI, 0.68 to 1.66).
Safety
In both treatment arms, most AEs were grade 1 or 2. Grade 3/4 AEs occurred in 15% of patients in the dasatinib arm and 11% of patients in the imatinib arm.
Rates of grade 3/4 hematologic AEs tended to be higher in the dasatinib arm than the imatinib arm.
But the rates of most drug-related, nonhematologic AEs were lower in the dasatinib arm than the imatinib arm or were comparable between the arms.
The exception was drug-related pleural effusion, which was more common with dasatinib (28%) than with imatinib (0.8%).
Drug-related AEs were largely manageable, although they led to treatment discontinuation in 16% of dasatinib-treated patients and 7% of imatinib-treated patients.
By 5 years, 26 patients (10%) in each treatment arm had died. Nine patients in the dasatinib arm died of disease progression, as did 17 patients in the imatinib arm. ![]()
Long-term results from the DASISION trial suggest that dasatinib and imatinib produce similar outcomes in patients with newly diagnosed chronic phase chronic myeloid leukemia (CP-CML).
Although patients who received dasatinib experienced faster and deeper molecular responses than patients who received imatinib, the overall survival and progression-free survival rates were similar between the treatment arms.
Overall, adverse events (AEs) were similar between the arms as well.
Researchers said these results suggest that dasatinib should continue to be considered an option for patients with newly diagnosed CP-CML.
The team reported the results of this study in the Journal of Clinical Oncology. The research was sponsored by Bristol-Myers Squibb.
The trial enrolled 519 patients with newly diagnosed CP-CML. They were randomized to receive dasatinib at 100 mg once daily (n=259) or imatinib at 400 mg once daily (n=260). Baseline characteristics were well-balanced between the arms.
At 5 years of follow-up, 61% of patients in the dasatinib arm and 63% of patients in the imatinib arm remained on treatment.
Response and survival
The cumulative 5-year rate of major molecular response was 76% in the dasatinib arm and 64% in the imatinib arm (P=0.0022). The rates of MR4.5 were 42% and 33%, respectively (P=0.0251).
The estimated 5-year overall survival was 91% in the dasatinib arm and 90% in the imatinib arm (hazard ratio=1.01; 95% CI, 0.58 to 1.73).
The estimated 5-year progression-free survival was 85% and 86%, respectively (hazard ratio=1.06; 95% CI, 0.68 to 1.66).
Safety
In both treatment arms, most AEs were grade 1 or 2. Grade 3/4 AEs occurred in 15% of patients in the dasatinib arm and 11% of patients in the imatinib arm.
Rates of grade 3/4 hematologic AEs tended to be higher in the dasatinib arm than the imatinib arm.
But the rates of most drug-related, nonhematologic AEs were lower in the dasatinib arm than the imatinib arm or were comparable between the arms.
The exception was drug-related pleural effusion, which was more common with dasatinib (28%) than with imatinib (0.8%).
Drug-related AEs were largely manageable, although they led to treatment discontinuation in 16% of dasatinib-treated patients and 7% of imatinib-treated patients.
By 5 years, 26 patients (10%) in each treatment arm had died. Nine patients in the dasatinib arm died of disease progression, as did 17 patients in the imatinib arm. ![]()
EC expands approved use of carfilzomib
Photo from Amgen
The European Commission (EC) has expanded the approved use of the proteasome inhibitor carfilzomib (Kyprolis).
The drug is now approved for use in combination with dexamethasone to treat adults with multiple myeloma (MM) who have received at least 1 prior therapy.
Carfilzomib was previously approved by the EC for use in combination with lenalidomide and dexamethasone to treat adult MM patients who have received at least 1 prior therapy.
The EC approved the extended indication for carfilzomib based on data from the phase 3 ENDEAVOR trial.
The trial included 929 MM patients whose disease had relapsed after 1 to 3 prior therapeutic regimens.
The patients received either carfilzomib plus dexamethasone (n=464) or bortezomib plus dexamethasone (n=465) until disease progression.
The primary endpoint was progression-free survival. The median progression-free survival was 18.7 months in the carfilzomib arm and 9.4 months in the bortezomib arm. The hazard ratio was 0.53 (P<0.0001).
Overall survival data were not yet mature at last follow-up.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the 2 treatment arms.
However, a number of known adverse events were reported at a higher rate in the carfilzomib arm than the bortezomib arm, including dyspnea (28% vs 13%), hypertension (25% vs 3%), pyrexia (27% vs 14%), cough (25% vs 15%), cardiac failure (8% vs 3%), and acute renal failure (8% vs 5%).
Carfilzomib is marketed as Kyprolis by Onyx Pharmaceuticals, Inc., a subsidiary of Amgen that holds development and commercialization rights to the drug globally, with the exception of Japan. ![]()
Photo from Amgen
The European Commission (EC) has expanded the approved use of the proteasome inhibitor carfilzomib (Kyprolis).
The drug is now approved for use in combination with dexamethasone to treat adults with multiple myeloma (MM) who have received at least 1 prior therapy.
Carfilzomib was previously approved by the EC for use in combination with lenalidomide and dexamethasone to treat adult MM patients who have received at least 1 prior therapy.
The EC approved the extended indication for carfilzomib based on data from the phase 3 ENDEAVOR trial.
The trial included 929 MM patients whose disease had relapsed after 1 to 3 prior therapeutic regimens.
The patients received either carfilzomib plus dexamethasone (n=464) or bortezomib plus dexamethasone (n=465) until disease progression.
The primary endpoint was progression-free survival. The median progression-free survival was 18.7 months in the carfilzomib arm and 9.4 months in the bortezomib arm. The hazard ratio was 0.53 (P<0.0001).
Overall survival data were not yet mature at last follow-up.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the 2 treatment arms.
However, a number of known adverse events were reported at a higher rate in the carfilzomib arm than the bortezomib arm, including dyspnea (28% vs 13%), hypertension (25% vs 3%), pyrexia (27% vs 14%), cough (25% vs 15%), cardiac failure (8% vs 3%), and acute renal failure (8% vs 5%).
Carfilzomib is marketed as Kyprolis by Onyx Pharmaceuticals, Inc., a subsidiary of Amgen that holds development and commercialization rights to the drug globally, with the exception of Japan. ![]()
Photo from Amgen
The European Commission (EC) has expanded the approved use of the proteasome inhibitor carfilzomib (Kyprolis).
The drug is now approved for use in combination with dexamethasone to treat adults with multiple myeloma (MM) who have received at least 1 prior therapy.
Carfilzomib was previously approved by the EC for use in combination with lenalidomide and dexamethasone to treat adult MM patients who have received at least 1 prior therapy.
The EC approved the extended indication for carfilzomib based on data from the phase 3 ENDEAVOR trial.
The trial included 929 MM patients whose disease had relapsed after 1 to 3 prior therapeutic regimens.
The patients received either carfilzomib plus dexamethasone (n=464) or bortezomib plus dexamethasone (n=465) until disease progression.
The primary endpoint was progression-free survival. The median progression-free survival was 18.7 months in the carfilzomib arm and 9.4 months in the bortezomib arm. The hazard ratio was 0.53 (P<0.0001).
Overall survival data were not yet mature at last follow-up.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the 2 treatment arms.
However, a number of known adverse events were reported at a higher rate in the carfilzomib arm than the bortezomib arm, including dyspnea (28% vs 13%), hypertension (25% vs 3%), pyrexia (27% vs 14%), cough (25% vs 15%), cardiac failure (8% vs 3%), and acute renal failure (8% vs 5%).
Carfilzomib is marketed as Kyprolis by Onyx Pharmaceuticals, Inc., a subsidiary of Amgen that holds development and commercialization rights to the drug globally, with the exception of Japan.
How multiple infections make malaria worse
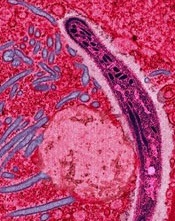
Image by Ute Frevert
and Margaret Shear
New research suggests that infections with 2 types of malaria parasite lead to greater health risks because 1 species helps the other thrive.
Investigators sought to understand what happens when the 2 most common malaria parasites cause infection at the same time, as they are known to attack the body in different ways.
The team found the first parasite helps provide the second with more of the resources it needs to prosper.
“Immune responses are assumed to determine the outcome of interactions between parasite species, but our study clearly shows that resources can be more important,” said Sarah Reece, of the University of Edinburgh in Scotland.
“Our findings also challenge ideas that 1 species will outcompete the other, which explains why infections involving 2 parasite species can pose a greater health risk to patients.”
Dr Reece and her colleagues recounted these findings in Ecology Letters.
In humans, the malaria parasite Plasmodium falciparum infects red blood cells of all ages, while the Plasmodium vivax parasite attacks only young red blood cells.
The current study, conducted in mice with equivalent malaria parasites (P chabaudi and P yoelii), showed that the body’s response to the first infection produces more of the type of red blood cell the second parasite needs.
In response to the first infection, millions of red blood cells are destroyed. The body responds by replenishing these cells.
The fresh cells then become infected by the second type of parasite, making the infection worse.
The investigators said these results appear to explain why infections with both P falciparum and P vivax often have worse outcomes for patients than infections with a single malaria parasite.
Image by Ute Frevert
and Margaret Shear
New research suggests that infections with 2 types of malaria parasite lead to greater health risks because 1 species helps the other thrive.
Investigators sought to understand what happens when the 2 most common malaria parasites cause infection at the same time, as they are known to attack the body in different ways.
The team found the first parasite helps provide the second with more of the resources it needs to prosper.
“Immune responses are assumed to determine the outcome of interactions between parasite species, but our study clearly shows that resources can be more important,” said Sarah Reece, of the University of Edinburgh in Scotland.
“Our findings also challenge ideas that 1 species will outcompete the other, which explains why infections involving 2 parasite species can pose a greater health risk to patients.”
Dr Reece and her colleagues recounted these findings in Ecology Letters.
In humans, the malaria parasite Plasmodium falciparum infects red blood cells of all ages, while the Plasmodium vivax parasite attacks only young red blood cells.
The current study, conducted in mice with equivalent malaria parasites (P chabaudi and P yoelii), showed that the body’s response to the first infection produces more of the type of red blood cell the second parasite needs.
In response to the first infection, millions of red blood cells are destroyed. The body responds by replenishing these cells.
The fresh cells then become infected by the second type of parasite, making the infection worse.
The investigators said these results appear to explain why infections with both P falciparum and P vivax often have worse outcomes for patients than infections with a single malaria parasite.
Image by Ute Frevert
and Margaret Shear
New research suggests that infections with 2 types of malaria parasite lead to greater health risks because 1 species helps the other thrive.
Investigators sought to understand what happens when the 2 most common malaria parasites cause infection at the same time, as they are known to attack the body in different ways.
The team found the first parasite helps provide the second with more of the resources it needs to prosper.
“Immune responses are assumed to determine the outcome of interactions between parasite species, but our study clearly shows that resources can be more important,” said Sarah Reece, of the University of Edinburgh in Scotland.
“Our findings also challenge ideas that 1 species will outcompete the other, which explains why infections involving 2 parasite species can pose a greater health risk to patients.”
Dr Reece and her colleagues recounted these findings in Ecology Letters.
In humans, the malaria parasite Plasmodium falciparum infects red blood cells of all ages, while the Plasmodium vivax parasite attacks only young red blood cells.
The current study, conducted in mice with equivalent malaria parasites (P chabaudi and P yoelii), showed that the body’s response to the first infection produces more of the type of red blood cell the second parasite needs.
In response to the first infection, millions of red blood cells are destroyed. The body responds by replenishing these cells.
The fresh cells then become infected by the second type of parasite, making the infection worse.
The investigators said these results appear to explain why infections with both P falciparum and P vivax often have worse outcomes for patients than infections with a single malaria parasite.
Study explains link between malignant hyperthermia and bleeding abnormalities

A new study helps explain why some patients with malignant hyperthermia may suffer from excessive bleeding.
The findings suggest a mutation that causes malignant hyperthermia can disrupt calcium signaling in vascular smooth muscle cells, leading to bleeding abnormalities.
What’s more, researchers found that a drug clinically approved to treat muscle-related symptoms in malignant hyperthermia helped stop bleeding.
Rubén Lopez, of Basel University Hospital in Switzerland, and his colleagues conducted this research and reported their findings in Science Signaling.
Patients with malignant hyperthermia experience dangerously high fever and severe muscle contractions when exposed to general anesthesia.
Malignant hyperthermia is often caused by mutations in the RYR1 gene, which encodes a calcium channel in skeletal muscle called ryanodine receptor type 1 (RyR1).
For some patients with these mutations, malignant hyperthermia is accompanied by a mild bleeding disorder, but whether the 2 conditions are connected has not been clear.
Working in a mouse model of malignant hyperthermia, researchers found that vascular smooth muscle cells with mutated RyR1 displayed frequent spikes in calcium levels, known as calcium sparks. These sparks led to excessive vasodilation and prolonged bleeding.
Blocking the receptor with dantrolene, a drug used to treat malignant hyperthermia, helped reduce bleeding in the mice and in a single human patient, pointing to an unexpected benefit from the drug.
The findings suggest that mutations in RyR1, which is also found in other types of smooth muscle cells such as those in the bladder and uterus, may have a wider range of effects than previously thought.
A new study helps explain why some patients with malignant hyperthermia may suffer from excessive bleeding.
The findings suggest a mutation that causes malignant hyperthermia can disrupt calcium signaling in vascular smooth muscle cells, leading to bleeding abnormalities.
What’s more, researchers found that a drug clinically approved to treat muscle-related symptoms in malignant hyperthermia helped stop bleeding.
Rubén Lopez, of Basel University Hospital in Switzerland, and his colleagues conducted this research and reported their findings in Science Signaling.
Patients with malignant hyperthermia experience dangerously high fever and severe muscle contractions when exposed to general anesthesia.
Malignant hyperthermia is often caused by mutations in the RYR1 gene, which encodes a calcium channel in skeletal muscle called ryanodine receptor type 1 (RyR1).
For some patients with these mutations, malignant hyperthermia is accompanied by a mild bleeding disorder, but whether the 2 conditions are connected has not been clear.
Working in a mouse model of malignant hyperthermia, researchers found that vascular smooth muscle cells with mutated RyR1 displayed frequent spikes in calcium levels, known as calcium sparks. These sparks led to excessive vasodilation and prolonged bleeding.
Blocking the receptor with dantrolene, a drug used to treat malignant hyperthermia, helped reduce bleeding in the mice and in a single human patient, pointing to an unexpected benefit from the drug.
The findings suggest that mutations in RyR1, which is also found in other types of smooth muscle cells such as those in the bladder and uterus, may have a wider range of effects than previously thought.
A new study helps explain why some patients with malignant hyperthermia may suffer from excessive bleeding.
The findings suggest a mutation that causes malignant hyperthermia can disrupt calcium signaling in vascular smooth muscle cells, leading to bleeding abnormalities.
What’s more, researchers found that a drug clinically approved to treat muscle-related symptoms in malignant hyperthermia helped stop bleeding.
Rubén Lopez, of Basel University Hospital in Switzerland, and his colleagues conducted this research and reported their findings in Science Signaling.
Patients with malignant hyperthermia experience dangerously high fever and severe muscle contractions when exposed to general anesthesia.
Malignant hyperthermia is often caused by mutations in the RYR1 gene, which encodes a calcium channel in skeletal muscle called ryanodine receptor type 1 (RyR1).
For some patients with these mutations, malignant hyperthermia is accompanied by a mild bleeding disorder, but whether the 2 conditions are connected has not been clear.
Working in a mouse model of malignant hyperthermia, researchers found that vascular smooth muscle cells with mutated RyR1 displayed frequent spikes in calcium levels, known as calcium sparks. These sparks led to excessive vasodilation and prolonged bleeding.
Blocking the receptor with dantrolene, a drug used to treat malignant hyperthermia, helped reduce bleeding in the mice and in a single human patient, pointing to an unexpected benefit from the drug.
The findings suggest that mutations in RyR1, which is also found in other types of smooth muscle cells such as those in the bladder and uterus, may have a wider range of effects than previously thought.
Team identifies potential therapeutic target for AML

New research suggests that E proteins and their antagonists, Id proteins, can play key roles in acute myeloid leukemia (AML).
The study showed that overexpression of the Id2 protein or knockdown of the E2-2 protein can suppress both mixed-lineage leukemia (MLL)-rearranged AML and t(8;21) AML.
These findings, published in Cancer Cell, suggest the Id2/E-protein axis may be a promising therapeutic target for AML.
“There is a particularly urgent need for new, targeted, drug-based therapies for AML, and with every discovery of what’s driving the cancer, we take a step closer to achieving that,” said study author Ricky Johnstone, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“What we found in this case was the suppression of Id2 protein plays an important, and previously unrecognized, role in allowing MLL re-arranged AML cancer cells to take hold and spread. Drugs that influence levels of this protein, or stop it being suppressed by the cancer, could provide a much-needed new avenue to combatting this disease.”
The researchers first found that Id2 regulates leukemia stem cell (LSC) potential. Specifically, low Id2 expression is associated with LSC enrichment, and Id2 overexpression hinders leukemia development.
Further investigation revealed that the fusion protein MLL-AF9 suppresses Id2 and activates E2-2 expression, while E2-2 depletion phenocopies Id2 overexpression in MLL-AF9-AML cells.
The team also found that Id2’s tumor-suppressive function is conserved in t(8;21) AML. And low expression of Id2 and its associated gene signature are associated with poor prognosis in patients with MLL-rearranged AML or t(8;21) AML.
New research suggests that E proteins and their antagonists, Id proteins, can play key roles in acute myeloid leukemia (AML).
The study showed that overexpression of the Id2 protein or knockdown of the E2-2 protein can suppress both mixed-lineage leukemia (MLL)-rearranged AML and t(8;21) AML.
These findings, published in Cancer Cell, suggest the Id2/E-protein axis may be a promising therapeutic target for AML.
“There is a particularly urgent need for new, targeted, drug-based therapies for AML, and with every discovery of what’s driving the cancer, we take a step closer to achieving that,” said study author Ricky Johnstone, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“What we found in this case was the suppression of Id2 protein plays an important, and previously unrecognized, role in allowing MLL re-arranged AML cancer cells to take hold and spread. Drugs that influence levels of this protein, or stop it being suppressed by the cancer, could provide a much-needed new avenue to combatting this disease.”
The researchers first found that Id2 regulates leukemia stem cell (LSC) potential. Specifically, low Id2 expression is associated with LSC enrichment, and Id2 overexpression hinders leukemia development.
Further investigation revealed that the fusion protein MLL-AF9 suppresses Id2 and activates E2-2 expression, while E2-2 depletion phenocopies Id2 overexpression in MLL-AF9-AML cells.
The team also found that Id2’s tumor-suppressive function is conserved in t(8;21) AML. And low expression of Id2 and its associated gene signature are associated with poor prognosis in patients with MLL-rearranged AML or t(8;21) AML.
New research suggests that E proteins and their antagonists, Id proteins, can play key roles in acute myeloid leukemia (AML).
The study showed that overexpression of the Id2 protein or knockdown of the E2-2 protein can suppress both mixed-lineage leukemia (MLL)-rearranged AML and t(8;21) AML.
These findings, published in Cancer Cell, suggest the Id2/E-protein axis may be a promising therapeutic target for AML.
“There is a particularly urgent need for new, targeted, drug-based therapies for AML, and with every discovery of what’s driving the cancer, we take a step closer to achieving that,” said study author Ricky Johnstone, PhD, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“What we found in this case was the suppression of Id2 protein plays an important, and previously unrecognized, role in allowing MLL re-arranged AML cancer cells to take hold and spread. Drugs that influence levels of this protein, or stop it being suppressed by the cancer, could provide a much-needed new avenue to combatting this disease.”
The researchers first found that Id2 regulates leukemia stem cell (LSC) potential. Specifically, low Id2 expression is associated with LSC enrichment, and Id2 overexpression hinders leukemia development.
Further investigation revealed that the fusion protein MLL-AF9 suppresses Id2 and activates E2-2 expression, while E2-2 depletion phenocopies Id2 overexpression in MLL-AF9-AML cells.
The team also found that Id2’s tumor-suppressive function is conserved in t(8;21) AML. And low expression of Id2 and its associated gene signature are associated with poor prognosis in patients with MLL-rearranged AML or t(8;21) AML.