User login
NOACs benefit early stage chronic kidney disease patients
Non–vitamin K oral anticoagulants (NOACs) significantly reduced the risk of stroke or systemic embolism compared to vitamin K antagonists (VKAs) for patients in the early stages of chronic kidney disease and comorbid atrial fibrillation, based on data from a meta-analysis of roughly 34,000 patients.
Chronic kidney disease increases the risk of complications including stroke, congestive heart failure, and death in patients who also have atrial fibrillation, but most trials of anticoagulant therapy to reduce the risk of such events have excluded these patients, wrote Jeffrey T. Ha, MBBS, of the George Institute for Global Health, Newtown, Australia, and colleagues.
To assess the benefits and harms of oral anticoagulants for multiple indications in chronic kidney disease patients, the researchers conducted a meta-analysis of 45 studies including 34,082 individuals. The findings were published in the Annals of Internal Medicine. The analysis included 8 trials of end stage kidney disease patients on dialysis; the remaining trials excluded patients with creatinine clearance less than 20 mL/min or an estimated glomerular filtration rate less than 15 mL/min per 1.73 m2. The interventional agents were rivaroxaban, dabigatran, apixaban, edoxaban, betrixaban, warfarin, and acenocoumarol.
A notable finding was the significant reduction in relative risk of stroke or systemic embolism (21%), hemorrhagic stroke (52%), and intracranial hemorrhage (51%) for early-stage chronic kidney disease patients with atrial fibrillation given NOACs, compared with those given VKAs.
The evidence for the superiority of NOACs over VKAs for reducing risk of venous thromboembolism (VTE) or VTE-related death was uncertain, as was the evidence to draw any conclusions about benefits and harms of either NOACs or VKAs for patients with advanced or end-stage kidney disease.
Across all trials, NOACs appeared to reduce the relative risk of major bleeding, compared with VKAs by roughly 25%, but the difference was not statistically significant, the researchers noted.
The findings were limited by the lack of evidence for oral anticoagulant use in patients with advanced chronic or end-stage kidney disease, as well as inability to assess differences among NOACs, the researchers noted. However, the results suggest that NOACs may be recommended over VKAs for the subgroup of early-stage chronic kidney disease patients with atrial fibrillation, they said.
Several additional trials are in progress, and future trials “should include not only participants with dialysis-dependent ESKD [end-stage kidney disease] but also those with CrCl [creatinine clearance of] less than 25 mL/min,” and compare NOACs with placebo as well, they noted.
Lead author Dr. Ha is supported by a University Postgraduate Award from University of New South Wales, Sydney, but had no financial conflicts to disclose; coauthors disclosed support from various organizations as well as pharmaceutical companies including Baxter, Amgen, Eli Lilly, Boehringer Ingelheim, Vifor Pharma, Janssen, Pfizer, Bristol-Myers Squibb, and GlaxoSmithKline.
SOURCE: Ha JT et al. Ann Intern Med. 2019 July 15. doi: 10.7326/M19-0087
The significant reduction in risk of hemorrhagic stroke, recurrent venous thromboembolism, and VTE-related deaths in patients with early-stage chronic kidney disease given a NOAC [non–vitamin K oral anticoagulants] in a meta-analysis supports clinical application, but is there a level of renal dysfunction for which clinicians should apply greater caution in extrapolating these findings? As the evidence supporting the safety and effectiveness of NOACs in the general population increases, there is a renewed interest in defining the role of anticoagulant therapy to prevent stroke and VTE in patients with chronic kidney disease and end-stage kidney disease. This interest is driven in part by uncertainty as to the benefits vs. harms of warfarin for patients with chronic kidney disease. The data in the meta-analysis by Ha and colleagues do not support any benefits for patients with end-stage disease, but the results of two ongoing clinical trials of patients with atrial fibrillation and end-stage kidney disease may offer insights.
Until the results of these trials become available, the decision to use anticoagulant therapy in patients with end-stage kidney disease will continue to require an individualized approach that balances potential benefits and harms.
Ainslie Hildebrand, MD, of University of Alberta, Edmonton; Christine Ribic, MD, of McMaster University, Hamilton, Ont.; and Deborah Zimmerman, MD, of the University of Ottawa, made these comments in an accompanying editorial (Ann Intern Med. 2019 July 15. doi:10.7326/M19-1504). Dr. Ribic disclosed grants from Pfizer, Leo Pharma, and Astellas Pharma. Dr. Hildebrand and Dr. Zimmerman had no financial conflicts to disclose.
The significant reduction in risk of hemorrhagic stroke, recurrent venous thromboembolism, and VTE-related deaths in patients with early-stage chronic kidney disease given a NOAC [non–vitamin K oral anticoagulants] in a meta-analysis supports clinical application, but is there a level of renal dysfunction for which clinicians should apply greater caution in extrapolating these findings? As the evidence supporting the safety and effectiveness of NOACs in the general population increases, there is a renewed interest in defining the role of anticoagulant therapy to prevent stroke and VTE in patients with chronic kidney disease and end-stage kidney disease. This interest is driven in part by uncertainty as to the benefits vs. harms of warfarin for patients with chronic kidney disease. The data in the meta-analysis by Ha and colleagues do not support any benefits for patients with end-stage disease, but the results of two ongoing clinical trials of patients with atrial fibrillation and end-stage kidney disease may offer insights.
Until the results of these trials become available, the decision to use anticoagulant therapy in patients with end-stage kidney disease will continue to require an individualized approach that balances potential benefits and harms.
Ainslie Hildebrand, MD, of University of Alberta, Edmonton; Christine Ribic, MD, of McMaster University, Hamilton, Ont.; and Deborah Zimmerman, MD, of the University of Ottawa, made these comments in an accompanying editorial (Ann Intern Med. 2019 July 15. doi:10.7326/M19-1504). Dr. Ribic disclosed grants from Pfizer, Leo Pharma, and Astellas Pharma. Dr. Hildebrand and Dr. Zimmerman had no financial conflicts to disclose.
The significant reduction in risk of hemorrhagic stroke, recurrent venous thromboembolism, and VTE-related deaths in patients with early-stage chronic kidney disease given a NOAC [non–vitamin K oral anticoagulants] in a meta-analysis supports clinical application, but is there a level of renal dysfunction for which clinicians should apply greater caution in extrapolating these findings? As the evidence supporting the safety and effectiveness of NOACs in the general population increases, there is a renewed interest in defining the role of anticoagulant therapy to prevent stroke and VTE in patients with chronic kidney disease and end-stage kidney disease. This interest is driven in part by uncertainty as to the benefits vs. harms of warfarin for patients with chronic kidney disease. The data in the meta-analysis by Ha and colleagues do not support any benefits for patients with end-stage disease, but the results of two ongoing clinical trials of patients with atrial fibrillation and end-stage kidney disease may offer insights.
Until the results of these trials become available, the decision to use anticoagulant therapy in patients with end-stage kidney disease will continue to require an individualized approach that balances potential benefits and harms.
Ainslie Hildebrand, MD, of University of Alberta, Edmonton; Christine Ribic, MD, of McMaster University, Hamilton, Ont.; and Deborah Zimmerman, MD, of the University of Ottawa, made these comments in an accompanying editorial (Ann Intern Med. 2019 July 15. doi:10.7326/M19-1504). Dr. Ribic disclosed grants from Pfizer, Leo Pharma, and Astellas Pharma. Dr. Hildebrand and Dr. Zimmerman had no financial conflicts to disclose.
Non–vitamin K oral anticoagulants (NOACs) significantly reduced the risk of stroke or systemic embolism compared to vitamin K antagonists (VKAs) for patients in the early stages of chronic kidney disease and comorbid atrial fibrillation, based on data from a meta-analysis of roughly 34,000 patients.
Chronic kidney disease increases the risk of complications including stroke, congestive heart failure, and death in patients who also have atrial fibrillation, but most trials of anticoagulant therapy to reduce the risk of such events have excluded these patients, wrote Jeffrey T. Ha, MBBS, of the George Institute for Global Health, Newtown, Australia, and colleagues.
To assess the benefits and harms of oral anticoagulants for multiple indications in chronic kidney disease patients, the researchers conducted a meta-analysis of 45 studies including 34,082 individuals. The findings were published in the Annals of Internal Medicine. The analysis included 8 trials of end stage kidney disease patients on dialysis; the remaining trials excluded patients with creatinine clearance less than 20 mL/min or an estimated glomerular filtration rate less than 15 mL/min per 1.73 m2. The interventional agents were rivaroxaban, dabigatran, apixaban, edoxaban, betrixaban, warfarin, and acenocoumarol.
A notable finding was the significant reduction in relative risk of stroke or systemic embolism (21%), hemorrhagic stroke (52%), and intracranial hemorrhage (51%) for early-stage chronic kidney disease patients with atrial fibrillation given NOACs, compared with those given VKAs.
The evidence for the superiority of NOACs over VKAs for reducing risk of venous thromboembolism (VTE) or VTE-related death was uncertain, as was the evidence to draw any conclusions about benefits and harms of either NOACs or VKAs for patients with advanced or end-stage kidney disease.
Across all trials, NOACs appeared to reduce the relative risk of major bleeding, compared with VKAs by roughly 25%, but the difference was not statistically significant, the researchers noted.
The findings were limited by the lack of evidence for oral anticoagulant use in patients with advanced chronic or end-stage kidney disease, as well as inability to assess differences among NOACs, the researchers noted. However, the results suggest that NOACs may be recommended over VKAs for the subgroup of early-stage chronic kidney disease patients with atrial fibrillation, they said.
Several additional trials are in progress, and future trials “should include not only participants with dialysis-dependent ESKD [end-stage kidney disease] but also those with CrCl [creatinine clearance of] less than 25 mL/min,” and compare NOACs with placebo as well, they noted.
Lead author Dr. Ha is supported by a University Postgraduate Award from University of New South Wales, Sydney, but had no financial conflicts to disclose; coauthors disclosed support from various organizations as well as pharmaceutical companies including Baxter, Amgen, Eli Lilly, Boehringer Ingelheim, Vifor Pharma, Janssen, Pfizer, Bristol-Myers Squibb, and GlaxoSmithKline.
SOURCE: Ha JT et al. Ann Intern Med. 2019 July 15. doi: 10.7326/M19-0087
Non–vitamin K oral anticoagulants (NOACs) significantly reduced the risk of stroke or systemic embolism compared to vitamin K antagonists (VKAs) for patients in the early stages of chronic kidney disease and comorbid atrial fibrillation, based on data from a meta-analysis of roughly 34,000 patients.
Chronic kidney disease increases the risk of complications including stroke, congestive heart failure, and death in patients who also have atrial fibrillation, but most trials of anticoagulant therapy to reduce the risk of such events have excluded these patients, wrote Jeffrey T. Ha, MBBS, of the George Institute for Global Health, Newtown, Australia, and colleagues.
To assess the benefits and harms of oral anticoagulants for multiple indications in chronic kidney disease patients, the researchers conducted a meta-analysis of 45 studies including 34,082 individuals. The findings were published in the Annals of Internal Medicine. The analysis included 8 trials of end stage kidney disease patients on dialysis; the remaining trials excluded patients with creatinine clearance less than 20 mL/min or an estimated glomerular filtration rate less than 15 mL/min per 1.73 m2. The interventional agents were rivaroxaban, dabigatran, apixaban, edoxaban, betrixaban, warfarin, and acenocoumarol.
A notable finding was the significant reduction in relative risk of stroke or systemic embolism (21%), hemorrhagic stroke (52%), and intracranial hemorrhage (51%) for early-stage chronic kidney disease patients with atrial fibrillation given NOACs, compared with those given VKAs.
The evidence for the superiority of NOACs over VKAs for reducing risk of venous thromboembolism (VTE) or VTE-related death was uncertain, as was the evidence to draw any conclusions about benefits and harms of either NOACs or VKAs for patients with advanced or end-stage kidney disease.
Across all trials, NOACs appeared to reduce the relative risk of major bleeding, compared with VKAs by roughly 25%, but the difference was not statistically significant, the researchers noted.
The findings were limited by the lack of evidence for oral anticoagulant use in patients with advanced chronic or end-stage kidney disease, as well as inability to assess differences among NOACs, the researchers noted. However, the results suggest that NOACs may be recommended over VKAs for the subgroup of early-stage chronic kidney disease patients with atrial fibrillation, they said.
Several additional trials are in progress, and future trials “should include not only participants with dialysis-dependent ESKD [end-stage kidney disease] but also those with CrCl [creatinine clearance of] less than 25 mL/min,” and compare NOACs with placebo as well, they noted.
Lead author Dr. Ha is supported by a University Postgraduate Award from University of New South Wales, Sydney, but had no financial conflicts to disclose; coauthors disclosed support from various organizations as well as pharmaceutical companies including Baxter, Amgen, Eli Lilly, Boehringer Ingelheim, Vifor Pharma, Janssen, Pfizer, Bristol-Myers Squibb, and GlaxoSmithKline.
SOURCE: Ha JT et al. Ann Intern Med. 2019 July 15. doi: 10.7326/M19-0087
FROM THE ANNALS OF INTERNAL MEDICINE
On second thought, lenalidomide does improve DLBCL outcomes
LUGANO, Switzerland – Hot on the heels of the phase 3 ROBUST study showing that adding lenalidomide to standard chemotherapy did not improve outcomes for patients with untreated diffuse large B-cell lymphoma come results of a different study showing a significant benefit with the therapy.

Although, as previously reported, adding lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone in ROBUST, results from the randomized phase 2 ECOG-ACRIN 1412 study showed that R2-CHOP was associated with a 34% reduction in the risk of disease progression or death, compared with R-CHOP alone.
“The efficacy endpoints are consistent, with trends toward higher PET complete response rate and improved overall survival with R-squared CHOP,” Grzegorz S. Nowakowski, MD, of the Mayo Clinic, Rochester, Minn., said at the International Conference on Malignant Lymphoma.
So what’s behind the conflicting findings?

The differences between the results of the two studies may be accounted for by the higher lenalidomide dose used in ECOG-ACRIN 1412, the patient populations – all comers in ECOG-ACRIN versus only patients with activated B-cell (ABC) type DLBCL in ROBUST – and by a 10-day shorter median time to treatment in ECOG-ACRIN 1412, said invited discussant Margaret A. Shipp, MD, of the Dana-Farber Cancer Institute in Boston.
The rationale for adding lenalidomide to R-CHOP came from in vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non-germinal center-like B (GCB) type DLBCL.
In a subanalysis of patients enrolled in MC078E, Dr. Nowakowski and colleagues found that using classification of patients by cell of origin with the NanoString Lymphoma Subtype assay, the addition of lenalidomide to R-CHOP “appears to mitigate the negative impact of an ABC molecular subtype on the outcome.”
ECOG-ACRIN 1412 details
Goals of the ECOG-ACRIN 1412 study were to evaluate the effect of lenalidomide both in all DLBCL subtypes and in the ABC subtype, maximize the synergy of the immunomodulator with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) while maintaining R-CHOP dose intensity, and facilitate the enrollment of patients with rapidly progressive disease.
To accomplish the last goal, the study was designed to allow enrollment based on local laboratory findings, scans, and diagnostic pathology, without required identification of the cell of origin. Built in to the design was the plan for final eligibility to be based on central pathology review. In other words, the trial design took into account the likelihood that some enrolled patients would not qualify for eligibility based on later pathology review.
The investigators enrolled 349 adults aged 18 years or older with pathologically confirmed DLBCL (regardless of the cell of origin), stage II bulky to stage IV disease, International Prognostic Index (IPI) scores of 2 or greater, and Eastern Cooperative Oncology Group performance status scores of 2 or less.
The patients were stratified by age (younger than 60 years vs. 60 years and older) and by IPI score (2/3 vs. 4/5), and then randomized to receive either six cycles of R-CHOP or R2-CHOP. Lenalidomide was given in a dose of 25 mg on days 1-10 of each cycle. In contrast, the dose used in ROBUST was 15 mg given on days 1-14 of each cycle.
In ECOG-ACRIN 1412, patients assigned to lenalidomide received mandatory neutropenia prophylaxis with granulocyte-colony stimulating factor.
The time from diagnosis to treatment was a median of 21 days, with only 61 of 280 evaluable patients starting treatment more than 31 days after diagnosis. In ROBUST, the median time to start therapy was 31 days.
Dr. Nowakowski and his colleagues had previously shown that time to treatment is an important prognostic factor in DLBCL.
The efficacy evaluation included 145 patients assigned to R2-CHOP, and 135 assigned to R-CHOP. Primary reasons for exclusion were ineligibity following central pathology review or lack of diagnostic material for review.
After a median follow-up of 2.5 years, R2-CHOP was associated with a 34% improvement in progression-free survival, the primary endpoint (hazard ratio [HR] 0.66, P = .03). The 1-year progression-free survival rates were 83% with R2-CHOP and 73% with R-CHOP. Respective 2-year progression-free survival rates were 76% and 70%.
There was no significant difference, however, in the secondary overall survival endpoints with 1-year and 2-year overall survival of 93% vs. 87% and 86% vs. 80%, respectively.
Similarly, there was no difference in rates of PET-ascertained complete response, at 72% with R2-CHOP and 67% with R-CHOP.
R2-CHOP showed greater benefit across most subgroups, including patients with lower IPI score, patients with bulky disease, patients younger than 60 years, women, and patients with shorter time to treatment. There were also nonsignificant trends hinting at better outcomes with R2-CHOP, regardless of cell of origin.
Toxicities were typical for R-CHOP, although patients on R2-CHOP had significantly higher rates of grade 3 or 4 diarrhea, febrile neutropenia, and thrombocytopenia.
In addition to the trial differences mentioned previously, the differences in outcomes might be explained by the possibility that lenalidomide activity is not restricted to ABC DLBCL, Dr. Shipp said.
“One of the things that’s important to remember about lenalidomide is that it’s an immunomodulating agent and it has also has effects on tumor-infiltrating T cells,” she said.
Differences in response to therapy may also be explained by recent findings showing genetic heterogeneity in transcription-defined ABC DLBCLs, she said.
ECOG-ACRIN 1412 was supported by the National Cancer Institute and by Celgene. Dr. Nowakowski reported consulting/advising for and research funding from Celgene and others. Dr. Shipp reported consulting/advising for Bristol-Myers Squibb, honoraria from BMS and AstraZeneca, and institutional research funding from BMS and Bayer.
SOURCE: Nowakowski GS et al. 15-ICML, Abstract 006.
LUGANO, Switzerland – Hot on the heels of the phase 3 ROBUST study showing that adding lenalidomide to standard chemotherapy did not improve outcomes for patients with untreated diffuse large B-cell lymphoma come results of a different study showing a significant benefit with the therapy.
Although, as previously reported, adding lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone in ROBUST, results from the randomized phase 2 ECOG-ACRIN 1412 study showed that R2-CHOP was associated with a 34% reduction in the risk of disease progression or death, compared with R-CHOP alone.
“The efficacy endpoints are consistent, with trends toward higher PET complete response rate and improved overall survival with R-squared CHOP,” Grzegorz S. Nowakowski, MD, of the Mayo Clinic, Rochester, Minn., said at the International Conference on Malignant Lymphoma.
So what’s behind the conflicting findings?
The differences between the results of the two studies may be accounted for by the higher lenalidomide dose used in ECOG-ACRIN 1412, the patient populations – all comers in ECOG-ACRIN versus only patients with activated B-cell (ABC) type DLBCL in ROBUST – and by a 10-day shorter median time to treatment in ECOG-ACRIN 1412, said invited discussant Margaret A. Shipp, MD, of the Dana-Farber Cancer Institute in Boston.
The rationale for adding lenalidomide to R-CHOP came from in vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non-germinal center-like B (GCB) type DLBCL.
In a subanalysis of patients enrolled in MC078E, Dr. Nowakowski and colleagues found that using classification of patients by cell of origin with the NanoString Lymphoma Subtype assay, the addition of lenalidomide to R-CHOP “appears to mitigate the negative impact of an ABC molecular subtype on the outcome.”
ECOG-ACRIN 1412 details
Goals of the ECOG-ACRIN 1412 study were to evaluate the effect of lenalidomide both in all DLBCL subtypes and in the ABC subtype, maximize the synergy of the immunomodulator with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) while maintaining R-CHOP dose intensity, and facilitate the enrollment of patients with rapidly progressive disease.
To accomplish the last goal, the study was designed to allow enrollment based on local laboratory findings, scans, and diagnostic pathology, without required identification of the cell of origin. Built in to the design was the plan for final eligibility to be based on central pathology review. In other words, the trial design took into account the likelihood that some enrolled patients would not qualify for eligibility based on later pathology review.
The investigators enrolled 349 adults aged 18 years or older with pathologically confirmed DLBCL (regardless of the cell of origin), stage II bulky to stage IV disease, International Prognostic Index (IPI) scores of 2 or greater, and Eastern Cooperative Oncology Group performance status scores of 2 or less.
The patients were stratified by age (younger than 60 years vs. 60 years and older) and by IPI score (2/3 vs. 4/5), and then randomized to receive either six cycles of R-CHOP or R2-CHOP. Lenalidomide was given in a dose of 25 mg on days 1-10 of each cycle. In contrast, the dose used in ROBUST was 15 mg given on days 1-14 of each cycle.
In ECOG-ACRIN 1412, patients assigned to lenalidomide received mandatory neutropenia prophylaxis with granulocyte-colony stimulating factor.
The time from diagnosis to treatment was a median of 21 days, with only 61 of 280 evaluable patients starting treatment more than 31 days after diagnosis. In ROBUST, the median time to start therapy was 31 days.
Dr. Nowakowski and his colleagues had previously shown that time to treatment is an important prognostic factor in DLBCL.
The efficacy evaluation included 145 patients assigned to R2-CHOP, and 135 assigned to R-CHOP. Primary reasons for exclusion were ineligibity following central pathology review or lack of diagnostic material for review.
After a median follow-up of 2.5 years, R2-CHOP was associated with a 34% improvement in progression-free survival, the primary endpoint (hazard ratio [HR] 0.66, P = .03). The 1-year progression-free survival rates were 83% with R2-CHOP and 73% with R-CHOP. Respective 2-year progression-free survival rates were 76% and 70%.
There was no significant difference, however, in the secondary overall survival endpoints with 1-year and 2-year overall survival of 93% vs. 87% and 86% vs. 80%, respectively.
Similarly, there was no difference in rates of PET-ascertained complete response, at 72% with R2-CHOP and 67% with R-CHOP.
R2-CHOP showed greater benefit across most subgroups, including patients with lower IPI score, patients with bulky disease, patients younger than 60 years, women, and patients with shorter time to treatment. There were also nonsignificant trends hinting at better outcomes with R2-CHOP, regardless of cell of origin.
Toxicities were typical for R-CHOP, although patients on R2-CHOP had significantly higher rates of grade 3 or 4 diarrhea, febrile neutropenia, and thrombocytopenia.
In addition to the trial differences mentioned previously, the differences in outcomes might be explained by the possibility that lenalidomide activity is not restricted to ABC DLBCL, Dr. Shipp said.
“One of the things that’s important to remember about lenalidomide is that it’s an immunomodulating agent and it has also has effects on tumor-infiltrating T cells,” she said.
Differences in response to therapy may also be explained by recent findings showing genetic heterogeneity in transcription-defined ABC DLBCLs, she said.
ECOG-ACRIN 1412 was supported by the National Cancer Institute and by Celgene. Dr. Nowakowski reported consulting/advising for and research funding from Celgene and others. Dr. Shipp reported consulting/advising for Bristol-Myers Squibb, honoraria from BMS and AstraZeneca, and institutional research funding from BMS and Bayer.
SOURCE: Nowakowski GS et al. 15-ICML, Abstract 006.
LUGANO, Switzerland – Hot on the heels of the phase 3 ROBUST study showing that adding lenalidomide to standard chemotherapy did not improve outcomes for patients with untreated diffuse large B-cell lymphoma come results of a different study showing a significant benefit with the therapy.
Although, as previously reported, adding lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone in ROBUST, results from the randomized phase 2 ECOG-ACRIN 1412 study showed that R2-CHOP was associated with a 34% reduction in the risk of disease progression or death, compared with R-CHOP alone.
“The efficacy endpoints are consistent, with trends toward higher PET complete response rate and improved overall survival with R-squared CHOP,” Grzegorz S. Nowakowski, MD, of the Mayo Clinic, Rochester, Minn., said at the International Conference on Malignant Lymphoma.
So what’s behind the conflicting findings?
The differences between the results of the two studies may be accounted for by the higher lenalidomide dose used in ECOG-ACRIN 1412, the patient populations – all comers in ECOG-ACRIN versus only patients with activated B-cell (ABC) type DLBCL in ROBUST – and by a 10-day shorter median time to treatment in ECOG-ACRIN 1412, said invited discussant Margaret A. Shipp, MD, of the Dana-Farber Cancer Institute in Boston.
The rationale for adding lenalidomide to R-CHOP came from in vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non-germinal center-like B (GCB) type DLBCL.
In a subanalysis of patients enrolled in MC078E, Dr. Nowakowski and colleagues found that using classification of patients by cell of origin with the NanoString Lymphoma Subtype assay, the addition of lenalidomide to R-CHOP “appears to mitigate the negative impact of an ABC molecular subtype on the outcome.”
ECOG-ACRIN 1412 details
Goals of the ECOG-ACRIN 1412 study were to evaluate the effect of lenalidomide both in all DLBCL subtypes and in the ABC subtype, maximize the synergy of the immunomodulator with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) while maintaining R-CHOP dose intensity, and facilitate the enrollment of patients with rapidly progressive disease.
To accomplish the last goal, the study was designed to allow enrollment based on local laboratory findings, scans, and diagnostic pathology, without required identification of the cell of origin. Built in to the design was the plan for final eligibility to be based on central pathology review. In other words, the trial design took into account the likelihood that some enrolled patients would not qualify for eligibility based on later pathology review.
The investigators enrolled 349 adults aged 18 years or older with pathologically confirmed DLBCL (regardless of the cell of origin), stage II bulky to stage IV disease, International Prognostic Index (IPI) scores of 2 or greater, and Eastern Cooperative Oncology Group performance status scores of 2 or less.
The patients were stratified by age (younger than 60 years vs. 60 years and older) and by IPI score (2/3 vs. 4/5), and then randomized to receive either six cycles of R-CHOP or R2-CHOP. Lenalidomide was given in a dose of 25 mg on days 1-10 of each cycle. In contrast, the dose used in ROBUST was 15 mg given on days 1-14 of each cycle.
In ECOG-ACRIN 1412, patients assigned to lenalidomide received mandatory neutropenia prophylaxis with granulocyte-colony stimulating factor.
The time from diagnosis to treatment was a median of 21 days, with only 61 of 280 evaluable patients starting treatment more than 31 days after diagnosis. In ROBUST, the median time to start therapy was 31 days.
Dr. Nowakowski and his colleagues had previously shown that time to treatment is an important prognostic factor in DLBCL.
The efficacy evaluation included 145 patients assigned to R2-CHOP, and 135 assigned to R-CHOP. Primary reasons for exclusion were ineligibity following central pathology review or lack of diagnostic material for review.
After a median follow-up of 2.5 years, R2-CHOP was associated with a 34% improvement in progression-free survival, the primary endpoint (hazard ratio [HR] 0.66, P = .03). The 1-year progression-free survival rates were 83% with R2-CHOP and 73% with R-CHOP. Respective 2-year progression-free survival rates were 76% and 70%.
There was no significant difference, however, in the secondary overall survival endpoints with 1-year and 2-year overall survival of 93% vs. 87% and 86% vs. 80%, respectively.
Similarly, there was no difference in rates of PET-ascertained complete response, at 72% with R2-CHOP and 67% with R-CHOP.
R2-CHOP showed greater benefit across most subgroups, including patients with lower IPI score, patients with bulky disease, patients younger than 60 years, women, and patients with shorter time to treatment. There were also nonsignificant trends hinting at better outcomes with R2-CHOP, regardless of cell of origin.
Toxicities were typical for R-CHOP, although patients on R2-CHOP had significantly higher rates of grade 3 or 4 diarrhea, febrile neutropenia, and thrombocytopenia.
In addition to the trial differences mentioned previously, the differences in outcomes might be explained by the possibility that lenalidomide activity is not restricted to ABC DLBCL, Dr. Shipp said.
“One of the things that’s important to remember about lenalidomide is that it’s an immunomodulating agent and it has also has effects on tumor-infiltrating T cells,” she said.
Differences in response to therapy may also be explained by recent findings showing genetic heterogeneity in transcription-defined ABC DLBCLs, she said.
ECOG-ACRIN 1412 was supported by the National Cancer Institute and by Celgene. Dr. Nowakowski reported consulting/advising for and research funding from Celgene and others. Dr. Shipp reported consulting/advising for Bristol-Myers Squibb, honoraria from BMS and AstraZeneca, and institutional research funding from BMS and Bayer.
SOURCE: Nowakowski GS et al. 15-ICML, Abstract 006.
REPORTING FROM 15-ICML
Crizanlizumab shows posttreatment effect in sickle cell
FORT LAUDERDALE, FLA. – Sickle cell patients who received high-dose crizanlizumab had fewer vaso-occlusive crises (VOCs) than patients on a low-dose regimen a year after stopping the treatment, findings from a real-world follow-up study suggest.

But hospitalization rates and use of other health care resources were similar during and after therapy, regardless of dose, Nirmish Shah, MD, of Duke Health in Durham, N.C., reported at the annual meeting of the Foundation for Sickle Cell Disease Research.
“This is our attempt at a real-world study of patients after being in a big study that has good results and what happens to them afterward in regard to VOCs and health care utilization,” Dr. Shah said.
He reported results from the SUCCESSOR trial – a multicenter, retrospective cohort study – that evaluated a subset of 48 adult patients up to a year after they completed the SUSTAIN placebo-controlled phase 2 trial of crizanlizumab, a P-selectin inhibitor designed to control sickle cell pain crises.
In SUSTAIN, researchers evaluated two different dosages of crizanlizumab: 5 mg/kg and 2.5 mg/kg.
In the follow-up study, researchers obtained data from medical records over the study period November 2014 to March 2017. Crizanlizumab was not administered in the 52 weeks following the SUSTAIN trial.
They found that the subset of patients on the high dose of crizanlizumab had annual VOC rates of 2.7, compared with 4.0 among patients on the low dose of the drug. By comparison, VOC rates in SUSTAIN were 1.7 per year for the 5-mg/kg–dose group and 3.2 per year for the 2.5-mg/kg–dose group.
Overall, at least 60% of patients in the follow-up study had at least one hospitalization in the year after SUSTAIN. In the higher-dose group, the rates of hospitalization were similar in both SUSTAIN and SUCCESSOR – 53%. In the lower-dose group, hospitalization rates were 67% and 61% in SUCCESSOR and SUSTAIN, respectively.
“Among the previously treated patients with crizanlizumab, the total number of emergency department visits did seem to be higher in SUCCESSOR,” Dr. Shah said.
The study was limited by its retrospective nature and the fact that full data sets and follow-up were available on only a small number of patients, Dr. Shah said. “It was underpowered for a statistical analysis between groups,” he said. The goal was to determine the drug’s effect after treatment is discontinued, he said.
Dr. Shah reported a financial relationship with Novartis, which is developing crizanlizumab.
SOURCE: Shah N et al. FSCDR 2019, Abstract JSCDH-D-19-00031.
FORT LAUDERDALE, FLA. – Sickle cell patients who received high-dose crizanlizumab had fewer vaso-occlusive crises (VOCs) than patients on a low-dose regimen a year after stopping the treatment, findings from a real-world follow-up study suggest.
But hospitalization rates and use of other health care resources were similar during and after therapy, regardless of dose, Nirmish Shah, MD, of Duke Health in Durham, N.C., reported at the annual meeting of the Foundation for Sickle Cell Disease Research.
“This is our attempt at a real-world study of patients after being in a big study that has good results and what happens to them afterward in regard to VOCs and health care utilization,” Dr. Shah said.
He reported results from the SUCCESSOR trial – a multicenter, retrospective cohort study – that evaluated a subset of 48 adult patients up to a year after they completed the SUSTAIN placebo-controlled phase 2 trial of crizanlizumab, a P-selectin inhibitor designed to control sickle cell pain crises.
In SUSTAIN, researchers evaluated two different dosages of crizanlizumab: 5 mg/kg and 2.5 mg/kg.
In the follow-up study, researchers obtained data from medical records over the study period November 2014 to March 2017. Crizanlizumab was not administered in the 52 weeks following the SUSTAIN trial.
They found that the subset of patients on the high dose of crizanlizumab had annual VOC rates of 2.7, compared with 4.0 among patients on the low dose of the drug. By comparison, VOC rates in SUSTAIN were 1.7 per year for the 5-mg/kg–dose group and 3.2 per year for the 2.5-mg/kg–dose group.
Overall, at least 60% of patients in the follow-up study had at least one hospitalization in the year after SUSTAIN. In the higher-dose group, the rates of hospitalization were similar in both SUSTAIN and SUCCESSOR – 53%. In the lower-dose group, hospitalization rates were 67% and 61% in SUCCESSOR and SUSTAIN, respectively.
“Among the previously treated patients with crizanlizumab, the total number of emergency department visits did seem to be higher in SUCCESSOR,” Dr. Shah said.
The study was limited by its retrospective nature and the fact that full data sets and follow-up were available on only a small number of patients, Dr. Shah said. “It was underpowered for a statistical analysis between groups,” he said. The goal was to determine the drug’s effect after treatment is discontinued, he said.
Dr. Shah reported a financial relationship with Novartis, which is developing crizanlizumab.
SOURCE: Shah N et al. FSCDR 2019, Abstract JSCDH-D-19-00031.
FORT LAUDERDALE, FLA. – Sickle cell patients who received high-dose crizanlizumab had fewer vaso-occlusive crises (VOCs) than patients on a low-dose regimen a year after stopping the treatment, findings from a real-world follow-up study suggest.
But hospitalization rates and use of other health care resources were similar during and after therapy, regardless of dose, Nirmish Shah, MD, of Duke Health in Durham, N.C., reported at the annual meeting of the Foundation for Sickle Cell Disease Research.
“This is our attempt at a real-world study of patients after being in a big study that has good results and what happens to them afterward in regard to VOCs and health care utilization,” Dr. Shah said.
He reported results from the SUCCESSOR trial – a multicenter, retrospective cohort study – that evaluated a subset of 48 adult patients up to a year after they completed the SUSTAIN placebo-controlled phase 2 trial of crizanlizumab, a P-selectin inhibitor designed to control sickle cell pain crises.
In SUSTAIN, researchers evaluated two different dosages of crizanlizumab: 5 mg/kg and 2.5 mg/kg.
In the follow-up study, researchers obtained data from medical records over the study period November 2014 to March 2017. Crizanlizumab was not administered in the 52 weeks following the SUSTAIN trial.
They found that the subset of patients on the high dose of crizanlizumab had annual VOC rates of 2.7, compared with 4.0 among patients on the low dose of the drug. By comparison, VOC rates in SUSTAIN were 1.7 per year for the 5-mg/kg–dose group and 3.2 per year for the 2.5-mg/kg–dose group.
Overall, at least 60% of patients in the follow-up study had at least one hospitalization in the year after SUSTAIN. In the higher-dose group, the rates of hospitalization were similar in both SUSTAIN and SUCCESSOR – 53%. In the lower-dose group, hospitalization rates were 67% and 61% in SUCCESSOR and SUSTAIN, respectively.
“Among the previously treated patients with crizanlizumab, the total number of emergency department visits did seem to be higher in SUCCESSOR,” Dr. Shah said.
The study was limited by its retrospective nature and the fact that full data sets and follow-up were available on only a small number of patients, Dr. Shah said. “It was underpowered for a statistical analysis between groups,” he said. The goal was to determine the drug’s effect after treatment is discontinued, he said.
Dr. Shah reported a financial relationship with Novartis, which is developing crizanlizumab.
SOURCE: Shah N et al. FSCDR 2019, Abstract JSCDH-D-19-00031.
REPORTING FROM FSCDR 2019
SC daratumumab deemed feasible for every multiple myeloma patient
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
REPORTING FROM ASCO 2019

Acalabrutinib extends PFS in advanced CLL
AMSTERDAM – For patients with relapsed or refractory chronic lymphocytic leukemia (CLL), monotherapy with the Bruton tyrosine kinase (BTK) inhibitor acalabrutinib (Calquence) was associated with better progression-free survival and a more tolerable safety profile than rituximab combined with either idelalisib (Zydelig) or bendamustine, an interim analysis from the phase 3 ASCEND trial showed.

Among 310 patients with previously treated CLL followed for a median of 16.1 months, the primary endpoint of median progression-free survival (PFS), as assessed by independent reviewers, had not been reached for patients treated with acalabrutinib, compared with 16.5 months for patients treated with idelalisib and rituximab (IdR) or bendamustine and rituximab (BR), reported Paolo Ghia, MD, PhD, from Università Vita-Salute San Raffaele in Milan.
“We show that acalabrutinib improved progression-free survival across all groups, including those with high-risk features,” he said at the annual congress of the European Hematology Association.
Acalabrutinib is approved in the United States for treatment of mantle cell lymphoma that has progressed on at least one prior therapy. It has been shown in preclinical studies to be more selective for BTK than the first-in-class agent ibrutinib (Imbruvica), with less off-target kinase inhibition, Dr. Ghia said.
ASCEND was designed to see whether acalabrutinib monotherapy could offer superior PFS to IdR or BR in patients with CLL who had progressed or were refractory to at least one prior line of therapy.
Patients were randomly assigned, with 155 patients in each arm, to either acalabrutinib 100 mg orally twice daily or the investigator’s choice of either idelalisib 150 mg orally twice daily plus IV rituximab at an initial dose of 375 mg/m2, followed by up to seven doses at 500 mg/m2 delivered every 2 weeks for four infusions, then every 4 weeks for the remaining three infusions or IV bendamustine 70 mg/m2 on days 1 and 2 of each cycle, plus rituximab at the 375 mg/m2 dose on day 1 for the first cycle, followed by 500 mg/m2 for up to six total cycles.
Dr. Ghia presented results of an interim analysis planned for when two-thirds of the predicted PFS events (approximately 79) had occurred.
The baseline patient characteristics were generally similar, with a median age of 68 years in the acalabrutinib arm and 67 years in the comparison arm. Almost half of all patients in each arm had bulky disease, defined as 5 cm or greater. The majority of patients had two or more prior lines of therapy.
The primary endpoint of PFS as assessed by independent review favored acalabrutinib, with a hazard ratio of 0.31 (P less than .0001). Results were similar when acalabrutinib was compared with each of the regimens in the comparison arm (HR, 0.29 vs. IdR, 0.36 vs. BR; P less than .001 for each comparison).
Acalabrutinib was also superior in patients with high-risk cytogenetic features, compared with the other two regimens combined (HR, 0.27; P less than .001).
The benefit of the BTK inhibitor was consistent across all subgroups, including age, sex, performance status, Rai stage at screening, bulky/nonbulky disease, number of prior therapies, presence or absence of deletion 17p or TP53 mutation, mutated or unmutated immunoglobulin heavy chain, and complex/noncomplex karyotype.
Reviewer-assessed objective response rates were similar, occurring in 81% of patients on acalabrutinib and 76% of patients on other regimens.
There were no complete responses in the acalabrutinib arm, compared with two complete responses in the comparison arm. The majority of responses in each arm were partial responses (81% and 74%, respectively).
The median duration of response was not reached with acalabrutinib, compared with 13.6 months with the other therapies (HR, 0.33; P less than .0001).
In all, 85% of patients on acalabrutinib had a response lasting at least 12 months, compared with 60% of patients on the other regimens. There was no difference in overall survival at the 16.1-month median follow-up.
Adverse events of any grade occurred in 94% of patients on acalabrutinib, 99% on IdR, and 80% on BR; the respective incidences of serious adverse events were 29%, 56%, and 26%. Grade 3-4 adverse events occurred in 45%, 86%, and 43% of patients, respectively.
There were 13 treatment-related deaths. Six deaths in the acalabrutinib arm were caused by brain neoplasm, cachexia, cerebral ischemia, malignant lung tumor, neuroendocrine carcinoma, and sepsis. Five deaths among IdR-treated patients included chronic heart failure, cardiopulmonary disease, interstitial lung disease, MI, and pseudomonal pneumonia. Two deaths in BR-treated patients were attributed to acute cardiac failure and a gastric neoplasm.
The results show that “acalabrutinib has demonstrated efficacy in previously untreated and relapsed/refractory CLL and may be considered as an option in the future treatment paradigm,” Dr. Ghia said.
Acalabrutinib monotherapy is currently being compared with ibrutinib monotherapy in patients with relapsed/refractory CLL; in addition, the phase 3 ELEVATE-TN study investigating acalabrutinib in combination with obinutuzumab (Gazyva) versus obinutuzumab plus chlorambucil has reached its primary PFS endpoint and will be reported soon, Dr. Ghia said.
The ASCEND trial is sponsored by Acerta Pharma; AstraZeneca holds majority shares in the company. Dr. Ghia reported consulting fees and honoraria from AstraZeneca and other companies, and research funding from several different companies.
SOURCE: Ghia P et al. EHA Congress, Abstract LB2606.
AMSTERDAM – For patients with relapsed or refractory chronic lymphocytic leukemia (CLL), monotherapy with the Bruton tyrosine kinase (BTK) inhibitor acalabrutinib (Calquence) was associated with better progression-free survival and a more tolerable safety profile than rituximab combined with either idelalisib (Zydelig) or bendamustine, an interim analysis from the phase 3 ASCEND trial showed.
Among 310 patients with previously treated CLL followed for a median of 16.1 months, the primary endpoint of median progression-free survival (PFS), as assessed by independent reviewers, had not been reached for patients treated with acalabrutinib, compared with 16.5 months for patients treated with idelalisib and rituximab (IdR) or bendamustine and rituximab (BR), reported Paolo Ghia, MD, PhD, from Università Vita-Salute San Raffaele in Milan.
“We show that acalabrutinib improved progression-free survival across all groups, including those with high-risk features,” he said at the annual congress of the European Hematology Association.
Acalabrutinib is approved in the United States for treatment of mantle cell lymphoma that has progressed on at least one prior therapy. It has been shown in preclinical studies to be more selective for BTK than the first-in-class agent ibrutinib (Imbruvica), with less off-target kinase inhibition, Dr. Ghia said.
ASCEND was designed to see whether acalabrutinib monotherapy could offer superior PFS to IdR or BR in patients with CLL who had progressed or were refractory to at least one prior line of therapy.
Patients were randomly assigned, with 155 patients in each arm, to either acalabrutinib 100 mg orally twice daily or the investigator’s choice of either idelalisib 150 mg orally twice daily plus IV rituximab at an initial dose of 375 mg/m2, followed by up to seven doses at 500 mg/m2 delivered every 2 weeks for four infusions, then every 4 weeks for the remaining three infusions or IV bendamustine 70 mg/m2 on days 1 and 2 of each cycle, plus rituximab at the 375 mg/m2 dose on day 1 for the first cycle, followed by 500 mg/m2 for up to six total cycles.
Dr. Ghia presented results of an interim analysis planned for when two-thirds of the predicted PFS events (approximately 79) had occurred.
The baseline patient characteristics were generally similar, with a median age of 68 years in the acalabrutinib arm and 67 years in the comparison arm. Almost half of all patients in each arm had bulky disease, defined as 5 cm or greater. The majority of patients had two or more prior lines of therapy.
The primary endpoint of PFS as assessed by independent review favored acalabrutinib, with a hazard ratio of 0.31 (P less than .0001). Results were similar when acalabrutinib was compared with each of the regimens in the comparison arm (HR, 0.29 vs. IdR, 0.36 vs. BR; P less than .001 for each comparison).
Acalabrutinib was also superior in patients with high-risk cytogenetic features, compared with the other two regimens combined (HR, 0.27; P less than .001).
The benefit of the BTK inhibitor was consistent across all subgroups, including age, sex, performance status, Rai stage at screening, bulky/nonbulky disease, number of prior therapies, presence or absence of deletion 17p or TP53 mutation, mutated or unmutated immunoglobulin heavy chain, and complex/noncomplex karyotype.
Reviewer-assessed objective response rates were similar, occurring in 81% of patients on acalabrutinib and 76% of patients on other regimens.
There were no complete responses in the acalabrutinib arm, compared with two complete responses in the comparison arm. The majority of responses in each arm were partial responses (81% and 74%, respectively).
The median duration of response was not reached with acalabrutinib, compared with 13.6 months with the other therapies (HR, 0.33; P less than .0001).
In all, 85% of patients on acalabrutinib had a response lasting at least 12 months, compared with 60% of patients on the other regimens. There was no difference in overall survival at the 16.1-month median follow-up.
Adverse events of any grade occurred in 94% of patients on acalabrutinib, 99% on IdR, and 80% on BR; the respective incidences of serious adverse events were 29%, 56%, and 26%. Grade 3-4 adverse events occurred in 45%, 86%, and 43% of patients, respectively.
There were 13 treatment-related deaths. Six deaths in the acalabrutinib arm were caused by brain neoplasm, cachexia, cerebral ischemia, malignant lung tumor, neuroendocrine carcinoma, and sepsis. Five deaths among IdR-treated patients included chronic heart failure, cardiopulmonary disease, interstitial lung disease, MI, and pseudomonal pneumonia. Two deaths in BR-treated patients were attributed to acute cardiac failure and a gastric neoplasm.
The results show that “acalabrutinib has demonstrated efficacy in previously untreated and relapsed/refractory CLL and may be considered as an option in the future treatment paradigm,” Dr. Ghia said.
Acalabrutinib monotherapy is currently being compared with ibrutinib monotherapy in patients with relapsed/refractory CLL; in addition, the phase 3 ELEVATE-TN study investigating acalabrutinib in combination with obinutuzumab (Gazyva) versus obinutuzumab plus chlorambucil has reached its primary PFS endpoint and will be reported soon, Dr. Ghia said.
The ASCEND trial is sponsored by Acerta Pharma; AstraZeneca holds majority shares in the company. Dr. Ghia reported consulting fees and honoraria from AstraZeneca and other companies, and research funding from several different companies.
SOURCE: Ghia P et al. EHA Congress, Abstract LB2606.
AMSTERDAM – For patients with relapsed or refractory chronic lymphocytic leukemia (CLL), monotherapy with the Bruton tyrosine kinase (BTK) inhibitor acalabrutinib (Calquence) was associated with better progression-free survival and a more tolerable safety profile than rituximab combined with either idelalisib (Zydelig) or bendamustine, an interim analysis from the phase 3 ASCEND trial showed.
Among 310 patients with previously treated CLL followed for a median of 16.1 months, the primary endpoint of median progression-free survival (PFS), as assessed by independent reviewers, had not been reached for patients treated with acalabrutinib, compared with 16.5 months for patients treated with idelalisib and rituximab (IdR) or bendamustine and rituximab (BR), reported Paolo Ghia, MD, PhD, from Università Vita-Salute San Raffaele in Milan.
“We show that acalabrutinib improved progression-free survival across all groups, including those with high-risk features,” he said at the annual congress of the European Hematology Association.
Acalabrutinib is approved in the United States for treatment of mantle cell lymphoma that has progressed on at least one prior therapy. It has been shown in preclinical studies to be more selective for BTK than the first-in-class agent ibrutinib (Imbruvica), with less off-target kinase inhibition, Dr. Ghia said.
ASCEND was designed to see whether acalabrutinib monotherapy could offer superior PFS to IdR or BR in patients with CLL who had progressed or were refractory to at least one prior line of therapy.
Patients were randomly assigned, with 155 patients in each arm, to either acalabrutinib 100 mg orally twice daily or the investigator’s choice of either idelalisib 150 mg orally twice daily plus IV rituximab at an initial dose of 375 mg/m2, followed by up to seven doses at 500 mg/m2 delivered every 2 weeks for four infusions, then every 4 weeks for the remaining three infusions or IV bendamustine 70 mg/m2 on days 1 and 2 of each cycle, plus rituximab at the 375 mg/m2 dose on day 1 for the first cycle, followed by 500 mg/m2 for up to six total cycles.
Dr. Ghia presented results of an interim analysis planned for when two-thirds of the predicted PFS events (approximately 79) had occurred.
The baseline patient characteristics were generally similar, with a median age of 68 years in the acalabrutinib arm and 67 years in the comparison arm. Almost half of all patients in each arm had bulky disease, defined as 5 cm or greater. The majority of patients had two or more prior lines of therapy.
The primary endpoint of PFS as assessed by independent review favored acalabrutinib, with a hazard ratio of 0.31 (P less than .0001). Results were similar when acalabrutinib was compared with each of the regimens in the comparison arm (HR, 0.29 vs. IdR, 0.36 vs. BR; P less than .001 for each comparison).
Acalabrutinib was also superior in patients with high-risk cytogenetic features, compared with the other two regimens combined (HR, 0.27; P less than .001).
The benefit of the BTK inhibitor was consistent across all subgroups, including age, sex, performance status, Rai stage at screening, bulky/nonbulky disease, number of prior therapies, presence or absence of deletion 17p or TP53 mutation, mutated or unmutated immunoglobulin heavy chain, and complex/noncomplex karyotype.
Reviewer-assessed objective response rates were similar, occurring in 81% of patients on acalabrutinib and 76% of patients on other regimens.
There were no complete responses in the acalabrutinib arm, compared with two complete responses in the comparison arm. The majority of responses in each arm were partial responses (81% and 74%, respectively).
The median duration of response was not reached with acalabrutinib, compared with 13.6 months with the other therapies (HR, 0.33; P less than .0001).
In all, 85% of patients on acalabrutinib had a response lasting at least 12 months, compared with 60% of patients on the other regimens. There was no difference in overall survival at the 16.1-month median follow-up.
Adverse events of any grade occurred in 94% of patients on acalabrutinib, 99% on IdR, and 80% on BR; the respective incidences of serious adverse events were 29%, 56%, and 26%. Grade 3-4 adverse events occurred in 45%, 86%, and 43% of patients, respectively.
There were 13 treatment-related deaths. Six deaths in the acalabrutinib arm were caused by brain neoplasm, cachexia, cerebral ischemia, malignant lung tumor, neuroendocrine carcinoma, and sepsis. Five deaths among IdR-treated patients included chronic heart failure, cardiopulmonary disease, interstitial lung disease, MI, and pseudomonal pneumonia. Two deaths in BR-treated patients were attributed to acute cardiac failure and a gastric neoplasm.
The results show that “acalabrutinib has demonstrated efficacy in previously untreated and relapsed/refractory CLL and may be considered as an option in the future treatment paradigm,” Dr. Ghia said.
Acalabrutinib monotherapy is currently being compared with ibrutinib monotherapy in patients with relapsed/refractory CLL; in addition, the phase 3 ELEVATE-TN study investigating acalabrutinib in combination with obinutuzumab (Gazyva) versus obinutuzumab plus chlorambucil has reached its primary PFS endpoint and will be reported soon, Dr. Ghia said.
The ASCEND trial is sponsored by Acerta Pharma; AstraZeneca holds majority shares in the company. Dr. Ghia reported consulting fees and honoraria from AstraZeneca and other companies, and research funding from several different companies.
SOURCE: Ghia P et al. EHA Congress, Abstract LB2606.
REPORTING FROM EHA CONGRESS
Ibrutinib tops chlorambucil against CLL
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.

The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
REPORTING FROM EHA CONGRESS
R2-CHOP doesn’t improve survival in DLBCL
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.

Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.
Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.
Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
REPORTING FROM 15-ICML
No reduction in PE risk with vena cava filters after severe injury
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
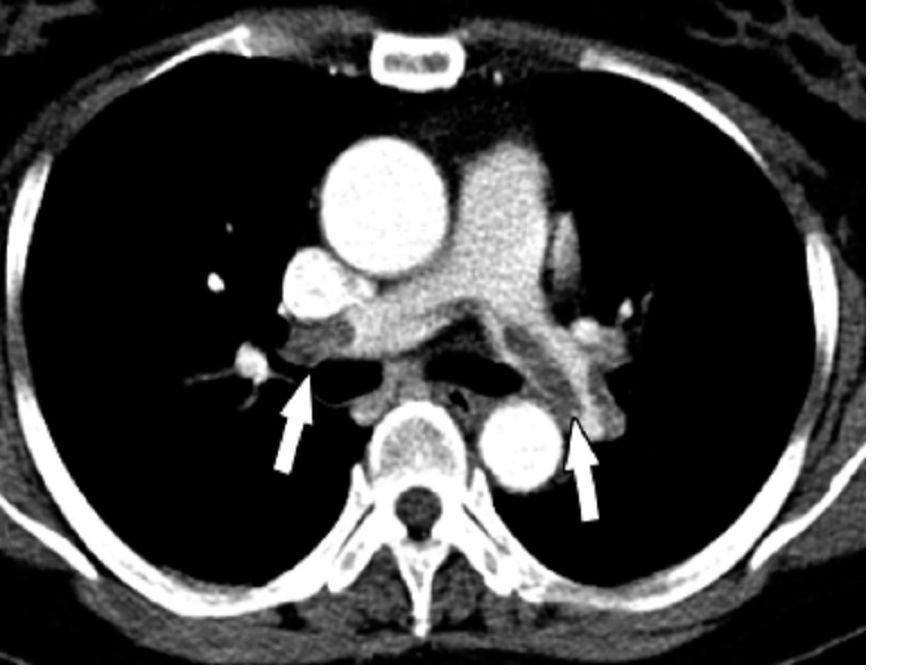
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
REPORTING FROM 2019 ISTH CONGRESS
CNS-directed therapy appears more effective for synDLBCL
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
FROM BRITISH JOURNAL OF HAEMATOLOGY
No benefit with rituximab after first CR in DLBCL
AMSTERDAM – Rituximab maintenance therapy did not significantly prolong disease-free survival among patients with diffuse large B-cell lymphoma in complete remission after induction chemotherapy, investigators in the HOVON-Nordic trial collaboration found.

Among 380 patients with diffuse large B-cell lymphoma (DLBCL) in complete remission following induction chemotherapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the median disease-free survival (DFS) after a median follow-up of 79.9 months had not been reached for patients randomized to either rituximab maintenance therapy or observation alone, reported Pieternella J. Lugtenburg, MD, PhD, from Erasmus Medical Center in Rotterdam, the Netherlands.
“In patients with diffuse large B-cell lymphoma in first complete remission after R-CHOP induction therapy, rituximab maintenance therapy does not prolong disease-free survival, and we could not identify a clinical subgroup that benefited,” she said at the annual congress of the European Hematology Association.
The randomized, phase 3 HOVON-Nordic LG trial compared R-CHOP-14 with rituximab delivered on day 1 for four cycles in one arm, and on days 1 and 8 in the comparison arm (R2-CHOP). Patients were assessed for response after four cycles, and those with progressive disease were taken off the study. The remaining patients continued on their assigned regimen for an additional four cycles and second assessment, and those who had complete responses then underwent a second randomization to rituximab maintenance or observation. Patients randomized to maintenance could receive up to 12 8-week cycles.
The second randomization was stratified by age (18-65 vs. 66-80 years), age-adjusted International Prognostic Index score, and R-CHOP regimen (one or two infusions per cycle in the first randomization).
The induction phase found no significant benefit for the additional rituximab infusion for the primary endpoint of progression-free survival, with 3-year PFS rates of 74% for R-CHOP and 71% for R2-CHOP. Respective 5-year PFS rates were 68% and 62%.
There were no differences between the arms by any of the stratification factors, and no difference in overall survival.
Dr. Lugtenburg presented results of the maintenance phase, which included all patients in complete remission at least 4 weeks after the last cycle of R-CHOP-14 who did not have rituximab-related adverse events or active infections.
A total of 195 patients were randomized to observation, and 185 to maintenance. Of this latter group, 149 patients received all 12 cycles, with a median duration of exposure of 22.5 months.
As noted, DFS, the primary endpoint for the maintenance phase, was not significantly different between the arms, with 136 events in the maintenance arm versus 134 in the observation arm. A total of 21 patients assigned to maintenance died, and 22 patients assigned to observation died during follow-up.
The respective 5-year DFS rates were 79% and 74%, a difference that was not statistically significant.
There were no significant differences between the arms in either time to relapse or death, and no difference in overall survival.

In the question-and-answer section following her talk, Marek Trnený, MD, from Charles University in Prague, pointed out that, in the NHL 13 study, which looked at rituximab maintenance for patients with DLBCL or follicular lymphoma in first remission, there was an apparent clinical benefit with rituximab maintenance for men with low International Prognostic Index scores, and asked whether Dr. Lugtenburg and colleagues had seen similar trends.
“We knew about this finding, and we really didn’t see any differences between males and females, not only in terms of efficacy, but also in terms of toxicity, they had actually the same rates of toxicity,” she said.
In NHL 13, women had a significantly higher incidence of grade 3-4 adverse events, compared with men.
The study was sponsored by HOVON, the HematoOncology Foundation for Adults in the Netherlands, with additional support from Roche. Dr. Lugtenburg reported consultancy for and research funding from Roche and others. Dr. Trnený reported advisory board activity, research support, and honoraria from Roche and others.
SOURCE: Lugtenburg PJ et al. EHA 2019, Abstract S1559.
AMSTERDAM – Rituximab maintenance therapy did not significantly prolong disease-free survival among patients with diffuse large B-cell lymphoma in complete remission after induction chemotherapy, investigators in the HOVON-Nordic trial collaboration found.
Among 380 patients with diffuse large B-cell lymphoma (DLBCL) in complete remission following induction chemotherapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the median disease-free survival (DFS) after a median follow-up of 79.9 months had not been reached for patients randomized to either rituximab maintenance therapy or observation alone, reported Pieternella J. Lugtenburg, MD, PhD, from Erasmus Medical Center in Rotterdam, the Netherlands.
“In patients with diffuse large B-cell lymphoma in first complete remission after R-CHOP induction therapy, rituximab maintenance therapy does not prolong disease-free survival, and we could not identify a clinical subgroup that benefited,” she said at the annual congress of the European Hematology Association.
The randomized, phase 3 HOVON-Nordic LG trial compared R-CHOP-14 with rituximab delivered on day 1 for four cycles in one arm, and on days 1 and 8 in the comparison arm (R2-CHOP). Patients were assessed for response after four cycles, and those with progressive disease were taken off the study. The remaining patients continued on their assigned regimen for an additional four cycles and second assessment, and those who had complete responses then underwent a second randomization to rituximab maintenance or observation. Patients randomized to maintenance could receive up to 12 8-week cycles.
The second randomization was stratified by age (18-65 vs. 66-80 years), age-adjusted International Prognostic Index score, and R-CHOP regimen (one or two infusions per cycle in the first randomization).
The induction phase found no significant benefit for the additional rituximab infusion for the primary endpoint of progression-free survival, with 3-year PFS rates of 74% for R-CHOP and 71% for R2-CHOP. Respective 5-year PFS rates were 68% and 62%.
There were no differences between the arms by any of the stratification factors, and no difference in overall survival.
Dr. Lugtenburg presented results of the maintenance phase, which included all patients in complete remission at least 4 weeks after the last cycle of R-CHOP-14 who did not have rituximab-related adverse events or active infections.
A total of 195 patients were randomized to observation, and 185 to maintenance. Of this latter group, 149 patients received all 12 cycles, with a median duration of exposure of 22.5 months.
As noted, DFS, the primary endpoint for the maintenance phase, was not significantly different between the arms, with 136 events in the maintenance arm versus 134 in the observation arm. A total of 21 patients assigned to maintenance died, and 22 patients assigned to observation died during follow-up.
The respective 5-year DFS rates were 79% and 74%, a difference that was not statistically significant.
There were no significant differences between the arms in either time to relapse or death, and no difference in overall survival.
In the question-and-answer section following her talk, Marek Trnený, MD, from Charles University in Prague, pointed out that, in the NHL 13 study, which looked at rituximab maintenance for patients with DLBCL or follicular lymphoma in first remission, there was an apparent clinical benefit with rituximab maintenance for men with low International Prognostic Index scores, and asked whether Dr. Lugtenburg and colleagues had seen similar trends.
“We knew about this finding, and we really didn’t see any differences between males and females, not only in terms of efficacy, but also in terms of toxicity, they had actually the same rates of toxicity,” she said.
In NHL 13, women had a significantly higher incidence of grade 3-4 adverse events, compared with men.
The study was sponsored by HOVON, the HematoOncology Foundation for Adults in the Netherlands, with additional support from Roche. Dr. Lugtenburg reported consultancy for and research funding from Roche and others. Dr. Trnený reported advisory board activity, research support, and honoraria from Roche and others.
SOURCE: Lugtenburg PJ et al. EHA 2019, Abstract S1559.
AMSTERDAM – Rituximab maintenance therapy did not significantly prolong disease-free survival among patients with diffuse large B-cell lymphoma in complete remission after induction chemotherapy, investigators in the HOVON-Nordic trial collaboration found.
Among 380 patients with diffuse large B-cell lymphoma (DLBCL) in complete remission following induction chemotherapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the median disease-free survival (DFS) after a median follow-up of 79.9 months had not been reached for patients randomized to either rituximab maintenance therapy or observation alone, reported Pieternella J. Lugtenburg, MD, PhD, from Erasmus Medical Center in Rotterdam, the Netherlands.
“In patients with diffuse large B-cell lymphoma in first complete remission after R-CHOP induction therapy, rituximab maintenance therapy does not prolong disease-free survival, and we could not identify a clinical subgroup that benefited,” she said at the annual congress of the European Hematology Association.
The randomized, phase 3 HOVON-Nordic LG trial compared R-CHOP-14 with rituximab delivered on day 1 for four cycles in one arm, and on days 1 and 8 in the comparison arm (R2-CHOP). Patients were assessed for response after four cycles, and those with progressive disease were taken off the study. The remaining patients continued on their assigned regimen for an additional four cycles and second assessment, and those who had complete responses then underwent a second randomization to rituximab maintenance or observation. Patients randomized to maintenance could receive up to 12 8-week cycles.
The second randomization was stratified by age (18-65 vs. 66-80 years), age-adjusted International Prognostic Index score, and R-CHOP regimen (one or two infusions per cycle in the first randomization).
The induction phase found no significant benefit for the additional rituximab infusion for the primary endpoint of progression-free survival, with 3-year PFS rates of 74% for R-CHOP and 71% for R2-CHOP. Respective 5-year PFS rates were 68% and 62%.
There were no differences between the arms by any of the stratification factors, and no difference in overall survival.
Dr. Lugtenburg presented results of the maintenance phase, which included all patients in complete remission at least 4 weeks after the last cycle of R-CHOP-14 who did not have rituximab-related adverse events or active infections.
A total of 195 patients were randomized to observation, and 185 to maintenance. Of this latter group, 149 patients received all 12 cycles, with a median duration of exposure of 22.5 months.
As noted, DFS, the primary endpoint for the maintenance phase, was not significantly different between the arms, with 136 events in the maintenance arm versus 134 in the observation arm. A total of 21 patients assigned to maintenance died, and 22 patients assigned to observation died during follow-up.
The respective 5-year DFS rates were 79% and 74%, a difference that was not statistically significant.
There were no significant differences between the arms in either time to relapse or death, and no difference in overall survival.
In the question-and-answer section following her talk, Marek Trnený, MD, from Charles University in Prague, pointed out that, in the NHL 13 study, which looked at rituximab maintenance for patients with DLBCL or follicular lymphoma in first remission, there was an apparent clinical benefit with rituximab maintenance for men with low International Prognostic Index scores, and asked whether Dr. Lugtenburg and colleagues had seen similar trends.
“We knew about this finding, and we really didn’t see any differences between males and females, not only in terms of efficacy, but also in terms of toxicity, they had actually the same rates of toxicity,” she said.
In NHL 13, women had a significantly higher incidence of grade 3-4 adverse events, compared with men.
The study was sponsored by HOVON, the HematoOncology Foundation for Adults in the Netherlands, with additional support from Roche. Dr. Lugtenburg reported consultancy for and research funding from Roche and others. Dr. Trnený reported advisory board activity, research support, and honoraria from Roche and others.
SOURCE: Lugtenburg PJ et al. EHA 2019, Abstract S1559.
REPORTING FROM EHA 2019