User login
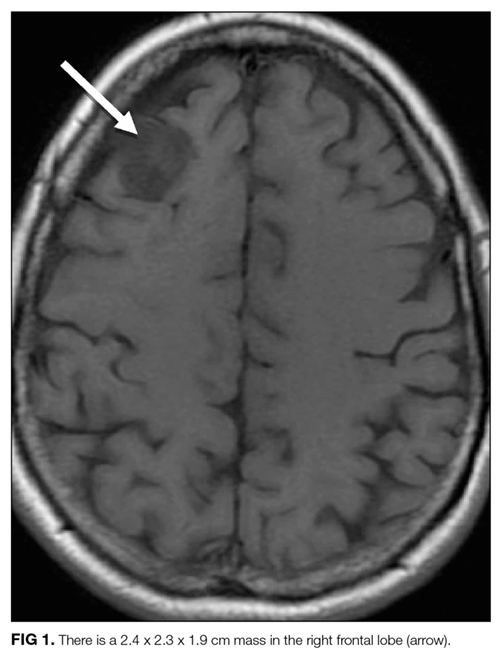
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5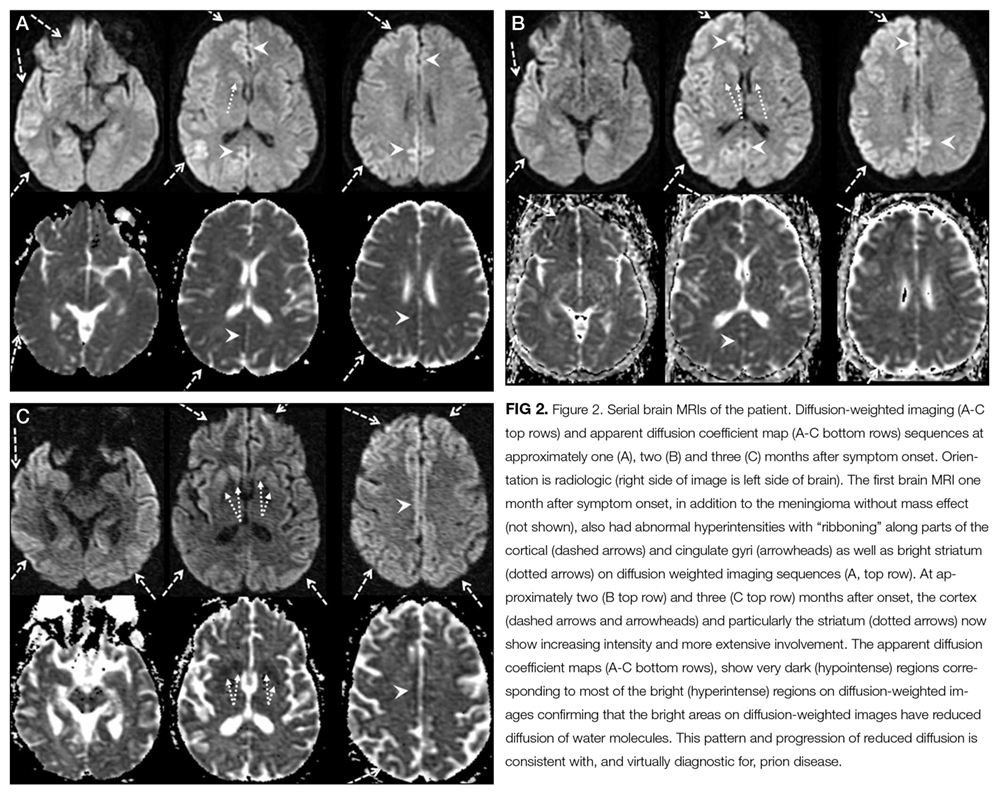
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
A 57-year-old woman presented to the emergency department of a community hospital with a 2-week history of dizziness, blurred vision, and poor coordination following a flu-like illness. Symptoms were initially attributed to complications from a presumed viral illness, but when they persisted for 2 weeks, she underwent magnetic resonance imaging (MRI) of the brain, which was reported as showing a 2.4 x 2.3 x 1.9 cm right frontal lobe mass with mild mass effect and contrast enhancement (Figure 1). She was discharged home at her request with plans for outpatient follow-up.
Brain masses are usually neoplastic, infectious, or less commonly, inflammatory. The isolated lesion in the right frontal lobe is unlikely to explain her symptoms, which are more suggestive of multifocal disease or elevated intracranial pressure. Although the frontal eye fields could be affected by the mass, such lesions usually cause tonic eye deviation, not blurry vision; furthermore, coordination, which is impaired here, is not governed by the frontal lobe.
Two weeks later, she returned to the same emergency department with worsening symptoms and new bilateral upper extremity dystonia, confusion, and visual hallucinations. Cerebrospinal fluid (CSF) analysis revealed clear, nonxanthochromic fluid with 4 nucleated cells (a differential was not performed), 113 red blood cells, glucose of 80 mg/dL (normal range, 50-80 mg/dL), and protein of 52 mg/dL (normal range, 15-45 mg/dL).
Confusion is generally caused by a metabolic, infectious, structural, or toxic etiology. Standard CSF test results are usually normal with most toxic or metabolic encephalopathies. The absence of significant CSF inflammation argues against infectious encephalitis; paraneoplastic and autoimmune encephalitis, however, are still possible. The CSF red blood cells were likely due to a mildly traumatic tap, but also may have arisen from the frontal lobe mass or a more diffuse invasive process, although the lack of xanthochromia argues against this. Delirium and red blood cells in the CSF should trigger consideration of herpes simplex virus (HSV) encephalitis, although the time course is a bit too protracted and the reported MRI findings do not suggest typical medial temporal lobe involvement.
The disparate neurologic findings suggest a multifocal process, perhaps embolic (eg, endocarditis), ischemic (eg, intravascular lymphoma), infiltrative (eg, malignancy, neurosarcoidosis), or demyelinating (eg, postinfectious acute disseminated encephalomyelitis, multiple sclerosis). However, most of these would have been detected on the initial MRI. Upper extremity dystonia would likely localize to the basal ganglia, whereas confusion and visual hallucinations are more global. The combination of a movement disorder and visual hallucinations is seen in Lewy body dementia, but this tempo is not typical.
Although the CSF does not have pleocytosis, her original symptoms were flu-like; therefore, CSF testing for viruses (eg, enterovirus) is reasonable. Bacterial, mycobacteria, and fungal studies are apt to be unrevealing, but CSF cytology, IgG index, and oligoclonal bands may be useful. Should the encephalopathy progress further and the general medical evaluation prove to be normal, then tests for autoimmune disorders (eg, antinuclear antibodies, NMDAR, paraneoplastic disorders) and rare causes of rapidly progressive dementias (eg, prion diseases) should be sent.
Additional CSF studies including HSV polymerase chain reaction (PCR), West Nile PCR, Lyme antibody, paraneoplastic antibodies, and cytology were sent. Intravenous acyclovir was administered. The above studies, as well as Gram stain, acid-fast bacillus stain, fungal stain, and cultures, were negative. She was started on levetiracetam for seizure prevention due to the mass lesion. An electroencephalogram (EEG) was reported as showing diffuse background slowing with superimposed semiperiodic sharp waves with a right hemispheric emphasis. Intravenous immunoglobulin (IVIG) 0.4 mg/kg/day over 5 days was administered with no improvement. The patient was transferred to an academic medical center for further evaluation.
The EEG reflects encephalopathy without pointing to a specific diagnosis. Prophylactic antiepileptic medications are not indicated for CNS mass lesions without clinical or electrophysiologic seizure activity. IVIG is often administered when an autoimmune encephalitis is suspected, but the lack of response does not rule out an autoimmune condition.
Her medical history included bilateral cataract extraction, right leg fracture, tonsillectomy, and total abdominal hysterectomy. She had a 25-year smoking history and a family history of lung cancer. She had no history of drug or alcohol use. On examination, her temperature was 37.9°C, blood pressure of 144/98 mm Hg, respiratory rate of 18 breaths per minute, a heart rate of 121 beats per minute, and oxygen saturation of 97% on ambient air. Her eyes were open but she was nonverbal. Her chest was clear to auscultation. Heart sounds were distinct and rhythm was regular. Abdomen was soft and nontender with no organomegaly. Skin examination revealed no rash. Her pupils were equal, round, and reactive to light. She did not follow verbal or gestural commands and intermittently tracked with her eyes, but not consistently enough to characterize extraocular movements. Her face was symmetric. She had a normal gag and blink reflex and an increased jaw jerk reflex. Her arms were flexed with increased tone. She had a positive palmo-mental reflex. She had spontaneous movement of all extremities. She had symmetric, 3+ reflexes of the patella and Achilles tendon with a bilateral Babinski’s sign. Sensation was intact only to withdrawal from noxious stimuli.
The physical exam does not localize to a specific brain region, but suggests a diffuse brain process. There are multiple signs of upper motor neuron involvement, including increased tone, hyperreflexia, and Babinski (plantar flexion) reflexes. A palmo-mental reflex signifies pathology in the cerebrum. Although cranial nerve testing is limited, there are no features of cranial neuropathy; similarly, no pyramidal weakness or sensory deficit has been demonstrated on limited testing. The differential diagnosis of her rapidly progressive encephalopathy includes autoimmune or paraneoplastic encephalitis, diffuse infiltrative malignancy, metabolic diseases (eg, porphyria, heavy metal intoxication), and prion disease.
Her family history of lung cancer and her smoking increases the possibility of paraneoplastic encephalitis, which often has subacute behavioral changes that precede complete neurologic impairment. Inflammatory or hemorrhagic CSF is seen with Balamuthia amoebic infection, which causes a granulomatous encephalitis and is characteristically associated with a mass lesion. Toxoplasmosis causes encephalitis that can be profound, but patients are usually immunocompromised and there are typically multiple lesions.
Laboratory results showed a normal white blood cell count and differential, basic metabolic profile and liver function tests, and C-reactive protein. Human immunodeficiency virus antibody testing was negative. Chest radiography and computed tomography of chest, abdomen, and pelvis were normal. A repeat MRI of the brain with contrast was reported as showing a 2.4 x 2.3 x 1.9 cm heterogeneously enhancing mass in the right frontal lobe with an enhancing dural tail and underlying hyperostosis consistent with a meningioma, and blooming within the mass consistent with prior hemorrhage. No mass effect was present.
The meningioma was resected 3 days after admission but her symptoms did not improve. Routine postoperative MRI was reported to show expected postsurgical changes but no infarct. Brain biopsy at the time of the operation was reported as meningioma and mild gliosis without encephalitis.
The reported MRI findings showing unchanged size and overall appearance of the mass, its connection to the dura and skull, and the pathology results all suggest that the mass is a meningioma. There is no evidence of disease outside of the CNS. Some cancers that provoke a paraneoplastic response can be quite small yet may incite an immune encephalitis; anti-NMDAR-mediated encephalitis can occur with malignancy (often ovarian), although it also arises in the absence of any tumor. Any inclination to definitively exclude conditions not seen on the brain biopsy must be tempered by the limited sensitivity of brain histology examination. Still, what was not seen warrants mention: vascular inflammation suggestive of CNS vasculitis, granulomas that might point to neurosarcoidosis, malignant cells of an infiltrating lymphoma or glioma, or inflammatory cells suggestive of encephalitis. Prion encephalopathy remains possible.
The patient remained unresponsive. A repeat EEG showed bilateral generalized periodic epileptiform discharges with accompanying twitching of the head, face, and left arm, which were suppressed with intravenous propofol and levetiracetam. Three weeks following meningioma resection, a new MRI was read as showing new abnormal signal in the right basal ganglia, abnormality of the cortex on the diffusion weighted images, and progressive generalized volume loss.
Among the aforementioned diagnoses, focal or diffuse periodic epileptiform discharges at 1-2 hertz are most characteristic of prion disease. Striatal and cortical transverse relaxation time (T2)-weighted and diffusion-weighted imaging (DWI) hyperintensities with corresponding restricted diffusion is characteristic of Creutzfeldt-Jakob disease (CJD), although metabolic disorders, seizures, and encephalitis can very rarely show similar MRI findings. The clinical course, the MRI and EEG findings, and nondiagnostic biopsy results, which were initially not assessed for prion disease, collectively point to prion disease. Detection of abnormal prion protein in the brain tissue by immunohistochemistry or molecular methods would confirm the diagnosis.
Review of the original right frontal cortex biopsy specimen at the National Prion Disease Pathology Surveillance Center, including immunostaining with 3F4, a monoclonal antibody to the prion protein, revealed granular deposits typical of prion disease. This finding established a diagnosis of prion disease, likely sporadic CJD. The patient was transitioned to palliative care and died shortly thereafter.
Brain autopsy showed regions with transcortical vacuolation (spongiform change), other cortical regions with varying degrees of vacuolation, abundant reactive astrocytes, paucity of neurons, and dark shrunken neurons. Vacuolation and gliosis were observed in the striatum and were most pronounced in the thalamus. There was no evidence of an inflammatory infiltrate or a neoplastic process. These findings with the positive 3F4 immunohistochemistry and positive Western blot from brain autopsy, as well as the absence of a mutation in the prion protein gene, were diagnostic for CJD.
An investigation was initiated to track the nondisposable surgical instruments used in the meningioma resection that may have been subsequently used in other patients. It was determined that 52 neurosurgical patients may have been exposed to prion-contaminated instruments. The instruments were subsequently processed specifically for prion decontamination. After 7 years, no cases of CJD have been diagnosed in the potentially exposed patients.
DISCUSSION
CJD is a rare neurodegenerative condition1 classified as one of the transmissible spongiform encephalopathies, so called because of the characteristic spongiform pattern (vacuolation) seen on histology, as well as the presence of neuronal loss, reactive gliosis in the gray matter, and the accumulation of the abnormal isoform of the cellular prion protein.2 It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. The most common form of human prion disease, sporadic CJD, is relentlessly progressive and invariably fatal, and in most cases, death occurs less than 5 months from onset.3 There is no cure, although temporizing treatments for symptoms can be helpful.
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. Dr Geschwind’s institution has received R01 grant funding from NIH/NIA; and Alliance Biosecure and the Michael J Homer Family Fund as paid money to his institution, Dr Geschwind has received consulting fees or honoraria from Best Doctors, Kendall Brill & Kelly, CJD Foundation, and Tau Consortium; Dr Geschwind is a consultant for Gerson Lehrman Group, Biohaven Pharmaceuticals, and Advance Medical, outside the submitted work; has grants/grantspending with Quest, Cure PSP, and Tau Consortium, and received payment for lectures from Multiple Grand Rounds Lectures, outside the submitted work. Dr Saint is on a medical advisory board of Doximity, has received honorarium for being a member of the medical advisory board; he is also on the scientifice advisory board of Jvion. Dr Safdar’s institution has received a grant from the VA Patient Safety Center.
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
1. Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529. PubMed
2. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:913-920. PubMed
3. Johnson RT, Gibbs CJ, Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004. PubMed
4. Will RG, Alpers MP, Dormont D, Schonberger LB. Infectious and sporadic prion diseases. In: Prusiner SB, ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:465-507. \
5. Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69:1578-1582. PubMed
6. Tschampa HJ, Kallenberg K, Kretzschmar HA, et al. Pattern of cortical changes in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2007;28:1114-1118. PubMed
7. Carswell C, Thompson A, Lukic A, et al. MRI findings are often missed in the diagnosis of Creutzfeldt-Jakob disease. BMC Neurol. 2012;12:153. PubMed
8. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-816. PubMed
9. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology. 2001;56:1528-1533. PubMed
10. Orrú CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6:pii: e02451-14 PubMed
11. Orrú CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371:519-529. PubMed
12. Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081. PubMed
13. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901-907. PubMed
14. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. http://www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed on July 10, 2017.
15. Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205. PubMed
© 2017 Society of Hospital Medicine